 Rut Mora-Buch
Rut Mora-Buch Shannon K. Bromley
Shannon K. Bromley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 19 March 2021
Sec. Immunological Memory
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.624199
This article is part of the Research TopicResident Memory T Cells: Guardians of the Balance of Local Immunity and PathologyView all 11 articles
Resident memory CD8+ T (TRM) cells are a lymphocyte lineage distinct from circulating memory CD8+ T cells. TRM lodge within peripheral tissues and secondary lymphoid organs where they provide rapid, local protection from pathogens and control tumor growth. However, dysregulation of CD8+ TRM formation and/or activation may contribute to the pathogenesis of autoimmune diseases. Intrinsic mechanisms, including transcriptional networks and inhibitory checkpoint receptors control TRM differentiation and response. Additionally, extrinsic stimuli such as cytokines, cognate antigen, fatty acids, and damage signals regulate TRM formation, maintenance, and expansion. In this review, we will summarize knowledge of CD8+ TRM generation and highlight mechanisms that regulate the persistence and responses of heterogeneous TRM populations in different tissues and distinct microenvironments.
Long-term memory to pathogens is a key feature of the adaptive immune system. The ability of memory T cells to mount rapid and potent responses against previously encountered antigens maintains human health by controlling infections and tumor growth; it also provides the rationale for designing vaccines against pathogens and immune therapies to treat cancer. By recirculating through blood and lymph, circulating memory T cells may provide broad tissue immune surveillance. However, recent findings demonstrated that long after the resolution of infection, the majority of memory CD8+ T cells are non-circulating (1). Rather, most CD8+ memory T cells are stably maintained in tissues as tissue resident memory T cells (TRM) that exhibit transcriptional and phenotypic characteristics distinct from circulating memory CD8+ T cells (2). Early studies identified TRM within the epithelial compartment of barrier tissues including skin, lung, and intestine (3–8). Later, TRM were identified in the tissue stroma as well as in non-barrier tissues such as liver, brain, and secondary lymphoid organs including spleen and lymph nodes (LN) (9–12). CD8+ TRM deliver highly effective, localized responses to pathogen challenge (4, 8). Additionally, CD8+ T cells with a TRM phenotype are a target candidate for anti-tumor immunotherapy (13–15) and predict an improved prognosis in several different cancers (16–23). Although TRM provide potent protection against pathogens and tumors, TRM dysregulation has been linked to immune-mediated diseases including psoriasis (24), vitiligo (24), and alopecia areata in the skin (25), and inflammatory bowel disease in the intestine (26). Additionally, TRM develop following sensitization to allergens and play a role in hypersensitivity reactions in allergic contact dermatitis (27, 28) and asthma (29). Finally, TRM have been linked to fixed drug eruptions (30), as well as rejection of solid organ transplants (31). This review will discuss intrinsic and extrinsic mechanisms that promote CD8+ TRM formation, maintenance and function for defense against invading pathogens, as well as mechanisms that limit TRM formation and effector response to prevent excessive inflammation and tissue damage (Figure 1).
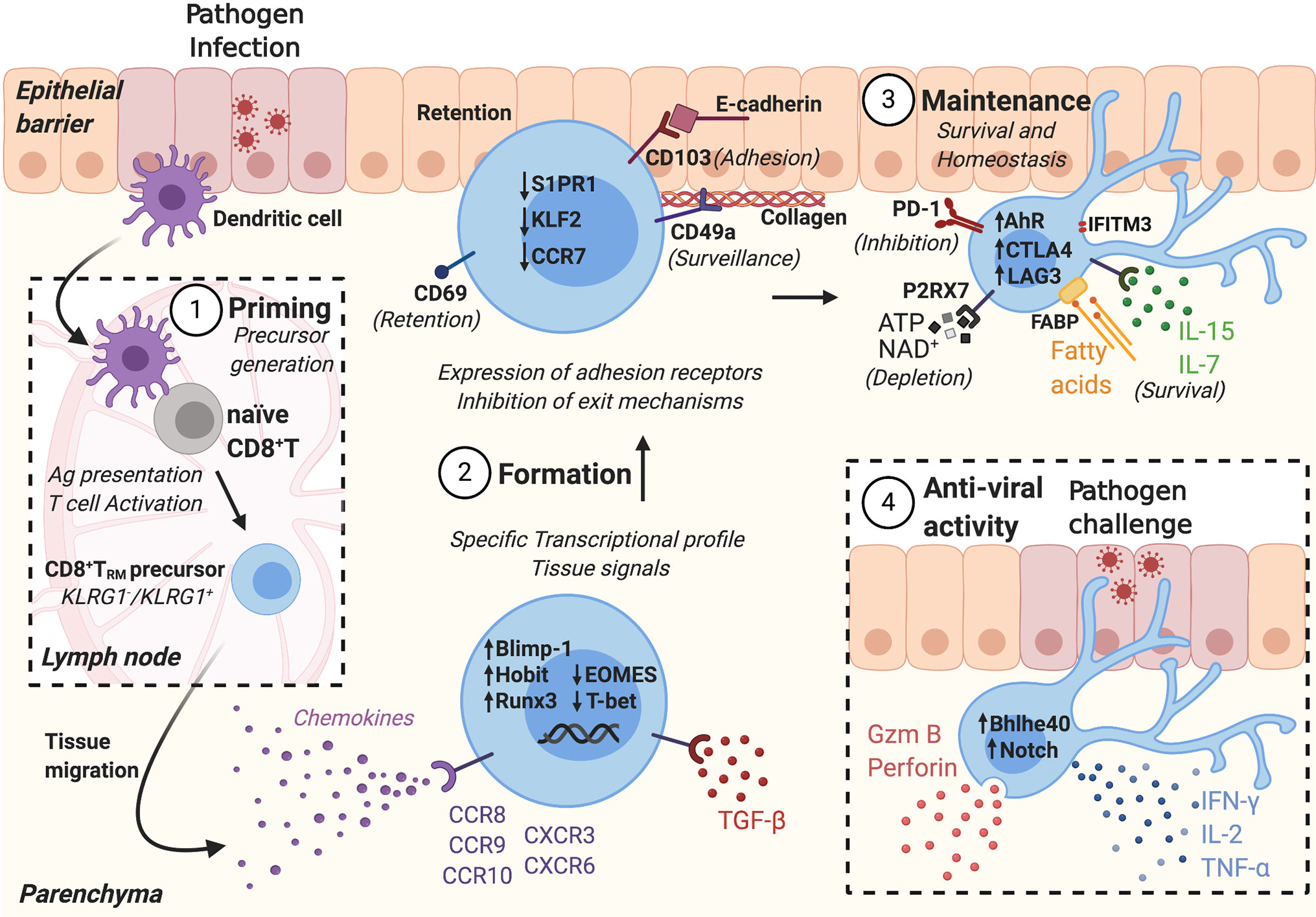
Figure 1 CD8+ TRM formation and anti-viral activity is tightly regulated in different stages. 1) Following pathogen infection, tissue dendritic cells (DCs) migrate to the draining lymph nodes and present antigens to naïve T cells. Antigen-specific naïve T cells are activated, generating CD8+ TRM precursors. 2) CD8+ TRM precursors migrate into peripheral tissues, following chemotactic signals. CD8+ TRM formation depends on tissue signals that activate a TRM transcriptional profile, including the expression of adhesion receptors and inhibition of exit mechanisms. 3) CD8+ TRM are maintained in the tissue where they receive survival signals and express inhibitory receptors to maintain tissue homeostasis. 4) During secondary infection, CD8+ TRM are activated, secrete effector molecules, and amplify the immune response.
Following cognate antigen recognition, naïve CD8+ T cells become activated, proliferate and give rise to heterogeneous progeny with distinct effector and memory cell fates. Recent experimental evidence suggests that extrinsic signals can influence CD8+ T cell fate even before antigen recognition (32) (Box 1). After antigen activation, the majority of activated T cells die by apoptosis during the contraction phase of the immune response, but a small minority survive to become memory CD8+ T cells. Whether activated T cells survive may depend on external signals, including growth factor availability, antigen, and inflammation, as well as internal signals such as transcription factor and growth factor receptor expression. Multiple, non-mutually exclusive models have been proposed to explain the development of diverse populations of effector and memory CD8+ T cells (34). For example, the fixed lineage model proposes that commitment to effector or memory T cell lineages occurs soon after T cell stimulation, as early as the first cell division and may result from the asymmetric division of effector fate-associated factors. On the other hand, the decreasing potential model posits that early effector cells have memory potential that is lost with increased or prolonged stimulation with antigen or cytokines. More recently, Rosato et al., have proposed an expanded model of decreasing potential to include CD8+ TRM. They propose that the differentiation of CD8+ T cells along a continuous axis of decreasing memory potential is irreversible. However, they also divide cells based on parallel paths of migration status-stationary or migratory, that may be altered by extrinsic stimuli including TCR signaling and inflammation (35), reflecting the cells’ plasticity.
Box 1. Pre-Programmed Naïve CD8+ T Cells: The Existence of a Stage 0.
Although current models suggest that a single naïve T cell has the potential to differentiate into all effector and memory subsets depending on the antigen, costimulatory, and cytokine stimulation they receive, recent experimental evidence suggests that extrinsic signals influence CD8+ T cell fate even before antigen recognition. Recent work by Mani et al. demonstrated that extrinsic cytokine signaling can imprint naïve CD8+ T cells for subsequent TRM formation. Migratory DCs expressing TGF-β-activating integrins in the LN activate TGF-β and epigenetically condition naïve CD8+ T cells, even before antigen stimulation, to form epithelial CD8+ TRM in the skin (32). These results suggest that during immune homeostasis, the LN environment affects future T cell fate. In addition, research using a tamoxifen-inducible fate-mapping mouse model to mark CD8+ T cells made in the thymus during fetal, neonatal, and adult stages, Smith et al. demonstrated that naïve CD8+ T cells generated during different developmental stages, fetal vs. adult, acquire different phenotypes upon antigen encounter. These results suggest that CD8+ T cell fate may be controlled by the timing of naïve precursor cell maturation in the thymus (33). These studies open the possibility of additional regulatory mechanisms and signals that impact future CD8+ TRM generation even before inflammatory or antigen insult. Future studies are needed to better understand how intrinsic and extrinsic signals during naïve CD8+ T cell generation and homeostasis influence CD8+ T cell fate.
Expression of KLRG1 and CD127 has been used to define the memory potential of effector CD8+ T cells around the peak of the immune response. Adoptive transfer studies suggest that KLRG1+ CD127− short-lived effector cells (SLEC) tend to die following clearance of antigen, whereas KLRG1− CD127+ memory precursor effector cells (MPEC) preferentially survive to give rise to memory CD8+ T cells (36). Using a single cell adoptive transfer approach, Stemberger et al. tracked the progeny of individual naïve CD8+ T cells. Using CD62L and CD127 as phenotypic markers, and IL-2, TNF-α, IFN-γ and CD107a expression as functional readouts, they demonstrated that diverse effector and memory CD8+ T cells can arise from the same naïve precursor T cell (37). Additionally, single cell tracing experiments using adoptive transfer of barcode labeled OT-I T cells and systemic or local infection models, confirmed that both effector and memory CD8+ T cell subsets derive from the same precursors in the naïve T cell pool (38). Moreover, TCR repertoire analysis of antigen-activated CD8+ T cells demonstrated that 35 days post-immunization, CD8+ memory T cells recovered from the skin share a common clonal origin with memory CD8+ T cells isolated from draining and distant LNs, suggesting that TRM and circulating memory T cells can develop from an individual naïve T cell (39). Together, these results suggest that memory T cell fate is not imprinted on naïve T cells, but rather that individual naïve T cells can give rise to all effector and memory CD8+ T cell subsets. However, recent data suggest that although the majority of naïve T cells contribute to both circulating memory and CD69+ CD103+ TRM cell populations, the contribution of individual clones to each memory pool varies (40). Additionally, analysis of individual T cell families (a naïve T cell and its progeny) demonstrated that clonal expansion and differentiation of T cells bearing the same TCR are heterogeneous, and so the contribution of the progeny of individual naïve T cells varies between primary versus recall responses (41).
Substantial effort has focused on identifying CD8+ TRM precursor cells and defining when CD8+ T cells commit to a TRM fate (Supplementary Table 1). Like circulating memory CD8+ T cells, CD8+ TRM can also differentiate from KLRG1− precursor cells. Mackay et al. demonstrated that KLRG1−, but not KLRG1+, HSV-specific gBT-I effector T cells sorted from the spleens of mice 6 days post-HSV infection, generated cutaneous CD103+ TRM cells following their adoptive transfer into HSV-infected recipient mice (42). Subsequent studies suggested that CD8+ TRM are derived from MPEC after their entry into peripheral tissues. For example, following infection with Listeria monocytogenes (LM), splenic MPEC and SLEC lack expression of the TRM receptors, CD69 and CD103. However, MPEC but not SLEC recovered from the intestine express CD103 and CD69 (43). Additionally, elegant work performed by Kurd et al. used single-cell RNA sequencing to define the gene expression patterns of individual CD8+ T cells in the spleen and small intestine intraepithelial lymphocyte (siIEL) compartments over the course of lymphocytic choriomeningitis virus (LCMV) infection. Four days post-infection, the earliest time-point that virus specific CD8+ T cells are detected within intestinal tissue, activated CD44hi small intestinal CD8+ T cells display a transcriptional profile distinct from splenic CD44hi CD8+ T cells. Even at day 3 following infection, splenic CD8+ T cells do not resemble siIEL, suggesting that circulating precursors are not committed to a TRM fate until after entry into the tissue (44). In contrast, using lineage tracing and single-cell transcriptome analysis, Kok et al. identified a subset of circulating effector CD8+ T cells at the peak of effector T cell expansion after skin DNA vaccination that are enriched for TRM fate-associated gene expression and have a higher propensity to form TRM (40). Because the clonal composition of TRM recovered from anatomically separate skin immunization sites is similar, they proposed that a committed TRM precursor pool exists in the circulation, before entry into the tissue. Although the nature, timing or location of the early signals that imprint the ability to form TRM before tissue entry were not defined by this study, work by Mani et al. suggests that during immune homeostasis, naïve CD8+ T cells are epigenetically preconditioned for TRM formation through their interaction with migratory dendritic cells (DCs) expressing TGF-β-activating integrins (32).
Recent studies suggest that effector cells may maintain plasticity to dedifferentiate and seed the memory pool. Using a KLRG1Cre reporter system that allows tracking of KLRG1+ T cells over time, Herndler-Brandstetter et al. demonstrated that early post infection, KLRG1+ effector CD8+ T cells can downregulate KLRG1 and differentiate into all memory T cell lineages, including CD8+ TRM in the lung, intestine, and skin, and mediate effective protective immunity (45). Additionally, work by Youngblood et al. examined the transcriptional and epigenetic changes in naïve CD8+ T cells during differentiation to effector and memory cells over the course of an acute LCMV infection. Whole genome bisulfite sequencing analysis demonstrated that epigenetic repression of naïve-associated genes in effector CD8+ T cells can be reversed in cells that develop into long-lived memory CD8+ T cells, while key effector genes including Gzmb and Prf1 remain demethylated (46). These studies suggest that effector CD8+ T cells may not have a fixed fate and contribute to the diversity of the memory T cell pool.
The finding that CD8+ TRM and circulating memory CD8+ T cells can express identical TCR sequences (37) counters the hypothesis that TCR affinity or signal strength determines CD8+ TRM differentiation. However, intrinsic signals, including TCR signal strength and antigen affinity can influence CD8+ memory T cell development. For example, a study using OT-I TCR transgenic mice with a point mutation in the conserved antigen receptor transmembrane (CART) motif suggests that effector and memory T cell differentiation require different signals. Both WT and mutant T cells differentiate comparably into effector T cells. However, mutant cells fail to polarize TCR to the immunological synapse, have decreased NFKB induction, and this impaired TCR signaling is correlated with decreased memory CD8+ T cell differentiation (47). Additionally, studies have demonstrated that higher affinity TCR interactions direct CD8+ T cells to a CD62L− TEM fate, whereas lower TCR affinities promote CD62L+ TCM formation (48). Several studies also support the idea that TCR affinity and signal strength have a direct and unique impact on CD8+ TRM formation. For example, in a mouse model of persistent polyomavirus (MPyV) infection, high-affinity CD8+ CD69+ TRM cells in the brain originate from high-affinity CD62L− effector cells present in the tissue during acute infection (49). In contrast, in a separate study again using a model of MPyV, the data instead suggested that lower TCR stimulation strength improves memory potential and generates functional brain CD62L− CD69+ TRM cells (50). Similarly, in an acute influenza infection model, lower affinity TCR stimulation is more likely than higher affinity interactions to induce TRM formation, suggesting that TCR affinity can influence TRM differentiation (51) and may provide a mechanism to regulate the diversity of antigen-specific TRM within tissues.
Additional intrinsic CD8+ T cell characteristics may also affect CD8+ T cell fate. For example, variation in expression levels of signaling proteins including CD8, ERK-1 and SHP-1 generates a range of CD8+ T cell responsiveness to antigen stimulation. However, co-regulation of signaling proteins limits this variability, potentially providing a mechanism to diversify cell fate, but control self-reactivity (52). Similarly, Marchingo et al. used a high-throughput clonal assay to simultaneously measure the expansion fate of multiple clonal families expressing identical TCR in a single culture well. Their results demonstrate that following stimulation, progeny from clonal families stop dividing and return to quiescence at or near the same generation, suggesting that regulation of CD8+ T cell expansion fate is at the level of the individual clone (53). Stochastic variation in costimulatory and cytokine receptor expression by naïve CD8+ T cells, for example differences in CD28 receptor expression, influences the generation at which an initial individual activated cell reverts to a quiescent state (53). Future in vivo research is required to determine whether stochastic variation in protein expression by naïve T cells, either before or during early priming, has an effect on subsequent T cell fate, including CD8+ TRM differentiation.
Contact between DCs and antigen-specific CD8+ T cells can influence the fate of responding T cells (54–57). DCs carrying pathogen-derived antigens migrate to draining LN and prime naïve CD8+ T cells. The interaction between DCs and T cells within the LN occurs in three stages initiated by brief encounters, followed by more stable contacts and concludes with a return to brief contacts and rapid T cell migration, accompanied by the commencement of T cell proliferation (58). Multiphoton intravital microscopy (MP-IVM) allowed for the analysis of how and when the interactions between naïve CD8+ T cells and DCs determine effector and memory CD8+ T cell differentiation, and suggested that stable contacts and a high antigen concentration are critical to induce memory T cell generation (59). Additionally, Ballesteros-Tato et al. showed that more abundant influenza epitopes are preferentially cross-presented at late times in the primary response, and responding T cells are favorably programmed toward a memory cell fate (60). More recently, studies have identified specific cross-priming DC populations that favor CD8+ TRM precursor differentiation. In a mouse model of vaccinia virus (VACV) infection, DNGR-1+ Batf3-dependent DCs prime naïve CD8+ T cells within the LN to form TRM within skin or lung (61). Further, human studies and experiments using a humanized mouse metastatic lung model identified a subset of activated CD88−CD1c+CD163+CD14+/− DCs, or DC3s, that prime naïve CD8+ T cells and induce TGF-β-triggered CD103 expression (62).
The gene expression profile and half-life of activated CD8+ T cells are determined by many signals during pathogen invasion, such as antigen presentation by mature DCs, T cell stimulation by receptor ligands and inflammatory cytokines (63). During T cell priming, different LN environments direct expression of distinct T cell homing receptors (5, 64, 65). For example, oral, but not intranasal mouse infection with LM induces efficient homing and precursor development of CD8+ TRM in the intestinal epithelium (43). In contrast, CD8+ T cells lodge within the skin following infection with herpes simplex virus (HSV) via either skin scarification or subcutaneous injection after controlling for priming efficiency (66).
Distinct patterns of cytokine expression within the LN environment during priming also modulate precursor formation and program CD8+ T cell fate (67, 68). For instance, IL-12 produced during LCMV infection induces T-bet expression in CD8+ T cells in a dose-dependent manner, and favors the development of SLEC over MPEC (69, 70). On the other hand, IL-10 plasma levels early following immunization with peptide antigen and adjuvant strongly correlates with the frequencies of antigen specific TRM in the lung of mice and non-human primates at a memory time point. Production of IL-10 by monocytes acts in an autocrine manner to release TGF-β during priming, increasing CD8+ T cell responsiveness to subsequent TGF-β stimulation, and thereby favors the formation of CD8+ CD103+ TRM (71).
Following CD8+ T cell activation and clonal expansion within draining LN, TRM precursors migrate to non-lymphoid tissues. Entry into peripheral tissues induces a unique TRM phenotype that promotes CD8+ T cell retention and prevents egress (Supplementary Table 2). More than a decade ago, Masopust et al. demonstrated that as early as 7 days following intestinal LCMV infection, the gut microenvironment induces a unique CD8+ T cell differentiation program; CD8+ IELs express both CD69 and CD103, while splenic circulating memory CD8+ T cells do not (72). Similarly, Ray and colleagues found that within 8 days following influenza infection, flu-specific CD8+ T cells recovered from the lung were predominantly CD49a+, while those recovered from the mediastinal LN were CD49a− (7). This phenotype persisted at memory timepoints. More recently, Mackay et al. performed microarray analysis of CD103+ CD8+ TRM isolated from the skin, gut, and lungs of mice and determined that CD8+ TRM express a unique TRM transcriptional signature that is distinct from circulating memory CD8+ T cells. This analysis identified 37 transcripts commonly regulated by TRM from all three tissues, including S1pr1, Itga1 and Itgae, encoding sphingosine 1-phosphate receptor-1 (S1P1), CD49a and CD103, respectively (42). A similar human CD8+ TRM core transcriptional profile was also later defined (73, 74).
CD69 is perhaps the most ubiquitous marker for CD8+ TRM cells in mouse and human tissues (74, 75). CD69 forms a complex with the chemoattractant receptor S1P1, inducing S1P1 internalization and thereby impairing S1P-directed lymphocyte exit via afferent lymphatic vessels (42, 75, 76). In parallel, downregulation of kruppel-like factor 2 (KLF2), the transcription factor that drives S1P1 gene expression, is necessary for the establishment of CD8+ TRM in tissues (77, 78). CD69 expression by CD8+ T cells is necessary for the generation of CD8+ TRM in the kidney (79) and skin (75). However, recent work demonstrated that CD69 expression is dispensable for the formation of CD8+ TRM in small intestine, lung, and female reproductive tract (79). Like CD69, the integrin, CD103 has also been used extensively as a marker for CD8+ TRM. CD103 is expressed by CD8+ TRM in the epithelial compartment of multiple tissues (4, 42, 80, 81) and is thought to mediate TRM retention through its interaction with e-cadherin. However, although CD103 is necessary for CD8+ TRM accumulation within epithelium, it is dispensable for TRM persistence in other tissue compartments (42, 43). For instance, Bergsbaken et al. demonstrated that following Yersinia pseudotuberculosis (Yptb) infection, a CD103− CD8+ TRM cell population persists long-term in the intestinal lamina propria (82). Additionally, CD49a, the α chain of integrin α1β1, is expressed by CD8+ TRM and promotes their accumulation within multiple mouse and human tissues (4, 7, 24, 74, 83, 84).
Comparison of CD8+ TRM and circulating memory CD8+ T cells transcriptomes has identified several transcription factors that are differentially expressed between memory CD8+ T cells subsets. Expression of Zfp683, encoding homolog of Blimp1 in T cells (Hobit) is upregulated in CD8+ TRM and is necessary for CD8+ TRM cell development in the skin, gut, liver and kidney of mice (83). Interestingly, Hobit has been described in several other cell lineages, including CD4+ T, Natural killer (NK), NKT, and Mucosal-associated invariant T (MAIT) cells, and acts as a transcriptional regulator of residency (83, 85–87). Hobit, together with the transcription factor Blimp1 coregulate genes required for tissue egress (83). In the absence of Hobit and Blimp1, Klf2, S1p1, and CCR7 are de-repressed. However, although human lung and liver CD69+ CD8+ T cells express Hobit, so do human circulating CD45RA+ CD27− and CD45RA−CD27− CD8+ T cells, suggesting that Hobit may not specifically promote human CD8+ TRM differentiation (88). Additionally, the requirement of Hobit for TRM differentiation may be tissue-specific. In the lung, Blimp1, but not Hobit, is required for the formation of virus-specific CD8+ TRM in a mouse influenza infection model (89). Moreover, Milner et al. used single-cell RNA sequencing (scRNA-seq) analysis to characterize CD8+ siIEL populations over time following LCMV infection. They demonstrated heterogeneity in the CD8+ siIEL TRM and identified distinct resident memory CD8+ T cell populations based on their expression of the transcription factors Blimp1 and Id3. Previous studies demonstrated that Blimp1hi expression favors an effector T cell fate (90). Accordingly, Milner et al. showed that compared to Blimp1lo Id3hi siIEL, Blimp1hi Id3lo siIEL CD8+ T cells dominate the early response and express increased effector-associated genes. Nonetheless, lower numbers of Blimp1hi Id3lo siIEL CD8+ T cells are still present in the tissue at memory timepoints. Although Blimp1 was expressed by a subset of CD8+ T cells across multiple non-lymphoid tissues, expression of Id3 was more restricted, raising the possibility that TRM transcriptional programs may be regulated by the local tissue microenvironment (91).
Two T-box transcription factors, Eomesodermin (Eomes) and T-bet, control CD8+ CD103+ TRM cell formation in lung, skin, and brain. Although TCM express both Eomes and T-bet (92), expression of these transcription factors must be downregulated for CD8+ TRM development. While extinguishment of Eomes expression is required for CD8+ CD103+ TRM cell formation (93, 94), residual T-bet expression maintains CD8+ T cell IL-15Rβ expression and IL-15 responsiveness for long-term TRM survival within lung and skin (94, 95). Additionally, recent data generated using ATAC-seq and transcriptional profiling identified the transcription factor, Runx3 as a central regulator of CD8+ TRM differentiation (32, 44, 73, 96). Runx3, previously described as a transcriptional regulator of CD8+ effector T cells (97), promotes expression of tissue residency genes while suppressing genes involved in tissue egress. Runx3−/− CD8+ T cells have elevated T-bet levels, suggesting that Runx3 represses T-bet expression; knockdown of T-bet expression in Runx3−/− CD8+ T cells increases CD8+ TRM numbers and restores CD69 and CD103 expression. Runx3 deficiency results in loss of CD8+ TRM in barrier (skin and lung) as well as non-barrier (salivary gland and kidney) tissues, suggesting that Runx3 may regulate CD8+ TRM formation independent of the local tissue milieu (96).
CD8+ TRM generation and long-term maintenance are also regulated by nuclear receptor subfamily 4 group A member 1 (NR4A1) (44, 98). Nr4a1, also known as Nur77, is rapidly induced following TCR stimulation and regulates CD8+ T cell proliferation and effector function (99). In a mouse model of influenza infection, similar numbers of co-adoptively transferred Nr4a1−/− and wild-type antigen-specific CD8+ TRM are recovered at the effector phase. However, fewer Nr4a1−/− CD8+ T cells are recovered from the liver and intestine at a memory time point, although similar numbers are recovered from lung (98). Finally, scRNA-seq analysis of siIEL and splenic CD8+ T cells over the course of LCMV infection demonstrated increased expression of Nr4a2, Junb proto-oncogene (Junb) and FOS-like 2 (Fosl2) in siIEL relative to splenic CD8+ T cells. Knockdown of these genes results in impaired formation of siIEL CD8+ TRM compared to circulating memory CD8+ T cells, although the mechanisms were not determined (44).
Following vesicular stomatitis virus (VSV) infection, local antigen presentation is required to drive CD103 expression by infiltrating CD8+ T cells that promotes their persistence within brain (9). Similarly, local antigen recognition is required for TRM formation in the lung (100, 101). Following influenza infection, viral antigen-bearing pulmonary monocytes interact with influenza-specific CD8+ T cells in vivo and can induce CD103 expression by CD8+ T cells in vitro (102). While localized inflammation can recruit CD8+ T cells into the lung, in the absence of local antigen recognition, memory CD8+ T cells fail to express the retention receptors CD69, CD103, and CD49a or persist long-term (103). However, the requirement of antigen recognition within peripheral tissues for CD8+ TRM formation is not absolute. CD8+ CD103+ TRM can be generated in the absence of antigen recognition in barrier tissues, including skin, intestine, and female reproductive tract (104–106). Nonetheless, subsequent studies demonstrated that local recognition of antigen dramatically increases the formation of CD8+ TRM in VACV-infected skin (107, 108). Moreover, local competition between CD8+ T cells of different specificities for different viral epitopes shapes the repertoire of cutaneous CD8+ TRM cells following VACV infection (107), underlining the importance of local antigen recognition in regulating the establishment of CD8+ TRM.
The local tissue cytokine microenvironment influences CD8+ TRM phenotype. TGF-β is critical for the formation of CD103+ CD8+ TRM in several tissues, including the siIEL compartment, skin epidermis, lung, and kidney (105, 109–111). CD8+ T cells expressing mutant TGF-β receptors fail to express CD103 or persist within multiple peripheral tissues (42, 43, 81, 105, 109). Recent data suggest that epidermal CD8+ TRM cells require transactivation of autocrine TGF-β for their long-term persistence, and competition for limited TGF-β influences which clones persist within the epidermis (112). CD8+ T cell TGF-β responsiveness is controlled by the transcription factors EOMES and T-bet, and downregulation of Eomes and T-bet is required for CD8+ T cell TGF-β responsiveness and CD8+ TRM formation (94). Additionally, recent research has identified a role for the transcriptional cofactor, SKI, in regulating CD8+ T cell CD103 expression. Using an LCMV infection model, Wu et al. demonstrated that ectopic expression of SKI proto-oncogene restricts CD103 expression by CD8+ T cells in vitro and in vivo. SKI is recruited to the Itgae locus to suppress CD103 transcription by preventing histone acetylation in a Smad4-dependent manner. Moreover, in the absence of Smad4, CD103 is constitutively expressed by CD8+ T cells even in the absence of TGF-β signaling, suggesting that modulation of TGF-β-SKI-Smad4 pathway could determine CD8+ CD103+ TRM generation (111).
Inflammatory cytokines produced in response to local infection, and the chemokines they induce also regulate TRM formation and phenotype. IFN-γ and the IFN-γ-induced chemokines, CXCL9 and CXCL10 have been shown to orchestrate CD8+ TRM precursor migration and localization within tissues in multiple infection models. For example, following influenza infection, IFN-γ produced by CD4+ T cells promotes the localization of CD8+ T cells to the airways, thereby controlling their exposure to TGF-β (95). Similarly, following genital HSV-2 infection, IFN-γ induces local expression of the CXCR3 ligands, CXCL9 and CXCL10 that promotes CD8+ T cell localization and long-term persistence within the tissue (113). Furthermore, local application of these chemokines is sufficient to recruit CD8+ T cells into the genital tract where they are retained long-term and enhance memory response to reinfection (106). Similarly, keratinocytes express CXCL9 and CXCL10 during HSV skin infection. KLRG1− CD8+ TRM precursors show preferential migration to these chemokines ex vivo compared to KLRG1+ effector CD8+ T cells. Moreover, following intradermal injection, CXCR3−/− CD8+ T cells generate fewer CD103+ TRM than adoptively transferred WT CD8+ T cells, suggesting that CXCR3 mediates TRM precursor entry into the epidermis where locally activated TGF-β may promote subsequent epidermal CD8+ CD103+ TRM generation (42, 114). Additionally, CXCR3-directed localization of type I Treg expressing the TGF-β activating integrin, αvβ8, within local inflammatory sites promotes CD8+ TRM generation in the intestine, liver, and lung. Positioning of these Treg adjacent to effector CD8+ T cells promotes CD8+ TRM generation via activated TGF-β availability (115). In contrast, generation of CD8+ CD103− TRM following oral Yptb infection is independent of TGF-β signaling, but requires CXCR3-dependent clustering of effector CD8+ T cells with CXCL10-producing CX3CR1+ intestinal cells in areas of inflammation within the intestinal lamina propria, suggesting that the microenvironment formed by immune cell aggregates supports CD8+ TRM formation (116). Indeed, IL-12 and IFN-β produced by intestinal macrophages during Yptb infection prevents TGF-β-induced CD103 expression by CD8+ T cells, favoring the differentiation of CD8+ CD103− TRM cells (82). Thus, inflammatory cytokines not only function to induce local chemokine expression to promote the recruitment of TRM precursors into tissues, but also influence the differentiation of CD8+ T cells within the tissue, providing a mechanism to promote TRM phenotypic diversity.
Several additional chemokine receptors may also participate in the formation of CD8+ TRM within peripheral tissues. For example, expression of the intestinal homing chemokine receptor CCR9 by CD8+ siIEL is increased compared to their circulating counterparts throughout their differentiation (5, 44). Additionally, expression of CXCR6 and CCR10 by mouse CD8+ T cells are required for optimal CD8+ TRM formation in the skin (117). Although CD8+ TRM formation in mouse skin appears to be CCR8-independent (117), human cutaneous CD69+ CD103+ TRM express CCR8, raising the possibility that CCR8 and its ligands may regulate human cutaneous CD8+ TRM generation or function (118, 119).
Competition for survival cytokines may also impact CD8+ TRM accumulation within tissues. A recent report using an LCMV infection model demonstrated that NK1.1+ innate lymphoid cells (ILCs) control the accumulation of memory CD8+ T cells in salivary glands. Specifically, establishment of CD8+ TRM is enhanced in anti-NK1.1+ antibody pretreated mice. The authors propose that ILCs might compete for survival signals such as IL-7, although no specific mechanism was determined (120). Similarly, following HSV skin infection, CD8+ TRM formation is accompanied by a concomitant local decrease in dendritic epidermal γδ T cells, suggesting possible competition for survival cytokines within the epidermal niche.
Costimulatory signals also play a role in the establishment of CD8+ TRM within tissues. During influenza infection, Zhou et al. showed that interaction of the costimulatory molecule, 4-1BB with its ligand 4-1BBL is necessary for the induction of long-lived lung-resident CD103+ and CD103− memory CD8+ T cell populations (121). In addition, glucocorticoid-induced TNFR-related protein ligand (GITRL), expressed by lung monocyte-derived inflammatory antigen presenting cells, provides a costimulatory signal for lung CD8+ T cells expressing GITR during influenza infection. GITRL/GITR interaction in the LN and lung is required for the differentiation of CD8+ TRM precursors and the formation of CD8+ TRM within the lung parenchyma (122).
Additional microenvironmental cues may also regulate the generation of CD8+ TRM. For example, microRNA-155 is upregulated during infection in response to TLR signaling and inflammatory cytokines (123). CD8+ TRM are established in the brain following infection of mice with neuroinvasive LM, and their accumulation is decreased in the absence of miR-155 (124). Also, CD8+ T cells require P2RX7 expression for CD8+ TRM formation in the siIEL, female reproductive tract, kidney, salivary glands, and liver. Extracellular ATP is released during inflammation and injury, and is sensed by the purinergic receptor, P2RX7. Upon CD8+ T cell activation, expression of TGF-β receptors is transiently down-regulated. Extracellular ATP derived from intestinal microbiota, activated cells and/or damaged tissue restores TGF-βRII expression and TGF-β responsiveness, resulting in CD8+ T cell CD103 upregulation, KLF2 downregulation, enhanced mitochondrial function and TRM formation (125). On the other hand, microbiota depletion by antibiotic treatment increases the antigen load following LM infection and promotes CXCR3-directed CD8+ T cell accumulation within the large intestinal lamina propria, resulting in increased mucosal CD8+ TRM accumulation and response (126).
CD8+ TRM persist long-term within several tissues, including intestinal IEL (105), vaginal mucosa (106), and skin (104, 127) independent of cognate antigen recognition. In contrast, lung CD8+ TRM are rapidly lost from the tissue. Several studies suggest that cognate antigen recognition is required for the persistence of lung CD8+ TRM. Residual local antigen persistence may promote continuous development of lung TRM and allow for the maintenance of CD8+ TRM within the tissue (128). Following influenza infection, CD8+ TRM receive chronic local TCR stimulation even weeks after the clearance of infectious influenza virus. Furthermore, tamoxifen-inducible H-2Db depletion or B7-CD28 blockade starting at least three weeks post-infection results in impaired maintenance of CD8+ TRM cells within the lung (129). Based on these findings, novel methods are being developed in attempt to prolong the persistence of CD8+ TRM within the lung. Combined subcutaneous and intranasal vaccination of mice with an adenovirus vector expressing influenza antigen is reported to induce persistent antigen expression in the lungs and maintains TRM within the lung for at least one year post-vaccination (130). Continual recruitment of circulating CD8+ TEM may convert into TRM following antigen recognition and help to sustain TRM within the interstitium.
However, the requirement of circulating memory CD8+ T cell recruitment for the long-term maintenance of lung CD8+ TRM has been questioned by a recent study using parabiosis and intravascular staining to exclude analysis of CD8+ T cells within the circulation. Takamura et al. demonstrated that CD8+ TRM can be retained in specific niches created at sites of tissue regeneration within the lung parenchyma, distant from lymph vessels, and independent of CD8+ T cell recruitment from the circulation (100). Still, the half-life of CD8+ TRM within lung airways is less than 14 days (131), and so they propose that maintenance of airway memory CD8+ T cells may require residual antigen-driven reactivation of CD8+ TRM in the lung parenchyma and recruitment into the airways (100, 132). More recently, an additional mechanism has been proposed to maintain regional immune memory specific for lung pathogens. Stolley et al. demonstrated that following influenza infection, CD8+ T cells migrate to draining mediastinal LN via lymphatic vessels. These cells express CD103 and CD69, are maintained long-term within the LN in an antigen-independent manner and maintain effector molecule expression. As such, repositioning and persistence of CD8+ TRM within the draining mediastinal LN may provide a means to maintain regional immune memory despite rapid attrition of lung CD8+ TRM (133).
Maintenance of CD8+ TRM is thought to require expression of retention receptors that act as adhesive anchors (Formation markers and transcriptional regulators in stage 2, Supplementary Table 2, and Supplementary Table 3). CD103 binds to E-cadherin, which is expressed in skin epidermis (134) and intestinal epithelium (5, 105). This interaction is thought to anchor CD8+ TRM within the epithelial compartment of tissues and facilitate their long-term residence (135). Similarly, CD49a binds collagen type I and IV, and also facilitates CD8+ TRM persistence within skin, lung, and intestine (7, 84, 136). In addition to its adhesive function, CD49a may also provide a pro-survival signal, limiting CD8+ memory T cell apoptosis (7).
Although CD69 is required for CD8+ TRM establishment in several tissues, it may not be required for their long-term maintenance. Following mouse influenza infection, CD8+ TRM are retained long-term within the lung independent of CD69 expression. Early after infection, CD69 is important for the accumulation of CD8+ T cells within the airways to inhibit strong S1P1-mediated exit signals. However, once CD8+ TRM are established, CD69 is dispensable even though the cells maintain residual S1P1 reactivity (100). Downregulation of KLF2, the transcription factor that drives S1P1 expression, may preclude the need for continued CD69 expression in TRM to inhibit any S1P-mediated exit signal. Moreover, physical separation of TRM from lymphatic vessels by their positioning within lung niches or within the epidermis may also facilitate their retention within tissues independent of CD69.
The expression patterns of several transcription factors that regulate CD8+ TRM formation are maintained long-term in established TRM (Transcriptional regulators in stage 2, Supplementary Table 2 and Supplementary Table 3). However, Milner et al. found divergent transcription factor expression patterns in CD8+ T cells with distinct phenotypic properties during different stages of TRM formation and maintenance. Specifically, while Blimp1hi Id3lo siIEL CD8+ T cells are abundant at the effector phase of the immune response, Blimp1lo Id3hi siIEL CD8+ T cells progressively accumulate over time, and are more abundant at the memory phase of the response. Moreover Blimp1lo Id3hi siIEL CD8+ T cells have higher recall proliferative capacity and multipotency than Blimp1hi siIEL CD8+ T cells (91). Additionally, Aryl hydrocarbon receptor (AhR) also regulates CD8+ TRM maintenance. Expression of AhR is increased in skin CD8+ TRM compared to naïve or circulating memory T cells. While Ahr−/− CD8+ T cells initially enter into sites of DNFB-induced skin inflammation, over time, they disappear from the skin but not spleen (134), suggesting that AhR is required for the long-term persistence of cutaneous CD8+ TRM. Accordingly, AhR expression is increased in mouse intestinal TRM compared to circulating memory CD8+ T cells following LCMV infection (44), as well as in human lung CD8+ CD103+ TRM compared to circulating memory T cells (73). Finally, Notch signaling regulates the maintenance of CD8+ CD103+ TRM in the lung by regulating both CD103 expression and CD8+ TRM metabolism (73).
TGF-β is not only required for the establishment of CD8+ TRM in multiple barrier tissues, but also to preserve their phenotype and long-term persistence in the intestine (109). Similarly, after cutaneous CD103+ CD8+ TRM have been established, neutralization of the TGF-β-activating integrin, αvβ6, results in reduced numbers of TRM in the epidermis but not LN or spleen over time (114). These results suggest that continuous TGF-β signaling is required for the long-term persistence of epidermal CD8+ TRM.
Survival cytokines also provide for the long-term sustenance of tissue-resident CD8+ T cells. Both IL-7 and IL-15 are required for the persistence of CD8+ TRM in the skin (94, 137). In contrast, maintenance of TRM in the lung and intestine is IL-15-independent (138, 139). On the other hand, IL-12 regulates Bcl-2 expression to promote the survival of CD8+ CD103− TRM within the intestinal lamina propria (82).
Although P2RX7 promotes CD8+ TRM formation within the intestine (125), Stark et al. demonstrated that sterile tissue damage led to loss of established WT, but not P2rx7−/− CD8+ TRM from the liver (140). They found that TCR triggering downregulates P2RX7 expression, and so proposed that tissue damage-induced depletion of established TRM might free space for the formation of new CD8+ TRM with infection-relevant specificities. In contrast, Wakim et al. determined that persistent expression of the anti-viral transmembrane protein, IFITM3 by lung CD103+ CD8+ T cells promotes the survival and maintenance of CD8+ TRM at sites of viral infection. Following influenza infection, cognate antigen induces persistent IFITM3 expression preferentially by lung CD8+ TRM compared to splenic memory CD8+ T cells. CD8+ TRM that lack IFITM3 expression exhibit increased susceptibility to influenza infection compared to IFITM3+ CD8+ TRM, and are selectively lost following virus challenge (141).
Finally, CD8+ TRM long-term survival and protective function require lipid uptake and oxidative metabolism. Fatty-acid-binding proteins 4 and 5 (FABP4 and FABP5) are required for the long-term maintenance of CD8+ TRM within the skin following VACV infection, and for CD8+ TRM-mediated protection from viral challenge (142). However, CD8+ TRM exhibit distinct patterns of FABP gene expression depending on their tissue of residence. An additional study demonstrated that following HSV infection, skin CD8+ TRM express Fabp4 and Fabp5, but lack expression of other FABP isoforms. However, following LCMV infection, liver CD8+ TRM highly express Fabp1, some Fabp4, but no Fabp5. In contrast, siIEL CD8+ TRM express Fabp1, Fabp2, and Fabp6, but negligible Fabp4 and Fabp5. These differences in FABP expression are determined by tissue-derived signals, and by altering FABP expression, CD8+ T cells can adapt to different host tissues (143).
CD8+ TRM are positioned to provide a first line of host defense in response to pathogen challenge. Recognition of cognate antigen stimulates CD8+ TRM to rapidly secrete cytokines that induce expression of anti-viral and anti-bacterial genes, activate innate immune cells, and enhance chemokine and adhesion receptor expression for increased recruitment of circulating immune cells (144–146). Following tissue entry, circulating memory CD8+ T cells can undergo antigen-dependent CD69+ CD103− TRM differentiation (147) as well as antigen-independent CD69+/− CD103+/− TRM differentiation (148, 149) in situ. Additionally, intravital microscopy studies revealed that established CD8+ TRM proliferate within the female reproductive tract and skin upon cognate antigen encounter. These cells dominate the recall response and contribute more than circulating memory CD8+ T cells to the pool of secondary TRM cells (148, 149).
At homeostasis, CD8+ TRM persist long-term within peripheral tissues, separate from the circulation. However, following antigen reencounter, CD8+ TRM exhibit plasticity. Beura et al. determined that CD8+ CD69+ TRM in the draining LNs derive from cells present in the upstream nonlymphoid tissue (11). Complementary studies by Behr et al. used Hobit reporter mice to demonstrate that CD69lo Hobit+ antigen specific T cells accumulate in the draining LNs in the effector phase after reinfection, and upregulate CD69 expression in the secondary memory phase, forming LN TRM. Virus challenge not only induces local proliferation of CD8+ TRM cells in peripheral tissues that can participate in the accumulation of secondary TRM in the draining LN, but also, formation of circulating memory CD8+ T cells downstream of CD8+ TRM. Studies using Hobit lineage tracer mice revealed that Hobit+ CD8+ TRM can downregulate Hobit expression upon antigen encounter and form KLRG1+ CXC3CR1+ circulating TEM with enhanced capacity to protect against reinfection (150). Similarly, Fonesca et al. demonstrated that following challenge, small intestinal iEL TRM give rise to circulating TCM and TEM. These ex-TRM cells are epigenetically poised for migration back to the tissue of origin and TRM re-differentiation (151).
Intravital confocal microscopy illustrated that CD8+ TRM actively patrol skin epithelium in search of cognate antigen, raising the possibility that TRM within barrier tissues do not depend on antigen delivery by professional APCs (152). In line with this hypothesis, Masopust et al. demonstrated that following depletion of ~90% of host DC in CD11c-DTR bone marrow chimeric mice, TRM still proliferate in response to challenge with cognate peptide antigen (149). In contrast, in the vaginal mucosa, TRM reactivation following HSV-2 challenge depends on CD301b+ DCs (153). In addition, transplantation of the dorsal root ganglia of HSV-infected mice under the kidney capsule of naive mice induces viral reactivation. Here, the CD8+ TRM proliferative response is initiated by recruitment of CD11b+ CD11c+ DCs. Together, these results suggest that the DC requirement for CD8+ TRM response to antigen challenge may be context dependent. Indeed, in models of LCMV and influenza infection, cDCs are dispensable for lung CD8+ TRM reactivation. Rather either hematopoietic or non-hematopoietic antigen presenting cells are sufficient, but they induce different TRM functional outputs. Whereas antigen presentation by hematopoietic cells reduces gene transcription of chemokines and cytokines such as Ccl1, Ccl3, Ccl9, and Ifng, activation by nonhematopoietic cells promote transcription of genes involved in cell cycle and proliferation but curbs type I interferon stimulated genes (154).
Although TRM remain resident long-term in peripheral tissues, they are not sessile cells; TRM continuously patrol the local area for invading pathogens. Upon cognate antigen recognition, CD8+ TRM become rounded and arrest their migration before undergoing proliferation in situ (148, 149). However, intravital microscopy studies demonstrated that depending on their tissue of residence, TRM display different migration speeds and morphologies. TRM migrate within skin epidermis, albeit slowly at a rate of ~1.3 µm/min, and extend dendrites laterally to probe their surroundings for cognate antigen (134). Imaging of the mouse uterus after acute LCMV infection revealed that CD8+ TRM migrate at different rates within the stroma of the female reproductive tract and this migratory speed correlates with collagen density. TRM within the collagen-rich perimetrium migrate more slowly than in the less collagen-rich myometrium where TRM exhibit motility rates that are similar to those of circulating lymphocytes in LNs (149). Interestingly, a recent study in influenza-infected mice suggests that the collagen receptor, CD49a promotes CD8+ T cell motility within the trachea to facilitate tissue surveillance (155). In contrast, CD103 restrains TRM motility in both trachea and skin (117, 155). How changes in the local microenvironment following challenge with distinct pathogens might affect CD8+ TRM phenotype and migratory behavior requires additional study.
CD8+ TRM provide immediate effector functions against secondary infections (Supplementary Table 4). The transcriptional profiles of both mouse and human CD8+ TRM exhibit higher expression of effector molecules compared to circulating memory CD8+ T cells (73, 74, 93, 105, 156). Constitutive expression of mRNAs encoding effector molecules may facilitate rapid TRM response. For example, notch signaling contributes to the maintenance of constitutive Ifng expression by lung TRM (73). Notch signaling transactivates Ifng, increasing Ifng expression by TRM independent of TCR stimulation. Following recognition of cognate antigen, CD8+ TRM secrete IFN-γ, IL-2 and TFN-α, inducing a rapid recall response at the site of pathogen invasion (146, 156–158). IFN-γ induces vascular cell adhesion molecule 1 (VCAM-1) expression by endothelial cells, as well as production of inflammatory chemokines that recruit circulating immune cells, resulting in amplification of the memory response (146). Additionally, resting lung CD8+ TRM constitutively express CCL3, CCL4, CCL20 and XCL1 (73), and intestinal CD8+ TRM express Ccl3 and Ccl4 (44), suggesting that CD8+ TRM themselves express genes to rapidly amplify the memory immune response.
CD8+ TRM targeted secretion of the cytotoxic proteins, perforin and granzyme B, destroy target cells. While circulating memory CD8+ T cells lack cytotoxic protein expression, TRM that form within intestinal IEL, liver, and brain following LCMV infection express granzyme B during quiescence (72, 156, 159). Constitutive expression of granzyme B might promote rapid control of pathogen infection. In contrast, airway CD8+ TRM are reported to be poorly cytolytic, even in the presence of antigen stimulation (157). The nutrient-poor airway environment induces cellular stress, limiting TRM effector function and survival at homeostasis, and perhaps providing a mechanism to prevent unnecessary epithelial damage (160).
Inhibitory molecule expression may be critical to prevent TRM-mediated damage in barrier tissues. The inhibitory surface protein programmed death protein 1 (PD-1), upregulated by exhausted T cells and tumor infiltrating lymphocytes (TILs), is also expressed by CD8+ TRM in mouse and human tissues (74, 161). Multiple studies suggest that PD-1 may provide TRM functional restraint. For example, PD-1 expression by T cells correlates with response to anti-PD-1 blockade treatment in patients with cancer (162). Additionally, CD8+ PD-1hi TRM cells in human pancreas may maintain immune homeostasis through interactions with resident macrophages; in samples from chronic pancreatitis, CD8+ T cells exhibit reduced PD-1 expression (163). Moreover, following influenza infection, antigen specific CD8+ T cells in the lung acquire both a memory and exhausted phenotype, including PD-1 surface expression. Blocking PD-1 ligand (PD-L1) promotes exhausted-like TRM cell expansion, and augments TRM cell function, enhancing TRM-mediated protection from reinfection. However, anti-PD-L1 treatment also causes chronic tissue fibrotic sequelae, suggesting that inhibitory receptors are important for balancing immune protection and fibrotic processes (129). Similarly, CD8+ TRM that form in the epidermis following acute contact hypersensitivity reaction express inhibitory checkpoint receptors that limit TRM reactivation. Treatment with inhibitory molecule antagonists increases the magnitude and severity of eczema exacerbations (27).
Human lung CD8+ TRM express not only PD-1, but also genes encoding inhibitory molecules such as CTLA4, BTLA, LAG3, SPRY1, and the adenosine receptor A2AR (73). Similarly, a recent study using sc-RNA seq demonstrated that inhibitory receptors including Ctla4, Lag3, Cd101, and Tigit, are upregulated early during formation of intestinal IEL CD8+ T cells in an acute LCMV infection model, suggesting a possible role in TRM differentiation (44). Moreover, following influenza infection, differences in TRM inhibitory molecule expression are observed depending on the T cell epitope, suggesting that initial TCR-MHCp interactions may determine not only T cell activation, but also inhibitory programs (161).
The balance between CD8+ TRM-mediated immune response and immune pathology may also be regulated by alterations in mitochondrial membrane composition. CD8+ TRM express early activation markers, contain cytolytic proteins, and have the capacity to release cytokines. However, epithelial TRM are metabolically arrested in a semi-activated state. Alterations in the mitochondrial membrane, including the cardiolipin composition, regulate IEL proliferation, and effector functions (164).
Finally, CD8+ TRM adaptation to the environment is regulated by mitochondrial gene expression. The transcription factor, Bhlhe40 is highly expressed in mouse and human CD8+ TRM compared to circulating memory CD8+ T cells (44, 165), and promotes TRM mitochondrial gene expression. Bhlhe40−/− CD8+ TRM exhibit decreased oxygen consumption and enhanced mitochondrial damage. Additionally, Bhlhe40 deficiency results in reduced acetyl-CoA and histone acetylation of TRM effector loci. Lack of Bhlhe40 reduces the production of IFN-γ, granzyme B and TNF by CD8+ TRM, suggesting that Bhlhe40 promotes epigenetic programs permissive for effector gene expression. PD-1 signaling inhibits Bhlhe40 expression. Importantly, however, targeting downstream epigenetic machinery rescues CD8+ TRM mitochondrial function and cytokine production in the absence of Bhlhe40, suggesting a possible mechanism for improved immunotherapy (165).
Over the last decade, scientists around the globe have contributed to the study of CD8+ TRM. Rapid progress has been achieved in understanding the generation, regulation, and protective or pathogenic functions of T cells that reside within tissues. Since the discovery of CD8+ TRM, much effort has focused on elucidating the transcriptional networks and mechanisms that regulate these cells. These studies have identified core transcriptional signatures for both mouse and human CD8+ TRM that promote their long-term retention and maintenance. However, with increasing data examining TRM formation and function in multiple tissues and infection models, it has become increasingly clear that TRM are a heterogeneous pool of cells with plastic properties. TRM formation and phenotype are influenced by extrinsic signals such as antigen, cytokines, nutrients, costimulatory, and inhibitory signals within the LN and tissue microenvironments, as well as by intrinsic receptor and signaling protein expression. These factors can shape TRM differentiation, maintenance and response, and their variability in different tissues and inflammatory settings promotes TRM diversity between organs, and even within the same tissue. Although a great deal has already been learned, an improved understanding of the mechanisms that regulate TRM formation and/or function in varied tissue environments is necessary not only to prevent autoimmune diseases, but also to improve cancer treatments and vaccine strategies.
RM-B drafted and edited the manuscript and figures. SB edited and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by NIH grant R01 AI121546 (SB) and by a National Eczema Association Catalyst Research Grant NEA19-CRG121 (RM-B).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Andrea Sama for helpful input on the manuscript. The figure was produced using Biorender.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.624199/full#supplementary-material
1. Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol (2010) 130(2):362–70. doi: 10.1038/jid.2009.247
2. Mackay LK, Kallies A. Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol (2017) 38(2):94–103. doi: 10.1016/j.it.2016.11.004
3. Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR. Dendritic Cell-Induced Memory T Cell Activation in Nonlymphoid Tissues. Sci (80- ) (2008) 319(5860):198–202. doi: 10.1126/science.1151869
4. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10(5):524–30. doi: 10.1038/ni.1718
5. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med (2010) 207(3):553–64. doi: 10.1084/jem.20090858
6. Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, et al. Cutting Edge: Intravascular Staining Redefines Lung CD8 T Cell Responses. J Immunol (2012) 189(6):2702–6. doi: 10.4049/jimmunol.1201682
7. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The Collagen Binding α1β1 Integrin VLA-1 Regulates CD8 T Cell-Mediated Immune Protection against Heterologous Influenza Infection. Immunity (2004) 20(2):167–79. doi: 10.1016/S1074-7613(04)00021-4
8. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8 + TRM cells providing global skin immunity. Nature (2012) 483(7388):227–31. doi: 10.1038/nature10851
9. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107(42):17872–9. doi: 10.1073/pnas.1010201107
10. Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, et al. Liver-Resident Memory CD8+ T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity (2016) 45(4):889–902. doi: 10.1016/j.immuni.2016.08.011
11. Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, et al. T Cells in Nonlymphoid Tissues Give Rise to Lymph-Node-Resident Memory T Cells. Immunity (2018) 48(2):327–38. doi: 10.1016/j.immuni.2018.01.015
12. Schenkel JM, Fraser KA, Masopust D. Cutting Edge: Resident Memory CD8 T Cells Occupy Frontline Niches in Secondary Lymphoid Organs. J Immunol (2014) 192(7):2961–4. doi: 10.4049/jimmunol.1400003
13. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103(+) Tumor-Resident CD8(+) T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin Cancer Res an Off J Am Assoc Cancer Res (2018) 24(13):3036–45. doi: 10.1158/1078-0432.CCR-17-2257
14. Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med (2018) 24(7):986–93. doi: 10.1038/s41591-018-0078-7
15. Corgnac S, Malenica I, Mezquita L, Auclin E, Voilin E, Kacher J, et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched with Tc17. Cell Rep Med (2020) 1(7):100127. doi: 10.1016/j.xcrm.2020.100127
16. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, et al. CD8+CD103+ Tumor–Infiltrating Lymphocytes Are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. J Immunol (2015) 194(7):3475–86. doi: 10.4049/jimmunol.1402711
17. Wang B, Wu S, Zeng H, Liu Z, Dong W, He W, et al. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J Urol [Internet] (2015) 194(2):556–62. doi: 10.1016/j.juro.2015.02.2941
18. Koh J, Kim S, Kim M-Y, Go H, Jeon YK, Chung DH. Prognostic implications of intratumoral CD103+ tumor-infiltrating lymphocytes in pulmonary squamous cell carcinoma. Oncotarget (2017) 8(8):13762–9. doi: 10.18632/oncotarget.14632
19. Komdeur FL, Prins TM, van de Wall S, Plat A, Wisman GBA, Hollema H, et al. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology (2017) 6(9):e1338230–e1338230. doi: 10.1080/2162402X.2017.1338230
20. Chu Y, Liao J, Li J, Wang Y, Yu X, Wang J, et al. CD103(+) tumor-infiltrating lymphocytes predict favorable prognosis in patients with esophageal squamous cell carcinoma. J Cancer (2019) 10(21):5234–43. doi: 10.7150/jca.30354
21. Workel HH, Komdeur FL, Wouters MCA, Plat A, Klip HG, Eggink FA, et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur J Cancer (2016) 60:1–11. doi: 10.1016/j.ejca.2016.02.026
22. Park MH, Kwon SY, Choi JE, Gong G, Bae YK. Intratumoral CD103-positive tumour-infiltrating lymphocytes are associated with favourable prognosis in patients with triple-negative breast cancer. Histopathology (2020) 77(4):560–9. doi: 10.1111/his.14126
23. Webb JR, Milne K, Watson P, deLeeuw RJ, Nelson BH. Tumor-Infiltrating Lymphocytes Expressing the Tissue Resident Memory Marker CD103 Are Associated with Increased Survival in High-Grade Serous Ovarian Cancer. Clin Cancer Res (2014) 20(2):434–44. doi: 10.1158/1078-0432.CCR-13-1877
24. Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity (2017) 46(2):287–300. doi: 10.1016/j.immuni.2017.01.009
25. Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med (2014) 20(9):1043–9. doi: 10.1038/nm.3645
26. Bottois H, Ngollo M, Hammoudi N, Courau T, Bonnereau J, Chardiny V, et al. KLRG1 and CD103 Expressions Define Distinct Intestinal Tissue-Resident Memory CD8 T Cell Subsets Modulated in Crohn’s Disease. Front Immunol (2020) 11:896. doi: 10.3389/fimmu.2020.00896
27. Gamradt P, Laoubi L, Nosbaum A, Mutez V, Lenief V, Grande S, et al. Inhibitory checkpoint receptors control CD8+ resident memory T cells to prevent skin allergy. J Allergy Clin Immunol (2019) 143(6):2147–2157.e9. doi: 10.1016/j.jaci.2018.11.048
28. Schmidt JD, Ahlström MG, Johansen JD, Dyring-Andersen B, Agerbeck C, Nielsen MM, et al. Rapid allergen-induced interleukin-17 and interferon-γ secretion by skin-resident memory CD8+ T cells. Contact Dermatitis (2017) 76(4):218–27. doi: 10.1111/cod.12715
29. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-Dependent Allergen-Specific Tissue-Resident Memory Cells Drive Asthma. Immunity (2016) 44(1):155–66. doi: 10.1016/j.immuni.2015.11.004
30. Shiohara T, Mizukawa Y, Teraki Y. Pathophysiology of fixed drug eruption: the role of skin-resident T cells. Curr Opin Allergy Clin Immunol (2002) 2(4):317–23. doi: 10.1097/00130832-200208000-00005
31. Lian CG, Bueno EM, Granter SR, Laga AC, Saavedra AP, Lin WM, et al. Biomarker evaluation of face transplant rejection: association of donor T cells with target cell injury. Mod Pathol (2014) 27(6):788–99. doi: 10.1038/modpathol.2013.249
32. Mani V, Bromley SK, Äijö T, Mora-Buch R, Carrizosa E, Warner RD, et al. Migratory DCs activate TGF-β to precondition naïve CD8+ T cells for tissue-resident memory fate. Sci (80-) (2019) 366(6462):eaav5728. doi: 10.1126/science.aav5728
33. Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, Watson NB, et al. Developmental Origin Governs CD8+ T Cell Fate Decisions during Infection. Cell (2018) 174(1):117–130.e14. doi: 10.1016/j.cell.2018.05.029
34. Kaech SM, Wherry EJ. Heterogeneity and Cell-Fate Decisions in Effector and Memory CD8+ T Cell Differentiation during Viral Infection. Immunity (2007) 27:393–405. doi: 10.1016/j.immuni.2007.08.007
35. Rosato PC, Wijeyesinghe S, Stolley JM, Masopust D. Integrating resident memory into T cell differentiation models. Curr Opin Immunol (2020) 63:35–42. doi: 10.1016/j.coi.2020.01.001
36. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol (2003) 164(12):1191–8. doi: 10.1038/ni1009
37. Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, et al. Single Naive CD8+ T Cell Precursor Can Develop into Diverse Effector and Memory Subsets. Immunity (2007) 27(6):985–97. doi: 10.1016/j.immuni.2007.10.012
38. Gerlach C, van Heijst JWJ, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med (2010) 207(6):1235–46. doi: 10.1084/jem.20091175
39. Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med (2015) 21(6):647–53. doi: 10.1038/nm.3860
40. Kok L, Dijkgraaf FE, Urbanus J, Bresser K, Vredevoogd DW, Cardoso RF, et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med (2020) 217(10). doi: 10.1084/jem.20191711
41. Gerlach C, Rohr JC, Perié L, van Rooij N, van Heijst JWJ, Velds A, et al. Heterogeneous Differentiation Patterns of Individual CD8+ T Cells. Sci (80-) (2013) 340(6132):635–9. doi: 10.1126/science.1235487
42. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14(12):1294–301. doi: 10.1038/ni.2744
43. Sheridan BS, Pham Q-M, Lee Y-T, Cauley LS, Puddington L, Lefrançois L. Oral Infection Drives a Distinct Population of Intestinal Resident Memory CD8+ T Cells with Enhanced Protective Function. Immunity (2014) 40(5):747–57. doi: 10.1016/j.immuni.2014.03.007
44. Kurd NS, He Z, Louis TL, Milner JJ, Omilusik KD, Jin W, et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci Immunol (2020) 5(47):eaaz6894. doi: 10.1126/sciimmunol.aaz6894
45. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity (2018) 48(4):716–729.e8. doi: 10.1016/j.immuni.2018.03.015
46. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature (2017) 552(7685):404–9. doi: 10.1038/nature25144
47. Teixeiro E, Daniels MA, Hamilton SE, Schrum AG, Bragado R, Jameson SC, et al. Different T Cell Receptor Signals Determine CD8+ Memory versus Effector Development. Sci (80-) (2009) 323(5913):502–5. doi: 10.1126/science.1163612
48. Smith-Garvin JE, Burns JC, Gohil M, Zou T, Kim JS, Maltzman JS, et al. T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood (2010) 116(25):5548–59. doi: 10.1182/blood-2010-06-292748
49. Frost EL, Kersh AE, Evavold BD, Lukacher AE. Cutting Edge: Resident Memory CD8 T Cells Express High-Affinity TCRs. J Immunol (2015) 195(8):3520–4. doi: 10.4049/jimmunol.1501521
50. Maru S, Jin G, Schell TD, Lukacher AE. TCR stimulation strength is inversely associated with establishment of functional brain-resident memory CD8 T cells during persistent viral infection. PloS Pathog (2017) 13(4):e1006318. doi: 10.1371/journal.ppat.1006318
51. Fiege JK, Stone IA, Fay EJ, Markman MW, Wijeyesinghe S, Macchietto MG, et al. The Impact of TCR Signal Strength on Resident Memory T Cell Formation during Influenza Virus Infection. J Immunol (2019) 203(4):936–45. doi: 10.4049/jimmunol.1900093
52. Feinerman O, Veiga J, Dorfman JR, Germain RN, Altan-Bonnet G. Variability and robustness in T cell activation from regulated heterogeneity in protein levels. Science (2008) 321(5892):1081–4. doi: 10.1126/science.1158013
53. Marchingo JM, Prevedello G, Kan A, Heinzel S, Hodgkin PD, Duffy KR. T-cell stimuli independently sum to regulate an inherited clonal division fate. Nat Commun (2016) 7:13540. doi: 10.1038/ncomms13540
54. Williams MA, Bevan MJ. Effector and Memory CTL Differentiation. Annu Rev Immunol (2007) 25(1):171–92. doi: 10.1146/annurev.immunol.25.022106.141548
55. Engelhardt JJ, Krummel MF. The importance of prolonged binding to antigen-presenting cells for T cell fate decisions. Immunity (2008) 28(2):143–5. doi: 10.1016/j.immuni.2008.01.006
56. Prlic M, Hernandez-Hoyos G, Bevan MJ. Duration of the initial TCR stimulus controls the magnitude but not functionality of the CD8+ T cell response. J Exp Med (2006) 203(9):2135–43. doi: 10.1084/jem.20060928
57. Zammit DJ, Cauley LS, Pham Q-M, Lefrançois L. Dendritic Cells Maximize the Memory CD8 T Cell Response to Infection. Immunity (2005) 22(5):561–70. doi: 10.1016/j.immuni.2005.03.005
58. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427(6970):154–9. doi: 10.1038/nature02238
59. Henrickson SE, Perro M, Loughhead SM, Senman B, Stutte S, Quigley M, et al. Antigen Availability Determines CD8+ T Cell-Dendritic Cell Interaction Kinetics and Memory Fate Decisions. Immunity (2013) 39(3):496–507. doi: 10.1016/j.immuni.2013.08.034
60. Ballesteros-Tato A, León B, Lee BO, Lund FE, Randall TD. Epitope-specific regulation of memory programming by differential duration of antigen presentation to influenza-specific CD8(+) T cells. Immunity (2014) 41(1):127–40. doi: 10.1016/j.immuni.2014.06.007
61. Iborra S, Martínez-López M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, et al. Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1+ Dendritic Cells. Immunity (2016) 45(4):847–60. doi: 10.1016/j.immuni.2016.08.019
62. Bourdely P, Anselmi G, Vaivode K, Ramos RN, Missolo-Koussou Y, Hidalgo S, et al. Transcriptional and Functional Analysis of CD1c+ Human Dendritic Cells Identifies a CD163+ Subset Priming CD8+CD103+ T Cells. Immunity (2020) 53(2):335–52. doi: 10.1016/j.immuni.2020.06.002
63. Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, et al. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev (2006) 211(1):81–92. doi: 10.1111/j.0105-2896.2006.00382.x
64. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to “program” T cell attraction to the epidermal chemokine CCL27. Nat Immunol (2007) 8(3):285–93. doi: 10.1038/ni1433
65. Mora JR, Bono MR, Manjunath N, Weninger W, et al. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature (2003) 424(6944):88–93. doi: 10.1038/nature01726
66. Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK. Cutting Edge: Tissue-Resident Memory T Cells Generated by Multiple Immunizations or Localized Deposition Provide Enhanced Immunity. J Immunol (2017) 198(6):2233–7. doi: 10.4049/jimmunol.1601367
67. Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham Q-M, Zickovich JM, et al. Pathogen-Induced Inflammatory Environment Controls Effector and Memory CD8+ T Cell Differentiation. J Immunol (2011) 187(10):4967–4978. doi: 10.4049/jimmunol.1102335
68. Plumlee CR, Sheridan BS, Cicek BB, Lefrançois L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity (2013) 39(2):347–56. doi: 10.1016/j.immuni.2013.07.014
69. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity (2007) 27(2):281–95. doi: 10.1016/j.immuni.2007.07.010
70. Cui W, Joshi NS, Jiang A, Kaech SM. Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine (2009) 27(15):2177–87. doi: 10.1016/j.vaccine.2009.01.088
71. Thompson EA, Darrah PA, Foulds KE, Hoffer E, Caffrey-Carr A, Norenstedt S, et al. Monocytes Acquire the Ability to Prime Tissue-Resident T Cells via IL-10-Mediated TGF-β Release. Cell Rep (2019) 28(5):1127–1135.e4. doi: 10.1016/j.celrep.2019.06.087
72. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting Edge: Gut Microenvironment Promotes Differentiation of a Unique Memory CD8 T Cell Population. J Immunol (2006) 176(4):2079–2083. doi: 10.4049/jimmunol.176.4.2079
73. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat Immunol (2016) 17(12):1467–78. doi: 10.1038/ni.3589
74. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep (2017) 20(12):2921–34. doi: 10.1016/j.celrep.2017.08.078
75. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting Edge: CD69 Interference with Sphingosine-1-Phosphate Receptor Function Regulates Peripheral T Cell Retention. J Immunol (2015) 194(5):2059. doi: 10.4049/jimmunol.1402256
76. Shiow LR, Rosen DB, Brdicková N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-[alpha]/[beta] to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature (2006) 440(7083):540–4. doi: 10.1038/nature04606
77. Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature (2006) 442(7100):299–302. doi: 10.1038/nature04882
78. Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14(12):1285–93. doi: 10.1038/ni.2745
79. Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, et al. The Functional Requirement for CD69 in Establishment of Resident Memory CD8+ T Cells Varies with Tissue Location. J Immunol (2019) 203(4):946–55. doi: 10.4049/jimmunol.1900052
80. Tang VA, Rosenthal KL. Intravaginal infection with herpes simplex virus type-2 (HSV-2) generates a functional effector memory T cell population that persists in the murine genital tract. J Reprod Immunol (2010) 87(1):39–44. doi: 10.1016/j.jri.2010.06.155
81. Lee Y-T, Suarez-Ramirez JE, Wu T, Redman JM, Bouchard K, Hadley GA, et al. Environmental and Antigen Receptor-Derived Signals Support Sustained Surveillance of the Lungs by Pathogen-Specific Cytotoxic T Lymphocytes. J Virol (2011) 85(9):4085–94. doi: 10.1128/JVI.02493-10
82. Bergsbaken T, Bevan MJ, Fink PJ. Local Inflammatory Cues Regulate Differentiation and Persistence of CD8+ Tissue-Resident Memory T Cells. Cell Rep (2017) 19(1):114–24. doi: 10.1016/j.celrep.2017.03.031
83. Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Sci (80- ) (2016) 352(6284):459. doi: 10.1126/science.aad2035
84. Bromley SK, Akbaba H, Mani V, Mora-Buch R, Chasse AY, Sama A, et al. CD49a Regulates Cutaneous Resident Memory CD8+ T Cell Persistence and Response. Cell Rep (2020) 32(9). doi: 10.1016/j.celrep.2020.108085
85. Zundler S, Becker E, Spocinska M, Slawik M, Parga-Vidal L, Stark R, et al. Hobit- and Blimp-1-driven CD4+ tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol (2019) 20(3):288–300. doi: 10.1038/s41590-018-0298-5
86. Lunemann S, Martrus G, Goebels H, Kautz T, Langeneckert A, Salzberger W, et al. Hobit expression by a subset of human liver-resident CD56bright Natural Killer cells. Sci Rep (2017) 7(1):6676. doi: 10.1038/s41598-017-06011-7
87. Salou M, Legoux F, Gilet J, Darbois A, du Halgouet A, Alonso R, et al. A common transcriptomic program acquired in the thymus defines tissue residency of MAIT and NKT subsets. J Exp Med (2018) 216(1):133–51. doi: 10.1084/jem.20181483
88. Vieira Braga FA, Hertoghs KML, Kragten NAM, Doody GM, Barnes NA, Remmerswaal EBM, et al. Blimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur J Immunol (2015) 45(10):2945–58. doi: 10.1002/eji.201545650
89. Behr FM, Kragten NAM, Wesselink TH, Nota B, van Lier RAW, Amsen D, et al. Blimp-1 Rather Than Hobit Drives the Formation of Tissue-Resident Memory CD8+ T Cells in the Lungs. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00400
90. Welsh RM. Blimp hovers over T cell immunity. Immunity (2009) 31(2):178–80. doi: 10.1016/j.immuni.2009.08.005
91. Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, et al. Heterogenous Populations of Tissue-Resident CD8+ T Cells Are Generated in Response to Infection and Malignancy. Immunity (2020) 52(5):808–824.e7. doi: 10.1016/j.immuni.2020.04.007
92. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol (2005) 6(12):1236–44. doi: 10.1038/ni1268
93. Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, et al. The Molecular Signature of Tissue Resident Memory CD8 T Cells Isolated from the Brain. J Immunol (2012) 189(7):3462–3471. doi: 10.4049/jimmunol.1201305
94. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity (2015) 43(6):1101–11. doi: 10.1016/j.immuni.2015.11.008
95. Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, et al. CD4+ T Cell Help Guides Formation of CD103+ Lung-Resident Memory CD8+ T Cells during Influenza Viral Infection. Immunity (2014) 41(4):633–45. doi: 10.1016/j.immuni.2014.09.007
96. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature (2017) 552(7684):253–7. doi: 10.1038/nature24993
97. Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, et al. The transcription factor Runx3 guards cytotoxic CD8+ effector T cells against deviation towards follicular helper T cell lineage. Nat Immunol (2017) 18(8):931–9. doi: 10.1038/ni.3773
98. Boddupalli CS, Nair S, Gray SM, Nowyhed HN, Verma R, Gibson JA, et al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J Clin Invest (2016) 126(10):3905–16. doi: 10.1172/JCI85329
99. Nowyhed HN, Huynh TR, Thomas GD, Blatchley A, Hedrick CC. Cutting Edge: The Orphan Nuclear Receptor Nr4a1 Regulates CD8<sup<+</sup< T Cell Expansion and Effector Function through Direct Repression of Irf4. J Immunol (2015) 195(8):3515–9. doi: 10.4049/jimmunol.1403027
100. Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med (2016) 213(13):3057–73. doi: 10.1084/jem.20160938
101. Pizzolla A, Nguyen THO, Smith JM, Brooks AG, Kedzierska K, Heath WR, et al. Resident memory CD8+ T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci Immunol (2017) 2(12):eaam6970. doi: 10.1126/sciimmunol.aam6970
102. Dunbar PR, Cartwright EK, Wein AN, Tsukamoto T, Tiger Li Z-R, Kumar N, et al. Pulmonary monocytes interact with effector T cells in the lung tissue to drive TRM differentiation following viral infection. Mucosal Immunol (2020) 13(1):161–71. doi: 10.1038/s41385-019-0224-7
103. McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, et al. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol (2018) 11(4):1071–8. doi: 10.1038/s41385-018-0003-x
104. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A (2012) 109(18):7037–42. doi: 10.1073/pnas.1202288109
105. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-Independent Differentiation and Maintenance of Effector-like Resident Memory T Cells in Tissues. J Immunol (2012) 188(10):4866. doi: 10.4049/jimmunol.1200402
106. Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491(7424):463–7. doi: 10.1038/nature11522
107. Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, König P-A, Tao S, et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med (2016) 213(13):3075–86. doi: 10.1084/jem.20160888
108. Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med (2016) 213(6):951–66. doi: 10.1084/jem.20151855
109. Zhang N, Bevan MJ. Transforming Growth Factor-β Signaling Controls the Formation and Maintenance of Gut-Resident Memory T Cells by Regulating Migration and Retention. Immunity (2013) 39(4):687–96. doi: 10.1016/j.immuni.2013.08.019
110. Ma C, Mishra S, Demel EL, Liu Y, Zhang N. TGF-β Controls the Formation of Kidney-Resident T Cells via Promoting Effector T Cell Extravasation. J Immunol (2017) 198(2):749–756. doi: 10.4049/jimmunol.1601500
111. Wu B, Zhang G, Guo Z, Wang G, Xu X, Li J, et al. The SKI proto-oncogene restrains the resident CD103+CD8+ T cell response in viral clearance. Cell Mol Immunol (2020). doi: 10.1038/s41423-020-0495-7.
112. Hirai T, Yang Y, Zenke Y, Li H, Chaudhri VK, De La Cruz Diaz JS, et al. Competition for Active TGFβ Cytokine Allows for Selective Retention of Antigen-Specific Tissue- Resident Memory T Cells in the Epidermal Niche. Immunity (2020) 54(1):84–98. doi: 10.1016/j.immuni.2020.10.022
113. Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8+T lymphocyte mobilization to virus-infected tissue requires CD4+T-cell help. Nature (2009) 462(7272):510–3. doi: 10.1038/nature08511
114. Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat Immunol (2016) 17(4):414–21. doi: 10.1038/ni.3396
115. Ferreira C, Barros L, Baptista M, Blankenhaus B, Barros A, Figueiredo-Campos P, et al. Type 1 T(reg) cells promote the generation of CD8(+) tissue-resident memory T cells. Nat Immunol (2020) 21(7):766–76. doi: 10.1038/s41590-020-0674-9
116. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol (2015):406–14. doi: 10.1038/ni.3108
117. Zaid A, Hor JL, Christo SN, Groom JR, Heath WR, Mackay LK, et al. Chemokine Receptor-Dependent Control of Skin Tissue-Resident Memory T Cell Formation. J Immunol (2017) 199(7):2451–9. doi: 10.4049/jimmunol.1700571
118. McCully ML, Ladell K, Hakobyan S, Mansel RE, Price DA, Moser B. Epidermis instructs skin homing receptor expression in human T cells. Blood (2012) 120(23):4591–8. doi: 10.1182/blood-2012-05-433037
119. McCully ML, Ladell K, Andrews R, Jones RE, Miners KL, Roger L, et al. CCR8 Expression Defines Tissue-Resident Memory T Cells in Human Skin. J Immunol (2018) 200(5):1639–1650. doi: 10.4049/jimmunol.1701377
120. Woyciechowski S, Weißert K, Ammann S, Aichele P, Pircher H. NK1.1(+) innate lymphoid cells in salivary glands inhibit establishment of tissue-resident memory CD8(+) T cells in mice. Eur J Immunol (2020) 50(12):1952–8. doi: 10.1002/eji.202048741
121. Zhou AC, Wagar LE, Wortzman ME, Watts TH. Intrinsic 4-1BB signals are indispensable for the establishment of an influenza-specific tissue-resident memory CD8 T-cell population in the lung. Mucosal Immunol (2017) 10(5):1294–309. doi: 10.1038/mi.2016.124
122. Chu K-L, Batista NV, Wang KC, Zhou AC, Watts TH. GITRL on inflammatory antigen presenting cells in the lung parenchyma provides signal 4 for T-cell accumulation and tissue-resident memory T-cell formation. Mucosal Immunol (2019) 12(2):363–77. doi: 10.1038/s41385-018-0105-5
123. O’Connell RM, Rao DS, Baltimore D. microRNA Regulation of Inflammatory Responses. Annu Rev Immunol (2012) 30(1):295–312. doi: 10.1146/annurev-immunol-020711-075013
124. Cassidy BR, Zhang M, Sonntag WE, Drevets DA. Neuroinvasive Listeria monocytogenes infection triggers accumulation of brain CD8+ tissue-resident memory T cells in a miR-155-dependent fashion. J Neuroinflammation (2020) 17(1):259. doi: 10.1186/s12974-020-01929-8
125. Borges da Silva H, Peng C, Wang H, Wanhainen KM, Ma C, Lopez S, et al. Sensing of ATP via the Purinergic Receptor P2RX7 Promotes CD8+ Trm Cell Generation by Enhancing Their Sensitivity to the Cytokine TGF-β. Immunity (2020) 53(1):158–71. doi: 10.1016/j.immuni.2020.06.010
126. Becattini S, Littmann ER, Seok R, Amoretti L, Fontana E, Wright R, et al. Enhancing mucosal immunity by transient microbiota depletion. Nat Commun (2020) 11(1):4475. doi: 10.1038/s41467-020-18248-4
127. Lauron EJ, Yang L, Harvey IB, Sojka DK, Williams GD, Paley MA, et al. Viral MHCI inhibition evades tissue-resident memory T cell formation and responses. J Exp Med (2018) 216(1):117–32. doi: 10.1084/jem.20181077
128. Zammit DJ, Turner DL, Klonowski KD, Lefrançois L, Cauley LS. Residual Antigen Presentation after Influenza Virus Infection Affects CD8 T Cell Activation and Migration. Immunity (2006) 24(4):439–49. doi: 10.1016/j.immuni.2006.01.015
129. Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, et al. PD-1hi CD8+resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol (2019) 4(36):eaaw1217. doi: 10.1126/sciimmunol.aaw1217
130. Uddbäck I, Cartwright EK, Schøller AS, Wein AN, Hayward SL, Lobby J, et al. Long-term maintenance of lung resident memory T cells is mediated by persistent antigen. Mucosal Immunol (2020) 14(1):92–9. doi: 10.1038/s41385-020-0309-3
131. Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T Cell Populations in the Lung Airways Are Maintained by Continual Recruitment. J Immunol (2006) 176(1):537–43. doi: 10.4049/jimmunol.176.1.537
132. Wein AN, McMaster SR, Takamura S, Dunbar PR, Cartwright EK, Hayward SL, et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J Exp Med (2019) 216(12):2748–62. doi: 10.1084/jem.20181308
133. Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J Exp Med (2020) 217(8):e20192197. doi: 10.1084/jem.20192197
134. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A (2014) 111(14):5307–12. doi: 10.1073/pnas.1322292111
135. Pauls K, Schön M, Kubitza RC, Homey B, Wiesenborn A, Lehmann P, et al. Role of integrin alphaE(CD103)beta7 for tissue-specific epidermal localization of CD8+ T lymphocytes. J Invest Dermatol (2001) 117(3):569–75. doi: 10.1046/j.0022-202x.2001.01481.x
136. Meharra EJ, Schön M, Hassett D, Parker C, Havran W, Gardner H. Reduced Gut Intraepithelial Lymphocytes in VLA1 Null Mice. Cell Immunol (2000) 201(1):1–5. doi: 10.1006/cimm.2000.1630
137. Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, et al. Hair follicle–derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med (2015) 21(11):1272–9. doi: 10.1038/nm.3962
138. Schenkel JM, Fraser KA, Casey KA, Beura LK, Pauken KE, Vezys V, et al. IL-15–Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J Immunol (2016) 196(9):3920–6. doi: 10.4049/jimmunol.1502337
139. Verbist KC, Field MB, Klonowski KD. Cutting Edge: IL-15–Independent Maintenance of Mucosally Generated Memory CD8 T Cells. J Immunol (2011) 186(12):6667–6671. doi: 10.4049/jimmunol.1004022
140. Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, et al. TRM maintenance is regulated by tissue damage via P2RX7. Sci Immunol (2018) 3(30):eaau1022. doi: 10.1126/sciimmunol.aau1022
141. Wakim LM, Gupta N, Mintern JD, Villadangos JA. Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nat Immunol (2013) 14(3):238–45. doi: 10.1038/ni.2525
142. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature (2017) 543(7644):252–6. doi: 10.1038/nature21379
143. Frizzell H, Fonseca R, Christo SN, Evrard M, Cruz-Gomez S, Zanluqui NG, et al. Organ-specific isoform selection of fatty acid–binding proteins in tissue-resident lymphocytes. Sci Immunol (2020) 5(46):eaay9283. doi: 10.1126/sciimmunol.aay9283
144. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Sci (80- ) (2014) 346(6205):101–5. doi: 10.1126/science.1254803
145. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Sci (80- ) (2014) 346(6205):98–101. doi: 10.1126/science.1254536
146. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8+ T cells. Nat Immunol (2013) 14(5):509–13. doi: 10.1038/ni.2568
147. Osborn JF, Hobbs SJ, Mooster JL, Khan TN, Kilgore AM, Harbour JC, et al. Central memory CD8+ T cells become CD69+ tissue-residents during viral skin infection independent of CD62L-mediated lymph node surveillance. PloS Pathog (2019) 15(3):e1007633. doi: 10.1371/journal.ppat.1007633
148. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol (2018) 19(2):183–91. doi: 10.1038/s41590-017-0027-5
149. Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, et al. Intravital mucosal imaging of CD8+ resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol (2018) 19(2):173–82. doi: 10.1038/s41590-017-0029-3
150. Behr FM, Parga-Vidal L, Kragten NAM, van Dam TJP, Wesselink TH, Sheridan BS, et al. Tissue-resident memory CD8(+) T cells shape local and systemic secondary T cell responses. Nat Immunol (2020) 21(9):1070–81. doi: 10.1038/s41590-020-0723-4
151. Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat Immunol (2020) 21(4):412–21. doi: 10.1038/s41590-020-0607-7
152. Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, et al. Tissue-resident memory CD8+T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci (2012) 109(48):19739–19744. doi: 10.1073/pnas.1208927109
153. Shin H, Kumamoto Y, Gopinath S, Iwasaki A. CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat Commun (2016) 7(1):13346. doi: 10.1038/ncomms13346
154. Low JS, Farsakoglu Y, Amezcua Vesely MC, Sefik E, Kelly JB, Harman CCD, et al. Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J Exp Med (2020) 217(8):e20192291. doi: 10.1084/jem.20192291
155. Reilly EC, Lambert Emo K, Buckley PM, Reilly NS, Smith I, Chaves FA, et al. TRM integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc Natl Acad Sci (2020) 117(22):12306–12314. doi: 10.1073/pnas.1915681117
156. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med (2016) 213(8):1571–87. doi: 10.1084/jem.20151916
157. McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. Airway-Resident Memory CD8 T Cells Provide Antigen-Specific Protection against Respiratory Virus Challenge through Rapid IFN-γ Production. J Immunol (2015) 195(1):203–209. doi: 10.4049/jimmunol.1402975
158. Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J Exp Med (2017) 214(6):1567–80. doi: 10.1084/jem.20162115
159. Kragten NAM, Behr FM, Vieira Braga FA, Remmerswaal EBM, Wesselink TH, Oja AE, et al. Blimp-1 induces and Hobit maintains the cytotoxic mediator granzyme B in CD8 T cells. Eur J Immunol (2018) 48(10):1644–62. doi: 10.1002/eji.201847771
160. Hayward SL, Scharer CD, Cartwright EK, Takamura S, Li Z-RT, Boss JM, et al. Environmental cues regulate epigenetic reprogramming of airway-resident memory CD8(+) T cells. Nat Immunol (2020) 21(3):309–20. doi: 10.1038/s41590-019-0584-x
161. Yoshizawa A, Bi K, Keskin DB, Zhang G, Reinhold B, Reinherz EL. TCR-pMHC encounter differentially regulates transcriptomes of tissue-resident CD8 T cells. Eur J Immunol (2018) 48(1):128–50. doi: 10.1002/eji.201747174
162. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment with Response to Anti–PD-1 Therapy. Clin Cancer Res (2014) 20(19):5064–74. doi: 10.1158/1078-0432.CCR-13-3271
163. Weisberg SP, Carpenter DJ, Chait M, Dogra P, Gartrell-Corrado RD, Chen AX, et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep (2019) 29(12):3916–32.e5. doi: 10.1016/j.celrep.2019.11.056
164. Konjar Š, Frising UC, Ferreira C, Hinterleitner R, Mayassi T, Zhang Q, et al. Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci Immunol (2018) 3(24):eaan2543. doi: 10.1126/sciimmunol.aan2543
Keywords: tissue resident memory T cell, T cell differentiation, recall response, microenvironment, transcriptional regulation
Citation: Mora-Buch R and Bromley SK (2021) Discipline in Stages: Regulating CD8+ Resident Memory T Cells. Front. Immunol. 11:624199. doi: 10.3389/fimmu.2020.624199
Received: 30 October 2020; Accepted: 31 December 2020;
Published: 19 March 2021.
Edited by:
Shiki Takamura, Kindai University, JapanReviewed by:
Klaas Van Gisbergen, Sanquin Diagnostic Services, NetherlandsCopyright © 2021 Mora-Buch and Bromley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shannon K. Bromley, c2Jyb21sZXlAbWdoLmhhcnZhcmQuZWR1
†Present address: Rut Mora-Buch, Cell Therapy Services, Blood and Tissue Bank, Barcelona, Spain
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.