 Chanjin Yoon
Chanjin Yoon Yu Seong Ham1†
Yu Seong Ham1† Chul-Su Yang
Chul-Su Yang- 1Department of Molecular and Life Science, Hanyang University, Ansan, South Korea
- 2Center for Bionano Intelligence Education and Research, Ansan, South Korea
Infection with the protozoan parasite Toxoplasma gondii (T. gondii) results in the activation of nucleotide-binding domain leucine-rich repeat containing receptors (NLRs), which in turn leads to inflammasome assembly and the subsequent activation of caspase-1, secretion of proinflammatory cytokines, and pyroptotic cell death. Several recent studies have addressed the role of the NLRP3 inflammasome in T. gondii infection without reaching a consensus on its roles. Moreover, the mechanisms of NLRP3 inflammasome activation in different cell types remain unknown. Here we review current research on the activation and specific role of the NLRP3 inflammasome in T. gondii infection.
Overview of NLRP3 inflammasome
The immune system has two subsystems, the innate and adaptive immune systems, that work together to protect the body from pathogens, such as bacteria, viruses, fungi, and parasites (1, 2). The innate immune system is the initial line of defense, and its quick response to pathogens involves the recognition of pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs) (3). Based upon their subcellular localization, PRRs are classified into (i) Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) located in the plasma membrane and (ii) cytoplasmic PRRs. TLRs and CLRs recognize extracellular PAMPs and DAMPs. Cytoplasmic PRRs, on the other hand, involve retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), AIM2-like receptors (ALRs), nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), and the cytosolic sensor cyclic GMP-AMP (cGAMP) synthase (cGAS) (4, 5). The signaling cascades triggered by PRRs play crucial roles in the immune response, including antigen presentation, cell death, and cytokine secretion (6–8).
The inflammasomes are large multi-protein complexes whose assembly is initiated by the different cytoplasmic PRRs, such as NLRs and ALRs (9). The proteins in the NLR family are derived from 22 human genes and share common structure motifs consisting of C-terminal leucine-rich repeat domains and central nucleotide-binding domains (NBDs) (10). (NBD is a component of the larger NACHT domain). According to their variable N-terminal domain, the NLR family has been classified into a variety of subfamilies, including NLRCs, whose N-terminal have one or more caspase-recruitment domains (CARDs), and NLRPs that have pyrin domains (PYD) instead of CARD. NLRC and NLRP are the two most characterized NLR subfamilies (11).
The NLRP3 is one of the most well-studied and characterized protein in the NLR family. The activation of the NLRP3 inflammasome requires signaling in two steps, with a priming signal in the first and an activation signal in the second. The first, priming, step is initiated through the recognition of the various PAMPs or DAMPs by PRRs, such as TLRs or nucleotide-binding oligomerization domain-containing protein 2 (NOD2) or interleukin 1 receptor, type I (IL1R1), that lead to nuclear factor-κB (NF-κB) activation (12). The NF-κB, once activated, boosts the transcription and expression of inflammatory cytokines, including pro-IL-1β and pro-IL-18 (13). The second, activation, step is triggered by not only bacteria, viruses, and fungi but also sterile inflammation mediated by DAMPs or various other stimuli, including ionic flux, mitochondrial dysfunction, production of reactive oxygen species (ROS), and lysosomal damage (14–16).
NLRP3 activation leads to the assembling of an oligomeric protein complex that includes an adaptor protein called apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) and an effector protein, pro-caspase-1 (17). Upon Inflammasome activation, the PRRs, such as NLRs and ALRs, are recruited to bind with the ASC through PYD (18). Then PRR-ACS complexes, interacting through their CARDs, recruit pro-caspase-1 (19), converting them into active caspase-1 by proteolytic cleavage (20). In turn, the activated caspase-1 promotes the cleavage of precursor cytokines, such as pro-IL-1β and pro-IL-18, to generate active cytokines (IL-1β and IL-18), and then facilitates their secretion (6, 9). Furthermore, the activated caspase-1 triggers pyroptosis, an inflammatory cell death, through the cleavage of the protein gasdermin D (GSDMD) (21).
Several studies have reported that the NLRP3 inflammasome responds to bacterial pathogens (22–24). More recent studies have reported that the NLRP3 inflammasome also plays a crucial role in the host’s response to protozoan parasite infection (25–27). This review focuses on the current understanding of the immune response mechanisms that involve the NLRP3 inflammasome in T. gondii infection.
Toxoplasma gondii
Toxoplasma gondii is a protozoan parasite that is estimated to infect at least one-third of the worldwide human population (28). T. gondii infection is typically asymptomatic in immunocompetent individuals. However, T. gondii infection may contribute to focal central nervous system disease and other severe diseases in immunodeficiency syndrome (AIDS) patients and poses a risk of congenital infection of a newborn baby by the mother who got infected during the pregnancy period (29). Because T. gondii can infect nearly all nucleated cell types in most species of warm-blooded animals, the range of T. gondii hosts is extremely broad.
The function of the NLRP3 inflammasome in various cell types infected with T. gondii
To resist T. gondii infection, components of the innate and adaptive immune system are recruited in various cell types. These include inflammatory monocyte, dendritic cells (DCs), neutrophils, macrophages, NK cells, and Th1 cells (30). TLRs are responsible for the initial detection of T. gondii. Resistance to T. gondii infection is mediated by the TLR-associated adaptor protein MyD88 and the induction of IL-12, interferon-γ (IFN-γ), and the synthesis of nitric oxide (NO) (31). TLRs induce the activation of MyD88 which leads to a downstream signaling cascade recruiting host resistance. Sher et al. reported that IL-12 and IFN-γ responses were reduced but not completely abolished and parasite infection was highly susceptible in MyD88-deficient mice (31). These results indicate that, in addition to the crucial role played by MyD88, there were other mechanisms involved in the detection of T. gondii that remain to be identified.
Recently, Grigg et al. showed that, compared with C57BL/6J mice, NLRP3−/− mice harbored greater parasite burdens and their IL-18 response was significantly reduced (32). In addition, Robson et al. reported that the P2X7 receptor (P2X7R) activated by extracellular ATP inhibited T. gondii growth through the NLRP3 inflammasome and produced reactive oxygen species (ROS) and IL-1β in murine macrophage (33).
Several studies reported that the sensitivities for activation of NLRP3 inflammasome were different in the human monocyte/macrophage and mouse macrophage. Chin and Kostura reported that, in humans, treatment with low-dose lipopolysaccharide (LPS) induced pro-IL-1β transcription, but treatment with a higher dose of LPS lead to the secretion of activated IL-1β (34). In contrast, both LPS priming and ATP induction are typically required for the NLRP3 inflammasome activation in mouse bone marrow-derived macrophages (BMDMs) (35). Additionally, Meng et al. showed that LPS treatment activated the NLRP3 inflammasome in the human monocyte cell line THP-1 but not in mouse BMDMs (36). Furthermore, despite the TLRs playing a crucial role in the detection of T. gondii, TLR11 is a pseudogene and TLR12 is not expressed in humans. Thus, NLRP3 inflammasome activation must involve different mechanisms in human and murine cells. Furthermore, in humans, T. gondii-induced NLRP3 inflammasome activation varies depending on the cell type.
Previous studies reported that T. gondii infection was implicated in the activation of NLRP3 inflammasomes and in stimulating IL-1β secretion in human monocytes. To directly address whether the regulation of IL-1β and IL-18 were responses to T. gondii infection in human monocytes, Lodoen et al. infected THP-1 and U937 cells, which are commonly used as models for human monocytes, with T. gondii and observed T. gondii-induced IL-1β response in both cell lines (37, 38). Primary human monocytes showed similar responses to those observed in the human monocytic cell lines (39). Specifically, T. gondii infection increases the rapid induction of NLRP3 transcription and IL-1β production. Conversely, treatment with MCC950, a selective small-molecule inhibitor of NLRP3, reduced IL-1β production compared with vehicle control (38). This study further suggested that potassium efflux was implicated in producing IL-1β since the production of IL-1β was significantly diminished in primary human monocytes after high potassium release (38). Another study reported that the increased IL-1β production in T. gondii-infected THP-1 cells depended on syk phosphorylation (40). The same study also showed that rapidly increased syk phosphorylation in human primary monocytes infected with T. gondii lead to a downstream cascade in the PKCδ-CARD9/MALT-1 pathway, subsequently leading to the activation of NF-κB, which in turn produces pro-IL-1β and NLRP3 (40). Taken together, these lines of evidence support the notion that IL-1β secretion in human monocytes induced by T. gondii infection requires the NLRP3 inflammasome through potassium efflux or the PKCδ-CARD9/MALT-1 signaling pathway.
Interestingly, the responses to the NLRP3 inflammasome in human macrophages were different from those in human monocyte during T. gondii infection. To investigate the role of the various inflammasome components in PMA-differentiated THP-1 macrophages infected with T. gondii, Quan et al. measured the expression of inflammasome-associated genes and the secretion of inflammatory cytokine IL-1β. The investigators reported that T. gondii infection significantly increased the expression of IL-1β in PMA-differentiated THP-1 macrophages. Furthermore, mRNA expressions levels for inflammasome sensors, including NLRP1, NLRP3, NLRC4, NLRP6, NLRP8, NLRP13, AIM2, NAID, ASC, and caspase-1, were significantly elevated over the levels in mock-infected THP-1 macrophages (41). In contrast, primary human macrophages infected with T. gondii did not secrete IL-1β, and NLRP3 was downregulated in these cells (40). Although these studies were unable to fully identify the difference in the mechanisms of T. gondii infection induced NLRP3 inflammasome activation between human macrophage cell lines and primary human macrophages, there are some clues for an explanation. Huang et al. showed that levels of NLRP3, caspase-1, and IL-1β were significantly elevated in PMA-differentiated THP-1 macrophages (42). In contrast, Karabina et al. reported that NLRP3, NLRP6, and NOD2, whose normal levels are already low in peripheral blood mononuclear cells, were further decreased during the differentiation of primary human monocytes to macrophages (43). This raises two possible explanations: either (i) T. gondii was not sufficient to trigger the expression of inflammasome components in primary human macrophages because these cells require a stronger signal to respond to T. gondii infection, or (ii) the primed state of human macrophage cell lines and primary human macrophages may be different (38).
T. gondii infection in FHs 74 Int cells, a human fetal small intestinal epithelial cell line, significantly increased NLRP3 activation, subsequently leading to IL-1β production. Furthermore, T. gondii infection of these cells activated P2X7R, whereas silencing P2X7R significantly reduced T. gondii-induced IL-1β secretion as well as T. gondii proliferation (44). In addition, T. gondii-induced NLRP3 inflammasome activation in FHs 74 Int cells involved the phosphorylation of both p38 MAPK and JNK1/2, even though the p38 MAPK pathway has a more crucial role than the JNK1/2 pathway (45).
Human neutrophils, which T. gondii manipulates to evade innate immunity, present a special case. Lodoen et al. showed that T. gondii infection in the human neutrophils inhibited LPS-induced IL-1β and NLRP3 transcription and decreased the expression of pro-IL-1β, mature IL-1β, and the NLRP3 inflammasome by inhibiting both NF-κB signaling and the activation of the NLRP3 inflammasome (46) (Table 1). However, as other mechanisms and pathways involving NLRP3 remain unclear, further experiments are required to fully understand the role of NLRP3 in T. gondii infection.

Table 1 Mechanisms of NLRP3 inflammasome regulation in various T. gondii-infected cell types.
The function of other NLR families in T. gondii infection
The study of NLR inflammasome activation related to the T. gondii infection was initiated by a report of the susceptibility alleles involved with human congenital toxoplasmosis. McLeod et al. reported that T. gondii progression was attenuated and proinflammatory cytokines, including IL-1β and IL-18, were not upregulated in the human monocyte with NLRP1 knockout engineered by RNA interference (47). These results implicated the NLRP1 inflammasome with a crucial role in limiting T. gondii replication and the production of pro-inflammatory cytokines.
Nuñez et al. reported that NOD2 deficiency results in a not fully functional immune response against T. gondii infection. Specifically, survival of T. gondii was impaired and IFN-γ secretion was reduced in Nod2−/− mice. In addition, Nod2 promoted the generation of an effective Th1 response through T cell-intrinsic signaling against T. gondii. In contrast, Th cell differentiation was impaired in association with reduced IL-2 production and nuclear accumulation of the transcription factor c-Rel in Nod2−/− mice (48).
AIM2 consists of a C-terminal HIN-200 domain, which interacts with cytosolic double-stranded DNA; it binds to a PYD domain of ASC and subsequently activates the AIM2 inflammasome (49). AIM2 detected GPT-promoted T. gondii and induced atypical apoptosis through ASC and Caspase-8 in PMA-differentiated THP-1 macrophages (50). Ahmadpour et al. reported that expression levels for NLRP12, caspase-3, caspase-1, IL-1β, IL-18, and ASC mRNA were significantly increased in mice injected with T. gondii live tachyzoites over levels in control mice. In addition, the intracellular ROS levels in tachyzoite-injected mice were significantly higher than in control mice. These results suggest that T. gondii infection may activate the NLRP12 inflammasome and increase the expression of proinflammatory cytokines such as IL-1β and IL-18 and activates the pyroptosis pathway (51).
In summary, despite the presence of several NLR families, only a few inflammasomes were identified. Thus, further investigations are required to understand the role of other NLR proteins, including NLRP1, NLRP3, NLRP12, AIM2, and NOD2, in T. gondii infection.
Mechanism of pyroptosis and potential application in T. gondii infection
Pyroptosis is an inflammatory programmed-necrotic cell death that is induced by various stimuli such as bacteria, viruses, fungi, and protozoa (52). Pyroptosis pathways are typically classified into caspase-1-dependent canonical pyroptosis and human caspase-4/5 or mouse caspase-11-induced non-canonical pyroptosis (53, 54). Several studies have reported that T. gondii is involved with pyroptosis through a series of inflammatory reactions. Kimberly et al. reported that knockdown of Nlrp1 increases replication of parasites and protects against cell death in pyroptosis-sensitive macrophages from rats such as Lewis and spontaneously hypertensive (SHR) (55). Furthermore, Wang et al. reported that T. gondii upregulated the level of ROS induced by GRA43 resulting in peritoneal macrophages (PMs) pyroptosis in iNOS−/−-SD rats (56), which indicated that iNOS is considered to be a key factor that leads to pyroptosis in SD rat PMs. In contrast, pyroptosis does not occur in macrophages from T. gondii-susceptible strains of rats such as Brown Norway (BN), Sprague Dawley (SD), and Fischer (CDF), or T. gondii-infected mouse macrophages (32, 33, 55). In addition, human monocytes infected with T. gondii were independent of GSDMD cleavage and pyroptosis, despite increased IL-1β production via the NLRP3 inflammasome. Moreover, GSDMD knockout THP-1 cells secreted a not statistically significant amount of IL-1β production compared with wild-type THP-1 cells after T. gondii infection (40). Taken together, T. gondii might manipulate the host cell niche to maintain the integrity of host cells and immune escape, although the host cell produces a protective cytokine.
Recently, an elevating incidence rate of clinical diseased, including atherosclerosis and cancer, have been reported to be involved with pyroptosis and play a crucial role in pyroptosis (57). Pyroptosis reveals not only a protective effect against pathogens but also beneficial effects on tumor suppression. Wang et al. reported that less amount of pyroptosis cell death was sufficient to clear the entire tumor graft (58). Furthermore, several studies reported that granzymes from cytotoxic lymphocytes cleave GSDMB or GSDME to trigger pyroptosis and potently suppress tumor growth (59, 60). However, the relationship between pyroptosis and T. gondii remains unclear, further experiments are required to understand and clinical application of pyroptosis will be one of the successful non-surgical treatments.
NLRP3 response to T. gondii effector proteins
T. gondii secretes numerous effector proteins from the rhoptry (ROP) and dense granule (GRA) organelles, which are required to maintain an equilibrium between host immune responses and the parasite immune evasion, resulting in the survival of both host and parasite. The ROPs are secreted by T. gondii and injected into the host cell during or immediately before the invasion to form the parasitophorous vacuole (PV) with ROP neck proteins (RON) (61). After the invasion and the establishment of the PV, T. gondii secretes GRA proteins that are involved in modifying the PV and creating an environment for intracellular survival and replication (62). Several recent reports implicated ROP and GRA secreted by T. gondii with the inflammasome complex. For example, Cheng et al. reported that T. gondii-infected PMA-differentiated THP-1 macrophages significantly up-regulate NF-κB and the secretion of IL-1β, which, depending on the presence of T. gondii effector protein ROP7, lead to the assembly of inflammasome. In addition, ROP7 was shown to interact with the NACHT domain of NLRP3 to induce inflammasome hyperactivation through the IL-1β/NF-κB/NLRP3-positive loop (63). GRA7 stimulation induced the expression of pro-inflammatory cytokine genes, including IL-1β in BMDM, in mice (64). Further, the T. gondii protein GRA7, after PKCα-mediated phosphorylation, interacted with the PYD domain of ACS, facilitating ASC oligomerization and inflammasome activation (65). Similarly, Yang et al. reported that GRA9, another T. gondii effector protein, interacted with NLRP3 to block the interaction between ASC and NLRP3, disrupting the assembly of the NLRP3 inflammasome. GRA9 also showed a potential to protect against sepsis by increasing anti-inflammatory and anti-bacterial effects through the polarization of M1 to M2 macrophages (66). Yet another T. gondii effector protein, GRA15, induced IL-1β production and secretion through the NLRP3 inflammasome. The IL-1β derived from THP-1 cells, together with IFN-γ, induce iNOS expression and NO production, resulting in the reduction of IDO1 expression and contributing to T. gondii growth in hepatocytes (67) (Figure 1). Three effector proteins, GRA35, GRA42, and GRA43, play an important role in T. gondii infection by inducing pyroptosis and IL-1β secretion in Lewis rat BMDMs (68). Although the involvement of these effector proteins with NLRP3 has not been studied, several studies reported that effector proteins of T. gondii induce an antitumor response. In particular, Shen et al. reported that GRA15 induced macrophage polarization into M1 that suppressed several hepatic carcinoma cell characteristics, including proliferation, invasion, metastasis, and reduced the expression of matrix metalloproteinases (MMP-9 and MMP-2). In addition, both spleen tumor tissues and tumor growth were decreased in GRA15-polarized macrophages-injected tumor-bearing C57BL/6 mice, and IL-6 expression was also decreased in tumor tissue (69). Another study reported that secretion of several ROPs and GRAs induced an antitumor immune response in C57BL/6 mice with ovarian tumors. Deletion of the genes for ROP5, ROP17, ROP18, ROP35 and ROP38, GRA2 or GRA12, and GRA24 markedly impaired the antitumor response against ovarian tumors in mice (70). Previous reports indicated that effector proteins resist tumor progression via enhancing the immune response of hosts. However, recent reports suggested T. gondii-infection regulated the expression of tumor-involved factors, which are able to enhance the anti-tumor ability of hosts. TP53 is a critical tumor-suppressor gene that is frequently mutated in most types of cancer (71). Lu et al. reported that the p53 signaling pathway was altered through up-regulated Gadd45 protein and down-regulated Fas protein after T. gondii infection (72). Further, the colorectal cancer pathway involved genes which are DCC, Smad2, Smad4, hMLH1, hMSH2, and hMSH3 protein expression were regulated after T. gondii infection. In particular, DCC, the colorectal cancer suppressor, is a prognostic marker in patients with stage II or stage III colorectal cancer (73). The DCC protein expression was elevated after T. gondii infection. The same study also reported that RASSF1, non-small cell lung cancer (NSCLC) tumor suppressor, expression was elevated after T. gondii infection, which indicated that T. gondii infection might be improved NSCLC. PTEN, BRCA1, BRCA2, PI3K, and CCND1 genes were involved with the breast cancer signaling pathway. T. gondii infection decreased CCND1 expression and increased BRCA2 expression, which suggests that T. gondii infection could suppress breast cancer growth (72).
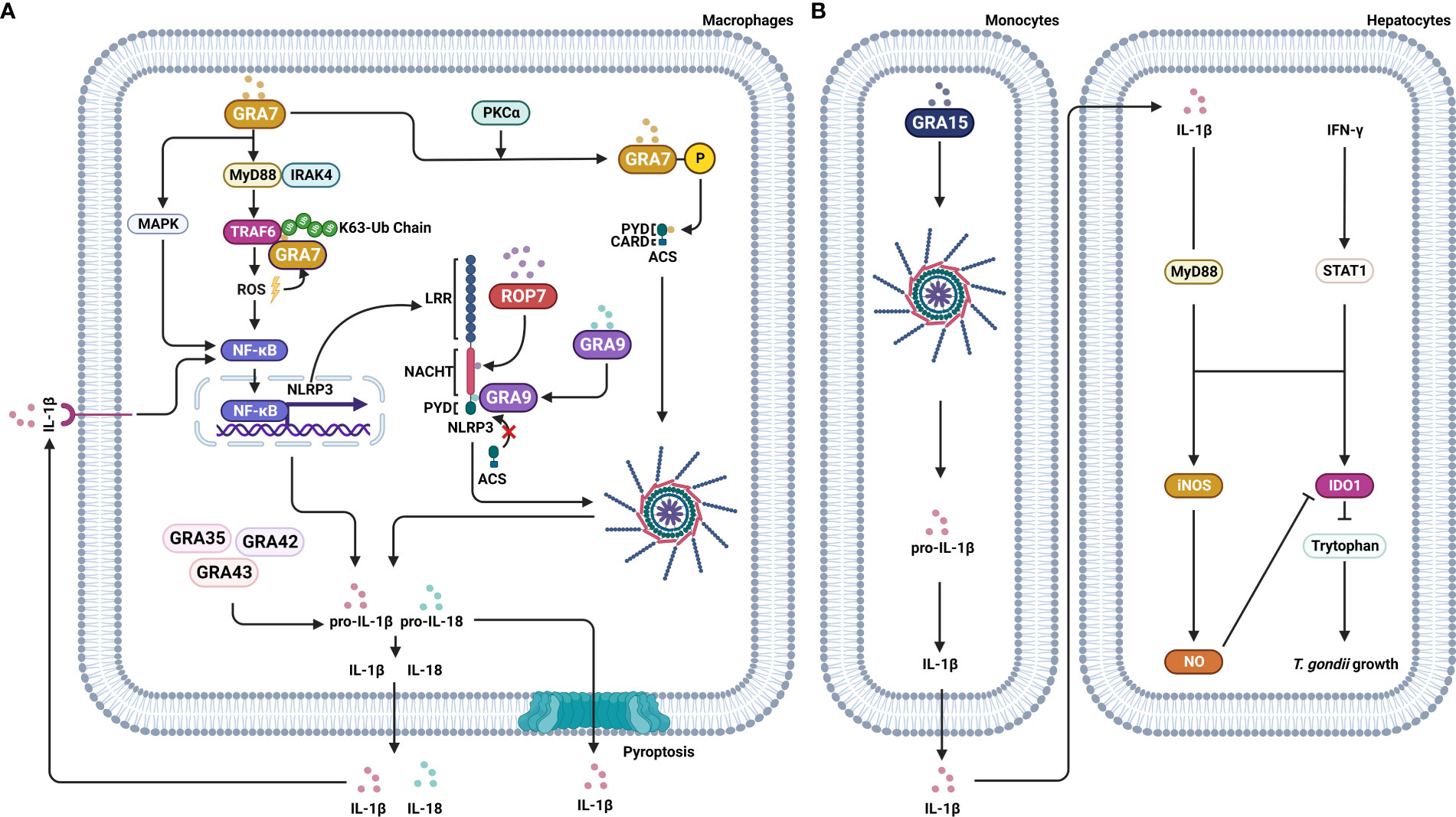
Figure 1 Effector proteins of T. gondii manipulate signaling pathways of the host cell. T. gondii secrets effector proteins, including ROP and GRA, into the host cell to influence the signaling pathways of the host. (A) ROP7 of T. gondii interacts with the NACHT domain of NLRP3 and assembles the NLRP3 inflammasome, leading to IL-1β secretion, which in turn induces an NF-κB/NLRP3-positive loop. GRA7 induces the expression of proinflammatory cytokine IL-1β through MyD88-dependent ROS generation and TRAF6 activation. GRA7, after PKCα-mediated phosphorylation, interacts with the PYD domain of ACS, inducing ASC oligomerization and inflammasome activation. GRA35, GRA42, and GRA43, induce pyroptosis and IL-1β secretion. In contrast, GRA9 binds with NLRP3, which results in blocking the interaction between ASC and NLRP3 and, in turn, the disrupting of the NLRP3 inflammasome assembly. (B) GRA15 induces IL-1β secretion through the NLRP3 inflammasome. Secreted IL-1β and IFN-γ induce iNOS expression and NO production, resulting in the reduction of IDO1 expression and contributing to T. gondii growth in hepatocytes.
In summary, although the effector proteins of T. gondii have various functions implicated in the host immune responses and the antitumor immune response, the associated mechanisms are unknown, and understanding them will require further studies.
Conclusion
Inflammasomes are involved with the immune response of the host combating T. gondii infection. Despite the considerable number of studies investigating inflammasomes implicated with T. gondii, only a few inflammasomes have been identified so far, pointing to the need for further investigations, focusing on additional inflammasome proteins involved in the detection of T. gondii by the host cells and the molecular mechanisms of the immune response. In addition, the responses of inflammasomes to T. gondii infection have been shown to depend on the host cell types. The reason for this is not well understood. It is hypothesized that NLRP3 inflammasome activation might have different requirements in different cell types. For instance, although ATP induces P2X7R signaling that implicates inflammasomes, ATP amount released differs with cell types (74). Moreover, several proteins involved in inflammasomes, such as CARD8, POP1, and POP2, are differentially expressed in species dependent way (36). Thus, future investigations will need new approaches using different perspectives, such as species and cellular characteristics.
T. gondii secretes effector proteins to invade the host cell and several these are known to be involved with inflammasomes. However, the function and mechanisms of action of the effector proteins of T. gondii remain unknown. Recent studies using transcriptome sequencing analysis of T. gondii-infected mice reported that differentially expressed genes involved in the p53 signaling pathway, colorectal cancer pathway, non-small cell lung cancer signaling pathway, and breast cancer signaling pathway were upregulated or downregulated (72). Other studies reported that effector proteins of T. gondii suppressed tumor growth and induced antitumor immune responses (69, 70). Others reported a protective effect of T. gondii effector proteins against sepsis through increasing anti-inflammatory and bacterial effects (66). Taken together, these results are consistent with the notion that the effector proteins of T. gondii act like a double-edged sword: support invasion during T. gondii infection but, depending on the application, they may be beneficial in the treatment of diseases, too. Therefore, future investigations should focus on the understanding of the effector proteins derived from T. gondii and their application to inflammasome-related diseases and cancer as well as other diseases. An understanding of inflammasome regulation in T. gondii infection suggests novel strategies for host immune response mechanisms and may provide opportunities to develop new treatments for various diseases.
Author contributions
CY, YH, WG, and C-SY designed, conceptualized, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a National Research Foundation of Korea grant funded by the Korea government (MSIP) (grant no. 2019R1I1A2A01064237 and 2021R1A4A5032463), by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI22C0884).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vivier E, Malissen B. Innate and adaptive immunity: Specificities and signaling hierarchies revisited. Nat Immunol (2005) 6(1):17–21. doi: 10.1038/ni1153
2. Flores-Villegas AL, Salazar-Schettino PM, Córdoba-Aguilar A, Gutiérrez-Cabrera AE, Rojas-Wastavino GE, Bucio-Torres MI, et al. Immune defence mechanisms of triatomines against bacteria, viruses, fungi and parasites. Bull Entomol Res (2015) 105(5):523–32. doi: 10.1017/s0007485315000504
3. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. Pamps and damps: Signal 0s that spur autophagy and immunity. Immunol Rev (2012) 249(1):158–75. doi: 10.1111/j.1600-065X.2012.01146.x
4. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the Nlrp3 inflammasome in cancer. Mol Cancer (2018) 17(1):158. doi: 10.1186/s12943-018-0900-3
5. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
6. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
7. Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
8. Wang Y, Zhu J, Cao Y, Shen J, Yu L. Insight into inflammasome signaling: Implications for toxoplasma gondii infection. Front Immunol (2020) 11:583193. doi: 10.3389/fimmu.2020.583193
9. Song N, Li T. Regulation of Nlrp3 inflammasome by phosphorylation. Front Immunol (2018) 9:2305. doi: 10.3389/fimmu.2018.02305
10. Zhao C, Zhao W. Nlrp3 inflammasome-a key player in antiviral responses. Front Immunol (2020) 11:211. doi: 10.3389/fimmu.2020.00211
11. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The nlr gene family: A standard nomenclature. Immunity (2008) 28(3):285–7. doi: 10.1016/j.immuni.2008.02.005
12. Qiao Y, Wang P, Qi J, Zhang L, Gao C. Tlr-induced nf-Kb activation regulates Nlrp3 expression in murine macrophages. FEBS Lett (2012) 586(7):1022–6. doi: 10.1016/j.febslet.2012.02.045
13. Gritsenko A, Yu S, Martin-Sanchez F, Diaz-Del-Olmo I, Nichols EM, Davis DM, et al. Corrigendum: Priming is dispensable for Nlrp3 inflammasome activation in human monocytes in vitro. Front Immunol (2021) 12:763899. doi: 10.3389/fimmu.2021.763899
14. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of Nlrp3 inflammasomes: Ros as trigger or effector? Antioxid Redox Signal (2015) 22(13):1111–29. doi: 10.1089/ars.2014.5994
15. Gong Z, Pan J, Shen Q, Li M, Peng Y. Mitochondrial dysfunction induces Nlrp3 inflammasome activation during cerebral Ischemia/Reperfusion injury. J Neuroinflamm (2018) 15(1):242. doi: 10.1186/s12974-018-1282-6
16. Zhang T, Zhao J, Liu T, Cheng W, Wang Y, Ding S, et al. A novel mechanism for Nlrp3 inflammasome activation. Metabol Open (2022) 13:100166. doi: 10.1016/j.metop.2022.100166
17. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the Nlrp3 inflammasome. Front Immunol (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
18. Platnich JM, Muruve DA. Nod-like receptors and inflammasomes: A review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
19. de Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a card (Asc). J Biol Chem (2009) 284(47):32932–41. doi: 10.1074/jbc.M109.024273
20. Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol (2009) 182(5):3173–82. doi: 10.4049/jimmunol.0802367
21. Shi Y. Caspase activation: Revisiting the induced proximity model. Cell (2004) 117(7):855–8. doi: 10.1016/j.cell.2004.06.007
22. Kim JJ, Jo EK. Nlrp3 inflammasome and host protection against bacterial infection. J Korean Med Sci (2013) 28(10):1415–23. doi: 10.3346/jkms.2013.28.10.1415
23. Beckwith KS, Beckwith MS, Ullmann S, Sætra RS, Kim H, Marstad A, et al. Plasma membrane damage causes Nlrp3 activation and pyroptosis during mycobacterium tuberculosis infection. Nat Commun (2020) 11(1):2270. doi: 10.1038/s41467-020-16143-6
24. Zhong Y, Lu Y, Yang X, Tang Y, Zhao K, Yuan C, et al. The roles of Nlrp3 inflammasome in bacterial infection. Mol Immunol (2020) 122:80–8. doi: 10.1016/j.molimm.2020.03.020
25. Clay GM, Sutterwala FS, Wilson ME. Nlr proteins and parasitic disease. Immunol Res (2014) 59(1-3):142–52. doi: 10.1007/s12026-014-8544-x
26. Zamboni DS, Lima-Junior DS. Inflammasomes in host response to protozoan parasites. Immunol Rev (2015) 265(1):156–71. doi: 10.1111/imr.12291
27. Gurung P, Kanneganti TD. Immune responses against protozoan parasites: A focus on the emerging role of nod-like receptors. Cell Mol Life Sci (2016) 73(16):3035–51. doi: 10.1007/s00018-016-2212-3
28. Halonen SK, Weiss LM. Toxoplasmosis. Handb Clin Neurol (2013) 114:125–45. doi: 10.1016/b978-0-444-53490-3.00008-x
29. Feleke DG, Gebreweld A, Zewde G. Toxoplasmosis in pregnant women and Hiv/Aids patients in Ethiopia: A systematic review and meta-analysis. J Parasitol Res (2019) 2019:4670397. doi: 10.1155/2019/4670397
30. Dupont CD, Christian DA, Hunter CA. Immune response and immunopathology during toxoplasmosis. Semin Immunopathol (2012) 34(6):793–813. doi: 10.1007/s00281-012-0339-3
31. Scanga CA, Aliberti J, Jankovic D, Tilloy F, Bennouna S, Denkers EY, et al. Cutting edge: Myd88 is required for resistance to toxoplasma gondii infection and regulates parasite-induced il-12 production by dendritic cells. J Immunol (2002) 168(12):5997–6001. doi: 10.4049/jimmunol.168.12.5997
32. Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, et al. Dual role for inflammasome sensors Nlrp1 and Nlrp3 in murine resistance to toxoplasma gondii. mBio (2014) 5(1):e01117–13. doi: 10.1128/mBio.01117-13
33. Moreira-Souza ACA, Almeida-da-Silva CLC, Rangel TP, Rocha GDC, Bellio M, Zamboni DS, et al. The P2x7 receptor mediates toxoplasma gondii control in macrophages through canonical Nlrp3 inflammasome activation and reactive oxygen species production. Front Immunol (2017) 8:1257. doi: 10.3389/fimmu.2017.01257
34. Chin J, Kostura MJ. Dissociation of il-1 beta synthesis and secretion in human blood monocytes stimulated with bacterial cell wall products. J Immunol (1993) 151(10):5574–85.
35. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity (2009) 30(6):860–74. doi: 10.1016/j.immuni.2009.04.012
36. Wang H, Mao L, Meng G. The Nlrp3 inflammasome activation in human or mouse cells, sensitivity causes puzzle. Protein Cell (2013) 4(8):565–8. doi: 10.1007/s13238-013-3905-0
37. Gov L, Karimzadeh A, Ueno N, Lodoen MB. Human innate immunity to toxoplasma gondii is mediated by host caspase-1 and asc and parasite Gra15. mBio (2013) 4(4):e00255–13. doi: 10.1128/mBio.00255-13
38. Gov L, Schneider CA, Lima TS, Pandori W, Lodoen MB. Nlrp3 and potassium efflux drive rapid il-1β release from primary human monocytes during toxoplasma gondii infection. J Immunol (2017) 199(8):2855–64. doi: 10.4049/jimmunol.1700245
39. Tosh KW, Mittereder L, Bonne-Annee S, Hieny S, Nutman TB, Singer SM, et al. The il-12 response of primary human dendritic cells and monocytes to toxoplasma gondii is stimulated by phagocytosis of live parasites rather than host cell invasion. J Immunol (2016) 196(1):345–56. doi: 10.4049/jimmunol.1501558
40. Pandori WJ, Lima TS, Mallya S, Kao TH, Gov L, Lodoen MB. Toxoplasma gondii activates a syk-Card9-Nf-Kb signaling axis and gasdermin d-independent release of il-1β during infection of primary human monocytes. PloS Pathog (2019) 15(8):e1007923. doi: 10.1371/journal.ppat.1007923
41. Chu JQ, Shi G, Fan YM, Choi IW, Cha GH, Zhou Y, et al. Production of il-1β and inflammasome with up-regulated expressions of nod-like receptor related genes in toxoplasma gondii-infected thp-1 macrophages. Korean J Parasitol (2016) 54(6):711–7. doi: 10.3347/kjp.2016.54.6.711
42. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses Nlrp3 inflammasome activation via Tlr4/Myd88/Nf-Kb signaling in pma-stimulated thp-1 monocytes. BioMed Pharmacother (2016) 82:167–72. doi: 10.1016/j.biopha.2016.04.043
43. Awad F, Assrawi E, Jumeau C, Georgin-Lavialle S, Cobret L, Duquesnoy P, et al. Impact of human monocyte and macrophage polarization on nlr expression and Nlrp3 inflammasome activation. PloS One (2017) 12(4):e0175336. doi: 10.1371/journal.pone.0175336
44. Quan JH, Huang R, Wang Z, Huang S, Choi IW, Zhou Y, et al. P2x7 receptor mediates Nlrp3-dependent il-1β secretion and parasite proliferation in toxoplasma gondii-infected human small intestinal epithelial cells. Parasit Vectors (2018) 11(1):1. doi: 10.1186/s13071-017-2573-y
45. Chu JQ, Gao FF, Wu W, Li C, Pan Z, Sun J, et al. Expression profiles of nod-like receptors and regulation of Nlrp3 inflammasome activation in toxoplasma gondii-infected human small intestinal epithelial cells. Parasit Vectors (2021) 14(1):153. doi: 10.1186/s13071-021-04666-w
46. Lima TS, Gov L, Lodoen MB. Evasion of human neutrophil-mediated host defense during toxoplasma gondii infection. mBio (2018) 9(1):e02027–17. doi: 10.1128/mBio.02027-17
47. Witola WH, Mui E, Hargrave A, Liu S, Hypolite M, Montpetit A, et al. Nalp1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of toxoplasma gondii-infected monocytic cells. Infect Immun (2011) 79(2):756–66. doi: 10.1128/iai.00898-10
48. Shaw MH, Reimer T, Sánchez-Valdepeñas C, Warner N, Kim YG, Fresno M, et al. T Cell-intrinsic role of Nod2 in promoting type 1 immunity to toxoplasma gondii. Nat Immunol (2009) 10(12):1267–74. doi: 10.1038/ni.1816
49. Man SM, Karki R, Kanneganti TD. Aim2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol (2016) 46(2):269–80. doi: 10.1002/eji.201545839
50. Fisch D, Bando H, Clough B, Hornung V, Yamamoto M, Shenoy AR, et al. Human Gbp1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J (2019) 38(13):e100926. doi: 10.15252/embj.2018100926
51. Rajabi S, Spotin A, Mahami-Oskouei M, Baradaran B, Babaie F, Azadi Y, et al. Toxoplasma gondii activates Nlrp12 inflammasome pathway in the Balb/C murine model. Acta Trop (2022) 225:106202. doi: 10.1016/j.actatropica.2021.106202
52. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev (2015) 265(1):130–42. doi: 10.1111/imr.12287
53. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, et al. Gsdmb promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol (2019) 11(6):496–508. doi: 10.1093/jmcb/mjy056
54. Zheng X, Chen W, Gong F, Chen Y, Chen E. The role and mechanism of pyroptosis and potential therapeutic targets in sepsis: A review. Front Immunol (2021) 12:711939. doi: 10.3389/fimmu.2021.711939
55. Cirelli KM, Gorfu G, Hassan MA, Printz M, Crown D, Leppla SH, et al. Inflammasome sensor Nlrp1 controls rat macrophage susceptibility to toxoplasma gondii. PloS Pathog (2014) 10(3):e1003927. doi: 10.1371/journal.ppat.1003927
56. Wang ZJ, Yu SM, Gao JM, Zhang P, Hide G, Yamamoto M, et al. High resistance to toxoplasma gondii infection in inducible nitric oxide synthase knockout rats. iScience (2021) 24(11):103280. doi: 10.1016/j.isci.2021.103280
57. Ju X, Yang Z, Zhang H, Wang Q. Role of pyroptosis in cancer cells and clinical applications. Biochimie (2021) 185:78–86. doi: 10.1016/j.biochi.2021.03.007
58. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature (2020) 579(7799):421–6. doi: 10.1038/s41586-020-2079-1
59. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin e suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579(7799):415–20. doi: 10.1038/s41586-020-2071-9
60. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme a from cytotoxic lymphocytes cleaves gsdmb to trigger pyroptosis in target cells. Science (2020) 368(6494):eaaz7548. doi: 10.1126/science.aaz7548
61. Rastogi S, Xue Y, Quake SR, Boothroyd JC. Differential impacts on host transcription by rop and gra effectors from the intracellular parasite toxoplasma gondii. mBio (2020) 11(3). doi: 10.1128/mBio.00182-20
62. Rastogi S, Cygan AM, Boothroyd JC. Translocation of effector proteins into host cells by toxoplasma gondii. Curr Opin Microbiol (2019) 52:130–8. doi: 10.1016/j.mib.2019.07.002
63. Zhu L, Qi W, Yang G, Yang Y, Wang Y, Zheng L, et al. Toxoplasma gondii rhoptry protein 7 (Rop7) interacts with Nlrp3 and promotes inflammasome hyperactivation in thp-1-Derived macrophages. Cells (2022) 11(10):1630. doi: 10.3390/cells11101630
64. Yang CS, Yuk JM, Lee YH, Jo EK. Toxoplasma gondii Gra7-induced Traf6 activation contributes to host protective immunity. Infect Immun (2016) 84(1):339–50. doi: 10.1128/iai.00734-15
65. Koh HJ, Kim YR, Kim JS, Yun JS, Jang K, Yang CS. Toxoplasma gondii Gra7-targeted asc and Pld1 promote antibacterial host defense Via pkcα. PloS Pathog (2017) 13(1):e1006126. doi: 10.1371/journal.ppat.1006126
66. Kim JS, Mun SJ, Cho E, Kim D, Son W, Jeon HI, et al. Toxoplasma gondii Gra9 regulates the activation of Nlrp3 inflammasome to exert anti-septic effects in mice. Int J Mol Sci (2020) 21(22):8437. doi: 10.3390/ijms21228437
67. Bando H, Lee Y, Sakaguchi N, Pradipta A, Ma JS, Tanaka S, et al. Inducible nitric oxide synthase is a key host factor for toxoplasma Gra15-dependent disruption of the gamma interferon-induced antiparasitic human response. mBio (2018) 9(5):e01738–18. doi: 10.1128/mBio.01738-18
68. Wang Y, Cirelli KM, Barros PDC, Sangaré LO, Butty V, Hassan MA, et al. Three toxoplasma gondii dense granule proteins are required for induction of Lewis rat macrophage pyroptosis. mBio (2019) 10(1):e02388–18. doi: 10.1128/mBio.02388-18
69. Li Y, Poppoe F, Chen J, Yu L, Deng F, Luo Q, et al. Macrophages polarized by expression of Toxogra15(Ii) inhibit growth of hepatic carcinoma. Front Immunol (2017) 8:137. doi: 10.3389/fimmu.2017.00137
70. Fox BA, Sanders KL, Rommereim LM, Guevara RB, Bzik DJ. Secretion of rhoptry and dense granule effector proteins by nonreplicating toxoplasma gondii uracil auxotrophs controls the development of antitumor immunity. PloS Genet (2016) 12(7):e1006189. doi: 10.1371/journal.pgen.1006189
71. Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting mutant P53 for cancer therapy: Direct and indirect strategies. J Hematol Oncol (2021) 14(1):157. doi: 10.1186/s13045-021-01169-0
72. Lu G, Zhou J, Zhao YH, Li QL, Gao YY, Wang L. Transcriptome sequencing investigated the tumor-related factors changes after T. Gondii Infection Front Microbiol (2019) 10:181. doi: 10.3389/fmicb.2019.00181
73. Shibata D, Reale MA, Lavin P, Silverman M, Fearon ER, Steele G Jr., et al. The dcc protein and prognosis in colorectal cancer. N Engl J Med (1996) 335(23):1727–32. doi: 10.1056/nejm199612053352303
Keywords: Toxoplasma gondii, NLRP3, inflammasome, rhoptry, dense granule
Citation: Yoon C, Ham YS, Gil WJ and Yang C-S (2022) The strategies of NLRP3 inflammasome to combat Toxoplasma gondii. Front. Immunol. 13:1002387. doi: 10.3389/fimmu.2022.1002387
Received: 25 July 2022; Accepted: 05 October 2022;
Published: 19 October 2022.
Edited by:
Dario S. Zamboni, University of São Paulo, BrazilReviewed by:
Celio Geraldo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilLulu Zheng, University of Shanghai for Science and Technology, China
Copyright © 2022 Yoon, Ham, Gil and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chul-Su Yang, Y2h1bHN1eWFuZ0BoYW55YW5nLmFjLmty
†These authors have contributed equally to this work