 Yu Gan
Yu Gan Xiaoying Li1
Xiaoying Li1 Qi Liang
Qi Liang Pengfei Rong
Pengfei Rong Wei Wang
Wei Wang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 January 2022
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.795401
This article is part of the Research Topic Impact of DNA Damage Response on Anti-tumor Immunity View all 6 articles
As a DNA receptor, cyclic GMP-AMP synthase (cGAS) plays a crucial role in the immune system by recognizing abnormal DNA in the cytoplasm and activating the stimulator of interferon genes (STING) signaling pathway. This signaling cascade reaction leads to an immune response produced by type I interferon and other immune mediators. Recent advances in research have enhanced our current understanding of the potential role of the cGAS/STING pathway in anticancer therapy; however, in some cases, chronic STING activation may promote tumorigenesis. The present review article discusses the biological mechanisms of the cGAS/STING pathway, its dichotomous role in tumors, and the latest advances with respect to STING agonists and antagonists.
Cancer is the leading cause of mortality globally and a prominent obstacle in prolonging human life expectancy. Classical therapies, such as surgical resection, radiotherapy and chemotherapy, remain the most common treatment regimens for cancer. However, not all cancers respond to classical treatments, which has prompted further research to discover novel treatment strategies (1). Over the past few decades, our understanding of immunology has increased, providing hope for success in cancer immunotherapy (2). Cyclic GMP-AMP (cGAMP) synthase (cGAS) is a cytoplasmic DNA sensor that can activate the stimulator of interferon (IFN) genes (STING) protein to subsequently induce a protective immune defense against various DNA-containing pathogens and provides antitumor immunity (3). The present review article aimed to summarize the immune response mediated by cGAS and discuss its dichotomous role in tumor development and the application of STING in antitumor therapy.
cGAS is an innate immune sensor that can recognize various cytoplasmic double-stranded (ds)DNAs, including viral, bacteria, mitochondrial, micronuclei and retroelement origin DNA. The C-terminal region of cGAS exhibits nucleotide transferase activity (4), binding to dsDNA can lead to profound conformational alterations in cGAS (5), primarily affecting catalytic pockets. Subsequently, ATP and GTP in this pocket can be used as substrates to synthesize GAMP (6). Another important N-terminal domain in cGAS is responsible for maintaining the liquid phase of dsDNA and cGAS. The non-specific ion interaction between the positively charged N-terminal of cGAS and the structural contact between the C site and dsDNA enforce cGAS/DNA liquid-liquid phase separation (LLPS) (7), which is beneficial for dimerization and protects DNA from the degradation of three-prime repair exonuclease 1 (TREX1) to promote cGAS activity (8).
As a second messenger, cGAMP is detected by the cyclic-dinucleotide sensor, STING (4), a ~40-kDa dimer transmembrane protein (9) in the endoplasmic reticulum (ER). Then STING is transported to the Golgi apparatus through the ER-Golgi intermediate compartment and initiates the downstream signaling cascade (10). After reaching the Golgi apparatus, STING is palmitoylated at two cysteine residues (Cys88 and Cys91) (11), recruiting TANK-binding kinase 1 (TBK1), which in turn phosphorylates the C-terminal of STING and recruits IFN regulatory factor 3 (IRF3). Moreover, STING can bind and stimulate IκB kinase to trigger the transcriptional activation of NF-κB (9), which eventually regulates the expression and secretion of pro-inflammatory cytokines, including IFN-α and IFN-β (12). Subsequently, IFN binding with heterodimeric IFN receptor IFNAR1/IFNAR2 activates Janus kinase 1 (JAK1), which can phosphorylate members of the signal transducer and activator of transcription (STAT) family and induces the expression of IFN-stimulated genes (ISGs) (Figure 1). A number of ISGs control viral, bacterial and parasite infections by directly targeting pathogen life cycle pathways and functions (13) .
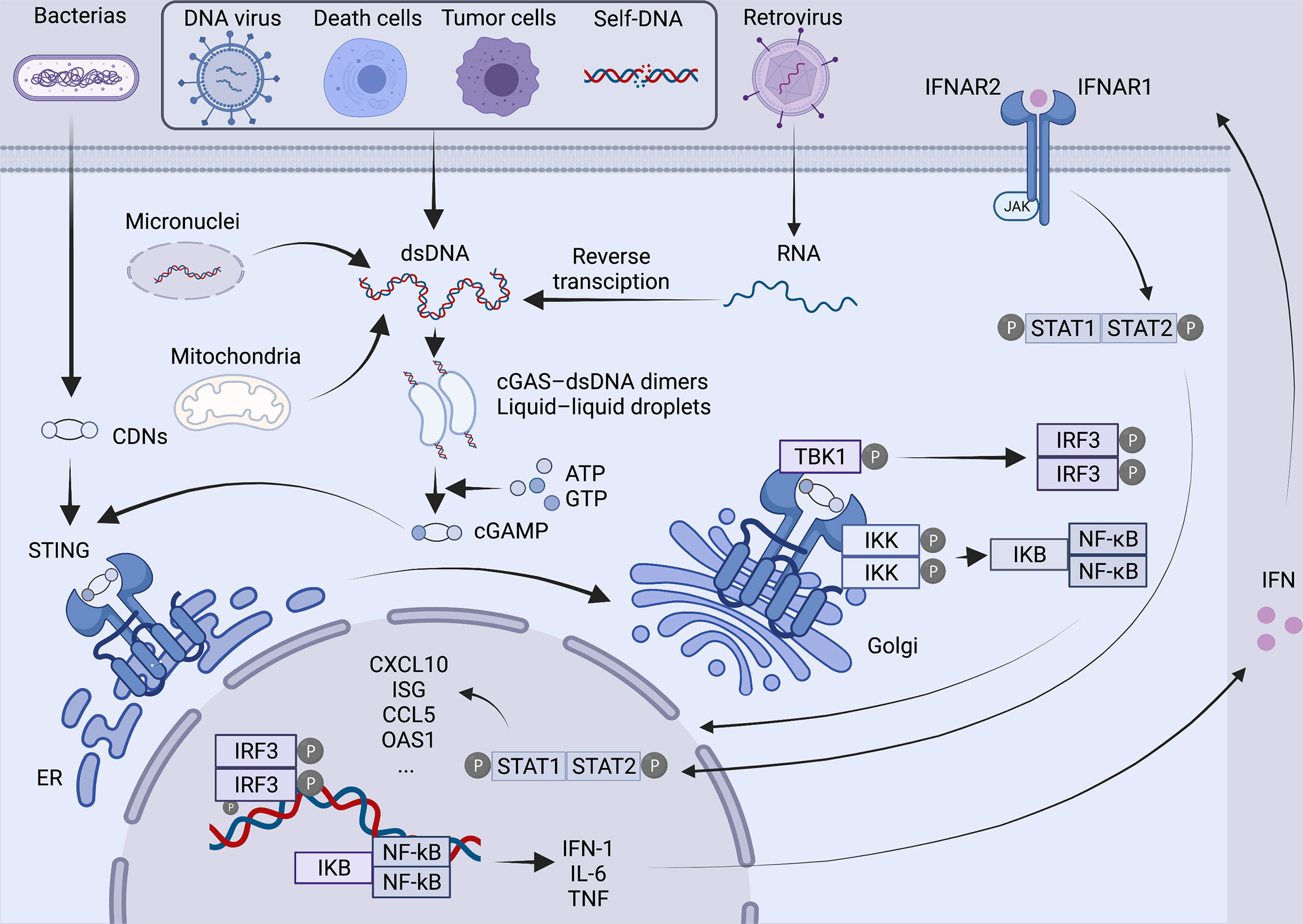
Figure 1 Overview of the cGAS-STING pathway. In the presence of various cytoplasmic DNAs, as a natural immunosensor, cGAS can bind to dsDNA to form a 2:2 cGAS-dsDNA complex, generating a second messenger cGAMP, which activates the STING protein at the endoplasmic reticulum. STING is then transferred from the endoplasmic reticulum via ERGIC to the Golgi apparatus, palmitoylated at two cysteine residues Cys88 and Cys91. Modified STING recruits TBK1 and IRF3. At the same time, STING also binds and stimulates IKK, triggering the transcriptional activation of NF-kB. Ultimately, it regulates the expression and secretion of pro-inflammatory cytokines such as IFN. Binding of IFN to IFNAR1/IFNAR2 activates JAK1, phosphorylates STAT, and induces the expression of ISG. Created with BioRender.com.
Pathogen infection, self-DNA damage and tumor DNA are three critical factors that induce cGAS/STING signal activation. The majority of DNA viruses, such as human cytomegalovirus (HCMV), Herpes simplex virus type 1 (HSV-1) (14) and hepatitis B virus, can activate the cGAS/STING pathway and initiate an antiviral immune response. The Dengue RNA virus has also been shown to induce cytoplasmic exposure of mitochondrial DNA (mtDNA) in infected cells to induce cGAS/STING signals (15). It has been shown that under conditions of chronic STING activation, high ISG expression in cancer cells may impose a transcriptional state on the tumor that is used to respond to aberrant dsRNA accumulation due to increased sensor levels (MDA5, RIG-I, and PKR), suggesting that cGAS may act as an indirect sensor of dsRNA and exert anti-tumor effects (16).
DNA is replicated with immense precision during each cell division cycle in normal cells, and genome maintenance systems will monitor and resolve DNA defects in real-time. The DNA damage response (DDR) can counteract endogenous and exogenous injuries and maintain genomic integrity by activating a complex signaling network that promotes transient cell cycle arrest and DNA repair. When DNA damage exceeds the ability of repair, DDR can trigger cell senescence or apoptosis (17). Micronuclei, small nuclear-like bodies composed of chromosome fragments wrapped in fragile nuclear membranes, are traditional biomarkers of DNA damage and chromosome instability (18). Due to the lack of a stable nuclear membrane, the micronucleus envelope can easily break and expose its genome contents to the cytoplasm (19), triggering the cGAS/cGAMP/STING pathway (20). However, during normal mitosis, when the nuclear membrane ruptures and the chromosome DNA expose to cytoplasmic, it is difficult for the chromosome to recognize cGAS. Existing evidence suggests that this is due to the N-terminal hyperphosphorylation and the inhibition of oligomerization caused by chromatin tethering (21).
Cancer cells have common characteristics. In addition to the classic 10 characteristics (22), a group of other markers has been proposed to describe the stress phenotype of cancer cells, including metabolic stress, mitotic stress, oxidative stress and DNA damage stress (23). Under these extreme stress conditions, nuclear and mtDNA are very fragile and are easily destroyed. At the sites of stalled replication forks, DNA structure-specific endonuclease MUS81 can cleave aberrant DNA structures, leading to the accumulation of DNA in the cytoplasm (24). In addition, the chromosome missegregation in cancer cells ultimately leads to the leakage of DNA in the form of micronuclei, chromatin fragments and/or free telomere DNA, triggering the cGAS/STING signaling pathway (25). Exposure to ionizing radiation (IR) or treatment with chemotherapeutic drugs, such as platinum drugs, can also induce DNA double-strand breaks (DSBs) according to a similar mechanism and can subsequently activate the cGAS/STING signaling pathway. In addition to nuclear DNA leakage, malignant tumor cells that have experienced oxidative stress and mitochondrial dysfunction can also release mtDNA into the cytoplasm (26). Under the inhibition of caspase, mtDNA can activate the cGAS/STING signaling by releasing it into the cytoplasm through BAX/BAK-dependent mitochondrial outer membrane permeabilization (27). Furthermore, tumors also acquire mtDNA from the extracellular environment to participate in the cGAS dsDNA-sensing cascade. Cancer cells without mtDNA exhibit a more delayed tumor growth compared with those with mtDNA. Thus, the transfer of mtDNA from host cells in the tumor microenvironment to tumor cells with impaired respiratory function can help reconstruct respiration and initiate the formation of the tumor (28). Genomic substrates from other sources, such as apoptosis-derived DNA, exosomes and transposons, may also induce cGAS/STING activation in tumors (29, 30).
Several studies have indicated that cancer-associated genomic instability can also modulate cGAS/STING signals. For example, BRAC1/2 promotes the repair of double-stranded DNA breaks (31). A statistical investigation pointed out that individuals with germline BRCA1/2 mutations have a higher risk of developing breast and ovarian cancer than bystanders (32). In such tumors, the cGAS-STING signaling pathway is activated due to the inability to cope with endogenous DNA damage or to follow exogenous DNA damage, resulting in massive lymphocytic infiltration (33). Interestingly, tumors with a BRCA1 mutation often carry amplifications in the MYC oncogene, and a recent study has shown that MYC can lead to immune escape by suppressing STING-dependent innate immunity (34). Besides, Neurofibromin 2 (NF2/Merlin/schwannomin) is a classical tumor suppressor and naturally occurring mutations in the FERM domain could transform it into a profound suppressor, which can inhibit the cGAS-STING pathway by blocking STING-mediated DNA sensing. This evidence may provide some clues to the pathogenesis of NF2-related tumors (35).
Although micronucleus cGAS can detect DNA damage and induce immune responses, nuclear cGAS has recently been found to inhibit DNA repair in a STING non-dependent manner. In normal cells, homologous recombination’s accurate repair of DNA double-stranded breaks preserve genome integrity and inhibit tumorigenesis. However, stimulation of DNA damaging agents leads to dephosphorylation of cGAS. It facilitates its shuttling to the nucleus, where cGAS is recruited to the DNA double-strand break site and interacts with PARP1 via poly(ADP-ribose), hindering the formation of the PARP1-Timeless complex, thereby inhibiting homologous recombination and promoting tumorigenesis (36).
Moreover, STING can also regulate cell cycle in a cGAS-STING axis-independent manner in some tumor models. In human colon cancer cell HCT116, STING was observed to downregulate the expression of proliferation-related genes BUB1 (Budding Uninhibited by Benzimidazoles related 1) and MAD2L1 (Mitotic Arrest Deficient 2-like 1), indicating that STING may also play a role in tumor suppression in cancer cells that lack cGAS expression (37).
cGAS/STING signaling plays an antitumor role in cancer cells in both an autonomous and non-autonomous manner (Figure 2). In the precancerous stage, oncogene-induced senescence signaling inhibits cancer by activating the p53 or p16-retinoblastoma protein pathway in response to carcinogenic stimuli, including carcinogenic stress, permanently inhibiting cell proliferation (38). It is characterized by the secretion of inflammatory mediators, consisting of various cytokines, chemokines, extracellular matrix proteins and growth factors, collectively known as the aging-related secretory phenotype (SASP) (39). Some studies have suggested that the cGAS/STING pathway regulates cell senescence mainly by suppressing soluble factors and inducing SASP autocrine and paracrine signal transduction. In turn, the rupture of the nuclear membrane and the appearance of cytoplasmic chromatin fragments (CCFs) caused by the loss of lamin B1 (40) in senescent cells can cause acute STING signal transduction and activation (41). The loss of ataxia telangiectasia-mutated (ATM) kinase leads to the accumulation of cytoplasmic DNA, which is partially released from the damaged mitochondria and triggers the phenotype of STING-dependent aging in the brain and in vitro (42). SASP may have both beneficial and disadvantageous effects. The reason for this is that some SASP components can maintain senescence stagnation through the autocrine cytokine network. When it exists for an extended period, the secretory activity may be harmful. Chronic SASP can induce epithelial-mesenchymal transformation and invasiveness through paracrine mechanisms dependent on the SASP factors IL-6 and IL-8 and the expression of RAS and the loss of function of p53 may significantly accelerate this phenomenon (43). The activation of the STING signaling pathway in cancer cells may also promote the apoptosis of tumor cells. When cells are affected by internal apoptosis-stimulating factors, such as the activation of oncogenes, DNA damage, hypoxia and the loss of cell growth factors, it can activate the mitochondrial apoptotic pathway, promoting apoptosis. In vertebrates, this mechanism contributes to mtDNA leakage and activation of the mtDNA-dependent cGAS-STING signaling pathway through mitochondrial outer membrane permeabilization (MOMP) induced by Bax and Bak, leading to I-IFN production. STING agonists can downregulate the anti-apoptotic protein BCL-2 and increase the proapoptotic factor Bax to drive apoptosis. It is worth noting that malignant T cells exhibit a high susceptibility to such death-induced characteristics of STING. Thus, STING agonists may help exert an antitumor effect on T cell malignant tumors (44). Besides, the activation of STING in tumor cells also promotes the transcription of downstream type I IFN, induces the maturation of dendritic cells (DCs) and recruits supportive immune cells to remove tumors (45).
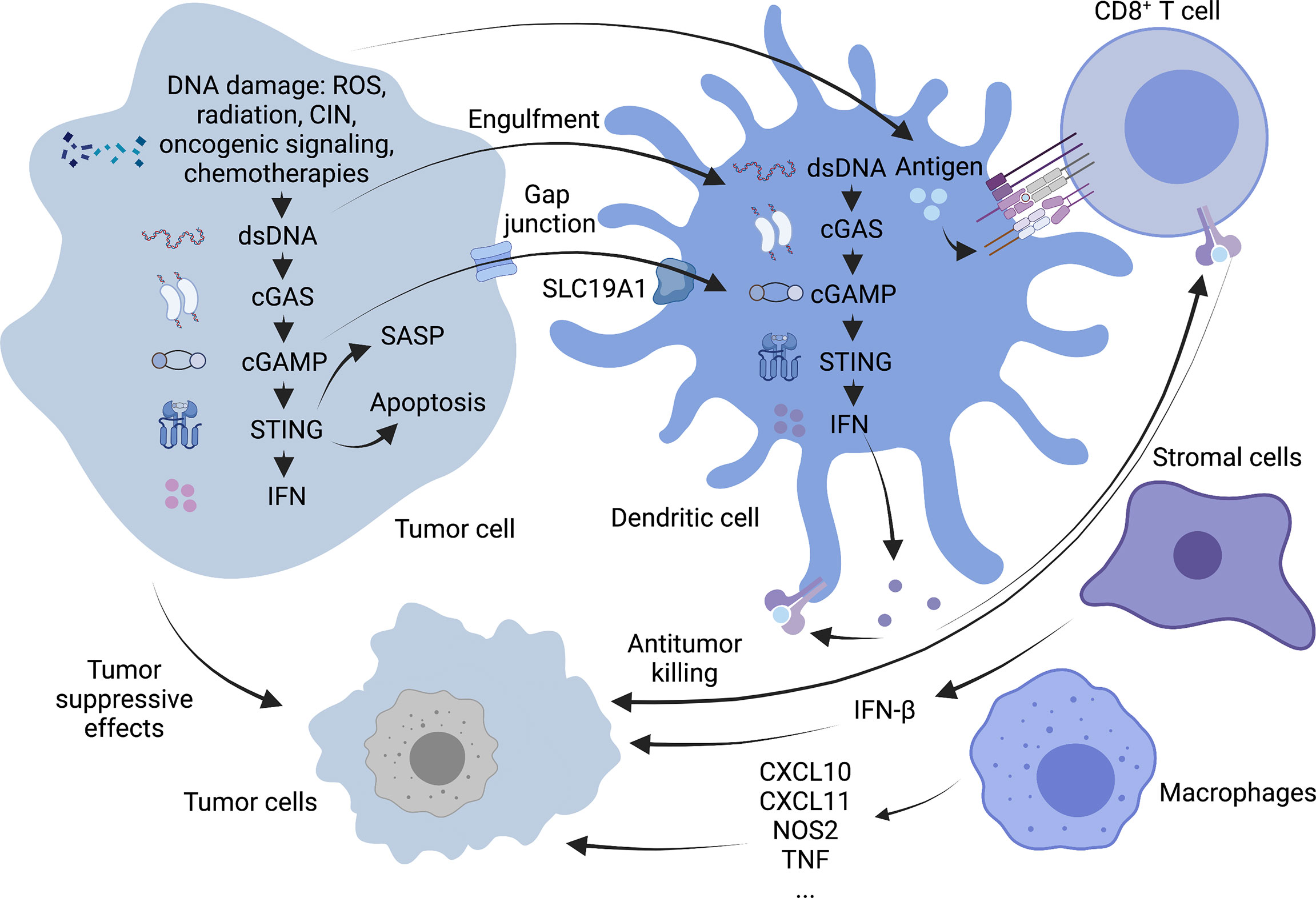
Figure 2 Antitumor mechanism of cGAS-STING pathway. cGAS-STING signaling exerts antitumor functions in cancer cells in an autonomous and non-autonomous manner. On the one hand, OIS secretes SASP in response to oncogenic stimuli to prevent cell proliferation, and activation of the STING signaling pathway in tumor cells may also promote apoptosis in tumor cells. On the other hand, STING activation in tumor cells promotes downstream type I IFN transcription and allows cross-talk between tumor and neighboring immune cells to modulate antitumor immunity. DCs can capture tumor-associated antigens and subsequently initiate tumor-killing by CD8+ T cells. Macrophages can produce TNF-α and express high levels of CxCl10, CxCl11, NOS2 and other molecules. Stromal cells can also be effective against tumor angiogenesis by expressing IFN-β. Created with BioRender.com.
In addition to tumor cells that can specifically activate the cGAS-STING pathway, some antigen-presenting cells (APCs), such as DCs and macrophages, can also initiate the body’s anti-tumor response via this pathway. In particular, activation of STING signaling in the DCs lineage driven by the basic leucine zipper transcription factor ATF-like 3 (BATF3) is a central step in the overall cancer immune cycle (46). Tumor cell DNA is a key inducer of innate immunity. It mainly includes tumor cell nuclear or mitochondrial DNA encapsulated in exosomes (47) and DNA released due to tumor cell turnover and death. Uptake of this DNA by the host APC activates the intracellular STING signaling pathway, leading to the secretion of type I interferons and chemokines, C-X-C motif chemokine ligands (CXCL)9 and CXCL10 (48). And then tumor cells with a T-cell inflammatory phenotype are phagocytosed by antigen-presenting cells which are activated by those cytokines mentioned above. Tumor-infiltrating Batf3 DCs take up tumor-associated antigens and migrate through lymphatic vessels to tumor-draining lymph nodes, where antigens are captured on major histocompatibility complex (MHC) I and MHC II molecules are presented to T cells and tumor-specific CD8+ T cells are cross-activated. Activated CD8+ T cells subsequently clone and expand in tumor-draining lymph nodes and kill cancer cells via vascular transport (49). Damaged cancer cells then release additional tumor-associated antigens (performing the first step again), increasing the breadth and depth of the response in subsequent cycles, which forms a positive feedback loop called the cancer-immune cycle (50). Although the role of intrinsic cGAS-STING activation in T cells is unknown, it has been shown that cGAS-STING-mediated type I interferon signaling enhances the stem cell-like CD8+ T cell differentiation program by inhibiting Akt activity (51). In addition, tumor cell-derived cGAMP can also transfer to host DCs through the folate transporter SLC19A1 and then directly bind to STING to activate it in DCs (52). CD8+ T cells can recognize target cells and eliminate them mainly via exocytosis-mediated apoptosis with granules containing the effector molecules, perforin and granzyme. Perforins form transmembrane pores in the cancer cells, allowing the extracellular environment (including granzymes) to directly diffuse into the cytoplasm, where they activate the apoptotic pathway through proteolytic substrate treatment (53).
Although DCs are considered the primary responders of tumor cGAMP (54), other cells, such as macrophages and stromal cells, may also have this function. In two tumor models that employed a mouse SCC cell line (mSCC1) that was generated from methylcholanthrene-induced carcinoma and a mouse CT26 colon cancer cell line, research shows that mature CD11bmid Ly6C+ F4/80+ MHC class II+ macrophages can instantly migrate to the tumor site through a STING-dependent signaling pathway in the tumor microenvironment. All these macrophages have familiar characteristics, such as the production of TNF-α; expressing CXCL10, CXCL11, NOS2, and IFNB1 at high levels; exert phagocytic activity but cannot produce IL-10, suggesting that they are effective antitumor effector cells (55).
As vital members of the tumor microenvironment, Stromal cells (such as endothelial cells and fibroblasts etc.) also express the STING gene. It is generally considered that DCs are the primary source of type I IFN in the immune response process (56). However, studies have identified endothelial cells as the most important IFN-producing cells responding to the spontaneous and enforced STING activation (57). Of note, in mouse melanoma models, researchers have found that IFN-β is expressed exclusively in the tumor microenvironment but have not detected IFN-α. This result may be related to the weak ability of endothelial cells to produce IFN-α following STING activation (57). IFN-β is an effective anti-angiogenic cytokine. It can suppress the proliferation or survival of endothelial cells and inhibit the formation of the capillary network. It has been demonstrated that intratumoral STING agonist therapy can activate type I IFN signaling and can induce the upregulation of vascular normalizing genes in Lewis lung carcinoma (LLC) models, such as angiopoietin 1 (ANGPT1), platelet-derived growth factor receptor beta (PDGFRB), melanoma cell adhesion molecule (MCAM) and cadherin 5 (CDH5). These changes lead to increased pericyte coverage, structural normalization of tumor blood vessels, and the basement membrane’s integrity, thereafter promoting the intratumoral infiltration of effector CD8+ T cells and alleviating hypoxia in the tumor microenvironment (58).
Type I INFs play multiple roles in inducing antitumor immunity. IFN-α and IFN-β produced by cancer cells bind to IFN receptors on the same cell, adjacent cells, or immune cells and induce autocrine or paracrine signals. The latter promotes the mobilization of immune cells, such as DCs and T cells, to eradicate tumors. IFN-1 is considered to increase the number of DCs and stimulate the maturation of DCs in the tumor microenvironment following cGAMP injection (57). It can also reduce the rate of endosomal-lysosomal system acidification and promote the storage of exogenous antigens in DCs. In addition, IFN-α can promote the localization of MHC I molecules in the antigen storage area of DCs and enhance the antigen presentation ability of DCs. Furthermore, IFN-1 can promote the production of chemokines (such as CXCL9 and CXCL10) and then enable the homing of antigen-presenting cells and the migration of CD8+ T and natural killer cells (48). Finally, IFN-1 suppresses the immunosuppression of regulatory T- (Treg) cells by downregulating phosphodiesterase 4 (PDE4) and upregulating cyclic adenosine monophosphate (CAMP) (45).
Based on The Cancer Genome Atlas (TCGA) dataset containing 18 types of malignant tumors, some scholars have compared the changes in the expression of critical molecules in the cGAS/STING signaling pathway between malignant and normal control tissues, including MB21D1 encoding cGAS and TMEM173 encoding STING, as well as TBK1 and IRF3. It has been found that the expression of these four essential molecular genes is significantly upregulated in almost all detected cancer types, suggesting that cGAS/STING signal transduction may be activated in all cancers (49). With further in-depth research and the presentation of new evidence (36, 59), it is generally considered that the cGAS/STING signaling cascade may play a dichotomous role in the development of tumors. In some cases, highly invasive and unstable tumors can paradoxically use cGAS/STING signals to stimulate carcinogenesis (60).
As a critical activator of the inflammatory response, NF-κB participates in regulating cell survival, proliferation and apoptosis. In addition, NF- κB promotes the occurrence and development of inflammation, immune diseases and tumors (61). Chromosome instability (CIN) can induce chronic inflammatory signal transduction by continuously activating cGAS/STING signaling and the downstream NF-κB pathway, resulting in the increased migration and invasion of cancer cells (62) (Figure 3). For example, 7,12-dimethylbenz(a)anthracene, a carcinogen of skin cancer, can cause nuclear DNA leakage into the cytoplasm to activate the STING signaling pathway, resulting in the production of inflammatory cytokines and skin inflammation and ultimately inducing skin carcinogenesis (63). Furthermore, from TCGA database analysis, it has been demonstrated that the expression level of STING in tumors is negatively correlated with the infiltration of immune cells in some tumor types, which means a high level of cGAS/STING signaling may predict a poor prognosis in patients with some cancers (49).
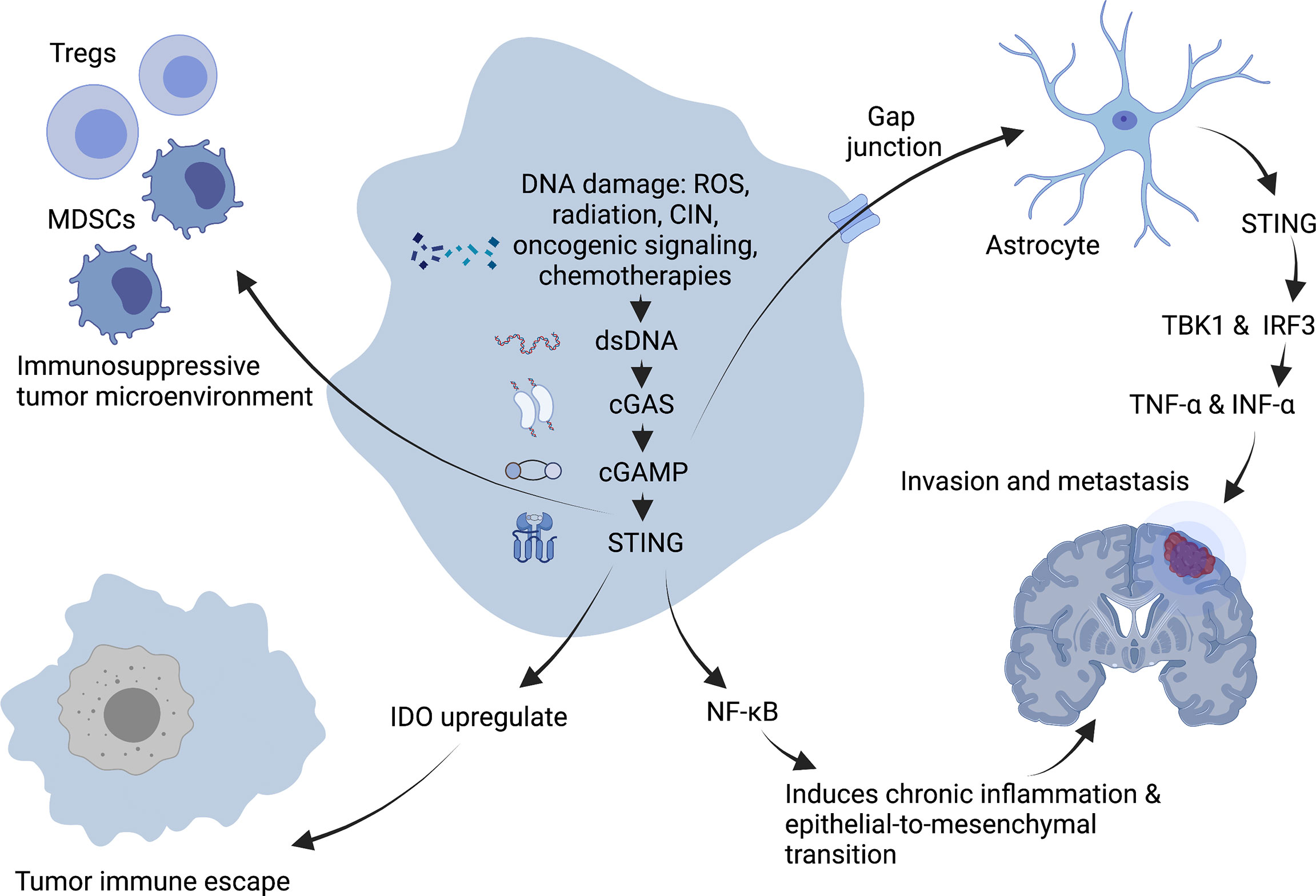
Figure 3 Tumor-promoting mechanism of cGAS-STING pathway. DNA damage induces chronic inflammatory signaling through sustained activation of NF-κB downstream of the cGAS-STING signaling pathway, facilitating epithelial-to-mesenchymal transition and leading to increased migration and invasion of cancer cells. Expression of STING in tumors promotes immunosuppressive cells such as Treg and MDSC to infiltrate into tumor sites, and upregulation of IDO expression promotes immune escape of tumors in the body, creating an immunosuppressive tumor microenvironment. In addition, the second messenger, cGAMP can be transferred through the cancer-astrocyte gap junction to activate STING protein in astrocytes, thus triggering TBK1 and IRF3 activation as well as IFNα and TNFα production to promote tumor brain metastasis. Created with BioRender.com.
A characteristic of an immunosuppressive tumor microenvironment is the upregulated expression of immune checkpoint indoleamine-2,3-dioxygenase (IDO). Existing study suggest that STING can upregulate IDO expression to directly or indirectly suppress T-cell numbers and function and then promoting immune immune escape (64).
The STING signal is closely related to tumor metastasis. CIN induces the production of a large number of micronuclei. When nuclear membrane breaks, genomic DNA will overflow into the cytoplasm, activating the cGAS/STING signaling pathway and triggering irregular NF-κB signals and inflammatory responses, conducive to the transformation of epithelial-to-mesenchymal transition. It has been demonstrated that cells with a high CIN have a higher migratory and invasive ability in vitro (59). Moreover, the metastasis of tumor cells may be driven by non-autonomous cGAS/STING pathway-related mechanisms (59). It has been demonstrated that some cancer cells can selectively promote the assembly of gap junctions between cancer cells and astrocytes composed of connexin 43 (Cx43) by expressing protocadherin 7 (PCDH7). These channels allow the transfer of the second messenger, cGAMP, from cancer cells to adjacent astrocytes to activate STING, thereby triggering TBK1 and IRF3 and producing IFN-α and TNF-α. As paracrine signals, these factors can further activate the STAT1 and NF-κB pathways in metastatic brain cells, finally promoting brain metastasis and chemotherapeutic resistance in breast and lung cancer (65).
cGAMP plays a crucial role in cGAS-STING signaling by binding and activating STING, which has a greater potential for immunotherapy. However, the role played by cGAMP varies among different cells.
In myeloid cells, cGAMP agonists can activate their cGAS-STING signaling pathway, thereby promoting their killing of tumor cells. For example, transient accumulation of CD11b + Ly6C + F4/80 + monocytes/macrophages at the tumor site by STING stimulation can be observed after cGAMP injection, enhance the phagocytic activity of cells for antitumor purpose (55). However, in tumor cells, cGAMP can play two quite different roles. In early preneoplastic cells, the cGAS-STING pathway acts as a tumor suppressor. Tumor-derived cGAMP can initiate antitumor immunity via downstream canonical NF-κB signaling pathway (66, 67). It can also activate cGAS-STING signaling through antigen-presenting cell, same as tumor DNA, to trigger immune cell-mediated tumor clearance. For instance, activated cGAMP within tumor cells can be transferred to host cells to trigger STING and subsequently activate the anti-tumor response of NK cells (68). In late tumor stages, sustained cGAMP expression by tumor cells leads to activation of downstream non-canonical NF-κB signaling pathways, releasing other pro-inflammatory cytokines such as IDO, thus creating an immunosuppressive tumor microenvironment that promotes tumor progression (69).
Overall, the impact of cGAMP on cancer depends on the period and type of tumorigenesis and the activated cell type, among others. This requires additional exploration to determine its exact role in oncology.
Cellular autophagy is a lysosome-based intracellular degradation process that aims to maintain the organism’s essential cell survival and homeostasis under various cellular stress conditions such as hypoxia and infection. In cells, activating the autophagy-related 1/autophagy activating kinase 1 (ATG1/ULK1) complex is essential. They can phosphorylate or recruit their downstream proteins to promote autophagosome formation and thus induce typical autophagy (70, 71). However, it was recently found that after stimulation of cGAMP production by cGAS from dsDNA generated by various causes of damage, STING can translocate to the endoplasmic reticulum-Golgi intermediate compartment (ERGIC), which could serve as a membrane source for LC3 lipidation. cGAMP can induce LC3 lipidation by relying on WIPI2 and ATG, 5 but not ULK and Vps34-Beclin kinase complexes. Subsequently, LC3-positive membranes wrap dsDNA, bacteria, or viruses and form autophagosomes, suggesting that STING can also activate atypical autophagy through a mechanism independent of interferon induction (72).
Similar to STING, autophagy plays a bidirectional role in tumor progression. A terminal response called replicative crisis occurs during early tumorigenesis, which leads to delayed mitosis, telomere deprotection amplification and cell death. Telomeric DNA damage activates the cGAS-STING pathway. The researchers found that cells with cGAS or STING removed bypassed the crisis to proliferate and exhibited a reduction in LC3-II and accumulation of P62 in the cells (73). This evidence suggests that STING-mediated autophagy may be an additional barrier to early tumor progression in normal cells. However, the endoplasmic reticulum stress response and autophagy may also allow cancer cells to survive in a stressful environment, thereby promoting the development of advanced cancer (74).
As the activation of the cGAS/STING pathway in the tumor microenvironment can induce the effective cross-priming of tumor-specific antigens and promote the infiltration of effector T cells (45), STING agonists have been developed to simulate this activation in order to enhance the anticancer effect. Thus far, several types of stimulants have been found, mainly divided into three categories: Cyclic dinucleotides (CDNs) and their derivatives, DMXAA and its analogs, and small molecule agonists.
CDNs are divided into several CDN families, which are composed of cyclic di-GMP (c-di-GMP), cyclic di-AMP (c-di-AMP), and cyclic AMP-GMP (cGAMP) molecules. cGAMP includes 3’,3’-cGAMP, 2’,3’-cGAMP, 3’,5’-cGAMP and 2’,5’-cGAMP (75). Their antitumor regulatory effect was first found in c-di-GMP. c-di-GMP was initially regarded as an allosteric regulator of cellulose synthase in Gluconacetobacter xylinus (formerly known as Acetobacter xylinum) and is currently considered to be a ubiquitous second messenger of bacteria (76). The injection of c-di-GMP into a newborn glioma model can improve the survival rate of glioma-bearing mice in a STING-dependent manner, such as enhancing the signal of IFN-1, CXCL10, and the migration of T cells to the brain (77). c-di-GMP can also combat metastatic breast cancer via numerous methods. A mouse metastatic breast cancer model observed that a low dose of c-di-GMP provided a potent adjuvant effect with the LM-Mb vaccine by promoting the production of IL-12 derived from MDSCs and increasing the response of CD8+ T cells to the tumor-associated antigen Mage-b. Moreover, a large dose of c-di-GMP can activate caspase-3 and kill cancer cells directly (78). 2’,3’-cGAMP is the second messenger of STING signal transduction in mammalian cells. In murine colon 26 adenocarcinoma, cGAMP inhibited tumor growth mainly by activating STING and its downstream STING-IRF3 signaling. Analysis of host serum and tumor tissues showed that cGAMP significantly upregulated anti-tumor cytokines such as IFN-β and IFN-γ and activated DCs. Interestingly, cGAMP still had some tumor-suppressive activity in STING-/- mice, suggesting that cGAMP may stimulate other STING-independent pathways to suppress tumor when STING is not present growth (79). A study using a model of mouse cervical cancer demonstrated that the use of the mutant type of HPV 16 E7 protein (E7GRG) as a therapeutic vaccine candidate antigen-stimulated the cell-mediated and humoral immune response and inhibited tumor growth. The co-administration of 2’,3’-cGAMP, E7GRG and CpG-C adjuvant exerted a synergistic effect, establishing a shift towards the Th1 type immune response and decreasing tumor growth (80). In malignant B-cell tumors, 3’3’-cGAMP induces the prolonged presence of STING in the endoplasmic reticulum or Golgi apparatus to form protein complexes to activate apoptosis, without any significant association with activated IFN. This suggests that STING agonists have potential therapeutic use for the treatment of B-cell malignancies such as chronic lymphatic leukemia, multiple myeloma, etc., in addition to their immunomodulatory activity against cancer (81). It was also shown that 2’,5’-cGAMP showed stronger complex formation with both human and mouse STING compared to 3’,5’-cGAMP and was unaffected by the species specificity of the bite, just like DNA. Moreover, cGAS-dependent two-step synthetic 2’,5’-cGAMP can be used to develop specific inhibitors to treat autoimmune diseases related to cGAS/STING signaling (82).
Based on natural CDNs, synthetic CDNs have been developed with optimal performance. ADU-S100 (ML RRS2 CDA or MIW815) was the first STING agonist used in human clinical trials for cancer immunotherapy. Compared with other CDNs, ADU-S100 exhibits higher stability and lipophilicity. It has been indicated that in B16 melanoma, CT26 colon cancer, or 4T1 breast cancer models, the injection of ADU-S100 effectively triggers CD8+ T cell responses in a STING-dependent manner and leads to significant tumor regression, exerting long-lasting antitumor effects (83). Intraperitoneal injection of ADU-S100 into mouse models with peritoneal carcinomatosis of ovarian cancer (ID8) and MC38 colon carcinoma observed inhibition of peritoneal carcinomatosis and malignant ascites progression.ADU-100 effectively reduced abnormal tumor vasculature formation by activating STING-mediated type I IFN and upregulated the number of CD8+ T cells, enhancing the remaining tumor vasculature of pericyte coverage, thereby inhibiting the formation of malignant ascites in the peritoneal cavity. During peritoneal metastasis, type I IFN signaling also inhibited the recruitment of CD206+ M2-like macrophages (84). Notably, the intratumoral injection of ADU-S100 was shown to be well-tolerated in patients with advanced solid tumors and lymphomas, and no DLT was reported (85). IACS-8779 and IACS-8803 are highly efficient CDN STING agonists that introduce a group of reasonably selected and targeted modifications into the nucleobase and ribose sections of the 2’,3’ CDA structure. These two drugs manifest a superior ability to activate the STING pathway in vitro and a more prominent systemic antitumor response than the clinical benchmark agonist, ADU-S100, in a B16 murine model of melanoma (86). MK-1454 is a synthetic CDN analog developed by Merck to treat advanced/metastatic solid tumors or lymphomas. The preliminary results showed good activity and safety in a study of the drug as a monotherapy and combined with pembrolizumab entering a phase I clinical trial. PRs observed in head and neck squamous cell carcinoma, triple-negative breast cancer and undifferentiated thyroid cancer (87).
Flavone 8-acetic acid (FAA) was initially synthesized as a non-steroidal anti-inflammatory drug (88). It induces extensive hemorrhagic necrosis associated with inhibiting tumor blood flow (89) and has exhibited significant antitumor activity in preclinical studies (90). However, the results obtained for FAA in clinical trials were not positive (91). Thus, a more active FAA analog, 5,6-dimethylxanthenone-4-acetic acid (DMXAA), was developed. DMXAA, also known as ASA404 and Vadimezan, can fight tumors directly and indirectly. First of all, DMXAA can induce apoptosis in tumor endothelial cells, increase specificity, and irreversibly directly destroy the established tumor vascular system and completely block the tumor blood flow. Secondly, DMXAA can indirectly activate the innate immune system and can induce the production of inflammatory cytokines. Clinical trials have been better results in selected patients with melanoma and squamous cell carcinoma (88).
At present, amidobenzimidazole (ABZI) represents a breakthrough for STING agonists in cancer treatment. By replacing its N1-hydroxyphenethyl moiety with a 4-carbon butane linker, researchers have derived a compound named diABZI. It has been demonstrated that the binding affinity of this new compound is significantly enhanced, and in vivo, it can lead to the adaptive response of CD8+ T cells. The intravenous administration of diABZI STING agonist to mice with a normal immune function and with established homologous colon tumors has been shown to exert potent antitumor effects, diminishing the tumor completely and persistently. The diABZI compound is the first effective non-nucleotide STING agonist used globally and has immense potential to improve the immunotherapy of human cancers (92).
Classical therapies, such as radiotherapy and chemotherapy, are the main methods for treating human cancers (93). Radiation-induced DNA damage leads to micronuclei formation or cell death and can activate immune cells’ cGAS/STING pathway (54). For example, exposure to gamma-rays can directly induce the production of type III IFN (mainly IFNL1) in human cancer cell lines through the cGAS/STING/TBK1/IRF1 signaling pathway (94). However, radiation may also exert an adverse inhibitory effect on the immune system. It has been demonstrated that radiation can activate the immunosuppressive TGF-β cytokine (95) and can promote the accumulation of Tregs (96) and pro-tumorigenic M2 macrophages (97). cGAS is activated by radiation in a dose-dependent manner. Evidence indicates that when the radiation dose exceeds 12-18 Gy, DNA exonuclease TREX1 helps degrade the DNA accumulated in the cytoplasm to attenuate its immunogenicity and downregulate the cGAS pathway. Conversely, radiation repeatedly below the threshold dose for the induction of TREX1 stimulates IFN-β in cancer cells (98). Chemotherapy can induce DNA damage and inhibit DNA repair simultaneously. The damaged DNA activates the cGAS/STING axis to enhance DC-mediated antigen presentation and T cell response (45). For example, one of the mechanisms of cisplatin therapy is to initiate the cGAS/STING pathway via the upregulation of the protein levels of cGAS and STING (99). In addition, the anthracycline antibiotics, adriamycin and daunorubicin, which can inhibit topoisomerase II, induce an IFN response and trigger the cGAS/STING pathway in an ATM kinase-dependent manner (100). Although radiotherapy and chemotherapy do not directly target the cGAS pathway, these treatments can activate the cGAS/STING axis in a ‘roundabout’ manner and can enhance the antitumor immune response. The co-administration of STING agonists with radiotherapy or chemical drugs can enhance the antitumor effect and reduce the side-effects caused by classical therapies. It has been demonstrated that the inhalation of phosphatidylserine-coated liposomes loaded with cGAMP (NP-cGAMP), a STING agonist, in mouse models of lung metastases can rapidly distribute NP-cGAMP to the lungs and stimulate STING signal transduction in antigen-presenting cells, promoting the production of type I IFN, and the synergistic effect of NP-cGAMP and graded IR (8 Gy x 3) can suppress tumor metastasis at the site of radiotherapy and lung metastasis without radiotherapy (101). 5-Fluorouracil (5-FU) is an effective clinical anti-neoplastic drug that exerts its effects by interfering with DNA synthesis; however, it also has noticeable side effects, such as nausea, mucosal necrosis, and hematochezia. When used in combination with cGAMP, a stimulant of innate immune antitumor agents, the antitumor effect of 5-FU is enhanced, while the side effects are reduced (79).
T cells are the primary therapeutic focus of endogenous antitumor immunity due to their ability to selectively recognize peptides from proteins in all cell compartments, directly recognize and kill cells expressing specific antigens, and coordinate various immune responses with the help of CD4+ helper T cells. Therefore, specific targeting agonists of co-stimulatory receptors or antagonists of inhibitory signals, such as targeting cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and programmed cell death protein 1 (PD1), can be leading to the enhancement of the antigen-specific T cell response (102–104). The activation of STING increases the critical Th1-recruiting cytokines, CXCL9 and CXCL10, and IFN-1. Type I IFN can promote the activation and maturation of DCs, improve the antigen presentation of CD4+ T lymphocytes and enhance antigen cross-presentation to CD8+ T lymphocytes, hence, T cell priming. Therefore, STING agonists seem to be ideal partners for immune checkpoint inhibitors (105). Compared with no treatment or treatment with immune-checkpoint inhibitors alone, the co-administration of STING agonists with CTLA4 and PD1 antibodies in a preclinical HPV+ oral tumor model displayed the most significant survival advantage (106). Moreover, in B16F10- and BRAF-mutated murine models, the group treated with cGAMP encapsulated into lipid nanoparticles conjugated with mannose (LP-cGAMP) and anti-programmed death-ligand 1 (PD-L1) exhibited a more long-lasting inhibition of tumor growth and achieved a more prolonged survival than others (107).
Due to central and peripheral tolerance, the immunogenicity of tumor-associated antigens is not potent enough to trigger a firm tumor antigen-specific CD8+ T cell response, which is essential for effective antitumor immunity (108). Therefore, appropriate adjuvants play an indispensable role in overcoming tolerance and enhancing tumor-specific immune responses. STINGVAX is the first designed STING-based tumor vaccine containing granulocyte-macrophage colony-stimulating factor (GM-CSF) and CDN ligands. In multiple syngeneic mouse models, the in vivo antitumor responses for STINGVAX were more prominent than those for the GM-vaccine. STINGVAX-treated mice exhibited increased tumor-infiltrating lymphocytes (TILs) and an upregulated PD-L1 expression, indicating that STINGVAX is suitable for co-administration with anti-PD-1 therapy (109). In a previously mentioned study using a metastatic breast cancer mouse model, the researchers used a Listeria monocytogenes (LM)-based vaccine with the expression of tumor-associated antigen Mage-b (LM-Mb) as the vaccine and c-di-GMP as the stimulating ligand. This model observed the enhanced presentation of tumor-associated antigens and increased tumor-associated antigen-specific T cell activation in vivo. The combination of c-di-GMP and LM-Mb also significantly reduced the number of MDSCs in the blood. The reduction in the MDSC population helps to reduce immunosuppression in the tumor microenvironment, reducing the growth of tumors and metastases. As MDSCs and immunosuppression exist in almost all cancers, using c-di-GMP as an adjuvant against other cancers is promising (78).
CAR T cell therapy is one of the most promising methods for anticancer treatment. T cells are redirected against a tumor following the enforced expression of CARs. CAR, as the core component of CAR T cells, enables T cells to recognize tumor antigens in an HLA-independent manner and identify a broader range of target antigens than natural T cell surface receptor (TCR) (110–112). In an orthotopic murine breast tumor model, a previous study proved that Th/Tc17 CAR T cells persist durably in the tumor microenvironment and play a prominent role in tumor growth control. However, the number of Th/Tc17 CAR T cells is only moderately increased in the tumor microenvironment, improving early tumor suppression but not leading to a long-term antitumor response. The study also compared the T cell population between two CAR T cell treatment groups combined with or without DMAXX. The results revealed that the combined treatment group expressed higher CXCL9 and CXCL10, which could recruit CXCR3-expressing Th/Tc17 T cells, leading to a more significant number of CAR T cells (113).
OV plays its antitumor role mainly via both direct and indirect mechanisms. On the one hand, it replicates in and lyses tumor cells selectively. On the other hand, OV acts as an ‘in situ’ vaccine and directs the host immune response to target tumors. Upon the lysis of tumor cells, viral offspring are released to infect neighboring cancer cells and finally induce the systemic antitumor response (114). In 2015, the US Food and Drug Administration (FDA) approved herpesvirus talimogene laherparepvec (T-VEC) for advanced melanoma, rendering it the first OV to be approved in the United States (115). Patients with metastatic melanoma present a high level of Tregs, Ts cells, and MDSCs within established tumors. Following the injection of OncovexGM-CSF, an oncolytic herpes simplex virus engineered to express GM-CSF, the researchers observed a decreased level of these suppressor cell populations and the production of local and systemic MART-1-specific CD8+ effector cells. which suggests that OncovexGM-CSF can induce potent local and systemic T cell immunity (116). The enhanced replication and intercellular spread of cancer cells due to STING deficiency make the effectiveness of oncolytic therapy in certain tumors can be significantly increased. For example, STING-defective melanoma cells are highly sensitive to DNA virus-mediated oncolytic effect. Therefore, it is now widely considered that STING/cGAS may be a more effective biomarker for prognosis (117).
Dysfunctional nucleases and abnormal mutations in STING can lead to abnormal activation of the cGAS-STING pathway, resulting in increased expression of IFN. Therefore STING inhibitors may be developed for use in a variety of interferon diseases. The existing inhibitors can be broadly classified into two categories, namely competitive antagonists that occupy CDN binding sites and inhibitors that covalently modify Csy88 and Csy91 to inhibit palmitoylation (118).
Based on biophysical and X-ray crystallography data, the STING C-terminal domain exists as a symmetric dimer with the ligand-binding site located at the interface between the two monomers, which is the binding site for the agonist CDN. Before binding to cGAMP, STING apolipoprotein adopts an “open” conformation, compared to a “closed” conformation after binding. It suggests that the “open” conformation may lead to inactivation. While the “closed” conformation may lead to protein activation. Tetrahydroisoquinolone can bind to the cGAMP binding site in the inactive “open” conformation. By taking advantage of the natural symmetry of the STING protein and utilizing a 2:1 binding stoichiometry, these compounds can fully occupy the binding pocket while mitigating the undesirable physicochemical properties associated with larger ligands. After incubation of THP1 cells with Tetrahydroisoquinolone, it was observed that this compound did not stimulate interferoβproduction and moderately inhibited cGAMP-induced interferonβproduction (119).
Astin C is a cyclic peptide isolated from the medicinal plant Aster tartaric that specifically targets STING and competes with CDN to bind the C-terminal activation pocket of STING. The exact mechanism of STING inhibition by astin C is still not fully determined, but it is known that this compound selectively inhibits the recruitment and activation of downstream IRF3 and does not affect the phosphorylation of STING and TBK1. It was demonstrated that astin C significantly attenuates the auto-inflammatory response in Trex1-/- bone marrow-derived macrophages (BMDM) and disease models while having a low cytotoxic side effect in cellular and animal models. This suggests that astin C may be used in STING-mediated cancer and autoimmune diseases (120).
As described above, Csy88 and Csy91 in the cGAS-STING pathway induce STING palmitoylation. Allowing their subsequent assembly into multimeric complexes at the Golgi as well as recruitment of downstream signaling factors.
Protein mass spectrometry studies have shown that the 3-acylamino indole derivative Indole ureas (H-151) can form covalent bonds with Cys91. Intraperitoneal H-151 administration was performed in a mouse model to which a specific STING agonist was applied. The investigators observed that H-151 achieved effective systemic levels and showed a short half-life significantly reduced serum levels of type I IFN and IL-6 in mice. This suggests that H-151 is a potent small molecule antagonist of STING that can effectively inhibit type I IFN response, TBK1 phosphorylation and hsSTING palmitoylation (121).
Nitro fatty acids (NO2-FAs) are formed in response to HSV-2 infection. It was shown that NO2-FAs could directly modify STING by nitroalkylation at Cys88 and Csy91 and two adjacent cysteines of the N-terminal histidine (His16).Pretreatment with NO2 -FA substantially reduced the induction of type I interferon in response to HSV-2 in both THP-1 cells. Treatment with NO2-FA after HSV-2 infection also reduced the release of the IFN-induced cytokine CXCL10. In addition, NO2-FA treatment with STING-associated vasculopathy with onset in infancy (SAVI) also resulted in a reduced STING-dependent type I interferon response in immortalized fibroblasts from patients. Therefore, it is suggested that Nitro fatty acids can target STING signaling and reduce type I interferon release in mice and human cells and may be used in the future to treat STING-dependent inflammatory diseases (122).
By detecting micronuclei or cytoplasmic chromatin fragments induced by DNA damage, the cGAS/STING pathway can mediate the interplay between cytotoxic effects and immune stimulation, exerting a dichotomous effect on tumor tissue. However, the underlying mechanisms responsible for this phenomenon are still poorly understood and require further research. Therefore, further research is required to obtain a more in-depth knowledge of the STING-related tumor microenvironment and biological mechanisms to reveal the potential mechanisms of the STING-mediated immune response, such as the effects of the canonical and non-canonical NF-κB pathway on tumors. Furthermore, exploring the molecular details of the cGAS/STING pathway may enhance the current understanding of the antitumor mechanism of innate immunity and may provide a theoretical basis for future drug design for the treatment of tumors. Thus, this pathway may play a more critical role in basic immunology, tumor biology, and clinical tumor therapy.
The co-administration of classical therapies and STING agonists has demonstrated an excellent synergistic antitumor effect in several preclinical tumor models. These agonists can reduce the side-effects of drug use, overcome the tolerance to tumor treatments and achieve an optimal therapeutic effect, thus rendering the development of a new variety of antitumor treatments promising. Therefore, the comparison of the effects of various combination therapies and the exploration of their mechanisms is of utmost significance. Likewise, the current exploration of STING inhibitors provides valuable clues for further clinical drug development in the future.
YG performed the analyses and wrote the manuscript; XL, SH contributed to the conception of the paper; QL, XM contributed to analysis; PR, WW, WL helped perform the analysis with constructive discussions. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 81972837, 82071986, 81771827, 81971721).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Yum S, Li M, Chen ZJ. Old Dogs, New Trick: Classic Cancer Therapies Activate cGAS. Cell Res (2020) 30(8):639–48. doi: 10.1038/s41422-020-0346-1
2. Makkouk A, Weiner GJ. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res (2015) 75(1):5–10. doi: 10.1158/0008-5472.CAN-14-2538
3. Chen Q, Sun L, Chen ZJ. Regulation and Function of the cGAS-STING Pathway of Cytosolic DNA Sensing. Nat Immunol (2016) 17(10):1142–9. doi: 10.1038/ni.3558
4. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (New York NY) (2013) 339(6121):786–91. doi: 10.1126/science.1232458
5. Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, et al. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex With DNA and Undergoes Switch-Like Conformational Changes in the Activation Loop. Cell Rep (2014) 6(3):421–30. doi: 10.1016/j.celrep.2014.01.003
6. Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, et al. Cyclic [G(2',5')pA(3',5')p] is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell (2013) 153(5):1094–107. doi: 10.1016/j.cell.2013.04.046
7. Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, et al. Human cGAS Catalytic Domain has an Additional DNA-Binding Interface That Enhances Enzymatic Activity and Liquid-Phase Condensation. Proc Natl Acad Sci USA (2019) 116(24):11946–55. doi: 10.1073/pnas.1905013116
8. Yu X, Zhang L, Shen J, Zhai Y, Jiang Q, Yi M, et al. The STING Phase-Separator Suppresses Innate Immune Signalling. Nat Cell Biol (2021) 23(4):330–40. doi: 10.1038/s41556-021-00659-0
9. Ishikawa H, Barber GN. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
10. Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca Sensor STIM1 Regulates the Type I Interferon Response by Retaining the Signaling Adaptor STING at the Endoplasmic Reticulum. Nat Immunol (2019) 20(2):152–62. doi: 10.1038/s41590-018-0287-8
11. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING Requires Palmitoylation at the Golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
12. Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered Recruitment of Chromatin Modifying and General Transcription Factors to the IFN-Beta Promoter. Cell (2000) 103(4):667–78. doi: 10.1016/s0092-8674(00)00169-0
13. Schneider WM, Chevillotte MD, Rice CM. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
14. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature (2015) 520(7548):553–7. doi: 10.1038/nature14156
15. Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue Virus NS2B Protein Targets cGAS for Degradation and Prevents Mitochondrial DNA Sensing During Infection. Nat Microbiol (2017) 2:17037. doi: 10.1038/nmicrobiol.2017.37
16. Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-Derived IFN Triggers Chronic Pathway Agonism and Sensitivity to ADAR Loss. Nat Med (2019) 25(1):95–102. doi: 10.1038/s41591-018-0302-5
17. d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature (2003) 426(6963):194–8. doi: 10.1038/nature02118
18. Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, et al. Molecular Mechanisms of Micronucleus, Nucleoplasmic Bridge and Nuclear Bud Formation in Mammalian and Human Cells. Mutagenesis (2011) 26(1):125–32. doi: 10.1093/mutage/geq052
19. Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell (2013) 154(1):47–60. doi: 10.1016/j.cell.2013.06.007
20. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic Progression Following DNA Damage Enables Pattern Recognition Within Micronuclei. Nature (2017) 548(7668):466–70. doi: 10.1038/nature23470
21. Li T, Huang T, Du M, Chen X, Du F, Ren J, et al. Phosphorylation and Chromatin Tethering Prevent cGAS Activation During Mitosis. Science (New York NY) (2021) 371(6535):eabc5386. doi: 10.1126/science.abc5386
22. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
23. Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and non-Oncogene Addiction. Cell (2009) 136(5):823–37. doi: 10.1016/j.cell.2009.02.024
24. Ho Samantha SW, Zhang Wendy YL, Tan Nikki Yi J, Khatoo M, Suter Manuel A, Tripathi S, et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity (2016) 44(5):1177–89. doi: 10.1016/j.immuni.2016.04.010
25. Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature (2017) 548(7668):461–5. doi: 10.1038/nature23449
26. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated With LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov (2019) 9(1):34–45. doi: 10.1158/2159-8290.CD-18-0689
27. Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, et al. Mitochondrial Inner Membrane Permeabilisation Enables mtDNA Release During Apoptosis. EMBO J (2018) 37(17):e99238. doi: 10.15252/embj.201899238
28. Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells Without Mitochondrial DNA. Cell Metab (2015) 21(1):81–94. doi: 10.1016/j.cmet.2014.12.003
29. Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov (2020) 10(1):26–39. doi: 10.1158/2159-8290.CD-19-0761
30. Sharma A, Johnson A. Exosome DNA: Critical Regulator of Tumor Immunity and a Diagnostic Biomarker. J Cell Physiol (2020) 235(3):1921–32. doi: 10.1002/jcp.29153
31. Schlacher K, Wu H, Jasin M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-Brca1/2. Cancer Cell (2012) 22(1):106–16. doi: 10.1016/j.ccr.2012.05.015
32. Parkes EE, Walker SM, Taggart LE, McCabe N, Knight LA, Wilkinson R, et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst (2017) 109(1):djw199. doi: 10.1093/jnci/djw199
33. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veize J, McMichael JF, et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
34. Wu S-Y, Xiao Y, Wei J-L, Xu X-E, Jin X, Hu X, et al. MYC Suppresses STING-Dependent Innate Immunity by Transcriptionally Upregulating DNMT1 in Triple-Negative Breast Cancer. J Immunother Cancer (2021) 9(7):e002528. doi: 10.1136/jitc-2021-002528
35. Meng F, Yu Z, Zhang D, Chen S, Guan H, Zhou R, et al. Induced Phase Separation of Mutant NF2 Imprisons the cGAS-STING Machinery to Abrogate Antitumor Immunity. Mol Cell (2021) 81(20):4147–64.e7. doi: 10.1016/j.molcel.2021.07.040
36. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature (2018) 563(7729):131–6. doi: 10.1038/s41586-018-0629-6
37. Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, et al. STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res (2019) 79(7):1465–79. doi: 10.1158/0008-5472.CAN-18-1972
38. Leon KE, Buj R, Lesko E, Dahl ES, Chen C-W, Tangudu NK, et al. DOT1L Modulates the Senescence-Associated Secretory Phenotype Through Epigenetic Regulation of IL1A. J Cell Biol (2021) 220(8):e202008101. doi: 10.1083/jcb.202008101
39. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
40. Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, et al. Lysosome-Mediated Processing of Chromatin in Senescence. J Cell Biol (2013) 202(1):129–43. doi: 10.1083/jcb.201212110
41. Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, et al. Innate Immune Sensing of Cytosolic Chromatin Fragments Through cGAS Promotes Senescence. Nat Cell Biol (2017) 19(9):1061–70. doi: 10.1038/ncb3586
42. Yang B, Dan X, Hou Y, Lee J-H, Wechter N, Krishnamurthy S, et al. NAD(+) Supplementation Prevents STING-Induced Senescence in Ataxia Telangiectasia by Improving Mitophagy. Aging Cell (2021) 20(4):e13329–e. doi: 10.1111/acel.13329
43. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol (2008) 6(12):2853–68. doi: 10.1371/journal.pbio.0060301
44. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling Strength Determines Proapoptotic Functions of STING. Nat Commun (2017) 8(1):427. doi: 10.1038/s41467-017-00573-w
45. Zheng J, Mo J, Zhu T, Zhuo W, Yi Y, Hu S, et al. Comprehensive Elaboration of the cGAS-STING Signaling Axis in Cancer Development and Immunotherapy. Mol Cancer (2020) 19(1):133. doi: 10.1186/s12943-020-01250-1
46. Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, et al. Host Type I IFN Signals Are Required for Antitumor CD8+ T Cell Responses Through CD8{alpha}+ Dendritic Cells. J Exp Med (2011) 208(10):2005–16. doi: 10.1084/jem.20101159
47. Diamond JM, Vanpouille-Box C, Spada S, Rudqvist N-P, Chapman JR, Ueberheide BM, et al. Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA From Irradiated Cancer Cells to DCs. Cancer Immunol Res (2018) 6(8):910–20. doi: 10.1158/2326-6066.CIR-17-0581
48. Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-Alpha2a Induces IP-10/CXCL10 and MIG/CXCL9 Production in Monocyte-Derived Dendritic Cells and Enhances Their Capacity to Attract and Stimulate CD8+ Effector T Cells. J Leukoc Biol (2002) 71(4):669–76. doi: 10.1189/jlb.71.4.669
49. An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J, et al. An Analysis of the Expression and Association With Immune Cell Infiltration of the cGAS/STING Pathway in Pan-Cancer. Mol Ther Nucleic Acids (2019) 14:80–9. doi: 10.1016/j.omtn.2018.11.003
50. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
51. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-Mediated DNA Sensing Maintains CD8 T Cell Stemness and Promotes Antitumor T Cell Therapy. Sci Transl Med (2020) 12(549):eaay9013. doi: 10.1126/scitranslmed.aay9013
52. Ritchie C, Cordova AF, Hess GT, Bassik MC, Li L. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell (2019) 75(2):372–81.e5. doi: 10.1016/j.molcel.2019.05.006
53. Lopez JA, Jenkins MR, Rudd-Schmidt JA, Brennan AJ, Danne JC, Mannering SI, et al. Rapid and Unidirectional Perforin Pore Delivery at the Cytotoxic Immune Synapse. J Immunol (2013) 191(5):2328–34. doi: 10.4049/jimmunol.1301205
54. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity (2014) 41(5):843–52. doi: 10.1016/j.immuni.2014.10.019
55. Ohkuri T, Kosaka A, Ishibashi K, Kumai T, Hirata Y, Ohara K, et al. Intratumoral Administration of cGAMP Transiently Accumulates Potent Macrophages for Anti-Tumor Immunity at a Mouse Tumor Site. Cancer Immunol Immunother (2017) 66(6):705–16. doi: 10.1007/s00262-017-1975-1
56. Veenhuis RT, Freeman ZT, Korleski J, Cohen LK, Massaccesi G, Tomasi A, et al. HIV-Antibody Complexes Enhance Production of Type I Interferon by Plasmacytoid Dendritic Cells. J Clin Invest (2017) 127(12):4352–64. doi: 10.1172/JCI95375
57. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc Natl Acad Sci USA (2015) 112(50):15408–13. doi: 10.1073/pnas.1512832112
58. Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, et al. STING Activation Reprograms Tumor Vasculatures and Synergizes With VEGFR2 Blockade. J Clin Invest (2019) 129(10):4350–64. doi: 10.1172/JCI125413
59. Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, et al. Chromosomal Instability Drives Metastasis Through a Cytosolic DNA Response. Nature (2018) 553(7689):467–72. doi: 10.1038/nature25432
60. Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity (2014) 41(5):830–42. doi: 10.1016/j.immuni.2014.10.017
61. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol (2021) 21(9):548–9. doi: 10.1038/s41577-021-00524-z
62. Li J, Duran MA, Dhanota N, Chatila WK, Bettigole SE, Kwon J, et al. Metastasis and Immune Evasion From Extracellular cGAMP Hydrolysis. Cancer Discov (2021) 11(5):1212–27. doi: 10.1158/2159-8290.CD-20-0387
63. Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-Driven Carcinogenesis Is Mediated Through STING. Nat Commun (2014) 5:5166. doi: 10.1038/ncomms6166
64. Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res (2016) 76(8):2076–81. doi: 10.1158/0008-5472.CAN-15-1456
65. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-Astrocyte Gap Junctions Promote Brain Metastasis by cGAMP Transfer. Nature (2016) 533(7604):493–8. doi: 10.1038/nature18268
66. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKepsilon and TBK1 Are Essential Components of the IRF3 Signaling Pathway. Nat Immunol (2003) 4(5):491–6. doi: 10.1038/ni921
67. Abe T, Barber GN. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-κb Activation Through TBK1. J Virol (2014) 88(10):5328–41. doi: 10.1128/JVI.00037-14
68. Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-Tumor Cells to Activate the NK Cell Response. Immunity (2018) 49(4):754–63.e4. doi: 10.1016/j.immuni.2018.09.016
69. Tas SW, Vervoordeldonk MJ, Hajji N, Schuitemaker JHN, van der Sluijs KF, May MJ, et al. Noncanonical NF-kappaB Signaling in Dendritic Cells Is Required for Indoleamine 2,3-Dioxygenase (IDO) Induction and Immune Regulation. Blood (2007) 110(5):1540–9. doi: 10.1182/blood-2006-11-056010
70. Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a Novel Protein Kinase Required for the Autophagic Process in Saccharomyces Cerevisiae. Gene (1997) 192(2):245–50. doi: 10.1016/S0378-1119(97)00084-X
71. Lazarus MB, Novotny CJ, Shokat KM. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex With Potent Inhibitors. ACS Chem Biol (2015) 10(1):257–61. doi: 10.1021/cb500835z
72. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy Induction via STING Trafficking Is a Primordial Function of the cGAS Pathway. Nature (2019) 567(7747):262–6. doi: 10.1038/s41586-019-1006-9
73. Nassour J, Radford R, Correia A, Fusté JM, Schoell B, Jauch A, et al. Autophagic Cell Death Restricts Chromosomal Instability During Replicative Crisis. Nature (2019) 565(7741):659–63. doi: 10.1038/s41586-019-0885-0
74. Song M, Sandoval TA, Chae C-S, Chopra S, Tan C, Rutkowski MR, et al. Ire1α-XBP1 Controls T Cell Function in Ovarian Cancer by Regulating Mitochondrial Activity. Nature (2018) 562(7727):423–8. doi: 10.1038/s41586-018-0597-x
75. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. cGAS-STING Pathway in Cancer Biotherapy. Mol Cancer (2020) 19(1):136. doi: 10.1186/s12943-020-01247-w
76. Ross P, Weinhouse H, Aloni Y, Michaeli D, Weinberger-Ohana P, Mayer R, et al. Regulation of Cellulose Synthesis in Acetobacter Xylinum by Cyclic Diguanylic Acid. Nature (1987) 325(6101):279–81. doi: 10.1038/325279a0
77. Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, et al. STING Contributes to Antiglioma Immunity via Triggering Type I IFN Signals in the Tumor Microenvironment. Cancer Immunol Res (2014) 2(12):1199–208. doi: 10.1158/2326-6066.CIR-14-0099
78. Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, et al. STING Ligand C-Di-GMP Improves Cancer Vaccination Against Metastatic Breast Cancer. Cancer Immunol Res (2014) 2(9):901–10. doi: 10.1158/2326-6066.CIR-13-0123
79. Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep (2016) 6:19049. doi: 10.1038/srep19049
80. Dorostkar F, Arashkia A, Roohvand F, Shoja Z, Navari M, Mashhadi Abolghasem Shirazi M, et al. Co-Administration of 2'3'-cGAMP STING Activator and CpG-C Adjuvants With a Mutated Form of HPV 16 E7 Protein Leads to Tumor Growth Inhibition in the Mouse Model. Infect Agent Cancer (2021) 16(1):7. doi: 10.1186/s13027-021-00346-7
81. Tang C-HA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res (2016) 76(8):2137–52. doi: 10.1158/0008-5472.CAN-15-1885
82. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS Produces a 2'-5'-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature (2013) 498(7454):380–4. doi: 10.1038/nature12306
83. Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep (2015) 11(7):1018–30. doi: 10.1016/j.celrep.2015.04.031
84. Lee SJ, Yang H, Kim WR, Lee YS, Lee WS, Kong SJ, et al. STING Activation Normalizes the Intraperitoneal Vascular-Immune Microenvironment and Suppresses Peritoneal Carcinomatosis of Colon Cancer. J Immunother Cancer (2021) 9(6):e002195. doi: 10.1136/jitc-2020-002195
85. Berry S, Giraldo N, Nguyen P, Green B, Xu H, Ogurtsova A, et al. Correction to: 33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018). J Immunother Cancer (2019) 7(1):46. doi: 10.1186/s40425-019-0519-y
86. Ager CR, Zhang H, Wei Z, Jones P, Curran MA, Di Francesco ME. Discovery of IACS-8803 and IACS-8779, Potent Agonists of Stimulator of Interferon Genes (STING) With Robust Systemic Antitumor Efficacy. Bioorg Med Chem Lett (2019) 29(20):126640. doi: 10.1016/j.bmcl.2019.126640
87. Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. LBA15 - Preliminary Results of the First-in-Human (FIH) Study of MK-1454, an Agonist of Stimulator of Interferon Genes (STING), as Monotherapy or in Combination With Pembrolizumab (Pembro) in Patients With Advanced Solid Tumors or Lymphomas. Ann Oncol (2018) 29:viii712. doi: 10.1093/annonc/mdy424.015
88. Daei Farshchi Adli A, Jahanban-Esfahlan R, Seidi K, Samandari-Rad S, Zarghami N. An Overview on Vadimezan (DMXAA): The Vascular Disrupting Agent. Chem Biol Drug Des (2018) 91(5):996–1006. doi: 10.1111/cbdd.13166
89. Pham MH, Dauzonne D, Chabot GG. Not Flavone-8-Acetic Acid (FAA) But its Murine Metabolite 6-OH-FAA Exhibits Remarkable Antivascular Activities. Vitro Anticancer Drugs (2016) 27(5):398–406. doi: 10.1097/CAD.0000000000000341
90. Zaharko DS, Grieshaber CK, Plowman J, Cradock JC. Therapeutic and Pharmacokinetic Relationships of Flavone Acetic Acid: An Agent With Activity Against Solid Tumors. Cancer Treat Rep (1986) 70(12):1415–21. doi: 10.1016/0304-3835(86)90075-3
91. Kerr DJ, Kaye SB. Flavone Acetic Acid–Preclinical and Clinical Activity. Eur J Cancer Clin Oncol (1989) 25(9):1271–2. doi: 10.1016/0277-5379(89)90072-2
92. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang S-Y, et al. Design of Amidobenzimidazole STING Receptor Agonists With Systemic Activity. Nature (2018) 564(7736):439–43. doi: 10.1038/s41586-018-0705-y
93. Lomax ME, Folkes LK, O'Neill P. Biological Consequences of Radiation-Induced DNA Damage: Relevance to Radiotherapy. Clin Oncol (R Coll Radiol) (2013) 25(10):578–85. doi: 10.1016/j.clon.2013.06.007
94. Chen J, Markelc B, Kaeppler J, Ogundipe VML, Cao Y, McKenna WG, et al. STING-Dependent Interferon-λ1 Induction in HT29 Cells, a Human Colorectal Cancer Cell Line, After Gamma-Radiation. Int J Radiat Oncol Biol Phys (2018) 101(1):97–106. doi: 10.1016/j.ijrobp.2018.01.091
95. Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA. Transforming Growth Factor-Beta Activation in Irradiated Murine Mammary Gland. J Clin Invest (1994) 93(2):892–9. doi: 10.1172/JCI117045
96. Schaue D, Comin-Anduix B, Ribas A, Zhang L, Goodglick L, Sayre JW, et al. T-Cell Responses to Survivin in Cancer Patients Undergoing Radiation Therapy. Clin Cancer Res an Off J Am Assoc Cancer Res (2008) 14(15):4883–90. doi: 10.1158/1078-0432.CCR-07-4462
97. Chiang C-S, Fu SY, Wang S-C, Yu C-F, Chen F-H, Lin C-M, et al. Irradiation Promotes an M2 Macrophage Phenotype in Tumor Hypoxia. Front Oncol (2012) 2:89. doi: 10.3389/fonc.2012.00089
98. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat Commun (2017) 8:15618. doi: 10.1038/ncomms15618
99. Grabosch S, Bulatovic M, Zeng F, Ma T, Zhang L, Ross M, et al. Cisplatin-Induced Immune Modulation in Ovarian Cancer Mouse Models With Distinct Inflammation Profiles. Oncogene (2019) 38(13):2380–93. doi: 10.1038/s41388-018-0581-9
100. Luthra P, Aguirre S, Yen BC, Pietzsch CA, Sanchez-Aparicio MT, Tigabu B, et al. Topoisomerase II Inhibitors Induce DNA Damage-Dependent Interferon Responses Circumventing Ebola Virus Immune Evasion. mBio (2017) 8(2):e00368–17. doi: 10.1128/mBio.00368-17
101. Liu Y, Crowe WN, Wang L, Lu Y, Petty WJ, Habib AA, et al. An Inhalable Nanoparticulate STING Agonist Synergizes With Radiotherapy to Confer Long-Term Control of Lung Metastases. Nat Commun (2019) 10(1):5108. doi: 10.1038/s41467-019-13094-5
102. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on Both Effector and Regulatory T Cell Compartments Contributes to the Antitumor Activity of Anti-CTLA-4 Antibodies. J Exp Med (2009) 206(8):1717–25. doi: 10.1084/jem.20082492
103. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847
104. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
105. Motedayen Aval L, Pease JE, Sharma R, Pinato DJ. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J Clin Med (2020) 9(10):3323. doi: 10.3390/jcm9103323
106. Dorta-Estremera S, Hegde VL, Slay RB, Sun R, Yanamandra AV, Nicholas C, et al. Targeting Interferon Signaling and CTLA-4 Enhance the Therapeutic Efficacy of Anti-PD-1 Immunotherapy in Preclinical Model of HPV Oral Cancer. J Immunother Cancer (2019) 7(1):252. doi: 10.1186/s40425-019-0728-4
107. Li K, Ye Y, Liu L, Sha Q, Wang X, Jiao T, et al. The Lipid Platform Increases the Activity of STING Agonists to Synergize Checkpoint Blockade Therapy Against Melanoma. Biomater Sci (2021) 9(3):765–73. doi: 10.1039/d0bm00870b
108. Shae D, Baljon JJ, Wehbe M, Christov PP, Becker KW, Kumar A, et al. Co-Delivery of Peptide Neoantigens and Stimulator of Interferon Genes Agonists Enhances Response to Cancer Vaccines. ACS Nano (2020) 14(8):9904–16. doi: 10.1021/acsnano.0c02765
109. Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to PD-1 Blockade. Sci Transl Med (2015) 7(283):283ra52. doi: 10.1126/scitranslmed.aaa4306
110. Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, et al. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3 Zeta Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and Can be Integrated Into One Combined CD28/CD3 Zeta Signaling Receptor Molecule. J Immunol (2001) 167(11):6123–31. doi: 10.4049/jimmunol.167.11.6123
111. Patel SD, Moskalenko M, Smith D, Maske B, Finer MH, McArthur JG. Impact of Chimeric Immune Receptor Extracellular Protein Domains on T Cell Function. Gene Ther (1999) 6(3):412–9. doi: 10.1038/sj.gt.3300831
112. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int J Biol Sci (2019) 15(12):2548–60. doi: 10.7150/ijbs.34213
113. Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING Agonist Promotes CAR T Cell Trafficking and Persistence in Breast Cancer. J Exp Med (2021) 218(2):e20200844. doi: 10.1084/jem.20200844
114. Xu W, Atkinson VG, Menzies AM. Intratumoural Immunotherapies in Oncology. Eur J Cancer (2020) 127:1–11. doi: 10.1016/j.ejca.2019.12.007
115. Broman KK, Zager JS. An Evaluation of Talimogene Laherparepvec for the Treatment of Melanoma. Expert Opin Biol Ther (2020) 20(1):9–14. doi: 10.1080/14712598.2020.1689951
116. Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and Distant Immunity Induced by Intralesional Vaccination With an Oncolytic Herpes Virus Encoding GM-CSF in Patients With Stage IIIc and IV Melanoma. Ann Surg Oncol (2010) 17(3):718–30. doi: 10.1245/s10434-009-0809-6
117. Xia T, Konno H, Barber GN. Recurrent Loss of STING Signaling in Melanoma Correlates With Susceptibility to Viral Oncolysis. Cancer Res (2016) 76(22):6747–59. doi: 10.1158/0008-5472.CAN-16-1404
118. Wu J-J, Zhao L, Hu H-G, Li W-H, Li Y-M. Agonists and Inhibitors of the STING Pathway: Potential Agents for Immunotherapy. Med Res Rev (2020) 40(3):1117–41. doi: 10.1002/med.21649
119. Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H, et al. Discovery of a Novel cGAMP Competitive Ligand of the Inactive Form of STING. ACS Med Chem Lett (2019) 10(1):92–7. doi: 10.1021/acsmedchemlett.8b00466
120. Li S, Hong Z, Wang Z, Li F, Mei J, Huang L, et al. The Cyclopeptide Astin C Specifically Inhibits the Innate Immune CDN Sensor STING. Cell Rep (2018) 25(12):3405–21.e7. doi: 10.1016/j.celrep.2018.11.097
121. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING With Covalent Small-Molecule Inhibitors. Nature (2018) 559(7713):269–73. doi: 10.1038/s41586-018-0287-8
Keywords: cGAS-STING, cancer, immunotherapy, STING agonists, combined therapy
Citation: Gan Y, Li X, Han S, Liang Q, Ma X, Rong P, Wang W and Li W (2022) The cGAS/STING Pathway: A Novel Target for Cancer Therapy. Front. Immunol. 12:795401. doi: 10.3389/fimmu.2021.795401
Received: 15 October 2021; Accepted: 08 December 2021;
Published: 03 January 2022.
Edited by:
Chaoyang Sun, Huazhong University of Science and Technology, ChinaReviewed by:
Geneviève Pépin, Université du Québec à Trois-Rivières, CanadaCopyright © 2022 Gan, Li, Han, Liang, Ma, Rong, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Wang, Y2pyLndhbmd3ZWlAdmlwLjE2My5jb20=; Pengfei Rong, cm9uZ3BlbmdmZWk2NkAxNjMuY29t; Wei Li, d2VpbGlseEBjc3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.