 Rebecca A. Marsh
Rebecca A. Marsh- Division of Bone Marrow Transplantation and Immune Deficiency, Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, United States
Epstein–Barr virus (EBV) is a ubiquitous virus that infects nearly all people worldwide without serious sequela. However, for patients who have genetic diseases which predispose them to the development of hemophagocytic lymphohistiocytosis (HLH), EBV infection is a life-threatening problem. As a part of a themed collection of articles on EBV infection and human primary immune deficiencies, we will review key concepts related to the understanding and treatment of HLH.
Introduction
Epstein–Barr virus (EBV) is a ubiquitous virus that infects nearly all people worldwide without serious sequela (1). However, EBV can cause serious disease complications in patients with primary immune deficiencies. In particular, for patients who have genetic diseases which predispose them to the development of hemophagocytic lymphohistiocytosis (HLH), EBV infection is often an immediately life-threatening problem due to the development of HLH. HLH is a syndrome of severe, life-threatening hyperinflammation (discussed below). Though it is difficult to quantify the association of EBV with HLH in North America and Europe, it is generally agreed that EBV is the most common infection observed to be associated with HLH. In one U.S. series, EBV was associated with HLH in approximately 1/3 of patients (2). For somewhat unclear reasons, EBV is highly associated with HLH in Asia, were it has been observed to be associated with HLH in almost 3/4 of patients in one report (3). It is important to note that HLH also develops in patients with genetic HLH in response to many other infections besides EBV, and it is also common for HLH to develop in these patients without an identified infectious trigger. Patients with X-linked lymphoproliferative disease type 1 (XLP1) are an exception to this statement. XLP1 is caused by mutations in SH2D1A (4–6), and HLH is these patients is nearly exclusively associated with EBV.
When to Suspect HLH in Patients with EBV
Hemophagocytic lymphohistiocytosis should be suspected when any of the characteristic signs and symptoms of HLH are present (Table 1), which include fever, splenomegaly, blood cytopenias, hepatitis and/or hepatomegaly, coagulopathy, central nervous system disturbances, and other more rare complications. However, the distinction between HLH and primary EBV infection can be very difficult, as patients with primary EBV infection may develop some of the hallmarks of HLH as part of natural infection. The timing and severity of these manifestations can help distinguish routine EBV infection from HLH complicating EBV infection. Most patients with routine EBV infection are non-toxic appearing. Fever typically dissipates over time, and splenomegaly should gradually improve. Thrombocytopenia develops in up to approximately 50% of patients, and neutropenia develops in up to 80% of patients with EBV, but cytopenias are generally mild and largely resolve by 4 weeks following infection (7). Mild hepatitis is common with EBV infection and develops in 50–80% of cases, but elevations of liver enzymes are generally mild and improve within a few weeks (7). Jaundice is observed in some cases, but coagulopathy is not typical. By contrast, patients who develop HLH in association with EBV, in association with other triggers, or with spontaneous HLH, are generally ill appearing. Fevers are typically profound and do not improve. Cytopenias are generally life-threatening. Patients usually need transfusion support and are often initially evaluated for hematologic malignancies. Hepatitis can be severe; coagulopathy is common and acute liver failure necessitating a liver transplant can occur. Central nervous system involvement can be profound. Patients may develop focal or global deficits, seizures, and altered mental status. There are not firm clinical diagnostic thresholds that can distinguish routine primary EBV infection from HLH, but good clinical judgment can often help identify patients who are experiencing more severe manifestations of disease and warrant further evaluations for possible HLH. Blood levels of fibrinogen, triglycerides, ferritin, and soluble IL-2 receptor can be measured to help differentiate HLH in appropriate cases, as these markers are typically used to support or refute a diagnosis of HLH.

Table 1. Commonly used diagnostic criteria for HLH, adapted from Henter et al. (8).
Diagnosing HLH
It is important to first recognize that HLH is a hyperinflammatory syndrome, which is clinically diagnosed. A clinical diagnosis of HLH should be suspected in patients with a variety of hyperinflammatory clinical presentations, such as patients who seem to be having a hyperinflammatory process in the setting of EBV infection. Most clinicians use the diagnostic criteria developed by the Histiocyte Society for the HLH-1994 and HLH-2004 clinical trials to help establish a clinical diagnosis of HLH (Table 1) (8). The “classic” clinical presentation is that of an infant or young child with unremitting fevers, pancytopenia, and hepatosplenomegaly. The patient may have a rash, jaundice, or bleeding problems. However, HLH can present at any age, and can present with a variety of other clinical manifestations including hepatitis or acute liver failure, or altered levels of consciousness or seizures if there is central nervous system involvement with HLH. Blood levels of the classic inflammatory markers ferritin and soluble IL-2 receptor are typically high in patients with HLH and are used to help make a diagnosis of HLH. Abnormalities in triglycerides (high) and fibrinogen (low) can also be supportive. Recent evidence strongly suggests that quantification of HLA-DR and other phenotypic markers on T cells can help distinguish patients with HLH (9). Of course, there are exceptions to these generalities, especially in the rare cases of isolated CNS disease in patients who lack any systemic illness. Newer markers of interferon gamma pathway activity or inflammasome activation such as CXCL9 and IL-18, respectively, are also starting to gain in use.
While the criteria in Table 1 can be very useful while considering a diagnosis of HLH, they should be considered to be a guideline only. Some patients with HLH lack 5/8 criteria at presentation, or even throughout their clinical course. Particularly, many patients lack hemophagocytosis in marrow or tissue samples. Additionally, the NK cell function assay has been recently found to be inferior to newer diagnostic screening tests for genetic HLH (10) (discussed later). Another shortfall of the criteria are that some patients who meet 5/8 criteria are ultimately found to have disorders other than HLH, and physicians should be careful to consider alternative diagnoses such as malignancies, infections, auto-immune, and rheumatologic diseases (though these problems can of course be complicated by HLH).
Primary Versus Secondary HLH
Once a clinical diagnosis of HLH is established, it is important to perform proper evaluations to check patients for genetic diseases which cause HLH (discussed below). HLH can be classified as “primary” HLH, in which case a patient has a proven genetic etiology or has repeatedly developed HLH or has a family history which supports that a genetic disease is very likely. These patients are typically infants or young children. HLH in patients who lack a known or strongly suspected genetic etiology can be classified as having “secondary” HLH. Patients with secondary HLH tend to be older, develop HLH in the setting of strong immunologic triggers such as infections (such as EBV) or malignancies, or in the setting of rheumatologic conditions. HLH that occurs in the setting of a rheumatologic disease is often termed macrophage activation syndrome. Sometimes, treating the underlying trigger of HLH in patients with secondary HLH will lead to resolution of HLH, but varying intensities of HLH-directed treatment are often needed.
Genetics and Defects of HLH
It was first recognized that HLH could have a hereditary basis in some patients in the 1950s (11, 12). Almost 50 years later, Stepp et al. reported that defects in perforin were responsible for HLH in eight unrelated families (13). The last few decades have seen a tremendous advance in the basic scientific understanding of HLH through the discovery of many additional genetic causes of HLH (Table 2). It is now clear that many genetic causes of HLH essentially cripple cytotoxic lymphocyte granule-mediated cytotoxicity (Figure 1). An exception to this is that patients with X-linked lymphoproliferative disease type 2 (XLP2) due to mutations in XIAP/BIRC4 appear to have normal cytotoxicity (14, 15), and instead may have dysregulated TNFR and inflammasome function (16). Very recently, activating mutations in NLRC4 have been found to cause HLH, which also represents another exception to the rule as pathophysiology relates to inflammasome activation.
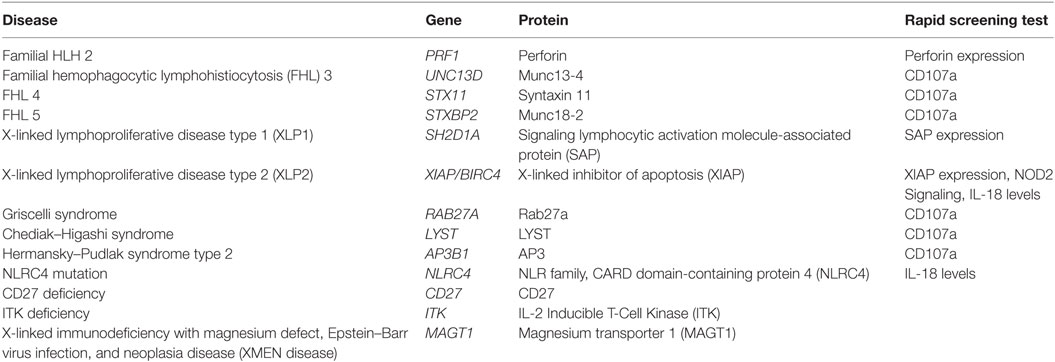
Table 2. Genetic causes of hemophagocytic lymphohistiocytosis (HLH) and associated rapid flow cytometric screening tests.
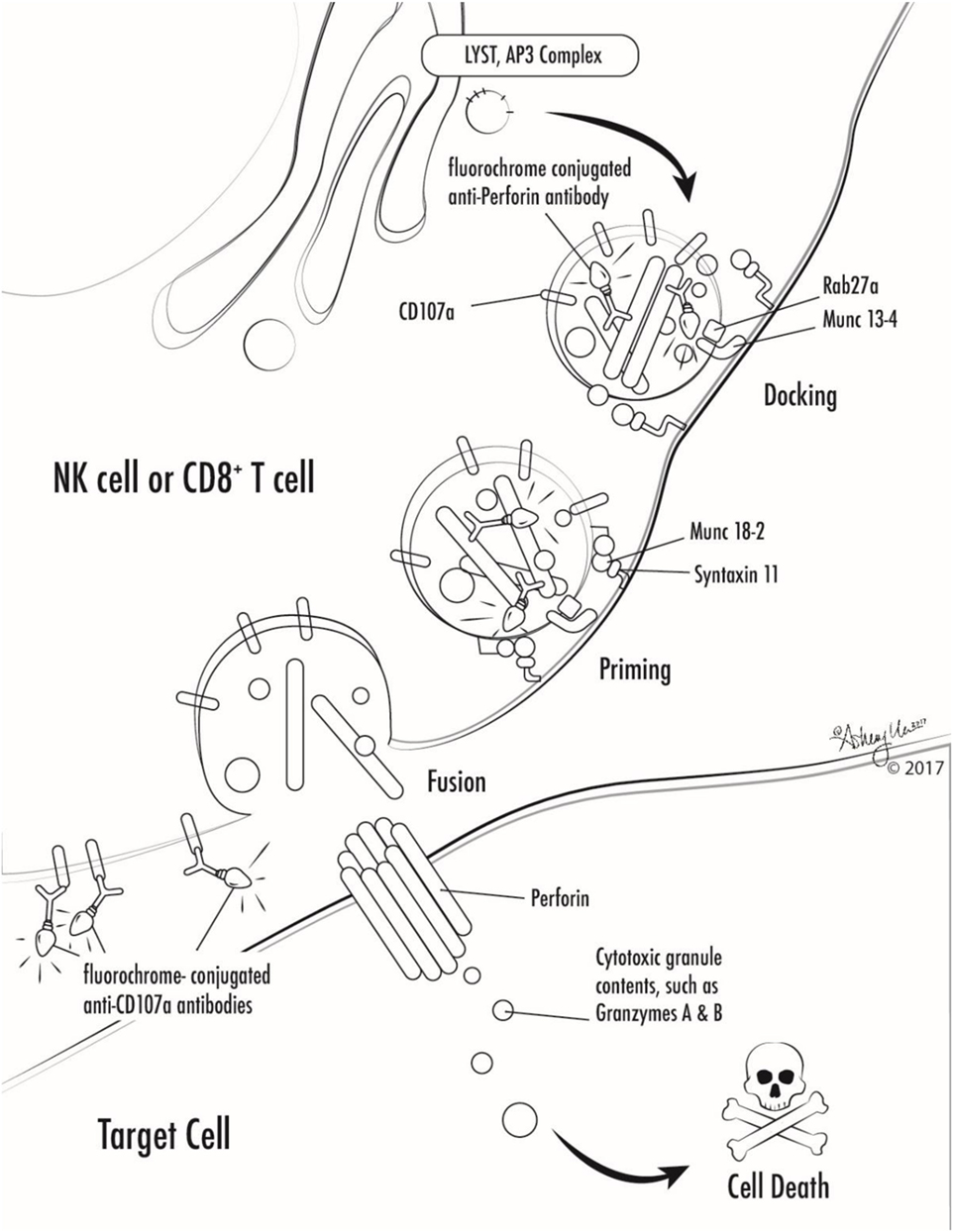
Figure 1. Illustration of involvement of selected primary hemophagocytic lymphohistiocytosis proteins in cytotoxic lymphocyte degranulation and target cell killing. Antibodies against markers used in selected screening diagnostics are also shown (perforin and CD107a).
Many genetic forms of HLH are classified together as familial HLH (FHL). FHL2 is due to mutations in PRF1. FHL3-5 are due to mutations in UNC13D, STX11, and STXBP2, respectively (17–20). Related genetic diseases that are associated with HLH and also pigmentary disorders are Griscelli syndrome, due to mutations in RAB27A, Chediak–Higashi syndrome, due to mutations in LYST, and Hermansky–Pudlak syndrome type 2, due to mutations in AP3B1 (21–23). Of note, patients with RAB27A mutations do not always have abnormal pigmentation. NK cells and T cells from patients with mutations in UNC13D, STX11, STXBP2, RAB27A, LYST, and AP3B1 all fail to degranulate normally, because these proteins are critical for the process of normal cytotoxic granule trafficking, docking, or fusion with the outer cell membrane (Figure 1). The extrusion of cytotoxic granule contents from NK cells and T cells toward their intended target is an important method of elimination of virus infected cells or malignant cells, and also serves to regulate immune homeostasis (24). When the machinery required for this process is broken, intended target cells fail to die and continue to stimulate immune cells, which leads to continued activation and proliferation of immune cells, and a vicious hyperinflammatory cycle ensues.
NK cells and T cells from patients with PRF1 mutations lack functional perforin. Perforin is normally contained within the cytotoxic granules of NK cells and T cells that were discussed above. Perforin is a unique protein that oligomerizes after release from cytotoxic granules and the complexes create pores in the surface membrane of the intended target cells, which allows cytotoxic granule contents to enter the target cell and ultimately results in target cell death (Figure 1) (25, 26). Lack of functional perforin results in the same pathophysiologic abnormality as other causes of FHL: defective cytotoxic lymphocyte granule-mediated cytotoxicity.
As mentioned above, there are other diseases which are associated with HLH that have different (and perhaps more complicated) mechanisms of disease. XLP1 is caused by mutations in SH2D1A, which leads to dysfunctional SLAM-associated protein (SAP) (4–6). SAP is a small SH2 domain-containing protein which is involved in signaling of the signaling lymphocytic activation molecule (SLAM) family of receptors. Lack of normal SAP function leads to several immunologic abnormalities including defective 2B4-mediated cytotoxicity, absence of invariant NKT cell development, defective T cell restimulation-induced cell death, and other humoral and cellular abnormalities (27–30). XLP2 is caused by mutations in X-linked inhibitor of apoptosis (XIAP)/baculoviral inhibitor of apoptosis repeat-containing 4 (BIRC4) (14). Defects in XIAP also lead to several immunologic abnormalities, but cytotoxicity is normal (14, 15). Many cells have an increased susceptibility to cell death, NOD2 signaling is defective, and TNF receptor signaling and inflammasome function are dysregulated (14–16, 31, 32). The development of HLH in these patients is multifactorial, but dysregulation of the NLRP3 inflammasome likely plays a key role in the development of disease. Patients with activating mutations in NLRC4 also develop HLH, associated with constitutive activation of the NLRC4 inflammasome (33, 34). Thus far, there does not seem to be a strong association with EBV infection, as reported patients have not had an identified trigger of hyperinflammation (33–35). Still other genetic diseases that can be associated with EBV-associated HLH, or chronic active EBV or lymphoproliferative diseases, include ITK deficiency, CD27 deficiency, CD70 deficiency, and magnesium transporter 1 (MAGT1) deficiency which is called X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia or XMEN disease (36–45).
Other Manifestations of HLH-Associated Diseases
The FHL disorders are primarily associated with HLH but are often also associated with atypical hyperinflammatory syndromes that lack the full spectrum of HLH, as well as acute liver failure or isolated central nervous system disease in the absence of systemic inflammation. They may uncommonly be associated with atypical chronic active EBV infection, hypogammaglobulinemia, vasculitis, gastrointestinal inflammation, recurrent infections, and very rarely, lymphoproliferative complications (46–48). XLP1 is commonly associated with lymphoma or hypogammaglobulinemia, and has more rare presentations that include aplastic anemia, vasculitis, and gastrointestinal inflammation. XLP2 is associated with a variety of other disease manifestations including atypical/mild HLH-like episodes, inflammatory bowel disease, recurrent infections, hypogammaglobulinemia, uveitis, fistulating skin disease, granulomatous hepatitis, granulomatous, and lymphocytic interstitial lung disease (14, 15, 49–52). CD70 deficiency, CD27 deficiency, ITK deficiency, and MAGT1 all share a strong predisposition to lymphoma.
Screening Tests for Genetic HLH
While the diagnosis of HLH is a clinical diagnosis based on clinical manifestations and laboratory findings, there are several specialized tests which can quickly screen patients for genetic forms of HLH (Table 2; Figure 1). Flow cytometric screening of perforin expression by NK cells and CD8+ T cells serves as a quick screening test for perforin deficiency, and has been found to be highly sensitive (53). A flow cytometric assay to detect abnormal degranulation of NK cells is also available, which quantifies the surface upregulation of CD107a following exposure of NK cells to K562 target cells (or upregulation of CD107a on NK cells or T cells following other appropriate triggers). CD107a is normally expressed within cytotoxic granule membranes, and very little is found on the surface of NK cells or CD8+ T Cells at rest. Hence, one can measure CD107a on the surface of NK cells before and following exposure to target cells as a marker of degranulation. This method has been shown to have good diagnostic accuracy for the detection of patients with mutations in the HLH genes associated with abnormal degranulation (UNC13D, STX11, STXBP2, RAB27A, LYST, and AP3B1) (54). Using both perforin and CD107a testing is more accurate for the identification of patients with genetic forms of HLH compared to traditional NK cell function testing (10). Of note, flow cytometric screening tests are also available to screen patients for XLP1 and XLP2 (55). XLP1 and XLP2 should be considered in male patients with HLH, and even in female patients in whom other genetic causes of HLH have been excluded, due to the observation that females with abnormal skewing of lionization toward XIAP-deficient cells can be symptomatic (56, 57). A functional screen for XIAP deficiency is available via evaluation of NOD2 signaling (58), and IL-18 levels can be helpful as a screening tool for patients with XIAP deficiency or NLRC4 mutations.
Treatment of HLH, Including EBV–HLH
Once HLH has been diagnosed, therapy should be started as soon as possible. Of note, however, therapy should not be started until complete evaluations for lymphoma and leukemia have been performed. The treatment of HLH generally includes a variety of potent immunosuppressive regimens. In North America and most of Europe, a regimen of dexamethasone and etoposide has been the mainstay of treatment, based on the HLH-1994 and HLH-2004 study protocols (8, 59, 60). The HLH-2004 protocol incorporated the addition of cyclosporine, but there has been no clear benefit related to early administration of cyclosporine (60), and it should be noted that cyclosporine can be associated with notable complications including hypertension, renal injury, and posterior reversible encephalopathy syndrome. CNS HLH is typically treated with targeted therapy if patients are stable enough to undergo lumbar punctures and administration of intrathecal steroids and methotrexate. Approximately 50% of patients can be expected to achieve a complete response, and approximately 30% of patients will experience a partial response (59). The incidence of death prior to allogeneic hematopoietic cell transplant was observed to be 19–27% in the HLH 2004 and 1994 studies, respectively.
In France, an alternative regimen containing steroids and ATG has been used. Seventy-three percent of patients were reported to achieve a complete response with the regimen, and 24% of patients achieved a partial response (61). In general, either approach is appropriate, though there has been more widespread experience with dexamethasone and etoposide treatment. The dexamethasone and etoposide regimen offers avoidance of the risk of severe reactions that can be associated with ATG, and may be less T cell immunosuppressive. ATG offers avoidance of chemotherapy exposure and the risk of marrow suppression associated with etoposide. There has been a recent trial of a Hybrid Immunotherapy approach for HLH1 and a European sister trial, but results are not yet available.
Many additional agents have been reported in single patients or small collections of patients. There have been several seemingly beneficial reports of anti-interleukin-1 directed therapies such as anakinra and canakinumab (62–71), and anti-tumor necrosis factor alpha-directed agents such as etanercept and infliximab (72–81), but there are no large data series on which to judge effectiveness. Likewise, plasma exchange has been reported in small numbers of patients, and while some authors report benefit (82, 83), it is difficult to draw conclusions about its effectiveness. More recently, newer agents are under formal investigation for the treatment of HLH. An anti-interferon gamma monoclonal antibody is actively being investigated in the U.S.A. and Europe.2 Ruxolitinib is being trialed at a single center in North America for secondary HLH.3 Both agents have strong mouse data to support their potential efficacy (84–86). Alemtuzumab is being trialed for up-front therapy in France4 and has had reasonable success when used in the salvage setting (below) (87).
For cases of refractory HLH, “salvage” therapy is sometimes needed. Unfortunately, there are very little data on which to base decisions about salvage therapy. The Histiocyte Society Salvage Therapy Working Group recently reviewed the literature, and found that there was evidence for only three agents which had been used in HLH refractory to steroids and either etoposide or ATG: anakinra, ATG, and alemtuzumab, as well as a regimen that combined liposomal doxorubicin with steroids and etoposide (88).
In patients with EBV–HLH, the addition of rituximab can be useful to deplete EBV-harboring B cells and improve HLH. In a retrospective multi-center study, Chellapandian et al. observed that rituximab (given with other HLH therapies) resulted in significant reductions in EBV load within 1 month of use and was also associated with significant decreases in ferritin levels (89).
In addition to these HLH-directed therapies, good supportive care, treatment of underlying triggers, anti-microbial prophylaxis, and close monitoring are usually needed. Anti-fungal prophylaxis, anti-pneumocystis jirovecii prophylaxis, anti-viral prophylaxis, and IVIG replacement during the active treatment period should all be considered. If patients have active virus infections that are associated with HLH such as EBV, CMV, adenovirus, influenza, etc., treatments targeting those infections should be initiated including rituximab for EBV, and anti-viral agents such ganciclovir, cidofovir, oseltamivir, and others as appropriate. The same is true for other infections such as histoplasmosis, tuberculosis, tick-borne diseases, etc. Routine monitoring of laboratory tests such as complete blood counts, liver panels, fibrinogen, and/or coagulation studies should be performed. Weekly or twice weekly monitoring of inflammatory markers such as soluble IL-2 receptor can help with identifying response to therapy or relapse of disease. Monitoring of ferritin can also be helpful, though it is often hindered by changes associated with blood transfusions, and is typically slow to normalize. Newer indicators of pathologic interferon gamma activity such as CXCL9 are gaining favor in use (90), and elevated levels of IL-18 have been found to be a good marker of XLP2/XIAP deficiency and disease activity in those patients (91). Markers of T cell activation such as HLA-DR can also be useful (9). For patients with EBV–HLH, EBV blood polymerase chain reaction (PCR) monitoring can be useful to monitor response to rituximab (89), and also watch for increasing viral loads following rituximab with B cell recovery. Persistently high EBV PCRs following rituximab in the setting of proven B cell depletion can suggest EBV infection of T and/or NK cells.
Definitive Treatment
For patients with secondary HLH, good medical management and follow-up following HLH resolution is needed. Depending on the underlying trigger of HLH in these patients, they may need indefinite care for management of chronic problems. For patients with proven or strongly suspected primary HLH, hematopoietic cell transplantation (HCT) is generally recommended. Historic outcomes of HCT using myeloablative regimens were poor due to high rates of toxicities that resulted in early deaths. Many groups reported survival of only 45–65% (59, 92–97). More recent reduced intensity conditioning (RIC) approaches have resulted in increased patient survival rates of 75% or higher (2, 98, 99). However, these approaches can be associated with the unique challenge of mixed donor and recipient chimerism, which somewhat limits this success of this approach. Patients with HLH do not require 100% donor chimerism for cure, but risk of HLH relapse increases as donor contribution to hematopoiesis (or cytotoxic lymphocyte development) decreases to less than 20–30% (100). Continued efforts to improve stable donor contribution to hematopoiesis will likely lead to increased success with RIC HCT approaches.
Conclusion
Hemophagocytic lymphohistiocytosis in response to EBV or otherwise remains a life-threatening problem for patients with genetic disorders that cause HLH. Discoveries made in recent decades have yielded extraordinary advances in our understanding of HLH. Early recognition and initiation of HLH-directed therapy remain key for patient survival. The next decade promises to yield even further advances in diagnostics and treatment breakthroughs which will continue to improve patient outcomes.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer TG and handling editor declared their shared affiliation.
Footnotes
References
1. Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer (2004) 4(10):757–68. doi:10.1038/nrc1452
2. Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation. Blood (2010) 116(26):5824–31. doi:10.1182/blood-2010-04-282392
3. Xu XJ, Wang HS, Ju XL, Xiao PF, Xiao Y, Xue HM, et al. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer (2017) 64(4). doi:10.1002/pbc.26264
4. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet (1998) 20(2):129–35. doi:10.1038/2424
5. Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature (1998) 395(6701):462–9. doi:10.1038/26683
6. Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A (1998) 95(23):13765–70. doi:10.1073/pnas.95.23.13765
7. Jenson HB. Acute complications of Epstein-Barr virus infectious mononucleosis. Curr Opin Pediatr (2000) 12(3):263–8. doi:10.1097/00008480-200006000-00016
8. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi:10.1002/pbc.21039
9. Ammann S, Lehmberg K, Zur Stadt U, Janka G, Rensing-Ehl A, Klemann C, et al. Primary and secondary hemophagocytic lymphohistiocytosis have different patterns of T-cell activation, differentiation and repertoire. Eur J Immunol (2017) 47(2):364–73. doi:10.1002/eji.201646686
10. Rubin TS, Zhang K, Gifford C, Lane A, Choo S, Bleesing JJ, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood (2017) 129(22):2993–9. doi:10.1182/blood-2016-12-753830
11. Reese AJ, Levy E. Familial incidence of non-lipoid reticuloendotheliosis (Letterer-Siwe disease). Arch Dis Child (1951) 26(130):578–81. doi:10.1136/adc.26.130.578
12. Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child (1952) 27(136):519–25. doi:10.1136/adc.27.136.519
13. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science (1999) 286(5446):1957–9. doi:10.1126/science.286.5446.1957
14. Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444(7115):110–4. doi:10.1038/nature05257
15. Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood (2010) 116(7):1079–82. doi:10.1182/blood-2010-01-256099
16. Yabal M, Muller N, Adler H, Knies N, Gross CJ, Damgaard RB, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep (2014) 7(6):1796–808. doi:10.1016/j.celrep.2014.05.008
17. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell (2003) 115(4):461–73. doi:10.1016/S0092-8674(03)00855-9
18. zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet (2005) 14(6):827–34. doi:10.1093/hmg/ddi076
19. zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet (2009) 85(4):482–92. doi:10.1016/j.ajhg.2009.09.005
20. Cote M, Menager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest (2009) 119(12):3765–73. doi:10.1172/JCI40732
21. Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet (2000) 25(2):173–6. doi:10.1038/76024
22. Barbosa MD, Nguyen QA, Tchernev VT, Ashley JA, Detter JC, Blaydes SM, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature (1996) 382(6588):262–5. doi:10.1038/382262a0
23. Enders A, Zieger B, Schwarz K, Yoshimi A, Speckmann C, Knoepfle EM, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood (2006) 108(1):81–7. doi:10.1182/blood-2005-11-4413
24. de Saint Basile G, Menasche G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol (2010) 10(8):568–79. doi:10.1038/nri2803
25. Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJ, et al. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity (2009) 30(5):684–95. doi:10.1016/j.immuni.2009.03.016
26. Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev (2010) 235(1):35–54. doi:10.1111/j.0105-2896.2010.00896.x
27. Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol (2011) 29:665–705. doi:10.1146/annurev-immunol-030409-101302
28. Nichols KE, Hom J, Gong SY, Ganguly A, Ma CS, Cannons JL, et al. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med (2005) 11(3):340–5. doi:10.1038/nm1189
29. Pasquier B, Yin L, Fondaneche MC, Relouzat F, Bloch-Queyrat C, Lambert N, et al. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med (2005) 201(5):695–701. doi:10.1084/jem.20042432
30. Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest (2009) 119(10):2976–89. doi:10.1172/JCI39518
31. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell (2012) 46(6):746–58. doi:10.1016/j.molcel.2012.04.014
32. Damgaard RB, Fiil BK, Speckmann C, Yabal M, Stadt UZ, Bekker-Jensen S, et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol Med (2013) 5(8):1278–95. doi:10.1002/emmm.201303090
33. Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet (2014) 46(10):1140–6. doi:10.1038/ng.3089
34. Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet (2014) 46(10):1135–9. doi:10.1038/ng.3066
35. Liang J, Alfano DN, Squires JE, Riley MM, Parks WT, Kofler J, et al. Novel NLRC4 mutation causes a syndrome of perinatal autoinflammation with hemophagocytic lymphohistiocytosis, hepatosplenomegaly, fetal thrombotic vasculopathy, and congenital anemia and ascites. Pediatr Dev Pathol (2017) 20(6):498–505. doi:10.1177/1093526616686890
36. Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest (2009) 119(5):1350–8. doi:10.1172/JCI37901
37. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia (2012) 26(5):963–71. doi:10.1038/leu.2011.371
38. Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, et al. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica (2011) 96(3):472–6. doi:10.3324/haematol.2010.033910
39. van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol (2012) 129(3):787–93.e6. doi:10.1016/j.jaci.2011.11.013
40. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica (2013) 98(3):473–8. doi:10.3324/haematol.2012.068791
41. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol (2015) 136(3):703–12.e10. doi:10.1016/j.jaci.2015.02.022
42. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med (2017) 214(1):91–106. doi:10.1084/jem.20160849
43. Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med (2017) 214(1):73–89. doi:10.1084/jem.20160784
44. Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature (2011) 475(7357):471–6. doi:10.1038/nature10246
45. Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood (2014) 123(14):2148–52. doi:10.1182/blood-2013-11-538686
46. Katano H, Ali MA, Patera AC, Catalfamo M, Jaffe ES, Kimura H, et al. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood (2004) 103(4):1244–52. doi:10.1182/blood-2003-06-2171
47. Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica (2010) 95(12):2080–7. doi:10.3324/haematol.2010.029389
48. Tesi B, Chiang SC, El-Ghoneimy D, Hussein AA, Langenskiold C, Wali R, et al. Spectrum of atypical clinical presentations in patients with biallelic PRF1 missense mutations. Pediatr Blood Cancer (2015) 62(12):2094–100. doi:10.1002/pbc.25646
49. Speckmann C, Lehmberg K, Albert MH, Damgaard RB, Fritsch M, Gyrd-Hansen M, et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol (2013) 149(1):133–41. doi:10.1016/j.clim.2013.07.004
50. Basiaga ML, Weiss PF, Behrens EM. BIRC4 mutation: an important rare cause of uveitis. J Clin Rheumatol (2015) 21(8):444–7. doi:10.1097/RHU.0000000000000327
51. Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood (2011) 117(5):1522–9. doi:10.1182/blood-2010-07-298372
52. Steele CL, Dore M, Ammann S, Loughrey M, Montero A, Burns SO, et al. X-linked inhibitor of apoptosis complicated by granulomatous lymphocytic interstitial lung disease (GLILD) and granulomatous hepatitis. J Clin Immunol (2016) 36(7):733–8. doi:10.1007/s10875-016-0320-3
53. Abdalgani M, Filipovich AH, Choo S, Zhang K, Gifford C, Villanueva J, et al. Accuracy of flow cytometric perforin screening for detecting patients with FHL due to PRF1 mutations. Blood (2015) 126(15):1858–60. doi:10.1182/blood-2015-06-648659
54. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood (2012) 119(12):2754–63. doi:10.1182/blood-2011-08-374199
55. Gifford CE, Weingartner E, Villanueva J, Johnson JJ, Zhang K, Filipovich AH, et al. Clinical flow cytometric screening of SAP and XIAP expression accurately identifies patients with SH2D1A and XIAP/BIRC4 mutations. Cytometry B Clin Cytom (2014) 86(4):263–71. doi:10.1002/cytob.21166
56. Yang X, Hoshino A, Taga T, Kunitsu T, Ikeda Y, Yasumi T, et al. A female patient with incomplete hemophagocytic lymphohistiocytosis caused by a heterozygous XIAP mutation associated with non-random X-chromosome inactivation skewed towards the wild-type XIAP allele. J Clin Immunol (2015) 35(3):244–8. doi:10.1007/s10875-015-0144-6
57. Holle JR, Marsh RA, Holdcroft AM, Davies SM, Wang L, Zhang K, et al. Hemophagocytic lymphohistiocytosis in a female patient due to a heterozygous XIAP mutation and skewed X chromosome inactivation. Pediatr Blood Cancer (2015) 62(7):1288–90. doi:10.1002/pbc.25483
58. Ammann S, Elling R, Gyrd-Hansen M, Duckers G, Bredius R, Burns SO, et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp Immunol (2014) 176(3):394–400. doi:10.1111/cei.12306
59. Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood (2002) 100(7):2367–73. doi:10.1182/blood-2002-01-0172
60. Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long term results of the cooperative HLH-2004 study. Blood (2017). doi:10.1182/blood-2017-06-788349
61. Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics (2007) 120(3):e622–8. doi:10.1542/peds.2006-3164
62. Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol (2011) 17(1):23–7. doi:10.1097/RHU.0b013e318205092d
63. Durand M, Troyanov Y, Laflamme P, Gregoire G. Macrophage activation syndrome treated with anakinra. J Rheumatol (2010) 37(4):879–80. doi:10.3899/jrheum.091046
64. Kahn PJ, Cron RQ. Higher-dose anakinra is effective in a case of medically refractory macrophage activation syndrome. J Rheumatol (2013) 40(5):743–4. doi:10.3899/jrheum.121098
65. Loh NK, Lucas M, Fernandez S, Prentice D. Successful treatment of macrophage activation syndrome complicating adult Still disease with anakinra. Intern Med J (2012) 42(12):1358–62. doi:10.1111/imj.12002
66. Simon DW, Aneja R, Carcillo JA, Halstead ES. Plasma exchange, methylprednisolone, IV immune globulin, and now anakinra support continued PICU equipoise in management of hyperferritinemia-associated sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome/secondary hemophagocytic lymphohistiocytosis syndrome*. Pediatr Crit Care Med (2014) 15(5):486–8. doi:10.1097/PCC.0000000000000098
67. Tayer-Shifman OE, Ben-Chetrit E. Refractory macrophage activation syndrome in a patient with SLE and APLA syndrome – successful use of PET-CT and anakinra in its diagnosis and treatment. Mod Rheumatol (2015) 25(6):954–7. doi:10.3109/14397595.2013.844403
68. Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol (2008) 4(11):615–20. doi:10.1038/ncprheum0919
69. Behrens EM, Kreiger PA, Cherian S, Cron RQ. Interleukin 1 receptor antagonist to treat cytophagic histiocytic panniculitis with secondary hemophagocytic lymphohistiocytosis. J Rheumatol (2006) 33(10):2081–4.
70. Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med (2014) 15(5):401–8. doi:10.1097/PCC.0000000000000078
71. Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford) (2011) 50(2):417–9. doi:10.1093/rheumatology/keq218
72. Gianella S, Schaer DJ, Schwarz U, Kurrer M, Heppner FL, Fehr J, et al. Retinal microangiopathy and rapidly fatal cerebral edema in a patient with adult-onset Still’s disease and concurrent macrophage activation syndrome. Am J Hematol (2008) 83(5):424–7. doi:10.1002/ajh.21084
73. Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, Gondo H, Imashuku S, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol (2006) 81(1):59–61. doi:10.1002/ajh.20462
74. Takahashi N, Naniwa T, Banno S. Successful use of etanercept in the treatment of acute lupus hemophagocytic syndrome. Mod Rheumatol (2008) 18(1):72–5. doi:10.3109/s10165-007-0006-z
75. Sellmer A, Stausbol-Gron B, Krag-Olsen B, Herlin T. Successful use of infliximab in macrophage activation syndrome with severe CNS involvement. Scand J Rheumatol (2011) 40(2):156–7. doi:10.3109/03009742.2010.508468
76. Makay B, Yilmaz S, Turkyilmaz Z, Unal N, Oren H, Unsal E. Etanercept for therapy-resistant macrophage activation syndrome. Pediatr Blood Cancer (2008) 50(2):419–21. doi:10.1002/pbc.21019
77. Cortis E, Insalaco A. Macrophage activation syndrome in juvenile idiopathic arthritis. Acta Paediatr Suppl (2006) 95(452):38–41. doi:10.1080/08035320600649713
78. Maeshima K, Ishii K, Iwakura M, Akamine M, Hamasaki H, Abe I, et al. Adult-onset Still’s disease with macrophage activation syndrome successfully treated with a combination of methotrexate and etanercept. Mod Rheumatol (2012) 22(1):137–41. doi:10.1007/s10165-011-0477-9
79. Ideguchi H, Ohno S, Takase K, Hattori H, Kirino Y, Takeno M, et al. Successful treatment of refractory lupus-associated haemophagocytic lymphohistiocytosis with infliximab. Rheumatology (Oxford) (2007) 46(10):1621–2. doi:10.1093/rheumatology/kem205
80. Kikuchi H, Yamamoto T, Asako K, Takayama M, Shirasaki R, Ono Y. Etanercept for the treatment of intractable hemophagocytic syndrome with systemic lupus erythematosus. Mod Rheumatol (2012) 22(2):308–11. doi:10.1007/s10165-011-0500-1
81. Prahalad S, Bove KE, Dickens D, Lovell DJ, Grom AA. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol (2001) 28(9):2120–4.
82. Bosnak M, Erdogan S, Aktekin EH, Bay A. Therapeutic plasma exchange in primary hemophagocytic lymphohistiocytosis: reports of two cases and a review of the literature. Transfus Apher Sci (2016) 55(3):353–6. doi:10.1016/j.transci.2016.09.015
83. Nakakura H, Ashida A, Matsumura H, Murata T, Nagatoya K, Shibahara N, et al. A case report of successful treatment with plasma exchange for hemophagocytic syndrome associated with severe systemic juvenile idiopathic arthritis in an infant girl. Ther Apher Dial (2009) 13(1):71–6. doi:10.1111/j.1744-9987.2009.00607.x
84. Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood (2016) 127(13):1666–75. doi:10.1182/blood-2015-12-684399
85. Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood (2016) 128(1):60–71. doi:10.1182/blood-2016-02-700013
86. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood (2004) 104(3):735–43. doi:10.1182/blood-2003-10-3413
87. Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer (2013) 60(1):101–9. doi:10.1002/pbc.24188
88. Marsh RA, Jordan MB, Talano JA, Nichols KE, Kumar A, Naqvi A, et al. Salvage therapy for refractory hemophagocytic lymphohistiocytosis: a review of the published experience. Pediatr Blood Cancer (2017) 64(4). doi:10.1002/pbc.26308
89. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol (2013) 162(3):376–82. doi:10.1111/bjh.12386
90. Buatois V, Chatel L, Cons L, Lory S, Richard F, Guilhot F, et al. Use of a mouse model to identify a blood biomarker for IFNgamma activity in pediatric secondary hemophagocytic lymphohistiocytosis. Transl Res (2017) 180:37–52.e2. doi:10.1016/j.trsl.2016.07.023
91. Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine (2014) 65(1):74–8. doi:10.1016/j.cyto.2013.09.007
92. Horne A, Janka G, Maarten Egeler R, Gadner H, Imashuku S, Ladisch S, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol (2005) 129(5):622–30. doi:10.1111/j.1365-2141.2005.05501.x
93. Ouachee-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics (2006) 117(4):e743–50. doi:10.1542/peds.2005-1789
94. Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, et al. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplant (2007) 39(7):411–5. doi:10.1038/sj.bmt.1705600
95. Baker KS, Filipovich AH, Gross TG, Grossman WJ, Hale GA, Hayashi RJ, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant (2008) 42(3):175–80. doi:10.1038/bmt.2008.133
96. Cesaro S, Locatelli F, Lanino E, Porta F, Di Maio L, Messina C, et al. Hematopoietic stem cell transplantation for hemophagocytic lymphohistiocytosis: a retrospective analysis of data from the Italian Association of Pediatric Hematology Oncology (AIEOP). Haematologica (2008) 93(11):1694–701. doi:10.3324/haematol.13142
97. Ohga S, Kudo K, Ishii E, Honjo S, Morimoto A, Osugi Y, et al. Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer (2010) 54(2):299–306. doi:10.1002/pbc.22310
98. Cooper N, Rao K, Gilmour K, Hadad L, Adams S, Cale C, et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis. Blood (2006) 107(3):1233–6. doi:10.1182/blood-2005-05-1819
99. Cooper N, Rao K, Goulden N, Webb D, Amrolia P, Veys P. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant (2008) 42(Suppl 2):S47–50. doi:10.1038/bmt.2008.283
Keywords: Epstein–Barr virus, hemophagocytic lymphohistiocytosis, primary immunodeficiency, X-linked Lymphoproliferative Disease, Mononucleosis
Citation: Marsh RA (2018) Epstein–Barr Virus and Hemophagocytic Lymphohistiocytosis. Front. Immunol. 8:1902. doi: 10.3389/fimmu.2017.01902
Received: 06 October 2017; Accepted: 13 December 2017;
Published: 08 January 2018
Edited by:
Jeffrey I. Cohen, National Institutes of Health (NIH), United StatesReviewed by:
Thomas Gross, National Institutes of Health (NIH), United StatesShigeaki Nonoyama, National Defense Medical College, Japan
Copyright: © 2018 Marsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca A. Marsh, cmViZWNjYS5tYXJzaEBjY2htYy5vcmc=