 Daria S. Chulpanova
Daria S. Chulpanova Kristina V. Kitaeva
Kristina V. Kitaeva Victoria James
Victoria James Albert A. Rizvanov
Albert A. Rizvanov Valeriya V. Solovyeva
Valeriya V. Solovyeva
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 03 July 2018
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01534
This article is part of the Research Topic Nano- and Microparticle-Induced Cell Death, Inflammation and Immune Responses View all 21 articles
Extracellular vesicles (EVs) are released by all cells within the tumor microenvironment, such as endothelial cells, tumor-associated fibroblasts, pericytes, and immune system cells. The EVs carry the cargo of parental cells formed of proteins and nucleic acids, which can convey cell-to-cell communication influencing the maintenance and spread of the malignant neoplasm, for example, promoting angiogenesis, tumor cell invasion, and immune escape. However, EVs can also suppress tumor progression, either by the direct influence of the protein and nucleic acid cargo of the EVs or via antigen presentation to immune cells as tumor-derived EVs carry on their surface some of the same antigens as the donor cells. Moreover, dendritic cell-derived EVs carry major histocompatibility complex class I and class II/peptide complexes and are able to prime other immune system cell types and activate an antitumor immune response. Given the relative longevity of vesicles within the circulation and their ability to cross blood–brain barriers, modification of these unique organelles offers the potential to create new biological-tools for cancer therapy. This review examines how modification of the EV cargo has the potential to target specific tumor mechanisms responsible for tumor formation and progression to develop new therapeutic strategies and to increase the efficacy of antitumor therapies.
Extracellular vesicles (EVs) are of particular interest due to their ability to mediate intercellular communication, influencing multiple cellular processes. EVs can be categorized based upon their biogenesis and divided into exosomes, microvesicles (MVs), and apoptotic bodies (ABs) (1, 2). Exosomes are small vesicles 40–100 nm in diameter, formed as part of the endocytic pathway. Exosomes carry the donor cell cargo, represented by various proteins and nucleic acids [DNA, mRNA, miRNA, and other non-coding RNAs (ncRNAs)] (Figure 1C) (3, 4). Exosomes are stable in biological fluids and small enough to pass through the blood–brain barrier (5). MVs have a diameter of 100–1,000 nm and are released by directly budding from the plasma membrane (6). MVs also carry cargos of proteins and nucleic acids, although their functional roles in cell-to-cell communication remains less well studied than the exosome population (7). In contrast to exosomes and MVs, which are formed continuously by cells, ABs are formed as part of the fragmentation process of cells undergoing apoptosis, the process of programmed cell death (1) (Figure 1A).
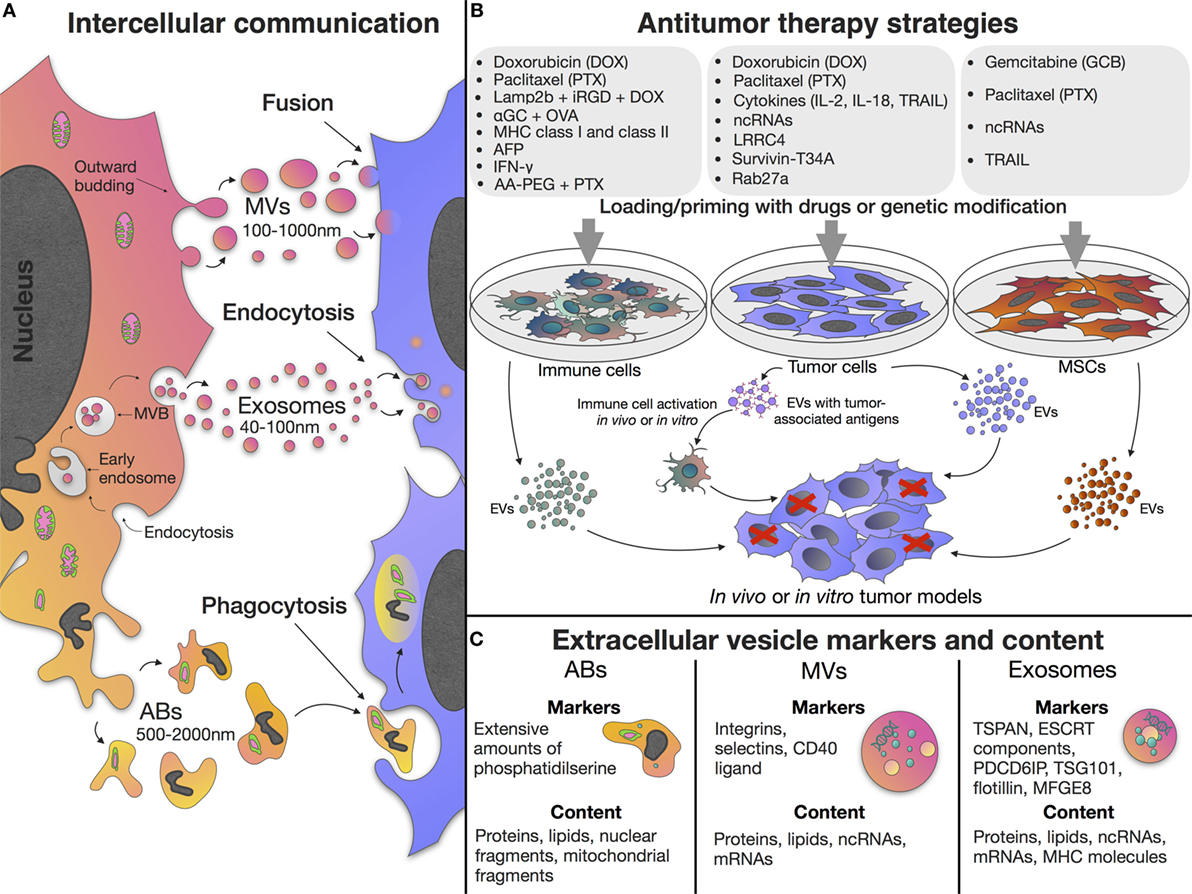
Figure 1. Extracellular vesicle (EV) properties and application in antitumor treatment. (A) EVs can be classified based upon their biogenesis and are divided into exosomes, microvesicles (MVs), and apoptotic bodies (ABs). Exosomes are formed as part of the endocytic pathway by inward budding of endosomal membranes, resulting in accumulation of early endosomes and formation of large multivesicular bodies (MVBs) which release their contents (exosomes) into the extracellular space. MVs are released by directly budding from the plasma membrane. ABs are formed as part of the fragmentation process of cells undergoing apoptosis. (B) EVs derived from native or primed/genetically modified cells can be used in antitumor treatment. (C) Different types of EVs contain various proteins, lipids, and nucleic acids and have specific membrane markers. Exosomes have tetraspanin (such as TSPAN29 or TSPAN30), endosomal sorting complex required for transport (ESCRT) components, milk fat globule-EGF factor 8 protein (MFGE8), programmed cell death 6 interacting protein (PDCD6IP), tumor susceptibility gene 101 protein (TSG101), and flotillin molecules on their surface. Exosome content include mRNAs, microRNAs, and other non-coding RNAs (ncRNAs), cytoplasmic and membrane proteins including receptors and major histocompatibility complex (MHC) molecules. MVs carry integrins, selectins, and CD40 ligand on their surface, and also contain mRNAs, microRNAs, ncRNAs, cytoplasmic and membrane proteins. ABs have extensive amounts of phosphatidylserine and contain various parts of the apoptotic cell such as proteins, lipids, nuclear fragments, and cell organelles. Cargo and biogenesis of EVs have been comprehensively discussed elsewhere (8, 9).
The tumor microenvironment is often a very complex and dynamic niche containing not only neoplastic cells but also a multitude of non-malignant stromal cells such as endothelial cells, tumor-associated fibroblasts, pericytes, and immune cells (10). In addition to stromal cells, the extracellular matrix and surrounding tumor adipose tissue also make an important contribution to tumor progression as they contain adipocytes and progenitor cells [preadipocytes and mesenchymal stem cells (MSCs)] (10, 11), as well as a variety of soluble cytokines, growth factors, and metabolites produced the stromal cells within the tumor microenvironment (10, 12). As EVs are believed to mediate cell-to-cell communication in the tumor microenvironment and induce phenotypic modification in recipient cells, there is a growing interest in the potential role of EVs as key mediators of tumor progression and the spread of malignant neoplasm (13–16). Since EV functions are related to the donor cell type and the imparted cargo of proteins and nucleic acids, EVs of different origins exhibit different features. However, as these have been comprehensively discussed elsewhere (17), this review focuses on the use and efficacy of EVs as antitumor therapies. For instance, as a result of the unique properties of MSCs, the EVs produced by stem cells retain the ability to migrate toward tumor niches (18), they also posses the same low immunogenicity of the donor MSCs (19). Therefore, the use of MSC-derived EVs as non-cell structures, in place of MSCs themselves, allows the avoidance of the risk of unlimited cell growth, undesirable transformation, and potential tumor formation (20). The ability to act as multi-signal messengers makes EVs a prospective new class of therapeutic agents to modulate the processes occurring in the tumor microenvironment (21) (Figure 1B).
Intercellular EV-mediated signaling by tumor cells has been linked with maintain angiogenesis, invasion, immune escape (22) and to develop an aggressive phenotype and chemo- and radiotherapy resistance (16, 23–25). The extent of the contribution of EVs in tumor maintenance has been demonstrated through the study of EV inhibition, following which malignancy is suppressed and cancer cells show enhanced sensitivity to proton-pump inhibitor (omeprazole) and cisplatin (26, 27). As EV traffic is regulated by an acidic microenvironment, a common feature of all solid tumors, altering intracellular pH is an effective means of modulating exosome release. Changes in intracellular pH alters the lipid composition of the cells membrane and subsequently modulates both exosome release and fusion/uptake (28). In addition, the lower extracellular pH can promote tumor resistance to cytotoxic drugs through neutralization of those antitumor drugs that are weak bases or isolating drugs in acidic vesicles and/or eliminating them through an exocytotic pathway (29).
Extracellular vesicles may also promote tumor progression through the transfer of their specific cargos, for example, during the formation of a pre-metastatic niche (PMN), where the transfer of EV-cargos to stromal cells, induce molecular and cellular changes that promote PMN development (30, 31). For example, the tumor exosomal transport of miR-494 and miR-542p to stromal cells and lung fibroblasts leads to cadherin-17 downregulation and matrix metalloproteinase upregulation (30), while proangiogenic RNAs contained within MVs trigger angiogenesis to promote PMN formation (32).
The ability of tumor cell-derived EVs to fuse with recipient cells through endocytosis and release their cargo into the recipient cell cytoplasm makes EVs a promising biological vector for targeted delivery of various antitumor agents (33). This is exemplified by the use of EVs derived from LNCaP and PC-3 prostate cancer cell lines modified to transport paclitaxel (PTX) into recipient cells through the endocytic pathway, significantly increasing PTX cytotoxicity in vitro (33). Furthermore, U-87 MG (brain neuronal glioblastoma–astrocytoma) derived EVs primed with doxorubicin (DOX) or PTX significantly decreased the viability of recipient U-87 MG cells by 70 and 50%, respectively, at the highest tested concentration of exosomes (200 µg/mL) in vitro (34).
Tumor-derived EVs can be used for therapeutic drug delivery to reduce systemic toxicity by targeting the tumor microenvironment. It was shown that in vitro and in vivo, doxorubicin-loaded exosomes (exoDOX) derived from MDA-MB-231 (breast adenocarcinoma) and HCT-116 (colorectal carcinoma) cell lines did not reduce DOX efficacy. Simultaneously, exoDOX treated nude mice did not show the cardiotoxicity observed in their free-DOX-treated counterparts. Mass spectrometry confirmed that DOX accumulation in the heart was reduced by approximately 40% when DOX was delivered via exosomes (exoDOX) (35). The reduced cardiotoxicity achieved when delivering DOX via modified exosomes would allow for a higher concentration of exoDOX to be used, thus offering the potential to increase DOX efficacy. Similar findings have also been reported for in vivo models of breast (MDA-MB-231) and ovarian (STOSE) cancer (36).
Tumor cell-derived EVs carry on their surface the same antigens as the cell that produced them (the donor cell), such as HER2/neu, melan-A, Silv, carcinoembryonic antigen (CEA), mesothelin, and others (37). Thus, they can act to prime immune cells by antigen presentation. The delivery of dendritic cells (DCs) in vitro primed with exosomes isolated from the mesothelioma cell line AB1 within a BALB/c mouse mesothelioma model, resulted in increased mean and overall survival times in vivo (38). Similarly, DCs primed with exosomes isolated from rat glioblastoma cells, induced a strong antitumor response and significantly increased median survival times in glioblastoma-bearing rats when used in combination with α-galactosylceramide (39).
The efficacy of priming immune cells can be improved by combining their use with immune cell stimulating drugs. For instance, exosomes derived from the pancreatic cancer cell line UNKC6141 were co-delivered with DCs (DCs/Exo) to UNKC16141 xenograft mice. Tumor onset was delayed in these animals and subsequently a significant increase in survival was observed. When the same assay was repeated, but with the inclusion of all-transretinoic acid (ATRA) alongside the delivery of DCs/Exo, increased lymphocyte proliferation within lymph nodes was reported which coincided with increased cytotoxic T-cell activity in comparison with untreated or DCs/Exo only treated animals. However, the inclusion of ATRA had no further effect on prolonging survival and only modest changes in metastasis to distant organs were observed. The combination of DCs/Exo with sunitinib in these animal models also led to an increase in cytotoxic activity which in these assays did lead to significantly prolonged survival times in DCs/Exo/sunitinib compared to animals treated only with free sunitinib therapy. Similar increases in survival time and a reduction in metastatic spread was also observed when DCs/Exo use was combined with gemcitabine treatment (40).
To increase the therapeutic potential and immunogenicity of EV-based tumor vaccines, tumor cells producing the EVs can be modified to express specific cytokine/chemokine genes that have an immunomodulating effect. Dai et al. reported that exosomes derived from LS-174T cells genetically modified to express IL-18 CEA (Exo/IL-18), had a more pronounced effect on specific antitumor immunity when compared with exosomes from native LS-174T cells. Exo/IL-18 promoted proliferation of peripheral blood mononuclear cells and induced cytokine secretion by T-lymphocytes and DC in vitro, as well as inducing the phenotypic and functional maturation of DCs (41). Similar results were obtained by Yang et al. using in vivo experiments, whereby exosomes were derived from IL-2-modified ovalbumin (OVA)-expressing EL-4 lymphoma cells (Exo/IL-2). Vaccination of C57BL/C mice with Exo/IL-2 more effectively inhibited tumor growth (42).
The modification of tumor cells through the aberrant expression of tumor suppressor genes, apoptosis inductors, and ncRNAs has also been shown to impart a potential therapeutic benefit to the resulting EVs. YUSAC 2 melanoma cells were engineered to overexpress a dominant-negative mutant form of Survivin (Survivin-T34A). Exosomes derived from Survivin-T34A-modified YUSAC 2 cells, in combination with gemcitabine, significantly increased apoptosis in pancreatic adenocarcinoma MIA PaCa-2 cells in comparison with gemcitabine alone (43). Rivoltini et al. showed that exosomes derived from K562 leukemia cells modified with TNF-related apoptosis-inducing ligand (TRAIL) [TRAIL(+) exosomes], induced apoptosis in TRAIL-death receptor (DR)5(+) SUDHL4 lymphoma and INT12 melanoma cells in vitro. In in vivo experiments of TRAIL(+) exosomes demonstrated homing of the exosomes to the tumor sites and significant suppression of tumor growth by 58% in SUDHL4-B-cell lymphoma bearing mice (44). Li et al. investigated exosomes derived from glioblastoma multiforme (GBM) cells with overexpression of the tumor suppressor gene LRRC4 (Exo/LRRC4). Exo/LRRC4 induced significant chemotaxis and expansion of CD4+CCR4+ T cells, inhibited the proportion of Ti-Treg cells, and promoted Ti-Teff cell expansion through cytokines release in vitro (45).
The Rab GTPases control many stages of membrane trafficking, including the formation and release of vesicles. Ostrowski et al. identified Rab GTPases Rab2b, Rab9a, Rab5a, Rab27a, and Rab27b that promote exosome secretion in HeLa cells (46), indicating the possibility of manipulating the secretion of Rab proteins to control exosome production. Exosomes, derived from Rab27a-overexpressing A549 cells (exo/Rab27a), exhibited the ability to regulate major histocompatibility complex (MHC) class II molecules and co-stimulatory molecules CD80 and CD86 on DCs. Furthermore, DCs primed with exosomes derived from Rab27-overexpressing A549 cells significantly increased CD4+ T cell proliferation in vitro. In vivo immunization with exo/Rab27a inhibited tumor growth in a tumor mouse model (47).
At present ncRNAs are actively being studied as potential antitumor agents. However, when developing miRNA-based therapies there are problems with specific targeting of tumor cells and target cells within the tumor microenvironment. Tumor-derived EVs can be used for delivering a variety of potentially therapeutic ncRNAs, for instance miR-134 (48), miR-29a, and miR-29c microRNAs (49), as well as short interfering RNAs (siRNAs) (50) (Table 1).
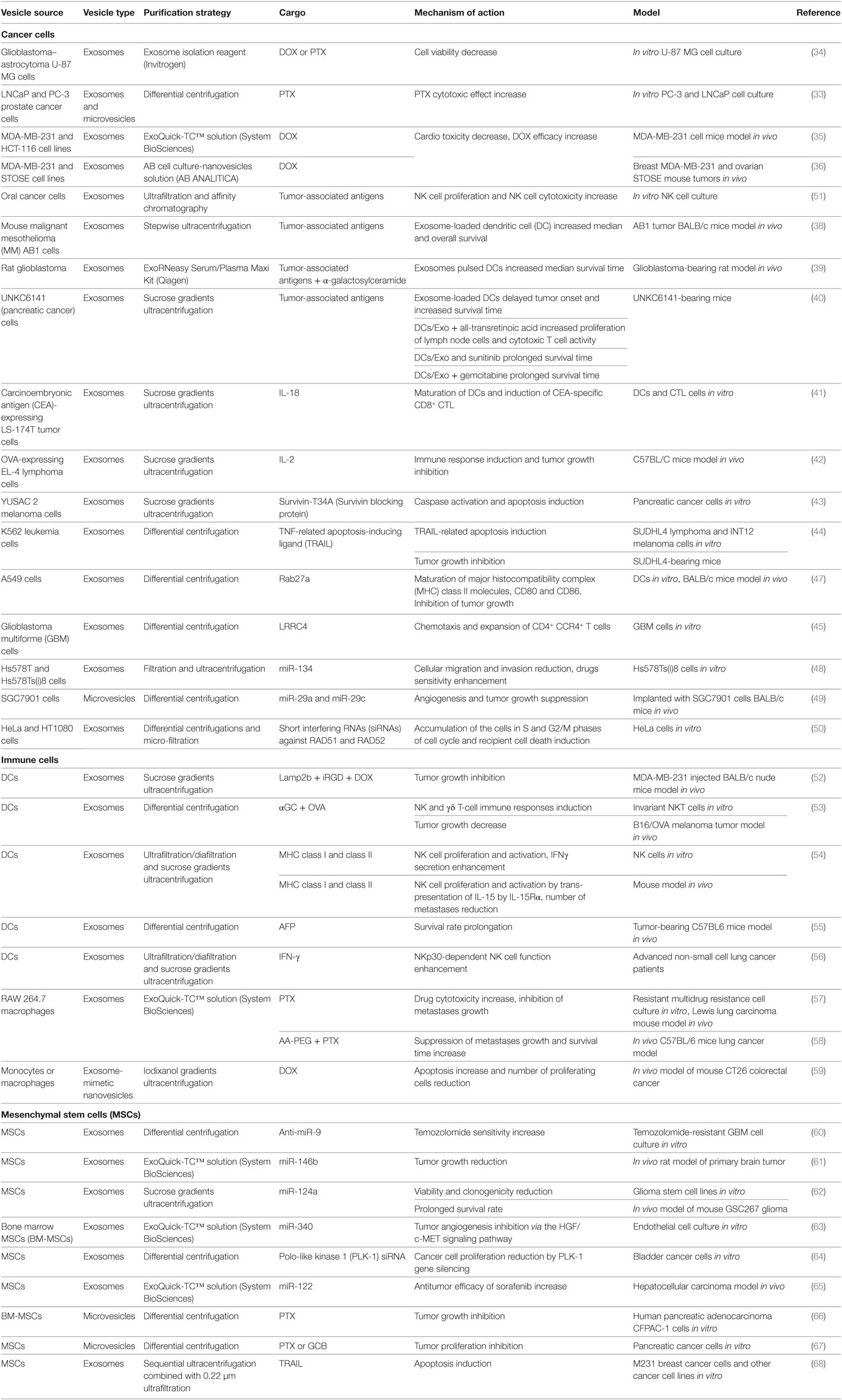
Table 1. The use of extracellular vesicles (EVs) with or without modified cargo for antitumor therapy.
Exosomes from immature dendritic cells (imDCs) can be used to deliver chemotherapeutic agents such as DOX. For instance, imDCs were modified to express lysosome-associated membrane protein 2 (Lamp2b) fused to the αv-integrin-specific iRGD peptide. It was shown that modified imDC-derived exosomes (Exo/iRGD) loaded with DOX, effectively targeted and delivered DOX to αv-integrin+ MDA-MB-231 breast cancer cells in vitro. Exo/iRGD intravenous injection in BALB/c mice led to inhibition of breast tumor cell growth without any apparent toxic effects (52).
A new approach for cancer immunotherapy is the combination of exosomes and the invariant NKT immune cell ligand α-galactosylceramide (αGC) (53). Loaded with αGC and OVA-model antigen exosomes induced potent NK and γδ T-cell innate immune responses in vitro and in vivo. In an OVA-expressing mouse model of melanoma treatment of tumor-bearing mice with αGC/OVA-loaded exosomes decreased tumor growth, increased antigen-specific CD8+ T-cell tumor infiltration, and increased median survival, relative to control mice immunized with soluble αGC + OVA alone (53). Similarly, exosomes derived from α-fetoprotein (AFP)-expressing DCs (DEXAFP) intravenously injected into hepatocarcinoma-bearing C57BL6 mice prolonged survival to 57 days in 100% of DEXAFP-treated mice (55).
Without modification, DC-derived exosomes alone carry MHC class I and class II/peptide complexes capable of leading to the priming of CD8+ and CD4+ T cells, respectively, and subsequent T cell-dependent tumor rejection (13, 54). DC-derived exosomes have also been reported to trigger NK cell proliferation and activation in vitro and in patients, by trans-presentation of IL-15 by IL-15Rα. This mechanism of action was shown to significantly reduce the number of lung metastases in vivo. Combination of DC-derived exosomes with IL-15Rα and rhIL-15 molecules led to NK cell proliferation and activation and significantly enhanced IFNγ secretion by NK cells in vitro (54).
Phase I clinical trials have demonstrated the safety of using DC-derived exosomes in patients with metastatic melanoma (69) and lung cancer (70). Phase II trials in non-small cell carcinoma patients using modified IFN-γ expressing DCs to produce exosomes have reported an increase in NKp30-dependent NK cell functions, and 32% of participants experienced stabilization for more than 4 months (56).
In addition to DCs, macrophages have also been studied as a source of EVs of potential therapeutic benefit. Derived from RAW 264.7 macrophages, vesicles loaded with PTX (exoPTX) were reported to significantly increase drug cytotoxicity (more than 50 times) in multidrug resistance (MDR) MDCKMDR1, MDCKwt, and 3LL-M27 cells in vitro. Furthermore, when delivered into the airway of mice modeling Lewis lung carcinoma pulmonary metastases, exoPTX were found to have a potent anticancer effect (57). For PTX targeted delivery macrophages can be modified with aminoethylanisamide-polyethylene glycol (AA-PEG) a vector moiety to target the σ-receptor which is overexpressed by lung cancer cells (58). Jang et al. developed a bioinspired exosome-mimetic nanovesicles that can be modified to deliver DOX, gemcitabine, or carboplatin to the tumor tissue after systemic administration. Chemotherapeutic-loaded nanovesicles, derived from monocytes or macrophages, induced TNF-α-stimulated endothelial cell (HUVECs) death in a dose-dependent manner in vitro. DOX-loaded nanovesicles increased apoptosis and reduced the number of proliferating cells in CT26 colorectal cancer murine models (59) (Table 1).
Extracellular vesicles released from MSCs have been reported to exhibit variable effects on tumor growth, indicating the influence of EVs is dependent on cargo and the donor cell type (71, 72). Delivered by MSC-derived exosomes molecules of different types of RNA can induce adipogenesis, angiogenesis, apoptosis, and proteolysis in recipient cells (15). Exosomes from gastric cancer-derived MSCs were found to deliver miR-221 to HGC-27 gastric cancer cells, promoting their proliferation and migration in vitro (73). Other biomolecules carried by exosomes such as oncogenic proteins, cytokines, adhesion molecules, and anti-apoptotic proteins can also promote tumor progression (74–76), as well as increase tumor resistance to chemotherapy drugs (77).
Exosomes from bone marrow MSCs (BM-MSCs) can transfer miRNAs from the BM, particularly miR-23b, which promote dormancy in bone marrow-metastatic human breast cancer through the suppression of a target gene, MARCKS in vivo (78). In support of this, Lee et al. showed that MSC-derived exosomes can suppress human breast cancer angiogenesis by downregulating the expression of VEGF in tumor cells in vitro and in vivo (79).
In addition to the endogenous effects of MSC-EVs, MSC-derived MVs can be used as delivery vehicles for a variety of potential therapeutic agents, in particular ncRNAs. For example, injection of exosomes derived from miR-146-expressing MSCs into xenograft gliomas in primary brain tumor rat models cause a significant reduction in tumor growth (61). Treatment with MSC-derived exosomes containing miR-124a reduce the viability and clonogenicity of glioma stem cell lines in vitro and increase the survival rate in glioma mouse models up to 50% by silencing FOXA2 (62), while the loading of MSC exosomes with miR-143 acts to significantly reduce the migration of 143B osteosarcoma cells (80). Transfection of bone marrow stromal cells with miR-340 generates exosomes capable of inhibiting tumor angiogenesis via the HGF/c-MET signaling pathway in endothelial cells (63). MSC-derived EVs can also be used to alter the chemosensitivity of tumor cells. Delivery of anti-miR-9 to temozolomide-resistant GBM cells increases cell sensitivity to this drug (60). The sensitivity of hepatocellular carcinoma cells to chemotherapeutic agents (5-fluorouracil and sorafenib) can similarly be altered through the use of miR-122 loaded MSC exosomes in vivo (65). MSC-derived MVs can also be loaded with various siRNAs that target key genes driving tumorigenesis, for example, MSC exosomes carrying siRNAs against polo-like kinase 1 significantly reduce bladder cancer cell proliferation in vitro (64).
In addition to biomolecules, MSC-derived vesicles can be loaded with chemotherapeutic drugs. BM-MSC-derived MVs primed with high-dose PTX inhibited cell growth by 50% in human CFPAC-1 pancreatic adenocarcinoma cells in vitro (66). This finding was supported by the recent studies of Cocce et al., which showed antitumor activity of MSCs MVs loaded with PTX or gemcitabine (GCB) on pancreatic cancer cells in vitro (67).
Recent studies have also highlighted the potential to deliver TRAIL by MSC-EVs (MSCT). MSCT-EVs induced apoptosis in 11 cancer cell lines in a dose-dependent manner but showed no cytotoxicity in human bronchial epithelial cells in vitro. Interestingly TRAIL-primed EVs that contain 3.88 ng TRAIL/mL induced significantly more apoptosis in M231 breast cancer cells compared with 100 ng/mL of recombinant TRAIL. TRAIL delivery by MSC-EVs induced significant apoptosis in TRAIL resistant A549 lung adenocarcinoma cells in a dose-dependent manner in vitro (68) (Table 1).
Extracellular vesicles, which include groups of differing origins such as exosomes and MVs, are released by all cells within the tumor microenvironment during normal cellular activity. EVs carry variable cargos that reflect the composition of the donor cells, these cargos can be transferred to neighboring cells and thus affect the processes occurring in those recipient cells and subsequently the tumor microenvironment as a whole. In addition to their endogenous ability to influence tumor progression, the ability to modify the EV content makes them a promising tool for cancer therapy. Surface antigens of tumor cell-derived vesicles can be used for immune cell priming. They can also be modified with various agents to directly affect tumor cells or modulate antitumor immunity. Genetic modifications can also be performed on MSC-derived vesicles, the main advantage of which is targeted cargo delivery to the tumor microenvironment. From priming the immune response to delivering ncRNAs and antitumor drugs, EVs provide a unique biological means of targeting tumors and their microenvironments, minimizing cytotoxic effects, and increasing the efficacy of treatments at lower drug doses (Table 1). However, despite these many advantages, EVs can have variable effects on tumor progression and the tumor microenvironment dependent upon their protein and nucleic acid cargos. One of the limitations of EV usage is the heterogeneity of the isolated population, since the size of exosomes and MVs overlap, and as yet it is not clear which population carries the greatest potential to elicit functional changes. Furthermore, the inconsistency of the EV cargo adds an additional caveat to their study and therapeutic use (81). In the case of drug loading, disadvantages include a low transfection efficiency, and, in the case of cell manipulation, there is a high dependence on cell division (82). Therefore, progressing their use as therapeutic tools requires full characterization of such disadvantages and limitations before the promise of MVs in clinical practice is achieved.
DC wrote the manuscript and made the table. KK created the figure. VJ edited the manuscript. DC, VS, and AR conceived the idea and edited the manuscript and table.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work is performed according to the Russian Government Program of Competitive Growth of Kazan Federal University. VS was supported by the Russian Foundation for Basic Research (RFBR) grant 16-34-60201. AR was supported by state assignment 20.5175.2017/6.7 of the Ministry of Education and Science of Russian Federation and RFBR grant 18-04-01133.
1. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35(4):495–516. doi:10.1080/01926230701320337
2. Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles (2013) 2(1):20389. doi:10.3402/jev.v2i0.20389
3. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 9(6):654–9. doi:10.1038/ncb1596
4. Wu K, Sharma S, Venkat S, Liu K, Zhou X, Watabe K. Non-coding RNAs in cancer brain metastasis. Front Biosci (Schol Ed) (2016) 8:187–202. doi:10.2741/s457
5. Li X, Tsibouklis J, Weng T, Zhang B, Yin G, Feng G, et al. Nano carriers for drug transport across the blood-brain barrier. J Drug Target (2017) 25(1):17–28. doi:10.1080/1061186X.2016.1184272
6. Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet (2012) 21(R1):R125–34. doi:10.1093/hmg/dds317
7. Wu K, Xing F, Wu SY, Watabe K. Extracellular vesicles as emerging targets in cancer: Recent development from bench to bedside. Biochim Biophys Acta (2017) 1868(2):538–63. doi:10.1016/j.bbcan.2017.10.001
8. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200(4):373–83. doi:10.1083/jcb.201211138
9. Kalra H, Drummen GP, Mathivanan S. Focus on extracellular vesicles: introducing the next small big thing. Int J Mol Sci (2016) 17(2):170. doi:10.3390/ijms17020170
10. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, et al. Role of tumor microenvironment in tumorigenesis. J Cancer (2017) 8(5):761–73. doi:10.7150/jca.17648
11. Jordan BF, Gourgue F, Cani PD. Adipose tissue metabolism and cancer progression: novel insights from gut microbiota? Curr Pathobiol Rep (2017) 5(4):315–22. doi:10.1007/s40139-017-0154-6
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
13. Andre F, Chaput N, Schartz NE, Flament C, Aubert N, Bernard J, et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol (2004) 172(4):2126–36. doi:10.4049/jimmunol.172.4.2126
14. Camussi G, Deregibus MC, Bruno S, Grange C, Fonsato V, Tetta C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res (2011) 1(1):98–110.
15. Eirin A, Riester SM, Zhu XY, Tang H, Evans JM, O’Brien D, et al. MicroRNA and mRNA cargo of extracellular vesicles from porcine adipose tissue-derived mesenchymal stem cells. Gene (2014) 551(1):55–64. doi:10.1016/j.gene.2014.08.041
16. Li XJ, Ren ZJ, Tang JH, Yu Q. Exosomal microRNA MiR-1246 promotes cell proliferation, invasion and drug resistance by targeting CCNG2 in breast cancer. Cell Physiol Biochem (2017) 44(5):1741–8. doi:10.1159/000485780
17. Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles (2015) 4:27066. doi:10.3402/jev.v4.27066
18. Kalimuthu S, Gangadaran P, Li XJ, Oh JM, Lee HW, Jeong SY, et al. In vivo therapeutic potential of mesenchymal stem cell-derived extracellular vesicles with optical imaging reporter in tumor mice model. Sci Rep (2016) 6:30418. doi:10.1038/srep30418
19. Chulpanova DS, Kitaeva KV, Tazetdinova LG, James V, Rizvanov AA, Solovyeva VV. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front Pharmacol (2018) 9:259. doi:10.3389/fphar.2018.00259
20. Gomzikova MO, Rizvanov AA. Current trends in regenerative medicine: from cell to cell-free therapy. BioNanoSci (2017) 7:240–5. doi:10.1007/s12668-016-0348-0
21. Han L, Xu J, Xu Q, Zhang B, Lam EW, Sun Y. Extracellular vesicles in the tumor microenvironment: therapeutic resistance, clinical biomarkers, and targeting strategies. Med Res Rev (2017) 37(6):1318–49. doi:10.1002/med.21453
22. Aubertin K, Silva AK, Luciani N, Espinosa A, Djemat A, Charue D, et al. Massive release of extracellular vesicles from cancer cells after photodynamic treatment or chemotherapy. Sci Rep (2016) 6:35376. doi:10.1038/srep35376
23. Feng Q, Zhang C, Lum D, Druso JE, Blank B, Wilson KF, et al. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat Commun (2017) 8:14450. doi:10.1038/ncomms14450
24. Zhang S, Zhang Y, Qu J, Che X, Fan Y, Hou K, et al. Exosomes promote cetuximab resistance via the PTEN/Akt pathway in colon cancer cells. Braz J Med Biol Res (2017) 51(1):e6472. doi:10.1590/1414-431X20176472
25. Khan FM, Saleh E, Alawadhi H, Harati R, Zimmermann WH, El-Awady R. Inhibition of exosome release by ketotifen enhances sensitivity of cancer cells to doxorubicin. Cancer Biol Ther (2018) 19(1):25–33. doi:10.1080/15384047.2017.1394544
26. Guan XW, Zhao F, Wang JY, Wang HY, Ge SH, Wang X, et al. Tumor microenvironment interruption: a novel anti-cancer mechanism of proton-pump inhibitor in gastric cancer by suppressing the release of microRNA-carrying exosomes. Am J Cancer Res (2017) 7(9):1913–25.
27. Qin X, Yu S, Zhou L, Shi M, Hu Y, Xu X, et al. Cisplatin-resistant lung cancer cell-derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100-5p-dependent manner. Int J Nanomedicine (2017) 12:3721–33. doi:10.2147/IJN.S131516
28. Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem (2009) 284(49):34211–22. doi:10.1074/jbc.M109.041152
29. Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst (2004) 96(22):1702–13. doi:10.1093/jnci/djh305
30. Rana S, Malinowska K, Zoller M. Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia (2013) 15(3):281–95. doi:10.1593/neo.122010
31. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol (2015) 17(6):816–26. doi:10.1038/ncb3169
32. Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res (2011) 71(15):5346–56. doi:10.1158/0008-5472.CAN-11-0241
33. Saari H, Lazaro-Ibanez E, Viitala T, Vuorimaa-Laukkanen E, Siljander P, Yliperttula M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J Control Release (2015) 220(Pt B):727–37. doi:10.1016/j.jconrel.2015.09.031
34. Yang T, Martin P, Fogarty B, Brown A, Schurman K, Phipps R, et al. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res (2015) 32(6):2003–14. doi:10.1007/s11095-014-1593-y
35. Toffoli G, Hadla M, Corona G, Caligiuri I, Palazzolo S, Semeraro S, et al. Exosomal doxorubicin reduces the cardiac toxicity of doxorubicin. Nanomedicine (Lond) (2015) 10(19):2963–71. doi:10.2217/nnm.15.118
36. Hadla M, Palazzolo S, Corona G, Caligiuri I, Canzonieri V, Toffoli G, et al. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine (Lond) (2016) 11(18):2431–41. doi:10.2217/nnm-2016-0154
37. Clayton A, Mason MD. Exosomes in tumour immunity. Curr Oncol (2009) 16(3):46–9. doi:10.3747/co.v16i3.367
38. Mahaweni NM, Kaijen-Lambers ME, Dekkers J, Aerts JG, Hegmans JP. Tumour-derived exosomes as antigen delivery carriers in dendritic cell-based immunotherapy for malignant mesothelioma. J Extracell Vesicles (2013) 2(1):22492. doi:10.3402/jev.v2i0.22492
39. Liu H, Chen L, Liu J, Meng H, Zhang R, Ma L, et al. Co-delivery of tumor-derived exosomes with alpha-galactosylceramide on dendritic cell-based immunotherapy for glioblastoma. Cancer Lett (2017) 411:182–90. doi:10.1016/j.canlet.2017.09.022
40. Xiao L, Erb U, Zhao K, Hackert T, Zoller M. Efficacy of vaccination with tumor-exosome loaded dendritic cells combined with cytotoxic drug treatment in pancreatic cancer. Oncoimmunology (2017) 6(6):e1319044. doi:10.1080/2162402X.2017.1319044
41. Dai S, Zhou X, Wang B, Wang Q, Fu Y, Chen T, et al. Enhanced induction of dendritic cell maturation and HLA-A*0201-restricted CEA-specific CD8(+) CTL response by exosomes derived from IL-18 gene-modified CEA-positive tumor cells. J Mol Med (Berl) (2006) 84(12):1067–76. doi:10.1007/s00109-006-0102-0
42. Yang Y, Xiu F, Cai Z, Wang J, Wang Q, Fu Y, et al. Increased induction of antitumor response by exosomes derived from interleukin-2 gene-modified tumor cells. J Cancer Res Clin Oncol (2007) 133(6):389–99. doi:10.1007/s00432-006-0184-7
43. Aspe JR, Diaz Osterman CJ, Jutzy JM, Deshields S, Whang S, Wall NR. Enhancement of gemcitabine sensitivity in pancreatic adenocarcinoma by novel exosome-mediated delivery of the Survivin-T34A mutant. J Extracell Vesicles (2014) 3(1):23244. doi:10.3402/jev.v3.23244
44. Rivoltini L, Chiodoni C, Squarcina P, Tortoreto M, Villa A, Vergani B, et al. TNF-related apoptosis-inducing ligand (TRAIL)-armed exosomes deliver proapoptotic signals to tumor site. Clin Cancer Res (2016) 22(14):3499–512. doi:10.1158/1078-0432.CCR-15-2170
45. Li P, Feng J, Liu Y, Liu Q, Fan L, Liu Q, et al. Novel therapy for glioblastoma multiforme by restoring LRRC4 in tumor cells: LRRC4 inhibits tumor-infitrating regulatory T cells by cytokine and programmed cell death 1-containing exosomes. Front Immunol (2017) 8:1748. doi:10.3389/fimmu.2017.01748
46. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol (2010) 12(1):19–30; sup 11–13. doi:10.1038/ncb2000
47. Li W, Mu D, Tian F, Hu Y, Jiang T, Han Y, et al. Exosomes derived from Rab27a-overexpressing tumor cells elicit efficient induction of antitumor immunity. Mol Med Rep (2013) 8(6):1876–82. doi:10.3892/mmr.2013.1738
48. O’Brien K, Lowry MC, Corcoran C, Martinez VG, Daly M, Rani S, et al. miR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivity. Oncotarget (2015) 6(32):32774–89. doi:10.18632/oncotarget.5192
49. Zhang H, Bai M, Deng T, Liu R, Wang X, Qu Y, et al. Cell-derived microvesicles mediate the delivery of miR-29a/c to suppress angiogenesis in gastric carcinoma. Cancer Lett (2016) 375(2):331–9. doi:10.1016/j.canlet.2016.03.026
50. Shtam TA, Kovalev RA, Varfolomeeva EY, Makarov EM, Kil YV, Filatov MV. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun Signal (2013) 11:88. doi:10.1186/1478-811X-11-88
51. Wang Y, Qin X, Zhu X, Chen W, Zhang J, Chen W. Oral cancer-derived exosomal NAP1 enhances cytotoxicity of natural killer cells via the IRF-3 pathway. Oral Oncol (2018) 76:34–41. doi:10.1016/j.oraloncology.2017.11.024
52. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials (2014) 35(7):2383–90. doi:10.1016/j.biomaterials.2013.11.083
53. Gehrmann U, Hiltbrunner S, Georgoudaki AM, Karlsson MC, Naslund TI, Gabrielsson S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res (2013) 73(13):3865–76. doi:10.1158/0008-5472.CAN-12-3918
54. Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One (2009) 4(3):e4942. doi:10.1371/journal.pone.0004942
55. Lu Z, Zuo B, Jing R, Gao X, Rao Q, Liu Z, et al. Dendritic cell-derived exosomes elicit tumor regression in autochthonous hepatocellular carcinoma mouse models. J Hepatol (2017) 67(4):739–48. doi:10.1016/j.jhep.2017.05.019
56. Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology (2016) 5(4):e1071008. doi:10.1080/2162402X.2015.1071008
57. Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine (2016) 12(3):655–64. doi:10.1016/j.nano.2015.10.012
58. Kim MS, Haney MJ, Zhao Y, Yuan D, Deygen I, Klyachko NL, et al. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine (2018) 14(1):195–204. doi:10.1016/j.nano.2017.09.011
59. Jang SC, Kim OY, Yoon CM, Choi DS, Roh TY, Park J, et al. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano (2013) 7(9):7698–710. doi:10.1021/nn402232g
60. Munoz JL, Bliss SA, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Delivery of functional anti-miR-9 by mesenchymal stem cell-derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol Ther Nucleic Acids (2013) 2:e126. doi:10.1038/mtna.2013.60
61. Katakowski M, Buller B, Zheng X, Lu Y, Rogers T, Osobamiro O, et al. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett (2013) 335(1):201–4. doi:10.1016/j.canlet.2013.02.019
62. Lang FM, Hossain A, Gumin J, Momin EN, Shimizu Y, Ledbetter D, et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro Oncol (2018) 20(3):380–90. doi:10.1093/neuonc/nox152
63. Umezu T, Imanishi S, Azuma K, Kobayashi C, Yoshizawa S, Ohyashiki K, et al. Replenishing exosomes from older bone marrow stromal cells with miR-340 inhibits myeloma-related angiogenesis. Blood Adv (2017) 1(13):812–23. doi:10.1182/bloodadvances.2016003251
64. Greco KA, Franzen CA, Foreman KE, Flanigan RC, Kuo PC, Gupta GN. PLK-1 silencing in bladder cancer by siRNA delivered with exosomes. Urology (2016) 91:242.e1–7. doi:10.1016/j.urology.2016.01.028
65. Lou G, Song X, Yang F, Wu S, Wang J, Chen Z, et al. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol (2015) 8:122. doi:10.1186/s13045-015-0220-7
66. Pascucci L, Cocce V, Bonomi A, Ami D, Ceccarelli P, Ciusani E, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release (2014) 192:262–70. doi:10.1016/j.jconrel.2014.07.042
67. Cocce V, Balducci L, Falchetti ML, Pascucci L, Ciusani E, Brini AT, et al. Fluorescent immortalized human adipose derived stromal cells (hASCs-TS/GFP+) for studying cell drug delivery mediated by microvesicles. Anticancer Agents Med Chem (2017) 17(11):1578–85. doi:10.2174/1871520617666170327113932
68. Yuan Z, Kolluri KK, Gowers KH, Janes SM. TRAIL delivery by MSC-derived extracellular vesicles is an effective anticancer therapy. J Extracell Vesicles (2017) 6(1):1265291. doi:10.1080/20013078.2017.1265291
69. Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med (2005) 3(1):10. doi:10.1186/1479-5876-3-10
70. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med (2005) 3(1):9. doi:10.1186/1479-5876-3-9
71. Huang WH, Chang MC, Tsai KS, Hung MC, Chen HL, Hung SC. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene (2013) 32(37):4343–54. doi:10.1038/onc.2012.458
72. Ramdasi S, Sarang S, Viswanathan C. Potential of mesenchymal stem cell based application in cancer. Int J Hematol Oncol Stem Cell Res (2015) 9(2):95–103.
73. Wang M, Zhao C, Shi H, Zhang B, Zhang L, Zhang X, et al. Deregulated microRNAs in gastric cancer tissue-derived mesenchymal stem cells: novel biomarkers and a mechanism for gastric cancer. Br J Cancer (2014) 110(5):1199–210. doi:10.1038/bjc.2014.14
74. Zhu W, Huang L, Li Y, Zhang X, Gu J, Yan Y, et al. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett (2012) 315(1):28–37. doi:10.1016/j.canlet.2011.10.002
75. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest (2013) 123(4):1542–55. doi:10.1172/JCI66517
76. Vallabhaneni KC, Penfornis P, Dhule S, Guillonneau F, Adams KV, Mo YY, et al. Extracellular vesicles from bone marrow mesenchymal stem/stromal cells transport tumor regulatory microRNA, proteins, and metabolites. Oncotarget (2015) 6(7):4953–67. doi:10.18632/oncotarget.3211
77. Ji R, Zhang B, Zhang X, Xue J, Yuan X, Yan Y, et al. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle (2015) 14(15):2473–83. doi:10.1080/15384101.2015.1005530
78. Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi RU, et al. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal (2014) 7(332):ra63. doi:10.1126/scisignal.2005231
79. Lee JK, Park SR, Jung BK, Jeon YK, Lee YS, Kim MK, et al. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One (2013) 8(12):e84256. doi:10.1371/journal.pone.0084256
80. Shimbo K, Miyaki S, Ishitobi H, Kato Y, Kubo T, Shimose S, et al. Exosome-formed synthetic microRNA-143 is transferred to osteosarcoma cells and inhibits their migration. Biochem Biophys Res Commun (2014) 445(2):381–7. doi:10.1016/j.bbrc.2014.02.007
81. Gilligan KE, Dwyer RM. Engineering exosomes for cancer therapy. Int J Mol Sci (2017) 18(6):1122. doi:10.3390/ijms18061122
Keywords: extracellular vesicles, tumor microenvironment, tumor cells, immune cells, stromal cells, vaccination, cancer therapy
Citation: Chulpanova DS, Kitaeva KV, James V, Rizvanov AA and Solovyeva VV (2018) Therapeutic Prospects of Extracellular Vesicles in Cancer Treatment. Front. Immunol. 9:1534. doi: 10.3389/fimmu.2018.01534
Received: 23 March 2018; Accepted: 21 June 2018;
Published: 03 July 2018
Edited by:
Martin Herrmann, Universitätsklinikum Erlangen, GermanyReviewed by:
Silvano Sozzani, University of Brescia, ItalyCopyright: © 2018 Chulpanova, Kitaeva, James, Rizvanov and Solovyeva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeriya V. Solovyeva, c29sb3Z5b3ZhdnZAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.