 Christian Chabannon1,2*
Christian Chabannon1,2* Bechara Mfarrej1,2
Bechara Mfarrej1,2 Sophie Guia3
Sophie Guia3 Sophie Ugolini3
Sophie Ugolini3 Raynier Devillier2
Raynier Devillier2 Didier Blaise2
Didier Blaise2 Eric Vivier3,4Boris Calmels1,2
Eric Vivier3,4Boris Calmels1,2
- 1CBT-1409: INSERM, Aix Marseille Univ, Institut Paoli-Calmettes, AP-HM, Marseille, France
- 2CRCM: INSERM, CNRS, Aix Marseille Univ, Institut Paoli-Calmettes, CRCM, Marseille, France
- 3UM2, INSERM, Centre d’Immunologie de Marseille-Luminy, U1104, CNRS UMR7280, Aix-Marseille University, Marseille, France
- 4Laboratoire d’Immunologie, Hôpital de la Conception, Assistance Publique – Hôpitaux de Marseille, Aix-Marseille University, Marseille, France
Natural Killer (NK) cells are innate lymphoid cells (ILC) with cytotoxic and regulatory properties. Their functions are tightly regulated by an array of inhibitory and activating receptors, and their mechanisms of activation strongly differ from antigen recognition in the context of human leukocyte antigen presentation as needed for T-cell activation. NK cells thus offer unique opportunities for new and improved therapeutic manipulation, either in vivo or in vitro, in a variety of human diseases, including cancers. NK cell activity can possibly be modulated in vivo through direct or indirect actions exerted by small molecules or monoclonal antibodies. NK cells can also be adoptively transferred following more or less substantial modifications through cell and gene manufacturing, in order to empower them with new or improved functions and ensure their controlled persistence and activity in the recipient. In the present review, we will focus on the technological and regulatory challenges of NK cell manufacturing and discuss conditions in which these innovative cellular therapies can be brought to the clinic.
Introduction
Cellular therapies are, nowadays, increasing in numbers and in diversity. Manufacturing of various types of immune cells is likely to provide additional therapeutic resources and complete the portfolio of immunotherapies, along with chemical molecules and engineered monoclonal antibodies. It is envisioned that combination of these different medicinal products, tailored to disease characteristics as well as to the host (immune) environment (1), will contribute to precision medicine and expectedly to higher rates of success in the cure of a variety of health disorders, including, but not restricted to, cancers. Among immune effectors amenable to cell and genetic manipulation prior to adoptive transfer, natural killer (NK) cells present with appealing biological characteristics. NK cells are innate lymphoid cells (ILC) and contribute to innate immunity (2). Their activities are regulated through the biological modulation of a large array of both inhibitory and activating receptors, including killer cell immunoglobulin-like receptors (KIR), NKp44, and NKp46. These receptors do not bind specific antigens on target cells as do T cells, but rather molecules induced by cellular stress that provide an activating signal, or human leukocyte antigen (HLA) molecules that predominantly provide inhibitory signals; already published material provides in-depth description of these pathways. Here, we review the existing literature that describes the rationale for various technological approaches to NK cell manufacturing, either autologous or allogeneic, as a prerequisite to adoptive transfer and clinical evaluation of these peculiar populations of immune effectors.
Medical Applications of NK Cell Manufacturing and Adoptive Transfer
Autologous NK Cells
Adoptive transfer of NK cells engineered to express new or augmented functions represent an interesting avenue for the treatment of various high-risk malignancies in which conventional options have failed (Figure 1). Examples include B-chronic lymphocytic leukemia (B-CLL) (3), multiple myeloma (4), and also tumors of non-hematopoietic origin such as breast cancer (5), melanoma, or renal cell carcinoma (6).
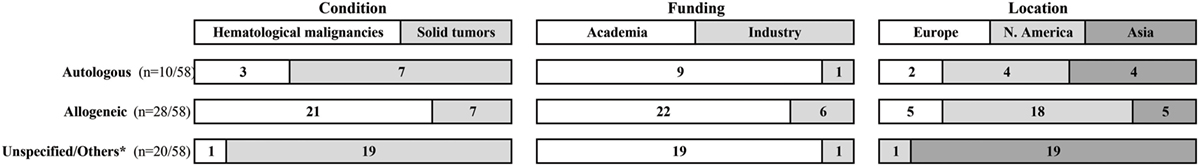
Figure 1. Ongoing clinical trials that evaluate NK cell-based cellular therapies. Data were collected at https://clinicaltrials.gov/ in September 2016, using the search terms: “recruiting studies,” “NK,” or “natural killer” in the “Title” field. Raw data are available in Data Sheet S1 in Supplementary Material. *Unspecified = 18 studies with no designation whether NK cells are autologous or allogeneic; others = 1 study tests NK cell line and 1 study compares autologous to allogeneic NK cells.
Early studies reporting the adoptive transfer of CD56+ bead-selected autologous NK cells into patients with metastatic cancers, in combination with high doses of IL-2 demonstrated feasibility yet poor clinical efficacy (7). Patients experienced severe toxic side effects due to the high doses of IL-2. In addition to activation-induced NK cell death, NK cell function may have been inhibited due to regulatory T cell expansion in response to high IL-2 doses (8, 9). Reducing the daily IL-2 doses after NK cell transfer resulted in limited clinical success (10). Several strategies are under investigation to overcome this hurdle [reviewed in Ref. (11)]. Another mechanism by which autologous NK cells are inhibited is by self-HLA molecules. Therefore “releasing the brakes” with anti-KIR antibodies such as Lirilumab® that targets the inhibitory KIR receptors on NK cells could be one approach (12–14).
Allogeneic NK Cells
A number of arguments support an important role for donor-derived NK cells in the context of allogeneic hematopoietic stem cell transplantation (allo-HSCT). Following the administration of either myelo-ablative (15) or reduced-intensity (15–17) conditioning regimen and allo–HSCT, the rapid reconstitution of high numbers of circulating and phenotypically defined NK cells is associated with better clinical outcome. The recovery of various functions for donor-derived cells may be further modulated and improved in vivo with additional intervention (18).
Transplantation of high doses of immune-selected CD34+ cells collected from haploidentical donors after myelo-ablative conditioning regimen has provided a setting which demonstrates that “KIR-incompatibility” was associated with lower incidence of disease relapses, at least for AML (19). Transplantation of T-replete marrow or blood cell grafts obtained from haploidentical donors, using modified immune-suppressive conditioning regimen such as those including posttransplant cyclophosphamide, represent a more widely applicable procedure, in which to further explore the potential contribution of alloreactive NK cells in posttransplant clinical events. Unexpectedly, a recently published report suggests that, in this context, the presence of recipient class I ligands to donor KIR receptors confers some protection to the recipient against leukemia relapse, an observation that needs further confirmation and would imply a role for killer activating receptors (KAR) as much as for KIR (20). The role of alloreactive NK cells remains more elusive in the context of HSCT performed from other categories of donors. Expression of specific KIR receptors in HLA-matched unrelated donors was demonstrated to produce superior or inferior clinical outcomes in recipients, depending on donor–recipient combinations (21–23).
Adoptive transfer of allogeneic NK cells either with a stem cell graft ex vivo depleted of immune effectors or as a substitute to posttransplant “donor lymphocyte infusions” (DLIs) is thus appealing as a way to improve engraftment, immune reconstitution, and antitumor activity with reduced chances of triggering graft-versus-host disease (GVHD) (24). Results of a small number of clinical trials have been reported so far, demonstrating the feasibility of manufacturing allogeneic NK cells from matched related, matched unrelated, or mostly from haploidentical donors (25–29). Although allogeneic NK cell infusions were generally reported as safe, a recent publication describes the clinical outcome of a small cohort of pediatric patients treated for non-hematological high-risk malignancies and a high proportion of aGVHD triggered by HLA-matched donor-derived NK cells (30). Mostly, these limited clinical results suggest that additional improvements are needed either during the ex-vivo manufacturing process (31) or after infusion of manufactured NK cells (25) to improve long-term persistence and activity in vivo.
Factors Affecting NK Cell Product Manufacturing
Many variables contribute to an efficient NK cell generation protocol (Table 1). Donor–recipient combinations, the source of starting material and culture conditions are factors that must be carefully selected to optimize the manufacturing process and potentially the clinical efficacy of the resulting medicinal product upon administration to the recipient.

Table 1. Factors affecting the ouctome of the manufacturing process of NK cell-based medicinal products.
Donor Selection
In the setting of allogeneic NK-DLI, donor selection can affect the clinical outcome of NK cell therapy, since certain KIR, HLA, and FcγR polymorphisms influence NK cell function (32, 33). KIR typing can be genotypic, classifying donors on the basis of gene expression of activating and inhibitory KIR (34), thereby assigning them scores to select “preferable donors” (35–37). Additionally, KIR typing can be phenotypic, assessing surface protein expression of KIRs (38), adding another layer of complexity to the selection of “preferable donors.” KIR “allele-typing” is a recent addition to the donor selection algorithm, whereby alleles that possess better functional properties (stronger licensing capability and more durable surface expression upon ligand binding) are favored (22, 39). Typing FcγR polymorphism is relevant in NK cell therapy settings that use monoclonal antibodies to enhance NK cell activation and consequently empower their antibody-dependent cytotoxicity (ADCC) properties (40, 41). All these strategies have helped define “preferable donor” profiles.
Source of Cells
Natural killer cell therapies can be manufactured from a variety of sources: these include peripheral blood, either steady-state or taking advantage of apheresis performed to collect hematopoietic stem and progenitor cells mobilized with growth factors such as granulocyte colony-stimulating factor (G-CSF), bone marrow, and cord blood.
Peripheral Blood Mononuclear Cells
Peripheral blood mononuclear cells (PBMC) can be collected in large numbers using apheresis. Nowadays, it is the preferred source for allo-HSCT; donor apheresis is collected after receiving a mobilization treatment that increases the percentage and number of circulating progenitor and stem cells (as evaluated by the number of circulating CD34+ cells); G-CSF is the only marketed agent for CD34+ cell mobilization that can be used in donors; the use of other mobilizing agents such as acutely myelo-suppressive drugs or plerixafor is restricted to patients undergoing autologous collection. Since CD34+ cells represent only a small proportion of collected PBMC, these collected cell products may also represent a source of immune effectors, and thus either an alternative to PBMC collected in homeostatic conditions for standard DLI or a starting material for further immune cell manufacturing, including NK cell manufacturing. One caveat to this approach is that studies looking at the effects of G-CSF on NK cell function have produced controversial results; some studies suggest minimal consequences (42, 43), while others suggest significant changes (44).
Non-mobilized apheresis products contain 5–15% NK cells. To isolate NK cells, the strategy commonly used is CD3+ cell depletion of PBMC, followed by CD56+ cell enrichment using immune-magnetic bead separation with medical devices and clinical-grade reagents.
Bone Marrow
Since bone marrow is nowadays used as the source of stem cells for a minority of recipients – in part due to the increased resource needed for the logistics of BM collection compared to aphereses – there is little preclinical or clinical experience in the manufacturing of NK cells from this starting material.
Cord Blood Cells
The use of cord blood (CB) as a source of stem cells has raised great hope in the field 30 years ago, when the first clinical transplants were reported (45). Cord blood unit (CBU) can be used even when not fully matched to the recipient, offering the opportunity to identify a “donor” even for patients who had no HLA-matched related or unrelated donor. CB transplantation is nowadays facing tough competition from the rapidly emerging field of related haploidentical transplantation, and is further hampered by the lengthy immune reconstitution and the lack of possibility to use pre-emptive or curative DLI posttransplant.
However, more than 600,000 CBU that are stored in public banks worldwide – not to mention the unknown cumulative number of CBU preserved in private CB banks – represent a unique source of human material to start with the manufacturing process. Indeed, preclinical validation studies reported the production of significant numbers of functional NK cells either from a complete CBU (46) or even from a minute sample of a CBU (47), the latter opening the way to posttransplant immune cellular therapies in recipients of CBU transplantation while the former offers the promise of “off-the-shelf” allogeneic NK cellular therapies.
Initial attempts to use CD56+ bead-selected NK cells from CB followed by culture on mesenchymal stromal cells in the presence of cytokines resulted in modest yields incompatible with therapeutic needs (48). Starting from immune-selected CB CD34+ cells and using refined protocols produced more interesting results (49–51). Such efforts culminated in a novel good manufacturing practice (GMP)-compliant technique that mimics the extracellular bone marrow environment: stromal cell-free/serum-free medium, heparin, and cytokine supplements (52, 53). Consequently, Glycostem Therapeutics (Oss, the Netherlands) and Radboud University Medical Center (Nijmegen, the Netherlands) are currently conducting a Phase I/II clinical trial in elderly AML patients using NK cell products generated using this method (CCMO nr. NL31699 and Dutch Trial Register nr. 2818) (see Figure 1 and Data Sheet S1 in Supplementary Material for a list of ongoing clinical trials).
ES and iPS Cells
Manufacturing of clinical-grade NK cells from either embryonic (ES) or induced pluripotent stem (iPS) cells nowadays appear as a futuristic option, although preclinical demonstrations that NK cells can be differentiated from these sources of pluripotent stem cells were already published (54–56). For iPS cells, several factors affect the pluripotency and differentiation abilities of reprogramed cells: the choice of target donor somatic cell type and the reprograming protocol, including the nature and combination of genes as well as the method used to deliver transcription factors into somatic cells (57).
An important step in the specific hematopoietic lineage-differentiating protocols starting from ES or iPS cells is the generation of CD34+ hematopoietic precursors, particularly CD34+CD45+ cells, preferred for their high content in hematopoietic progenitors (58). A 30-day culture protocol of sorted ES cells-derived CD34+ cells together with feeder cells (murine fetal-liver-derived stromal cell line) and cytokines generates NK cells with typical maturation markers and target cell lysis capabilities (58, 59). Another expansion method has been described for ES or iPS cells, using an embryonic body assay followed by culture with feeder cells and cytokines (60, 61).
Similar to what has been mentioned for CBU, progress in the development of safe, efficient, and standardized clinical-grade manufacturing protocols will offer an opportunity to develop off-the-shelf personalized and non-immunogenic cellular therapies.
NK Cell Lines
Ex vivo-expanded primary NK cells persist in vivo for short periods of time after adoptive transfer. In an attempt to take advantage of the long lifetime of established cell lines, several groups have evaluated their therapeutic potential. Although other cell lines exist (NKG, YT, NK-YS, YTS cells, HANK-1, and NKL cells), the NK-92 cell line (NantKWest Inc., Culver City, CA, USA) characterized by good cytotoxicity and expansion kinetics (62, 63) has been predominantly evaluated in preclinical investigations and clinical trials (NCT00900809 and NCT00990717) (64). It has been tested in a small number of clinical contexts, yet with minimal efficacy (65–67). Recently, chimeric antigen receptor (CAR) modification by gene transfer for NK cells has opened a new avenue to explore (68, 69). NK cell lines represent a more homogeneous population for CAR modification, compared to peripheral blood NK cells; however, this advantage is largely offset by the need to additionally transfect CD16 to gain ADCC function and the necessary irradiation before infusion for safety reasons, rendering them unable to expand in vivo. Choice of the CAR construct adds another layer of complexity (69).
Culture Conditions: Medium, Cytokines, and Cell Culture Systems
As already described, NK cells are generally isolated through immune-selection techniques, using the canonical CD3−/CD56+ phenotype (42, 70), then cultured for functional activation and possibly expansion. Furthermore, NK cells can be genetically engineered to express natural or chimeric molecules empowering them with improved immune functions (5, 64).
Expansion and activation of potent cytotoxic NK cells require several signals for survival, proliferation, and activation. Culture conditions, thus, incorporate media and serum supplements, together with clinical-grade cytokines, monoclonal antibodies, or other soluble molecules, and possibly native or engineered cell feeders. Culture conditions can further be improved through the substitution of bioreactors to static conditions (Table 1).
As already mentioned, and since most protocols that use only cytokines result in limited NK cell expansion, the introduction of feeder cells in the culture protocol has been extensively tested. Feeder cells provide additional stimulatory signals necessary for NK cell proliferation. Monocytes provide humoral signals and cell-to-cell contacts hence can serve as feeder cells (71); irradiated autologous PBMC have been used as feeder cells to produce sufficient numbers of NK cells with acceptable purity (6, 72, 73). Alternatively, irradiated allogeneic cells have been evaluated: Epstein–Barr virus-transformed lymphoblastoid B cell lines (EBV-TM-LCL), K562 cells (leukemic cell line) engineered to express a membrane-bound form of IL-15 fused to the T-cell receptor CD8α and the 41BB ligand (74–78), or K562 cells transduced with IL-21 (79). Such feeder cells proved effective in preclinical validation of the production of clinically relevant numbers of NK cells, however, raise regulatory issues when it comes to manufacture medicinal products.
Since the presence of residual feeder cells in the final product is of major concern for clinical applications (80), alternative approaches are evaluated as substitutes. Anti-CD3 (OKT3) antibodies in addition to IL-2, with or without IL-15, produced substantial although lower fold expansion of CD3−/CD56+-enriched cells (81–86) than protocols that use feeder cells.
Expansion strategies of clinical-grade NK cells usually require 7–21 days of culture; up to 28 days of culture have been reported (87). There is an incentive to substitute animal or human serum-replete medium with animal and human component-free medium. The most commonly used media for CB-derived NK cells is glycostem basal growth medium (Clear Cell Technologies, Beernem, Belgium), preferred for being free of animal-derived components (52, 53). For PBMC-derived NK cells, preferred media are: X-Vivo™ serum-free media (Biowhittaker, Verviers, Belgium), AIM V® serum-free medium (Thermo Fisher Scientific, Grand Island, NY, USA), Stem Cell Growth medium (Cell Genix, Freiburg, Germany), or complete Roswell Park Memorial Institute 1640 (Biowhittaker, Verviers, Belgium).
Media supplements still being used by some groups include GMP-grade human AB serum, pooled human AB plasma, or fetal bovine serum (FBS). GMP-grade cytokines (recombinant human IL-2 and IL-15), antibodies (anti-CD3-OKT3), and other ancillary reagents (nicotinamide-NAM) commonly serve as medium supplements for PBMC-derived NK cell generation. Additional growth factors and cytokines are necessary for CB-derived NK cells since the starting material is commonly CD34+ stem cells [stem cell factor (SCF), IL-7, IL-15, IL-2, IL-6, flt3 ligand (Flt3-L), thrombopoietin (TPO), G-CSF, low molecular weight heparin (LMWH), granulocyte-macrophage colony-stimulating factor (GM-CSF), leukemia inhibitory factor (LIF), MIP1α].
In addition to using T75 cell culture flasks, several groups have used culture bags (Baxter LifeCell® or VueLife®) (76, 83). On larger scales, gas-permeable static cell culture (G-Rex®) flasks (Wilson Wolf Manufacturing, New Brighton, MN, USA) (78) or WAVE Bioreactor™ (GE Healthcare Life Sciences, Chicago, IL, USA) (83) served as expansion platforms.
Amplitude of NK Cells Expansion and Definition of an Optimal Cell Dose
Numbers of infused NK cells in clinical trials typically range from 5 to 50 × 106 NK cells/kg, but infusion of as many as 108 NK cells/kg has been reported (88). Based on percentages of NK cells in the starting materials, manufacturing the higher doses implies significant expansion during in vitro cultures. This raises a practical issue, since, in the absence of feeder cells, NK cells expansion is modest if any. Using autologous irradiated PBMC as feeder cells, up to 2,500-fold expansion of functionally active NK cells at day 17 has been reported (89). The use of genetically modified cell lines as feeder leads to a 30,000-fold expansion of NK cells after 21 days of culture (79).
A recent study took advantage of the introduction of anti-CD3 and anti-CD52 monoclonal antibodies over a period of 14 days and reports a median 1500-fold increase in NK cell numbers; however, it must be emphasized that T cells represent up to 40% of the final cell product and that NK cells were not obtained through a cGMP protocol (90).
Quality Controls and Release Criteria for Engineered NK Cell Cells
Tools for assessing the efficacy of NK cell generation protocols are necessary for comparing technical results from different NK cell therapy studies. Furthermore, European Medicine Agency (EMA), Food and Drug Administration (FDA), and several guidelines require the characterization of the final product to define release criteria in order to ensure safety and efficacy.
Basic, yet essential, criteria are generally used to characterize the final product: these include purity and viability of the target cell population, contamination with undesirable cells such as residual T and B cells, and sterility. These are commonly used as release criteria although their relevance may vary for different clinical conditions: T cell contamination for instance is most important in an allogeneic, but not so much in an autologous setting. More sophisticated testing may provide additional information: a reduction in telomere length indicates cell senescence due to extensive long-term culturing.
Phenotype and function (tumor cytotoxicity) are additional characteristics that should help identify the most effective NK cell products. When expanded NK cells were compared with freshly isolated and IL-2-activated NK cells, a higher expression of NKG2D, TNF-related apoptosis-inducing ligand (TRAIL), and natural cytotoxicity receptors NKp30, NKp44, and NKp46 was reported (91), in addition to higher cytotoxicity to K562 cells. Efforts are, however, much needed to harmonize technical protocols and identify a panel of phenotypic and functional biomarkers that would allow comparisons between protocols that evaluate adoptive transfer of NK cells (92). It is essential to mention that such a panel needs to be run within a reasonably short time to release the product in time for a “fresh infusion.”
Cryopreservation and conservation of cytolytic activity of thawed NK cells would render multiple rounds of adoptive NK cell infusions feasible. Lapteva et al. and Berg et al. reported that an overnight activation with IL-2 would rescue the reduced cytolytic activity of thawed NK cells, yet at the cost of a diminished recovery (74, 78). Efforts to optimize cryopreservation and thawing methods are in progress.
Regulatory Status of Engineered NK Cells and Commercial Perspectives
Autologous and allogeneic NK cells engineered from primary human cells are individually produced for a unique and designated individual, rather than manufactured as batches. Since manufacturing incorporates ex-vivo culture and activation of immune-selected cells from the primary material, these will be considered as substantially manipulated or more-than minimally manipulated cell products, and thus will qualify as “advanced therapy medicinal products” (ATMPs) and somatic cell therapy products as defined in EC regulation 1394/2007. Before the regulation was released, such cell therapies were engineered as part of clinical research protocols by cell processing facilities, usually supported and operated by academia, no differently from minimally manipulated cell transplants. Since the regulation has been published, the view is that somatic cell therapy products and gene therapy products will be manufactured in compliance with good manufacturing practices (85, 86, 93), and eventually marketed by industry, when a marketing authorization is granted by competent authorities at a European level, i.e., by the EMA or by the FDA in the USA.
Beyond academic investigations, NK cells have now aroused the interest of a significant number of pharma companies (94) (see Figure 1), although no NK-based cellular therapy has so far been authorized as an ATMP in Europe, nor a somewhat comparable status in the USA. However, in December 2014, orphan designation (EU/3/14/1395) was granted by the European Commission to Glycostem, for the GCT-NK cell product, made of allogeneic ex vivo-generated natural killer (NK) cells from CD34+ CB progenitor cells for the treatment of acute myeloid leukemia (EMA/COMP/730059/2014 Committee for Orphan Medicinal Products).
Conclusion
Over the last 30 years, enormous progress has been made in our understanding of the biology of NK cells. New agents targeting their activity in vivo have been evaluated, and technological improvements in NK cell manufacturing have been introduced in the clinic. However, the demonstration that modulation of NK cell activity by any of these means can achieve therapeutic activity over a wide range of diseases is still awaited. The ability to follow and image in vivo adoptively transferred autologous or allogeneic NK cells would represent a major advantage to understand the “pharmacokinetics” and mechanisms of action of these immune effectors, as illustrated in preclinical (95) as well as in clinical (96) studies. It would help to understand the consequences of culture conditions on in vivo persistence and activity. Ongoing developments for innovative cellular therapies in the academic sector as well as in the commercial sector suggest that such progress may result in broader clinical applications in the near future.
Author Contributions
All authors are members of a Marseille-based consortium that aims at translation of scientific discoveries to clinically applicable innovations, with a focus on innovative cellular therapies for patients affected with hematological diseases. All authors have contributed to the writing of this manuscript and had the opportunity to review the draft and final versions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by Institut Paoli-Calmettes, by grant INCa-DGOS-Inserm 6038 (Site Intégré de Recherches et d’Innovations en Cancérologie, SIRIC Marseille) from Institut National du cancer (INCa), by Programme Hospitalier de Recherche Clinique (PHRC, to DB), by Fondation ARC pour la recherche sur le cancer and by Cancéropôle PACA. EV’s laboratory is supported by the European Research Council (THINK Advanced Grant), the Ligue Nationale contre le Cancer (“Equipe Labellisée”), and by institutional grants from INSERM, CNRS, and Aix-Marseille University to CIML. EV is a scholar of the Institut Universitaire de France. SU is supported by grants from the European Research Council, l’Agence nationale de la recherche (ANR) and la Fondation ARC pour la recherche sur le cancer, la région PACA.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00504/full#supplementary-material.
Abbreviations
ADCC, antibody-dependent cytotoxicity; ATMP, advanced therapy medicinal product; CAR, chimeric antigen receptor; CB, cord blood, placental blood; CBU, cord blood unit; CLL, chronic lymphocytic leukemia; DLI, donor lymphocyte infusion; EMA, European Medicines Agency; ES, embryonic stem (cells); FDA, Food and Drug Agency; Flt3-L, flt3 ligand; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GMP, good manufacturing practices; GvHD, graft-versus-host disease; HLA, human leukocyte antigens; HSCT, hematopoietic stem cell transplantation; IL-x, interleukin x; ILC, innate lymphoid cells; iPS, induced pluripotent stem (cells); KAR, killer activating receptors; KIR, killer cell Immunoglobulin-like receptors or killer inhibitory receptors; LIF, leukemia inhibitory factor; LMWH, low molecular weight heparin; MIP1α, Macrophage Inflammatory Protein-1α; NK, natural killer; PBMC, peripheral blood mononuclear cells; SCF, stem cell factor; TPO, thrombopoietin; TRAIL, TNF-related apoptosis-inducing ligand.
References
1. Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The “cancer immunogram”. Science (2016) 352(6286):658–60. doi:10.1126/science.aaf2834
2. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi:10.1038/nri3365
3. Guven H, Gilljam M, Chambers BJ, Ljunggren HG, Christensson B, Kimby E, et al. Expansion of natural killer (NK) and natural killer-like T (NKT)-cell populations derived from patients with B-chronic lymphocytic leukemia (B-CLL): a potential source for cellular immunotherapy. Leukemia (2003) 17(10):1973–80. doi:10.1038/sj.leu.2403083
4. Garg TK, Szmania SM, Khan JA, Hoering A, Malbrough PA, Moreno-Bost A, et al. Highly activated and expanded natural killer cells for multiple myeloma immunotherapy. Haematologica (2012) 97(9):1348–56. doi:10.3324/haematol.2011.056747
5. Schonfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther (2015) 23(2):330–8. doi:10.1038/mt.2014.219
6. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res (2011) 17(19):6287–97. doi:10.1158/1078-0432.CCR-11-1347
7. Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med (1985) 313(23):1485–92. doi:10.1056/NEJM198512053132327
8. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med (2005) 202(8):1075–85. doi:10.1084/jem.20051511
9. Rodella L, Zamai L, Rezzani R, Artico M, Peri G, Falconi M, et al. Interleukin 2 and interleukin 15 differentially predispose natural killer cells to apoptosis mediated by endothelial and tumour cells. Br J Haematol (2001) 115(2):442–50. doi:10.1046/j.1365-2141.2001.03055.x
10. Hayakawa M, Hatano T, Ogawa Y, Gakiya M, Ogura H, Osawa A. Treatment of advanced renal cell carcinoma using regional arterial administration of lymphokine-activated killer cells in combination with low doses of rIL-2. Urol Int (1994) 53(3):117–24. doi:10.1159/000282651
11. Abdel-Azim H, Heisterkamp N. Potential of autologous NK cell therapy to eradicate leukemia: “Education is [not] the best provision for old age” – Aristotle. Oncoimmunology (2015) 4(2):e984549. doi:10.4161/2162402X.2014.984549
12. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123(5):678–86. doi:10.1182/blood-2013-08-519199
13. Terszowski G, Klein C, Schmied L, Stern M. How to outsmart NK cell tolerance. Oncoimmunology (2015) 4(9):e1016708. doi:10.1080/2162402X.2015.1016708
14. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120(22):4317–23. doi:10.1182/blood-2012-06-437558
15. Kheav VD, Busson M, Scieux C, Peffault de Latour R, Maki G, Haas P, et al. Favorable impact of natural killer cell reconstitution on chronic graft-versus-host disease and cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation. Haematologica (2014) 99(12):1860–7. doi:10.3324/haematol.2014.108407
16. Dunbar EM, Buzzeo MP, Levine JB, Schold JD, Meier-Kriesche HU, Reddy V. The relationship between circulating natural killer cells after reduced intensity conditioning hematopoietic stem cell transplantation and relapse-free survival and graft-versus-host disease. Haematologica (2008) 93(12):1852–8. doi:10.3324/haematol.13033
17. Pical-Izard C, Crocchiolo R, Granjeaud S, Kochbati E, Just-Landi S, Chabannon C, et al. Reconstitution of natural killer cells in HLA-matched HSCT after reduced-intensity conditioning: impact on clinical outcome. Biol Blood Marrow Transplant (2015) 21(3):429–39. doi:10.1016/j.bbmt.2014.11.681
18. Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, et al. NK cell education after allogeneic transplantation: dissociation between recovery of cytokine-producing and cytotoxic functions. Blood (2011) 118(10):2784–92. doi:10.1182/blood-2011-04-347070
19. Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science (2002) 295(5562):2097–100. doi:10.1126/science.1068440
20. Zhao XY, Chang YJ, Xu LP, Zhang XH, Liu KY, Li D, et al. HLA and KIR genotyping correlates with relapse after T-cell-replete haploidentical transplantation in chronic myeloid leukaemia patients. Br J Cancer (2014) 111(6):1080–8. doi:10.1038/bjc.2014.423
21. Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med (2012) 367(9):805–16. doi:10.1056/NEJMoa1200503
22. Bari R, Rujkijyanont P, Sullivan E, Kang G, Turner V, Gan K, et al. Effect of donor KIR2DL1 allelic polymorphism on the outcome of pediatric allogeneic hematopoietic stem-cell transplantation. J Clin Oncol (2013) 31(30):3782–90. doi:10.1200/JCO.2012.47.4007
23. Gagne K, Busson M, Bignon JD, Balere-Appert ML, Loiseau P, Dormoy A, et al. Donor KIR3DL1/3DS1 gene and recipient Bw4 KIR ligand as prognostic markers for outcome in unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2009) 15(11):1366–75. doi:10.1016/j.bbmt.2009.06.015
24. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi:10.1038/nri3174
25. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood (2014) 123(25):3855–63. doi:10.1182/blood-2013-10-532531
26. Choi I, Yoon SR, Park SY, Kim H, Jung SJ, Jang YJ, et al. Donor-derived natural killer cells infused after human leukocyte antigen-haploidentical hematopoietic cell transplantation: a dose-escalation study. Biol Blood Marrow Transplant (2014) 20(5):696–704. doi:10.1016/j.bbmt.2014.01.031
27. Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transplant (2013) 48(3):433–8. doi:10.1038/bmt.2012.162
28. Curti A, Ruggeri L, D’Addio A, Bontadini A, Dan E, Motta MR, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood (2011) 118(12):3273–9. doi:10.1182/blood-2011-01-329508
29. Curti A, Ruggeri L, Parisi S, Bontadini A, Dan E, Motta MR, et al. Larger size of donor alloreactive NK cell repertoire correlates with better response to NK cell immunotherapy in elderly acute myeloid leukemia patients. Clin Cancer Res (2016) 22(8):1914–21. doi:10.1158/1078-0432.CCR-15-1604
30. Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood (2015) 125(5):784–92. doi:10.1182/blood-2014-07-592881
31. Miller JS, Rooney CM, Curtsinger J, McElmurry R, McCullar V, Verneris MR, et al. Expansion and homing of adoptively transferred human natural killer cells in immunodeficient mice varies with product preparation and in vivo cytokine administration: implications for clinical therapy. Biol Blood Marrow Transplant (2014) 20(8):1252–7. doi:10.1016/j.bbmt.2014.05.004
32. Leung W. Infusions of allogeneic natural killer cells as cancer therapy. Clin Cancer Res (2014) 20(13):3390–400. doi:10.1158/1078-0432.CCR-13-1766
33. Lim O, Jung MY, Hwang YK, Shin EC. Present and future of allogeneic natural killer cell therapy. Front Immunol (2015) 6:286. doi:10.3389/fimmu.2015.00286
34. Leung W. Use of NK cell activity in cure by transplant. Br J Haematol (2011) 155(1):14–29. doi:10.1111/j.1365-2141.2011.08823.x
35. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood (2010) 116(14):2411–9. doi:10.1182/blood-2010-05-283051
36. Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Marsh SG, et al. Donor killer cell Ig-like receptor B haplotypes, recipient HLA-C1, and HLA-C mismatch enhance the clinical benefit of unrelated transplantation for acute myelogenous leukemia. J Immunol (2014) 192(10):4592–600. doi:10.4049/jimmunol.1302517
37. Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol (2004) 172(1):644–50. doi:10.4049/jimmunol.172.1.644
38. Leung W, Iyengar R, Triplett B, Turner V, Behm FG, Holladay MS, et al. Comparison of killer Ig-like receptor genotyping and phenotyping for selection of allogeneic blood stem cell donors. J Immunol (2005) 174(10):6540–5. doi:10.4049/jimmunol.174.10.6540
39. Bari R, Bell T, Leung WH, Vong QP, Chan WK, Das Gupta N, et al. Significant functional heterogeneity among KIR2DL1 alleles and a pivotal role of arginine 245. Blood (2009) 114(25):5182–90. doi:10.1182/blood-2009-07-231977
40. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood (2002) 99(3):754–8. doi:10.1182/blood.V99.3.754
41. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood (1997) 90(3):1109–14.
42. Leung W, Iyengar R, Leimig T, Holladay MS, Houston J, Handgretinger R. Phenotype and function of human natural killer cells purified by using a clinical-scale immunomagnetic method. Cancer Immunol Immunother (2005) 54(4):389–94. doi:10.1007/s00262-004-0609-6
43. Pascal V, Brunet C, Pradel V, Thirion X, Andre P, Faucher C, et al. Analysis of donor NK and T cells infused in patients undergoing MHC-matched allogeneic hematopoietic transplantation. Leukemia (2002) 16(11):2259–66. doi:10.1038/sj.leu.2402670
44. Su YC, Li SC, Hsu CK, Yu CC, Lin TJ, Lee CY, et al. G-CSF downregulates natural killer cell-mediated cytotoxicity in donors for hematopoietic SCT. Bone Marrow Transplant (2012) 47(1):73–81. doi:10.1038/bmt.2011.22
45. Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood (2013) 122(4):491–8. doi:10.1182/blood-2013-02-453175
46. Shah N, Martin-Antonio B, Yang H, Ku S, Lee DA, Cooper LJ, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS One (2013) 8(10):e76781. doi:10.1371/journal.pone.0076781
47. Vasu S, Berg M, Davidson-Moncada J, Tian X, Cullis H, Childs RW. A novel method to expand large numbers of CD56(+) natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy (2015) 17(11):1582–93. doi:10.1016/j.jcyt.2015.07.020
48. Boissel L, Tuncer HH, Betancur M, Wolfberg A, Klingemann H. Umbilical cord mesenchymal stem cells increase expansion of cord blood natural killer cells. Biol Blood Marrow Transplant (2008) 14(9):1031–8. doi:10.1016/j.bbmt.2008.06.016
49. Beck RC, Padival M, Yeh D, Ralston J, Cooke KR, Lowe JB. The Notch ligands Jagged2, Delta1, and Delta4 induce differentiation and expansion of functional human NK cells from CD34+ cord blood hematopoietic progenitor cells. Biol Blood Marrow Transplant (2009) 15(9):1026–37. doi:10.1016/j.bbmt.2009.06.002
50. Vitale C, Cottalasso F, Montaldo E, Moretta L, Mingari MC. Methylprednisolone induces preferential and rapid differentiation of CD34+ cord blood precursors toward NK cells. Int Immunol (2008) 20(4):565–75. doi:10.1093/intimm/dxn014
51. Kao IT, Yao CL, Kong ZL, Wu ML, Chuang TL, Hwang SM. Generation of natural killer cells from serum-free, expanded human umbilical cord blood CD34+ cells. Stem Cells Dev (2007) 16(6):1043–51. doi:10.1089/scd.2007.0033
52. Spanholtz J, Preijers F, Tordoir M, Trilsbeek C, Paardekooper J, de Witte T, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One (2011) 6(6):e20740. doi:10.1371/journal.pone.0020740
53. Spanholtz J, Tordoir M, Eissens D, Preijers F, van der Meer A, Joosten I, et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS One (2010) 5(2):e9221. doi:10.1371/journal.pone.0009221
54. Lan T, Wang L, Xu L, Jin N, Yan G, Xia J, et al. Induced pluripotent stem cells can effectively differentiate into multiple functional lymphocyte lineages in vivo with negligible bias. Stem Cells Dev (2016) 25(6):462–71. doi:10.1089/scd.2015.0248
55. Bock AM, Knorr D, Kaufman DS. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and and induced pluripotent stem cells (iPSCs). J Vis Exp (2013) 74:e50337. doi:10.3791/50337
56. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood (2009) 113(24):6094–101. doi:10.1182/blood-2008-06-165225
57. Eguizabal C, Zenarruzabeitia O, Monge J, Santos S, Vesga MA, Maruri N, et al. Natural killer cells for cancer immunotherapy: pluripotent stem cells-derived NK cells as an immunotherapeutic perspective. Front Immunol (2014) 5:439. doi:10.3389/fimmu.2014.00439
58. Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol (2005) 175(8):5095–103. doi:10.4049/jimmunol.175.8.5095
59. Knorr DA, Bock A, Brentjens RJ, Kaufman DS. Engineered human embryonic stem cell-derived lymphocytes to study in vivo trafficking and immunotherapy. Stem Cells Dev (2013) 22(13):1861–9. doi:10.1089/scd.2012.0608
60. Tabatabaei-Zavareh N, Vlasova A, Greenwood CP, Takei F. Characterization of developmental pathway of natural killer cells from embryonic stem cells in vitro. PLoS One (2007) 2(2):e232. doi:10.1371/journal.pone.0000232
61. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med (2013) 2(4):274–83. doi:10.5966/sctm.2012-0084
62. Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia (1994) 8(4):652–8.
63. Tam YK, Martinson JA, Doligosa K, Klingemann HG. Ex vivo expansion of the highly cytotoxic human natural killer-92 cell-line under current good manufacturing practice conditions for clinical adoptive cellular immunotherapy. Cytotherapy (2003) 5(3):259–72. doi:10.1080/14653240310001523
64. Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol (2015) 6:21. doi:10.3389/fphar.2015.00021
65. Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res (2001) 10(4):535–44. doi:10.1089/15258160152509145
66. Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy (2013) 15(12):1563–70. doi:10.1016/j.jcyt.2013.06.017
67. Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy (2008) 10(6):625–32. doi:10.1080/14653240802301872
68. Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology (2013) 2(10):e26527. doi:10.4161/onci.26527
69. Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol (2015) 6:195. doi:10.3389/fimmu.2015.00195
70. Carlens S, Gilljam M, Chambers BJ, Aschan J, Guven H, Ljunggren HG, et al. A new method for in vitro expansion of cytotoxic human CD3-CD56+ natural killer cells. Hum Immunol (2001) 62(10):1092–8. doi:10.1016/S0198-8859(01)00313-5
71. Miller JS, Oelkers S, Verfaillie C, McGlave P. Role of monocytes in the expansion of human activated natural killer cells. Blood (1992) 80(9):2221–9.
72. Luhm J, Brand JM, Koritke P, Hoppner M, Kirchner H, Frohn C. Large-scale generation of natural killer lymphocytes for clinical application. J Hematother Stem Cell Res (2002) 11(4):651–7. doi:10.1089/15258160260194794
73. Siegler U, Meyer-Monard S, Jorger S, Stern M, Tichelli A, Gratwohl A, et al. Good manufacturing practice-compliant cell sorting and large-scale expansion of single KIR-positive alloreactive human natural killer cells for multiple infusions to leukemia patients. Cytotherapy (2010) 12(6):750–63. doi:10.3109/14653241003786155
74. Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, Tawab A, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy (2009) 11(3):341–55. doi:10.1080/14653240902807034
75. Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J Cancer (2011) 2:383–5. doi:10.7150/jca.2.383
76. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res (2009) 69(9):4010–7. doi:10.1158/0008-5472.CAN-08-3712
77. Granzin M, Soltenborn S, Muller S, Kollet J, Berg M, Cerwenka A, et al. Fully automated expansion and activation of clinical-grade natural killer cells for adoptive immunotherapy. Cytotherapy (2015) 17(5):621–32. doi:10.1016/j.jcyt.2015.03.611
78. Lapteva N, Durett AG, Sun J, Rollins LA, Huye LL, Fang J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy (2012) 14(9):1131–43. doi:10.3109/14653249.2012.700767
79. Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One (2012) 7(1):e30264. doi:10.1371/journal.pone.0030264
80. Geraghty RJ, Capes-Davis A, Davis JM, Downward J, Freshney RI, Knezevic I, et al. Guidelines for the use of cell lines in biomedical research. Br J Cancer (2014) 111(6):1021–46. doi:10.1038/bjc.2014.166
81. Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, Nahi H, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood (2008) 111(6):3155–62. doi:10.1182/blood-2007-09-110312
82. Klingemann HG, Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy (2004) 6(1):15–22. doi:10.1080/14653240310004548
83. Sutlu T, Stellan B, Gilljam M, Quezada HC, Nahi H, Gahrton G, et al. Clinical-grade, large-scale, feeder-free expansion of highly active human natural killer cells for adoptive immunotherapy using an automated bioreactor. Cytotherapy (2010) 12(8):1044–55. doi:10.3109/14653249.2010.504770
84. Iyengar R, Handgretinger R, Babarin-Dorner A, Leimig T, Otto M, Geiger TL, et al. Purification of human natural killer cells using a clinical-scale immunomagnetic method. Cytotherapy (2003) 5(6):479–84. doi:10.1080/14653240310003558
85. Lim O, Lee Y, Chung H, Her JH, Kang SM, Jung MY, et al. GMP-compliant, large-scale expanded allogeneic natural killer cells have potent cytolytic activity against cancer cells in vitro and in vivo. PLoS One (2013) 8(1):e53611. doi:10.1371/journal.pone.0053611
86. Koehl U, Brehm C, Huenecke S, Zimmermann SY, Kloess S, Bremm M, et al. Clinical grade purification and expansion of NK cell products for an optimized manufacturing protocol. Front Oncol (2013) 3:118. doi:10.3389/fonc.2013.00118
87. Suck G, Oei VY, Linn YC, Ho SH, Chu S, Choong A, et al. Interleukin-15 supports generation of highly potent clinical-grade natural killer cells in long-term cultures for targeting hematological malignancies. Exp Hematol (2011) 39(9):904–14. doi:10.1016/j.exphem.2011.06.003
88. Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother (2015) 38(1):24–36. doi:10.1097/CJI.0000000000000059
89. Ahn YO, Kim S, Kim TM, Song EY, Park MH, Heo DS. Irradiated and activated autologous PBMCs induce expansion of highly cytotoxic human NK cells in vitro. J Immunother (2013) 36(7):373–81. doi:10.1097/CJI.0b013e3182a3430f
90. Masuyama J, Murakami T, Iwamoto S, Fujita S. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies. Cytotherapy (2016) 18(1):80–90. doi:10.1016/j.jcyt.2015.09.011
91. Childs RW, Berg M. Bringing natural killer cells to the clinic: ex vivo manipulation. Hematology Am Soc Hematol Educ Program (2013) 2013:234–46. doi:10.1182/asheducation-2013.1.234
92. Koehl U, Kalberer C, Spanholtz J, Lee DA, Miller JS, Cooley S, et al. Advances in clinical NK cell studies: donor selection, manufacturing and quality control. Oncoimmunology (2016) 5(4):e1115178. doi:10.1080/2162402X.2015.1115178
93. McKenna DH Jr, Sumstad D, Bostrom N, Kadidlo DM, Fautsch S, McNearney S, et al. Good manufacturing practices production of natural killer cells for immunotherapy: a six-year single-institution experience. Transfusion (2007) 47(3):520–8. doi:10.1111/j.1537-2995.2006.01145.x
94. Garber K. Natural killer cells blaze into immuno-oncology. Nat Biotech (2016) 34(3):219–20. doi:10.1038/nbt0316-219
95. Mallett CL, McFadden C, Chen Y, Foster PJ. Migration of iron-labeled KHYG-1 natural killer cells to subcutaneous tumors in nude mice, as detected by magnetic resonance imaging. Cytotherapy (2012) 14(6):743–51. doi:10.3109/14653249.2012.667874
Keywords: natural killer cells, innate lymphoid cells, immunotherapy, immuno-oncology, cellular therapy, cell transplant, manufacturing
Citation: Chabannon C, Mfarrej B, Guia S, Ugolini S, Devillier R, Blaise D, Vivier E and Calmels B (2016) Manufacturing Natural Killer Cells as Medicinal Products. Front. Immunol. 7:504. doi: 10.3389/fimmu.2016.00504
Received: 28 July 2016; Accepted: 27 October 2016;
Published: 15 November 2016
Edited by:
Lutz Walter, Leibniz-Institute for Primate Research, GermanyReviewed by:
Markus Uhrberg, University of Düsseldorf, GermanyUlrike Koehl, Hannover Medical School, Germany
Copyright: © 2016 Chabannon, Mfarrej, Guia, Ugolini, Devillier, Blaise, Vivier and Calmels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Chabannon, Y2hhYmFubm9uY0BpcGMudW5pY2FuY2VyLmZy, Y2hyaXN0aWFuLmNoYWJhbm5vbkB1bml2LWFtdS5mcg==, dGhlcmNlbGxAaXBjLnVuaWNhbmNlci5mcg==