 Els Lebegge1,2
Els Lebegge1,2 Sana M. Arnouk1,2
Sana M. Arnouk1,2 Pauline M. R. Bardet1,2
Pauline M. R. Bardet1,2 Máté Kiss1,2
Máté Kiss1,2 Geert Raes1,2†
Geert Raes1,2† Jo A. Van Ginderachter1,2*†
Jo A. Van Ginderachter1,2*†- 1Laboratory of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Brussels, Belgium
- 2Myeloid Cell Immunology Laboratory, VIB Center for Inflammation Research, Brussels, Belgium
Over the past decade, cancer immunotherapy has been steering immune responses toward cancer cell eradication. However, these immunotherapeutic approaches are hampered by the tumor-promoting nature of myeloid cells, including monocytes, macrophages, and neutrophils. Despite the arsenal of defense strategies against foreign invaders, myeloid cells succumb to the instructions of an established tumor. Interestingly, the most primordial defense responses employed by myeloid cells against pathogens, such as complement activation, antibody-dependent cell cytotoxicity and phagocytosis, actually seem to favor cancer progression. In this review, we discuss how rudimentary defense mechanisms deployed by myeloid cells can promote tumor progression.
Introduction
Immune cells abundantly infiltrate tumors, creating a complex environment mediated by repetitive cycles of antitumor responses and immune evasion (1). Myeloid innate immune cells, such as granulocytes, monocytes, macrophages and dendritic cells (DCs), play an important role in cancer-cell recognition, initiation of inflammation and antitumor responses (2). Chronic inflammation, however, can initiate tumorigenesis and can drive cancer progression in some cancer types (3, 4). Hence, myeloid cells play a dual role in cancer as they can initiate antitumor responses and communicate with cells of the adaptive immune system, but also promote local inflammation leading to chronic cancer-associated inflammation (5, 6).
In the tumor microenvironment, tumor-associated macrophages (TAMs) display an array of phenotypes beyond the M1/M2 paradigm, ranging from antitumoral to immunosuppressive, proangiogenic, immunomodulatory and tissue-remodeling phenotypes (7–9). The presence of TAMs in most solid tumors is correlated with poor prognosis and overall survival of patients (10). In addition to TAMs, solid tumors are also infiltrated by immunosuppressive, immature myeloid progenitor cells, commonly referred to as monocytic or polymorphonuclear myeloid-derived suppressor cells (M/PMN-MDSC) (11–13). Similarly, an increased infiltration of MDSCs has been associated with poor prognosis for a variety of cancer types (14). Neutrophils also contribute to tumor progression, yet establishing the difference between PMN-MDSCs and tumor-associated neutrophils (TAN) remains challenging (11, 15, 16). Although tumor-promoting functions have been attributed to other granulocytes, like eosinophils (17), basophils (18) and mast cells (19), further research is required to fully elucidate their role in cancer, as antitumoral roles have also been described (20, 21). Another myeloid population in the tumor microenvironment (TME) are DCs, that originate from different precursors and display various phenotypes, ranging from immunosuppressive monocyte-derived DCs (Mo-DCs) to immunocompetent cDC1 and cDC2 subsets (22). Altogether, the myeloid compartment in the TME is heterogenous and varies across tumor types, individuals and tumor stage (23). Nevertheless, the majority of scientific discoveries points toward a more tumor-supporting role for myeloid cells in the TME.
Rudimentary Myeloid Defense Strategies as Tumor Promoters
The innate immune response by myeloid cells occurs as a succession of events starting at signaling through cytosolic or surface PRRs, followed by effector responses including the release of cytokines, reactive oxygen species (ROS), reactive nitrogen species (RNS), antibacterial peptides and degranulation (Figure 1). PRR on myeloid cells can be triggered by pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), but also indirectly by secondary mechanisms such as complement activation and circulating antibodies (Abs), resulting in cytolytic and phagocytic effector mechanisms. Pathogen clearance is mediated by mechanisms such as phagocytosis, respiratory burst with the production of ROS and RNS and release of bacteriostatic peptides, but also through the cell-extrinsic initiation of inflammation via the release of proinflammatory cytokines and chemokines (24). However, this succession of events does not always appear to be a linear cascade, as feedforward loops and interactions exist between different effector mechanisms (Figure 2). Yet, even such early, innate effector mechanisms performed by myeloid cells surprisingly seem capable of promoting tumor progression.
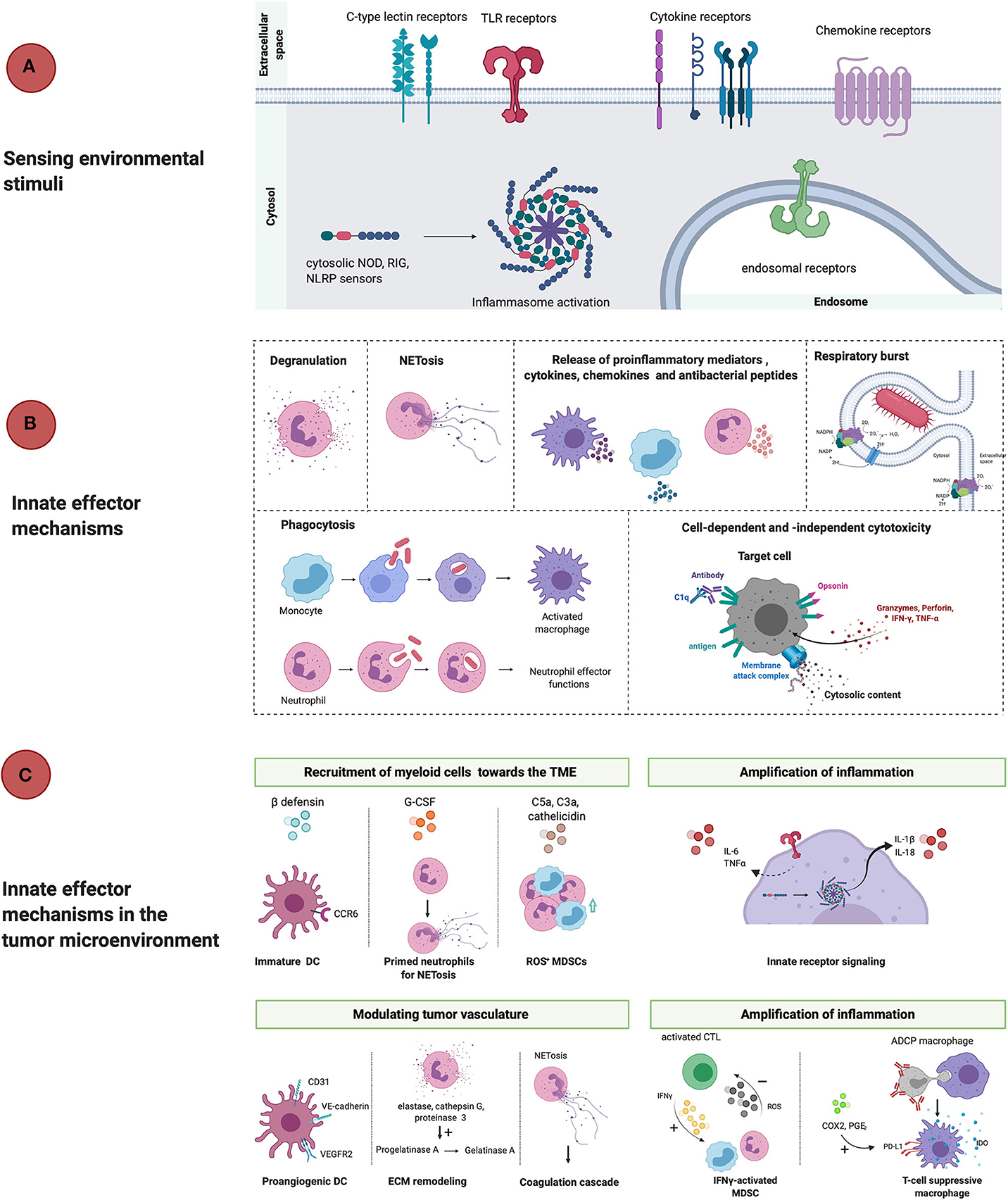
Figure 1. Linear representation of classical innate immunity in response to threats and in the TME. (A) PAMPs and DAMPs are recognized by surface-expressed, endosomal and cytosolic pattern recognition receptors (TLR, CLR, cytokine, chemokine receptors, NLRP3) which results in phenotypical changes that counteract ongoing threats or tissue damage. (B) Effector mechanisms that take place during inflammation are degranulation, NETosis, release of proinflammatory mediators, respiratory burst, phagocytosis and cell-dependent and -independent cytotoxicity. The net result is the recruitment of immunocompetent cells that mount an inflammatory reaction and potentially resolve the infection. (C) However, in the tumor microenvironment innate myeloid cells promote tumor progression through active recruitment to the TME in response to ß-defensins, cathelicidin, G-CSF, complement factors and chemokines. Once arrived in the TME, myeloid cells are activated and release proinflammatory mediators, which empowers tumor-associated inflammation. Activation of myeloid cells also allows for remodeling of the tissue vasculature and extracellular matrix, which also allows for cancer-cell invasion and metastasis. Furthermore, myeloid cells contribute to immunosuppression once activated by for example, upregulation of PD-L1 and IDO release during antibody-dependent phagocytosis of target cells or stimulatory cytokines (IFNγ). DC, dendritic cell; ECM, extracellular matrix; VEGFR2, vascular endothelial growth factor receptor 2; IFNγ, interferon gamma; ROS, reactive oxygen species; MDSC, myeloid-derived suppressor cell; ADCP, antibody-dependent cell-mediated phagocytosis; IDO, indoleamine 2,3-dioxygenase; COX2, cyclooxygenase 2; PGE2, prostaglandin E2; TNFα, tumor necrosis factor alpha; G-CSF, granulocyte colony stimulating factor; NOD, nucleotide-binding oligomerization domain; RIG, retinoic acid-inducible gene; NLRP, nucleotide-binding oligomerization domain; leucine-rich repeat and pyrin domain containing.
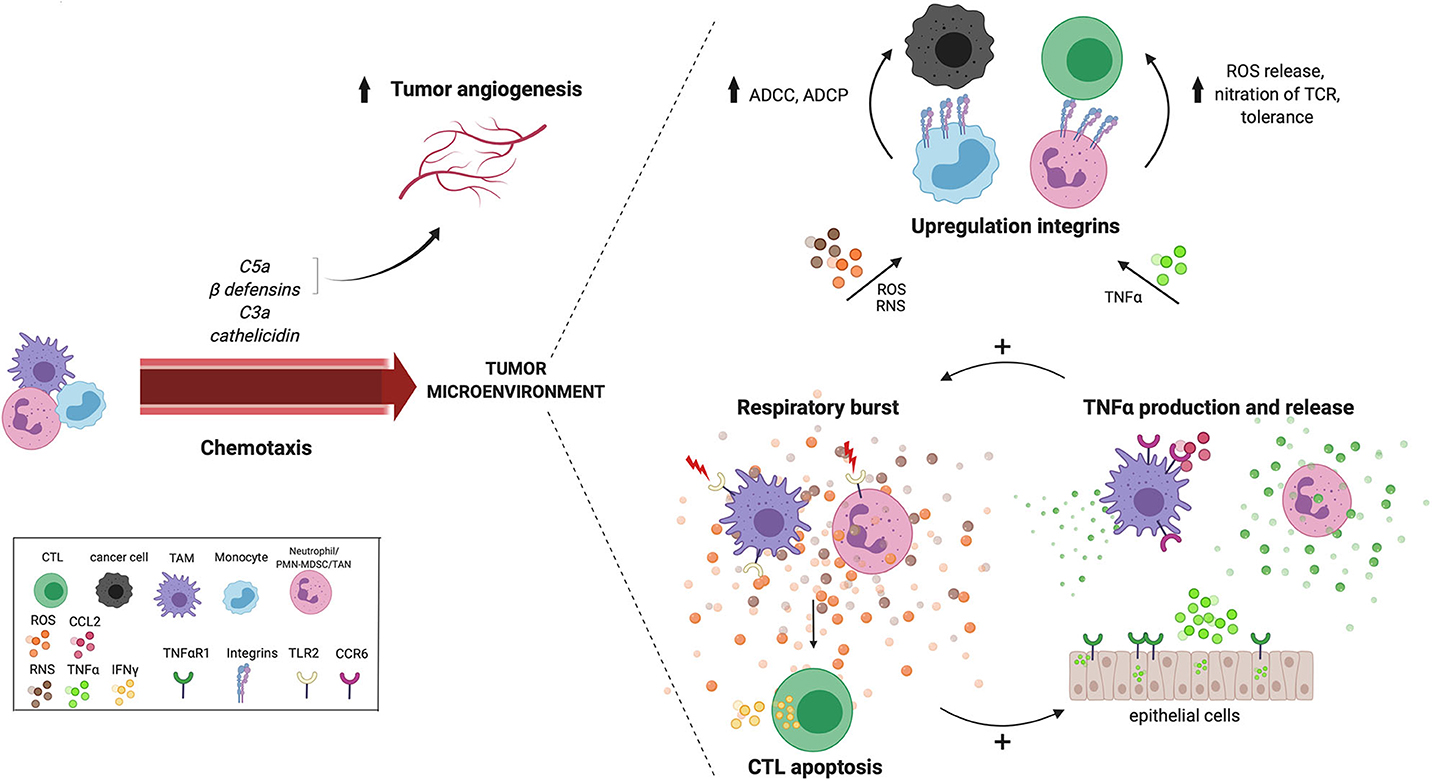
Figure 2. Cross talk between reoccurring innate effector mechanisms in the TME. Tumor-derived chemokines that are produced as a result of innate effector mechanisms including C5a, C3a, cathelicidin and ß-defensin, recruit myeloid cells to the TME. Tissue vasculature during chronic inflammation is maintained by complement anaphylatoxin C5a and beta-defensins. Anaphylatoxin C5a also recruits MDSCs with increased ROS and NRS production in the TME. Many innate pathways converge at the production of ROS and NOS in the TME. For example, TLR2 signaling increases the antigen presenting capacity of TAMs, which activates CTLs resulting in IFNγ release and subsequent ROS and NO release by TAMs. Neutrophil-derived ROS induces CTL apoptosis, while hydrogen peroxide released by TAMs, induces the expression of TNFα and TNFαR1 in surrounding epithelial cells. A positive feedback loop seems to exist between the respiratory burst and TNFα release, creating a potential cross talk between TAMs, neutrophils and epithelial cells in the TME. Furthermore, both ROS and TNFα also increases the expression of integrins, which increases cell-cell contact and facilitates cell-mediated killing via ADCC and ADCP either by performed by monocytes to kill cancer cells, or by MDSCs to suppress CTLs. ADCC, antibody-dependent cell-mediated cytotoxicity; MDSC, myeloid-derived suppressor cell; ADCP, antibody-dependent cell-mediated phagocytosis; ROS, reactive oxygen species; RNS, reactive nitrogen species; TNFα, tumor necrosis factor alpha; TNFαR1, tumor necrosis factor alpha receptor 1; CTL, cytotoxic T lymphocyte; IFNγ, interferon gamma; TCR, T-cell receptor; TAM, tumor-associated macrophage; CTL, cytotoxic T lymphocyte; TME, tumor microenvironment; TLR2, Toll-like receptor 2; TAM, tumor-associated macrophage.
Pathogen and Tissue Damage Recognition Mechanisms as Tumor Promoters
Toll-like receptors (TLRs), C-type lectin receptors (CLRs), the retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) and NOD-like receptors (NLRs) are PRR families expressed by macrophages and DCs, but also by non-immune cells, such as endothelial cells and fibroblasts (25). Based on current literature, it seems that PRR signaling can both contribute to cancer progression and is capable of steering antitumor responses. Here, we will focus on the tumor-promoting role of PRRs, where TLR signaling and inflammasome activation will serve as an example to demonstrate the effect of PRR signaling in tumor-infiltrating myeloid cells.
In response to the tumor-derived proteoglycan versican V1, TLR2- and TLR6-signaling in TAMs induces the expression of cathelicidin (hCAP18/LL-37), an antimicrobial peptide which in turn promotes the proliferation of human ovarian cancer cells in vitro (26). When a TLR2-agonist, lipoprotein Pam2CSK4, is administered intravenously, TLR2-expressing PMN-MDSCs accumulate and proliferate systemically in EG7 lymphoma-bearing mice (27). Moreover, Pam2CSK4-mediated TLR2 signaling promotes the survival of M-MDSCs and mediates the differentiation of M-MDSCs into macrophages. These macrophages are capable of presenting tumor antigens to CTLs, resulting in interferon gamma (IFNγ) release upon T-cell activation and the subsequent expression of inducible nitric oxide synthase (iNOS) and nitric oxide (NO) release by surrounding macrophages, which in turn leads to T-cell suppression (28). In the presence of bacterial lipopolysaccharides in the colonic lumen, TLR4 signaling in TAMs promotes chronic inflammation through increased production of cyclo-oxygenase 2 (COX2) and prostaglandin E2 (PGE2) (29). Damage-associated high mobility group box-1 protein (HMGB1), released from necrotic keratinocytes in the skin upon irradiation, interacts with TLR4 on bone marrow-derived immune cells (30). The resulting signaling facilitates papilloma progression through an increase in the recruitment of proinflammatory immune cells (30). Moreover, HMBG1-mediated TLR4 signaling causes an increased infiltration of radiation-resistant cells upon radiotherapy.
Upon intracellular PAMP or DAMP recognition by cytosolic sensors like NLRP3, inflammasomes are assembled, which results in the release of the proinflammatory cytokines IL-1ß and IL-18 and leads to a proinflammatory form of cell death, also referred to as pyroptosis (31). In different murine tumor models, NLRP3 plays a role in the migration of MDSCs to the TME, where MDSCs suppress antitumor CTL responses independent of NLRP3 and induce unresponsiveness to DC vaccination (32). The role of inflammasome activation in tumor progression is also demonstrated in obese mice, where obesity-associated NLRC4 inflammasome activation in tumor-infiltrating myeloid cells promotes breast cancer progression (33). Importantly, the release or administration of PRR agonists may give rise to therapy resistance in patients that underwent radiotherapy (34), chemotherapy (35, 36) or cancer vaccination (32). For example, myeloid Gr1-negative cells accumulate in murine B16 melanoma and CT26 colon adenocarcinoma tumors after local irradiation, where mitochondrial DNA of dead, irradiated cancer cells induces TLR9 signaling, which mediates revascularization and immune evasion in an interleukin (IL)-6- and STAT3-dependent manner (34, 37). Paclitaxel-induced TLR4 signaling in murine and human breast cancer cells results in the production of the proinflammatory cytokines IL-1ß and IL-6, which promotes the expansion of MDSCs in the bone marrow and spleen as well as their recruitment to the TME (36). In response to gemcitabine and 5-fluorouracil chemotherapy, cathepsin B is released in the cytosol of MDSCs which induces NLRP3-dependent IL-1ß release (35). In return, IL-1ß drives the polarization of CD4+ T cells into Th17 cells that promote tumor angiogenesis in the TME, which hampers the antitumor response of gemcitabine and 5-fluorouracil.
Altogether, it seems that the tumor microenvironment can be a source of PRR agonists, stimulating PRR signaling in myeloid cells that in turn perform tumor-promoting functions. Alternatively, PRR signaling can also directly affect cancer cells. TLR4 expression and signaling in gastric cancer cells results in mitochondrial ROS production, which induces secondary signaling cascades in response to oxidative stress that may regulate cancer-cell survival (38). TLR4 signaling in colorectal cancer and breast cancer cells promotes invasion and metastasis of these cells (36, 39). Therefore, PRR signaling is not strictly a myeloid cell-restricted, tumor-promoting mechanism.
Release of Proinflammatory Mediators as Tumor Promoters
A common downstream effect of PRR signaling is the release of proinflammatory cytokines, like IL-12, IL-6, IL-1β and tumor necrosis factor alpha (TNFα). In the TME, cytokines like IL-10 and transforming growth factor beta (TGF-ß) play an important role in suppressing antitumor responses, so it is within expectation that strongly opposing, proinflammatory mediators would be capable of eliciting and sustaining antitumor responses. However, a number of key proinflammatory cytokines, such as IL-1β and IL-6, have been reported to promote tumor progression through the mobilization of MDSCs (40, 41), the contribution to chronic inflammation (40, 42) and the stimulation of angiogenesis (43, 44). For example, in murine models of pancreatic ductal adenocarcinoma, neutralization of tumor-derived IL-1β enhances CTL-infiltration and ameliorates the response to anti-PD-1 immune checkpoint blockade (45). In accordance, IL-1β-blockade synergizes with anti-PD-1 immune checkpoint blockade in 4T1 breast cancers by restoring the cytotoxic capacity of CTLs without inducing systemic inflammation (46).
Other proinflammatory cytokines, such as TNFα and IFNγ, seem to have an ambiguous effect on cancer progression. For example, neutrophil-derived TNFα promotes the production of NO in an autocrine manner, which in turn induces apoptosis of non-activated CTLs in murine models of thoracic malignancies (47). Subcutaneous Tnfr1-deficient fibrosarcoma FB61 tumors are rejected in Tnfr-deficient mice, while tumor growth is reestablished via an adoptive transfer of Tnfr1-expressing MDSCs. Mechanistically, MDSCs of Tnfr-deficient mice displayed increased caspase-8 cleavage which induces apoptosis, and lower levels of c-FLIP, a natural caspase-8 inhibitor, which causes reduced accumulation of MDSCs in the TME along with a reduced tumor-suppressive capacity (48). These data suggest that endogenous and persistent TNFR signaling promotes tumor growth by maintaining survival of MDSCs (48). In accordance, a study by Sade-Feltman et al. demonstrated that TNFα is required to maintain the immature and immunosuppressive phenotype of MDSCs (49). Hence, TNFα blockade using Etanercept, a biological compound composed of the extracellular domain of TNFR fused to an IgG1 Fc fragment, restores NK-cell cytotoxicity and T-cell proliferation, reduces splenic MDSC accumulation and enhances the maturation of MDSC into CD11b+CD11c+ and CD11b+ F4/80+ cells (49). In addition, TNFα induced upon anti-PD-1 immune checkpoint blockade, increases PD-L1 and TIM-3 expression on tumor-infiltrating T cells and promotes their cell death upon TNFα binding to TNFRs (50). TNFα blockade increases the infiltration of tumor-specific CTLs, reduces the proliferation of immunosuppressive, regulatory T cells (Tregs) and minimizes toxicity of immune checkpoint blockade (51–53). These tumor-promoting effects of TNFα in the TME are in contrast to its inhibition of breast cancer-cell proliferation by blocking the G1/S phase transition of the cell cycle (54). Furthermore, TNFα may hamper the polarization and differentiation of monocytes into M2-like TAMs, instead steering the macrophage phenotype toward an anti-tumoral M1-like TAM in the TME (55). Altogether, TNFα also carries the potential to mount antitumoral responses in cancer therapy, as described elsewhere (56).
The role of the proinflammatory cytokine IFNγ in tumor progression appears to be concentration- (57, 58) and context-dependent (28, 59). He et al. demonstrated that, at low local levels, IFNγ promotes tumor progression of several murine tumor models, including hepatic, mammary and skin cancer, through increased gene expression of Cd274 (PD-L1), Ctla4 and Foxp3, whereas at higher levels, IFNγ reduces the gene expression of Foxp3 and co-inhibitory molecules (58). If either TNFα or IFNγ signaling in tumor-infiltrating CD4+ T cells is absent upon antigen recognition, tumor progression is stimulated, whereas combined TNFα and IFNγ signaling in CD4+ T cells prevents tumor angiogenesis and tumor-cell proliferation (59). Hence, cytokines like IFNγ and TNFα can play dual roles in cancer progression and the internal complexity of combined receptor signaling strongly affects antitumor responses (59).
Besides cytokines, other inflammatory mediators influence tumor progression. Indeed, proinflammatory enzymes and products of the prostaglandin production pathway, including COX2 and PGE2, have been associated with enhanced tumor progression, as they induce the expression of PD-L1 on macrophages and MDSCs (60). A tumor-promoting feedback loop has been discovered between MDSCs, colorectal cancer cells and T cells, that all release PGE2 and express receptor-interacting protein kinase 3 (RIPK3) (61). PGE2-induced RIPK3 signaling in MDSCs results in the expression of COX2 that catalyzes PGE2 synthesis, which is then released in the TME. PGE2 promotes proliferation of cancer cells and suppresses T-cell activation through RIPK3 signaling. Macrophage-derived IL-1β induces ROS-dependent COX2 production and activity in breast cancer cells, leading to PGE2 release in vitro (62). Culturing blood-derived monocytes with PGE2 induces the expression of COX2, which inhibits differentiation of monocytes into monocyte-derived DCs. Instead, the expression of indoleamine 2,3-dioxygenase (IDO), IL-4 receptor, iNOS and IL-10 is upregulated and drives the suppressive phenotype of M-MDSCs in vitro (63). Hence, PGE2 contributes to polarizing the phenotype of myeloid cells in the TME.
In conclusion, two trends are observed regarding proinflammatory cytokines or mediators; (1) either their role in cancer progression is generally protumoral, such as IL-6, IL-1β, or PGE2, or (2) their function in cancer progression is ambiguous, such as for TNFα and IFNγ. The severity of inflammation may play an important role here; to a certain extent, proinflammatory mediators are required to stimulate anti-tumoral T-cell responses, whereas prolonged exposure or exposure to high levels of inflammatory mediators can lead to unresponsiveness. In addition, it is not clear whether cancer cells or myeloid cells initiate the expression and release of tumor-promoting inflammatory mediators in the TME.
Respiratory Burst as Tumor Promoter
Upon PAMP recognition through PRR signaling, neutrophils and macrophages engulf pathogens via phagocytosis, which activates phagosome- and surface membrane-bound NADPH oxidase, resulting in the production of superoxide () and derivatives, hydrogen peroxide (H2O2) and hypochlorous acid (HOCl), through downstream processing by superoxide dismutase (SOD) and myeloperoxidase (MPO) (64–66). The release of ROS in phagosomes and the extracellular space is referred to as the respiratory burst, which is a primary antimicrobial and antifungal defense mechanism deployed by phagocytes (64). MDSCs are a major source of ROS in the TME, where ROS and peroxynitrite () abrogate antigen recognition by CTLs and instead induce tolerance (67, 68). This depends on direct contact between T cells and MDSCs, mediated by the integrins CD11b, CD18, and CD29 (68). Mechanistically, nitration and oxidation of amino acids in the T-cell receptor (TCR) and CD8 co-receptor molecules prevents interaction with major histocompatibility complex (MHC) molecules, which in turn induces tolerance (67). Constitutive upregulation of STAT3 in MDSCs directly regulates the expression of NOX2 components necessary for the formation of the NADPH protein complex, which is followed by a subsequent increase in production and release of ROS (69). MDSCs are unable to suppress T cells in the absence of NOX2 activity, and instead differentiate into mature macrophages and DCs (69). ROS also mediate the polarization of macrophages, as inhibition of impedes the differentiation of monocytes into M2 macrophages while differentiation into M1 macrophages remains unaltered (70, 71). Thus, while ROS production in MDSCs maintains their immature phenotype, MDSC-derived ROS in the TME mediates the differentiation of tumor-infiltrating monocytes. Furthermore, H2O2 released by macrophages and neutrophils induces the expression of Tnfa and Tnfr1 in epithelial cells, that in turn release TNFα leading to the upregulation of other proinflammatory and angiogenic factors, hence, sustaining tumor progression in a paracrine loop (72). Aside from myeloid-derived ROS, Xia et al. demonstrated that ROS can also be produced by cancer cells themselves. They showed that ROS production by ovarian cancer cells promotes angiogenesis and tumor growth through in vivo transcriptional activation of Vegf and Hif1a (72, 73). The above-mentioned studies provide evidence for the protumoral role of ROS in tumor progression, by suppressing T-cell responses, supporting angiogenesis and maintaining the phenotypical identity of MDSCs, regardless of the strong pathogen-killing potential of the respiratory burst in mature myeloid cells.
Release of Antibacterial Peptides as Tumor Promoter
In addition to ROS, myeloid cells release a vast array of antimicrobial peptides such as defensins and cathelicidins, representing two major families of mammalian antibacterial peptides. In leukocytes, α- and ß-defensins are stored in cytoplasmic granules that fuse with the phagosome upon microbe phagocytosis, while epithelial cells can secrete defensins to maintain their barrier integrity (74). Yang et al. demonstrated that ß-defensins act as a chemoattractant for immature DCs and memory T cells by binding chemokine receptor CCR6, which bridges the innate recognition of microbes and the initiation of an adaptive immune response (75). As such, it is not surprising that in a similar fashion immature DCs are recruited to the TME in response to tumor-derived ß-defensins. Indeed, Conejo-Garcia et al. discovered a subset of immature DCs, that is recruited to murine and human ovarian tumors in response to ß-defensins through CCR6 signaling and that acquires epithelial features, including surface expression of CD31 and VE-cadherin. These cells support vasculogenesis in a VEGFR-2-dependent manner which leads to enhanced tumor progression (76). CCR6 signaling also promotes murine transplantable colon cancer by recruiting macrophages to the TME through a CCL2-CCR6 axis, which results in the release of IL-1β, IL-6, and TNFα, further enhancing tumor progression (77).
Holterman et al. reported that α-defensins overexpressed by cancer cells, stimulate the proliferation and migration of bladder cancer cell lines in vitro, most-likely in an autocrine and calcium-dependent manner (78). Similarly, Xu et al. showed that human ß-defensin 3 promoted in vitro proliferation, migration and invasion of cervical cancer through the NF-κB signaling pathway, demonstrating that cancer cells are also able to release defensins (79). It is important to note that defensin-secreting cancer cells are of epithelial origin, since epithelial cells are known to secrete defensins as part of their barrier function. In addition, it should be remarked that the role of defensins in tumor progression also seems ambiguous and may vary according to the cancer type or defensin molecule, as several studies showed a potential antitumoral role of defensins in cancer (80, 81).
The release of cathelicidins, human LL-37 and murine CRAMP, in the TME has been described in several studies, whereby macrophages and neutrophils are the main sources. Li et al. demonstrated that CD68+ macrophages in tumor tissue of colorectal cancer patients stained positive for cathelicidin, whereas weak to unmeasurable signal was picked up for cathelicidins in colon epithelial cells (82). The importance of cathelicidins in tumor progression was demonstrated by a slower tumor growth in Lewis lung carcinoma-bearing, cathelicidin-deficient mice, along with a reduced infiltration of myeloid cells (83). These studies suggest that cathelicidins are chemoattractants that recruit myeloid cells to the TME (84), where, in turn, myeloid-derived cathelicidins directly enhance cancer-cell proliferation, creating a self-sustaining loop of cathelicidin production. In contrast, antitumoral roles of cathelicidins, independent of myeloid cells, have also been described. For example, cathelicidins could be involved in potentiating the cytotoxic capacity of tumor-infiltrating NK cells (85) and impairing the tumor-supportive role of cancer-associated fibroblasts (CAFs) in colon cancer (86).
Neutrophil Degranulation as Tumor Promoter
Neutrophils carry heterogenous primary, secondary and tertiary granules that contain different enzymes and modulatory proteins, such as elastase, gelatinase, MPO, cathepsins, ficolin-1, and lactoferrin (87). Neutrophil degranulation occurs in a calcium-dependent manner in response to proinflammatory mediators like TNFα (88), lipopolysaccharides (LPS) (89) and IL-8 (90). The majority of neutrophil-derived granule contents promote tumor progression, such as elastase, cathepsin D, cathepsin B, and proteinase 3.
Neutrophil-derived elastase hydrolyses insulin receptor substrate-1 (IRS1) in the cytosol of lung cancer cells, leading to an altered regulation of phosphoinositide 3-kinase (PI3K). IRS1 degradation indirectly increases the interaction between the p85 protein of PI3K and platelet-derived growth factor receptor (PDGFR), which enhances cancer-cell proliferation through signaling downstream of the PDGFR (91). Elastase released by PMN-MDSCs in lymphangioleiomyomatosis patients, a condition where estrogen-sensitive metastatic tumors grow in the lungs, stimulate the proliferation, migration and invasion of these tumor cells in vitro (92). Cathepsin-D stimulates cancer-cell proliferation as well, but also stimulates tumor angiogenesis and could protect cancer cells from apoptosis (93). Hepsin, a transmembrane serine protease involved in cell motility and shape, is degraded by the proteasome through cathepsin D-stimulated ubiquitination (94). By downregulating hepsin, cathepsin D contributes to enhanced migration and invasion of breast cancer. Cathepsin B cleaves cell cycle inhibitor p27Kip1 in the lysosomes of colorectal cancer cells, which contributes to tumorigenicity and metastasis of colorectal cancer cells (95). Extracellular matrix (ECM) and intracellular collagen IV can be degraded by cathepsin B, stimulating tumor invasion, metastasis, and the formation of vessel-like structures in vivo (96).
Although proteinase 3 can be secreted by myeloid cells, neutrophils carry a membrane-bound proteinase 3 that seems to play a role in cellular interactions. Neutrophils in acute myeloid leukemia inhibit T-cell proliferation in a contact-dependent manner. Antibody-based blockade of membrane-bound proteinase 3 on the surface of neutrophils partially restores proliferation of CD4+ and CD8+ T cells (97). The resulting signaling cascade caused by the interaction between membrane-bound proteinase 3 on neutrophils and receptor for advanced glycation end-products (RAGE) on prostate cancer cells promotes tumor-cell migration and metastasis to the bone barrow, independent of the proteolytic activity of proteinase 3 (98). Combined efforts of neutrophil elastase, cathepsin G and proteinase 3 activate progelatinase A, that degrades the extracellular matrix followed by the subsequent release of growth factors, tumor-cell invasion and angiogenesis in the TME (99). In conclusion, the majority of enzymes released or upregulated upon neutrophil degranulation can remodel the extracellular matrix, which stimulates tumor-cell invasion, metastasis and tumor growth, but also promotes tumor angiogenesis.
Neutrophil Extracellular Trap Formation (NETosis) as Tumor Promoter
Neutrophil extracellular traps (NETs) are extracellular strands composed of granule content and nuclear fragments that entrap and kill bacteria through granule proteases and DNA histones (100, 101). Various studies have demonstrated that the formation of NETs is ROS-dependent (64, 102, 103), but can also occur through CXCR2 signaling during chronic inflammation, and through TLR2 and C3 signaling (101, 104). A study unraveling the role of high sensitivity troponin T (hsTnT) plasma levels in the onset of ischemic stroke, revealed an unexpected high prevalence of cancer among patients with elevated hsTnT plasma levels in the post mortem analysis (105). In these patients, the elevated hsTnT plasma level was associated with an increased plasma level of NET-associated citrullinated histone H3, a marker for NETosis, as well as increased plasma levels of G-CSF and coagulation factors. This study demonstrates that NETosis can take place in cancer patients with elevated citrullinated histone H3 levels (105). In fact, tumor-derived G-CSF primes neutrophils to form NETs, which could also contribute to a systemic, prothrombic state in these cancer patients (105, 106).
Furthermore, a study by Miller-Ocuin et al. correlated circulating neutrophil DNA, resulting from NETosis, to the cancer stage of pancreatic ductal adenocarcinoma patients (107). They demonstrated that neutrophil DNA activates pancreatic stellate cells that support tumor progression, and propose that NET DNA acts as a DAMP capable of stimulating tumor progression (107). In patients that underwent major liver resection of metastatic colorectal cancer, in which ischemia and reperfusion is inevitable, NET formation was increased compared to cancer patients that underwent minor liver resection, in which ischemia and reperfusion is limited, demonstrating that surgery-induced stress promotes NET formation (108). These authors further demonstrated that NETs in the liver provide an anchoring site for circulating cancer cells, that supports metastases and cancer-cell growth after resection of the primary tumor. Hence, NETs may support tumor progression through various mechanisms.
Surrounding macrophages deal with the aftermath of NETosis by digesting cellular debris. Interestingly, M1-like macrophages have been shown to release uncoiled or uncondensed DNA upon interaction with NETs in vitro, suggesting a possible contribution to NETosis through their own form of extracellular trap formation (METosis) (109). It remains to be seen whether such a mechanism contributes to the tumor-promoting effects of macrophages.
Complement Activation as Tumor Promoter
Complement is an innate defense mechanism that detects and eliminates pathogens from the circulation and tissues, clears cellular debris and stimulates adaptive immunity.
Complement activation through the classical, alternative or lectin-mediated pathway ultimately results in the formation of a cytolytic membrane attack complex (MAC) in the membrane of target cells or microorganisms (110) and the production of anaphylatoxins C3a and C5a (111). Anaphylatoxins can be involved in T-cell homeostasis (112, 113) and in the recruitment of granulocytes (114–116), monocytes (117) and DCs (118) to the site of inflammation through chemotaxis via C3a and C5a receptors (C3aR, C5aR).
Aside from anaphylatoxin production during complement activation, opsonin C3b and its cleavage products (iC3b, C3c, C3d) are deposited on the surface of target cells or microorganisms, when C3 is cleaved by C3 convertase (119). Myeloid cells express complement receptors that bind C3-derivatives, leading to phagocytosis, cell-cell adhesion and adhesion to the extracellular matrix (120). Complement can also steer the adaptive immunity by activating B and T cells through combined engagement of complement receptors and the B-cell receptor or TCR, respectively (121).
Overall, the resulting effector mechanisms of complement activation are (1) cell-mediated phagocytosis (complement-dependent cellular phagocytosis or CDCP) and (2) cytotoxicity (complement-dependent cellular cytotoxicity or CDCC), initiated by the interaction between opsonized target cells or microbes and CR-expressing myeloid cells, as well as (3) complement-dependent cytotoxicity (CDC) through the formation of the MAC in the membrane of target cells or microorganisms, and (4) the recognition and clearance of dying cells (122) (Figure 3). However, distinguishing the different effector mechanisms that contribute to cancer-cell eradication as a result of complement activation remains challenging up to now. Furthermore, complement-induced cytolytic effector mechanisms on the surface of host cells is prevented through the expression of complement regulatory proteins (CRPs), such as CD46, CD55, CD59 and factor H. Several cancer types overexpress CRPs and make use of this defense mechanism against complement-induced cytolysis (123–125), whereas downregulation or blockade of CRPs sensitizes cancer cells to complement- and antibody-mediated cytotoxicity (126, 127).
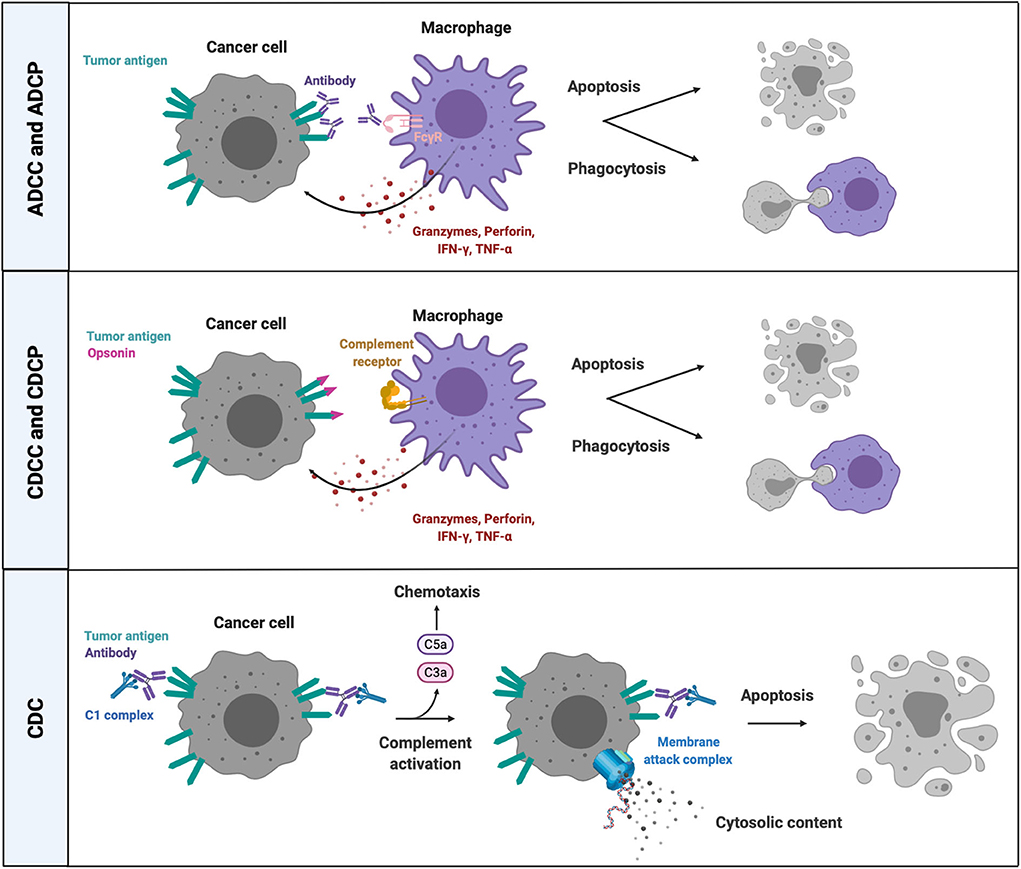
Figure 3. Cell-dependent and -independent effector mechanisms of complement activation and FcR-mediated killing. Complement factor- and antibody-opsonized cancer cells can be eliminated through cell-dependent and cell-independent effector mechanisms. CRs and FcRs on phagocytes bind opsonins and antibodies, respectively, on the surface of targeted cancer cells, followed by phagocytosis and/or release of lytic enzymes (granzyme B, perforins) and proinflammatory mediators (TNFα, IFNγ). The classical pathway of complement activation mediates a cell-independent form of lytic cell death by introducing a MAC in the membrane of antibody opsonized target cells that are recognized by complement C1 complex. ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cell-mediated phagocytosis; CDC, complement-dependent cytotoxicity; CDCC, complement-dependent cell-mediated cytotoxicity; CDCP, complement-dependent cell-mediated phagocytosis; CRs, complement receptors; IFNγ, interferon gamma; TNFα, tumor necrosis factor alpha; C5a, complement factor 5a; C3a, complement factor C3a; FcγR, crystallizable fragment receptor gamma; C1, complement factor.
Complement Anaphylatoxins as Tumor Promoters
Complement activation has been reported to promote tumor progression through the recruitment of immune suppressive macrophages, MDSCs and neutrophils, while on the other hand, there are also reports of its capacity to stimulate antitumoral T-cell responses and the recruitment of NK cells (128–130). Recruitment of MDSCs in response to anaphylatoxins has been demonstrated in several studies (130–132). Markiewski et al. revealed that aside from increased recruitment of MDSCs to the tumor in response to C5a, the latter also enhances the production of ROS and RNS in MDSCs via C5aR signaling (132). As mentioned earlier, ROS and RNS release by MDSCs in the TME abrogates antigen recognition by CTLs and instead induces tolerance (67, 68). Moreover, C5a is also implicated in the formation of new blood vessels. Corrales et al. demonstrated that human umbilical vein endothelial cells treated with C5a form vessel-like structures. They further elaborated on the vessel-like structures in a murine 3LL lung cancer model, where they showed that the number of newly formed microvessels in the tumor is reduced upon C5aR antagonism (133). While the role of C5a in cancer progression has been extensively studied, less is known about the implication of C3a in cancer. In the absence of C3aR signaling, murine B16 melanoma tumor growth is reduced, along with an increased tumor infiltration of CD4+ T cells and neutrophils (134). Similar results were observed in orthotopic mouse models of lung cancer (CMT167, LLC), where flow cytometry and immunohistochemistry analysis revealed an increased abundance of activated CD4+ and CD8+ T cells in tumors grown in C3-deficient mice (135). Interestingly, depletion of CD4+ T cells, but not CD8+ T cells, restored tumor growth in C3-deficient mice.
Tumor-infiltrating macrophages and neutrophils also carry the potential to suppress the detrimental effects of complement activation through IL-1ß-induced expression of pentraxin 3 (PTX3) (136). Surface-expressed PTX3 recruits complement factor H that inhibits the C3 cleavage upstream of the complement cascade and prevents complement-induced inflammation and recruitment of immunosuppressive myeloid cells to the TME. However, Ptx3 is epigenetically silenced at the gene level in murine and human colorectal cancer through hypermethylation (136). Altogether, the above-mentioned studies provide evidence for the role of complement in cancer that seems to promote tumor progression by recruiting MDSCs to the tumor, reducing the infiltration of activated CD4+ T cells and stimulating new vessel formation. The effect of anaphylatoxins on tumor-infiltrating CTLs remains unresolved, whereas several studies highlight the importance of CD4+ T cells in response to anaphylatoxins.
Complement in Cancer Immunotherapy
Despite the intrinsic protumoral functions of complement in cancer, it should not be forgotten that complement can be useful in the context of antibody-mediated cancer immunotherapy. Indeed, the classical pathway of complement activation, initiated by antibody-opsonized target cells, is one of the effector mechanisms of therapeutic monoclonal antibodies (mAb) (137, 138). This was demonstrated in a study by Lee et al., who designed therapeutic mAbs capable of discerning complement-mediated and Fc receptor (FcR)-mediated killing mechanisms (139). Aglycosylated, anti-CD20 IgG1 mAb, engineered with a C1q-selective Fc-part that does not bind FcRs, demonstrated similar potency in clearing CD20+ Raji and Ramos lymphoblastic cells compared to antibodies that rely on FcR-mediated functions (139). Along the same line, the therapeutic anti-CD20 mAb Rituximab at least partially relies on the classical complement activation pathway for destruction of neoplastic CD20+ B cells (140). However, the release of proinflammatory mediators (IL-6, TNFα) and degranulation by granulocytes in response to complement anaphylatoxins contribute to the toxic side effects of anti-CD20 therapy, such as fever, dyspnea, chills and flushes (141). Similarly, the in vivo effector functions of Cetuximab, an anti-EGFR mAb, have been attributed to complement activation in several murine models of non-small cell lung carcinoma (142). However, it should be remarked that the efficacy of mAb-mediated complement activation is likely to be cancer type-dependent and may be influenced by the characteristics of the cancer cells and/or factors present in the tumor microenvironment.
Moreover, the efficacy of antibody-based therapy, that relies on the cytotoxic effector mechanisms of complement and FcR-mediated cytotoxicity, is restricted by the limited availability of suitable antigens for therapeutic targeting. In addition, the dual role of complement in cancer must be taken into account when using complement as an effector mechanism of antibody-based therapy. It appears that complement can promote tumor growth through high C5a concentrations, sublytic MACs levels and high CRP levels on the surface of cancer cells, while intermediate concentrations of C5a, increased MAC formation in the membrane of cancer cells and low surface expression of CRPs could eliminate cancer cells (129, 143). Future therapeutic strategies should take this delicate balance between tumor promotion and tumor eradication into account.
FcR-Mediated Killing
When the Fc part of an antibody interacts with cognate surface-expressed FcRs, this may result in ADCC, ADCP, antigen presentation, degranulation and an altered cytokine production profile (Figure 3) (144). NK cells are thought to be the main effector cells of ADCC, yet studies have shown that antibody-based cellular destruction mechanisms can also take place in the absence of NK cells (145). The relevance for therapeutic mAbs is shown by mice deficient in the common gamma chain of the FcγR. These mice do not engage ADCC or ADCP in the presence of Trastuzumab and Rituximab (145, 146). Members of the mononuclear phagocyte system, including monocytes and macrophages, are responsible for the working mechanism of Rituximab (145). Indeed, CD20-targeted B-cell depletion seems to be dependent on FcγRI and FcγRIII expressed by monocytes and macrophages and is absent in colony stimulating factor 1-deficient mice, which lack tissue macrophage subsets (145). Biburger et al. discovered a murine subset of Ly6Clow non-classical monocytes capable of autoantibody-mediated platelet depletion and antibody-dependent B-cell depletion via ADCC and ADCP mediated by FcγRIV, a low affinity FcγR that is not expressed by NK cells or tissue-resident macrophages (147). Human CD16+ (FcγRI) monocytes similarly perform ADCC, almost as efficiently as NK cells. TNFα release by these CD16+ monocytes upregulates type 2 beta integrins (CD11a, CD11b), which facilitate the interaction between CD16+ monocytes and antibody-coated cancer cells (148). The number of murine B16 melanoma metastases in the lung of FcγRIIb-deficient mice significantly decreased when treated with a mAb targeting melanoma differentiation antigen gp75 (146). FcγRIIb is an inhibitory Fc receptor which is not expressed by NK cells. Therefore, an enhanced ADCC response cannot be attributed to increased NK-cell activation in FcγRIIb-deficient mice and is likely monocyte/macrophage-mediated. Moreover, a synergistic effect was observed when combining FcγRIIb deficiency and a therapeutic mAb against mouse and human HER2 (4D5, Trastuzumab) (146).
However, not all FcR-mediated effects are beneficial in the context of mAb-mediated therapy. For example, phagocytosis of antibody-opsonized cancer cells by TAMs was shown to activate the inflammasome AIM2, which results in the subsequent release of IL-1ß, hence increasing PD-L1 surface expression and cytosolic IDO production in TAMs (149). As a result, TAMs that underwent ADCP display an immunosuppressive phenotype, which is relieved upon PD-L1 and IDO blockade (149). Furthermore, in vivo imaging by Arlauckas et al. (150) demonstrated that PD-1-negative TAMs take up anti-PD-1 antibodies that were initially bound to PD-1+ CTLs, in an FcR-mediated way. Hence, TAMs could serve as a sink for anti-PD-1 antibodies and possibly also other mAbs, strongly diminishing the efficacy of mAb-dependent therapies such as immune checkpoint blockade (150).
Entangled Network of Innate Responses
Innate immune responses are often regarded as the default first-line defense responses, that become less significant once a more complex, adaptive and antigen-directed response is initiated. With this review, we provide evidence for the detrimental effects of innate effector mechanisms performed by myeloid cells during cancer development and progression. Noteworthy, effector mechanisms that are initially deployed by innate myeloid cells, such as ROS production, release of inflammatory mediators and response to PRR signaling, can be adopted by cancer cells. However, contradicting literature studies are available on the role of several innate defense mechanisms in cancer, and this duality between tumor-promoting and -eradicating roles seems to be linked to the presence of persisting, tumor-associated inflammation. Inflammation is required to mount anti-tumor immune responses, while chronic tumor-associated inflammation promotes tumor progression. This duality can even be extended to the response of so called “hot tumors” and “cold tumors” to immunotherapy. Interestingly, mAb therapy targeting immune checkpoints seem to be effective in “hot tumors,” abundantly infiltrated by T cells, whereas “cold tumors” that lack proper T-cell responses remain largely unresponsive to mAb therapy (151). Cold tumors, however, are still infiltrated by myeloid cells, that create an immune suppressive environment, which impedes T-cell infiltration and tumor eradication. Therefore, innate defense strategies might play a more important role in cancers with an inflammatory nature or origin, for example in organs like the liver, stomach, lungs and skin due to alcohol abuse, H. pylori infection, tobacco and asbestos, UV irradiation and even obesity. In any case, due to the abundance of tumor-infiltrating myeloid cells in multiple solid tumor types, their effector mechanisms should be investigated in depth and exploited in cancer therapy, perhaps alongside T-cell stimulatory immunotherapy to improve therapy outcome.
Author Contributions
EL has conceptualized, written, reviewed, and edited the content of this review, with contributions from SA, PB, and MK. JV and GR contributed to the conceptualization and reviewing of this review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants of the Research Foundation Flanders (FWO-Vlaanderen), Kom op tegen Kanker (Stand up to Cancer), and Stichting tegen Kanker (Foundation against Cancer) granted to GR and JV. EL, SA, PB, and MK received a doctoral grant from FWO-Vlaanderen. MK was also supported by a doctoral finishing grant from Kom op Tegen Kanker.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank all researchers in the Cellular and Molecular Immunology lab for critical discussions and insightful ideas.
References
1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
2. Woo S-R, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. (2015) 33:445–74. doi: 10.1146/annurev-immunol-032414-112043
3. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
4. Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. (2005) 7:211–17. doi: 10.1016/j.ccr.2005.02.013
5. De Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. (2006) 6:24–37. doi: 10.1038/nrc1782
6. Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
7. Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, et al. M-CSF and GM-CSF receptor signaling differentially regulate monocyte maturation and macrophage polarization in the tumor microenvironment. Cancer Res. (2016) 76:35–42. doi: 10.1158/0008-5472.CAN-15-0869
8. Bolli E, Movahedi K, Laoui D, Ginderachter JAV. Novel insights in the regulation and function of macrophages in the tumor microenvironment. Curr Opin Oncol. (2017) 29:55–61. doi: 10.1097/CCO.0000000000000344
9. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van Bossche JD, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.CAN-09-4672
10. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. (2012) 7:e50946. doi: 10.1371/journal.pone.0050946
11. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
12. Ostrand-Rosenberg S, Fenselau C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol. (2018) 200:422–31. doi: 10.4049/jimmunol.1701019
13. Movahedi K, Guilliams M, Van Den Bossche J, Van Den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell suppressive activity. Blood. (2008) 11:4233–44. doi: 10.1182/blood-2007-07-099226
14. Ai L, Mu S, Wang Y, Wang H, Cai L, Li W, et al. Prognostic role of myeloid-derived suppressor cells in cancers: a systematic review and meta-analysis. BMC Cancer. (2018) 18:1220. doi: 10.1186/s12885-018-5086-y
15. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS ONE. (2012) 7:e31524. doi: 10.1371/journal.pone.0031524
16. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
17. Reichman H, Karo-Atar D, Munitz A. Emerging roles for eosinophils in the tumor microenvironment. Trends Cancer. (2016) 2:664–75. doi: 10.1016/j.trecan.2016.10.002
18. Marone G, Gambardella AR, Mattei F, Mancini J, Schiavoni G, Varricchi G. Basophils in tumor microenvironment and surroundings. Adv Exp Med Biol. (2020) 1224:21–34. doi: 10.1007/978-3-030-35723-8_2
19. Ribatti D, Tamma R, Crivellato E. The dual role of mast cells in tumor fate. Cancer Lett. (2018) 433:252–8. doi: 10.1016/j.canlet.2018.07.005
20. Reichman H, Itan M, Rozenberg P, Yarmolovski T, Brazowski E, Varol C, et al. Activated eosinophils exert antitumorigenic activities in colorectal cancer. Cancer Immunol Res. (2019) 7:388–400. doi: 10.1158/2326-6066.CIR-18-0494
21. Sektioglu IM, Carretero R, Bulbuc N, Bald T, Tüting T, Rudensky AY, et al. Basophils promote tumor rejection via chemotaxis and infiltration of CD8+ T cells. Cancer Res. (2017) 77:291–302. doi: 10.1158/0008-5472.CAN-16-0993
22. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. (2016) 7:13720. doi: 10.1038/ncomms13720
23. Kiss M, Van Gassen S, Movahedi K, Saeys Y, Laoui D. Myeloid cell heterogeneity in cancer: not a single cell alike. Cell Immunol. (2018) 330:188–201. doi: 10.1016/j.cellimm.2018.02.008
24. Kaufmann SHE, Dorhoi A. Molecular determinants in phagocyte-bacteria interactions. Immunity. (2016) 44:476–91. doi: 10.1016/j.immuni.2016.02.014
25. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
26. Li D, Wang X, Wu JL, Quan WQ, Ma L, Yang F, et al. Tumor-Produced Versican V1 enhances hCAP18/LL-37 expression in macrophages through activation of TLR2 and vitamin D3 signaling to promote ovarian cancer progression in vitro. PLoS ONE. (2013) 8:e56616. doi: 10.1371/journal.pone.0056616
27. Maruyama A, Shime H, Takeda Y, Azuma M, Matsumoto M, Seya T. Pam2 lipopeptides systemically increase myeloid-derived suppressor cells through TLR2 signaling. Biochem Biophys Res Commun. (2015) 457:445–50. doi: 10.1016/j.bbrc.2015.01.011
28. Shime H, Maruyama A, Yoshida S, Takeda Y, Matsumoto M, Seya T. Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. Oncoimmunology. (2017) 7:e1373231. doi: 10.1080/2162402X.2017.1373231
29. Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. (2007) 133:1869–81. doi: 10.1053/j.gastro.2007.09.008
30. Mittal D, Saccheri F, Vénéreau E, Pusterla T, Bianchi ME, Rescigno M. TLR4-mediated skin carcinogenesis is dependent on immune and radioresistant cells. EMBO J. (2010) 29:2242–52. doi: 10.1038/emboj.2010.94
31. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev. (2015) 265:130–42. doi: 10.1111/imr.12287
32. Van Deventer HW, Burgents JE, Wu QP, Woodford RMT, Brickey WJ, Allen IC, et al. The inflammasome component Nlrp3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. (2010) 70:10161–9. doi: 10.1158/0008-5472.CAN-10-1921
33. Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun. (2016) 7:13007. doi: 10.1038/ncomms13007
34. Gao C, Kozlowska A, Nechaev S, Li H, Zhang Q, Hossain DMS, et al. TLR9 signaling in the tumor microenvironment initiates cancer recurrence after radiotherapy. Cancer Res. (2013) 73:7211–21. doi: 10.1158/0008-5472.CAN-13-1314
35. Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. (2013) 19:57–64. doi: 10.1038/nm.2999
36. Volk-Draper L, Hall K, Griggs C, Rajput S, Kohio P, DeNardo D, et al. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. (2014) 74:5421–34. doi: 10.1158/0008-5472.CAN-14-0067
37. Roda JM, Parihar R, Carson WE. CpG-containing oligodeoxynucleotides act through TLR9 to enhance the NK cell cytokine response to antibody-coated tumor cells. J Immunol. (2005) 175:1619–27. doi: 10.4049/jimmunol.175.3.1619
38. Yuan X, Zhou Y, Wang W, Li J, Xie G, Zhao Y, et al. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. (2013) 4:e794. doi: 10.1038/cddis.2013.334
39. Hsu RYC, Chan CHF, Spicer JD, Rousseau MC, Giannias B, Rousseau S, et al. LPS-induced TLR4 signaling in human colorectal cancer cells increases β1 integrin-mediated cell adhesion and liver metastasis. Cancer Res. (2011) 71:1989–98. doi: 10.1158/0008-5472.CAN-10-2833
40. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. (2008) 14:408–19. doi: 10.1016/j.ccr.2008.10.011
41. Chen MF, Kuan FC, Yen TC, Lu MS, Lin PY, Chung YH, et al. IL-6-stimulated CD11b+CD14+HLA-DR- myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget. (2014) 5:8716–28. doi: 10.18632/oncotarget.2368
42. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. (2011) 19:456–69. doi: 10.1016/j.ccr.2011.03.009
43. Voronov E, Carmi Y, Apte RN. The role IL-1 in tumor-mediated angiogenesis. Front Physiol. (2014) 5:114. doi: 10.3389/fphys.2014.00114
44. Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB, Lee CN, et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. (2003) 22:1517–27. doi: 10.1038/sj.onc.1206226
45. Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. (2020) 80:1088–101. doi: 10.1158/0008-5472.CAN-19-2080
46. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc Natl Acad Sci USA. (2019) 116:1361–69. doi: 10.1073/pnas.1812266115
47. Michaeli J, Shaul ME, Mishalian I, Hovav AH, Levy L, Zolotriov L, et al. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology. (2017) 6:e1356965. doi: 10.1080/2162402X.2017.1356965
48. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. (2012) 122:4094–104. doi: 10.1172/JCI64115
49. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity. (2013) 38:541–54. doi: 10.1016/j.immuni.2013.02.007
50. Bertrand F, Montfort A, Marcheteau E, Imbert C, Gilhodes J, Filleron T, et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat Commun. (2017) 8:2256. doi: 10.1038/s41467-017-02358-7
51. Chang LY, Lin YC, Chiang JM, Mahalingam J, Su SH, Huang CT, et al. Blockade of TNF-α signaling benefits cancer therapy by suppressing effector regulatory T cell expansion. Oncoimmunology. (2015) 4:e1040215. doi: 10.1080/2162402X.2015.1040215
52. Perez-Ruiz E, Minute L, Otano I, Alvarez M, Ochoa MC, Belsue V, et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature. (2019) 569:428–32. doi: 10.1038/s41586-019-1162-y
53. Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell. (2019) 178:585–99.e15. doi: 10.1016/j.cell.2019.06.014
54. Pusztai L, Lewis CE, McGee JOD. Growth arrest of the breast cancer cell line, T47D, by TNFα cell cycle specificity and signal transduction. Br J Cancer. (1993) 67:290–6. doi: 10.1038/bjc.1993.55
55. Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep. (2015) 12:1902–14. doi: 10.1016/j.celrep.2015.08.033
56. Josephs SF, Ichim TE, Prince SM, Kesari S, Marincola FM, Escobedo AR, et al. Unleashing endogenous TNF-alpha as a cancer immunotherapeutic. J Transl Med. (2018) 16:242. doi: 10.1186/s12967-018-1611-7
57. Barth RJ, Mule JJ, Spiess PJ, Rosenberg SA. Interferon γ and tumor necrosis factor have a role in tumor regressions mediated by murine CD8+ tumor-infiltrating lymphocytes. J Exp Med. (1991) 173:647–58. doi: 10.1084/jem.173.3.647
58. He YF, Wang XH, Zhang GM, Chen HT, Zhang H, Feng ZH. Sustained low-level expression of interferon-γ promotes tumor development: potential insights in tumor prevention and tumor immunotherapy. Cancer Immunol Immunother. (2005) 54:891–7. doi: 10.1007/s00262-004-0654-1
59. Müller-Hermelink N, Braumüller H, Pichler B, Wieder T, Mailhammer R, Schaak K, et al. TNFR1 signaling and IFN-γ signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. (2008) 13:507–18. doi: 10.1016/j.ccr.2008.04.001
60. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci USA. (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
61. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res. (2018) 78:5586–99. doi: 10.1158/0008-5472.CAN-17-3962
62. Hou Z, Falcone DJ, Subbaramaiah K, Dannenberg AJ. Macrophages induce COX-2 expression in breast cancer cells: role of IL-1β autoamplification. Carcinogenesis. (2011) 32:695–702. doi: 10.1093/carcin/bgr027
63. Obermajer N, Kalinski P. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplant Res. (2012) 1:15. doi: 10.1186/2047-1440-1-15
64. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. (2017) 7:373. doi: 10.3389/fcimb.2017.00373
65. Iles KE, Forman HJ. Macrophage signaling and respiratory burst. Immunol Res. (2002) 26:95–105. doi: 10.1385/IR:26:1-3:095
66. McCloskey PS, Salo RJ. Flow cytometric analysis of group B streptococci phagocytosis and oxidative burst in human neutrophils and monocytes. FEMS Immunol Med Microbiol. (2000) 27:59–65. doi: 10.1111/j.1574-695X.2000.tb01412.x
67. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. (2007) 13:828–35. doi: 10.1038/nm1609
68. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8 + T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
69. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
70. Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. (2013) 23:898–914. doi: 10.1038/cr.2013.75
71. Ghosh S, Mukherjee S, Choudhury S, Gupta P, Adhikary A, Baral R, et al. Reactive oxygen species in the tumor niche triggers altered activation of macrophages and immunosuppression: role of fluoxetine. Cell Signal. (2015) 27:1398–412. doi: 10.1016/j.cellsig.2015.03.013
72. Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, Pesic M, et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell. (2017) 32:869–83. doi: 10.1016/j.ccell.2017.11.004
73. Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. (2007) 67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783
74. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. (2003) 3:710–20. doi: 10.1038/nri1180
75. Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, et al. β-Defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. (1999) 286:525–8. doi: 10.1126/science.286.5439.525
76. Conejo-Garcia J-R, Benencia F, Coukos G. Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of VEGF-A in ovarian cancer. Nat Med. (2004) 10:950–8. doi: 10.1038/nm1097
77. Nandi B, Shapiro M, Samur MK, Pai C, Frank NY, Yoon C, et al. Stromal CCR6 drives tumor growth in a murine transplantable colon cancer through recruitment of tumor-promoting macrophages. Oncoimmunology. (2016) 5:e1189052. doi: 10.1080/2162402X.2016.1189052
78. Holterman DA, Diaz JI, Blackmore PF, Davis JW, Schellhammer PF, Corica A, et al. Overexpression of α-defensin is associated with bladder cancer invasiveness. Urol Oncol Semin Orig Investig. (2006) 24:97–108. doi: 10.1016/j.urolonc.2005.07.010
79. Xu D, Zhang B, Liao C, Zhang W, Wang W, Chang Y, et al. Human beta-defensin 3 contributes to the carcinogenesis of cervical cancer via activation of NF-κB signaling. Oncotarget. (2016) 7:75902–13. doi: 10.18632/oncotarget.12426
80. Uraki S, Sugimoto K, Shiraki K, Tameda M, Inagaki Y, Ogura S, et al. Human β-defensin-3 inhibits migration of colon cancer cells via downregulation of metastasis-associated 1 family, member 2 expression. Int J Oncol. (2014) 45:1059–64. doi: 10.3892/ijo.2014.2507
81. Hubert P, Herman L, Maillard C, Caberg J-H, Nikkels A, Pierard G, et al. Defensins induce the recruitment of dendritic cells in cervical human papillomavirus-associated (pre)neoplastic lesions formed in vitro and transplanted in vivo. FASEB J. (2007) 21:2765–75. doi: 10.1096/fj.06-7646com
82. Li D, Liu W, Wang X, Wu J, Quan W, Yao Y, et al. Cathelicidin, an antimicrobial peptide produced by macrophages, promotes colon cancer by activating the Wnt/ß-catenin pathway. Oncotarget. (2015) 6:2939–50. doi: 10.18632/oncotarget.2845
83. Li D, Beisswenger C, Herr C, Schmid RM, Gallo RL, Han G, et al. Expression of the antimicrobial peptide cathelicidin in myeloid cells is required for lung tumor growth. Oncogene. (2014) 33:2709–16. doi: 10.1038/onc.2013.248
84. De Yang B, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. (2000) 192:1069–74. doi: 10.1084/jem.192.7.1069
85. Büchau AS, Morizane S, Trowbridge J, Schauber J, Kotol P, Bui JD, et al. The host defense peptide cathelicidin is required for NK cell-mediated suppression of tumor growth. J Immunol. (2010) 184:369–78. doi: 10.4049/jimmunol.0902110
86. Cheng M, Ho S, Yoo JH, Tran DHY, Bakirtzi K, Su B, et al. Cathelicidin suppresses colon cancer development by inhibition of cancer associated fibroblasts. Clin Exp Gastroenterol. (2014) 8:13–29. doi: 10.2147/CEG.S70906
87. Häger M, Cowland JB, Borregaard N. Neutrophil granules in health and disease. J Intern Med. (2010) 268:25–34. doi: 10.1111/j.1365-2796.2010.02237.x
88. Richter J, Ng-Sikorski J, Olsson I, Andersson T. Tumor necrosis factor-induced degranulation in adherent human neutrophils is dependent on CD11b/CD18-integrin-triggered oscillations of cytosolic free Ca2+. Proc Natl Acad Sci USA. (1990) 87:9472–6. doi: 10.1073/pnas.87.23.9472
89. Naegelen I, Beaume N, Plançon S, Schenten V, Tschirhart EJ, Bréchard S. Regulation of neutrophil degranulation and cytokine secretion: a novel model approach based on linear fitting. J Immunol Res. (2015) 2015:817038. doi: 10.1155/2015/817038
90. Lacy P. Mechanisms of degranulation in neutrophils. Allergy, Asthma Clin Immunol. (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
91. Houghton AMG, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. (2010) 16:219–23. doi: 10.1038/nm.2084
92. Taya M, Garcia-Hernandez M, Rangel-Moreno J, Minor B, Gibbons E, Hammes SR. Neutrophil elastase from myeloid cells promotes TSC2-null tumor growth. Endocr Relat Cancer. (2020) 27:261–74. doi: 10.1530/ERC-19-0431
93. Berchem G, Glondu M, Gleizes M, Brouillet JP, Vignon F, Garcia M, et al. Cathepsin-D affects multiple tumor progression steps in vivo: proliferation, angiogenesis and apoptosis. Oncogene. (2002) 21:5951–5. doi: 10.1038/sj.onc.1205745
94. Zhang C, Zhang M, Song S. Cathepsin D enhances breast cancer invasion and metastasis through promoting hepsin ubiquitin-proteasome degradation. Cancer Lett. (2018) 438:105–15. doi: 10.1016/j.canlet.2018.09.021
95. Bian B, Mongrain S, Cagnol S, Langlois MJ, Boulanger J, Bernatchez G, et al. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol Carcinog. (2016) 55:671–87. doi: 10.1002/mc.22312
96. Mirkovic B, Markelc B, Butinar M, Mitrovic A, Sosic I, Gobec S, et al. Nitroxoline impairs tumor progression in vitro and in vivo by regulating cathepsin B activity. Oncotarget. (2015) 6:19027–42. doi: 10.18632/oncotarget.3699
97. Yang T-H St, John LS, Garber HR, Kerros C, Ruisaard KE, Clise-Dwyer K, et al. Membrane-associated proteinase 3 on granulocytes and acute myeloid leukemia inhibits T cell proliferation. J Immunol. (2018) 201:1389–99. doi: 10.4049/jimmunol.1800324
98. Kolonin MG, Sergeeva A, Staquicini DI, Smith TL, Tarleton CA, Molldrem JJ, et al. Interaction between tumor cell surface receptor RAGE and proteinase 3 mediates prostate cancer metastasis to bone. Cancer Res. (2017) 77:3144–50. doi: 10.1158/0008-5472.CAN-16-0708
99. Shamamian P, Schwartz JD, Pocock BJZ, Monea S, Whiting D, Marcus SG, et al. Activation of progelatinase A (MMP-2) by neutrophil elastase, cathepsin G, and proteinase-3: a role for inflammatory cells in tumor invasion and angiogenesis. J Cell Physiol. (2001) 189:197–206. doi: 10.1002/jcp.10014
100. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (80). (2004) 303:1532–5. doi: 10.1126/science.1092385
101. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 19:1386–93. doi: 10.1038/nm.2847
102. Kirchner T, Mller S, Klinger M, Solbach W, Laskay T, Behnen M. The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators Inflamm. (2012) 2012:849136. doi: 10.1155/2012/849136
103. Remijsen Q, Berghe TV, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
104. Marcos V, Zhou Z, Yildirim AÖ, Bohla A, Hector A, Vitkov L, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med. (2010) 16:1018–23. doi: 10.1038/nm.2209
105. Thålin C, Demers M, Blomgren B, Wong SL, Von Arbin M, Von Heijne A, et al. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb Res. (2016) 139:56–64. doi: 10.1016/j.thromres.2016.01.009
106. Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA. (2012) 109:13076–81. doi: 10.1073/pnas.1200419109
107. Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D, et al. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology. (2019) 9:e1605822. doi: 10.1080/2162402X.2019.1605822
108. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. (2016) 76:1367–80. doi: 10.1158/0008-5472.CAN-15-1591
109. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun. (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
110. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. (2016) 7:10587. doi: 10.1038/ncomms10587
111. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol. (2009) 46:2753–66. doi: 10.1016/j.molimm.2009.04.027
112. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. (2008) 28:425–35. doi: 10.1016/j.immuni.2008.02.001
113. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
114. DiScipio RG, Schraufstatter IU. The role of the complement anaphylatoxins in the recruitment of eosinophils. Int Immunopharmacol. (2007) 7:1909–23. doi: 10.1016/j.intimp.2007.07.006
115. Hartmann K, Henz BM, Krüger-Krasagakes S, Köhl J, Burger R, Gurtl S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood. (1997) 89:2863–70. doi: 10.1182/blood.V89.8.2863
116. Riedemann NC, Guo RF, Bernacki KD, Reuben JS, Laudes IJ, Neff TA, et al. Regulation by C5a of neutrophil activation during sepsis. Immunity. (2003) 19:193–202. doi: 10.1016/S1074-7613(03)00206-1
117. Soruri A, Kiafard Z, Dettmer C, Riggert J, Köhl J, Zwirner J. IL-4 down-regulates anaphylatoxin receptors in monocytes and dendritic cells and impairs anaphylatoxin-induced migration in vivo. J Immunol. (2003) 170:3306–14. doi: 10.4049/jimmunol.170.6.3306
118. Gutzmer R, Köther B, Zwirner J, Dijkstra D, Purwar R, Wittmann M, et al. Human plasmacytoid dendritic cells express receptors for anaphylatoxins C3a and C5a and are chemoattracted to C3a and C5a. J Invest Dermatol. (2006) 126:2422–9. doi: 10.1038/sj.jid.5700416
119. Gros P, Milder FJ, Janssen BJC. Complement driven by conformational changes. Nat Rev Immunol. (2008) 8:48–58. doi: 10.1038/nri2231
120. van Lookeren Campagne M, Wiesmann C, Brown EJ. Macrophage complement receptors and pathogen clearance. Cell Microbiol. (2007) 9:2095–102. doi: 10.1111/j.1462-5822.2007.00981.x
121. Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol. (2018) 40:37–48. doi: 10.1007/s00281-017-0644-y
122. Martin M, Blom AM. Complement in removal of the dead – balancing inflammation. Immunol Rev. (2016) 274:218–32. doi: 10.1111/imr.12462
123. Kesselring R, Thiel A, Pries R, Fichtner-Feigl S, Brunner S, Seidel P, et al. The complement receptors CD46, CD55 and CD59 are regulated by the tumour microenvironment of head and neck cancer to facilitate escape of complement attack. Eur J Cancer. (2014) 50:2152–61. doi: 10.1016/j.ejca.2014.05.005
124. Okroj M, Holmquist E, Nilsson E, Anagnostaki L, Jirström K, Blom AM. Local expression of complement factor I in breast cancer cells correlates with poor survival and recurrence. Cancer Immunol Immunother. (2015) 64:467–78. doi: 10.1007/s00262-015-1658-8
125. Wang Y, Yang YJ, Wang Z, Liao J, Liu M, Zhong XR, et al. CD55 and CD59 expression protects HER2-overexpressing breast cancer cells from trastuzumab-induced complement-dependent cytotoxicity. Oncol Lett. (2017) 14:2961–69. doi: 10.3892/ol.2017.6555
126. Mamidi S, Höne S, Teufel C, Sellner L, Zenz T, Kirschfink M. Neutralization of membrane complement regulators improves complement-dependent effector functions of therapeutic anticancer antibodies targeting leukemic cells. Oncoimmunology. (2015) 4:e979688. doi: 10.4161/2162402X.2014.979688
127. Ajona D, Hsu Y-F, Corrales L, Montuenga LM, Pio R. Down-regulation of human complement factor H sensitizes non-small cell lung cancer cells to complement attack and reduces in vivo tumor growth. J Immunol. (2007) 178:5991–8. doi: 10.4049/jimmunol.178.9.5991
128. Roumenina LT, Daugan MV, Petitprez F, Sautès-Fridman C, Fridman WH. Context-dependent roles of complement in cancer. Nat Rev Cancer. (2019) 19:698–715. doi: 10.1038/s41568-019-0210-0
129. Gunn L, Ding C, Liu M, Ma Y, Qi C, Cai Y, et al. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J Immunol. (2012) 189:2985–94. doi: 10.4049/jimmunol.1200846
130. Hsu BE, Roy J, Mouhanna J, Rayes RF, Ramsay L, Tabariès S, et al. C3a elicits unique migratory responses in immature low-density neutrophils. Oncogene. (2020) 39:2612–23. doi: 10.1038/s41388-020-1169-8
131. Han X, Zha H, Yang F, Guo B, Zhu B. Tumor-derived tissue factor aberrantly activates complement and facilitates lung tumor progression via recruitment of myeloid-derived suppressor cells. Int J Mol Sci. (2017) 18:22. doi: 10.3390/ijms18010022
132. Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. (2008) 9:1225–35. doi: 10.1038/ni.1655
133. Corrales L, Ajona D, Rafail S, Lasarte JJ, Riezu-Boj JI, Lambris JD, et al. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J Immunol. (2012) 189:4674–83. doi: 10.4049/jimmunol.1201654
134. Nabizadeh JA, Manthey HD, Steyn FJ, Chen W, Widiapradja A, Md Akhir FN, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4 + T cell responses. J Immunol. (2016) 196:4783–92. doi: 10.4049/jimmunol.1600210
135. Kwak JW, Laskowski J, Li HY, McSharry MV, Sippel TR, Bullock BL, et al. Complement activation via a C3a receptor pathway alters CD4+ T lymphocytes and mediates lung cancer progression. Cancer Res. (2018) 78:143–56. doi: 10.1158/0008-5472.CAN-17-0240
136. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. (2015) 160:700–14. doi: 10.1016/j.cell.2015.01.004
137. Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. (2010) 2:181–9. doi: 10.4161/mabs.2.2.11158
138. Weiner LM, Murray JC, Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. (2012) 148:1081–4. doi: 10.1016/j.cell.2012.02.034
139. Lee CH, Romain G, Yan W, Watanabe M, Charab W, Todorova B, et al. IgG Fc domains that bind C1q but not effector Fc3 receptors delineate the importance of complement-mediated effector functions. Nat Immunol. (2017) 18:889–98. doi: 10.1038/ni.3770
140. Di Gaetano N, Cittera E, Nota R, Vecchi A, Grieco V, Scanziani E, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. (2003) 171:1581–7. doi: 10.4049/jimmunol.171.3.1581
141. Van Der Kolk LE, Grillo-López AJ, Baars JW, Hack CE, Van Oers MHJ. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. (2001) 115:807–11. doi: 10.1046/j.1365-2141.2001.03166.x
142. Hsu YF, Ajona D, Corrales L, Lopez-Picazo JM, Gurpide A, Montuenga LM, et al. Complement activation mediates cetuximab inhibition of non-small cell lung cancer tumor growth in vivo. Mol Cancer. (2010) 9:139. doi: 10.1186/1476-4598-9-139
143. Kraus S, Seger R, Fishelson Z. Involvement of the ERK mitogen-activated protein kinase in cell resistance to complement-mediated lysis. Clin Exp Immunol. (2001) 123:366–74. doi: 10.1046/j.1365-2249.2001.01477.x
144. Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. (2002) 2:580–92. doi: 10.1038/nri856
145. Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. (2004) 199:1659–69. doi: 10.1084/jem.20040119
146. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. (2000) 6:443–6. doi: 10.1038/74704
147. Biburger M, Aschermann S, Schwab I, Lux A, Albert H, Danzer H, et al. Monocyte subsets responsible for immunoglobulin G-dependent effector functions in vivo. Immunity. (2011) 35:932–44. doi: 10.1016/j.immuni.2011.11.009
148. Yeap WH, Wong KL, Shimasaki N, Teo ECY, Quek JKS, Yong HX, et al. CD16 is indispensable for antibodydependent cellular cytotoxicity by human monocytes. Sci Rep. (2016) 6:34310. doi: 10.1038/srep34310
149. Su S, Zhao J, Xing Y, Zhang X, Liu J, Ouyang Q, et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell. (2018) 175:442–57. doi: 10.1016/j.cell.2018.09.007
150. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. (2017) 9:eaal3604. doi: 10.1126/scitranslmed.aal3604
Keywords: cancer immunotherapy, tumor-associated myeloid cells, tumor microenvironment, innate immune response, immune suppression, immunotherapy resistance
Citation: Lebegge E, Arnouk SM, Bardet PMR, Kiss M, Raes G and Van Ginderachter JA (2020) Innate Immune Defense Mechanisms by Myeloid Cells That Hamper Cancer Immunotherapy. Front. Immunol. 11:1395. doi: 10.3389/fimmu.2020.01395
Received: 06 April 2020; Accepted: 01 June 2020;
Published: 09 July 2020.
Edited by:
Sergei Kusmartsev, University of Florida, United StatesReviewed by:
Tsukasa Seya, Aomori University, JapanJosé Mordoh, IIBBA-CONICET Leloir Institute Foundation, Argentina
Copyright © 2020 Lebegge, Arnouk, Bardet, Kiss, Raes and Van Ginderachter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jo A. Van Ginderachter, am8udmFuLmdpbmRlcmFjaHRlckB2dWIuYmU=
†These authors share senior authorship