 Jun Wei
Jun Wei Jana Raynor
Jana Raynor Thanh-Long M. Nguyen
Thanh-Long M. Nguyen Hongbo Chi
Hongbo Chi- Department of Immunology, St. Jude Children’s Research Hospital, Memphis, TN, USA
T cells play pivotal roles in shaping host immune responses in infectious diseases, autoimmunity, and cancer. The activation of T cells requires immune and growth factor-derived signals. However, alterations in nutrients and metabolic signals tune T cell responses by impinging upon T cell fates and immune functions. In this review, we summarize how key nutrients, including glucose, amino acids, and lipids, and their sensors and transporters shape T cell responses. We also briefly discuss regulation of T cell responses by oxygen and energy sensing mechanisms.
Introduction
T cells express specific T cell receptors (TCRs) and play pivotal roles in cell-mediated immunity during infection, autoimmune diseases, and cancer. Conventional αβ T cells are divided into two major lineages based on the exclusive expression of the co-receptor CD8 or CD4 (1). Upon cognate antigen stimulation, naïve CD8+ T cells expand and differentiate into cytotoxic effector cells to clear infected or malignant cells. After this rapid expansion, effector CD8+ T cells undergo contraction, and only a small subset of them eventually forms the memory population (2). Likewise, naïve CD4+ T cells are activated by cognate antigens and undergo proliferation and functional specialization. In response to an instructive cytokine milieu, naïve CD4+ T cells differentiate into functionally distinct T helper cells, including TH1, TH2, TH17, and TFH effector cells, and immunosuppressive Treg cells (3). Although immune receptors, signaling proteins and transcriptional factors allow T cells to sense and transduce antigenic and inflammatory signals, emerging studies highlight that cellular metabolism is dynamically regulated to control T cell survival, proliferation, and function (4). Nutrients are the basis of cellular metabolism, with glucose, amino acids, and lipids playing crucial roles in immune signaling and immune cell functions (5). Because T cell priming and function occur within discrete microenvironments, they must adapt to changes in nutrient and environmental signals to mount effective adaptive immune responses.
Nutrient-sensing systems, composed of sensors, transporters, and signaling proteins, are utilized by cells to monitor and respond to fluctuations in environmental nutrient levels. Several evolutionarily conserved nutrient-sensing systems have been identified (6, 7). Recently, extensive efforts have been made to dissect mechanisms of nutrient sensing in T cells. This review article first discusses the regulation and function of glucose metabolism in T cell responses. Second, we summarize recent findings on how specific amino acids and amino acid transporters control T cell responses. Third, we summarize the contributions of fatty acid (FA) and cholesterol sensing to T cell-mediated immunity. Finally, we briefly discuss how T cells adapt to oxygen levels and energy stress to maintain their functions.
Glucose Sensing
Glucose is a major cellular nutrient that serves as a critical fuel for cellular adenosine triphosphate (ATP) generation and precursor for biosynthetic pathways. T cell activation, subset differentiation, and effector function are tightly regulated by glucose metabolism as detailed below (Figure 1).
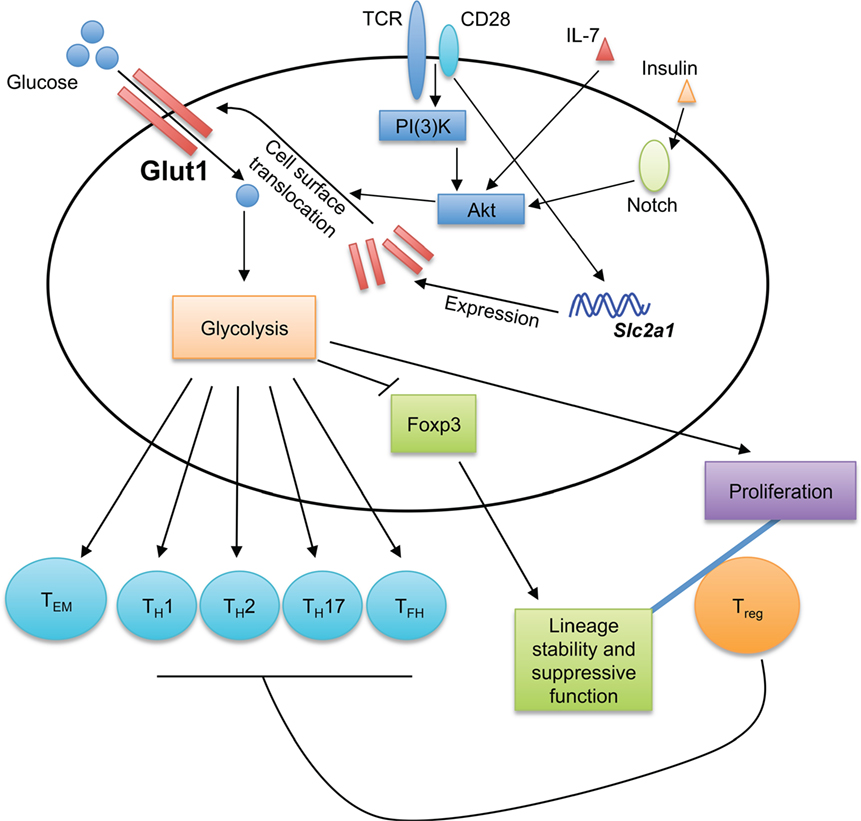
Figure 1. Glucose sensing shapes T cell immune responses. Glucose transporter 1 (Glut1) transports glucose into T cells to fuel glycolysis. The generation of effector T cells, including TH1, TH2, TH17, and TFH cells, is dependent on Glut1 expression and glycolytic metabolism. While Glut1 expression and glycolysis promote Treg cell proliferation, they impair Treg cell lineage stability and suppressive function. Thus, Glut1 expression provides a key mechanism to shape T cell responses. Glycolysis also promotes the formation of effector memory CD8+ T cells (TEM). TCR and CD28 co-stimulation potently induces Glut1 expression, while the translocation of Glut1 from the cytoplasm to T cell surface is induced by various stimuli, including TCR-CD28, IL-7, and insulin.
Glucose Metabolism in Conventional T Cell Activation, Differentiation, and Memory Formation
Activated T cells use glucose metabolism to fulfill the metabolic requirements for rapid proliferation and biosynthesis supporting cellular growth and differentiation (4). Such demands are met, in part, by upregulation of glucose transporter 1 (Glut1) during T cell activation. Naïve T cells express low levels of Glut1 on the cell surface, but Glut1 expression and surface localization are significantly increased in activated T cells (8–10). Several upstream signals regulate Glut1 expression in T cells. TCR and CD28 co-stimulation induces the expression and surface localization of Glut1 in T cells (10). The phosphoinositol-3 kinase (PI(3)K)-Akt pathway triggers the translocation of Glut1 from cytoplasm to T cell surface (9, 10). Other stimuli also increase surface Glut1 levels on T cells; for example, IL-7-induced STAT5 signaling and downstream Akt activation induce Glut1 trafficking to the cell surface in both naïve and activated T cells (11). Increased surface expression of Glut1 is observed on memory CD4+ T cells upon insulin stimulation through a mechanism that requires Notch-Akt signaling (12). In human CD4+ T cells, the SLC2a1 (encodes for human Glut1) mRNA transcript is targeted by miR-150, whose expression is downregulated by co-stimulation with the complement receptor CD46 (13). CD46 signaling is also important for effective TH1 cell differentiation by potentiating TCR-driven Glut1 expression and surface translocation (14).
Extensive efforts have been made to determine the role of Glut1 in T cell responses. While Glut1 overexpression does not alter T cell development, it enhances activated T cell proliferation and growth (10, 15), and generation of TFH cells (16). Also, TH1, TH2, and TH17 cells express high levels of Glut1 and are highly glycolytic (15, 17). Consistent with this observation, conditional deletion of Slc2a1 (encodes for mouse Glut1) in the T cell compartment leads to defective generation of TH1, TH2, and TH17 cells both in vitro and in vivo (8). How Glut1 expression and glucose metabolism specifically contribute to the functional specialization of effector CD4+ T cell subsets requires further investigation. Transgenic expression of Glut1 leads to an accumulation of activated/memory phenotype T cells in vivo, suggesting a role of glucose metabolism in T cell memory responses (10, 15). Memory T cells have reduced glycolysis as compared to effector T cells (18), but Glut1 expression and glucose uptake are essential for CD4+ memory T cell maintenance (12). A recent study demonstrates that constitutive glycolysis promotes the formation of effector memory CD8+ T cells (TEM) in a murine genetic model with T cell-specific deletion of a key hypoxia-inducible factor 1α (HIF1α) negative regulator, Von Hippel-Lindau (VHL) (19). Consistent with these observations, Glut1 expression favors human TEM formation (20). Moreover, the rapid acquisition of effector functions by human TEM requires immediate-early glycolysis upon activation (21). Thus, Glut1 and glucose metabolism are essential for effector and TEM responses. Further studies are required to reveal the underlying molecular mechanisms.
Recent studies have uncovered important roles of glucose competition between tumor cells and effector T cells within the tumor microenvironment (22–24). Similar to T cells, tumor cells rewire their metabolism to greatly increase glucose uptake and aerobic glycolysis, which is known as the Warburg Effect (25). The functional state of tumor-infiltrating T cells is positively correlated with their glycolytic activity and glucose availability within tumor tissues (23, 24). Due to the limited glucose levels in the tumor microenvironment, tumor-infiltrating T cells produce less IFN-γ than T cells taken from glucose-replete environments (23). Mechanistically, this is linked to the ability of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to serve as both a glycolytic enzyme and RNA-binding protein (26, 27). When aerobic glycolysis is inhibited, GAPDH binds to the Ifng (encodes for IFN-γ) mRNA transcript to suppress its translation (28). Aerobic glycolysis also plays a pivotal role in sustaining TCR-mediated calcium-NFAT signaling to maintain T cell effector functions (24). Specifically, phosphoenolpyruvate (PEP) generated during glycolysis maintains cytosolic calcium levels by suppressing sacro/endoplasmic reticulum calcium ATPase activity (24). Notably, increasing PEP production enhances anti-tumor T cell responses (24). Removing the metabolic restrictions in T cells may also contribute to the therapeutic effects of checkpoint blockade therapies, including anti-CTLA4, anti-PD-1, and anti-PD-L1 antibody administrations, since those treatments restore glucose levels within tumors and glycolytic metabolism in T cells (23). More research is needed to determine the therapeutic potential of targeting the components of glucose sensing and metabolism in T cells in cancer patients.
Glucose Metabolism in Treg Cells
The roles of glycolytic metabolism have also been investigated in suppressive Foxp3+ Treg cells. Murine Treg cells express comparable levels of Glut1 as naïve T cells but lower levels of Glut1 than effector T cells (8, 15). Such regulation of Glut1 expression is partially dependent on elevated AMP-activated protein kinase (AMPK) activation in Treg cells (15). Foxp3, the master transcription factor that governs Treg cell differentiation and function, limits Glut1 expression through inhibiting Akt (29). Glut1 deficiency does not affect Treg cell suppressive function but increases the proportion of Treg cells in the peripheral CD4+ T cell compartment (8). In contrast, Treg cells with aberrant increases in glucose metabolism tend to lose their lineage stability. Indeed, murine Treg cells with elevated Glut1 expression have reduced CD25 and Helios expression and are unable to maintain Foxp3 expression and suppressive function in a murine inflammatory bowel disease model, indicative of reduced Treg cell stability (30). These results are consistent with recent findings that aberrant glycolysis is detrimental to Treg cell lineage stability and functional integrity (31–33). Of note, proliferating human and murine Treg cells have elevated glucose uptake and glycolysis than non-dividing Treg cells, and glycolysis contributes to the functional differentiation of human Treg cells by inducing FOXP3 expression (34, 35). These studies highlight a pivotal role of glucose metabolism in balancing the proliferation and suppressive function of Treg cells, which is likely important for controlling effector and suppressive T cell responses during infection and inflammation.
Amino Acid Sensing
Amino acids are the building blocks for protein synthesis, and their uptake into cells is critical for cellular function. During cellular division, the influx of amino acids is especially critical to meet the increased demands for protein synthesis. Furthermore, amino acids can serve as sources for metabolites that enter into metabolic processes, such as the tricarboxylic acid (TCA) cycle. Such energy-demanding cellular processes must be tightly regulated, requiring the sensing of extracellular and intracellular amino acid abundance. Recent studies have begun to identify specific amino acids and amino acid transporters that are critical in regulating T cell homeostasis and function (Figure 2).
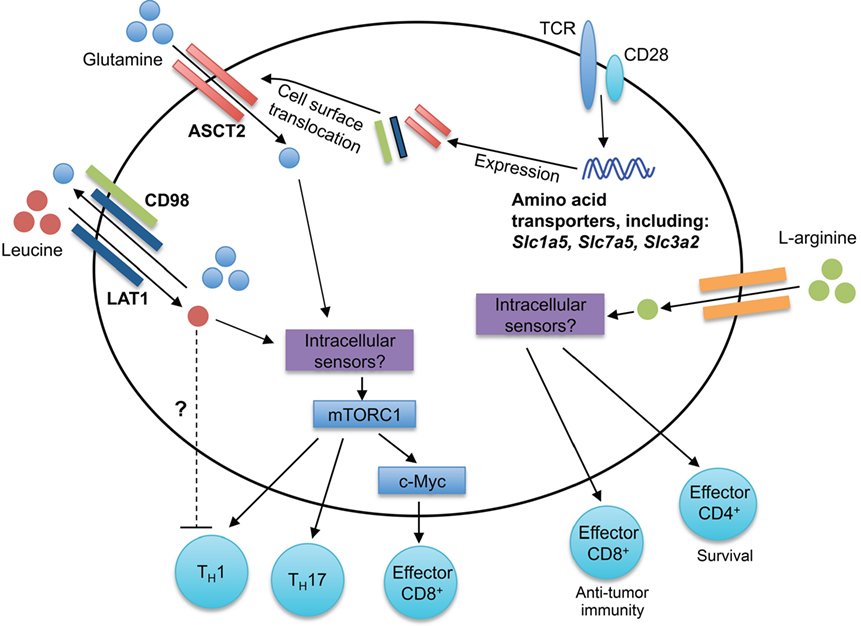
Figure 2. Amino acid sensing modulates T cell responses. Antigen-driven activation of T cells through TCRs upregulates expression of many amino acid transporters, including the leucine and glutamine transporters LAT1, ASCT2, and CD98. LAT1 associates with CD98, forming a bidirectional transporter for leucine and glutamine. The intracellular sensors of leucine and glutamine in T cells remain unknown. mTORC1 is activated downstream of intracellular amino acids, leading to the regulation of CD4+ T cell differentiation (TH1 and TH17) and CD8+ T cell effector responses. Elevated intracellular l-arginine levels promote effector CD8+ T cell anti-tumor immunity and effector CD4+ T cell IFN-γ production and survival. Although the intracellular mediators critical for arginine sensing are unclear, three potential sensors are BAZ1B, PSIP1, and TSN.
Leucine and Glutamine
Amino acids are critical for efficient T cell activation and proliferative responses (36). The SLC7 amino acid transporter family contributes to the uptake of amino acids by T cells. The SLC7 amino acid transporter family is comprised of the cationic amino acid transporters (SLC7A1-4, CAT1-4, and SLC7A14) and the glycoprotein-associated amino acid transporters (SLC7A5-13, SLC7A15) (37). Antigen signaling through the TCR increases phenylalanine, leucine, and glutamine uptake into CD8+ T cells (38, 39). Furthermore, the expression of many SLC7 transporters, including Slc7a5 (encodes for LAT1), is upregulated during activation in both mouse and human T cells (38, 40), which may be mediated by TCR-induced ERK/MAPK signaling (39). LAT1 associates with CD98 (encoded by Slc3a2) to form a high-affinity transporter that facilitates the bidirectional influx of leucine and efflux of glutamine from cells (38, 41). LAT1 is not required for normal T cell development; however, LAT1-deficient CD8+ T cells have impaired primary effector expansion following antigenic stimulation (38). Mechanistically, leucine uptake induces activation of mechanistic target of rapamycin complex 1 (mTORC1) in effector CD8+ T cells and promotes c-Myc expression (38). Furthermore, the loss in c-Myc expression in CD8+ T cells correlates with impaired glucose, glutamine, and arginine uptake and impaired glycolytic metabolism, which promote proper CD8+ T cell effector function (38, 42). Thus, leucine is a positive regulator of peripheral CD8+ T cell functionality. There is much left to be understood how amino acid sensing regulates CD8+ T cell responses. For instance, while glutamine receptor ASCT2 (encoded by Slc1a5) is dispensable for CD8+ T cell development (41), how ASCT2 regulates CD8+ T cell effector and memory responses has not been explored.
Both leucine and glutamine are positive regulators of CD4+ T cell differentiation into TH1 and TH17 cells. Indeed, the absence of LAT1 expression impairs TH1 and TH17 cell differentiation in vitro (38), and elevated glutamine levels favor TH1 and TH17 cell differentiation in vitro (41). Consistent with these observations, ASCT2 is important for TH1 and TH17 cell differentiation (41). Likely due to the bidirectional transport of glutamine and leucine via LAT1, activated ASCT2-deficient CD4+ T cells also have decreased leucine uptake (41, 43). Interestingly, highly elevated leucine levels favor TH17 differentiation, but impair TH1 differentiation in vitro, indicating that high concentrations of leucine may negatively regulate TH1 differentiation (41). Glutaminolysis plays crucial roles in replenishing the TCA cycle under conditions of aerobic glycolysis and supports T cell activation (42). Furthermore, the glutamine-derived metabolite alpha-ketoglutarate promotes TH1 differentiation, potentially through enhancing mTORC1 signaling (44). Unlike TH1 and TH17 cells, TH2 and Treg cells do not require the expression of LAT1 or ASCT2 for their differentiation in vitro (38, 41), likely due to their altered dependency on mTOR signaling (45). Indeed, Treg cell differentiation is favored during glutamine deprivation (44). Thus, leucine and glutamine likely act in concert to regulate the threshold of mTORC1 signaling and subsequent CD4+ T cell differentiation.
Arginine
Arginine is transported into cells through the cationic transporters CAT1-4 (46), where it is metabolized by arginase 1 or nitric-oxide synthase 2 (46). Myeloid-derived suppressor cells, which are elevated in tumor microenvironment and other disease states, consume and deplete extracellular arginine (46). These arginine-depleted environments impair T cell proliferation (47–50). Arginine is also directly metabolized in T cells (51). Activated T cells have increased l-arginine metabolism mediated by arginase 2, which contributes to enhanced CD4+ and CD8+ T cell survival and anti-tumor immunity (51). Interestingly, Arg2-deficient CD4+ and CD8+ T cells have elevated levels of intracellular l-arginine, which correlates with increased survival after IL-2 withdrawal (51), suggesting that additional arginine-sensing pathways contribute to T cell survival. While the mechanisms of how arginine is sensed within T cells remain elusive, BAZ1B, PSIP1, and Translin are potential arginine sensors that promote T cell survival (51). Interestingly, these survival effects appear to be independent of modulating mTORC1 activation, which is a known downstream sensor of arginine in other cell lineages (52–54). Thus, regulation of arginine availability and sensing is critical for T cell functionality and combating environmental insults, such as infections and tumors. Future studies can explore how lysine and ornithine, whose uptake is also CAT dependent, contribute to T cell responses.
Serine
Serine is a non-essential amino acid that is synthesized intracellularly from the glycolytic intermediate 3-phosphoglycerate, or directly transported into cells (55). Recent work reveals that serine metabolism is enhanced in activated T cells and provides intracellular glycine and one-carbon units for purine nucleotide biosynthesis, thereby supporting T cell proliferation (56). Serine uptake is critical for effector T cell responses, as serine and glycine-limiting conditions impair anti-CD3/CD28 antibody-driven CD4+ and CD8+ T cell proliferation in vitro (56). Furthermore, mice on a serine and glycine-restricted diet have impaired antigen-driven CD8+ effector T cell expansion and pathogen clearance (56). These findings elucidate a novel role for serine metabolism in effector T cell responses.
Tryptophan
Indoleamine 2,3-dioxygenase (IDO) catabolizes the amino acid tryptophan, resulting in localized tryptophan depletion. Tryptophan depletion is then sensed by general control non-derepressible 2 (GCN2), which has a high affinity for uncharged tRNAs that accumulate when intracellular amino acid abundance is low. GCN2 binding to uncharged tRNAs induces GCN2 kinase activity, which subsequently inhibits protein translation via the phosphorylation of eIF2α (6). It has been demonstrated that GCN2 sensing of amino acid deprivation in activated T cells promotes T cell survival, limits T cell proliferation, and induces T cell anergy (36, 57). In contrast to these findings, a recent study has shown that both control and GCN2-deficient CD4+ and CD8+ T cells have impaired proliferation when tryptophan, arginine, lysine, leucine, or asparagine levels are low, suggesting additional pathways contribute to sensing amino acid deprivation in T cells (58). IDO may also influence T cell responses independently of GCN2 kinase activity by altering the inflammatory environment. Indeed, tryptophan metabolites promote Treg cell differentiation indirectly by increasing TGF-β production by dendritic cells (DCs) (59). Furthermore, blocking IDO promotes IL-6 production by plasmacytoid DCs, which enables skewing of CD4+ T cells toward the TH17 cell subset and suppression of Treg cell development (60, 61). Consequently, IDO deficiency promotes TH17 differentiation and exacerbates experimental autoimmune encephalomyelitis (EAE), a murine model of human multiple sclerosis (59). Thus, the IDO-tryptophan degradation pathway plays a regulatory role in local inflammation driving TH17 cell responses.
Lipid Sensing
Lipid molecules, including FAs and sterols, are essential for cellular function, serving important functions in membrane biosynthesis and intracellular signaling. Several studies indicate roles for cholesterol and FAs in activation, differentiation, and function of T cells as discussed below (Figure 3).
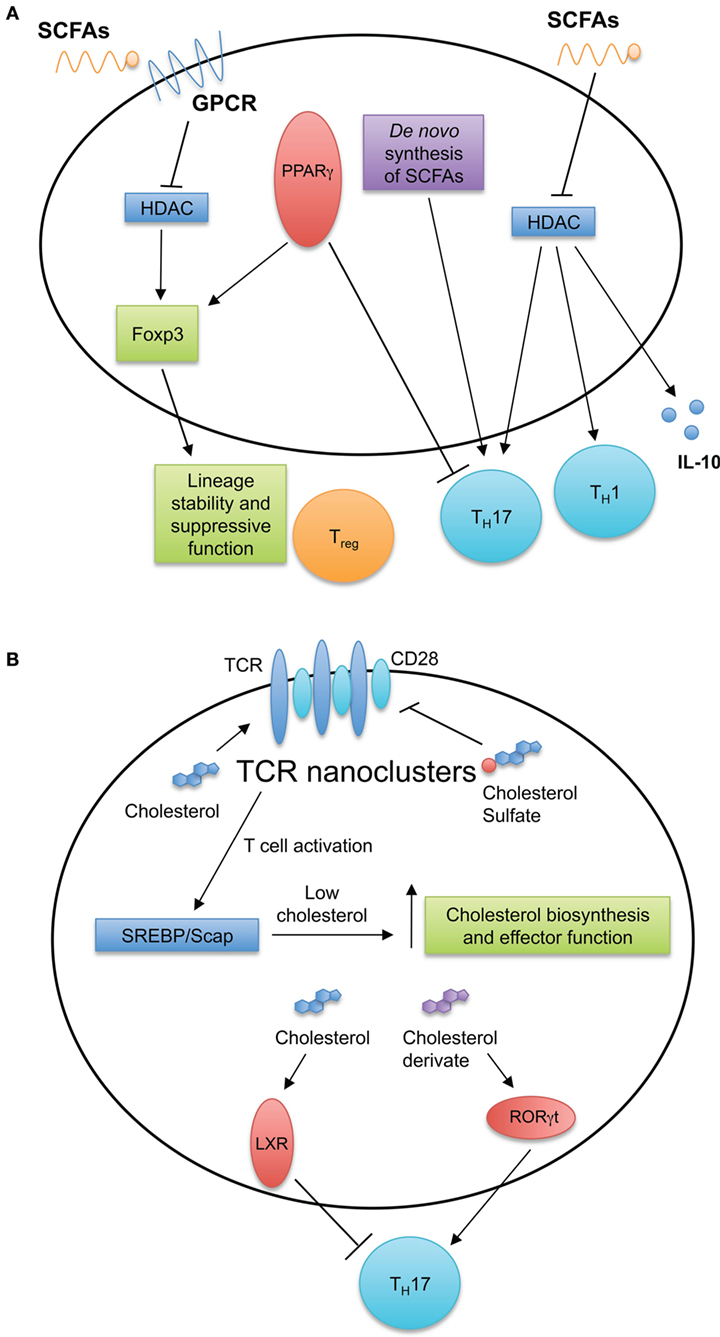
Figure 3. Fatty acid (FA) and cholesterol sensing in T cell immune responses. (A) FA metabolism and sensing serve multiple roles in T cell biology. Short-chain fatty acids (SCFAs) diffuse passively or are transported into cells by G-protein-coupled receptors. SCFAs can inhibit histone deacetylases and induce TH1 and TH17 cell differentiation and IL-10 production. De novo FA synthesis is required for TH17 generation. Intracellular FAs are recognized by peroxisome proliferator-activated receptors (PPARs). Expression of PPARγ is important for visceral adipose tissue Treg cell accumulation and function, while antagonizing PPARγ by chemical drug promotes TH17 development. (B) Cholesterol and cholesterol sulfates regulate T cell receptor (TCR) nanoclustering and signaling within the T cell. After stimulation of TCRs, T cells upregulate the cholesterol biosynthesis pathway through the SREBP/SCAP axis. This increase in cholesterol biosynthesis is important for acquiring T cell effector function. Additionally, intracellular sterols serve as ligands for intracellular receptors and transcription factors, such as liver X receptor (LXR) and RORγt to modulate TH17 development.
Fatty Acids
The basic structure of a FA is a carboxylic acid linked to a saturated or unsaturated aliphatic chain. FAs are categorized broadly into groups based on the length of the aliphatic chain. Short-chain fatty acids (SCFAs) have 2–6 carbons; medium chain fatty acids have 7–12 carbons; and long chain fatty acids (LCFAs) have more than 12 carbons (62). Some FAs are generated within the cell through de novo synthesis; however, many important FAs cannot be synthesized and are obtained only through dietary sources. For instance, when dietary fats and fibers are catabolized in the liver and intestines, respectively, free fatty acids (FFAs) are generated and circulate through the blood (62). Although certain FFAs can diffuse across the plasma membrane into the cytosol, most require transport facilitated by surface receptors. LCFAs and oxidized FAs are transported across the membrane by fatty acid translocase (FAT or CD36) (63). Additionally, several G protein receptors bind FAs of various lengths—GPCR40 (free fatty acid receptor 1) and GPCR120 bind to LCFAs; GPCR84 recognizes MCFAs; GPCR43 (FFA2) and GPCR41 (FFA3) bind to SFCAs (62).
Several studies have revealed roles for SFCAs and their transporters in T cell responses. The breakdown of dietary fibers into acetate, butyrate, and propionate by intestinal bacteria plays an important role in controlling T cell inflammatory responses. For instance, within the intestines, these SCFAs bind the GPCR43 receptor on Treg cells and promote their differentiation and function to maintain intestinal homeostasis (64, 65). Mechanistically, butyrate inhibits histone deacetylase (HDAC) activity, subsequently enhancing histone acetylation in the Foxp3 promoter to promote stable Foxp3 expression (64, 66, 67). Interestingly, these intestine-derived SCFAs also promote the generation of Treg cells in distal tissues (66) and shape inflammatory responses within the central nervous system, kidneys, and lungs (68–71). For example, during an acute Listeria monocytogenes infection, serum levels of acetate increase, which is required for optimal memory CD8+ T cell formation (72). The increase in intracellular acetate enhances glycolytic activity necessary for a rapid recall response by CD8+ T cells (72). Mechanistically, the accumulation of acetate drives GAPDH acetylation to increase glycolytic flux (72). Furthermore, SCFA-mediated inhibition of HDAC activity increases the acetylation of S6K. This process promotes TH1 and TH17 cell differentiation and induces IL-10 production by CD4+ T cells (70). These studies show the pleiotropic functions of SCFAs in immune responses and provide evidence for targeting FA sensing pathways to modulate T cell responses.
Intracellular FAs are also recognized by peroxisome proliferator-activated receptors (PPARs). The PPAR family of nuclear receptors has three isoforms: PPARα, PPARδ, and PPARγ. After recognizing their ligands, PPARs associate with the retinoid X receptor heterodimer, which binds PPAR-responsive elements to promote gene transcription (73). PPARγ is an important regulator of effector T cell functions, as PPARγ-deficient CD4+ T cells cannot proliferate and survive in a lymphopenic environment and hence are less capable of inducing autoimmunity and graft-versus-host disease in mice (74). Antagonizing PPARγ promotes TH17 cell differentiation and increases the severity of EAE (75, 76). Excessive TH17 cell differentiation might also be linked to the role of PPARγ in Treg cell differentiation. Indeed, PPARγ is essential for the maintenance and accumulation of Treg cells in adipose tissue (77). Thus, PPARγ serves as a key regulator of CD4+ T cell function and differentiation.
Cholesterol
To facilitate ligand recognition and initiate efficient signaling, several TCRs must oligomerize into nanoclusters (78). This process is controlled by cholesterol and its derivatives. Cholesterol-facilitated nanoclustering of TCRs enhances the avidity of the TCR toward its ligand (79). Conversely, cholesterol sulfate and cholesterol esters antagonize TCR function by displacing cholesterol and disrupting the formation of nanoclusters (80). Interestingly, cholesterol may serve temporal or spatial roles in TCR signaling. Cholesterol is an allosteric inhibitor of the TCRβ transmembrane domain and hence limits spontaneous TCR activation; however, the spontaneous unbinding of cholesterol in this region switches the conformation from inactive to active, allowing for the transmission of TCR signals (81). Therefore, cholesterol level in the plasma membrane of T cells is important for regulating TCR activity, which may contribute to T cell development and functions under different conditions.
Intracellular concentrations of cholesterol play key roles in both development and effector functions of T cells. Inhibition of the esterification enzyme acetyl-coenzyme A acetyltransferases 1 increases cholesterol content in the plasma membrane of CD8+ T cells, which augments TCR nanoclustering and subsequently increases effector CD8+ T cell functions (82). Furthermore, deletion of ATP-binding cassette sub-family G member 1 (ABCG1), a protein promoting cholesterol efflux from cell, leads to an accumulation of intracellular cholesterol in Treg cells and increase in Treg cell frequency. Mechanistically, loss of ABCG1 and subsequent disruption in intracellular cholesterol distribution decreases mTOR activity and increases STAT5 signaling, thereby promoting Treg cell formation (83). These data demonstrate that intracellular cholesterol levels are essential for T cell function.
Cholesterol and its derivatives can also serve as intracellular ligands to influence T cell differentiation and function. Specifically, cholesterol derivatives control TH17 cell differentiation by modulating liver X receptor (LXR) and retinoic acid responsive-related orphan receptor γt (RORγt) activity. LXR engagement antagonizes TH17 cell development by mediating the binding of sterol response element-binding protein (SREBP)1 to the Il17a locus and blocking the upregulation of aryl hydrocarbon receptor (AHR) necessary to induce Il17a transcription (84). Additionally, many cholesterol biosynthetic intermediates and derivatives are natural ligands for RORγt, the master transcription factor for TH17 cell development (85–87). These derivatives include oxysterol, desmosterol, and many cholesterol intermediates generated between lanosterol and zymosterol biosynthesis. Pharmaceutical or genetic disruption of these metabolites limits TH17 cell generation and ameliorates the severity of EAE (85–87). If and how cholesterol intermediates influence other effector CD4+ T cell differentiation require further investigation.
FA and Cholesterol Biosynthesis
The ability to sense cholesterol and generate de novo cholesterol is also critical for the activation and effector functions of T cells. Cholesterol synthesis is regulated within the cell by the transcription factor, SREBP2, and its chaperone SREBP cleavage activating protein (SCAP), which helps sense intracellular cholesterol levels (88). T cell activation induces the activation of SULT2B1, an oxysterol-metabolizing enzyme, which indirectly enhances SREBP2-dependent cholesterol synthesis by limiting oxysterols that activate LXR. Thus, SULT2B1 limits cholesterol export from the cell by blocking the LXR-dependent upregulation of cholesterol-modifying enzymes and transporters (89). Intrinsic SCAP expression is required for T cell proliferation and effector function (90). While SCAP-deficient T cells develop normally and are capable of undergoing homeostatic proliferation, they do not generate the requisite lipids for cell growth induced by TCR stimulation (90). Whether SCAP deficiency affects TCR nanonclustering is unknown, but early TCR signaling is not impaired in the absence of SCAP despite these cells having reduced total cholesterol.
TCR stimulation-induced lipid biosynthesis is dependent on mTORC1 activation (90). Naïve CD4+ T cells deficient in Raptor, an obligatory component of mTORC1, neither efficiently upregulate the expression of SREBP or other cholesterol-related genes nor appropriately grow and proliferate upon TCR and CD28 co-stimulation. These defects in cholesterol synthesis, along with compromised glycolysis and oxidative phosphorylation, are correlated with a reduced capacity to differentiate into effector CD4+ T cells (91). Raptor-deficient Treg cells also have reduced cholesterol-related gene expression, which severely compromises Treg cell proliferation and suppressive function (92).
Of interest, the energy production and suppressive capacity of Treg cells are also linked to FA oxidation (FAO) (15), similar as memory CD8+ T cells (18). Memory CD8+ T cells fuel FAO via de novo lipid synthesis to support their homeostasis (18, 93). Additionally, de novo FA synthesis is crucial for the regulation of Treg and TH17 cell differentiation. Inhibition of acetyl-CoA carboxylase-1 (ACC1), a key enzyme for FA synthesis, promotes Treg cell and inhibits TH17 cell differentiation. Mechanistically, ACC1 deficiency limits FA synthesis and glycolysis needed for TH17 cell development (94). While the low level of exogenous FA intake may be enough to sustain Treg cells (94), future studies can explore if such interplay between FAO and de novo lipid synthesis and/or cholesterol catabolism contributes to Treg cell functions. Furthermore, identifying and characterizing the physiological function of additional sterol-related metabolites in T cell responses may provide a novel approach to treat inflammatory diseases.
Oxygen Sensing
Oxygen is required for aerobic respiration and mitochondrial energy generation, and the ability for cells to sense and respond to changing oxygen levels promotes their homeostasis. The cellular mechanisms and signaling cascades of oxygen sensing are reviewed elsewhere (95, 96). Reactive oxygen species (ROS), such as peroxides, superoxide, hydroxyl radicals, and singlet oxygen, are byproducts of aerobic metabolism, and the accumulation of ROS causes cellular oxidative stress. ROS plays an important role in regulating T cell responses (97), which will not be discussed. Here, we focus on recent findings on the functions of the oxygen sensing components in T cells (Figure 4).
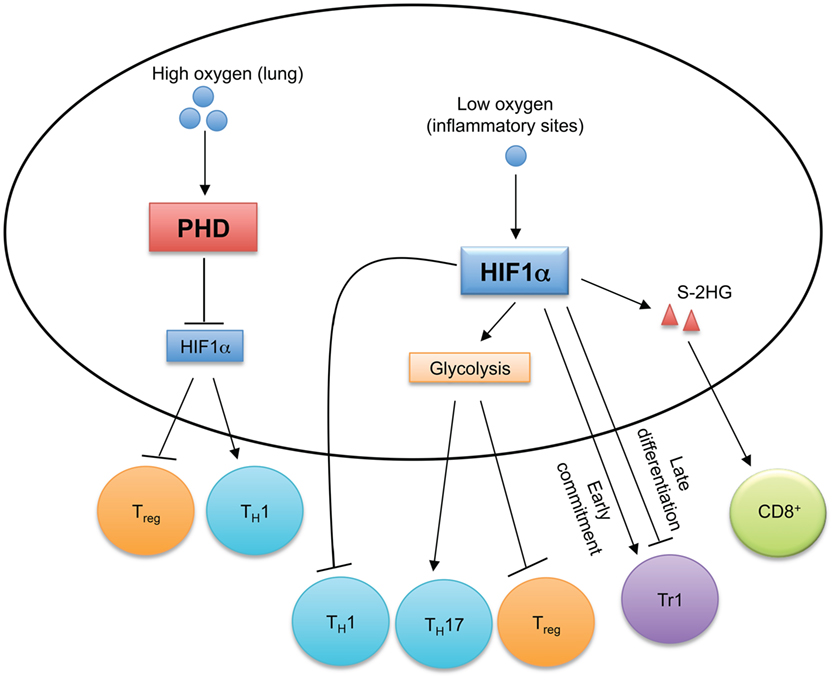
Figure 4. Oxygen sensing orchestrates T cell immune responses. In oxygen-replete environments, such as in the lung, prolyl-hydroxylase domain (PHD) proteins sense high levels of cellular oxygen. Activated PHD proteins establish immune tolerance in the lung by inducing hypoxia-inducible factor 1α (HIF1α) degradation to promote Treg cell and inhibit TH1 cell differentiation. Hypoxia induces HIF1α expression, which regulates the reciprocal generation of TH17 and Treg cells and antagonizes TH1 cell differentiation. HIF1α also temporally regulates Tr1 cell differentiation. HIF1α-dependent upregulation of S-2-hydroxyglutarate (S-2HG) enhances anti-tumor CD8+ T cell responses.
Prolyl-hydroxylase domain (PHD) proteins are expressed in T cells and function as intracellular oxygen sensors (98). In the lungs, high oxygen levels activate PHD proteins in T cells to suppress TH1 responses and promote Treg cell induction (98). However, PHD protein-mediated pulmonary tolerance licenses tumor metastasis to the lungs, and inactivation of PHD proteins promotes anti-tumor T cell responses (98). Mechanistically, activated PHD proteins induce the degradation of HIF1α through post-translational hydroxylation (99), which changes cellular metabolism to promote Treg cell generation and inhibit TH1 cell differentiation (98). Sensing of decreased oxygen levels (termed hypoxia) activates HIF1α, which induces glycolysis and provides T cells with the metabolic plasticity to adapt to environmental stress. T cells are recruited to inflammatory sites that are often hypoxic (100), so how HIF1α controls T cell responses has been investigated. HIF1α is dispensable for T cell activation and early metabolic reprogramming in vitro (42), and HIF1α-deficient T cells also develop normally in vivo (101, 102). However, HIF1α-dependent glycolysis is crucial for promoting TH17 cell differentiation and inhibiting Treg cell generation (102). HIF1α also directly interacts with RORγt and Foxp3 to regulate the reciprocal differentiation of TH17 and Treg cells (101). HIF1α not only suppresses Treg cell generation (101, 102), but augmented HIF1α activity also impairs Treg cell lineage stability and suppressive function in a VHL or Deltex1-deficient background (103, 104). Although increased HIF1α activity is associated with enhanced TH1 cell differentiation in PHD-deficient T cells (98), forced expression of a non-degradable mutant of HIF1α in TH1 cells inhibits IFN-γ expression, and TH1 cell differentiation is enhanced in HIF1α-deficient T cells after in vivo priming (105). Interestingly, HIF1α has differential effects on different stages of type 1 regulatory (Tr1) cell differentiation. Tr1 cells are Foxp3 negative cells that produce IL-10 to control inflammation (106). While HIF1α is required for the metabolic reprogramming during the early commitment of Tr1 cells, its expression reduces at later time points to boost Tr1 cell differentiation by stabilizing AHR expression (107). Augmented HIF1α enhances the effector function of CD8 T cells (108). In the hypoxic tumor microenvironment, the VHL-HIF1α axis regulates CD8+ T cell responses by controlling the accumulation of S-2-hydroxyglutarate (S-2HG) in response to TCR stimulation (109). HIF1α promotes the expression of lactate dehydrogenase A that is the enzymatic source of S-2HG (109). S-2HG regulates the differentiation, in vivo proliferation and persistence of CD8+ T cells, and enhances their anti-tumor efficacy (109). Therefore, by sensing oxygen levels, HIF1α orchestrates T cell responses in given contexts.
Regulation of Energy and Redox Homeostasis
Energy homeostasis is regulated by dynamic changes in cellular catabolic and anabolic pathways, which generate or consume ATP, respectively. Under conditions of energy and nutrient stress, T cells depend on sensors, such as AMPK, and catabolic pathways, such as autophagy, to replenish energy and cellular resources. Moreover, the redox balance mediated by NAD+/NADH is also implicated in T cell responses.
AMP-Activated Protein Kinase
Energy homeostasis is the basis of cell survival and function. AMPK is directly activated by an increased cellular AMP to ATP concentration during energy deprivation. AMPK functions as a fundamental regulator of T cell metabolism to inhibit anabolic pathways and to promote catabolic pathways to preserve cellular energy homeostasis (110). While the activation, regulation, and signaling of AMPK has been reviewed elsewhere (7, 111), here we will briefly discuss the functions of AMPK in T cells (Figure 5). AMPK is activated in glucose-deprived CD8+ T cells experiencing energy stress, such as during infection and inflammation. AMPK is critical for both primary and memory CD8+ T cell responses to infections (112, 113). In nutrient-limiting tumor microenvironment, CD8+ T cells also require AMPK activation for their survival to defend against tumors (114). The pro-survival functions of AMPK could also be linked to the induction of macroautophagy, which is discussed in detail below (115).
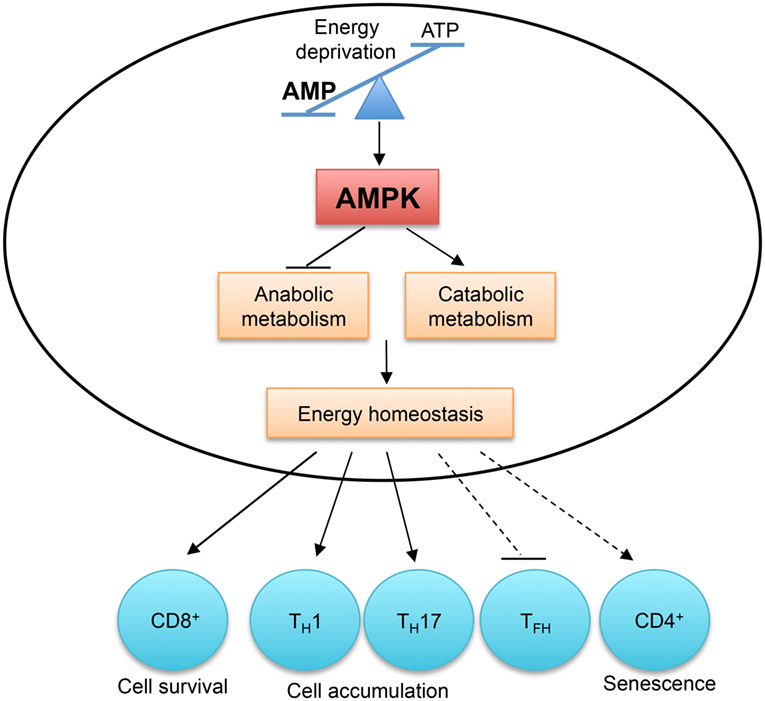
Figure 5. AMP-activated protein kinase (AMPK) and energy sensing dictate T cell immunity. AMPK is activated by the increased AMP to ATP ratio, which indicates energy deprivation. AMPK functions as a master regulator of cellular metabolism to preserve cellular energy homeostasis. AMPK activity is required for CD8+ T cell survival during infection and in tumor microenvironment. The accumulation of TH1 and TH17 cells in the colon is also dependent on AMPK activity. Aberrant AMPK activation is linked to impaired TFH cell generation and CD4+ T cell senescence.
In activated T cells, AMPK is a master regulator of cellular metabolism. It preserves energy homeostasis by suppressing mRNA translation and promoting glutamine-fueled mitochondrial oxidization in response to glucose deprivation (112). The differentiation of TH1 and TH17 cells from naïve CD4+ T cells does not require AMPK activity, but their accumulation and their ability to induce inflammation in the colon are severely impaired in the absence of AMPK (112). However, AMPK activation is detrimental to T cell responses in certain contexts. In response to nutrient stress and DNA damage, active AMPK induces p38-dependent senescence in human CD4+ T cells, and silencing of AMPK restores telomerase activity and proliferation in senescent T cells (116). Aberrant AMPK activation is linked to impaired TFH cell generation in T cells deficient in the ROQUIN RING domain (117). The AMPK agonist, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), inhibits TH17 cell induction and promotes Treg cell generation in vitro (118), but these results should be interpreted cautiously due to the reported off-target effects of AICAR (119). Therefore, by sensing energy stress, AMPK plays a pivotal role in metabolic reprogramming to regulate T cell responses.
NAD+/NADH and Redox Balance
The coenzyme β-nicotinamide adenine dinucleotide (NAD+) and its reduced form, NADH, are critical regulators of T cell biology. Early studies demonstrated that NAD+ induces apoptosis in T cells in a process called NAD+-induced cell death (NICD). When cells die as a result of tissue damage or inflammation, intracellular NAD+ is released into microenvironment, which then activates signaling to induce cell death. In T cells, the accumulation of NAD+ increases ADP-ribosylation mediated by mono-ADP-ribosyltransferase 2 (ART2) (120). The purinergic receptor P2X7 is one substrate of ART2, and its ADP-ribosylation increases its sensitivity for activation by extracellular ATP, which initiates the cell death program (120). Consistent with this observation, elevated NAD+ levels generated during acute inflammation result in ART2-dependent decrease in peripheral CD4+ and CD8+ T cell numbers (121). Interestingly, peripheral T cells are more sensitive to NICD than thymocytes (120), suggesting a more dominant role for NAD+ in regulating peripheral T cell homeostasis. Compared to conventional T cells, Treg cells express higher levels of ART2 and P2X7 and are more sensitive to NICD (122). In vivo NAD+ administration preferentially depletes Treg cells and enhances anti-tumor T cell responses (122). Thus, localized changes in NAD+ levels likely influence the balance between conventional and Treg cell responses.
The flux of electrons via mitochondrial electron transport chain (ETC) regulates NAD+/NADH redox balance important for maintaining mitochondrial function and energy metabolism (123). Furthermore, perturbations in ETC alter NAD+/NADH levels and attenuate T cell proliferation. Recently, mechanisms linking ETC dysfunction and impaired proliferation of Jurkat leukemic T cells were uncovered. Disruption of ETC alters NAD+/NADH redox balance and limits mitochondrial aspartate synthesis, a process linked to purine and pyrimidine biosynthesis and protein synthesis (124). Pyruvate supplementation restores NAD+ levels and increases aspartate aminotransferase (GOT1)-dependent aspartate synthesis (124). However, when GOT1 (encodes for GOT1) is deleted from Jurkat cells, NAD+ levels and aspartate levels are further depleted, leading to increased Jurkat cell arrest following ETC inhibition (124). Thus, aspartate synthesis is limiting for proliferation when ETC is inhibited. The regulatory role of aspartate biosynthesis in primary T cell development and function is unexplored; however, because aspartate synthesis is linked to purine nucleotide biosynthesis important for T cell proliferation (56), this pathway likely plays an important role in these cells.
Recent studies highlight the role of NAD+ in regulating CD4+ T cell differentiation. NAD+ regeneration through mitochondrial respiration is essential for proper lysosome function, which restrains excessive TH1 cell generation both in vivo and in vitro (125). NAD+ treatment blocks the induction of EAE in mice by promoting the generation of immunosuppressive IFN-γ+IL-10+ CD4+ T cells (126). Collectively, these studies highlight the potential for novel NAD+-based immune modulation.
Autophagy
Macroautophagy, hereafter referred to as autophagy, is an evolutionarily conserved cellular process that breaks down endogenous cytosolic materials and organelles (127). Autophagy helps regulate energy consumption and cellular resources during cellular stress. Indeed, amino acid starvation and other energy-stressing conditions induce autophagy, which in part depends upon AMPK and mTORC1 signaling (128). How nutrient availability regulates autophagy in T cells remains elusive. Earlier in vitro studies showed evidence for increased autophagy in T cells after TCR and cytokine stimulation (129–131). However, in vivo, virus-specific CD8+ T cells downregulate autophagy during clonal expansion (132). In contrast, during the contraction phase, autophagy is upregulated in virus-specific CD8+ T cells (132). Furthermore, upregulation of autophagy is critical for CD8+ T cell memory formation, as acute deletion of Atg7 or Atg5 in peripheral CD8+ T cells impairs survival during the contraction phase, resulting in a decreased memory CD8+ T cell pool (132). Thus, the regulation of autophagy during the kinetics of T cell responses is dynamic. The contribution of cellular stress conditions, such as nutrient levels, growth factor withdrawal, energy, and oxidative stress, in regulating autophagy and effector T cell responses remains to be further elucidated.
Recent studies have shown that autophagy regulates CD4+ T cell lineage differentiation and functionality. Treg cells have increased autophagosomes compared to conventional CD4+ T cells (33, 133, 134). Treg cell-specific deletion of genes required for autophagy (Atg7, Atg5, Atg16l1) reduces Treg cell survival, stability of Foxp3 expression and ability to control pro-inflammatory CD4+ T cell responses (33, 133). Furthermore, loss of autophagy in Treg cells aberrantly elevates mTORC1 and c-Myc expression, which enhances glycolysis and limits Treg cell stability and function (33). This is in contrast to autophagy-dependent reactivation of mTORC1 signaling under conditions of sustained nutrient deprivation (135). Interestingly, the microenvironment may alter the role of autophagy in T cells, as Atg16l1-deficient CD4+ T cells had elevated TH2 cell responses selectively within the intestinal environment (133). How autophagy regulates mTORC1 signaling is not completely understood. In Treg cells, autophagy may selectively target PI(3)K-related signaling proteins for degradation (33). In effector T cells, autophagy selectively degrades Bcl-10, affecting the signaling between TCR and NF-κB (136). Interestingly, the CARMA1/Bcl-10/MALT1 complex contributes to the upregulation of ASCT2 expression and glutamine uptake in TCR activated T cells, which controls mTORC1 activation (41). Thus, the role of autophagy in T cells is complex, as it not only regulates cellular degradation in times of nutrient deprivation but also serves to modulate TCR signaling.
Concluding Remarks
While it is clear that the interplay of nutrient sensing, signaling proteins, and cellular metabolism controls T cell functional fitness, it remains an open question as to how T cells sense dynamic nutrient cues in complex inflammatory contexts. Recent studies have begun to identify specific nutrients, nutrient sensors, and nutrient transporters that are crucial for proper T cell immune responses. However, the importance of additional sensors identified in other cellular systems remains to be explored in T cells. For example, SLC38A9 and Castor1 are proteins associated with the lysosomes that directly bind intracellular arginine, leading to downstream regulation of mTORC1 signaling (52–54). Recent studies have also shown that ion sensing regulates T cell immunity (137, 138), but how ion and other nutrients cooperate to control T cell responses remains to be established. One major challenge in studying nutrient and energy sensing is to identify key molecules involved in these processes. The development of cutting-edge, gene-targeting technologies, such as CRISPR, will likely allow us to identify additional molecules that specifically control selective aspects of T cell functions. Furthermore, the complex interplay between nutrient and cellular stress signals should also be explored in the context of cancer, autoimmunity, and infectious diseases to gain a better understanding of how T cell functional responses adapt to different physiological and pathological conditions. A detailed understanding of nutrient sensing in T cells has the potential to be translated into innovative therapies for immune-mediated diseases and cancer.
Author Contributions
JW wrote the manuscript and organized the review. JR and T-LN wrote part of the manuscript. HC edited the manuscript and provided direction.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge N. Chapman, H. Zeng, and Y. Wang for carefully reading and editing the manuscript.
Funding
This work was supported by grants from the National Institutes of Health (NIH AI105887, AI101407, CA176624, and NS064599 to HC), American Asthma Foundation (to HC), and a St. Jude Gephardt Postdoctoral Fellowship (to JW).
References
1. Ellmeier W, Sawada S, Littman DR. The regulation of CD4 and CD8 coreceptor gene expression during T cell development. Annu Rev Immunol (1999) 17:523–54. doi:10.1146/annurev.immunol.17.1.523
2. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12:749–61. doi:10.1038/nri3307
3. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
4. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
5. Walls J, Sinclair L, Finlay D. Nutrient sensing, signal transduction and immune responses. Semin Immunol (2016) 28:396–407. doi:10.1016/j.smim.2016.09.001
6. Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature (2015) 517:302–10. doi:10.1038/nature14190
7. Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell (2015) 161:67–83. doi:10.1016/j.cell.2015.02.041
8. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20:61–72. doi:10.1016/j.cmet.2014.05.004
9. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi:10.1016/S1074-7613(02)00323-0
10. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180:4476–86. doi:10.4049/jimmunol.180.7.4476
11. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111:2101–11. doi:10.1182/blood-2007-06-096297
12. Maekawa Y, Ishifune C, Tsukumo S, Hozumi K, Yagita H, Yasutomo K. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat Med (2015) 21:55–61. doi:10.1038/nm.3758
13. King BC, Esguerra JL, Golec E, Eliasson L, Kemper C, Blom AM. CD46 activation regulates miR-150-mediated control of GLUT1 expression and cytokine secretion in human CD4+ T cells. J Immunol (2016) 196:1636–45. doi:10.4049/jimmunol.1500516
14. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity (2015) 42:1033–47. doi:10.1016/j.immuni.2015.05.024
15. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
16. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity (2016) 45:540–54. doi:10.1016/j.immuni.2016.08.017
17. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125:194–207. doi:10.1172/JCI76012
18. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi:10.1016/j.immuni.2011.12.007
19. Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, et al. Constitutive glycolytic metabolism supports CD8+ T cell effector memory differentiation during viral infection. Immunity (2016) 45:1024–37. doi:10.1016/j.immuni.2016.10.017
20. Cretenet G, Clerc I, Matias M, Loisel S, Craveiro M, Oburoglu L, et al. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci Rep (2016) 6:24129. doi:10.1038/srep24129
21. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol (2013) 14:1064–72. doi:10.1038/ni.2687
22. Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol (2016) 17:364–8. doi:10.1038/ni.3415
23. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi:10.1016/j.cell.2015.08.016
24. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162:1217–28. doi:10.1016/j.cell.2015.08.012
26. Singh R, Green MR. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science (1993) 259:365–8. doi:10.1126/science.8420004
27. Nagy E, Rigby WF. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J Biol Chem (1995) 270:2755–63. doi:10.1074/jbc.270.6.2755
28. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi:10.1016/j.cell.2013.05.016
29. Basu S, Hubbard B, Shevach EM. Foxp3-mediated inhibition of Akt inhibits Glut1 (glucose transporter 1) expression in human T regulatory cells. J Leukoc Biol (2015) 97:279–83. doi:10.1189/jlb.2AB0514-273RR
30. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol (2016) 17:1459–66. doi:10.1038/ni.3577
31. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol (2015) 16:178–87. doi:10.1038/ni.3076
32. Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16:188–96. doi:10.1038/ni.3077
33. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol (2016) 17:277–85. doi:10.1038/ni.3365
34. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16:1174–84. doi:10.1038/ni.3269
35. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity (2016) 44:406–21. doi:10.1016/j.immuni.2016.01.028
36. Cobbold SP, Adams E, Farquhar CA, Nolan KF, Howie D, Lui KO, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A (2009) 106:12055–60. doi:10.1073/pnas.0903919106
37. Fotiadis D, Kanai Y, Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med (2013) 34:139–58. doi:10.1016/j.mam.2012.10.007
38. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14:500–8. doi:10.1038/ni.2556
39. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2010) 185:1037–44. doi:10.4049/jimmunol.0903586
40. Hayashi K, Jutabha P, Endou H, Sagara H, Anzai N. LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol (2013) 191:4080–5. doi:10.4049/jimmunol.1300923
41. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi:10.1016/j.immuni.2014.04.007
42. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
43. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136:521–34. doi:10.1016/j.cell.2008.11.044
44. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal (2015) 8:ra97. doi:10.1126/scisignal.aab2610
45. Chapman NM, Chi H. mTOR links environmental signals to T cell fate decisions. Front Immunol (2015) 5:686. doi:10.3389/fimmu.2014.00686
46. Bronte V, Zanovello P. Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol (2005) 5:641–54. doi:10.1038/nri1668
47. Rodriguez PC, Quiceno DG, Ochoa AC. l-Arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi:10.1182/blood-2006-06-031856
48. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi:10.1158/0008-5472.CAN-04-0465
49. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. l-Arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol (2003) 171:1232–9. doi:10.4049/jimmunol.171.3.1232
50. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res (2005) 65:3044–8. doi:10.1158/0008-5472.CAN-04-4505
51. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. l-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–42.e813. doi:10.1016/j.cell.2016.09.031
52. Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature (2016) 536:229–33. doi:10.1038/nature19079
53. Rebsamen M, Pochini L, Stasyk T, de Araújo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature (2015) 519:477–81. doi:10.1038/nature14107
54. Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science (2015) 347:188–94. doi:10.1126/science.1257132
55. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454. doi:10.1126/science.1242454
56. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab (2017) 25(2):482. doi:10.1016/j.cmet.2017.01.014
57. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22:633–42. doi:10.1016/j.immuni.2005.03.013
58. Van de Velde LA, Guo XJ, Barbaric L, Smith AM, Oguin TH III, Thomas PG, et al. Stress kinase GCN2 controls the proliferative fitness and trafficking of cytotoxic T cells independent of environmental amino acid sensing. Cell Rep (2016) 17:2247–58. doi:10.1016/j.celrep.2016.10.079
59. Yan Y, Zhang GX, Gran B, Fallarino F, Yu S, Li H, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol (2010) 185:5953–61. doi:10.4049/jimmunol.1001628
60. Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, et al. IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol (2009) 183:2475–83. doi:10.4049/jimmunol.0900986
61. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood (2009) 113:6102–11. doi:10.1182/blood-2008-12-195354
62. Alvarez-Curto E, Milligan G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol (2016) 114:3–13. doi:10.1016/j.bcp.2016.03.017
63. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr (2014) 34:281–303. doi:10.1146/annurev-nutr-071812-161220
64. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi:10.1126/science.1241165
65. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi:10.1038/nature12721
66. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
68. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity (2015) 43:817–29. doi:10.1016/j.immuni.2015.09.007
69. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med (2014) 20:159–66. doi:10.1038/nm.3444
70. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol (2015) 8:80–93. doi:10.1038/mi.2014.44
71. Park J, Goergen CJ, HogenEsch H, Kim CH. Chronically elevated levels of short-chain fatty acids induce T cell-mediated ureteritis and hydronephrosis. J Immunol (2016) 196:2388–400. doi:10.4049/jimmunol.1502046
72. Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, et al. Memory CD8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity (2016) 44:1312–24. doi:10.1016/j.immuni.2016.03.016
73. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med (2013) 19:557–66. doi:10.1038/nm.3159
74. Housley WJ, Adams CO, Vang AG, Brocke S, Nichols FC, LaCombe M, et al. Peroxisome proliferator-activated receptor gamma is required for CD4+ T cell-mediated lymphopenia-associated autoimmunity. J Immunol (2011) 187:4161–9. doi:10.4049/jimmunol.1101731
75. Li B, Reynolds JM, Stout RD, Bernlohr DA, Suttles J. Regulation of Th17 differentiation by epidermal fatty acid-binding protein. J Immunol (2009) 182:7625–33. doi:10.4049/jimmunol.0804192
76. Raikwar HP, Muthian G, Rajasingh J, Johnson C, Bright JJ. PPARgamma antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J Neuroimmunol (2005) 167:99–107. doi:10.1016/j.jneuroim.2005.06.026
77. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature (2012) 486:549–53. doi:10.1038/nature11132
78. Pageon SV, Tabarin T, Yamamoto Y, Ma Y, Bridgeman JS, Cohnen A, et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc Natl Acad Sci U S A (2016) 113:E5454–63. doi:10.1073/pnas.1607436113
79. Molnár E, Swamy M, Holzer M, Beck-García K, Worch R, Thiele C, et al. Cholesterol and sphingomyelin drive ligand-independent T-cell antigen receptor nanoclustering. J Biol Chem (2012) 287:42664–74. doi:10.1074/jbc.M112.386045
80. Wang F, Beck-Garcia K, Zorzin C, Schamel WW, Davis MM. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat Immunol (2016) 17:844–50. doi:10.1038/ni.3462
81. Swamy M, Beck-Garcia K, Beck-Garcia E, Hartl FA, Morath A, Yousefi OS, et al. A cholesterol-based allostery model of T cell receptor phosphorylation. Immunity (2016) 44:1091–101. doi:10.1016/j.immuni.2016.04.011
82. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531:651–5. doi:10.1038/nature17412
83. Cheng HY, Gaddis DE, Wu R, McSkimming C, Haynes LD, Taylor AM, et al. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J Clin Invest (2016) 126:3236–46. doi:10.1172/JCI83136
84. Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest (2011) 121:658–70. doi:10.1172/JCI42974
85. Santori FR, Huang P, van de Pavert SA, Douglass EF Jr, Leaver DJ, Haubrich BA, et al. Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell Metab (2015) 21:286–97. doi:10.1016/j.cmet.2015.01.004
86. Hu X, Wang Y, Hao L-Y, Liu X, Lesch CA, Sanchez BM, et al. Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol (2015) 11:141–7. doi:10.1038/nchembio.1714
87. Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, et al. Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proc Natl Acad Sci U S A (2014) 111:12163–8. doi:10.1073/pnas.1322807111
88. Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab (2012) 16:414–9. doi:10.1016/j.cmet.2012.09.002
89. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell (2008) 134:97–111. doi:10.1016/j.cell.2008.04.052
90. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14:489–99. doi:10.1038/ni.2570
91. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56. doi:10.1016/j.immuni.2013.09.015
92. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499:485–90. doi:10.1038/nature12297
93. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41:75–88. doi:10.1016/j.immuni.2014.06.005
94. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20:1327–33. doi:10.1038/nm.3704
95. Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev (2004) 18:2183–94. doi:10.1101/gad.1243304
96. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5:343–54. doi:10.1038/nrm1366
97. Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol (2013) 13:349–61. doi:10.1038/nri3423
98. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell (2016) 166:1117–31.e1114. doi:10.1016/j.cell.2016.07.032
99. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi:10.1126/science.1059796
100. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med (2011) 364:656–65. doi:10.1056/NEJMra0910283
101. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
102. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
103. Lee JH, Elly C, Park Y, Liu YC. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42:1062–74. doi:10.1016/j.immuni.2015.05.016
104. Hsiao HW, Hsu TS, Liu WH, Hsieh WC, Chou TF, Wu YJ, et al. Deltex1 antagonizes HIF-1alpha and sustains the stability of regulatory T cells in vivo. Nat Commun (2015) 6:6353. doi:10.1038/ncomms7353
105. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting edge: hypoxia-inducible factor 1 negatively regulates Th1 function. J Immunol (2015) 195:1372–6. doi:10.4049/jimmunol.1402552
106. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
107. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med (2015) 21:638–46. doi:10.1038/nm.3868
108. Mascanfroni AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol (2013) 14:1173–82. doi:10.1038/ni.2714
109. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Veliça P, et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature (2016) 540:236–41. doi:10.1038/nature20165
110. Yang K, Chi H. AMPK helps T cells survive nutrient starvation. Immunity (2015) 42:4–6. doi:10.1016/j.immuni.2014.12.029
111. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol (2012) 13:251–62. doi:10.1038/nrm3311
112. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity (2015) 42:41–54. doi:10.1016/j.immuni.2014.12.030
113. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA, et al. AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol (2013) 43:889–96. doi:10.1002/eji.201243008
114. Rao E, Zhang Y, Zhu G, Hao J, Persson XM, Egilmez NK, et al. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget (2015) 6:7944–58. doi:10.18632/oncotarget.3501
115. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi:10.1038/ncb2152
116. Lanna A, Henson SM, Escors D, Akbar AN. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB 1 drives the senescence of human T cells. Nat Immunol (2014) 15:965–72. doi:10.1038/ni.2981
117. Ramiscal RR, Parish IA, Lee-Young RS, Babon JJ, Blagih J, Pratama A, et al. Attenuation of AMPK signaling by ROQUIN promotes T follicular helper cell formation. Elife (2015) 4:e08698. doi:10.7554/eLife.08698
118. Gualdoni GA, Mayer KA, Göschl L, Boucheron N, Ellmeier W, Zlabinger GJ. The AMP analog AICAR modulates the Treg/Th17 axis through enhancement of fatty acid oxidation. FASEB J (2016) 30:3800–9. doi:10.1096/fj.201600522R
119. Rao E, Zhang Y, Li Q, Hao J, Egilmez NK, Suttles J, et al. AMPK-dependent and independent effects of AICAR and compound C on T-cell responses. Oncotarget (2016) 7:33783–95. doi:10.18632/oncotarget.9277
120. Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, et al. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity (2003) 19:571–82. doi:10.1016/S1074-7613(03)00266-8
121. Adriouch S, Hubert S, Pechberty S, Koch-Nolte F, Haag F, Seman M. NAD+ released during inflammation participates in T cell homeostasis by inducing ART2-mediated death of naive T cells in vivo. J Immunol (2007) 179:186–94. doi:10.4049/jimmunol.179.1.186
122. Hubert S, Rissiek B, Klages K, Huehn J, Sparwasser T, Haag F, et al. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J Exp Med (2010) 207:2561–8. doi:10.1084/jem.20091154
123. Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab (2012) 23:420–8. doi:10.1016/j.tem.2012.06.005
124. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162:540–51. doi:10.1016/j.cell.2015.07.016
125. Baixauli F, Acín-Pérez R, Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez E, et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab (2015) 22:485–98. doi:10.1016/j.cmet.2015.07.020
126. Tullius SG, Biefer HR, Li S, Trachtenberg AJ, Edtinger K, Quante M, et al. NAD+ protects against EAE by regulating CD4+ T-cell differentiation. Nat Commun (2014) 5:5101. doi:10.1038/ncomms6101
127. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi:10.1038/nature09782
128. Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol (2012) 32:2–11. doi:10.1128/MCB.06159-11
129. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med (2007) 204:25–31. doi:10.1084/jem.20061303
130. Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol (2010) 185:7349–57. doi:10.4049/jimmunol.1000576
131. Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol (2006) 177:5163–8. doi:10.4049/jimmunol.177.8.5163
132. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15:1152–61. doi:10.1038/ni.3025
133. Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife (2016) 5:e12444. doi:10.7554/eLife.12444
134. Le Texier L, Lineburg KE, Cao B, McDonald-Hyman C, Leveque-El Mouttie L, Nicholls J, et al. Autophagy-dependent regulatory T cells are critical for the control of graft-versus-host disease. JCI Insight (2016) 1:e86850. doi:10.1172/jci.insight.86850
135. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature (2010) 465:942–6. doi:10.1038/nature09076
136. Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity (2012) 36:947–58. doi:10.1016/j.immuni.2012.04.008
137. Feske S, Skolnik EY, Prakriya M. Ion channels and transporters in lymphocyte function and immunity. Nat Rev Immunol (2012) 12:532–47. doi:10.1038/nri3233
Keywords: glucose, amino acid, lipid, oxygen, energy, T cell response
Citation: Wei J, Raynor J, Nguyen TL and Chi H (2017) Nutrient and Metabolic Sensing in T Cell Responses. Front. Immunol. 8:247. doi: 10.3389/fimmu.2017.00247
Received: 05 January 2017; Accepted: 20 February 2017;
Published: 09 March 2017
Edited by:
Ping-Chih Ho, University of Lausanne, SwitzerlandReviewed by:
Chia-Lin Hsu, National Yang-Ming University, TaiwanMaria Mittelbrunn, Hospital Universitario 12 De Octubre, Spain
Copyright: © 2017 Wei, Raynor, Nguyen and Chi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbo Chi, aG9uZ2JvLmNoaUBzdGp1ZGUub3Jn