
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 March 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.631279
This article is part of the Research Topic Harnessing the Complexity of Normal and Pathological Hematopoietic Supportive Niches View all 13 articles
 Alice Pievani1†
Alice Pievani1† Roberto Savoldelli2,3†
Roberto Savoldelli2,3† Juliane Poelchen4,5†Elisa Mattioli2,3†Giorgio Anselmi6Alice Girardot7Jochen Utikal4,5Pierre Bourdely7Marta Serafini1‡
Juliane Poelchen4,5†Elisa Mattioli2,3†Giorgio Anselmi6Alice Girardot7Jochen Utikal4,5Pierre Bourdely7Marta Serafini1‡ Pierre Guermonprez7*‡
Pierre Guermonprez7*‡Tissue engineering opens multiple opportunities in regenerative medicine, drug testing, and modeling of the hematopoiesis in health and disease. Recapitulating the organization of physiological microenvironments supporting leukocyte development is essential to model faithfully the development of immune cells. Hematopoietic organs are shaped by spatially organized niches defined by multiple cellular contributions. A shared feature of immune niches is the presence of mesenchymal stromal cells endowed with unique roles in organizing niche development, maintenance, and function. Here, we review challenges and opportunities in harnessing stromal cells for the engineering of artificial immune niches and hematopoietic organoids recapitulating leukocyte ontogeny both in vitro and in vivo.
Mechanistic studies of the human hematopoiesis, drug testing, immunization and regenerative medicine purposes share a need for immune organoids recapitulating physiological immune niches. Hematopoietic organs supporting leukocyte development are shaped by spatially organized areas defined by multiple cellular contributions. All immune niches contain mesenchymal stromal cells (MSCs) endowed with unique and specific roles in organizing niche development, maintenance and function. MSCs were first described in murine bone marrow (BM) as a population of cells containing fibroblast progenitors and capable to differentiate into cartilage, bones, adipocytes, and to recapitulate a hematopoiesis supporting microenvironment upon transplantation (1, 2). MSC features were subsequently identified in non-hematopoietic cells of the human BM niche (2–6). Here the term “MSCs” will be used from a practical perspective, referring to an often heterogenous populations of mesodermal origin, regardless of their multilineage potential and self-renewal capabilities at the single cell level.
In this review we highlight the challenges and opportunities in harnessing MSCs for the engineering of artificial immune niches recapitulating the physiological bone marrow (BM) or thymic niche.
The BM is the primary lymphoid organ deputed to the maintenance, self-renewal and lineage commitment of adult hematopoietic stem cells (HSCs). HSCs originate from the dorsal aorta in the aorta-gonad-mesonephron region of the embryo. Embryonic HSCs seed and expand first in the fetal liver before colonizing specific areas of the trabecular region of the BM called niches where they become proper mature HSCs. Niches are complex 3D environments composed of multiple cell types and factors, such as extracellular matrix (ECM), oxygen tension, soluble molecules, and shear forces, all of which govern the fate of HSCs. Most of our knowledge regarding the components of the BM niche and how they interact to modulate HSC fate has been attained by findings in murine models due to the limited possibilities to access and harness information from human specimens. MSCs are critical contributors of the HSC niche. A key feature of MSCs is the delivery of chemokines and hematopoietic growth factors essential for the maintenance and differentiation of HSCs (2). The chemokine CXCL12 engages the CXCR4 chemokine receptor on HSCs. As a consequence, the CXCR4/CXCL12 axis is essential for HSC retention within the BM niche and maintaining HSC quiescence (7). SCF/KITL engages the KIT tyrosine kinase receptor on HSC which is crucial for their maintenance. Proteolytic cleavage and alternative splicing mechanisms generate membrane bound and secreted forms of SCF/KITL. Selective inactivation of the membrane bound form of KITL in Sl/Sld mutant mice impairs long term HSCs despite the presence of soluble KITL (8, 9). In addition, adult deletion of SCF/KITL in inducible knock-out mice depletes HSCs (10). This highlights the importance of cell-to-cell contacts in the delivery of hematopoietic growth factors to HSCs for their maintenance. The production of niche factors has been an important feature guiding the identification of MSC subpopulations relevant for the HSCs niche. For instance, CXCL12gfp mice allowed to identify CXCL12 abundant fibroblastic reticular cells (CAR) (7), while SCFgfp reporter mice showed SCF/KITL expression in both MSCs and endothelial cells (ECs) (10). In addition to ECs and MSCs, multiple other lineages have been reported to also contribute to the maintenance of the niche: for instance, osteolineage cells, adipocytes, neurons and hematopoietic cells. The endosteal niche is found in proximity of the endocortical region of the internal bone shell. It is formed mainly by MSCs together with osteoclasts, osteoblasts, and tissue resident macrophages (11, 12). Multiple lines of evidence locate the HSCs niches mainly in the perivascular areas of BM. The perivascular niche is formed by endothelial and other MSCs. SCFgfp reporter mice have enabled a systematic evaluation of SCF/KITL-expressing cells and revealed a crucial contribution of endothelial and perivascular MSCs compartments (10).
The precise location of HSCs with respect to sinusoidal ECs (sECs) or arterial ECs (aECs) has been controversial (13–18). It is unclear if sinusoids represent a “proliferative” niche whereas arteriole would represent a “quiescent” niche (15) or if sinusoids are the main HSCs localization regardless of their proliferation status (16). The late conclusion has been provided by examining thoroughly HSCs localization defined by KIT+ GFP-labeled cells in alpha-catulingfp reporter mice (16). The development of a fluorescent reporter mouse line fully specific for long term Flt3− HSCs (MFG mice) has enabled to address their localization in un-manipulated environments by intravital imaging (18). This breakthrough revealed that quiescent HSCs seat mostly in vicinity to sECs (and the endosteum), in mildly hypoxic environment (18). Despite the preferential localization of HSCs next to sECs, sECs have recently been reported to express lower levels of SCF/KITL and CXCL12 as compared to aECs, thereby questioning the involvement of aECs in the niche (19–22). Identifying the MSC population of the HSC niche and defining its developmental potential within mesenchymal lineages has proven to be a major challenge. Early work has evidenced BM mesenchymal osteoprogenitors (i) can be engrafted and regenerate bones (2, 23, 24); (ii) are endowed with a multi-lineage potential for osteoblasts, chondrocytes, adipocytes, and fibroblasts (2, 4). Later studies have attempted to prospectively identify self-renewing and multi-potent bona fide MSCs (2). A breakthrough in the field has been afforded by studies which reported that murine CD45-Tie2-CD105+Thy1.1-fetal BM MSCs are able to recapitulate a functional HSCs niche upon transfer within the renal capsule (25). In addition, Morikawa et al. have shown that PDGFRa+Sca1+CD45−TER119− MSCs from mouse BM give rise to osteoblasts, reticular cells and adipocytes after in vivo transplantation (26, 27). Taken together, these studies have defined the so-called “two stem cells” paradigm in which multipotent MSCs are both organizers and components, besides HSCs, of the BM niche (25, 28, 29). The Frenette lab has shown that Nestingfp reporter mice could be instrumental to identify MSCs with stem cell properties: (i) high CFU-F activity; (ii) a multi-lineage potential to generate fibroblasts, osteoblasts, chondrocytes and adipocytes (in both mice and humanized mice); (iii) a self-renewal potential upon transfer (28). More recently, MSCs activity has been tracked using the LepRcre mice targeting MSCs expressing the leptin receptor (30, 31). Recent scRNAseq studies have re-evaluated the heterogeneity among LepR+ cells and highlighted the existence of two subsets, one expressing adipocyte-related genes (Adipo-CAR) while the other expressing osteogenic genes (Osteo-CAR) (19, 21). Micro-dissection analysis suggests that Adipo-CAR would be more associated to sinusoids, as compared to Osteo-CAR, preferring arteriole and other regions (21, 22). Multiple line of evidences, including the inference of developmental trajectories in silico (32), suggest that the Adipo-CAR fraction of LepR+ cells contains the most primitive MSC progenitors activity giving rise to multiple adipocytes, osteoblasts and chondrocytes (22, 32). Studies from the Morrison lab have established that LepR+ cells account for most BM CFU-F activity, represent the main source of bone and adipocytes in adult BM and can give rise to cartilage (30). Various technical approaches have identified multiple cellular entities within the MSCs compartment proposed to play a major role in shaping of the perivascular niche (10, 15, 28, 33):
i) Arteriole-associated pericytes. Pioneer work from the Frenette lab identified arterioles-associated pericytes expressing NG2/CSPG4 coupled with quiescent HSCs in the endosteum region (15). NG2+ pericytes are rare and display bright GFP levels in Nestingfp reporter mice (Nestinbright) (15). Conditional and inducible cellular deletion of NG2 pericytes (using NG2-creERTMxROSAiDTR mice) induces HSCs cycling and reduces long-term repopulating activity (15). Nestinbright pericytes express high levels of CXCL12. This feature is relevant for HSCs niche, as conditional inactivation of CXCL12 -but not SCF- in NG2+ pericytes reduces HSCs numbers, perturbs their localization within the BM and induces their peripheral mobilization (34).
ii) Sinusoid-associated pericytes. Sinusoid associated MSCs display high level of GFP in Cxcl12gfp mice (7), intermediate level of GFP in Nestingfp reporter mice (15), and are labeled in LepRcre mice (10, 30). From the niche point of view, LepR+ positive cells (labeled in LepRcre mice) express high levels of SCF/KITL and some levels of CXCL12 (10, 33). Conditional deletion of SCF in LepR+ cells depletes quiescent HSCs (10). Conditional inactivation of CXCL12 in LepR+ cells triggers HSCs mobilization in periphery (33). In addition to SCF and CXCL12, Adipo-CAR produce multiple hematopoietic factors like IL-15, IL-34, Csf1, Bmp4, Ccl19, and Ccl2 (22). Furthermore, LepR+ cells act also as a major source of IL-7, important for the homeostasis of lymphoid-committed progenitors which also express CXCR4, ensuring their correct positioning by responding to CXCL12 from LepR+ MSCs (35).
Compared with the mouse system, much less knowledge exists regarding the architecture of the human BM niche and the function of its different cellular components. Using human bone biopsy specimens, Guezguez et al. provided evidence of HSC propensity to localize to endosteal regions of the trabecular bone area (36). CD34+ cells follow a spatial gradient within the marrow cavities with a maximal concentration in the first 50 μm from the bone trabecula surfaces where the blood vessels are most concentrated (37). Efforts in recent years have also been made to identify the specific human MSC subpopulation that supports HSC activity. In the human BM, a multi-potent CD45−CD105+CD146+ sinusoid-associated fraction of MSCs was shown to be able to recapitulate a functional HSCs niche upon transfer under the mouse skin (5). Human pericytes CD146+ express nestin, CXCL12, and LepR similar to mouse perivascular MSCs and directly support the ex vivo maintenance of human HPSCs through cell-to-cell contact and activation of Notch signaling (38). A fraction of human CD146+ perivascular MSCs expressing PDGFRα, CD51, and multiple niche factors (e.g., CXCL12, SCF, and angiopoietin1) would correspond to an ortholog of GFP-positive cells in Nestingfp mice. Upon transplant in immunodeficient mice, this CD146+ MSC subset is mainly localized in close proximity to mouse sinusoids and recruit hematopoietic cells (39). All these evidences seem to suggest that CD146+ perivascular cells could represent the human counterpart of the CAR cells or nestin+ cells described in the mouse. More recently, a population of CD146− CD271+ MSCs localized in the trabecular region of the human BM has been identified. Like the CD146+ perivascular cells, these CD271+ MSCs showed high clonogenicity, trilineage differentiation capacity in vitro, and ability to transfer a HSC microenvironment upon transplantation (40). CD271+SSEA4+ MSCs have also been shown to express high levels of HSC-supportive genes and to support HSC engraftment potential (41). These data indicate that different subtypes of MSCs exist in the human BM niche and interact with HSCs in specific regions.
HSCs in vitro generation is instrumental to understand hematopoiesis as well as to model genetical disorders and cancers. Moreover, large scale manufacturing of HSCs could represent a valuable therapeutic option for many patients. To date, two different approaches have been attempted in this way: (i) the expansion of large number of HSCs from BM or umbilical cord blood (CB) (42, 43); (ii) the de novo generation of HSCs from induced pluripotent stem cells/embryonic stem cells or via somatic cell reprogramming (44–48).
Standard culture protocols supporting the proliferation of long term bona fide engraftable, self-renewing hematopoietic stem and progenitor cells (HSPCs) with multi-lineage potential remain a challenge. Given the natural role of MSCs in the HSC niche, co-culture with MSCs is a very popular approach to maintain and expand HSCs in vitro. However, these conventional systems fail at reproducing the complexity of the BM niche.
The first step in mimicking the physiological HSC niche consist on generating a 3D environment using different biomaterials, such as hydrogels, silicate structures, and human bone-derived scaffolds (49) (Table 1).
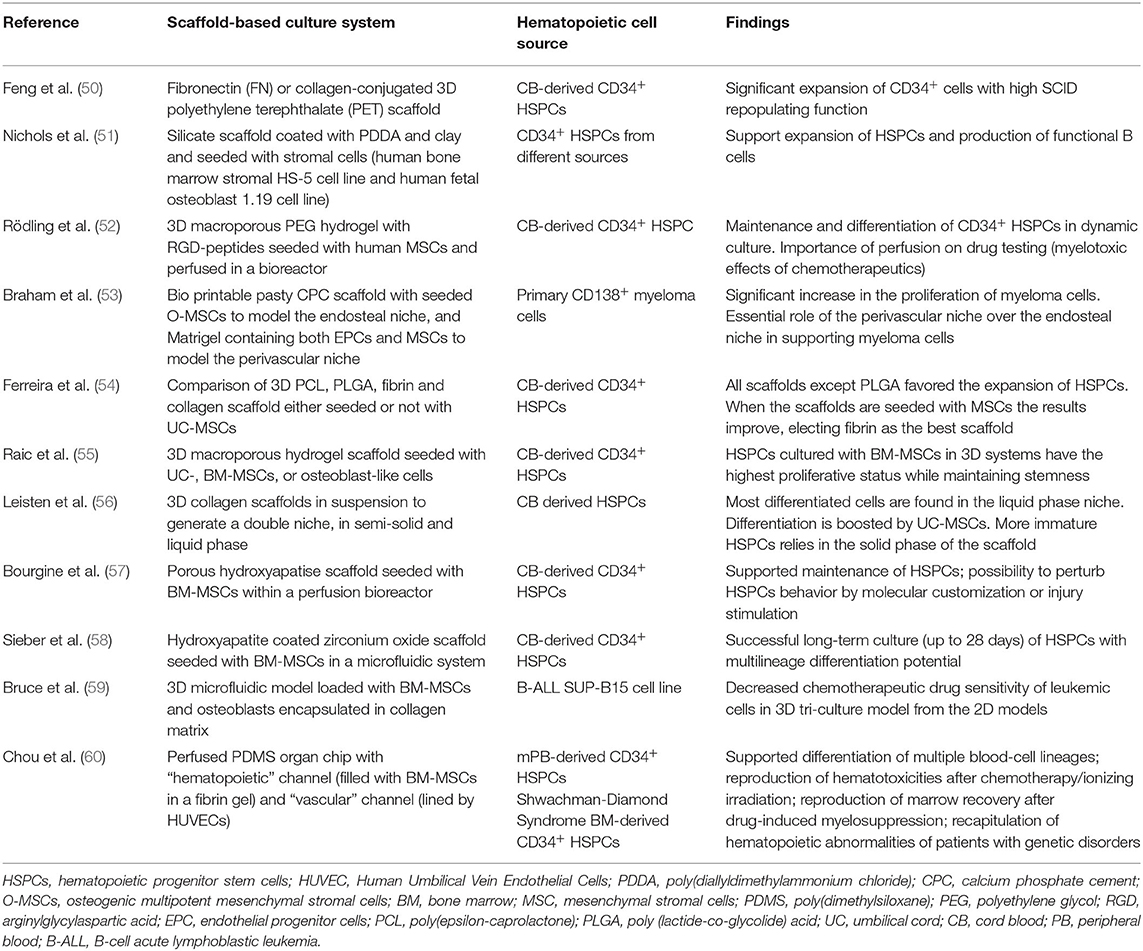
Table 1. Modeling the BM niche in vitro using human MSCs.
Those polymers provide structure and support for cell proliferation but also a spatial control of the cell interactions. This is achieved by physical limitation in cell-to-cell contact and controlled availability of soluble factors. Integration of ECM within these scaffolds has been explored. Feng et al. demonstrated how a polyethylene terephthalate (PET) scaffold can be engineered with ECM proteins, such as collagen and fibronectin, to support the expansion and differentiation of CB-CD34+ cells (50). Importantly, those synthetic scaffolds can be colonized with different types of MSCs together with ECs to mimic in vivo niches. Ferreira et al. and Raic et al. (54, 55) have developed two independents models based on porous 3D scaffold for the expansion of HSCs in vitro. Ferreira et al. tested several natural polymers as scaffolds in combination with MSCs as support and found that 3D fibrin scaffold seeded with MSCs is the most efficient system to expand CD34+ cells. Importantly, expanded HSPCs maintain a more primitive immunophenotype and exhibit strong engraftment and multi-organ repopulation capability (54). Similarly, Raic et al. showed that the positive effect of MSCs on preservation of HSPCs stemness was more pronounced in a porous 3D hydrogel scaffold in comparison to standard 2D culture systems (55). Moreover, phenotypically immature HSPCs (CD34+CD38−) with self-renewal and repopulation capacity are shown to be maintained in 3D collagen co-culture with MSCs, in close proximity to the collagen fibers (56). Nichols et al. demonstrated how a silicate and clay 3D structure seeded with heterogeneous stromal cells and human HSCs is able to expand the HSCs and promote B cell development after 28 days, with a significant increase compared to its 2D counterpart (51).
Latest development in the field is the combination of 3D organoids with perfusion-based bioreactor systems, the so-called “4D system,” to further increase the amount of resemblance to in vivo niches. The BM microenvironment is indeed strictly regulated by the concentration of soluble factors, oxygen levels, and the mechanical stress applied by blood flow. The use of bioreactors and microfluidics devices can than allow modeling the niche situation more closely. Rödling et al. developed a bioreactor system for perfusion of 3D scaffolds seeded with MSCs mimicking the BM in vivo and demonstrated the importance of perfusion during drug treatment as results are different with and without perfusion (52). Indeed, while under static conditions the more mature CD34− subpopulation was more sensitive to 5-fluorouracil treatment, under dynamic conditions both CD34− and CD34+ cells responded similarly. Bourgine et al. reported the engineering of BM-like tissues in a perfusion bioreactor system partially recapitulating structural, compositional and organizational features of the native human osteoblastic niche environment, resulting in the support of HSPC functions. Their approach consists in the use of bone-like porous hydroxyapatite scaffold functionalized by MSCs and osteoblastic cells and by the ECM they deposited during perfusion culture in bioreactors (57). 3D scaffold-based microfluidic chips have been introduced for the generation of a “BM-on-a-chip.” Torisawa et al. in a pioneer murine study showed that PDMS (poly-dimethyl-siloxan) device loaded with bone forming factors could be seeded in vivo by BM-MSCs, such as CXCL12+ CAR and Nestin+ LepR+ perivascular MSCs. Ex vivo culture of the organoid can be achieved in a microfluidics chip (61). Sieber et al. loaded hydroxyapatite-coated zirconium oxide scaffold with human MSCs in a microfluidics chip. This enables HSPCs to maintain multilineage potential up to 28 days in vitro (58). Chou et al. seeded a fibrin scaffold with MSCs and CD34+ cells in a microfluidics device embedding vascular channel seeded with human ECs. Upon 4 weeks of culture the vascularized chip recapitulated the ontogeny of multiple blood cell lineages while maintaining undifferentiated HSPCs. Moreover, this system has been used to model the impact of a genetic disorder on myelopoiesis (60).
MSCs in microfluidic devices have also been instrumental in modeling the leukemic niche in vitro for the sake of studying chemotherapeutics and immunotherapy (53, 59, 62).
By virtue of their role in HSC niche, engineering of MSCs offers a window of opportunity to control HSC self-renewal or drive hematopoietic differentiation. Several factors regulate the HSC-supporting activity of MSCs. For instance, primary Nestin+ murine MSCs rapidly lose their hematopoietic supporting potential upon ex vivo culture (63). The transcriptional down-regulation of key niche factors (SCF, ANGPT1, CXCL12, VCAM1) is underlying this process (63). An elegant genetic screen has revealed that overexpression of defined transcription factors (Klf7, Ostf1, Xbp1, Irf3, Irf7) can “revitalize” the niche promoting activity of ex vivo cultured primary MSCs. MSCs overexpressing those transcription factors become able to maintain transplantable HSCs (63). This important study opens new avenues in the engineering of MSCs. Some niche factors involved in the hematopoietic hierarchy present a limited sequence homology between mouse and human genes. This is limiting the reactivity across species thereby rendering mouse MSCs sub-optimal for applications involving human HSPCs (64, 65). For these reasons, multiple groups have attempted the expression of defined human hematopoietic factors in murine MSCs lines (Table 2). For instance, co-expression of IL-7 and FLT3L synergizes with DLL1 expression in OP9 to induce the proliferation of T cell progenitors (66). Building on the MS5 mouse MSC line, Anselmi et al. have developed a screening for combination of human niche factors promoting the efficient generation of dendritic cells (DCs) from human CB-CD34+ cells (68). They have found that a combination of membrane-bound FLT3L and SCF and soluble CXCL12 is efficient in promoting the differentiation of DCs resembling their circulating counterparts (68). Importantly, transwell experiments indicate that this system relies on the establishment of cell-to-cell contacts. Subcutaneous engraftment of engineered MSCs in basement membrane matrix (Matrigel®) in NSG mice defines a niche supporting both the maintenance of a pool of undifferentiated CD34+ cells and the differentiation of DCs (68). Within this niche, it was found that poorly differentiated human CD34+ cells would develop cell-to-cell contact with engineered MSCs. Previous reports have established that membrane-bound forms of hematopoietic growth factors like SCF would be specifically required for niche function in vivo (8, 9, 69–71). A practical consequence of this is that engineering of MSCs for the over-expression of membrane-bound SCF (or FLT3L) is an attractive strategy to improve the niche-promoting activity of MSCs in vitro or in vivo (68, 72) (Figure 1). In the same vein, human BM-MSCs have been engineered by Carretta et al. to over-express IL-3 and thrombopoietin (TPO). IL-3/TPO over-expressing MSCs displayed an increased ability to drive the in vitro expansion of CD34+ cells and improved capacity to support in vivo growth of CD34+ progenitors expressing the MLL-AF9 fusion gene in a humanized scaffold xenograft model (67). In vivo delivery of engineering stromal cells could be improved by the implementation of chemically defined scaffold. For instance, Tavakol et al. have shown that collagen coated carboxyl methyl cellulose micro scaffold (CCMs) seeded with OP9 and HSPCs in vitro supports the long term maintenance, over 12 weeks, upon engraftment in immunodeficient mice (73).
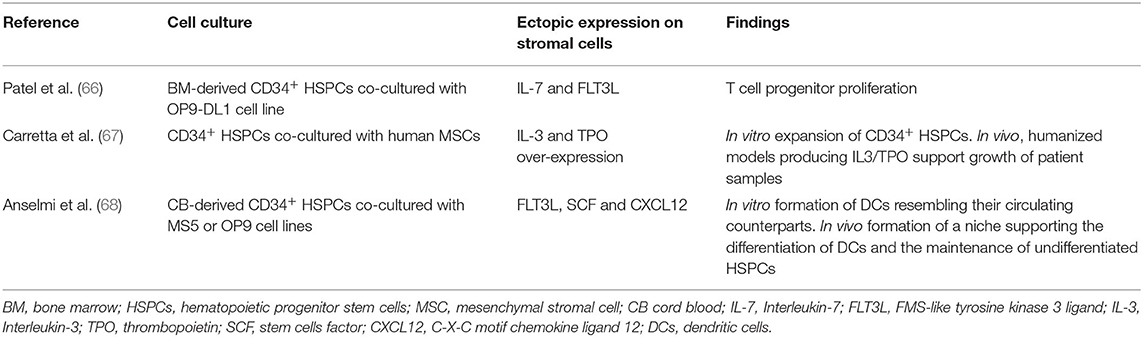
Table 2. Engineering of MSCs for enhanced human niche activity.
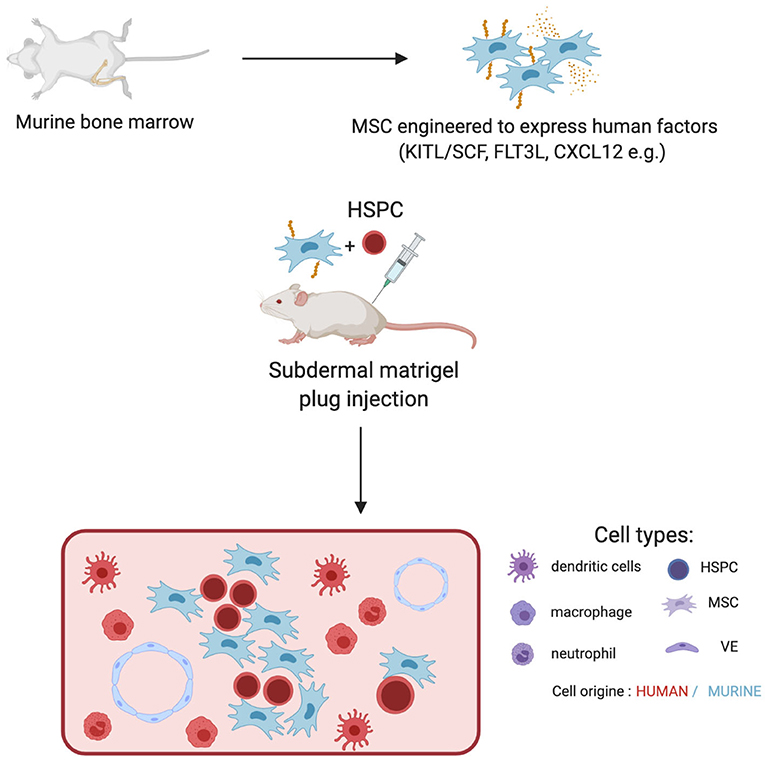
Figure 1. Engineering of murine MSCs to recapitulate human hematopoiesis. MSC from murine bone marrow are isolated and expanded ex-vivo. Those are subsequentially engineered to express human factors and injected together with human HSPCs into the back of an NSG mice in Matrigel plugs. The plug is retreated 2 weeks later. It displays an unorganized structure containing murine MSCs and vasculature together with human hematopoietic progeny. Progeny is dependent on the factors expressed by MSCs.
Altogether those approaches highlight the versatility of MSC engineering to control and direct HSPCs fate in health and diseases.
Pioneer studies showing that human MSCs can establish a hematopoietic microenvironment upon transplantation in rodents at non-skeletal sites date back nearly 50 years (74–76). The evolution of bone tissue engineering strategies together with the identification of the human specific osteoprogenitor subpopulations associated with the formation of ectopic bone and bone marrow have been fundamental steps for the generation of humanized BM tissues in mice, the so-called “ossicle” model (5, 25, 28, 29). These human MSC–generated organoids are tissue-specific chimeras, as bone, myelo-supportive stroma, and adipocytes derived from donor MSCs, while blood vessels and hematopoietic tissues are derivatives of host tissues, and they are harmoniously integrated into an overall tissue structure. Several versions of heterotopic transplantation assays have been used, which differ from one another concerning the site of grafting, such as kidney, subcapsular space (25, 77), intramuscular (78), and subcutaneous tissue (79) or type of osteoconductive scaffold/material employed as a carrier (79).
The next level of humanization of human MSC–generated ossicles has been the introduction of human HSCs. In 2010, Vaiselbuh et al. for the first time reported the successful engraftment of human hematopoietic cells in an ectopic humanized niche obtained implanting subcutaneously in immunocompromised mice polyurethane scaffolds coated with human MSCs, giving rise to the first fully humanized bone/BM organoid model (80). The establishment of a functional human BM niche that could support the maintenance of human blood cells opened the possibility to bridge gaps between the interspecies divergence at a molecular and cellular level in the hematopoietic niche and aspires to become an advanced model to investigate human hematopoiesis and leukemogenesis. Although many aspects of the hematopoiesis are conserved between mice and humans, several differences need to be taken into consideration before applying results obtained in the mouse to humans, specifically for studies aimed at preclinical testing of new therapies (81). Several humanized ossicle models have been reported to date and each has its specificity, as extensively reviewed recently (Figure 2) (82, 83). Current strategies differ for human stromal cell types, carrier material scaffold, human hematopoietic transplantation protocols, and experimental time frames (Table 3). Most protocols use stromal cells derived from the BM of healthy donors. Reinisch et al. suggest that only MSCs, as opposed to the umbilical cord-, skin-, or white adipose tissue-derived MSCs, possess the capabilities to form ectopic bone and BM in vivo (77). However, other studies reported the formation of ectopic BM niches through endochondral ossification using stromal cells from different sources, such as cord blood (94) and adipose tissue (97), when primed toward chondrogenesis in the presence of transforming growth factor-β in vitro before implant. In most protocols, cells are seeded onto ceramic, collagen, calcium phosphate, or hydroxyapatite-based scaffolds or hydrogels before implantation. These scaffolds provide instructive cues to ensure osteogenesis and represent a 3D template that supports the formation of a bone organ. According to their composition and degradation properties, scaffolds can be entirely remodeled during the ossicle formation or remain part of the organoid structure. The persistence within heterotopic ossicles of artificial, mineralized scaffold material that are not resorbable, is not desirable. It prevents the establishment of the completely normal architecture of bone marrow and complicates the analysis of stromal and hematopoietic cell populations contained within the ossicle, particularly their quantitative assessments. The transplantation of cartilage pellets made ex vivo by MSCs consent to avoid these limitations due to the use of exogenous scaffold (91, 93). Stimulating factors, such as BMP2 (96), BMP7 (95), or parathyroid hormone (PTH) (87) can be used to promote osteoblast differentiation of MSCs for the successful in vivo formation of mature bone and BM tissues. Furthermore, MSCs have also been genetically modified to express BMP2 (98) or BMP7 (99) generating new bone in vivo.
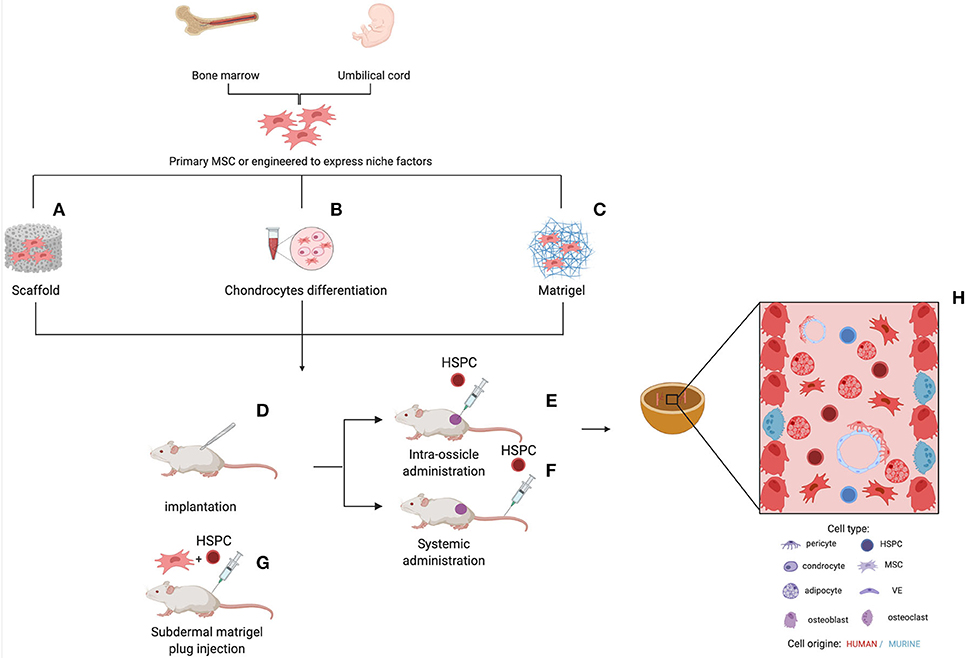
Figure 2. Different strategies for the generation of humanized ossicles. In vitro expanded human MSCs (from healthy donors or patients) are seeded onto a scaffold, (A) primed to differentiate in cartilage (B) or cultured on Matrigel® (C) Some protocols include the co-seeding of human ECs and the supplement of osteogenic factors, such as BMPs and PTH. Following the first in vitro step, the implantable structures are transplanted subcutaneously into the back of immunodeficient mice for the ossicle formation (D). Aberrant/normal human HSPCs can be added to the system by direct intra-ossicle transplantation (E) or intravenous injection (F) either before or after the implantation of the ossicle. Irradiation is usually performed to promote engraftment. As an alternative, HSPCs can be seeded onto the Matrigel® plug before the in vivo implant (G). The whole process can take several months. The resulting ossicle collected from the mouse is composed of both myelo-supportive marrow stroma and hematopoietic tissues and its progeny of human origin (H). Sinusoidal endothelium, nerve fibers, residual hematopoietic cells, and osteoclasts are derivatives of mouse host.
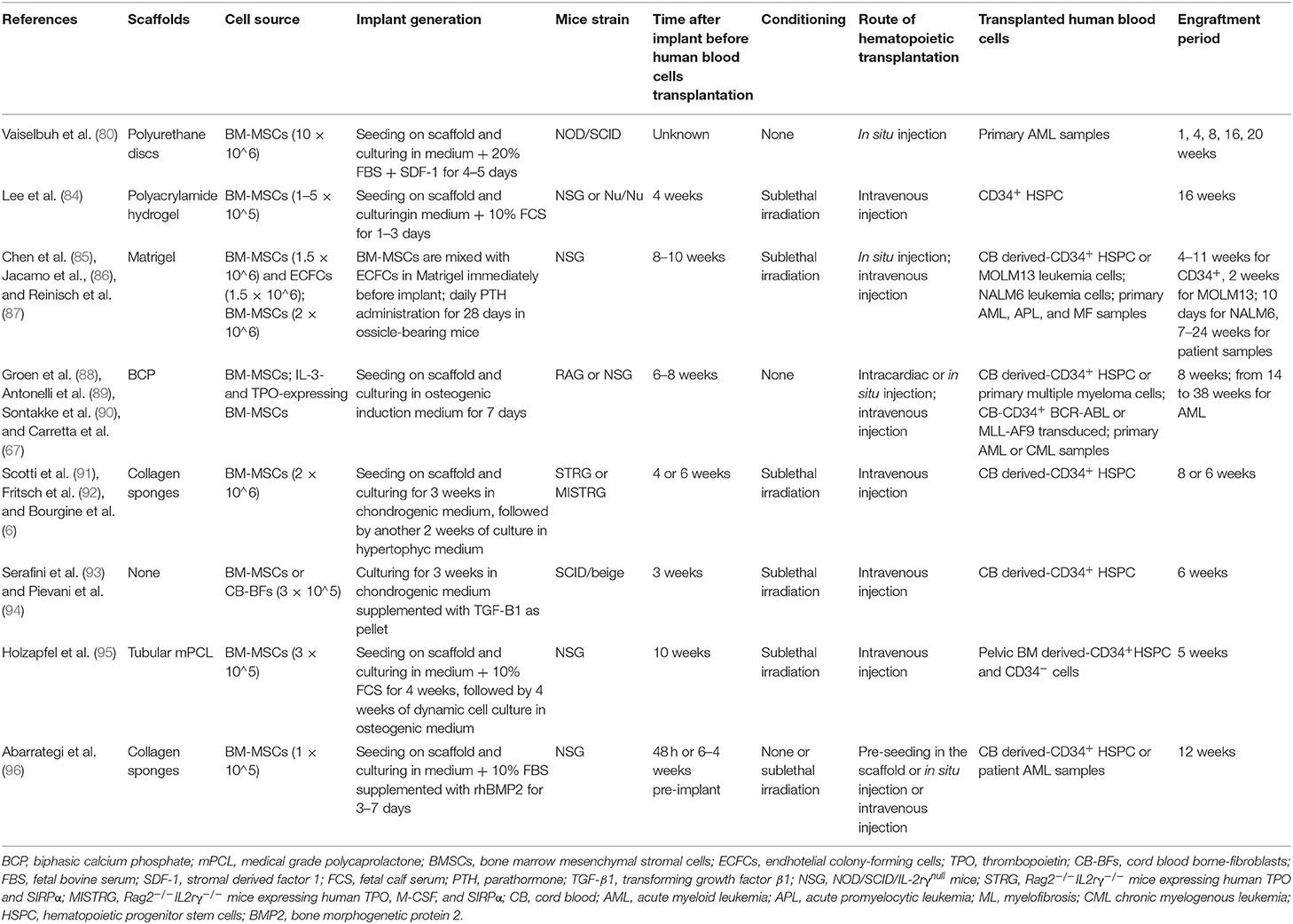
Table 3. Humanized ossicle models.
Human blood cells (healthy or malignant) can be administered either by peripheral (tail vein or retro-orbital) or intra-ossicle infusion for organoids of larger dimensions. By contrast, Abarrategi et al. proposed co-implantation of MSCs and human CD34+ cells within a collagen sponge, thus avoiding the requirement for subsequent transplantation (96). Conditioning before transplantation does not seem to be a requirement for the successful engraftment of ossicles within immunodeficient mice. The human hematopoietic cell transplantation was generally performed within 3–10 weeks after the in vivo implant of the ossicles. Four weeks represent the minimum period required for the formation of marrow cavities allowing human engraftment, through osteoclasts resorption of mineralized tissue within the ossicles (93). Engraftment assessment post-transplantation was achieved from 4 to 38 weeks, depending on the type of cells transplanted (malignant cells with fast/slow engraftment) and the functional readout targeted (short/long-term HSCs engraftment). However, numerous challenges remain to be solved and the variability of different protocols does not allow to do a comparison between different systems in terms of accurate reconstitution of the human niche and potential for human blood engraftment sustaining. The topic has been recently well-reviewed by Dupard et al. (82). Despite the optimization efforts, the ossicles remain largely chimeric as both the nervous system and blood vessels are of mouse origin, although human mesenchymal perivascular cells were detected (6). This implies that investigations on the role of specific niche cellular factors or cytokines should consider the influence of their murine counterparts. Human vascular structure can be generated by seeding human ECs (e.g., HUVEC) together with MSCs on carrier materials (85, 96). Stringent quantification of both human MSCs-derived stroma and human blood populations in humanized ossicles is difficult to be performed and required the application of most sophisticated imaging strategies. Most of the reported ossicle approaches are based on the use of primary MSCs isolated from BM of healthy donors. Specifically, these cells are very heterogeneous in terms of proliferation and differentiation capacity and this reflects not only the source- and the donor-dependent variability, but also specific differences in isolation/expansion procedures (100). Furthermore, there is a striking batch-to-batch variability in their ability to form ossicles in vivo. All these aspects highlight some limits in the full exploitation of these models although this does not diminish the potential of humanized ossicle approaches for studying human healthy and pathological hematopoiesis.
The establishment of a human BM microenvironment within heterotopic ossicles was associated with enhanced long-term engraftment of human HSCs, as compared to mouse bones (87, 95, 96). The human HSC engraftment was successfully supported also by BM niches generated from cord blood stromal cells (94). Notably, human MSCs, included in the ossicle, release an increased level of cytokines, such as VEGF and IL-6, that accelerates vascularization and enhances the recruitment of human HSCs (84). Moreover, an improved myeloid development was obtained compared with lymphoid-biased human engraftment usually observed in the BM of intravenously transplanted NSG mice (85, 87, 96). Furthermore, some studies have demonstrated that humanized ossicles maintain the quiescence and the self-renewal potential of human HSCs, which can successfully engraft secondary recipient mice, with a higher efficiency compared to murine BM (87, 92). Within the ossicle, human HSCs CD45+CD34+CD90+ have been found in close proximity to human MSCs, suggesting that direct cell-cell contact is fundamental to regulate their fate (6). The ossicle system allows genetic manipulation of human niche components to better understand directly in vivo the role of specific factors critical for human hematopoietic reconstitution. Overexpression of CXCL12 by human MSCs in the ossicle results in a specific enrichment in common myeloid progenitors, megakaryocyte/erythrocyte progenitors, multipotent progenitors, and HSC populations expressing the CXCR4 receptor (6).
Patient-derived xenograft (PDX) mouse models are currently the gold standard for studying the development of human leukemia. However, engraftment and expansion of human acute myeloid leukemia (AML) in vivo remain challenging as a substantial number of samples fail to engraft also the most optimized host mice, particularly in the case of malignancies less aggressive (101). The reason may be that some subtypes of AML have low progenitor cell frequency or some samples may be particularly sensitive to the lack of a specific cell type in the mouse BM or a factor that is poorly or not at all cross-reactive between mice and humans. Hence, MSC-derived humanized ossicle models have raised great interest in the leukemia field, as recently extensively discussed in the review of Abarrategi et al. (83). The first study of AML engraftment in humanized microenvironment was reported by Vaiselbuh et al., who demonstrated that primary AML cells injected directly in pre-implanted scaffolds coated with human MSCs or intravenously in mice after implant, successfully engrafted in the ectopic niche (80). Further studies demonstrated that AML samples non-engrafting in mouse BM, such as acute promyelocytic leukemia (APL), were able to efficiently engraft in the humanized microenvironment (87, 96). Importantly, ossicles maintained the clonal heterogeneity in xenografted AML cells and their stem cell self-renewal capacity better than murine BM, as demonstrated by serial transplantation assays (87, 89, 90). Humanized BM ossicles are useful also for the engraftment of small myeloid clones, such as TP53 mutated AML subclones (102). Battula et al. developed a different approach called “human bone implant” that consists of subcutaneous transplants in NSG mice of fragments from freshly collected human BM biopsies using Matrigel® as a carrier (103). The implanted human BM tissue undergoes vascularization and bone restoration in mice, providing a functional human BM microenvironment capable of supporting the human AML engraftment. In addition to AML, a humanized ossicle system has been used to engraft primary multiple myeloma patient samples, which are known to be highly dependent on the human BM microenvironment for their survival and growth (88). Furthermore, humanized ossicles facilitated robust engraftment of myelofibrosis specimens, which has previously shown only limited engraftment with transplantation of large numbers of patient-derived CD34+ cells in conventional xenograft models (87). Genetical manipulation of human niche components can likely help in better understanding the role of factors critical for leukemia engraftment/progression. Deletion of hypoxia-inducible factor (HIF)-1α in human MSCs impaired leukemia engraftment in BM organoids by decreasing CXCL12 expression (85). Another pilot study demonstrated that the blockade of NF-kB activation through IkBα-SR overexpression in MSCs in the humanized ossicle model reduces leukemia burden following chemotherapy, diminishing the stroma-mediated chemoresistance (86). Carretta et al. improved the development of the myeloid compartment from leukemic samples by genetically engineering human MSCs to express IL-3 and TPO (67). Recently, a fully humanized hematopoietic niche system has been exploited to investigate the multidirectional crosstalk among AML, HSCs and the microenvironment and allowed to identify stanniocalcin 1 and its transcriptional regulator HIF-1α as specific mediators whereby AML impairs normal hematopoiesis by remodeling the mesenchymal niche (104). Of note, current 3D models use MSCs isolated from BM of healthy donors, which are molecularly and functionally different from disease-exposed ones. The use of patient-derived niche components may further improve these models and help unravel the role of the niche in the development of hematopoietic diseases. We recently reported an AML stromal niche model obtained using MSCs derived from BM of AML patients (105). AML-MSCs derived ossicles contained a significantly increased fraction occupied by adipocyte and represent an osteoprogenitor-rich niche with the presence of osterix+/osteocalcin− pre-osteoblasts and osteocalcin+/Dentin matrix acid phosphoprotein (DMP) 1− immature osteocytes that correlated with the reduced mature bone formation. However, the generation of humanized ossicles from MSCs and hematopoietic cells from the same patient in an autologous setting has yet to be demonstrated but it would provide a personalized in vivo model to test new therapies.
T cells originate from BM derived lymphoid progenitors differentiating in the thymus. The 3D organization of the thymus is provided by different cell types and creates a complex unique environment for T cell development (106). There are numerous reasons and motivations to recapitulate T cell education in the context of thymic organoids.
i) Primary immunodeficiencies constitute a major cause of deficiencies. BM or umbilical cord transplant represent a clinical approach that is potentially limited by the onset of graft vs. host disease (GvHD) and slow reconstitution of the T cell compartment (107). Also, patients who underwent thymectomy or suffer from the DiGeorge syndrome, a genetic disorder underpinning thymus hypoplasia resulting from microdeletion on chromosome 22, would benefit from thymus bioengineering.
ii) Thymic involution. After reaching the maximum size during adolescence, the thymus begins to shrink and T cell generation decreases in a process called “thymus involution” (108). Thymic involution is associated with aging and exacerbated by several pathological and environmental influences including viral and bacterial infections, drugs or irradiation, affecting its functionality and leading to a decline in naïve T cell output (109, 110). Defective thymus structure and dysfunction negatively influences the adaptive immune system (110). Therefore, regenerating thymic function through replacing a defective thymus by an artificial thymus organoid is of high clinical interest for overcoming potential immunodeficiency or malignancies and maintaining the adaptive immune system.
iii) Adoptive T cell therapy using infusion of antigen-specific T cells is a promising approach in personalized medicine for the treatment of cancer or chronic viral infections. Engineering TCR-specific T cells starting from CD34+ HSPCs (111–113) or T cell precursors derived from induced pluripotent stem cells (iPSCs) (49, 114–116) represent a promising approach that necessitates to recapitulate T cell ontogeny.
iv) The engraftment of thymic organoids into humanized mice is a promising approach for the induction of T cell tolerance against transplanted tissue (110).
v) Mechanistic studies. In vitro models of thymic education offer unique advantage to study and mechanistically dissect thymic selection. For instance, in vitro systems are particularly amenable to live imaging approaches (117).
T cells differentiate from BM derived Lin−CD34+/intCD38−CD45RA+ progenitors seeding the thymus (118, 119), within the thymus T cell progenitors upregulate CD7, CD1a, and CD4 to generate immature single positive (ISP) cells. ISP cells further develop to CD4+CD8+ double positive (DP) cells that ultimately differentiate into CD8−CD4+ or CD8+CD4− single positive mature T cells (SP). Thymic epithelial cells mediate positive and negative selection of T cell progenitors cells (TEC). TEC can be classified in cortical (cTEC) and medullary (mTEC) epithelial cells. cTEC deliver chemotactic (e.g., CCL25, CXCL12), differentiation (e.g., DLL4), and survival (e.g., IL-7, SCF) signals to developing T-cells undergoing positive selection (120, 121). mTEC express AIRE, together with dendritic cells, present self-antigens ensuring the deletion of high affinity self-reactive T-cells (122).
Most experimental systems aiming at modeling thymus function relies on the manipulation of TECs and fall outside of the scope of this review focusing on mesodermal components of hematopoietic niches (123). In brief, 2D culture of TECs provided disappointing results in generating lymphocyte progenitors in line with the loss of primary phenotype upon in vitro culture (124–126) (Table 4). First attempts of 3D cultures were based on murine fetal thymic organ culture (FTOC) (140, 141) (Figure 3B). Despite the successes in supporting T cell development, FTOC evolved into reaggregate thymus organ culture (RTOC) (141). RTOC allows to manipulate the cellular composition and thereby to study the role of specific cell types, pathways or key signals including the Notch Delta like ligand (123) (Figure 3B).
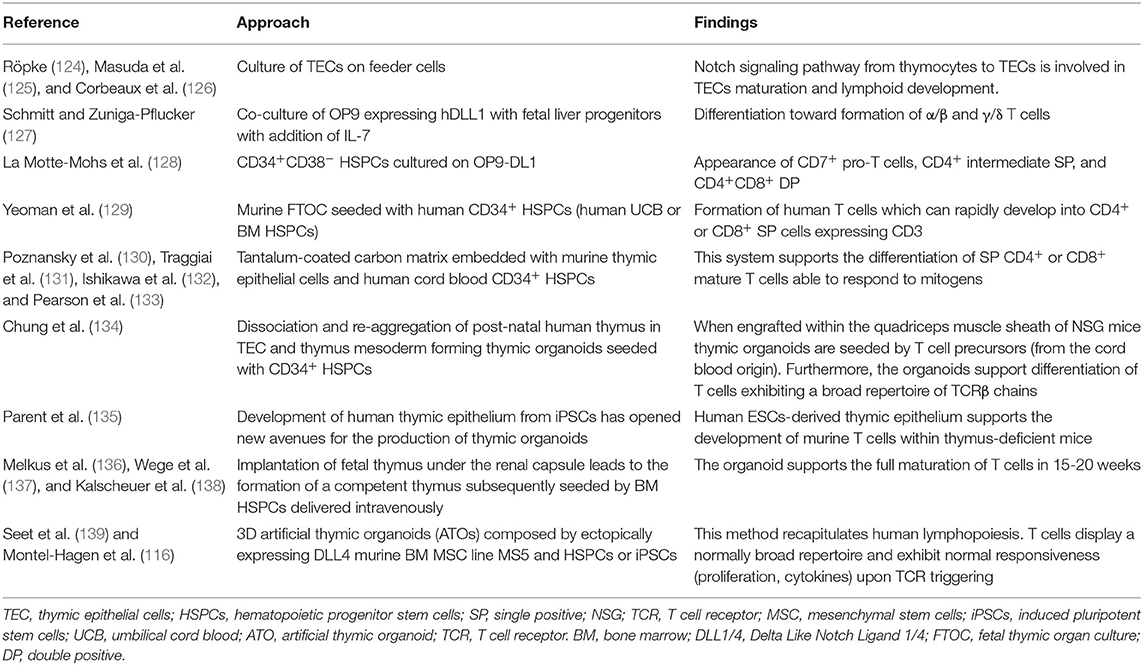
Table 4. Stromal systems recapitulating T cell maturation.
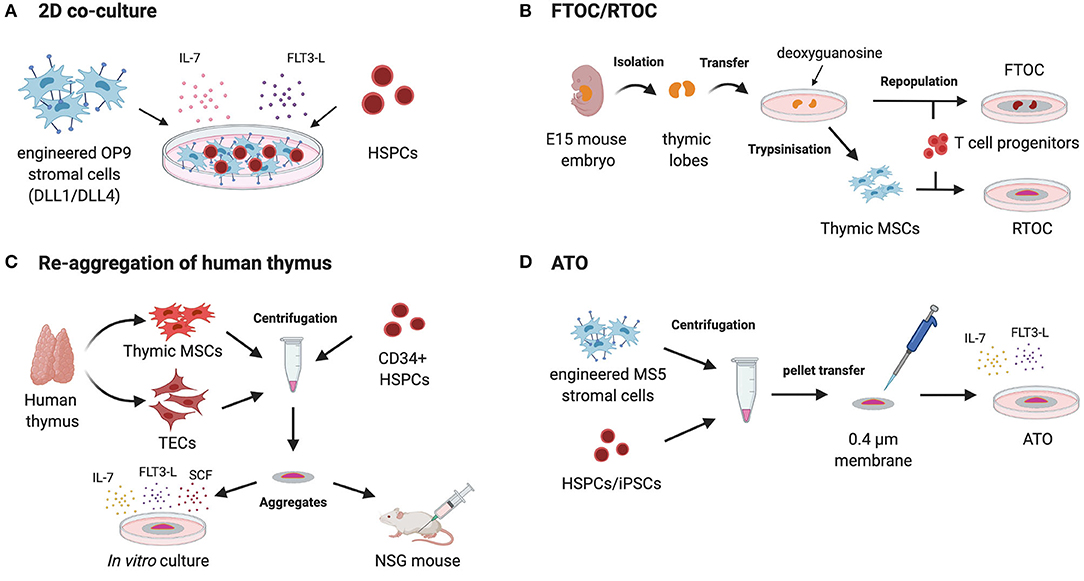
Figure 3. Different strategies to recapitulate T cell ontogeny using MSCs. 2D co-culture of HPCs (hematopoietic progenitor cells) on bioengineered MSC-derived OP9 cells expressing Notch ligands DLL1/DLL4 with recombinant IL-7 and FLT3-L (A). Isolation of murine thymic lobes from day 14 to 15 old mouse embryos followed by their in vitro culture with deoxyguanosine for 5–7 days to deplete intra-thymic T cells. The thymic lobes are then repopulated by different T cell progenitors and cultured on the surface of membranes as fetal thymic organ culture (FTOC). For reaggregation thymic organ culture (RTOC) thymic stromal cells (thymic SCs) are extracted from the lobes and reaggregated with T cell progenitors by centrifugation. The cell suspension is cultured on a filter membrane in vitro (B). Post-natal human thymus is dissociated in TECs and thymus mesoderm (TM) and further reaggregated with human CD34+ cells leading to the formation of thymic organoids. These organoids support the development of mature human T cells when kept in culture or when engrafted within the quadriceps muscle sheath of NSG mice (C). Artificial thymic organoids (ATOs) are generated by the centrifugation of bioengineered MS5 cells expressing Notch ligands DLL1/DLL4 with HSPCs or iPSCs, respectively. After resuspension in a small amount of culture medium the cell suspension is placed on a membrane at the air-liquid interface to form 3D aggregates. This method recapitulates human lymphopoiesis and offers the ability to generate conventionally naïve T cells from HSPCs or iPSCs in vitro (D).
Mesenchymal stromal cells are part of the physiological thymic microenvironment. Recent single cell studies have highlighted the complexity of the mesenchymal compartment of the thymus. Thymic fibroblasts can be distinguished in type 1 (Fb1) and type 2 (Fb2) fibroblasts (142). Fb1 cells are characterized by the expression of an important key player in innate immunity termed COLEC11 as well as by the expression of the enzyme ALDH1A2 that controls the production of retinoic acid functioning as an epithelial growth regulator. On the other hand, Fb2 cells are characterized by ECM genes as well as semaphorins that regulate vascular development. For that reason Fb2 cells are mainly found close to large blood vessels lined with VSMCs (142). ECM produced by thymic MSCs might play a crucial role in the maintenance of TEC phenotypes. For instance, human fibroblast/MSCs uniquely provides an environment supporting promiscuous gene expression by mTECs associated to AIRE and FoxN1 expression (143).
Here we provide examples of the implementation of MSCs in modeling T-cell education and thymic function.
The Zúñiga-Pflücker lab has pioneered the implementation of murine MSCs engineered to express NOTCH ligands to drive T-cell maturation from HSPCs. OP9 is MSC line derived from CSF1-deficient mice with a broad hematogenic activity dependent on SCF/KITL expression (144) but unable to sustain the generation of T cells (127). Schmitt et al. have shown that ectopic expression of the Delta-Like-1 (DLL1) NOTCH ligand is sufficient to confer the ability to support ontogeny of α/β and γ/δ T cells from fetal liver progenitors in the presence of FLT3L and IL-7 growth factors (127) (Figure 3A). DLL1 provides a key signal for T cell specification at the expense of B cell development. This approach was later found to also recapitulate the ontogeny of human T cells from umbilical cord blood (UCB) CD34+ HSPCs via the ordered appearance of CD7+ pro-T cells, CD4+ intermediate SP, and CD4+CD8+ DP (128). Importantly, the OP9 approach (using DLL4) could also be implemented to generate T cells from human embryonic stem cells (145). However, one major drawback of the OP9-DLL system is that T cell differentiation and maturation of TCR+ SP CD4+ or CD8+ T cells remained inefficient for most sources of human pre or post-natal HSPCs with the exception of UCB (146). Similar to the in vitro TECs culture, the culture of Notch ligand expressing OP9 cell lines shows the drawback of lacking a 3D architecture (147). The BLT (BM, liver, thymus) model of humanized mice provides an option to model human thymus function in vivo. Implantation of human fetal thymus under the renal capsule leads to the formation of a competent thymus that can be seeded by BM HSPCs delivered intravenously to support the full maturation of T cells in 15–20 weeks (136–138) (Table 4).
Bypassing the need for fetal thymus mesodermal components have been shown to help building 3D models of human thymus. Using the dissociation and re-aggregation of post-natal humans TECs and thymus mesoderm Chung et al. have recapitulated thymus function (134) (Figure 3C). Those thymic organoids supported the development of mature human T cells when seeded with human CD34+ HSPCs (134). This system offers the advantage to be amenable to lentiviral transduction for manipulation of the thymic environment and bypass the use of xenogenic (murine) thymic epithelium (134). When engrafted within the quadriceps muscle sheath of NSG mice previously reconstituted with human CB-CD34+ HSPCs, thymic organoids are seeded by T cell precursors (from the CB origin). Furthermore, the organoids support differentiation of T cells exhibiting a broad repertoire of TCRβ chains (134).
More recently, Seet et al. have shown that MSCs can help modeling thymus function (139). Seet et al. report a method for the formation of artificial thymic organoids (ATOs) in which TECs were replaced by the ectopically expressing DLL4 murine MSC line MS5 (MS5_DLL4) and centrifuged with HSPCs or iPSCs, respectively (116, 139) (Figure 3D). ATO simulate the 3D structure of the thymus and can be compared to RTOC (139). After resuspension in a small amount of culture medium the cell suspension was dropwise placed on a membrane at the air-liquid interface to form 3D aggregates. This method recapitulates human lymphopoiesis and offers the ability to generate conventionally naïve T cells from ESCs or iPSCs, respectively (116, 139). Furthermore, this system enables long-term culture and provides improved positive selection due to the 3D organization. T cells developing in ATOs display a normally broad repertoire and exhibit normal responsiveness (proliferation, cytokines) upon TCR triggering. Of crucial relevance for clinical application, this method is amenable to generate TCR-transduced T cells generated after efficient allelic exclusion at the Vβ locus (116, 139). This has been exemplified using TCR specific for NY-ESO or MART1 tumor-associated antigens (116, 139). In sum, the ATO system demonstrates the high versatility of engineered MSCs to recapitulate cellular interactions underlying T cell development.
This review highlights the potentially vast range of application for MSCs in the engineering of immune niches supporting leukocyte development. Among the multiple technological challenges raised by the implementation of MSCs, some salient topics emerge defining possible future directions in the field.
Recent developments in high dimensional approaches, such as unbiased scRNAseq have brought a fresh look on the heterogeneity of MSCs associated to immune niches (19–22, 148–151). For instance, whole genome expression analysis reveals an exquisite specificity in the distribution of niche factors among the diverse MSC types (22). Important development in spatial transcriptomics and live imaging should unravel the spatial organization of intracellular interactions supporting the function of MSCs within niches (21, 152). Deciphering the functional impact of MSCs heterogeneity and the division of labor between different MSC types should substantially assist tissue engineering purposes.
MSCs can be genetically manipulated ex-vivo to modulate the expression of key molecules before they are embedded within immune niches. This approach has been developed with success for the establishment of immune niches supporting AML engraftment (67) or supporting the development of dendritic cells (68) in humanized mice. Further programming of MSC transcriptional landscapes (63) broadly impacting on their function might open new avenues for the engineering of immune niches.
Understanding the developmental pathways of MSCs populations is of major relevance for tissue engineering. The phenotype and function of differentiated MSC types is potentially difficult to maintain in ex vivo cultures (63). Therefore, approaches co-opting the physiological developmental pathway of MSCs are of particular interest. For instance, disentangling the developing hierarchies within early MSC progenitors underpinning the BM niche (30–32, 153) should facilitate the technological implementation of MSCs to build synthetic niches and organoids.
An essential biological feature of immune niche is their ability to respond dynamically to immune perturbations. BM, for instance, also respond to acute inflammation, often by increasing myelopoiesis, a process termed as “emergency myelopoiesis” (154). In both cases dynamic changes of hematopoietic organs rely on adaptation of the stromal network (19, 22, 150, 151, 155, 156). Assessing this responsiveness feature should be of interest to recapitulate leukocyte development associated to inflammatory settings.
Genetic variation is likely to impact on physiological niches function. One key feature of the BM niche ossicle model is the possibility to transfer in the mouse the human BM microenvironment, either normal or pathological. For instance, the ossicle models represent a valuable tool to unravel the role of cellular and molecular mechanisms underlying the interactions between the hemopoietic and stromal compartment in normal or pathological niche. Ultimately, this analysis could be performed using MSCs and malignant cells from the same patient thereby defining a platform for drug screening. This approach could be applied to targeted therapies interfering with stromal support.
RS, AP, EM, and JP contributed equally to the manuscript writing and figure generation. AG, GA, JU, and PB helped during the manuscript writing and revision. MS and PG supervised the work and took part in the writing process. All authors contributed to the article and approved the submitted version. All figures were created by RS with BioRender.com.
The research was supported by the BBSRC, NC3RS, WWCR, CRUK, Rosetrees Trust, CRUK King's Health Partners Cancer Centre; IDEX (ANR-18-IDEX-0001) and INCA (PL-BIO-ICR1), Institut Curie, Institut National de la Santé et de la Recherche Médicale, Labex DCBIOL (ANR-10-IDEX-0001-02 PSL, ANR-11-LABX0043). RS was founded by Cancer research UK (C604/A29283). MS research was supported by AIRC-5x1000 2018-21147. JP was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project number 259332240/RTG 2099.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Friedenstein AJ, Gorskaja JF, Kulagina NN. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol. (1976) 4:267–74.
2. Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. (2015) 16:239–53. doi: 10.1016/j.stem.2015.02.019
4. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. (1999) 284:143–7. doi: 10.1126/science.284.5411.143
5. Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. (2007) 131:324–36. doi: 10.1016/j.cell.2007.08.025
6. Bourgine PE, Fritsch K, Pigeot S, Takizawa H, Kunz L, Kokkaliaris KD, et al. Fate distribution and regulatory role of human mesenchymal stromal cells in engineered hematopoietic bone organs. iScience. (2019) 19:504–13. doi: 10.1016/j.isci.2019.08.006
7. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. (2006) 25:977–88. doi: 10.1016/j.immuni.2006.10.016
9. Barker JE. Early transplantation to a normal microenvironment prevents the development of Steel hematopoietic stem cell defects. Exp Hematol. (1997) 25:542–7.
10. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. (2012) 481:457–62. doi: 10.1038/nature10783
11. Parmar K, Mauch P, Vergilio J, Sackstein R, Down J. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. (2007) 104:5431–6. doi: 10.1073/pnas.0701152104
12. Askmyr M, Sims N, Martin T, Purton L. What is the true nature of the osteoblastic hematopoietic stem cell niche? Trends Endocrinol Metab. (2009) 20:303–9. doi: 10.1016/j.tem.2009.03.004
13. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. (2005) 121:1109–21. doi: 10.1016/j.cell.2005.05.026
14. Kiel MJ, Radice GL, Morrison SJ. Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell. (2007) 1:204–17. doi: 10.1016/j.stem.2007.06.001
15. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. (2013) 502:637–43. doi: 10.1038/nature12612
16. Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. (2015) 526:126–30. doi: 10.1038/nature15250
17. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. (2016) 532:323–8. doi: 10.1038/nature17624
18. Christodoulou C, Spencer JA, Yeh SA, Turcotte R, Kokkaliaris KD, Panero R, et al. Live-animal imaging of native haematopoietic stem and progenitor cells. Nature. (2020) 578:278–83. doi: 10.1038/s41586-020-1971-z
19. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, et al. The bone marrow microenvironment at single-cell resolution. Nature. (2019) 569:222–8. doi: 10.1038/s41586-019-1104-8
20. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. (2019) 177:1915–32 e1916. doi: 10.1016/j.cell.2019.04.040
21. Baccin C, Al-Sabah J, Velten L, Helbling PM, Grunschlager F, Hernandez-Malmierca P, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. (2020) 22:38–48. doi: 10.1038/s41556-019-0439-6
22. Tikhonova AN, Lasry A, Austin R, Aifantis I. Cell-by-cell deconstruction of stem cell niches. Cell Stem Cell. (2020) 27:19–34. doi: 10.1016/j.stem.2020.06.013
23. Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. (1968) 6:230–47. doi: 10.1097/00007890-196803000-00009
24. Tavassoli M, Crosby WH. Transplantation of marrow to extramedullary sites. Science. (1968) 161:54–6. doi: 10.1126/science.161.3836.54
25. Chan CK, Chen CC, Luppen CA, Kim JB, DeBoer AT, Wei K, et al. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature. (2009) 457:490–4. doi: 10.1038/nature07547
26. Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. (2009) 206:2483–96. doi: 10.1084/jem.20091046
27. Houlihan DD, Mabuchi Y, Morikawa S, Niibe K, Araki D, Suzuki S, et al. Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-alpha. Nat Protoc. (2012) 7:2103–11. doi: 10.1038/nprot.2012.125
28. Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. (2010) 466:829–34. doi: 10.1038/nature09262
29. Bianco P. Bone and the hematopoietic niche: a tale of two stem cells. Blood. (2011) 117:5281–8. doi: 10.1182/blood-2011-01-315069
30. Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. (2014) 15:154–68. doi: 10.1016/j.stem.2014.06.008
31. Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell. (2016) 18:782–96. doi: 10.1016/j.stem.2016.02.015
32. Wolock SL, Krishnan I, Tenen DE, Matkins V, Camacho V, Patel S, et al. mapping distinct bone marrow niche populations and their differentiation paths. Cell Rep. (2019) 28:302–11 e305. doi: 10.1016/j.celrep.2019.06.031
33. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. (2013) 495:231–5. doi: 10.1038/nature11885
34. Asada N, Kunisaki Y, Pierce H, Wang Z, Fernandez NF, Birbrair A, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. (2017) 19:214–23. doi: 10.1038/ncb3475
35. Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, et al. Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity. (2016) 45:1219–31. doi: 10.1016/j.immuni.2016.11.004
36. Guezguez B, Campbell CJ, Boyd AL, Karanu F, Casado FL, Di Cresce C, et al. Regional localization within the bone marrow influences the functional capacity of human HSCs. Cell Stem Cell. (2013) 13:175–89. doi: 10.1016/j.stem.2013.06.015
37. Watchman CJ, Bourke VA, Lyon JR, Knowlton AE, Butler SL, Grier DD, et al. Spatial distribution of blood vessels and CD34+ hematopoietic stem and progenitor cells within the marrow cavities of human cancellous bone. J Nucl Med. (2007) 48:645–54. doi: 10.2967/jnumed.106.035337
38. Corselli M, Chin CJ, Parekh C, Sahaghian A, Wang W, Ge S, et al. Perivascular support of human hematopoietic stem/progenitor cells. Blood. (2013) 121:2891–901. doi: 10.1182/blood-2012-08-451864
39. Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, et al. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med. (2013) 210:1351–67. doi: 10.1084/jem.20122252
40. Tormin A, Li O, Brune JC, Walsh S, Schütz B, Ehinger M, et al. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood. (2011) 117:5067–77. doi: 10.1182/blood-2010-08-304287
41. Matsuoka Y, Nakatsuka R, Sumide K, Kawamura H, Takahashi M, Fujioka T, et al. Prospectively isolated human bone marrow cell-derived MSCs support primitive human CD34-negative hematopoietic stem cells. Stem Cells. (2015) 33:1554–65. doi: 10.1002/stem.1941
42. Kumar S, Geiger H. HSC niche biology and HSC expansion ex vivo. Trends Mol Med. (2017) 23:799–819. doi: 10.1016/j.molmed.2017.07.003
43. Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature. (2019) 571:117–21. doi: 10.1038/s41586-019-1244-x
44. Ng ES, Azzola L, Bruveris FF, Calvanese V, Phipson B, Vlahos K, et al. Differentiation of human embryonic stem cells to HOXA(+) hemogenic vasculature that resembles the aorta-gonad-mesonephros. Nat Biotechnol. (2016) 34:1168–79. doi: 10.1038/nbt.3702
45. Ditadi A, Sturgeon CM, Keller G. A view of human haematopoietic development from the Petri dish. Nat Rev Mol Cell Biol. (2017) 18:56–67. doi: 10.1038/nrm.2016.127
46. Ivanovs A, Rybtsov S, Ng ES, Stanley EG, Elefanty AG, Medvinsky A. Human haematopoietic stem cell development: from the embryo to the dish. Development. (2017) 144:2323–37. doi: 10.1242/dev.134866
47. Lis R, Karrasch CC, Poulos MG, Kunar B, Redmond D, Duran JGB, et al. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature. (2017) 545:439–45. doi: 10.1038/nature22326
48. Sugimura R, Jha DK, Han A, Soria-Valles C, da Rocha EL, Lu YF, et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. (2017) 545:432–8. doi: 10.1038/nature22370
49. Kim S, Shah SB, Graney PL, Singh A. Multiscale engineering of immune cells and lymphoid organs. Nat Rev Mater. (2019) 4:355–78. doi: 10.1038/s41578-019-0100-9
50. Feng Q, Chai C, Jiang XS, Leong KW, Mao HQ. Expansion of engrafting human hematopoietic stem/progenitor cells in three-dimensional scaffolds with surface-immobilized fibronectin. J Biomed Mater Res A. (2006) 78:781–91. doi: 10.1002/jbm.a.30829
51. Nichols JE, Cortiella J, Lee J, Niles JA, Cuddihy M, Wang S, et al. In vitro analog of human bone marrow from 3D scaffolds with biomimetic inverted colloidal crystal geometry. Biomaterials. (2009) 30:1071–9. doi: 10.1016/j.biomaterials.2008.10.041
52. Rödling L, Schwedhelm I, Kraus S, Bieback K, Hansmann J, Lee-Thedieck C. 3D models of the hematopoietic stem cell niche under steady-state and active conditions. Sci Rep. (2017) 7:4625. doi: 10.1038/s41598-017-04808-0
53. Braham MVJ, Ahlfeld T, Akkineni AR, Minnema MC, Dhert WJA, Oner FC, et al. Endosteal and perivascular subniches in a 3D bone marrow model for multiple myeloma. Tissue Eng Part C Methods. (2018) 24:300–12. doi: 10.1089/ten.tec.2017.0467
54. Ferreira MS, Jahnen-Dechent W, Labude N, Bovi M, Hieronymus T, Zenke M, et al. Cord blood-hematopoietic stem cell expansion in 3D fibrin scaffolds with stromal support. Biomaterials. (2012) 33:6987–97. doi: 10.1016/j.biomaterials.2012.06.029
55. Raic A, Rödling L, Kalbacher H, Lee-Thedieck C. Biomimetic macroporous PEG hydrogels as 3D scaffolds for the multiplication of human hematopoietic stem and progenitor cells. Biomaterials. (2014) 35:929–40. doi: 10.1016/j.biomaterials.2013.10.038
56. Leisten I, Kramann R, Ventura Ferreira MS, Bovi M, Neuss S, Ziegler P, et al. 3D co-culture of hematopoietic stem and progenitor cells and mesenchymal stem cells in collagen scaffolds as a model of the hematopoietic niche. Biomaterials. (2012) 33:1736–47. doi: 10.1016/j.biomaterials.2011.11.034
57. Bourgine PE, Klein T, Paczulla AM, Shimizu T, Kunz L, Kokkaliaris KD, et al. In vitro biomimetic engineering of a human hematopoietic niche with functional properties. Proc Natl Acad Sci USA. (2018) 115:E5688–95. doi: 10.1073/pnas.1805440115
58. Sieber S, Wirth L, Cavak N, Koenigsmark M, Marx U, Lauster R, et al. Bone marrow-on-a-chip: Long-term culture of human haematopoietic stem cells in a three-dimensional microfluidic environment. J Tissue Eng Regen Med. (2018) 12:479–89. doi: 10.1002/term.2507
59. Bruce A, Evans R, Mezan R, Shi L, Moses BS, Martin KH, et al. Three-dimensional microfluidic tri-culture model of the bone marrow microenvironment for study of acute lymphoblastic leukemia. PLoS ONE. (2015) 10:e0140506. doi: 10.1371/journal.pone.0140506
60. Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat Biomed Eng. (2020) 4:394–406. doi: 10.1038/s41551-019-0495-z
61. Torisawa YS, Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, et al. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat Methods. (2014) 11:663–9. doi: 10.1038/nmeth.2938
62. Reagan MR, Mishima Y, Glavey SV, Zhang Y, Manier S, Lu ZN, et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood. (2014) 124:3250–9. doi: 10.1182/blood-2014-02-558007
63. Nakahara F, Borger DK, Wei Q, Pinho S, Maryanovich M, Zahalka AH, et al. Engineering a haematopoietic stem cell niche by revitalizing mesenchymal stromal cells. Nat Cell Biol. (2019) 21:560–7. doi: 10.1038/s41556-019-0308-3
64. Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, Flavell RA, et al. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. (2013) 31:635–74. doi: 10.1146/annurev-immunol-032712-095921
65. Theocharides AP, Rongvaux A, Fritsch K, Flavell RA, Manz MG. Humanized hemato-lymphoid system mice. Haematologica. (2016) 101:5–19. doi: 10.3324/haematol.2014.115212
66. Patel ES, Okada S, Hachey K, Yang LJ, Durum SK, Moreb JS, et al. Regulation of in vitro human T cell development through interleukin-7 deprivation and anti-CD3 stimulation. BMC Immunol. (2012) 13:46. doi: 10.1186/1471-2172-13-46
67. Carretta M, de Boer B, Jaques J, Antonelli A, Horton SJ, Yuan H, et al. Genetically engineered mesenchymal stromal cells produce IL-3 and TPO to further improve human scaffold-based xenograft models. Exp Hematol. (2017) 51:36–46. doi: 10.1016/j.exphem.2017.04.008
68. Anselmi G, Vaivode K, Dutertre CA, Bourdely P, Missolo-Koussou Y, Newell E, et al. Engineered niches support the development of human dendritic cells in humanized mice. Nat Commun. (2020) 11:2054. doi: 10.1038/s41467-020-15937-y
69. Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. (2006) 12:657–64. doi: 10.1038/nm1417
70. Brehm MA, Racki WJ, Leif J, Burzenski L, Hosur V, Wetmore A, et al. Engraftment of human HSCs in nonirradiated newborn NOD-scid IL2rgamma null mice is enhanced by transgenic expression of membrane-bound human SCF. Blood. (2012) 119:2778–88. doi: 10.1182/blood-2011-05-353243
71. Takagi S, Saito Y, Hijikata A, Tanaka S, Watanabe T, Hasegawa T, et al. Membrane-bound human SCF/KL promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood. (2012) 119:2768–77. doi: 10.1182/blood-2011-05-353201
72. Toksoz D, Zsebo KM, Smith KA, Hu S, Brankow D, Suggs SV, et al. Support of human hematopoiesis in long-term bone marrow cultures by murine stromal cells selectively expressing the membrane-bound and secreted forms of the human homolog of the steel gene product, stem cell factor. Proc Natl Acad Sci USA. (1992) 89:7350–4. doi: 10.1073/pnas.89.16.7350
73. Tavakol D, Tratwal J, Bonini F, Genta M, Campos V, Burch P, et al. Injectable, scalable 3D tissue-engineered model of marrow hematopoiesis. Biomaterials. (2020) 232:119665. doi: 10.1016/j.biomaterials.2019.119665
74. Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation. (1974) 17:331–40. doi: 10.1097/00007890-197404000-00001
75. Krebsbach PH, Kuznetsov SA, Satomura K, Emmons RV, Rowe DW, Robey PG. Bone formation in vivo: comparison of osteogenesis by transplanted mouse and human marrow stromal fibroblasts. Transplantation. (1997) 63:1059–69. doi: 10.1097/00007890-199704270-00003
76. Kuznetsov SA, Krebsbach PH, Satomura K, Kerr J, Riminucci M, Benayahu D, et al. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res. (1997) 12:1335–47. doi: 10.1359/jbmr.1997.12.9.1335
77. Reinisch A, Etchart N, Thomas D, Hofmann NA, Fruehwirth M, Sinha S, et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood. (2015) 125:249–60. doi: 10.1182/blood-2014-04-572255
78. Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. (2008) 3:301–13. doi: 10.1016/j.stem.2008.07.003
79. Mankani MH, Kuznetsov SA, Marshall GW, Robey PG. Creation of new bone by the percutaneous injection of human bone marrow stromal cell and HA/TCP suspensions. Tissue Eng Part A. (2008) 14:1949–58. doi: 10.1089/ten.tea.2007.0348
80. Vaiselbuh SR, Edelman M, Lipton JM, Liu JM. Ectopic human mesenchymal stem cell-coated scaffolds in NOD/SCID mice: an in vivo model of the leukemia niche. Tissue Eng Part C Methods. (2010) 16:1523–31. doi: 10.1089/ten.tec.2010.0179
81. Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. (2012) 10:120–36. doi: 10.1016/j.stem.2012.01.006
82. Dupard S, Grigoryan A, Farhat S, Coutu D, Bourgine P. Development of humanized ossicles: bridging the hematopoietic gap. Trends Mol Med. (2020) 26:552–69. doi: 10.1016/j.molmed.2020.01.016
83. Abarrategi A, Mian S, Passaro D, Rouault-Pierre K, Grey W, Bonnet D. Modeling the human bone marrow niche in mice: From host bone marrow engraftment to bioengineering approaches. J Exp Med. (2018) 215:729–43. doi: 10.1084/jem.20172139
84. Lee J, Li M, Milwid J, Dunham J, Vinegoni C, Gorbatov R, et al. Implantable microenvironments to attract hematopoietic stem/cancer cells. Proc Natl Acad Sci USA. (2012) 109:19638–43. doi: 10.1073/pnas.1208384109
85. Chen Y, Jacamo R, Shi YX, Wang RY, Battula VL, Konoplev S, et al. Human extramedullary bone marrow in mice: a novel in vivo model of genetically controlled hematopoietic microenvironment. Blood. (2012) 119:4971–80. doi: 10.1182/blood-2011-11-389957
86. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood. (2014) 123:2691–702. doi: 10.1182/blood-2013-06-511527
87. Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med. (2016) 22:812–21. doi: 10.1038/nm.4103
88. Groen RW, Noort WA, Raymakers RA, Prins HJ, Aalders L, Hofhuis FM, et al. Reconstructing the human hematopoietic niche in immunodeficient mice: opportunities for studying primary multiple myeloma. Blood. (2012) 120:e9–16. doi: 10.1182/blood-2012-03-414920
89. Antonelli A, Noort WA, Jaques J, de Boer B, de Jong-Korlaar R, Brouwers-Vos AZ, et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood. (2016) 128:2949–59. doi: 10.1182/blood-2016-05-719021
90. Sontakke P, Carretta M, Jaques J, Brouwers-Vos AZ, Lubbers-Aalders L, Yuan H, et al. Modeling BCR-ABL and MLL-AF9 leukemia in a human bone marrow-like scaffold-based xenograft model. Leukemia. (2016) 30:2064–73. doi: 10.1038/leu.2016.108
91. Scotti C, Piccinini E, Takizawa H, Todorov A, Bourgine P, Papadimitropoulos A, et al. Engineering of a functional bone organ through endochondral ossification. Proc Natl Acad Sci USA. (2013) 110:3997–4002. doi: 10.1073/pnas.1220108110
92. Fritsch K, Pigeot S, Feng X, Bourgine PE, Schroeder T, Martin I, et al. Engineered humanized bone organs maintain human hematopoiesis in vivo. Exp Hematol. (2018) 61:45–51 e45. doi: 10.1016/j.exphem.2018.01.004
93. Serafini M, Sacchetti B, Pievani A, Redaelli D, Remoli C, Biondi A, et al. Establishment of bone marrow and hematopoietic niches in vivo by reversion of chondrocyte differentiation of human bone marrow stromal cells. Stem Cell Res. (2014) 12:659–72. doi: 10.1016/j.scr.2014.01.006
94. Pievani A, Sacchetti B, Corsi A, Rambaldi B, Donsante S, Scagliotti V, et al. Human umbilical cord blood-borne fibroblasts contain marrow niche precursors that form a bone/marrow organoid in vivo. Development. (2017) 144:1035–44. doi: 10.1242/dev.142836
95. Holzapfel BM, Hutmacher DW, Nowlan B, Barbier V, Thibaudeau L, Theodoropoulos C, et al. Tissue engineered humanized bone supports human hematopoiesis in vivo. Biomaterials. (2015) 61:103–14. doi: 10.1016/j.biomaterials.2015.04.057
96. Abarrategi A, Foster K, Hamilton A, Mian SA, Passaro D, Gribben J, et al. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J Clin Invest. (2017) 127:543–8. doi: 10.1172/JCI89364
97. Osinga R, Di Maggio N, Todorov A, Allafi N, Barbero A, Laurent F, et al. Generation of a bone organ by human adipose-derived stromal cells through endochondral ossification. Stem Cells Transl Med. (2016) 5:1090–7. doi: 10.5966/sctm.2015-0256
98. Dragoo JL, Lieberman JR, Lee RS, Deugarte DA, Lee Y, Zuk PA, et al. Tissue-engineered bone from BMP-2-transduced stem cells derived from human fat. Plast Reconstr Surg. (2005) 115:1665–73. doi: 10.1097/01.PRS.0000161459.90856.AB
99. Kang Y, Liao WM, Yuan ZH, Sheng PY, Zhang LJ, Yuan XW, et al. In vitro and in vivo induction of bone formation based on adeno-associated virus-mediated BMP-7 gene therapy using human adipose-derived mesenchymal stem cells. Acta Pharmacol Sin. (2007) 28:839–49. doi: 10.1111/j.1745-7254.2007.00583.x
100. Sacchetti B, Funari A, Remoli C, Giannicola G, Kogler G, Liedtke S, et al. No identical “mesenchymal stem cells” at different times and sites: human committed progenitors of distinct origin and differentiation potential are incorporated as adventitial cells in microvessels. Stem Cell Rep. (2016) 6:897–913. doi: 10.1016/j.stemcr.2016.05.011
101. Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood. (2015) 125:2630–40. doi: 10.1182/blood-2014-11-570218
102. Pabst G, Lind K, Graf R, Zebisch A, Stölzel F, Döhner K, et al. TP53 mutated AML subclones exhibit engraftment in a humanized bone marrow ossicle mouse model. Ann Hematol. (2020) 99:653–5. doi: 10.1007/s00277-020-03920-y
103. Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. (2017) 2:e90036. doi: 10.1172/jci.insight.90036
104. Waclawiczek A, Hamilton A, Rouault-Pierre K, Abarrategi A, Albornoz MG, Miraki-Moud F, et al. Mesenchymal niche remodeling impairs hematopoiesis via stanniocalcin 1 in acute myeloid leukemia. J Clin Invest. (2020) 130:3038–50. doi: 10.1172/JCI133187
105. Pievani A, Donsante S, Tomasoni C, Corsi A, Dazzi F, Biondi A, et al. Acute myeloid leukemia shapes the bone marrow stromal niche in vivo. Haematologica. (2020). doi: 10.3324/haematol.2020.247205
106. van Ewijk W, Wang B, Hollander G, Kawamoto H, Spanopoulou E, Itoi M, et al. Thymic microenvironments, 3-D versus 2-D? Semin Immunol. (1999) 11:57–64. doi: 10.1006/smim.1998.0158
107. Brauer PM, Singh J, Xhiku S, Zuniga-Pflucker JC. T cell genesis: in vitro veritas est? Trends Immunol. (2016) 37:889–901. doi: 10.1016/j.it.2016.09.008
108. Palmer DB. The effect of age on thymic function. Front Immunol. (2013) 4:316. doi: 10.3389/fimmu.2013.00316
109. Savino W. The thymus is a common target organ in infectious diseases. PLoS Pathog. (2006) 2:e62. doi: 10.1371/journal.ppat.0020062
110. Tajima A, Pradhan I, Trucco M, Fan Y. Restoration of thymus function with bioengineered thymus organoids. Curr Stem Cell Rep. (2016) 2:128–39. doi: 10.1007/s40778-016-0040-x
111. Zhao Y, Parkhurst MR, Zheng Z, Cohen CJ, Riley JP, Gattinoni L, et al. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via Notch signaling. Cancer Res. (2007) 67:2425–9. doi: 10.1158/0008-5472.CAN-06-3977
112. Giannoni F, Hardee CL, Wherley J, Gschweng E, Senadheera S, Kaufman ML, et al. Allelic exclusion and peripheral reconstitution by TCR transgenic T cells arising from transduced human hematopoietic stem/progenitor cells. Mol Ther. (2013) 21:1044–54. doi: 10.1038/mt.2013.8
113. Snauwaert S, Verstichel G, Bonte S, Goetgeluk G, Vanhee S, Van Caeneghem Y, et al. In vitro generation of mature, naive antigen-specific CD8(+) T cells with a single T-cell receptor by agonist selection. Leukemia. (2014) 28:830–41. doi: 10.1038/leu.2013.285
114. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. (2013) 31:928–33. doi: 10.1038/nbt.2678
115. Montel-Hagen A, Crooks GM. From pluripotent stem cells to T cells. Exp Hematol. (2019) 71:24–31. doi: 10.1016/j.exphem.2018.12.001
116. Montel-Hagen A, Seet CS, Li S, Chick B, Zhu Y, Chang P, et al. Organoid-induced differentiation of conventional T cells from human pluripotent stem cells. Cell Stem Cell. (2019) 24:376–89 e378. doi: 10.1016/j.stem.2018.12.011
117. Bousso P, Bhakta NR, Lewis RS, Robey E. Dynamics of thymocyte-stromal cell interactions visualized by two-photon microscopy. Science. (2002) 296:1876–80. doi: 10.1126/science.1070945
118. Haddad R, Guimiot F, Six E, Jourquin F, Setterblad N, Kahn E, et al. Dynamics of thymus-colonizing cells during human development. Immunity. (2006) 24:217–30. doi: 10.1016/j.immuni.2006.01.008
119. Farley AM, Morris LX, Vroegindeweij E, Depreter ML, Vaidya H, Stenhouse FH, et al. Dynamics of thymus organogenesis and colonization in early human development. Development. (2013) 140:2015–26. doi: 10.1242/dev.087320
120. Rossi FM, Corbel SY, Merzaban JS, Carlow DA, Gossens K, Duenas J, et al. Recruitment of adult thymic progenitors is regulated by P-selectin and its ligand PSGL-1. Nat Immunol. (2005) 6:626–34. doi: 10.1038/ni1203
121. Calderón L, Boehm T. Three chemokine receptors cooperatively regulate homing of hematopoietic progenitors to the embryonic mouse thymus. Proc Natl Acad Sci USA. (2011) 108:7517–22. doi: 10.1073/pnas.1016428108
122. Metzger TC, Anderson MS. Control of central and peripheral tolerance by Aire. Immunol Rev. (2011) 241:89–103. doi: 10.1111/j.1600-065X.2011.01008.x
123. Piccinini E, Bonfanti P. Disassembling and reaggregating the thymus: the pros and cons of current assays. Methods Mol Biol. (2019) 1899:129–42. doi: 10.1007/978-1-4939-8938-6_10
124. Röpke C. Thymic epithelial cell culture. Microsc Res Tech. (1997) 38:276–86. doi: 10.1002/(SICI)1097-0029(19970801)38:3<276::AID-JEMT8>3.0.CO
125. Masuda K, Germeraad WT, Satoh R, Itoi M, Ikawa T, Minato N, et al. Notch activation in thymic epithelial cells induces development of thymic microenvironments. Mol Immunol. (2009) 46:1756–67. doi: 10.1016/j.molimm.2009.01.015
126. Corbeaux T, Hess I, Swann JB, Kanzler B, Haas-Assenbaum A, Boehm T. Thymopoiesis in mice depends on a Foxn1-positive thymic epithelial cell lineage. Proc Natl Acad Sci USA. (2010) 107:16613–8. doi: 10.1073/pnas.1004623107
127. Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. (2002) 17:749–56. doi: 10.1016/S1074-7613(02)00474-0
128. La Motte-Mohs RN, Herer E, Zuniga-Pflucker JC. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood. (2005) 105:1431–9. doi: 10.1182/blood-2004-04-1293
129. Yeoman H, Gress RE, Bare CV, Leary AG, Boyse EA, Bard J, et al. Human bone marrow and umbilical cord blood cells generate CD4+ and CD8+ single-positive T cells in murine fetal thymus organ culture. Proc Natl Acad Sci USA. (1993) 90:10778–82. doi: 10.1073/pnas.90.22.10778
130. Poznansky MC, Evans RH, Foxall RB, Olszak IT, Piascik AH, Hartman KE. Efficient generation of human T cells from a tissue-engineered thymic organoid. Nat Biotechnol. (2000) 18:729–34. doi: 10.1038/77288
131. Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. (2004) 304:104–7. doi: 10.1126/science.1093933
132. Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. (2005) 106:1565–73. doi: 10.1182/blood-2005-02-0516
133. Pearson T, Shultz LD, Miller D, King M, Laning J, Fodor W, et al. Non-obese diabetic-recombination activating gene-1 (NOD-Rag1 null) interleukin (IL)-2 receptor common gamma chain (IL2r gamma null) null mice: a radioresistant model for human lymphohaematopoietic engraftment. Clin Exp Immunol. (2008) 154:270–84. doi: 10.1111/j.1365-2249.2008.03753.x
134. Chung B, Montel-Hagen A, Ge S, Blumberg G, Kim K, Klein S, et al. Engineering the human thymic microenvironment to support thymopoiesis in vivo. Stem Cells. (2014) 32:2386–96. doi: 10.1002/stem.1731
135. Parent AV, Russ HA, Khan IS, LaFlam TN, Metzger TC, Anderson MS. Generation of functional thymic epithelium from human embryonic stem cells that supports host T cell development. Cell Stem Cell. (2013) 13:219–29. doi: 10.1016/j.stem.2013.04.004
136. Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. (2006) 12:1316–22. doi: 10.1038/nm1431
137. Wege AK, Melkus MW, Denton PW, Estes JD, Garcia JV. Functional and phenotypic characterization of the humanized BLT mouse model. Curr Top Microbiol Immunol. (2008) 324:149–65. doi: 10.1007/978-3-540-75647-7_10
138. Kalscheuer H, Danzl N, Onoe T, Faust T, Winchester R, Goland R, et al. A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med. (2012) 4:125ra130. doi: 10.1126/scitranslmed.3003481
139. Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat Methods. (2017) 14:521–30. doi: 10.1038/nmeth.4237
140. Hare KJ, Jenkinson EJ, Anderson G. In vitro models of T cell development. Semin Immunol. (1999) 11:3–12. doi: 10.1006/smim.1998.0151
141. Anderson G, Jenkinson EJ. Fetal thymus organ culture. CSH Protoc. (2007) 2007:pdb.prot4808. doi: 10.1101/pdb.prot4808
142. Park JE, Botting RA, Domínguez Conde C, Popescu DM, Lavaert M, Kunz DJ, et al. A cell atlas of human thymic development defines T cell repertoire formation. Science. (2020) 367:eaay3224. doi: 10.1126/science.aay3224
143. Kadouri N, Nevo S, Goldfarb Y, Abramson J. Thymic epithelial cell heterogeneity: TEC by TEC. Nat Rev Immunol. (2020) 20:239–53. doi: 10.1038/s41577-019-0238-0
144. Kodama H, Nose M, Niida S, Nishikawa S, Nishikawa S. Involvement of the c-kit receptor in the adhesion of hematopoietic stem cells to stromal cells. Exp Hematol. (1994) 22:979–84.
145. Schmitt TM, de Pooter RF, Gronski MA, Cho SK, Ohashi PS, Zuniga-Pflucker JC. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat Immunol. (2004) 5:410–7. doi: 10.1038/ni1055
146. Patel E, Wang B, Lien L, Wang Y, Yang LJ, Moreb JS, et al. Diverse T-cell differentiation potentials of human fetal thymus, fetal liver, cord blood and adult bone marrow CD34 cells on lentiviral Delta-like-1-modified mouse stromal cells. Immunology. (2009) 128:e497–505. doi: 10.1111/j.1365-2567.2008.03013.x
147. Mohtashami M, Zúñiga-Pflücker JC. Three-dimensional architecture of the thymus is required to maintain delta-like expression necessary for inducing T cell development. J Immunol. (2006) 176:730–4. doi: 10.4049/jimmunol.176.2.730
148. Rodda LB, Lu E, Bennett ML, Sokol CL, Wang X, Luther SA, et al. Single-cell RNA sequencing of lymph node stromal cells reveals niche-associated heterogeneity. Immunity. (2018) 48:1014–28.e1016. doi: 10.1016/j.immuni.2018.04.006
149. Cheng HW, Onder L, Novkovic M, Soneson C, Lutge M, Pikor N, et al. Origin and differentiation trajectories of fibroblastic reticular cells in the splenic white pulp. Nat Commun. (2019) 10:1739. doi: 10.1038/s41467-019-09728-3
150. Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, et al. Stress-induced changes in bone marrow stromal cell populations revealed through single-cell protein expression mapping. Cell Stem Cell. (2019) 25:570–83 e577. doi: 10.1016/j.stem.2019.06.003
151. Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Dominguez A, et al. Author Correction: the bone marrow microenvironment at single-cell resolution. Nature. (2019) 572:E6. doi: 10.1038/s41586-019-1394-x
152. Lucas D, Salomonis N, Grimes HL. Unraveling bone marrow architecture. Nat Cell Biol. (2020) 22:5–6. doi: 10.1038/s41556-019-0447-6
153. Hu X, Garcia M, Weng L, Jung X, Murakami JL, Kumar B, et al. Identification of a common mesenchymal stromal progenitor for the adult haematopoietic niche. Nat Commun. (2016) 7:13095. doi: 10.1038/ncomms13095
154. Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. (2017) 38:345–57. doi: 10.1016/j.it.2017.01.004
155. Acton SE, Reis e Sousa C. Dendritic cells in remodeling of lymph nodes during immune responses. Immunol Rev. (2016) 271:221–9. doi: 10.1111/imr.12414
Keywords: hematopoiesis, organoids model, bone marrow niche, mesenchymal stroma cell, thymus epithelial cell, 3D culture
Citation: Pievani A, Savoldelli R, Poelchen J, Mattioli E, Anselmi G, Girardot A, Utikal J, Bourdely P, Serafini M and Guermonprez P (2021) Harnessing Mesenchymal Stromal Cells for the Engineering of Human Hematopoietic Niches. Front. Immunol. 12:631279. doi: 10.3389/fimmu.2021.631279
Received: 19 November 2020; Accepted: 10 February 2021;
Published: 15 March 2021.
Edited by:
César Nombela Arrieta, University of Zurich, SwitzerlandReviewed by:
Paul Emile Bourgine, Lund University, SwedenCopyright © 2021 Pievani, Savoldelli, Poelchen, Mattioli, Anselmi, Girardot, Utikal, Bourdely, Serafini and Guermonprez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre Guermonprez, cGllcnJlLmd1ZXJtb25wcmV6QGNucnMuZnI=
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.