 Gangyu Long
Gangyu Long Rui Gong
Rui Gong Qian Wang
Qian Wang Dingyu Zhang
Dingyu Zhang Chaolin Huang
Chaolin Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 August 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.973089
This article is part of the Research Topic Immune Regulation of Mitochondria in Lung Diseases View all 4 articles
Acute lung injury(ALI)/acute respiratory distress syndrome(ARDS) is a form of acute-onset hypoxemic respiratory failure characterised by an acute, diffuse, inflammatory lung injury, and increased alveolar-capillary permeability, which is caused by a variety of pulmonary or nonpulmonary insults. Recently, aberrant mitochondria and mitochondrial DNA(mtDNA) level are associated with the development of ALI/ARDS, and plasma mtDNA level shows the potential to be a promising biomarker for clinical diagnosis and evaluation of lung injury severity. In mechanism, the mtDNA and its oxidised form, which are released from impaired mitochondria, play a crucial role in the inflammatory response and histopathological changes in the lung. In this review, we discuss mitochondrial outer membrane permeabilisation (MOMP), mitochondrial permeability transition pore(mPTP), extracellular vesicles (EVs), extracellular traps (ETs), and passive release as the principal mechanisms for the release of mitochondrial DNA into the cytoplasm and extracellular compartments respectively. Further, we explain how the released mtDNA and its oxidised form can induce inflammatory cytokine production and aggravate lung injury through the Toll-like receptor 9(TLR9) signalling, cytosolic cGAS-stimulator of interferon genes (STING) signalling (cGAS-STING) pathway, and inflammasomes activation. Additionally, we propose targeting mtDNA-mediated inflammatory pathways as a novel therapeutic approach for treating ALI/ARDS.
The Lung is a complex branched organ that terminates in a highly vascularised alveolus designed to efficiently exchange inspired oxygen with carbon dioxide (1). The alveolus, which takes up 90 percent of the total lung volume, consists of an epithelial layer of simple squamous epithelium, and an extracellular matrix surrounded by capillaries and is vulnerable to various extrapulmonary/indirect (e.g. sepsis, pancreatitis, trauma) or pulmonary/direct (e.g. pneumonia, aspiration) insults (2, 3). There has been growing evidence of diffuse alveolar damage in alveolar specimens taken from patients suffering from ARDS, and laboratory studies have documented epithelial and endothelial damage, which leads to the accumulation of protein-rich inflammatory fluids in the alveolar space (4). Excessive inflammatory fluids in the interstitium and alveoli cause impaired gas exchange, decreased compliance, and increased pulmonary arterial pressure. The annual rate of ARDS is generally 13–23 people per 100,000 in the general population (5), and is higher in intensive care units; approximately 10 to 15 percent of admitted patients and up to 23 percent of mechanically ventilated patients meet the criteria for ARDS (6), which may be underestimated seriously due to the ongoing COVID-19 pandemic. Over the past decades, great progress has been made in understanding the epidemiology, pathogenesis, and pathophysiology of ARDS, which is identified as a heterogeneous syndrome. Additionally, randomized trials have identified improved outcomes for patients with ARDS following the optimization of mechanical ventilation, fluid therapy, prone position ventilation, and neuromuscular blockade. However, none of these supportive therapies can reduce inflammation in the lungs nor have efficiently decreased the mortality of ALI/ARDS. According to the severity of lung injury (mild, moderate, and severe), the mortality rate remains high, approximately 27% to 45% (6), and has caused worldwide concern, especially in the COVID-19 pandemic, to establish more effective treatment strategies based on the pathogenesis to reduce the ALI mortality is urgently needed.
Mitochondria are the powerhouses of cells, which generate a large portion of adenosine triphosphate (ATP) through oxidative phosphorylation depending on the electron transport chain (ETC). In addition to their canonical function, mitochondria also play a role in immune modulation against infectious and sterile insults (7). Furthermore, mitochondria are closely associated with cellular integrity, alveolar surfactant secretion, and repair of alveolar epithelial and endothelial barriers (8–10). Mitochondrial dysfunction is a crucial causative factor that leads to pathological features of lung injury, such as inflammatory cell infiltration, explosive release of inflammatory cytokines, microvascular hyperpermeability, release of mitochondrial damage-associated molecular patterns (mtDAMPs), increased reactive oxygen species (ROS) production, and mitochondrial DNA damage (8, 11–16). However, several studies demonstrate that mitochondrial DNA (mtDNA), a mtDAMP, is significantly associated with ALI development under various stimuli (17–21). Furthermore, mtDNA obtained from bronchoalveolar lavage fluid (BALF) in animal models, and patients is associated with ALI development (22–25). Although evidences suggest some accessible ways to effectively protect against ALI by eliminating mitochondrial mtDNA (26–30), the mechanisms by which mitochondrial mtDNA promotes ALI and ARDS are complex and require further study. Thus, in this review, we summarize the possible mechanisms of mtDNA release, the pathogenic mechanisms of mtDNA in ALI/ARDS, as shown in Figure 1, and prospective therapeutic directions for ALI/ARDS.
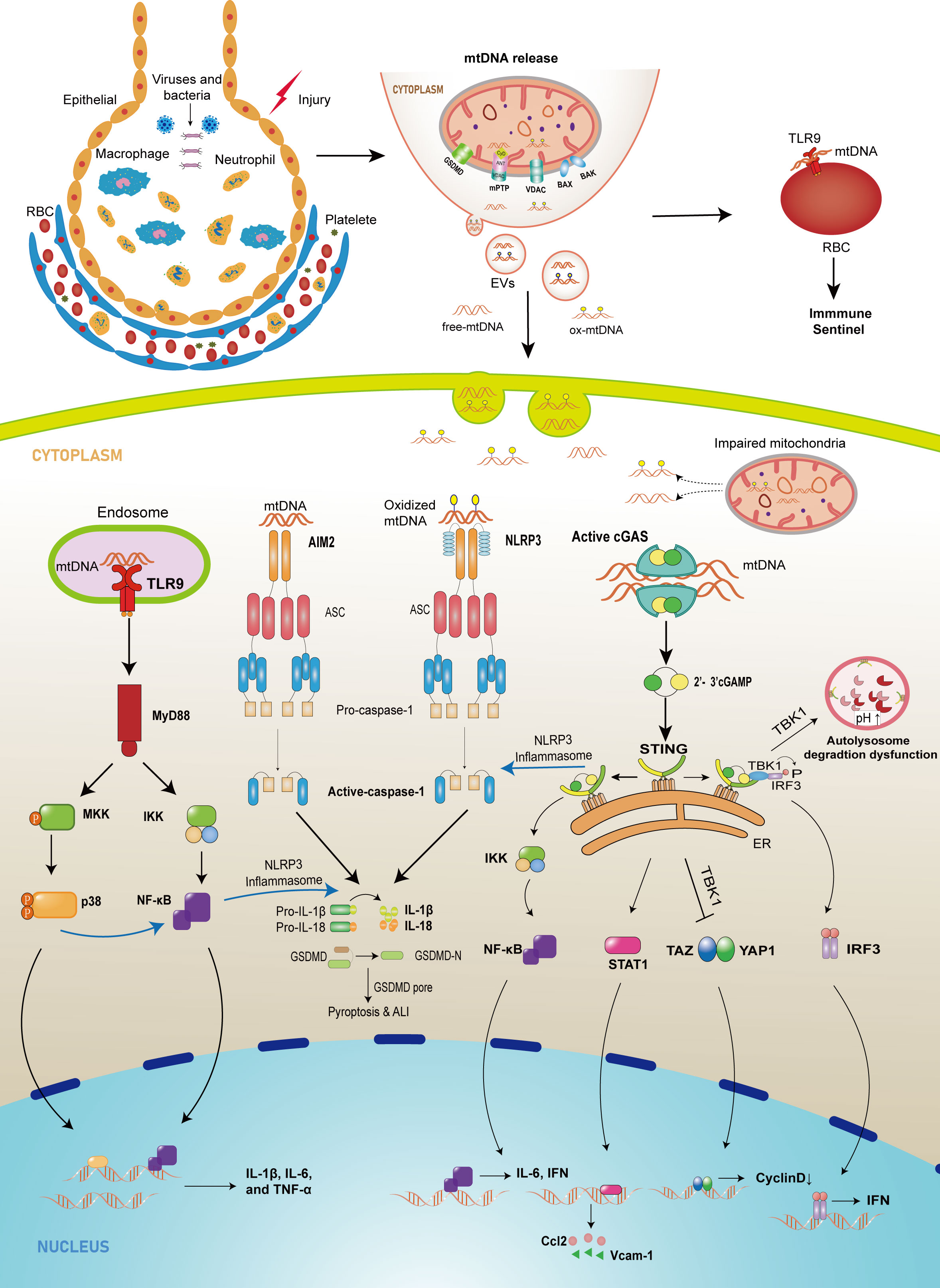
Figure 1 The schematic diagram of mtDNA-releasing and sensing pathways mediating injury in lung tissue. In cell injury or infection conditions, abnormal mtDNA can be released from mitochondria into cytosol through mPTP, GSDMD pore, MOMP mediated by BAX/BAK and VDAC, or from cytosol to extracellular environment by EVs. Once in the cytosol, mtDNA, which comes from intracellular injured mitochondria or extracellular space, can be recognized by three major sensors that drive the innate immune response. First, the released mtDNA can bind to TLR9 on the surface of erythrocytes to induce an immune response or bind to TLR9 in the endosome, which promotes the expression of downstream NF-κB or p38 MAPK, leading to an upregulation of pro-inflammatory factor expression. Cytoplasmic mtDNA is also recognized by cGAS and leads to increased expression of cytokines, adhesion molecules, and chemokines through different pathways and can inhibit autophagy and endothelial cell proliferation. In addition, the released mtDNA activates PRRs such as NLRP3 and AIM2, recruiting ASC and procaspase-1 to form inflammasomes and promoting IL-1β and IL-18 maturation and pyroptosis.
Human mitochondrial DNA (mtDNA), localised in the mitochondrial matrix, comprises 16.5 kb circular dsDNA lacking introns and codes for 22 tRNAs, two rRNAs, and 13 mRNAs that encode the essential oxidative phosphorylation (OXPHOS) protein subunits (31). However, according to the mitochondrial endosymbiotic theory, which explains mitochondrial morphology (structurally similar to bacteria) and that mitochondria have their genetic code (32, 33), the unmethylated CpG and formylated peptides from mitochondrial DNA can be recognized as “foreign molecules” by our bodies after escaping from mitochondria (34–36). Thus, mtDNA is gradually recognized as a damage-associated molecular pattern (DAMP), a cellular component of dying cells, which contributes to initiating and propagating inflammation (37, 38). Moreover, under various stimuli such as bacterial or viral infections, trauma, and burns, mtDNA can be released into the cytoplasm and extracellular environment. As a newly identified mitochondrial DAMP in the last decade, mtDNA has attracted increasing attention in clinical and basic research, including ALI studies.
In the early stages of ALI/ARDS, mitochondrial dysfunction is observed in different types of ALI/ARDS, including infection, trauma, and burns (39, 40). Extracellular mtDNA in the blood and BALF of patients with ALI/ARDS has recently been investigated. As reported, mtDNA copies measured in the peripheral blood were associated with 28-day survival in patients with ARDS and poor outcomes in COVID-19 patients (41, 42). After multiple transfusions, mtDNA can also enter the receiver’s circulation and probably contributes to ALI/ARDS (43, 44).
Furthermore, circulating mtDNA in the blood has been suggested as a promising biomarker in the clinical context of ALI/ARDS (38, 45–48). Apart from circulating mtDNA, BALF mtDNA levels were significantly increased in patients with ALI/ARDS and were positively correlated with ALI progression (25). Moreover, numerous in vivo and in vitro experiments have shown the critical role of mtDNA in promoting ALI progression (49–52). Animal experiments have confirmed that mtDNA plays a critical role in ALI/ARDS models by activating the TLR9/MyD88/NF‐κB signalling pathway (18). Moreover, the administration of isolated mtDNA into the lungs or circulation induces the production of pro-inflammatory mediators (22, 29, 53). To further prove the hazardous potential of mtDNA, suppressing mtDNA levels by cyclosporine-A has been shown to alleviate ALI in LPS-induced ALI models (26).
The above evidence suggests that extracellular mtDNA is a significant damage factor during ALI progression and could serve as a promising predictor of ALI. Furthermore, strategies targeting mtDNA-related methods have been prospectively tested for treating ALI (26, 53).
The innate immune system is crucial in mediating injuries to alveolar epithelial cells and capillary endothelial cells in ALI/ARDS. During ALI/ARDS, elevated cytokines produced by innate immune pathways are pivotal factors in the deterioration of histopathological changes and lung function (54–56). Due to the unique features of mtDNA, it can trigger the activation of the innate immune system in numerous pathological conditions such as sepsis, ALI/ARDS, kidney injury, heart failure, and liver failure (18, 57–60). In terms of the wide distribution of mtDNA and its hazard potential, there is an urgent need to determine the comprehensive role of mtDNA in ALI/ARDS.
Given the important role of mtDNA in the development of ALI/ARDS, we review the possible molecular mechanisms triggering the release of the mitochondrial genome into the cytoplasm and extracellular space as follows.
MOMP is a highly regulated process responsible for activating caspases and initiating apoptosis, usually controlled through the interactions between pro- and anti-apoptotic members of the B cell lymphoma 2 (BCL-2) family (61). This process is initiated by intrinsic apoptotic stimuli, such as DNA damage or endoplasmic reticulum (ER) stress, and mediated by BCL-2-associated X (BAX) and BCL-2 antagonist killer (BAK) to release intermembrane proteins into the cytosol (61). Then BAX/BAK oligomers induce the formation of macropores in mitochondria, which allows the herniation of the inner mitochondrial membrane (IMM) from these pores, resulting in the loss of membrane integrity and the subsequent release of the mitochondrial content, including mtDNA, into cytosol (62). Additionally, research has found that following MOMP, when the mitochondrial inner membrane is extruded through the permeabilized outer membrane, mitochondrial inner membrane permeabilization (MIMP) can then occur, allowing mtDNA egress into the cytosol (63). Recently, Li and colleagues found that severe fever with thrombocytopenia syndrome virus (SFTSV) infection triggers BAK upregulation and BAX activation in THP-1 cells, leading to mtDNA oxidization and subsequent cytosolic release (64). Additionally, ALI models induced by LPS in mice are characterized by an increased expression of BAK and BAX, indicating the participation of MOMP mediated by BAK/BAX in ALI/ARDS (65).
In addition, the mechanism of mtDNA release can also be mediated by voltage-dependent anion channels (VDACs) oligomers. VDACs are a family of pore-forming proteins discovered in the mitochondrial outer membrane and have the ability to form large mitochondrial outer membrane(MOM) pores, which are BAX/BAK-independent MOMP pathway. In mammals, three different isoforms have been described, namely VDAC-1, VDAC-2, and VDAC-3, which have different functions (66). Human VDAC-1 appears to be the most abundant isoform, and its oligomerization is associated with Cyto c release and apoptosis induction (67). In MEFs, Kim et al. reported that mtDNA fragment release requires VDAC oligomerization, and direct mtDNA–VDAC1 interactions are likely to increase VDAC oligomerization and mtDNA fragment release (68). Furthermore, in rats’ ALI models induced by paraquat, VDAC and caspase-3, -8, and -9 are significantly increased, together with acute diffuse damage of alveolar capillary endothelial cells, alveolar epithelial cells, and interstitial cells, indicating that VDAC plays a role in MOMP (69).
mPT is a phenomenon in which the accumulation of Ca2+ in the mitochondrial matrix makes IMM permeable and poorly selective (70). The mPTP is comprised of VDAC, adenine nucleotide translocase (ANT), and cyclophilin D (CyD) which is a permanent ingredient and modulator of mPTP (71). Moreover, alternating mitochondrial membrane potential, elevated Ca2+ level, oxidative stress, thiol oxidation, or altered pyridine nucleotide status could modulate mPTP opening (71). In 2004, Patrushev et al. firstly showed that the opening of the mPT pore could trigger the release of DNA fragments from mitochondria (72). Subsequently, a similar result was confirmed, the opening of the mPTP allowing the release of mtDNA fragments into the cytoplasm in multiple models, including immune cells, induced by irradiation, oxidative stress, lipopolysaccharide and extracellular ATP (70). Moreover, Fonai and colleagues discovered that the loss of this essential protein improves survival rate and intensely ameliorates LPS-induced lung injury in CyD(-/-) mice through inhibition of pro-inflammatory cytokine release, redox-sensitive cellular pathways such as MAPKs, Akt, and NF-κB activation, and the production of ROS (73). These data demonstrate that CyD-dependent mPT plays a crucial role in mitochondrial DAMPs (including mtDNA) release to inhibit inflammation and disease progression in ALI/ARDS.
EVs, a heterogeneous group of cell-derived membranous structures comprising exosomes, microvesicles (MVs) (74, 75), and apoptotic EVs (ApoEVs) (76), are considered a mode of intercellular communication, acting as transport cargo of different messengers, including nucleic acids (mitochondrial and genomic DNA, non-coding RNA) and proteins (74, 77)
Exosomes are a subset of EVs with a size range of 40 to 160 nm (average 100 nm around) in diameter with an endosomal origin (78).Lázaro-Ibáñez and colleagues identified the existence of mtDNA in exosomes from the human mast cell line HMC-1 and erythroleukemic cell line TF-1 with sequencing analysis (79). Microvesicles (MVs), whose dimensions generally range from 50 to 500 nm, are circular fragments of membranes released from the endosomal compartment or shed from the surface membranes of most cell types (80), which have been shown to contain mtDNA from some contexts such as Streptococcus pneumonia (81). Apoptotic bodies (ApoBDs) with dimensions ranging between 1~5µm, a major subset of ApoEVs generated by apoptotic cells during the final stages of apoptosis (76), are formed through a process termed apoptotic cell disassembly characterized by a series of tightly regulated morphological steps including plasma membrane blebbing, apoptotic membrane protrusion formation, and fragmentation into ApoBDs (76)[05]. Although a recent study has demonstrated the existence of mtDNA in apoptotic bodies from acute myeloid leukaemia cells (82), more investigations in ALI/ARDS are urgently demanded.
Till now, growing evidence shows EVs not only participate in many physiological processes but also contribute to the pathogenesis of some diseases (83). In ALI/ARDS, alveolar macrophages (AMs)-derived MVs may play a significant role in the regulation of ALI-associated inflammation since macrophages were the major source of MVs in bronchoalveolar lavage fluid (84). Additionally, a recent study demonstrated that lung epithelial EVs or AM-EVs are responsible for the development of lung inflammation after sterile (hyperoxia) or infectious(P. pneumonia and S. pneumonia) stimuli in mice, respectively (85). However, due to the complex composition of the Evs, it is still difficult to confirm whether all components(including mtDNA) of the EVs can exacerbate lung damage, which needs further investigation.
Recently, growing evidence has associated Extracellular Traps (ETs) with mtDNA, indicating ETs may participate in the process of mtDNA exteriorization. Neutrophil extracellular traps (NETs) are studied with great enthusiasm. Following strong activation signals, neutrophils release granule proteins and chromatin that form extracellular fibres that bind Gram-positive and -negative bacteria, shaping the so-called NETs (86). Moreover, NETs formation can be found in trauma or operation patients and are made of mtDNA without nuclear DNA component (87). However, the origin of mtDNA is a mystery. In LPS-induced ALI mice models, NETs produced by the recruited neutrophils can promote lung injury and inflammation, which can be rescued by NET inhibitors (88). Moreover, in ALI/ARDS models caused by influenza pneumonitis, neutrophils from infected lungs strongly induced NETs generation and augmented endothelial damage (89). In patients with severe pneumonia or COVID-19 acute respiratory distress syndrome, the plasma NETs level is also increased and associated with ARDS severity and mortality (90, 91). Although some evidence confirms the mtDNA release from immunocytes to form ETs, the release mechanisms are still unclear. Moreover, due to the direct evidence of mtDNA in ALI/ARDS ETs being scarce, the role of mtDNA from ETs in ALI/ARDS models and patients demands further investigation.
Passive release of mtDNA is mainly induced by cell death. In current knowledge, the main forms of cell death associated with ALI/ARDS can be concluded into apoptosis, necrosis, and pyroptosis (92). The most well-characterized and prevalent form of controlled cell death is apoptosis. According to different initiators in apoptosis pathways, the process can be summarized as the extrinsic and intrinsic pathways, the latter known as the mitochondrial pathway of apoptosis. During the intrinsic pathway, stimuli such as toxins, hypoxia, viral infections, and free radicals cause changes in the IMM and open the mPTP, causing mitochondrial components, including mtDNA and cytochrome c, to be released into the cytoplasm (93). ApoBDs are formed during the final stages of apoptosis and then released into the extracellular space with mtDNA and other materials inside.
Unlike apoptosis, necrosis is an alternative uncontrolled cell death induced by external injury, such as hypoxia or inflammation. This process often involves upregulating various pro-inflammatory proteins and compounds, such as nuclear factor-κB. The rupture of the cell membrane causes the spillage of the cell contents(including mtDNA) into surrounding areas during necrosis, resulting in a cascade of inflammation and tissue damage (92). Numerous studies have shown that circulation-free mtDNA level is associated with the extent of necrosis or damage, indicating that mtDNA is released from the necrotic cells into plasma (94, 95). In addition, in gastric aspiration-induced mice ALI models, mtDNA from cell-free bronchoalveolar lavage fluid increased 120-fold, indicating acid-induced cellular necrosis release numerous mtDNA and may augment lung injury (95).
Except for apoptosis and necrosis, pyroptosis also participates in the process of mtDNA release. Pyroptosis is a pro-inflammatory form of regulated cell death that relies on the enzymatic activity of inflammatory proteases that belong to the cysteine-dependent aspartate-specific protease (caspase) family and the subsequent cleavage of gasdermins (GSDMs) by different caspases (96). The latest study has found that GSDMs mediate the cellular release of mtDNA during pyroptosis or intrinsic apoptosis (97). Carlos and colleagues confirmed that GSDMDNT (amino-terminal of gasdermins D) contributes to mitochondrial DNA relocalization to the cytosol and GSDMENT (amino-terminal of gasdermins E) facilitates the cellular release of mtDNA in bone marrow-derived macrophages, HEK293T cells, and mice. In the development of ALI/ARDS, pyroptosis may be more common than other types of cell death due to uncontrolled progressive lung inflammation, and pyroptosis in different types of cells such as macrophages, neutrophils, alveolar epithelial cells, and capillary endothelial cells may play vital roles in lung injury and excessive inflammation (97).
During the development of ALI/ARDS, dysfunctional mitochondria in the injured endothelium, epithelium, and recruited leukocytes generate large amounts of reactive oxygen species (ROS) and release mtDNA. Excessive ROS can disturb protein folding and structure and cause mitochondrial DNA mutations (98). Damaged mitochondrial genomes may disrupt aerobic respiration, cause cellular dysfunction, and even cell death. Moreover, the released mtDNA can contribute to the activation of the innate immune response through mtDNA sensors. Accumulating evidences suggest that mtDNA can be recognized by different mtDNA sensors (34, 99). Various mtDNA sensors and damaged mtDNA have been reported to play vital roles in ALI progression, as discussed below.
Toll-like receptors (TLRs) are critical components of innate pattern recognition receptors. To date, 12 TLRs have been identified in mice, and ten have been found in humans. Among these receptors, TLR9 is currently the only receptor that recognizes mtDNA with hypomethylated CpG motifs (100). TLR9 belongs to the type I integral membrane glycoprotein family with an N-terminal ligand recognition domain, a single transmembrane helix, and a C-terminal cytoplasmic signalling domain. It is mainly present in the endoplasmic reticulum (ER) membrane of unstimulated cells. When stimulated by CpG DNA, TLR9 translocates to the membrane of endosomes, where it recognizes its ligands and initiates cellular activation (101). After binding to mtDNA, TLR9 signalling can proceed through the adaptor myeloid differentiation primary response protein 88 (MyD88) to activate nuclear factor-κB (NF-κB) and through mitogen-activated protein kinases (MAPKs) to trigger inflammatory responses (34, 102). MAPKs include extracellular signal-regulated kinases (ERKs), c-Jun amino-terminal kinases (JNKs), and p38. p38 MAPKs are vital regulators of inflammatory cytokine expression and are activated by various stimuli (103). The transcription factor NF-κB plays its most important and evolutionarily conserved role in the immune system by regulating the expression of inducers and effectors at multiple points in the immune response (104).
Previous studies have revealed that mtDNA activates p38 MAPK after CpG-TLR9 interaction to induce inflammation in neutrophils (105, 106). Injury caused by cell-free mtDNA via the TLR9-p38 MAPK pathway has also been confirmed in some organs (59, 107, 108). In ALI, in vivo experiments demonstrated that TLR9 contributes to the development of ALI and systemic inflammation, and inhibition of TLR9 prominently attenuated histopathological changes and pro-inflammatory mediators, including interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor–α (TNF-α) (50, 109–114). Meanwhile, enhanced levels of mtDNA have been found in BALF and plasma from ALI patients and animal models (22, 25, 115). Additional administrations of mitochondrial DNA into the trachea or peritoneum aggravate systemic inflammation and lung injury in a TLR9-dependent manner via P38 MAPK activation (109, 110). Moreover, inhibition of TLR9 or p38 MAPK attenuated this effect, proving that extracellular mtDNA could mediate lung injury through the TLR9-p38 MAPK pathway (52, 110, 116, 117).
In addition, studies have found that mtDNA can induce NF-κB activity through TLR9 to regulate the expression of pro-inflammatory cytokine genes. According to these studies, mtDNA can increase NF-κB, IκB-α, and TLR9 expression in macrophage (118), and CPG-TLR9 activates NF-κB by regulating MyD88 to increase the production of pro-inflammatory mediators, such as TNF, IL-6, and adhesion molecules in lung epithelial cells (101, 119, 120). Furthermore, MyD88 knockout markedly reduced the levels of inflammatory cytokines and NF-κB and attenuated ALI (121). Suppression of TLR9 also partly decreases the production of inflammatory mediators by the TLR9-NF-κB pathway in alveolar epithelial cells (102, 122, 123). In addition to the single mechanisms described above, CpG DNA can promote collaborative activation of p38 MAPK and NF-κB in type I IFN-independent induction of IRF-7, CXCL10, and CCL3 to enhance IFN-α gene expression in human plasmacytoid dendritic cells (124). Additionally, although exogenous DNA from bacteria and viruses can activate interferon regulatory factors in a TLR9-dependent manner to enhance type I IFN responses, the role of mtDNA in this pathway still lacks strong evidence.
Interestingly, as conventional oxygen deliverers, red blood cells have been found to have a novel function: immune sentinels. Hotz et al. in 2018 found that red blood cells harbor a type of molecular sensor, TLR9, that recognizes and sticks to DNA molecules containing CpG motifs (125). In vivo experiments revealed a critical role of red blood cells in regulating systemic inflammation by scavenging cell-free mtDNA and demonstrated the importance of RBC-mediated DNA scavenging in limiting lung injury (125). Mangalmurti et al. in 2021 then explored the mechanisms by which RBCs binding with mtDNA through TLR9 appeared beneficial during quiescent states, where it promotes the scavenging of trace CpG to prevent nonspecific inflammation. During conditions characterized by excess circulating CpG, such as sepsis and COVID-19, binding CpG by RBC-TLR9 may lead to inflammation, and anaemia (126). Altogether, mtDNA recognition by TLR9 on RBCs provides evidence that RBCs are immune sentinels (127). However, the exact mechanism of this pathway requires further investigation.
Cyclic GMP-AMP synthase (cGAS) is a cytoplasmic DNA sensor that detects self-and non-self-DNA to synthesize cGAMP in a DNA-dependent manner (128, 129). cGAMP, a cyclic dinucleotide, is a second messenger usually generated by cells following DNA transfection or DNA virus infection (128). These cGAMPs bind to and activate STING to induce type I IFNs production. Stimulator of interferon genes (STING, also known as MITA, MPYS, ERIS, and TMEM173), a transmembrane protein located in the endoplasmic reticulum (ER) membrane, functions as a direct sensor of cGAMP and an essential signalling adaptor linking the interferon response to cytosolic DNA (130). Initially, studies illustrated that cytosolic DNA induces type I IFNs through the endoplasmic reticulum membrane protein STING, which subsequently activates the transcription factors NF-kB and IRF3 through the kinases IKK and TBK1, respectively (131–133). However, the mechanism upstream of STING activation is not fully understood. Subsequently, the cGAS-cGAMP-STING pathway was found to be essential for DNA-mediated immune response irrespective of cell type or DNA sequence, filling the gaps missing upstream of STING (134–137).
Activation of the cGAS-STING pathway begins with DNA recognition. DNA binds cyclic GMP-AMP synthase (cGAS) in a nonsequence-dependent manner through its phosphate backbone to form a 2:2 cGAS: DNA complex (138). Subsequently, cGAS generates a unique cGAMP, characterized by two phosphodiester bonds: one between the 2’OH of GMP and the 5’phosphate of AMP and the other between the 3’ OH of AMP and the 5’ phosphate of GMP, hereafter referred to as cGAMP (139). This novel class of second messengers is recognized by the ligand-binding pocket of STING and binds with hydrophobic interactions and hydrogen bonds (138), inducing an extensive conformational change in STING that is postulated to release a carboxy-terminal tail (CTT) that recruits and activates TBK1 (137). After binding to cGAMP, STING exits the ER in the form of vesicles and is transferred to the Golgi, where two cysteine residues of STING (Cys88 and Cys91) undergo palmitoylation, essential for STING oligomerization (140). STING recruits TANK-binding kinase (TBK1) and activates transcription factors IRF3 and NF-κB, inducing the production of type I interferons and pro-inflammatory cytokines (137, 141–143). Additionally, substantial evidence indicates that the cGAS-STING pathway also leads to dysfunctions in autophagy and endothelial proliferation (144–146).
Previous studies on STING have mainly focused on the immune response mediated by foreign nuclear acids from DNA viruses, retroviruses, bacteria, and parasites. Owing to the lack of pathogen-specific attributes, cGAS can participate in the antimicrobial defense and recognize self-DNA, including genomic and mitochondrial DNA. Cellular DNA is usually confined to the nucleus or mitochondria. However, in response to specific cellular stress or environmental insults, mitochondrial DNA is more susceptible to oxidative damage than nuclear DNA (nDNA) because of the incomplete DNA repair capacity in mitochondria and the proximity of mtDNA to the respiratory chain, which is the source of ROS production (34). Recent evidence has shown that mtDNA release and the cGAS-STING pathway can drive lung injury, making this pathway a promising therapeutic target for ALI (147). Activation of the cGAS-STING pathway plays a significant role in ALI/ARDS models and patients (51, 148, 149). Specifically, CMA (10-carboxymethyl-9-acridanone), a species-specific agonist of STING in mice, can promote type I IFN production by recognizing STING (150). DMXAA (5,6-dimethylxanthenone-4-acetic acid), another STING agonist in mice, can bind to STING and activate the TBK1-IRF-3 signalling pathway inducing IFN-β production (151). Recently, Togbe et al. found that the synthetic, non-nucleotidyl STING agonist diABZI induced type I IFN and pro-inflammatory mediators in bone marrow-derived macrophages and human airway epithelial cells.
Furthermore, endotracheal administration of diABZI also increased IFNα and IFNβ levels in BALF and triggered neutrophilic inflammation, accompanied by a substantial increase in mtDNA (149). Then, they treated mice receiving local airway diABZI with DNase I, efficiently abrogating dsDNA in the BALF and reducing neutrophil recruitment and neutrophil extracellular trap (NET) formation in the airways, while the levels of type I IFN and pro-inflammatory mediators were barely affected (149). In addition, antagonists of STING, such as C-178 and C-176 for mouse STING and H-151 for human STING, have been shown to inhibit the palmitoylation of STING, reduce TBK1 phosphorylation, and abrogate type I IFNs and IL-6 (152, 153). In addition, recent studies have revealed that recruited macrophages in the lung suppress alveolar macrophage-STING signalling via sphingosine kinase-2 (SPHK2)-mediated generation of sphingosine-1-phosphate (S1P), thereby mitigating inflammatory signalling and vascular injury (154, 155). Furthermore, as upstream stimulators of the cGAS-STING pathway, mtDNA and oxidized mtDNA were confirmed to play an essential role in enhancing immune and inflammatory responses (156–158). Li Liu et al. revealed that endogenous mtDNA and oxidized mtDNA could induce the formation of NETs depending on the TLR9 and STING pathways in bone marrow neutrophils from C57BL/6 mice then, the effect was abrogated by TLR9 knockout, STING knockout and reactive oxygen species scavenger (156). Additionally, Qing et al. reported that mtDNA in the cytoplasm triggered pyroptosis in a cGAS-STING-dependent manner, and the NLRP3 inflammasome participated in downstream effects to induce the production of IL-1β and IL-18 in mice (158). However, they did not reveal a connection between TBK1-IRF3 and NLRP3 activation in the downstream effects of the cGAS-STING pathway.
Besides the above mechanism, autophagy dysfunction has reached a stage recently (159–161). Ren et al. confirmed that autophagy dysfunction triggered by mtDNA could promote sepsis-related ALI via STING. They revealed that mtDNA contributed to disordered lysosomal acidification via STING, which caused deficient autophagic flux, and aggravated the disease in mice macrophages. Moreover, they revealed that STING-mediated autophagic flux blockade is partially associated with TBK1 downstream signalling (51). In addition, downstream signalling of the cGAS-STING pathway can suppress vascular regeneration to promote inflammatory lung injury. Huang et al. discovered that cGAS/STING activated by the released mtDNA inhibited endothelial proliferation by suppressing YAP signalling in HEK293 cells, human and mouse lung microvascular endothelial cells. Moreover, mtDNA-induced activation of TBK1 activated LATS1, inducing YAP phosphorylation and degradation, thereby preventing downstream YAP-mediated endothelial proliferation and repair (162). Interestingly, STING influences the mechanical barrier repair of endothelial cells and their paracrine function by over-expressing adhesion molecules and chemokines, which can organise the recruitment of immune cells and regulate leukocyte extravasation at places of inflammation. Wu et al. discovered that STING inhibition alleviated LPS-induced ALI in mice by suppressing vascular endothelial cell-mediated immune cell chemotaxis and adhesion. They revealed that a STING inhibitor or STING knockdown downregulated the expression levels of adhesion molecules and chemokines by decreasing the phosphorylation of the transcription factor STAT1 in HMEC-1cells (153).
Inflammasomes are large multimolecular complexes consisting of receptor and sensor molecules, an adaptor protein (apoptosis-associated speck-like protein containing a CARD [ASC]), and the inflammatory cysteine protease caspase-1. There are five central receptors contributing to the formation of an inflammasome, including NOD, LRR, pyrin domain-containing protein 1 (NLRP1), NLRP3, NLR family CARD domain-containing protein 4 (NLRC4), absent in melanoma 2 (AIM2), and pyrin (163). Most of these receptors are activated by exogenous pathogen-associated molecular patterns (PAMPs) that gain access to the cytosol during microbial infection and endogenous DAMPs released from damaged or dying cells. Activated inflammasomes then activate the proteolytic enzyme caspase-1, which conversely regulates the proteolytic maturation of IL-1β and IL-18, as well as a quick, noxious, and inflammatory form of cell death termed pyroptosis (164). Recent studies have revealed the role of mtDNA in inducing caspase-1-mediated innate immune responses through the NLRP3 and AIM2 inflammasomes.
NLRP3 inflammasome is one of the most studied inflammasomes and is generally located in the cytoplasm. The NLRP3 inflammasome can be activated by a wide range of exogenous and endogenous stimuli such as lipopolysaccharide, nigericin, asbestos, alum, and extracellular ATP. In 2011, mtDNA was first revealed to contribute to NLRP3 inflammasome activation, confirming that upon LPS and ATP stimulation, mitochondrial ROS and mitochondrial membrane permeability increase prominently, causing the release of mtDNA into the cytoplasm. The translocated mtDNA requires activation of NLRP3 and could directly induce downstream activation of caspase-1 in response to treatment with LPS and ATP to enhance the production of IL-1β and IL-18 (165). Then, Shimada et al. discovered that oxidized mtDNA, generated and released into the cytosol during apoptosis, can bind to NLRP3 and activate the NLRP3 inflammasome, which 8-OH-dG competitively inhibits. They also demonstrated that NLRP3 is preferentially activated by oxidized DNA and AIM2 by normal DNA (166). Subsequently, Zhong and colleagues also confirmed that different activators of NLRP3 cause a specific form of mitochondrial damage that results in fragmented mtDNA release and increased production of ROS that is responsible for the conversion of mtDNA to an oxidized form, to be used as the ultimate NLRP3 ligand (167). However, the process of NLPR3 inflammasome activation induced by cytosolic mtDNA and extracellular mtDNA may differ. Li Ning et al. discovered that the cystolic mtDNA released from self-mitochondria contributed to LPS-induced ALI by upregulating cGAS, activating STING, and activating NLRP3 in mice. They also revealed that cGAS or STING deficiency relieved NLRP3-mediated pyroptosis of macrophages, and NLRP3 overexpression abolished the protective roles of STING deficiency in macrophages, indicating that the activation of NLRP3 is essential for the proinflammatory responses of STING (158). Nevertheless, the mechanism by which extracellular mtDNA induces the activation of the NLRP3 inflammasome is slightly different. Wu et al. first revealed the specific mechanism by which extracellular mtDNA in the lung induces NLRP3 inflammasome priming and activation through TLR9- p38 MAPK- NF-κB in THP-1 macrophages. They showed that knockdown of TLR9 offsets mtDNA-induced NLRP3 upregulation, decreases extracellular mtDNA-induced NLRP3 inflammasome transcription, and suppresses the production of mtDNA-induced IL-1β and TNF-α. This is consistent with the inhibition of p38 MAPK or NF-κB (52). Together with these studies, this shows that after the body is subjected to various external stresses, mitochondria are damaged and mtDNA is released into the cytoplasm. When cystolic mtDNA further activates the NLRP3 inflammasome in the cell, pyroptosis may be induced, and more mtDNA can be released outside the cell, causing the activation of the NLRP3 inflammasome in the peripheral cells, which may aggravate diseases such as ALI/ARDS.
AIM2 functions as a cytoplasmic dsDNA sensor and has been studied for over a decade. It binds DNA, recruits the inflammasome adaptor ASC, and activates caspase-1 (168, 169). Studies have found that the C-terminal HIN-200 domain interacts with the dsDNA sugar-phosphate backbone in a non-sequence-specific DNA recognition pathway, whereas the PYD domain of AIM2 associates with the adapter molecule ASC to activate both NF-κB and caspase-1 (170–172). Furthermore, activated caspase-1 promotes the maturation of IL-1β and IL-18 and cleaves gasdermin D, which can induce pyroptosis by forming pores on cellular membranes. Recently, Li et al. revealed that DNA from NETs, which accumulates in the lysosomes of alveolar macrophages under LPS treatment, binds with the AIM2 sensor to trigger the AIM2 inflammasome inducing the production of proinflammatory cytokines and macrophage pyroptosis (173). The study also found that the degradation of NET DNA or silencing of the AIM2 gene can alleviate pyroptosis in alveolar macrophages co-stimulated with LPS and NETs. Although NETs have been identified as an important source of extracellular mtDNA (87, 174, 175), the interaction between mtDNA from NETs and AIM2 sensors in lung injury has not been specifically illustrated. Further studies are needed to explore the interactions involved in ALI pathogenesis.
In addition, some studies have provided evidence of mtDNA binding to NLRC4 complexes. For example, in 2015, Jabir et al. discovered that mitochondrial DNA could bind specifically to NLRC4 immunoprecipitates and that the transfected mtDNA could directly activate the NLRC4 inflammasome in BMDMs from mice and HEK cells (176). They also revealed that inflammasome activation was enhanced by DNA oxidation and downregulated by autophagy. However, it is still challenging to demonstrate the role of the mtDNA-NLRC4 interaction in lung injury owing to the lack of direct evidence.
Bacterial and viral pneumonia account for the major cases of ARDS in recent clinical trials. Increasing evidence showed bacterial and viral infection could induce mitochondria damage and mtDNA release (64, 81, 162, 177–181). Once leaving its original location, the mtDNA can be recognized as foreign, eliciting a potent innate immune response. Nerlich et al. found that the pneumolysin produced by Streptococcus pneumoniae (S.pn.) could cause mtDNA to release through mPTP opening and transfer into extracellular space via microvesicles, which may contribute to S.pn. related inflammatory lung injury (81). Meanwhile, Huang et al. demonstrated that LPS(Lipopolysaccharide), the main pathogenic component of Gram-negative bacteria, activated the pore-forming protein Gasdermin D, which formed mitochondrial pores and induced mitochondrial DNA (mtDNA) release into the cytosol of endothelial cells, suppressing endothelial cell proliferation by cGAS-YAP signalling pathway and promoting inflammatory injury (162). Additionally, upon different viral infections, including cytomegalovirus (MCMV) and Sendai virus (SeV), both of which are associated with the ALI/ARDS development (182, 183), vaccinia virus-related kinase two is associated with voltage-dependent anion channel 1 (VDAC1) and promoted VDAC1 oligomerization and mtDNA release, leading to the cGAS-mediated innate immune response (178). Moreover, Moriyama and colleagues discovered that the cytosolic mtDNA release induced by Influenza A virus can be partially inhibited by the knockdown of Bax and induce cGAS/STING-dependent IFN-β gene expression
(177), indicating the critical role of Bax/Bak for mtDNA release into the cytosol after the influenza virus infection which can lead to ARDS, a fatal complication (184). Although studies have now elucidated the release of mitochondrial DNA and the immune response generated by some bacteria and viruses, further exploration of common pathogens causing ALI/ARDS is still urgently needed due to the large heterogeneity between different pathogens.
As the release of mtDNA and its oxidized form are the common priming step for mtDNA-sensing pathways mediating excessive immune responses, protective strategies specific to mtDNA or mitochondria may be preferred choices for the treatment of ALI/ARDS. Xian et al. demonstrated Metformin inhibits mtDNA synthesis, abrogating NLRP3 inflammasome activation and pulmonary inflammation in LPS and SARS-CoV-2 induced mice ARDS models (185). Additionally, prevention of mtDNA release can also prominently alleviate lung injury. Reversely, exogenous administration of mtDNA can provoke lung injury and systemic inflammation (109).
Based on the essential role of mtDNA at the onset and progression of ALI/ARDS, accessible therapeutic targets can be divided into three main aspects: inhibiting the release of mtDNA, clearing the released mtDNA, and impeding mtDNA-mediated pathways. We conclude with the following possible therapeutic methods.
In terms of mtDNA release mechanisms, the reduction of mtDNA release primarily focuses on scavenging ROS and maintaining mitochondrial homeostasis. For example, Chao et al. found that peroxyauraptenol, isolated from the seeds of Cnidium monnieri, could inhibit inflammation and NLRP3 inflammasome activation by reducing mitochondrial ROS levels, indicating a possible candidate for anti-inflammatory agents (186). Recently, Sok et al. found that 1′-cetoxychavicol acetate, a natural compound from the tropical ginger Alpinia species, is a promising inhibitor of the NLRP3 inflammasome via mitochondrial ROS suppression (187). Additionally, dexmedetomidine and tyrosine phosphatase SHP2 have been shown to preserve mitochondrial dynamic equilibrium and homeostasis respectively, to attenuate inflammation (188, 189). In addition, some possible agents can limit mtDNA release with unknown mechanisms, such as cyclosporine A, epigallocatechin gallate, α7 nicotinic acetylcholine receptor signalling, and metformin (26, 53, 185, 190, 191). Some of these agents can inhibit inflammasome activation to alleviate inflammation by limiting the release of mtDNA.
Autophagy is the most studied target in the process of eliminating the released mtDNA. Several studies support the crucial role of autophagy in eliminating and degrading mitochondrial DNA (59, 165, 192, 193). Moreover, many studies have revealed potent agents that can reinforce autophagy to mitigate inflammatory injuries, such as Apelin-13, carbon monoxide, genipin, and transcription factor EB (194–197). Furthermore, some studies have found that enhanced mitophagy can have similar effects in alleviating lung injury. These agents include polydatin, redox, and Sestrin 2 (198–200). In addition, DNaseI targeting mtDNA shows treatment potential in paraquat-induced ALI and pulmonary fibrosis, which needs further investigation in other kinds of ALI (201).
Regarding mtDNA-mediated pathways, regulating the levels of released mtDNA can indirectly abrogate downstream pathways. The possible agents are principally studied in the innate immune pathogenic pathways described above to explore the direct inhibiting mechanisms, which may not always be completely explained by the downstream pathways and require further investigation. Such agents, including carbon monoxide, epigallocatechin-3-gallate, riboflavin, Tanreqing, and ethyl pyruvate, need to be explored further (28, 202–205).
For ALI/ARDS development, growing evidence associates mitochondrial dysfunction with impaired metabolism and pathological changes, leading to different levels of alveolar inflammation and pulmonary edema (11, 12, 23, 206). Among the mitochondrial DAMPs from impaired mitochondria, mtDNA has shown a high correlation with the severity of ALI/ARDS and has great potential to promote lung inflammatory responses in clinical and basic research. Therefore, We summarize the main mechanisms for releasing mitochondrial DNA into the cytoplasm and extracellular compartments, such as MOMP, mPTP, Evs, ETs and passive release. Moreover, we also discuss the released mtDNA and its oxidized form can bind to DNA sensors, such as TLR9, cGAS, NLRP3, AIM2, and NLRC4, to induce lung injury and the production of proinflammatory cytokines. According to the vital role of mtDNA in ALI/ARDS, eliminating released mtDNA, inhibiting the release of mtDNA, or disturbing mtDNA-mediated pathways have shown prominent therapeutic effects, indicating that therapies targeting released mtDNA are promising. However, the mechanisms underlying mtDNA-mediated pathways in ALI/ARDS must be explored. Moreover, strategies targeting mtDNA-associated lung injury must be optimized and made more efficient.
CH and DZ generated the research idea and finally revised the manuscript. GL, RG and QW wrote the general manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the National Natural Science Foundation of China (92169107).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Yee M, Gelein R, Mariani TJ, Lawrence BP, O'Reilly MA. The oxygen environment at birth specifies the population of alveolar epithelial stem cells in the adult lung. Stem Cells (Dayton Ohio) (2016) 34(5):1396–406. doi: 10.1002/stem.2330
2. Knudsen L, Ochs M. The micromechanics of lung alveoli: Structure and function of surfactant and tissue components. Histochem Cell Biol (2018) 150(6):661–76. doi: 10.1007/s00418-018-1747-9
3. Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: Advances in diagnosis and treatment. JAMA (2018) 319(7):698–710. doi: 10.1001/jama.2017.21907
4. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers (2019) 5(1):18. doi: 10.1038/s41572-019-0069-0
5. Lewandowski K, Lewandowski M. Epidemiology of Ards. Minerva anestesiologica (2006) 72(6):473–7. doi: 10.1016/j.jclinane.2006.05.001
6. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA (2016) 315(8):788–800. doi: 10.1001/jama.2016.0291
7. Banoth B, Cassel SL. Mitochondria in innate immune signaling. Trans Res (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
8. Hough RF, Islam MN, Gusarova GA, Jin G, Das S, Bhattacharya J. Endothelial mitochondria determine rapid barrier failure in chemical lung injury. JCI Insight (2019) 4(3):e124329. doi: 10.1172/jci.insight.124329
9. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial transfer from Bone-Marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med (2012) 18(5):759–65. doi: 10.1038/nm.2736
10. Zhang J-R, Lin Q, Liang F-Q, Xie T. Dexmedetomidine attenuates lung injury by promoting mitochondrial fission and oxygen consumption. Med Sci Monitor (2019) 25:1848–56. doi: 10.12659/msm.913239
11. Cloonan SM, Kim K, Esteves P, Trian T, Barnes PJ. Mitochondrial dysfunction in lung ageing and disease. Eur Respir Rev (2020) 29(157):200165. doi: 10.1183/16000617.0165-2020
12. Piantadosi CA, Suliman HB. Mitochondrial dysfunction in lung pathogenesis. Annu Rev Physiol (2017) 79(1):495–515. doi: 10.1146/annurev-physiol-022516-034322
13. Moreno Fernández-Ayala DJ, Navas P, López-Lluch G. Age-related mitochondrial dysfunction as a key factor in covid-19 disease. Exp Gerontology (2020) 142:111147. doi: 10.1016/j.exger.2020.111147
14. Zhu L-L, Li M-Q, He F, Zhou S-B, Jiang W. Mitochondria targeted peptide attenuates mitochondrial dysfunction, controls inflammation and protects against spinal cord injury-induced lung injury. Cell Physiol Biochem (2017) 44(1):388–400. doi: 10.1159/000484919
15. Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated e. coli. FASEB J (2005) 19(11):1531–3. doi: 10.1096/fj.04-3500fje
16. Zhao Y-Y, Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, et al. Mitochondrial damps increase endothelial permeability through neutrophil dependent and independent pathways. PloS One (2013) 8(3):e59989. doi: 10.1371/journal.pone.0059989
17. Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, et al. Plasma mitochondrial DNA levels are associated with Ards in trauma and sepsis patients. Chest (2020) 157(1):67–76. doi: 10.1016/j.chest.2019.09.028
18. Zeng Z, Li D, Liu F, Zhou C, Shao Q, Ding C, et al. Mitochondrial DNA plays an important role in lung injury induced by sepsis. J Cell Biochem (2018) 120(5):8547–60. doi: 10.1002/jcb.28142
19. Liu R, Xu F, Bi S, Zhao X, Jia B, Cen Y. Mitochondrial DNA-induced inflammatory responses and lung injury in thermal injury murine model: Protective effect of cyclosporine-a. J Burn Care Res (2019) 40(3):355–60. doi: 10.1093/jbcr/irz029
20. Gan L, Chen X, Sun T, Li Q, Zhang R, Zhang J, et al. Significance of serum mtdna concentration in lung injury induced by hip fracture. Shock (2015) 44(1):52–7. doi: 10.1097/shk.0000000000000366
21. Mao J-Y, Li D-K, Zhang H-M, Wang X-T, Liu D-W. Plasma mitochondrial DNA levels are associated with acute lung injury and mortality in septic patients. BMC Pulmonary Med (2021) 21(1):66. doi: 10.1186/s12890-021-01437-2
22. Szczesny B, Marcatti M, Ahmad A, Montalbano M, Brunyánszki A, Bibli SI, et al. Mitochondrial DNA damage and subsequent activation of z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci Rep (2018) 8(1):914. doi: 10.1038/s41598-018-19216-1
23. Hepokoski M, Wang J, Li K, Li Y, Gupta P, Mai T, et al. Altered lung metabolism and mitochondrial damps in lung injury due to acute kidney injury. Am J Physiology-Lung Cell Mol Physiol (2021) 320(5):L821–L31. doi: 10.1152/ajplung.00578.2020
24. Fehrenbach H, Grazioli S, Dunn-Siegrist I, Pauchard L-A, Blot M, Charles P-E, et al. Mitochondrial alarmins are tissue mediators of ventilator-induced lung injury and Ards. PloS One (2019) 14(11):e0225468. doi: 10.1371/journal.pone.0225468
25. Pabon M, Ma KC, Nakahira K, Salvatore M, Choi AMK. Mitochondrial dna in bronchoalveolar lavage fluid is associated with mortality in patients with viral lower respiratory tract infections. Am J Respir Crit Care Med (2016) 193.
26. Xiao Z, Jia B, Zhao X, Bi S, Meng W. Attenuation of lipopolysaccharide-induced acute lung injury by cyclosporine-a Via suppression of mitochondrial DNA. Med Sci Monitor (2018) 24:7682–8. doi: 10.12659/msm.909909
27. Xiao M-J, Zou X-F, Li B, Li B-L, Wu S-J, Zhang B. Simulated aeromedical evacuation exacerbates burn induced lung injury: Targeting mitochondrial DNA for reversal. Military Med Res (2021) 8(1):30. doi: 10.1186/s40779-021-00320-9
28. He Y-Q, Zhou C-C, Deng J-L, Wang L, Chen W-S. Tanreqing inhibits lps-induced acute lung injury in vivo and in vitro through downregulating sting signaling pathway. Front Pharmacol (2021) 12:746964. doi: 10.3389/fphar.2021.746964
29. Liu R, Xu F, Si S, Zhao X, Bi S, Cen Y. Mitochondrial DNA-induced inflammatory responses and lung injury in thermal injury rat model. J Burn Care Res (2017) 38(5):304–11. doi: 10.1097/bcr.0000000000000501
30. Fredenburgh LE, Perrella MA, Barragan-Bradford D, Hess DR, Peters E, Welty-Wolf KE, et al. A phase I trial of low-dose inhaled carbon monoxide in sepsis-induced Ards. JCI Insight (2018) 3(23):e124039. doi: 10.1172/jci.insight.124039
31. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature (1981) 290(5806):457–65. doi: 10.1038/290457a0
32. Dyall SD, Brown MT, Johnson PJ. Ancient invasions: From endosymbionts to organelles. Science (2004) 304(5668):253–7. doi: 10.1126/science.1094884
33. Supinski GS, Schroder EA, Callahan LA. Mitochondria and critical illness. Chest (2020) 157(2):310–22. doi: 10.1016/j.chest.2019.08.2182
34. West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol (2017) 17(6):363–75. doi: 10.1038/nri.2017.21
35. Groot GS, Kroon AM. Mitochondrial DNA from various organisms does not contain internally methylated cytosine in -ccgg- sequences. Biochim Biophys Acta (1979) 564(2):355–7. doi: 10.1016/0005-2787(79)90233-8
36. Carp H. Mitochondrial n-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med (1982) 155(1):264–75. doi: 10.1084/jem.155.1.264
37. Gentile LF, Moldawer LL. Damps, pamps, and the origins of sirs in bacterial sepsis. Shock (2013) 39(1):113–4. doi: 10.1097/SHK.0b013e318277109c
38. Simmons JD, Lee Y-L, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA damps are linked to clinical outcome in severely injured human subjects. Ann Surg (2013) 258(4):591–8. doi: 10.1097/SLA.0b013e3182a4ea46
39. Yang H, Sun R, Ma N, Liu Q, Sun X, Zi P, et al. Inhibition of nuclear factor-kb signal by pyrrolidine dithiocarbamate alleviates lipopolysaccharide-induced acute lung injury. Oncotarget (2017) 8(29):47296–304. doi: 10.18632/oncotarget.17624
40. Dutra Silva J, Su Y, Calfee CS, Delucchi KL, Weiss D, McAuley DF, et al. Mesenchymal stromal cell extracellular vesicles rescue mitochondrial dysfunction and improve barrier integrity in clinically relevant models of Ards. Eur Respir J (2021) 58(1):2002978. doi: 10.1183/13993003.02978-2020
41. Hernández-Beeftink T, Guillen-Guio B, Rodríguez-Pérez H, Marcelino-Rodríguez I, Lorenzo-Salazar JM, Corrales A, et al. Whole-blood mitochondrial DNA copies are associated with the prognosis of acute respiratory distress syndrome after sepsis. Front Immunol (2021) 12:737369. doi: 10.3389/fimmu.2021.737369
42. Scozzi D, Cano M, Ma L, Zhou D, Zhu JH, O’Halloran JA, et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from covid-19. JCI Insight (2021) 6(4):e143299. doi: 10.1172/jci.insight.143299
43. Simmons JD, Lee Y-l, Pastukh VM, Capley G, Muscat CA, Muscat DC, et al. Potential contribution of mitochondrial DNA damage associated molecular patterns in transfusion products to the development of acute respiratory distress syndrome after multiple transfusions. J Trauma Acute Care Surg (2017) 82(6):1023–9. doi: 10.1097/ta.0000000000001421
44. Lee Y-L, King MB, Gonzalez RP, Brevard SB, Frotan MA, Gillespie MN, et al. Blood transfusion products contain mitochondrial DNA damage-associated molecular patterns: A potential effector of transfusion-related acute lung injury. J Surg Res (2014) 191(2):286–9. doi: 10.1016/j.jss.2014.06.003
45. Vincent J-L, Nakahira K, Kyung S-Y, Rogers AJ, Gazourian L, Youn S, et al. Circulating mitochondrial DNA in patients in the icu as a marker of mortality: Derivation and validation. PloS Med (2013) 10(12):e1001577. doi: 10.1371/journal.pmed.1001577
46. Huang L, Chang W, Huang Y, Xu X, Yang Y, Qiu H. Prognostic value of plasma mitochondrial DNA in acute respiratory distress syndrome (Ards): A single-center observational study. J Thorac Dis (2020) 12(4):1320–8. doi: 10.21037/jtd.2020.02.49
47. Hernandez-Beeftink T, Guillen-Guio B, Rodriguez-Perez H, Marcelino-Rodriguez I, Lorenzo-Salazar J, Corrales A, et al. Mitochondrial DNA in peripheral blood as a prognostic biomarker of acute respiratory distress syndrome. Eur J Hum Genet (2020) 28(SUPPL 1):980–1.
48. Faust HE, Reilly JP, Anderson BJ, Mangalmurti NS, Zhang P, Dunn TG, et al. Plasma mitochondrial DNA is associated with acute respiratory distress syndrome in sepsis. Am J Respir Crit Care Med (2019) 199. doi: 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A2715
49. Ruchko M, Gorodnya O, LeDoux SP, Alexeyev MF, Al-Mehdi A-B, Gillespie MN. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am J Physiology-Lung Cell Mol Physiol (2005) 288(3):L530–L5. doi: 10.1152/ajplung.00255.2004
50. Deng S, Ai Y, Zhang L, Pan P, Wu D. Lps-induced mitochondrial DNA release causes acute lung injury and systemic inflammation through toll-like receptor 9 in mice. Chest (2016) 149(4):A168. doi: 10.1016/j.chest.2016.02.174
51. Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, et al. Circulating mitochondrial DNA-triggered autophagy dysfunction Via sting underlies sepsis-related acute lung injury. Cell Death Dis (2021) 12(7):673. doi: 10.1038/s41419-021-03961-9
52. Wu G, Zhu Q, Zeng J, Gu X, Miao Y, Xu W, et al. Extracellular mitochondrial DNA promote Nlrp3 inflammasome activation and induce acute lung injury through Tlr9 and nf-kb. J Thorac Dis (2019) 11(11):4816–28. doi: 10.21037/jtd.2019.10.26
53. Qin C-Y, Gu J, Fan J-X, Zhang H-W, Xu F, Liang H-M, et al. Epigallocatechin gallate attenuates mitochondrial DNA-induced inflammatory damage in the development of ventilator-induced lung injury. Phytomedicine (2018) 48:120–8. doi: 10.1016/j.phymed.2018.05.017
54. Tsushima K, King LS, Aggarwal NR, De Gorordo A, D'Alessio FR, Kubo K. Acute lung injury review. Internal Med (2009) 48(9):621–30. doi: 10.2169/internalmedicine.48.1741
55. Han S, Mallampalli RK. The acute respiratory distress syndrome: From mechanism to translation. J Immunol (2015) 194(3):855–60. doi: 10.4049/jimmunol.1402513
56. Fan J, Ye RD, Malik AB. Transcriptional mechanisms of acute lung injury. Am J Physiology-Lung Cell Mol Physiol (2001) 281(5):L1037–L50. doi: 10.1152/ajplung.2001.281.5.L1037
57. van der Slikke EC, Star BS, van Meurs M, Henning RH, Moser J, Bouma HR. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-aki. Crit Care (2021) 25(1):36. doi: 10.1186/s13054-020-03424-1
58. Jin L, Yu B, Armando I, Han F, Immenschuh S. Mitochondrial DNA-mediated inflammation in acute kidney injury and chronic kidney disease. Oxid Med Cell Longevity (2021) 2021:1–12. doi: 10.1155/2021/9985603
59. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature (2012) 485(7397):251–5. doi: 10.1038/nature10992
60. He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/Microrna-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology (2017) 66(1):220–34. doi: 10.1002/hep.29153
61. Tait SW, Green DR. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol (2010) 11(9):621–32. doi: 10.1038/nrm2952
62. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. Bak/Bax macropores facilitate mitochondrial herniation and mtdna efflux during apoptosis. Science (2018) 359(6378):eaao6047. doi: 10.1126/science.aao6047
63. Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtdna release during apoptosis. EMBO J (2018) 37(17):e99238. doi: 10.15252/embj.201899238
64. Li S, Li H, Zhang YL, Xin QL, Guan ZQ, Chen X, et al. Sftsv infection induces Bak/Bax-dependent mitochondrial DNA release to trigger Nlrp3 inflammasome activation. Cell Rep (2020) 30(13):4370–85.e7. doi: 10.1016/j.celrep.2020.02.105
65. Lv X, Lu X, Zhu J, Wang Q. Lipopolysaccharide-induced acute lung injury is associated with increased ran-binding protein in microtubule-organizing center (Ranbpm) molecule expression and mitochondria-mediated apoptosis signaling pathway in a mouse model. Med Sci monitor Int Med J Exp Clin Res (2020) 26:e923172. doi: 10.12659/msm.923172
66. Messina A, Reina S, Guarino F, De Pinto V. Vdac isoforms in mammals. Biochim Biophys Acta (2012) 1818(6):1466–76. doi: 10.1016/j.bbamem.2011.10.005
67. Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol (2010) 30(24):5698–709. doi: 10.1128/mcb.00165-10
68. Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. Vdac oligomers form mitochondrial pores to release mtdna fragments and promote lupus-like disease. Science (2019) 366(6472):1531–6. doi: 10.1126/science.aav4011
69. Lai D, Xia J, Wang J, Wei X, Qian J, Lou Q, et al. [the effect of paraquat on voltage-dependent anion channel and caspase-3, 8, 9 in the mitochondria of rat lung]. Zhonghua lao dong wei sheng zhi ye bing za zhi = Zhonghua laodong weisheng zhiyebing zazhi = Chin J Ind hygiene Occup Dis (2015) 33(5):363–5.
70. Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol (2022) 23(4):266–85. doi: 10.1038/s41580-021-00433-y
71. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol (2013) 87(7):1157–80. doi: 10.1007/s00204-013-1034-4
72. Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A. Mitochondrial permeability transition triggers the release of mtdna fragments. Cell Mol Life Sci CMLS (2004) 61(24):3100–3. doi: 10.1007/s00018-004-4424-1
73. Fonai F, Priber JK, Jakus PB, Kalman N, Antus C, Pollak E, et al. Lack of cyclophilin d protects against the development of acute lung injury in endotoxemia. Biochim Biophys Acta (2015) 1852(12):2563–73. doi: 10.1016/j.bbadis.2015.09.004
74. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol (2018) 19(4):213–28. doi: 10.1038/nrm.2017.125
75. Cocucci E, Meldolesi J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol (2015) 25(6):364–72. doi: 10.1016/j.tcb.2015.01.004
76. Santavanond JP, Rutter SF, Atkin-Smith GK, Poon IKH. Apoptotic bodies: Mechanism of formation, isolation and functional relevance. Sub-cellular Biochem (2021) 97:61–88. doi: 10.1007/978-3-030-67171-6_4
77. Esquivel-Ruiz S, González-Rodríguez P, Lorente JA, Pérez-Vizcaíno F, Herrero R, Moreno L. Extracellular vesicles and alveolar epithelial-capillary barrier disruption in acute respiratory distress syndrome: Pathophysiological role and therapeutic potential. Front Physiol (2021) 12:752287. doi: 10.3389/fphys.2021.752287
78. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science (2020) 367(6478):eaau6977. doi: 10.1126/science.aau6977
79. Lázaro-Ibáñez E, Lässer C, Shelke GV, Crescitelli R, Jang SC, Cvjetkovic A, et al. DNA Analysis of low- and high-density fractions defines heterogeneous subpopulations of small extracellular vesicles based on their DNA cargo and topology. J extracellular vesicles (2019) 8(1):1656993. doi: 10.1080/20013078.2019.1656993
80. Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L. Exosomes/Microvesicles as a mechanism of cell-to-Cell communication. Kidney Int (2010) 78(9):838–48. doi: 10.1038/ki.2010.278
81. Nerlich A, Mieth M, Letsiou E, Fatykhova D, Zscheppang K, Imai-Matsushima A, et al. Pneumolysin induced mitochondrial dysfunction leads to release of mitochondrial DNA. Sci Rep (2018) 8(1):182. doi: 10.1038/s41598-017-18468-7
82. Moore JA, Mistry JJ, Hellmich C, Horton RH, Wojtowicz EE, Jibril A, et al. Lc3-associated phagocytosis in bone marrow macrophages suppresses acute myeloid leukemia progression through sting activation. J Clin Invest (2022) 132(5):e153157. doi: 10.1172/jci153157
83. Marostica G, Gelibter S, Gironi M, Nigro A, Furlan R. Extracellular vesicles in neuroinflammation. Front Cell Dev Biol (2020) 8:623039. doi: 10.3389/fcell.2020.623039
84. Ye C, Li H, Bao M, Zhuo R, Jiang G, Wang W. Alveolar macrophage - derived exosomes modulate severity and outcome of acute lung injury. Aging (2020) 12(7):6120–8. doi: 10.18632/aging.103010
85. Lee H, Zhang D, Laskin DL, Jin Y. Functional evidence of pulmonary extracellular vesicles in infectious and noninfectious lung inflammation. J Immunol (2018) 201(5):1500–9. doi: 10.4049/jimmunol.1800264
86. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
87. McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care (2014) 29(6):1133.e1–5. doi: 10.1016/j.jcrc.2014.07.013
88. Song C, Li H, Li Y, Dai M, Zhang L, Liu S, et al. Nets promote Ali/Ards inflammation by regulating alveolar macrophage polarization. Exp Cell Res (2019) 382(2):111486. doi: 10.1016/j.yexcr.2019.06.031
89. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol (2011) 179(1):199–210. doi: 10.1016/j.ajpath.2011.03.013
90. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight (2018) 3(3):e98178. doi: 10.1172/jci.insight.98178
91. Teluguakula N. Neutrophils set extracellular traps to injure lungs in coronavirus disease 2019. J Infect Dis (2021) 223(9):1503–5. doi: 10.1093/infdis/jiab053
92. D'Arcy MS. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int (2019) 43(6):582–92. doi: 10.1002/cbin.11137
93. Elmore S. Apoptosis: A review of programmed cell death. Toxicologic Pathol (2007) 35(4):495–516. doi: 10.1080/01926230701320337
94. Wang L, Xie L, Zhang Q, Cai X, Tang Y, Wang L, et al. Plasma nuclear and mitochondrial DNA levels in acute myocardial infarction patients. Coronary artery Dis (2015) 26(4):296–300. doi: 10.1097/mca.0000000000000231
95. Bindi E, Li B, Zhou H, Janssen Lok M, Alganabi M, Angotti R, et al. Mitochondrial DNA: A biomarker of disease severity in necrotizing enterocolitis. Eur J Pediatr Surg (2020) 30(1):85–9. doi: 10.1055/s-0039-1697910
96. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol CB (2016) 26(13):R568–r72. doi: 10.1016/j.cub.2016.02.019
97. de Torre-Minguela C, Gómez AI, Couillin I, Pelegrín P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J Off Publ Fed Am Societies Exp Biol (2021) 35(8):e21757. doi: 10.1096/fj.202100085R
98. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis (2007) 12(5):913–22. doi: 10.1007/s10495-007-0756-2
99. De Gaetano A, Solodka K, Zanini G, Selleri V, Mattioli AV, Nasi M, et al. Molecular mechanisms of mtdna-mediated inflammation. Cells (2021) 10(11):2898. doi: 10.3390/cells10112898
100. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
101. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. Tlr9 signals after translocating from the er to cpg DNA in the lysosome. Nat Immunol (2004) 5(2):190–8. doi: 10.1038/ni1028
102. Qi Z, He Z, Chen J, Ma M, Deng M, Zhang Y, et al. Protection of toll-like receptor 9 against lipopolysaccharide-induced inflammation and oxidative stress of pulmonary epithelial cells Via Myd88-mediated pathways. Physiol Res (2022) 71(12):259–73. doi: 10.33549/physiolres.934741
103. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by erk, jnk, and P38 protein kinases. Science (2002) 298(5600):1911–2. doi: 10.1126/science.1072682
104. Hayden MS, Ghosh S. Shared principles in nf-kb signaling. Cell (2008) 132(3):344–62. doi: 10.1016/j.cell.2008.01.020
105. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial damps cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi: 10.1038/nature08780
106. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils Via P38 map kinase. Shock (2010) 34(1):55–9. doi: 10.1097/SHK.0b013e3181cd8c08
107. Xie L, Liu S, Cheng J, Wang L, Liu J, Gong J. Exogenous administration of mitochondrial DNA promotes ischemia reperfusion injury Via Tlr9-P38 mapk pathway. Regul Toxicol Pharmacol (2017) 89:148–54. doi: 10.1016/j.yrtph.2017.07.028
108. Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of Tlr9. J Clin Invest (2016) 126(3):859–64. doi: 10.1172/jci83885
109. Zhang L, Deng S, Zhao S, Ai Y, Zhang L, Pan P, et al. Intra-peritoneal administration of mitochondrial DNA provokes acute lung injury and systemic inflammation Via toll-like receptor 9. Int J Mol Sci (2016) 17(9):1425. doi: 10.3390/ijms17091425
110. Gu X, Wu G, Yao Y, Zeng J, Shi D, Lv T, et al. Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the Tlr9–P38 mapk pathway. Free Radical Biol Med (2015) 83:149–58. doi: 10.1016/j.freeradbiomed.2015.02.034
111. Shen H, Wu N, Wang Y, Zhang L, Hu X, Chen Z, et al. Toll-like receptor 9 mediates paraquat-induced acute lung injury: An in vitro and in vivo study. Life Sci (2017) 178:109–18. doi: 10.1016/j.lfs.2017.03.021
112. Han WZ, Xu SW, Wang L. Impact of ketamine intervention for acute lung injury on rage and Tlr9. Eur Rev Med Pharmacol Sci (2018) 22(13):4350–4. doi: 10.26355/eurrev_201807_15432
113. Chen X, Wang T, Song L, Liu X. Activation of multiple Toll−Like receptors serves different roles in Sepsis−Induced acute lung injury. Exp Ther Med (2019) 18(1):443–50. doi: 10.3892/etm.2019.7599
114. Suresh MV, Thomas B, Dolgachev VA, Sherman MA, Goldberg R, Johnson M, et al. Toll-like receptor-9 (Tlr9) is requisite for acute inflammatory response and injury following lung contusion. Shock (2016) 46(4):412–9. doi: 10.1097/shk.0000000000000601
115. Wang H, Wang T, Yuan Z, Wen F. Role of mitochondrial Dna/Toll-like receptor 9 axis in alveolar fluid balance in acute lung injury. Am J Respir Crit Care Med (2017) 195.
116. Hu D, Yang X, Xiang Y, Li H, Yan H, Zhou J, et al. Inhibition of toll-like receptor 9 attenuates sepsis-induced mortality through suppressing excessive inflammatory response. Cell Immunol (2015) 295(2):92–8. doi: 10.1016/j.cellimm.2015.03.009
117. Li J, Ma Z, Tang ZL, Stevens T, Pitt B, Li S. Cpg DNA-mediated immune response in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol (2004) 287(3):L552–8. doi: 10.1152/ajplung.00436.2003
118. Zhang J-Z, Liu ZHI, Liu JIA, Ren J-X, Sun T-S. Mitochondrial DNA induces inflammation and increases Tlr9/Nf-kb expression in lung tissue. Int J Mol Med (2014) 33(4):817–24. doi: 10.3892/ijmm.2014.1650
119. Chao W. Toll-like receptor signaling: A critical modulator of cell survival and ischemic injury in the heart. Am J Physiology-Heart Circulatory Physiol (2009) 296(1):H1–H12. doi: 10.1152/ajpheart.00995.2008
120. Jing R, Hu Z-K, Lin F, He S, Zhang S-S, Ge W-Y, et al. Mitophagy-mediated mtdna release aggravates stretching-induced inflammation and lung epithelial cell injury Via the Tlr9/Myd88/Nf-kb pathway. Front Cell Dev Biol (2020) 8:819. doi: 10.3389/fcell.2020.00819
121. Shen H, Wu N, Wang Y, Guo F, Chen L, Zhang Z, et al. Myd88 gene knockout attenuates paraquat-induced acute lung injury. Toxicol Lett (2017) 269:41–6. doi: 10.1016/j.toxlet.2017.01.015
122. Knuefermann P, Baumgarten G, Koch A, Schwederski M, Velten M, Ehrentraut H, et al. Cpg oligonucleotide activates toll-like receptor 9 and causes lung inflammation in vivo. Respir Res (2007) 8(1):72. doi: 10.1186/1465-9921-8-72
123. Yang C, Song Y, Wang H. Suppression of rage and Tlr9 by ketamine contributes to attenuation of lipopolysaccharide-induced acute lung injury. J Invest Surg (2016) 30(3):177–86. doi: 10.1080/08941939.2016.1232448
124. Osawa Y, Iho S, Takauji R, Takatsuka H, Yamamoto S, Takahashi T, et al. Collaborative action of nf-kb and P38 mapk is involved in cpg DNA-induced ifn-α and chemokine production in human plasmacytoid dendritic cells. J Immunol (2006) 177(7):4841–52. doi: 10.4049/jimmunol.177.7.4841
125. Hotz MJ, Qing D, Shashaty MGS, Zhang P, Faust H, Sondheimer N, et al. Red blood cells homeostatically bind mitochondrial DNA through Tlr9 to maintain quiescence and to prevent lung injury. Am J Respir Crit Care Med (2018) 197(4):470–80. doi: 10.1164/rccm.201706-1161OC
126. Lam LKM, Murphy S, Kokkinaki D, Venosa A, Sherrill-Mix S, Casu C, et al. DNA Binding to Tlr9 expressed by red blood cells promotes innate immune activation and anemia. Sci Trans Med (2021) 13(616):eabj1008. doi: 10.1126/scitranslmed.abj1008
127. Leslie M. Red blood cells may be immune sentinels. Science (2021) 374(6566):383. doi: 10.1126/science.acx9389
128. Cai X, Chiu Y-H, Chen Zhijian J. The cgas-Cgamp-Sting pathway of cytosolic DNA sensing and signaling. Mol Cell (2014) 54(2):289–96. doi: 10.1016/j.molcel.2014.03.040
129. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic gmp-amp synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2012) 339(6121):786–91. doi: 10.1126/science.1232458
130. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. Sting is a direct innate immune sensor of cyclic di-gmp. Nature (2011) 478(7370):515–8. doi: 10.1038/nature10429
131. Ishikawa H, Barber GN. Sting is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
132. Ishikawa H, Ma Z, Barber GN. Sting regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461(7265):788–92. doi: 10.1038/nature08476
133. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an Irf3-dependent innate immune response. Immunity (2006) 24(1):93–103. doi: 10.1016/j.immuni.2005.12.003
134. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cgas. Nature (2013) 498(7454):332–7. doi: 10.1038/nature12305
135. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic gmp-amp is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2012) 339(6121):826–30. doi: 10.1126/science.1229963
136. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic gmp-amp containing mixed phosphodiester linkages is an endogenous high-affinity ligand for sting. Mol Cell (2013) 51(2):226–35. doi: 10.1016/j.molcel.2013.05.022
137. Chen Q, Sun L, Chen ZJ. Regulation and function of the cgas–sting pathway of cytosolic DNA sensing. Nat Immunol (2016) 17(10):1142–9. doi: 10.1038/ni.3558
138. Kato K, Omura H, Ishitani R, Nureki O. Cyclic gmp–amp as an endogenous second messenger in innate immune signaling by cytosolic DNA. Annu Rev Biochem (2017) 86(1):541–66. doi: 10.1146/annurev-biochem-061516-044813
139. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. Cgas produces a 2′-5′-Linked cyclic dinucleotide second messenger that activates sting. Nature (2013) 498(7454):380–4. doi: 10.1038/nature12306
140. Zhang X, Bai X-C, Chen ZJ. Structures and mechanisms in the cgas-sting innate immunity pathway. Immunity (2020) 53(1):43–53. doi: 10.1016/j.immuni.2020.05.013
141. de Oliveira Mann CC, Orzalli MH, King DS, Kagan JC, Lee ASY, Kranzusch PJ. Modular architecture of the sting c-terminal tail allows interferon and nf-kb signaling adaptation. Cell Rep (2019) 27(4):1165–75.e5. doi: 10.1016/j.celrep.2019.03.098
142. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32(1):461–88. doi: 10.1146/annurev-immunol-032713-120156
143. Abe T, Barber GN. Cytosolic-DNA-Mediated, sting-dependent proinflammatory gene induction necessitates canonical nf-kb activation through Tbk1. J Virol (2014) 88(10):5328–41. doi: 10.1128/jvi.00037-14
144. Wan D, Jiang W, Hao J. Research advances in how the cgas-sting pathway controls the cellular inflammatory response. Front Immunol (2020) 11:615. doi: 10.3389/fimmu.2020.00615
145. Hopfner K-P, Hornung V. Molecular mechanisms and cellular functions of cgas–sting signalling. Nat Rev Mol Cell Biol (2020) 21(9):501–21. doi: 10.1038/s41580-020-0244-x
146. Decout A, Katz JD, Venkatraman S, Ablasser A. The cgas–sting pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol (2021) 21(9):548–69. doi: 10.1038/s41577-021-00524-z
147. Ma R, Ortiz Serrano TP, Davis J, Prigge AD, Ridge KM. The cgas-sting pathway: The role of self-DNA sensing in inflammatory lung disease. FASEB J (2020) 34(10):13156–70. doi: 10.1096/fj.202001607R
148. Comish PB, Liu M-M, Huebinger R, Carlson D, Kang R, Tang D. The cgas-sting pathway connects mitochondrial damage to inflammation in burn-induced acute lung injury in rat. Burns (2022) 48(1):168–75. doi: 10.1016/j.burns.2021.04.007
149. Messaoud-Nacer Y, Culerier E, Rose S, Maillet I, Rouxel N, Briault S, et al. Sting agonist diabzi induces panoptosis and DNA mediated acute respiratory distress syndrome (Ards). Cell Death Dis (2022) 13(3):269. doi: 10.1038/s41419-022-04664-5
150. Cavlar T, Deimling T, Ablasser A, Hopfner K-P, Hornung V. Species-specific detection of the antiviral small-molecule compound cma by sting. EMBO J (2013) 32(10):1440–50. doi: 10.1038/emboj.2013.86
151. Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, et al. Mouse, but not human sting, binds and signals in response to the vascular disrupting agent 5,6-Dimethylxanthenone-4-Acetic acid. J Immunol (2013) 190(10):5216–25. doi: 10.4049/jimmunol.1300097
152. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting sting with covalent small-molecule inhibitors. Nature (2018) 559(7713):269–73. doi: 10.1038/s41586-018-0287-8
153. Wu B, Xu M-m, Fan C, Feng C-L, Lu Q-K, Lu H-M, et al. Sting inhibitor ameliorates lps-induced Ali by preventing vascular endothelial cells-mediated immune cells chemotaxis and adhesion. Acta Pharmacologica Sin (2021) 43(8):2055–66. doi: 10.1038/s41401-021-00813-2
154. Joshi JC, Joshi B, Rochford I, Rayees S, Akhter MZ, Baweja S, et al. Sphk2-generated S1p in Cd11b+ macrophages blocks sting to suppress the inflammatory function of alveolar macrophages. Cell Rep (2020) 30(12):4096–109.e5. doi: 10.1016/j.celrep.2020.02.112
155. Joshi JC, Joshi B, Rochford I, Mehta D. S1p generation by sphingosine kinase-2 in recruited macrophages resolves lung inflammation by blocking sting signaling in alveolar macrophages. J Cell Signaling (2021) 2(1):47–51. doi: 10.33696/SIGNALING.2.034
156. Liu L, Mao Y, Xu B, Zhang X, Fang C, Ma Y, et al. Induction of neutrophil extracellular traps during tissue injury: Involvement of sting and toll-like receptor 9 pathways. Cell Proliferation (2019) 52(3):e12579. doi: 10.1111/cpr.12579
157. Hu Q, Wu J, Ren Y, Wu X, Gao L, Wang G, et al. Degree of sting activation is associated with disease outcomes. Gut (2020) 69(4):792–4. doi: 10.1136/gutjnl-2019-318597
158. Ning L, Wei W, Wenyang J, Rui X, Qing G. Cytosolic DNA-Sting-Nlrp3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin Trans Med (2020) 10(7):e228. doi: 10.1002/ctm2.228
159. Sun Y, Yao X, Zhang Q-J, Zhu M, Liu Z-P, Ci B, et al. Beclin-1-Dependent autophagy protects the heart during sepsis. Circulation (2018) 138(20):2247–62. doi: 10.1161/circulationaha.117.032821
160. Nassour J, Radford R, Correia A, Fusté JM, Schoell B, Jauch A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature (2019) 565(7741):659–63. doi: 10.1038/s41586-019-0885-0
161. Kumar V. Sepsis roadmap: What we know, what we learned, and where we are going. Clin Immunol (2020) 210:108264. doi: 10.1016/j.clim.2019.108264
162. Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. Mtdna activates cgas signaling and suppresses the yap-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity (2020) 52(3):475–86.e5. doi: 10.1016/j.immuni.2020.02.002
163. Man SM, Kanneganti T-D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol (2015) 16(1):7–21. doi: 10.1038/nri.2015.7
164. Rathinam Vijay AK, Fitzgerald Katherine A. Inflammasome complexes: Emerging mechanisms and effector functions. Cell (2016) 165(4):792–800. doi: 10.1016/j.cell.2016.03.046
165. Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the Nalp3 inflammasome. Nat Immunol (2010) 12(3):222–30. doi: 10.1038/ni.1980
166. Shimada K, Crother Timothy R, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the Nlrp3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
167. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables Nlrp3 inflammasome activation. Nature (2018) 560(7717):198–203. doi: 10.1038/s41586-018-0372-z
168. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies Aim2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10(3):266–72. doi: 10.1038/ni.1702
169. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi: 10.1038/nature07710
170. Jin T, Perry A, Jiang J, Smith P, Curry James A, Unterholzner L, et al. Structures of the hin Domain:DNA complexes reveal ligand binding and activation mechanisms of the Aim2 inflammasome and Ifi16 receptor. Immunity (2012) 36(4):561–71. doi: 10.1016/j.immuni.2012.02.014
171. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. Hin-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323(5917):1057–60. doi: 10.1126/science.1169841
172. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. Aim2 recognizes cytosolic dsdna and forms a caspase-1-Activating inflammasome with asc. Nature (2009) 458(7237):514–8. doi: 10.1038/nature07725
173. Li H, Li Y, Song C, Hu Y, Dai M, Liu B, et al. Neutrophil extracellular traps augmented alveolar macrophage pyroptosis Via Aim2 inflammasome activation in lps-induced Ali/Ards. J Inflammation Res (2021) 14:4839–58. doi: 10.2147/jir.s321513
174. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differentiation (2009) 16(11):1438–44. doi: 10.1038/cdd.2009.96
175. Balogh ZJ, McIlroy DJ, Smith DW, Hansbro PM. The origin and the role of mitochondrial DNA in postinjury inflammation. J Crit Care (2013) 28(6):1099–100. doi: 10.1016/j.jcrc.2013.08.027
176. Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, et al. Mitochondrial damage contributes topseudomonas aeruginosaactivation of the inflammasome and is downregulated by autophagy. Autophagy (2015) 11(1):166–82. doi: 10.4161/15548627.2014.981915
177. Moriyama M, Koshiba T, Ichinohe T. Influenza a virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat Commun (2019) 10(1):4624. doi: 10.1038/s41467-019-12632-5
178. He WR, Cao LB, Yang YL, Hua D, Hu MM, Shu HB. Vrk2 is involved in the innate antiviral response by promoting mitostress-induced mtdna release. Cell Mol Immunol (2021) 18(5):1186–96. doi: 10.1038/s41423-021-00673-0
179. Storci G, Bonifazi F, Garagnani P, Olivieri F, Bonafè M. The role of extracellular DNA in covid-19: Clues from inflamm-aging. Ageing Res Rev (2021) 66:101234. doi: 10.1016/j.arr.2020.101234
180. Zhou L, Zhang YF, Yang FH, Mao HQ, Chen Z, Zhang L. Mitochondrial DNA leakage induces odontoblast inflammation Via the cgas-sting pathway. Cell communication Signaling CCS (2021) 19(1):58. doi: 10.1186/s12964-021-00738-7
181. Hu Q, Ren H, Li G, Wang D, Zhou Q, Wu J, et al. Sting-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine (2019) 41:497–508. doi: 10.1016/j.ebiom.2019.02.055
182. van Daal GJ, So KL, Gommers D, Eijking EP, Fiévez RB, Sprenger MJ, et al. Intratracheal surfactant administration restores gas exchange in experimental adult respiratory distress syndrome associated with viral pneumonia. Anesth analgesia (1991) 72(5):589–95. doi: 10.1213/00000539-199105000-00004
183. Ong DSY, Spitoni C, Klein Klouwenberg PMC, Verduyn Lunel FM, Frencken JF, Schultz MJ, et al. Cytomegalovirus reactivation and mortality in patients with acute respiratory distress syndrome. Intensive Care Med (2016) 42(3):333–41. doi: 10.1007/s00134-015-4071-z
184. Short KR, Kroeze E, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis (2014) 14(1):57–69. doi: 10.1016/s1473-3099(13)70286-x
185. Xian H, Liu Y, Rundberg Nilsson A, Gatchalian R, Crother TR, Tourtellotte WG, et al. Metformin inhibition of mitochondrial atp and DNA synthesis abrogates Nlrp3 inflammasome activation and pulmonary inflammation. Immunity (2021) 54(7):1463–77.e11. doi: 10.1016/j.immuni.2021.05.004
186. Chao LK, Lin C-H, Chiu H-W, Wong W-T, Chiu H-W, Tasi Y-L, et al. Peroxyauraptenol inhibits inflammation and Nlrp3 inflammasome activation by inhibiting reactive oxygen species generation and preserving mitochondrial integrity. J Agric Food Chem (2015) 63(4):1210–9. doi: 10.1021/jf5054436
187. Sok SPM, Ori D, Wada A, Okude H, Kawasaki T, Momota M, et al. 1′-acetoxychavicol acetate inhibits Nlrp3-dependent inflammasome activation Via mitochondrial ros suppression. Int Immunol (2021) 33(7):373–86. doi: 10.1093/intimm/dxab016
188. Shi J, Yu T, Song K, Du S, He S, Hu X, et al. Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the hif-1a/Ho-1 signaling pathway. Redox Biol (2021) 41:101954. doi: 10.1016/j.redox.2021.101954
189. Guo W, Liu W, Chen Z, Gu Y, Peng S, Shen L, et al. Tyrosine phosphatase Shp2 negatively regulates Nlrp3 inflammasome activation Via Ant1-dependent mitochondrial homeostasis. Nat Commun (2017) 8(1):2168. doi: 10.1038/s41467-017-02351-0
190. Zhao X-D, Liu H, Li T, Gong Q, Zhang W-L. Epigallocatechin gallate attenuates hip fracture-induced acute lung injury by limiting mitochondrial DNA (Mtdna) release. Med Sci Monitor (2017) 23:3367–72. doi: 10.12659/msm.902477
191. Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, et al. α7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med (2014) 20(1):350–8. doi: 10.2119/molmed.2013.00117
192. Peng W, Peng F, Lou Y, Li Y, Zhao N, Shao Q, et al. Autophagy alleviates mitochondrial damp-induced acute lung injury by inhibiting Nlrp3 inflammasome. Life Sci (2021) 265:118833. doi: 10.1016/j.lfs.2020.118833
193. Shu L, Hu C, Xu M, Yu J, He H, Lin J, et al. Atad3b is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J (2021) 40(8):e106283. doi: 10.15252/embj.2020106283
194. Kong X, Lin D, Lu L, Lin L, Zhang H, Zhang H. Apelin-13-Mediated ampk ameliorates endothelial barrier dysfunction in acute lung injury mice Via improvement of mitochondrial function and autophagy. Int Immunopharmacol (2021) 101:108230. doi: 10.1016/j.intimp.2021.108230
195. Lee S-J, Ryter SW, Xu J-F, Nakahira K, Kim HP, Choi AMK, et al. Carbon monoxide activates autophagy Via mitochondrial reactive oxygen species formation. Am J Respir Cell Mol Biol (2011) 45(4):867–73. doi: 10.1165/rcmb.2010-0352OC
196. Zhang Z, Wang X, Ma C, Li Z, Chen H, Zhang Z, et al. Genipin protects rats against lipopolysaccharide-induced acute lung injury by reinforcing autophagy. Int Immunopharmacol (2019) 72:21–30. doi: 10.1016/j.intimp.2019.03.052
197. Liu W, Li C-C, Lu X, Bo L-Y, Jin F-G. Overexpression of transcription factor eb regulates mitochondrial autophagy to protect lipopolysaccharide-induced acute lung injury. Chin Med J (2019) 132(11):1298–304. doi: 10.1097/cm9.0000000000000243
198. Li T, Liu Y, Xu W, Dai X, Liu R, Gao Y, et al. Polydatin mediates parkin-dependent mitophagy and protects against mitochondria-dependent apoptosis in acute respiratory distress syndrome. Lab Invest (2019) 99(6):819–29. doi: 10.1038/s41374-019-0191-3
199. Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine staphylococcus aureus sepsis. Free Radical Biol Med (2015) 78:179–89. doi: 10.1016/j.freeradbiomed.2014.10.582
200. Wu D, Zhang H, Wu Q, Li F, Wang Y, Liu S, et al. Sestrin 2 protects against lps-induced acute lung injury by inducing mitophagy in alveolar macrophages. Life Sci (2021) 267:118941. doi: 10.1016/j.lfs.2020.118941
201. Li G, Yuzhen L, Yi C, Xiaoxiang C, Wei Z, Changqing Z, et al. Dnasei protects against paraquat-induced acute lung injury and pulmonary fibrosis mediated by mitochondrial DNA. BioMed Res Int (2015) 2015:1–10. doi: 10.1155/2015/386952
202. Jung S-S, Moon J-S, Xu J-F, Ifedigbo E, Ryter SW, Choi AMK, et al. Carbon monoxide negatively regulates Nlrp3 inflammasome activation in macrophages. Am J Physiology-Lung Cell Mol Physiol (2015) 308(10):L1058–L67. doi: 10.1152/ajplung.00400.2014
203. Luo Z-L, Sun H-Y, Wu X-B, Cheng L, Ren J-D. Epigallocatechin-3-Gallate attenuates acute pancreatitis induced lung injury by targeting mitochondrial reactive oxygen species triggered Nlrp3 inflammasome activation. Food Funct (2021) 12(12):5658–67. doi: 10.1039/d1fo01154e
204. Ahn H, Lee G-S. Riboflavin, vitamin B2, attenuates Nlrp3, Nlrc4, Aim2, and non-canonical inflammasomes by the inhibition of caspase-1 activity. Sci Rep (2020) 10(1):19091. doi: 10.1038/s41598-020-76251-7
205. Li S, Liang F, Kwan K, Tang Y, Wang X, Tang Y, et al. Identification of ethyl pyruvate as a Nlrp3 inflammasome inhibitor that preserves mitochondrial integrity. Mol Med (2018) 24(1):8. doi: 10.1186/s10020-018-0006-9
Keywords: ALI, ARDS, mtDNA, STING, TLR9, inflammasomes activation
Citation: Long G, Gong R, Wang Q, Zhang D and Huang C (2022) Role of released mitochondrial DNA in acute lung injury. Front. Immunol. 13:973089. doi: 10.3389/fimmu.2022.973089
Received: 19 June 2022; Accepted: 01 August 2022;
Published: 18 August 2022.
Edited by:
Guochang Hu, University of Illinois at Chicago, United StatesReviewed by:
Vijay Kumar, Louisiana State University, United StatesCopyright © 2022 Long, Gong, Wang, Zhang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingyu Zhang, WmhhbmdkaW5neXUyMDIxQDEyNi5jb20=; Chaolin Huang, aHVhbmdjaGFvbGluMjAyMUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.