
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Hortic. , 06 November 2023
Sec. Breeding and Genetics
Volume 2 - 2023 | https://doi.org/10.3389/fhort.2023.1250875
 Silvia Farinati
Silvia Farinati Francesco Scariolo
Francesco Scariolo Fabio Palumbo
Fabio Palumbo Alessandro Vannozzi
Alessandro Vannozzi Gianni Barcaccia*
Gianni Barcaccia* Margherita Lucchin
Margherita LucchinHeterosis in plants has been among the challenging topics for plant scientists worldwide. The production of F1 hybrid varieties of seed-propagated horticultural species is one of the most successful applications of plant breeding techniques. The exploitation of the heterosis phenomenon promotes homogeneity and maximizes crop yields and is a way for breeders to legally control and protect their commercial products. In the past heterosis has been largely studied and explored in cereal crop systems, considering maize as a model for understanding the genetic bases of this phenomenon. To date, crossbreeding in horticultural vegetables has also rapidly progressed. F1 hybrid varieties are available for many horticultural crops, including both allogamous and autogamous species. Several genetic and nongenetic mechanisms have been applied to facilitate the large-scale production of F1 hybrid seeds in vegetable crops to prevent undesirable selfing. Although the development and commercialization of F1 hybrids is currently common in agriculture, this phenomenon is still being investigated at different levels. With the rapid accumulation of knowledge on plant genome structures and gene activities and the advancement of new genomics platforms and methodologies, significant progress has been achieved in recent years in the study of the genetic and molecular bases of heterosis. This paper provides a brief overview of current theoretical advances and practical predictions of the molecular mechanisms underlying heterosis in plants. The aim is to carefully summarize the fundamental mechanisms of heterosis in plants, focusing on horticultural plant breeding, to improve the existing knowledge in this research area. We describe the quantitative genetic model of phenotypic variation and combine evolutionary, phenotypic and molecular genetic views to explain the origin and manifestation of heterosis and its significance for breeding F1 hybrid varieties in horticultural crops. The principles of genomic prediction and its applications in genomic selection are then covered.
The phenomenon of heterosis has long been among the most popular topics for plant scientists worldwide. As largely described in the past, the term heterosis refers to the increase in vigour seen in progeny generated from specific crossings between pure or inbred lines (Lippman and Zamir, 2007). The resulting offspring have a hybrid genotypic structure and a phenotypic value higher than those of parental lines. This phenomenon is usually known as “hybrid vigour” and is most prominent in allogamous species. It can also be thought of as the inverse of inbreeding depression, represented by a reduction in vigour and observed after self-pollination and/or cross-pollination between sibling lines.
The employment of heterosis in agriculture has been one of the most significant successes in the application of plant breeding programs over the last century. Manifesting as increased vigour of plant traits related to vegetative and reproductive processes, heterosis has always been considered similar to a ‘miraculous’ agricultural occurrence. In particular, breeding programs aimed at taking advantage of this phenomenon have significant commercial implications for several reasons, first of all the constitution and cultivation of elite F1 hybrid varieties with specific properties. Indeed, the yield of F1 hybrid varieties is higher than that of other types of varieties (e.g., synthetic varieties) (Patella et al., 2019), and the development of F1 hybrid seeds also increases the possibility of obtaining different agronomically interesting traits at the genome level combined in a single genotype (Fu et al., 2014). Furthermore, the genetic uniformity of F1 hybrids and parental line secrecy facilitate the protection of seed companies’ or plant breeders’ rights, thus stimulating partnerships of the private sector in breeding programs, which are usually expensive and time-consuming (Scariolo et al., 2023). Finally, the constitution of F1 hybrid varieties does not reduce the need to preserve the genetic resources of agronomically attractive species, since research on highly relevant inbred lines suitable for specific crossings requires advanced research within germplasm collections, to persevere their continuous maintenance.
The most striking examples of varieties having a very high level of heterosis range from maize (Zea mays) to oil palm [Elaeis guineensis; (Duvick, 2005; Fu et al., 2014; Cros et al., 2015)], in which F1 hybrid crop cultivars vastly outperform their inbred progenitors. In general, the production of F1 hybrid crops is unquestionably one of the key developments in the global seed market and results in a large boost in crop yields and crop husbandry revenue per se: for example, hybrid rice has improved China’s rice production by 44.1% (Cheng et al., 2007), the European market has favoured F1 hybrid seeds for several of its important crops, such as sugar beet, rapeseed, and rye (Hochholdinger and Hoecker, 2007), and in America an increase in yields of at least eightfold has been achieved through the cultivation of maize hybrid varieties (Tian et al., 2011). It is precisely on corn that, already in the early 1900s, Shull (Shull, 1908) observed phenotypic traits related to a heterotic effect in hybrid offspring of corn, proposing the concept of heterosis, and since the 1930s maize heterosis-based genetic breeding started to be commonly applied, providing several excellent maize hybrids.
Although cereal crops, primarily maize, have always been considered models for understanding heterotic phenomena, crossbreeding in horticultural vegetables has rapidly progressed, exploiting, in this agronomical sector, the genetic basis underlying heterosis. F1 hybrid varieties are now available for many horticultural crops, both allogamous (sunflower, melons, pepper, etc.) and autogamous (tomato, lettuce, etc.). For most vegetables, the global hybrid seed production is increasing by 8%–10% per year (Silva Dias, 2014). The application of heterosis to horticultural cultivation was first proposed by Hayes and Jones (Hayes and Jones, 1916) in cucumber systems. Since the discovery of the male sterility and self-incompatibility mechanisms in onion (Pearson, 1933) and cabbage (Jones and Clarke, 1943), respectively, a number of genetic- and nongenetic-related strategies have been developed and applied to prevent undesirable selfing for the development of experimental and commercial F1 hybrid seeds (Kumar and Singh, 2004; Colombo and Galmarini, 2017). Improved phenotypes related to heterosis phenomena have been observed and described also in Solanaceae (Spaldon et al., 2015; Tamta and Singh, 2017), Cruciferae (Behr et al., 2018; Kong et al., 2020), and Cucurbitaceae vegetables (Am et al., 2014; Sharma et al., 2016). The various traits that exhibit remarkable vigour in F1 hybrids include yield, earliness, growth, and stress tolerance (Krieger et al., 2010; Liu et al., 2020a; Zhang et al., 2020), all aspects that justify the increase in research on horticultural crops, since a more productive hybrid will actually reduce the time to the first harvest. Currently, the most important vegetables cultivated with economic relevance in European areas are represented by hybrid cultivars and include species such as tomato, eggplant, chicory, sunflower and rapeseed. In particular, the top five F1 hybrid varieties of vegetables registered in Europe are tomato, pepper, melon, cucumber, and onion, which are represented almost exclusively by hybrid genotypes, which are replacing open-pollinated varieties (Santamaria and Signore, 2021, Yu et al., 2021).
Although the constitution and commercialization of F1 hybrids are currently common in agriculture and hybrid vigour has been largely exploited by breeders for many decades, this phenomenon is still currently being investigated at different levels by geneticists. Although much information is currently available for most model cereals and in an increasing number of horticultural crops, the genetic and molecular mechanisms remain of great interest yet not completely unravelled. However, with the rapid accumulation of knowledge on gene function and the advancement of new genomics methodologies, considerable progress has been made in recent years to highlight new frontiers for the genetic and molecular foundation of heterosis.
Starting from these presumptions, the following manuscript provides a general overview of current scientific advances and related practical prospects, explaining the molecular mechanisms of heterosis in plants, focusing on horticultural species. Beginning with relative theoretical bases, the main aim is to improve exiting knowledge in the horticultural plant breeding research sector. We describe the quantitative genetic model of phenotype and combine the old theoretical bases with modern quantitative, evolutionary, phenotypic, and molecular genetic views to explain the origins of heterosis and its significance in hybrid breeding. The principles of genomic prediction and its applications in genomic selection are then covered.
Over the years, a vast amount of heterosis literature has been published for major crops, mainly cereals and herbaceous species, which makes its description and appreciation difficult due to the several implied variables influencing this phenomenon. However, among the most important aspects, the reproductive system is a key starting point for understanding the genetic and molecular basis of heterosis. In fact, several reproductive systems imply that the establishment of heterosis occurs differently, as reported in Table 1, which describes how heterosis varies primarily in relation to this aspect.
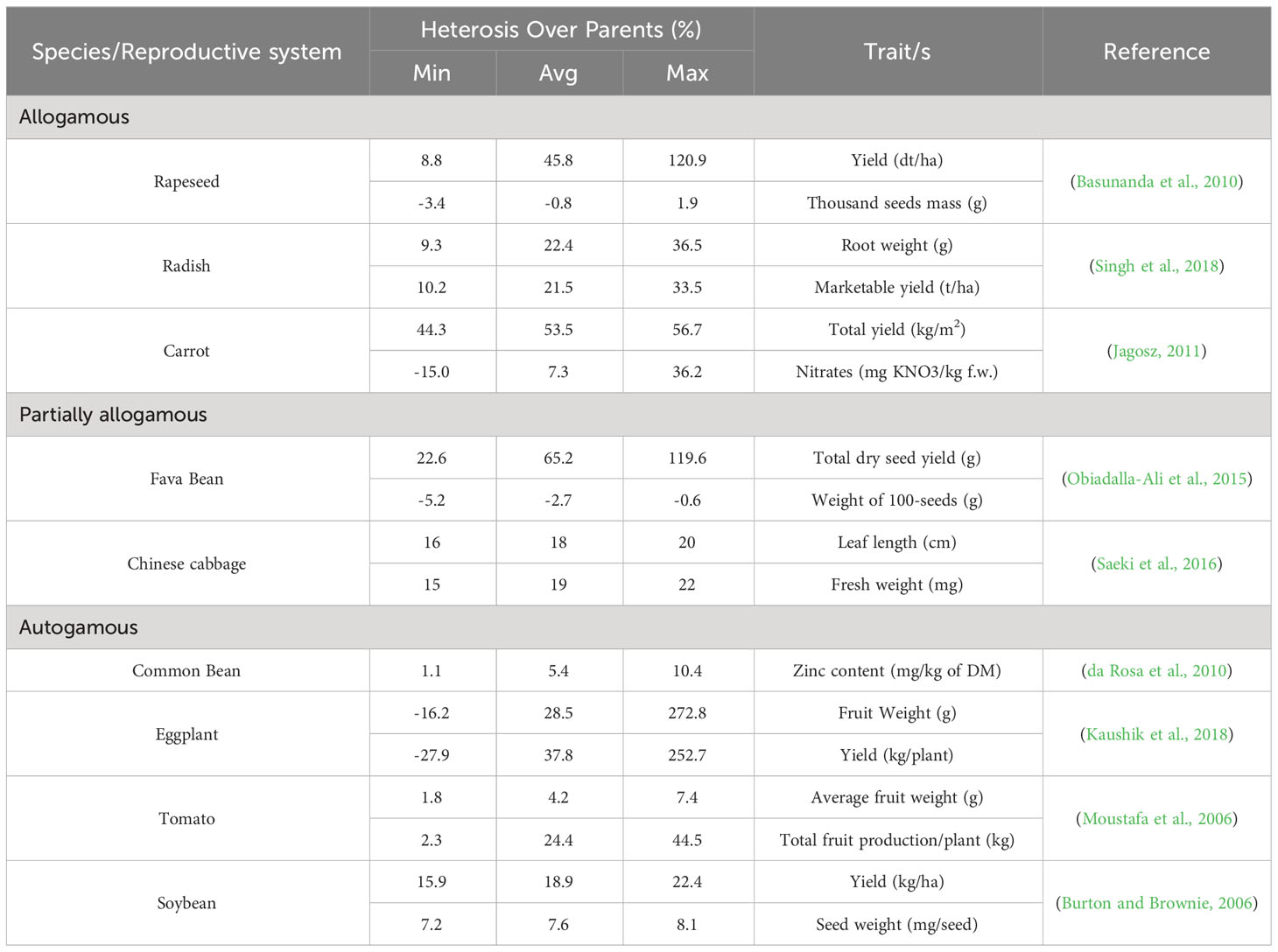
Table 1 Heterosis estimates of F1 hybrids yields for different traits.
As shown in Table 1, the degree of heterosis in progenies over the parents is well represented in allogamous species. The genetic basis in allogamous plant populations under natural conditions refers to the genetic equilibrium described by the Hardy–Weinberg law. It is stated that, in a larger population presenting random crosses (panmixis) and the absence of evolutionary pressures (selections, mutations or/and migrations), at a specific locus having alleles A and a, genotypic frequencies are fixed at values equal to p2(AA), 2pq(Aa) and q2(aa), where p and q are the allelic frequencies in the population. The Hardy–Weinberg law presumes that the mating of genotypes depends on their frequency: the probability that two genotypes cross together is thus equal to the product of their frequencies. Whenever genetically similar individuals, thus possibly related to each other, cross together (selfing or full-sibling) with higher frequencies than would be expected in the case of random mating, genetic equilibrium is disturbed. Notably, this kind of crossing drives an increase in homozygosity, a reduction in the genetic variability of the population and a decrease in vigour in the obtained progenies (inbreeding depression). Indeed, in these situations, the probability that progenies inherit the same alleles from both parental plants increases, bringing about a fixed homozygous condition. Conversely, crossings between genetically different individuals (outbreeding) reduce homozygosity and maintain or even increase genetic variability in the population. Consequently, in outcrossing populations, the probability that progenies inherit different alleles from the parents increases, leading to a heterozygous condition in the population.
In applicative fields (breeding), controlled crossing represents a very useful strategy for developing pure or inbred lines homozygous at all loci (selfing or full-sibling) or for developing heterozygous hybrids at most loci (outcrossing). The reproductive system of a species, therefore, represents the first determinant able to influence the genotypic composition of both natural and breeding populations and the phenotypic manifestation of heterosis. The intercrossing between individuals of the same population, as well as crossing plants belonging to different populations, have marked consequences for crop productivity and fitness. In model maize, the existence of strong correlation between vigour depression and, on the one hand, heterosis and, on the other hand, the genetic distance of the parents, calculated using both morpho-physiological traits (Moll et al., 1965) and molecular marker data (Reif et al., 2003), has been proven. In horticultural crops, the relationship between heterosis and the genetic distance between parents is in some cases controversial: yield traits have been examined in fruit vegetables such as tomato (Fortuny et al., 2021), eggplant (Rodríguez-Burruezo et al., 2008; Kaushik et al., 2018), cucumber (Liu et al., 2022), pepper (Geleta et al., 2004) and melon (Luan et al., 2010; Dafna et al., 2021), where a negative or no correlation between them was observed, suggesting that the genetic distance between parents is not a strong predictor in these fruit vegetables. In contrast, heterosis in root biomass in carrot and radish was observed, and moderate positive relationships between yield, heterosis and the genetic distance of parental lines were observed. Even though F1 varieties have already been developed for these species without clear and focused reports related to this phenomenon, research on heterosis is still lacking for many crops, as it is mainly limited to leafy and fruit vegetables (Jagosz, 2011; Turner et al., 2018).
However, opposite findings were also reported regarding the relationship between genetic distance and heterosis in horticultural crops. Figueiredo et al. (Figueiredo et al., 2016) described that when two parents that are strongly genetically divergent are crossed together, the chances of obtaining highly heterotic hybrids are higher than if the parents are closely related. This effect is due to the recombination events that occur when mating gametes from individuals share a small number of common alleles, thus increasing the possibility of transferring those with a positive influence on progeny vigour. In fact, through the recombination of positive alleles, meaning those that have a positive effect on the phenotype, genetic mechanisms of dominance, epistasis and quantitative additivity influence the phenotypic manifestation of useful traits for agronomic purposes in crops.
These findings agreed with what was reported in other studies in tomato, potato, sweet pepper, onion and sesame (Buso et al, 2003; Figueiredo et al., 2016; Li et al., 2022; Geleta et al., 2004; Shapturenko et al., 2013; Shapturenko et al., 2014; Pandey et al., 2018; Faria et al., 2019).
Horticultural hybrids are better represented varieties than their inbred parents and the original open-pollinated population. The reason for this is their potential in integrating dominantly inherited disease resistances, their higher quality and disease-free hybrid seeds (in contrast to locally grown open-pollinated ones), or the fact that hybrid agronomic performance is superior, and these are only some of the possible explanations for their spread in the market (Havey, 1998).
In particular, similar to what emerged in cereals, such as maize (Yang et al., 2017; Liu et al., 2020a; Birdseye et al., 2021), rice (Dan et al., 2014; Shen et al., 2015; Zhu et al., 2016) and wheat (Song et al., 2009), multiple morphological features associated with vegetative tissue also showed different levels of heterosis in vegetable F1 hybrids. These insights, which have been observed depending on the crop and developmental stage, from post-germination to final yield, were derived from Brassicaceae crops, such as Chinese cabbage (Kawamura et al., 2016; Fujimoto et al., 2018; Shiraki et al., 2023), cabbage (Jeong et al., 2017), cauliflower (Verma and Kalia, 2017), rapeseed or canola (Brassica napus L.) (Basunanda et al., 2010; Wolko et al., 2019; Wang et al., 2021; Hu et al., 2022); Solanaceae crops, such as tomato (Chandel et al., 2021; Rajendran et al., 2022), potato (Buso et al, 2003), and eggplant (Kaushik, 2019; Kumar et al., 2020); Apiaceae crops, such as carrot, coriander and caraway (Jagosz, 2011; von Maydell et al., 2021; Hanifei et al., 2022); and Leguminosae crops, such as common bean (Gonçalves-Vidigal et al., 2008) and pea (Sharma et al., 2023).
Regarding these findings, it must be reported that genetic distance based on phenotypic data analysis was less informative than that calculated using molecular data. In most cases, no strong correlation was observed between molecular distance and specific phenotypic traits, although the use of molecular markers was demonstrated to be useful in the determination of suitable parental genotypes for breeding purposes and heterosis exploitation. Supporting this, higher heterozygosity and uniformity, computed using molecular information, were observed in hybrid progenies obtained by crossing strongly homozygous and dissimilar lines than in those obtained by crossing highly similar and heterozygous parents (Reif et al., 2003; Jagosz, 2011; Scariolo et al., 2022).
For a long time, the evolutionary and genetic bases of heterosis have been investigated, and several studies have deeply described the genetic foundation to shed light on this intriguing scenario. First, it is necessary to anticipate that in conventional breeding and genetic research, we work with chromosome blocks rather than individual genes. Thus, heterosis and estimates of gene action are supported by the chromosome block structure notion. Chromosome blocks fluctuate in size depending on the degree of linkage (frequency of recombination) and the number of sexual generations (the direction towards linkage equilibrium); however, the only factor we often know is the number of sexual generations. We frequently do not know how much genetic material is linked to the gene of interest, not even in cases of backcrossing of single gene features.
Second, it is plausible that biologically additive, dominant, and epistatic gene action can fully account for heterosis and that no single gene, class of genes, or physiological phenomenon is responsible for heterosis (Birchler et al., 2010; Fievet et al., 2018). If this is the case, looking for the genes underlying heterosis would reveal the genes underlying the particular trait under consideration in a given experimental sample, and heterosis would be brought about by biological dominance, over-dominance, or epistasis of the genes controlling the trait (Fievet et al., 2018). Even for single traits within a species, attempts to map heterosis for different phenotypes typically fail to identify loci for which the connection holds consistently across genotypes (Huang et al., 2016; Liu et al., 2020b). From an evolutionary perspective, heterosis in quantitative genetics ultimately rests on assumptions of biological dominance and biological epistasis, even though the additive model captures most of the effects of biological dominance and epistasis (Huang and Mackay, 2016). For biological dominance to affect heterosis, dominant alleles should have directional effects on fitness (Lynch and Walsh, 1998). In particular, biologically dominant mutations in a population alter phenotype and are subject to selection. Because they are not selected as heterozygotes, recessive mutations are only subject to phenotypic selection in their homozygous condition, where they can spread throughout populations. Negative dominant alleles are therefore more likely than negative recessive alleles to be removed from populations by natural selection, and over evolutionary time, it is expected that biologically dominant alleles tend to be favoured, and deleterious alleles tend to be recessive (Falconer, 1996). The formation of dominance has been explained by evolutionary processes other than directional selection, such as stabilizing selection (Manna et al., 2011). Overdominance, a form of biological dominance in which alleles persist in populations at intermediate frequencies and heterozygotes have more extreme phenotypes than both homozygotes, can also result in heterosis (Crow, 1999). However, it can be challenging to detect over-dominance because it necessitates inbred parents to be identical at all loci except the one of interest. If they are not, parents may have biologically dominant alleles with opposing fitness effects tightly linked in repulsion phase, leading to pseudo over-dominance, in which the loci never occur in their uncoupled condition and are instead combined to form a single over-dominant locus.
As interactions among several loci contribute to fitness, biological epistasis may also contribute to heterosis. There is an abundance of evidence supporting biological epistasis, such as the physical binding of genes encoding transcription factor proteins to DNA sequence motifs to activate or repress other genes that influence phenotypes (Phillips, 2008; Lehner, 2011). However, it is sometimes not possible to identify all types of statistical and biological epistasis in normal experimental samples since there are many more allele combinations than there are different genotypes in a population (Wei et al., 2014). Furthermore, if none of the interacting genes in the experimental sample segregate, epistasis cannot be detected in the population (Stitzer and Ross-Ibarra, 2018).
The hypothesis formulated by Alfred H. Sturtevant and Thomas H. Morgan at the beginning of the last century, according to which crossing-over events take place randomly throughout the chromosomes, is currently considered only partially acceptable. In fact, many data acquired over the years from model organisms of different kingdoms (fungi, plants, insects and mammals) demonstrate the existence of chromosomal hot spots, namely regions characterized by higher crossing-over frequencies. (Lichten and Goldman, 1995; Schnable and Wise, 1998; Steiner and Smith, 2005; Choi et al., 2013; Bhakta et al., 2015; Si et al., 2015). In general, the size of the chromosomes reflects the frequency of crossing-over events that may occur. The relationship between the mean recombination frequency and the genetic distance between loci along the chromosome, as well as the relationship between the average frequency of the chiasmata and the genetic dimension of the chromosomes, allows us to estimate the overall length of the genetic maps (Table 2).
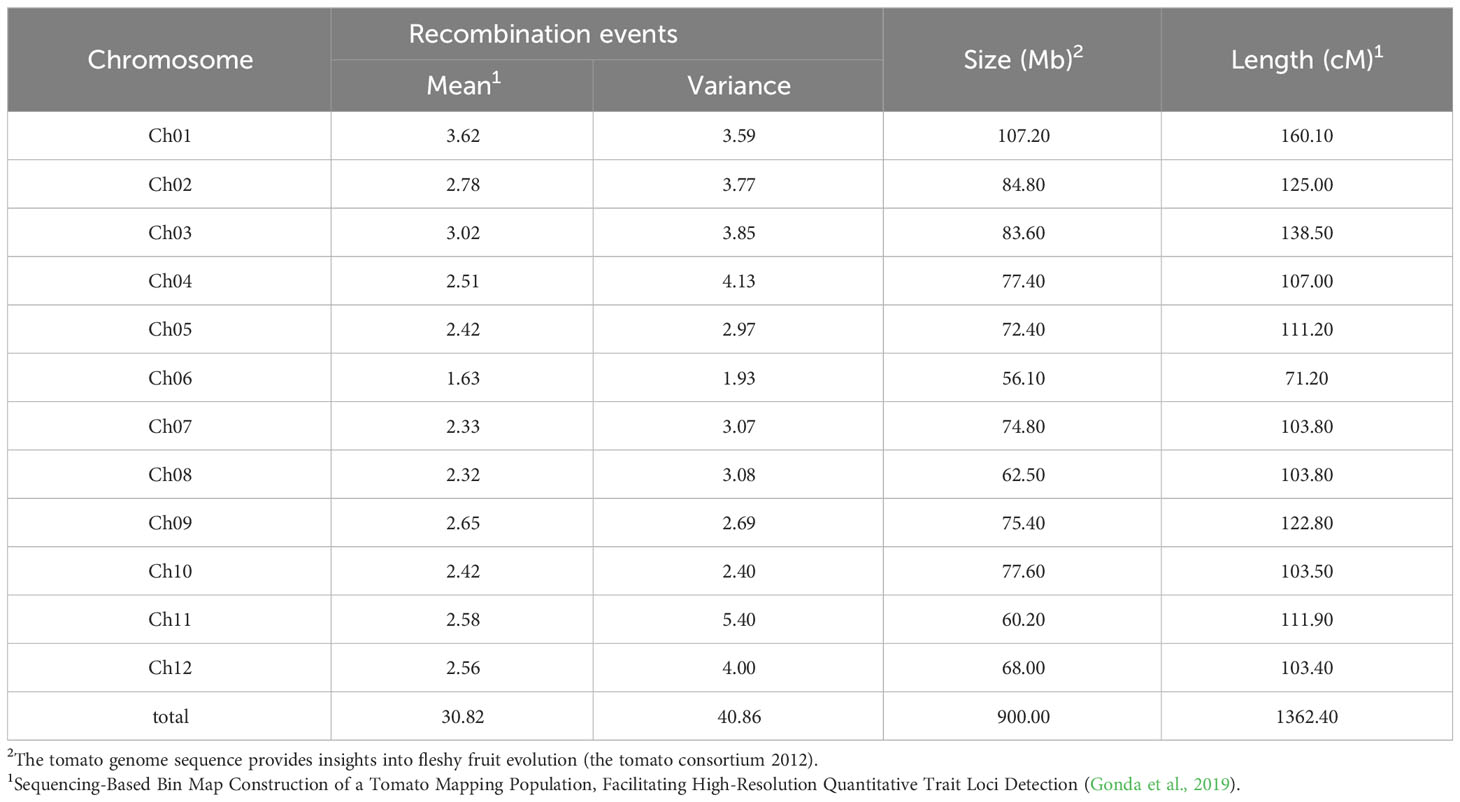
Table 2 Recombination events frequencies and chromosomes length estimates in tomato.
However, those chromosomal regions more frequently subject to crossing-over negatively condition the correspondence between a genetic and physical map (Rafalski and Morgante, 2004; Wang et al., 2015) and, vice versa, in regions with reduced or even suppressed recombination events (cold spots), small genetic distances can be translated as large physical distances (Bustamante et al., 2017). Consequently, the genetic distances, measured in cM (recombination units), do not always correspond exactly to the physical distances, measured instead in Mb (millions of base pairs). The comparison between the physical map thus obtained and the genetic map previously determined revealed considerable discrepancies in terms of reciprocal distances between loci. In fact, in all species, the chromosome ends (telomeres) as well as the regions adjacent to the centromere are, as a general rule, less affected by exchange events than other regions scattered along the chromosomal arms (Aguilar, 2021; Talbert and Henikoff, 2010). In a genetic map, physical regions scarcely affected by recombination events are compressed, while those most affected by crossing-over appear expanded. However, even if the relationship between the physical map and the genetic map of the chromosomes cannot be considered uniform, the genetic and physical maps of a chromosome are generally collinear, in the sense that loci retain the same order. The sequencing of whole plant genomes allowed the acquisition of important information on the synteny between maps of different species. This means that, in addition to the gene order, the gene composition along the chromosomes and within genomes can also be analyzed.
In the genomes of plants as well as other eukaryotes, the existence of genetic elements, called cis- and trans-genetic modifiers, that are able to regulate meiotic recombination events, has been demonstrated over the years (Wright et al., 2003; Pan et al., 2018). In addition to specific sequences found in the cis-phase within or close to recombination hot spots and corresponding to crossing-over regulatory elements, polymorphic sequences were also found to be dislocated in the trans-phase, probably due to consistent rearrangements such as deletions, insertions, and translocations (Pan et al., 2018) or to changes in the chromatin structure (Choi and Henderson, 2015). These two types of genetic modifiers affect the recombination rate per unit of chromosome physical length and the distribution of the recombination sites on each chromosome (Stapley et al., 2017). In model maize, for example, a non-uniform distribution of recombination events was found in two hotspot regions (a1-sh2 and sh1-bz1) more than 20 years ago (Civardi et al., 1994; Dooner and Martinez-Ferez, 1997). Timmermans and Berg (Timmermans and Berg, 1997) demonstrated the presence of a genetic element, in the region between the loci sh1 and bz1, that locally determines an increase in recombination events without affecting the adjacent regions. Moreover, analysis of recombination along chromosomes 7 and 5 also suggested the presence of heritable factors controlling recombination in these intervals and acting independently and in trans. Since then, tens of novel recombination hot spots have been identified, along with their regulatory elements (Pan et al., 2018).
The fact that the crossing-over processes do not involve random regions along the chromosomes, but instead preferential sites for the establishment of chiasmata and for the exchange of corresponding parts between chromatids of homologous chromosomes corroborated the hypothesis advanced a few decades ago that inheritance depends on the recombination of “chromosomal blocks” and not on the recombination of single genes.
In sexually reproducing species, chromosomal blocks represent the genomic units of hereditary transmission. Therefore, genetic improvement activity acts not on single genes but on chromosomal blocks, namely, complexes of associated genes in the segments resulting from crossing-over. According to this interpretation, each crossover chromosome consists of a succession of blocks of maternal and paternal origin, whose number depends on the number of exchange events that occur between counterparts during meiosis. In each chromosomal block, it is possible to find genes controlling qualitative traits and complexes of polygenes involved in the expression of quantitative characters (QTLs, quantitative trait loci). The improvement of complex characters, controlled by many genes, implies simultaneous and laborious action on more chromosomal blocks (Gitonga et al., 2016). Additionally, the introgression of characters controlled by a single gene requires special attention since the desired gene will be inherited along with the entire chromosomal block of which it is a part. This situation, known as linkage drag, is particularly undesirable, especially if the quality and the quantity of unwanted genetic material that is associated with the gene of interest are unknown (Ward et al., 2017; Li et al., 2018).
The number of chromosomal blocks depends on the number of chromosome pairs and on their exchange frequencies. Both factors, in turn, depend on the species, but it is likely that the number of chromosomal blocks is limited in each genome (Table 3). From a practical point of view, understanding the number of chromosomal blocks on which it is possible to act in a genetic improvement program is crucial. In fact, whereas in pure lines (homozygous at all the loci) as well as in inbred lines, the position of crossing-over has no effect on the associations among genes or the combinations of genes in the gametes, in heterozygous lines, both the gene order in the association groups and the allele composition in the gametes are influenced by the position of crossing-over.
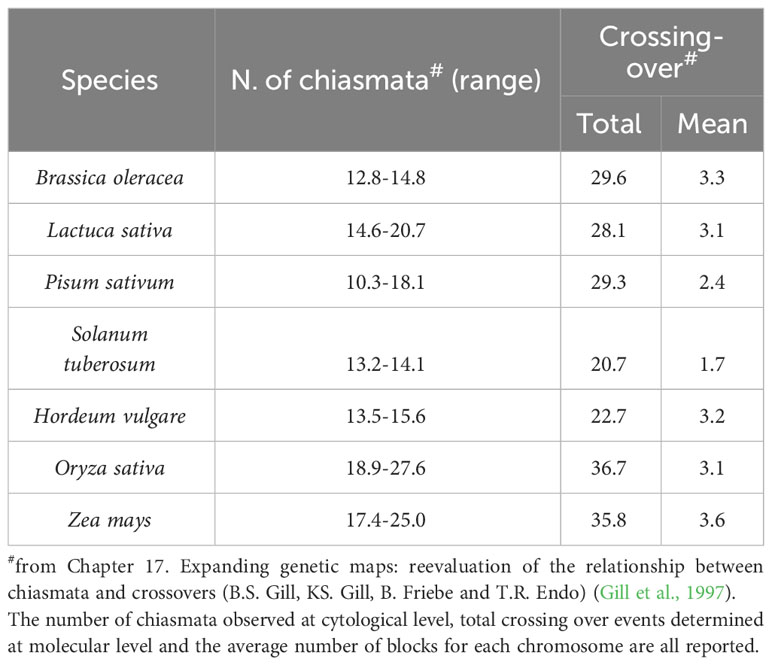
Table 3 Frequency of recombination events, in terms of number of chiasmata, for model cereal crops and some of the most important horticultural species.
Chromosomal blocks influence the manifestation of heterosis, conditioning both gene action and gene interaction. In 1917, Jones was the first to hypothesize and recognize the role of chromosomal blocks, proposing the dominance theory of associated factors to explain the heterosis phenomenon (Jones, 1917). In genetic terms, this theory relies on the cumulative effect of associated dominant genes in chromosomal blocks. In the following years there was intense debate about the type of gene action, and the heterosis phenomenon started to be considered an expression of real over-dominance (see section below).
To complicate things further, it must be acknowledged that an allele having complete dominance sometimes could also have an additive effect compared to the recessive one (i.e., the phenotypic value of the recessive allele is not always null). Furthermore, the effects of multiple alleles of different loci responsible for the same quantitative character are cumulative at the phenotypic level. Based on these considerations, in 1988 Hallauer and Miranda stated that “heterosis is the result of the cumulative effect of favorable alleles having partial or complete dominance” (Hallauer et al., 2010).
As previously mentioned, the first observations and the genetic basis of heterosis have been widely discussed in model maize (Shull, 1908; Bruce, 1910; Jones, 1917). The experimental evidence available at that time with which to formulate genetic hypotheses was substantial: i) heterosis was a consequence of the union between genetically different individuals, and ii) two individuals homozygous for different alleles were able to produce an F1 hybrid showing a phenotypic value than the average one calculated between the two parental lines or equal to the average phenotypic value of the “best” parental line (partial or complete dominance) and in some cases even higher than the average phenotypic value of the best parental line (over-dominance). Two theories based on these hypotheses were therefore advanced to explain the heterotic behaviour. According to the first one, elaborated by East in 1908 and Shull in 1912, a genetically differentiated germplasm could determine a “physiological stimulus directly proportional to the diversity of gametes involved in fertilization”. According to this interpretation, known as the over-dominance hypothesis, heterosis relates to the heterozygosity of the hybrid, which confers an advantage with respect to the homozygous parental lines. In a second hypothesis, heterosis was considered the consequence of the total or partial suppression of a deleterious recessive allele effect, transmitted by one of the parents, through the dominant allele transmitted by the other parent. This interpretation, known as the dominance hypothesis, was formulated by Bruce in 1910, with the following statement: “the effects of deleterious recessive genes could be nullified by a dominance effect in heterozygotes”. It must be recognized that Keeble and Pellew, in the same year, were actually the first to speak explicitly of the dominance hypothesis. Currently, heterosis is considered a biological phenomenon that is extremely difficult to explain in genetic molecular terms, mainly due to its multigenic nature. The classical theories of quantitative genetics formulated around the concepts of dominance and over-dominance were reviewed and refined by Crow (Crow, 1948) and Dobzhansky (Dobzhansky, 1952). According to the original dominance hypothesis, a hybrid displays heterosis when the favourable, dominant allele transmitted by one of the two parental lines masks the effect of the unfavourable, recessive allele transmitted by the other parent. This means that some lines can produce high-performing hybrids, while others can not due to overlapping allelic states (i.e. homozygous at the same loci). According to the over-dominance hypothesis, heterosis is instead attributable to specific allelic interactions at single loci in the hybrid and not possible in the parental lines. The contrast between the theory of dominance and that of over-dominance is not yet resolved. Although the over-dominance theory has become more popular, many researchers have pointed out that the gap (attributable to dominance) between genetic variance measurements and theoretical estimates could instead be due to both over-dominance and linkage disequilibrium between favourable alleles of loci associated in trans (pseudo over-dominance) (Gardner and Lonnquist, 1959; Lonnquist).
The genetic basis of heterosis has long been debated, but with the advent of the genomic era and new technologies in molecular biology, the tools for establishing its molecular basis are more accessible, allowing us to understand some important evidence. The genetic process of heterosis is complicated, especially that of yield heterosis, because of the numerous loci involved and the potential significance of the cumulative effects of dominance, over-dominance, pseudo-over-dominance, and epistasis (Guo et al., 2014; Huang and Mackay, 2016). These theories have been studied for a century, but despite their limitations, they continue to be a keystone of heterosis research itself.
Studies at the genome, transcriptome and proteome levels in model species have led to unexpected and unpredicted results. Despite the fact that allelic sequences in a particular genome might vary greatly, it was assumed that every gene in one individual should have an allelic counterpart in a different member of the same species. Violations in genetic microcolinearity were reported in model maize and potentially related both to the classification of heterotic groups and to the occurrence of hybrid vigour in this model species. Furthermore, allelic expression variation of fundamental genes has recently been uncovered in maize hybrids. Most of the investigated genes showed differences at the messenger level, ranging from unequal expression of the two alleles (biallelic) to expression of a single allele (monoallelic). Changes in mRNA expression levels and allele-specific transcript ratios are attributable mainly to differences in noncoding DNA sequences (i.e., cis- and trans-acting elements) (Yu et al., 2021). One of the most important findings was that genetically improved modern maize hybrids express both alleles at each locus, whereas less improved old hybrids frequently show monoallelic expression. Interestingly, the two alleles in the hybrids responded differently depending on plant tissue and environmental conditions, suggesting inequivalent functions of the parental alleles in the hybrids. Additional studies on gene dosage effects in either diploids or polyploids revealed that the expression of many genes, both in terms of transcripts and proteins, does not exhibit the mid-parent value expected in the case of additive gene action. Overall, the results indicated that allele dosage effects can play important roles in determining phenotypic diversity and have important impacts on hybrid vigour. Since housekeeping genes that encode metabolic functions usually show a greater degree of dominant/recessive behaviour between allelic alternatives and are believed to be less influenced by dosage effects, it has been argued that dosage effects reflect the dosage dependence of most regulatory genes. Following this train of thought, researchers have proposed that heterosis might be the result of different allele combinations being present at loci that contribute hierarchically to the regulatory networks controlling quantitative traits. With the support of next-generation sequencing (NGS), it is possible to create sizable sequence datasets that can be used for molecular breeding, genetic mapping, gene identification, and genetic diversity analysis as well as to characterize sequence diversity, and, additionally, might be able to provide some answers about the spectrum of genes that show regulated changes in expression patterns in hybrids. Thanks to the rapid advancement of genome sequencing technologies, genome-wide association studies (GWASs) have allowed the identification of gene loci connected to heterosis and associated phenotypes (Huang and Han, 2014).
Despite lots of progress has been made in understanding the phenomenon of heterosis at the genetic and genomics levels, the new molecular omics techniques are still developed and set on major model species (i.e., cereals) and Arabidopsis (Liu et al., 2022). However a recent growing number of horticultural crops have been examined using integrated omics (transcriptome, metabolome, proteome, and epigenome) approaches that are becoming more widely available (Li et al., 2017; Natalini et al., 2021; Pott et al., 2021; Yu et al., 2021). Moreover, the detection of the genomic divergence between the hybrid’s two sets of genomes, which is the cause of heterosis, made easier thanks to high-quality genome assemblies or whole genome resequencing of significant crop accessions (Sun et al., 2018; Zhang et al., 2008; Lai et al., 2010). These characteristics include allelic divergences like single nucleotide polymorphisms (SNPs) and minor insertions/deletions (INDELs), copy number variations (CNVs), and presence/absence variations (PAVs). The identification of heterosis-related loci and the clarification of the selection and integration of superior alleles during hybrid breeding in crops have been accomplished using quantitative trait locus (QTL) mapping or genome-wide association studies (GWAS) based on genome variation in populations (Lin et al., 2020; Huang et al., 2015; Xiao et al., 2021).
Until a few years ago, due to the limited number of genetic markers available, it was extremely difficult to determine the mechanisms of heredity underlying the transmission and expression of quantitative traits.
The success of molecular breeding for instance, has been greatly attributed to the use of marker-assisted selection (MAS) for many traits related to resistance (Mehraj et al., 2020; Baby et al., 2023). Furthermore, the prediction of high-yielding parental combinations with high levels of heterosis has been investigated to combine agronomic traits utilizing diallel breeding and to learn about yield-related QTLs (Rahman et al., 2022). More loci associated with yield should be discovered as high-throughput genotyping advances, which may promote the use of MAS for F1 breeding. Currently, the availability of genetic maps saturated with molecular markers allows the identification and precise location of the chromosomal regions where those genes that determine a quantitative character reside. Almost all QTLs described in plant species do not reflect a single Mendelian locus but rather a chromosomal tract that is likely to be identified, in all or in part, as a chromosomal block. Moreover, based on recent acquisitions concerning a number of molecular markers closely associated with QTLs in different species, the same chromosomal region is able to control multiple quantitative traits (Paterson et al., 1988; Tanksley, 1993; Melchinger et al., 2000; Morgante and Salamini, 2003).
To create plots of the impacts on a trait and their significance along the linkage groups, it is possible to perform a “one marker at a time” comparison of the mean values for a trait in the different genotypic groups in segregating populations or in GWAS. With this strategy, MAS became a requirement in breeding programs for disease resistance and improved crop quality. Although we can currently score thousands of SNPs, we still primarily study them “one at a time” along the genome. Conducting genome-wide scans for epistatic QTLs, which have little to no effect on a trait but have unexpected results when paired with QTLs at other positions, provides a more in-depth perspective on the examination of complex traits. According to Weinreich et al., epistasis can be characterized as a surprising phenotype when QTLs are combined, given the individual effects of the constituent QTLs (Weinreich et al., 2013).
Results supporting the pleiotropy of some QTL, able to influence two or more different and unrelated characters, suggest that natural selection led to the formation of chromosomal regions with closely associated genes that rarely recombine and that probably determine adaptive and reproductive capacity. The most surprising observation is that for most of the quantitative traits studied the number of QTLs is rather low, between 2 and 18, with an average number equal to 8 (Melchinger, 1999). Since it is unlikely that complex characters (like many quantitative traits) depend on so few genes, it can be hypothesized that in the currently available genetically improved materials many favourable alleles are fixed in all the lines examined. The gene relations evoked to explain the action of the genes included in QTLs range from additivity to dominance and epistasis; it is possible that they all contribute to heterotic manifestations, but it remains a responsibility of molecular biology to clarify the biochemical processes behind these terms. In maize, the identification of a plurality of QTLs that control a single character, made possible by the mapping carried out with the use of molecular markers closely associated with these loci, has highlighted the existence of different genic actions, mainly intralocus due to complete and partial dominance, but also to over-dominance and pseudo-overdominance (Stuber et al., 1992). However, the existence of over-dominance has often been traced to pseudo-over-dominance phenomena, derived from dominant alleles associated in the trans phase (Crow, 1999). Finally, in a limited number of cases, effects of epistasis were found, attributable above all to interactions between alleles of associated loci (Melchinger and Gumber, 1998), and qKWPE9, a mid-parent heterosis QTL regulating yield-related characteristics, was identified (Yi et al., 2019). In rice, the analysis of QTLs using molecular markers has shown that in this species the main genetic basis of heterosis is constituted by dominance, both total and partial: hybrid individuals with heterozygous loci have shown, for the individual quantitative trait examined, a phenotype equal to or intermediate to those of the respective pure parental lines (Xiao et al., 1995). Other additional examples have been described in rice, in which the QTL RH8 played a major role in yield heterosis by affecting spikelet number per panicle and effective panicle number in hybrid rice (Li et al., 2016). Furthermore, GW3p6 was detected as a heterotic QTL with considerable functions in increasing grain yield in rice (Wang et al., 2019). Finally, epistasis has never been detected between the chromosomal regions where strictly associated QTL molecular markers have been mapped. In wheat, another autogamous species, the experimental data on the QTLs acquired using molecular markers suggest that heterosis is compatible with the dominance hypothesis, with interaction effects between genes of associated loci (Pickett and Galwey, 1997).
In addition to cereal crops, important examples are also available in horticultural crops. Recently, Yu et al. (Yu et al., 2021) summarized QTL effects on heterosis based on 35 studies that mainly addressed 6 canonical crops and horticultural species (Yu et al., 2021), confirming that, among the types of QTL effects, dominance and epistasis had equal proportions (19% and 23%, respectively). It is interesting to note that out of all the effects, the over-dominance effect accounted for the highest percentage (42%). This indicates that even though the plant genome has a large number of gene loci, they combine to produce a variety of complicated, challenging-to-replicate effects that lead to heterosis. Of these effects, over-dominance effects frequently occurred and considerably aided in the development of heterosis. Additionally, the tomato market has successfully harnessed the over-dominance effect for artificial breeding (Krieger et al., 2010), and the role of epistasis between two independently segregating homologous MADS box mutations was investigated (Soyk et al., 2017). In addition, most commercial cabbage varieties that are frequently sold on the market are hybrids, as cabbage clearly displays heterosis. The major focus in recent studies on heterosis in cabbage has been on the use of male sterility for hybrid breeding and the relationship of genetic distance with heterosis (Dafna et al., 2021; Yu et al., 2021). Agronomically relevant QTL discovery was the main topic of other papers on cabbage yield. These investigations found 66 and 73 QTLs linked to mid-parent heterosis and transgressive heterosis of twelve yield-related characteristics, respectively (Li et al., 2023).
In fruits and vegetables, yield-related traits are also quantitative traits: in the wild species Solanum pennellii introgressed into the elite inbred line M82, the over-representation of over-dominant QTLs for reproductive traits was observed, while this was not observed for non-reproductive traits (Gur et al., 2004; Semel et al., 2006; Gur and Zamir, 2015; Shiraki et al., 2023). Furthermore, in fruit horticultural species, the yield is directly affected by flowering time, as well described in the tomato system, in which increased yield is regulated by the balance between the antagonistic influences of SFT (a floral activator), and SELF PRUNING (SP, a repressor) (Thouet et al., 2008). Increased fruit output and over-dominance of yield heterosis involving a single gene were observed in plants with sft/SFT heterozygosity and an sp/sp homozygous background (Krieger et al., 2010). For horticultural melon crops, the fruit size and shape traits are the most relevant agronomically important traits, and QTL analysis for both allowed the identification of multiple QTLs in F2 populations derived from crosses between wild and cultivated lines (Diaz et al., 2017; Zhao et al., 2019; Lian et al., 2021), with additive gene action at these QTLs for fruit size (Diaz et al., 2017) and a dominance effect for fruit shape (Fernandez-Silva et al., 2009).
Regarding gene expression in hybrids, two distinct levels can be imagined in relation to the genetic theories previously discussed in this review. In the first model, it can be considered that when two different alleles of a given gene, each transmitted by a parental line, are inherited together in the hybrid, expression is similar to that of one of the homozygous genotypes (complete dominance) or between the expression levels of homozygous genotypes (additivity or incomplete dominance). In the second model, on the other hand, the presence of two different alleles at the same locus leads to their interaction, which is capable of triggering deviant gene expression with respect to that expected on the basis of data from homozygous parental lines (over-dominance). The difficulties listed below appear to be of particular importance for the effects they may have on hybrids among those that arose at the molecular level. What are the levels of transcriptional efficiency and how much do alternative allelic variants of a gene differ from a nucleotide perspective? How functionally advanced are the proteins encoded by the potential allelic variations at a certain locus? Is it possible to say that all alleles are transcriptionally active and capable of controlling the production of proteins with metabolic functions? Or is it more likely that some alleles of particular genes will not be translated or transcribed or that their offspring will not function?
To date, studies of comparative expression between inbred lines and hybrids have been limited to a reduced number of genes and to one or a few organs. For the genetic molecular understanding of the influence of gene expression levels on heterosis, the possibility of evaluating a number of genes representative of the entire genome and several vegetative and reproductive organs at different levels of development is crucial. The experimental challenge is to define which changes in gene expression are crucial to promote the heterozygote advantage. Since the characters that indicate heterosis in hybrids are under complex genetic control and only rarely increase more than twice in terms of phenotypic value, relatively modest changes in many genes can play an important role compared to substantial changes in a few genes (Birchler et al., 2003). Significant in this regard are also the data that are accumulating on the structure of the genome that highlighted unexpected differences between crossed materials. For example, the comparison of the genomic structure of the locus bronze1 (Bz1) in several maize inbred lines showed that the intergenic regions contain different types and combinations of transposons and retrotransposons, that are in different positions. This comparison also showed that inbred lines are different in terms of gene composition: several loci of a line present genes not found at the corresponding loci of another line, thus violating intraspecific collinearity (Fu and Dooner, 2002). Brunner et al. (Brunner et al., 2005), through genomic comparison of the genetic content of BAC allelic contigs related to the inbred lines B73 and Mo17 documented the degree and type of intraspecific diversity existing in maize and reported that the polymorphisms involve at least 10,000 sequences and are mainly due to DNA insertions. This work also showed that the corn genome is constantly evolving because certain transposable elements are capable of causing variations at the level of the coding regions and at the level of the regulatory regions through duplication mechanisms of single genes and integration of portions of different genes.
Taking into account these new acquisitions, in addition to an effect due to gene dosage as a possible hypothesis underlying heterosis, another possible hypothesis for maize is based on the existence of regulatory regions in transposons and retrotransposons able to provide ample and diversified possibilities for gene expression and/or to determine gene silencing due to the presence or absence of repeated sequences. Acquired data on gene expression indicate that in F1 hybrids the transcript level of many genes is significantly different from the average of the transcript levels observed in the corresponding parental lines (Osborn et al., 2003; Song and Messing, 2003). Similar data were also found for the level of proteins (Romagnoli et al., 1990; Leonardi et al., 1991), demonstrating that in the hybrid, substantial changes actually occur with respect to the inbred lines. In maize, expression analysis of a dozen genes coding for endosperm zeins has shown in most hybrids a level of gene expression twice as high as or lower than the average level of parental lines (Romagnoli et al., 1990). Similar results were obtained in maize by Guo et al. (Guo et al., 2003), who studied the expression profiles of thousands of genes in the endosperm. More recently, analyses carried out on the expression of 30 genes in maize hybrids confirmed that a substantial proportion of these genes do not show transcription levels attributable to an additive model and that in hybrids the expression of genes is frequently repressed or enhanced (Auger et al., 2005).
In pepper, a comparison between transcriptomes derived from parental and F1 lines at the seedling and blooming stages, has shown that non-additive gene expression, i.e., over-dominance, underdominance, high-parental dominance, and low-parental dominance, is the most common effect. Additionally, “primary metabolic process,” “photosystem,” “phosphotransferase activity,” and “kinase activity” were revealed to be overrepresented in the DEG analysis between the F1generation and its parental lines utilizing gene ontology (Shu et al., 2021). Furthermore, in both the seedling and flowering stages, different noncoding RNAs, including miRNAs, long noncoding RNAs, and circular RNAs, were detected in F1s compared to the parental lines, indicating their connection to heterosis (Shu et al., 2021) and suggesting that epigenetic variation might be associated with heterosis (Miyaji and Fujimoto, 2018; Tonosaki et al., 2023).
In clonally propagated horticultural crops, such as potato, sales of seeds are not major, and F1 varieties have not been developed, leading limited heterosis studies. However, strategies through seed sales of potato (hybrid seeds) are also being considered for this kind of crop (Zhang et al., 2021; Li et al., 2022). The reciprocal hybrids from the heterosis study of homozygous diploid potato parental lines showed heterosis in vegetative size, grow size, and tuber yield (Li et al., 2022). A metabolome investigation using the same tissues revealed non-additive accumulation of metabolites that varied among the three tissues. Transcriptome analysis utilizing seedling leaves, flowers, and growing tubers revealed that 4–15% of genes were non-additively expressed. Mid-parent heterosis in seedling leaves and flowers was positive for primary metabolites, whereas it was negative for secondary metabolites. Both primary and secondary metabolites in tubers showed negative mid-parent heterosis (Li et al., 2022).
The expression levels of the sucrose synthase sh1 (Shrunken1) and cyclin δ3 (Cyclin delta-3) genes in inbred lines and their hybrids are reported. Adams et al. (Adams et al., 2003) also highlighted unequal contributions of parental genomes in newly synthesized cotton allopolyploids, suggesting that when two different genomes are combined together in a hybrid, the gene expression levels are not predictable based on their expression levels in parents. Guo et al. (Guo et al., 2004) studied mRNA levels transcribed by specific alleles in seedlings and immature ears of corn hybrids at different developmental times, under normal and water stress conditions and under different cultivation densities. Among the 15 genes studied, 11 showed significant differences in mRNA levels, ranging from the unequal expression of the two alleles (biallelic expression) to the expression of a single allele (monoallelic expression). Maternal or paternal origin did not influence, or only modestly influenced the ratio between the number of transcribed alleles. One of the most important results of this study is the one that emerged from the comparison between hybrid varieties developed at different times: more modern hybrids, deriving from intense genetic improvement activity, almost exclusively showed biallelic-type expression, while the older ones frequently showed monoallelic-type expression. Even more interesting are the results found under the different abiotic stress conditions (density and drought): in hybrids, the two alleles at different loci always showed different expression (Guo et al., 2004).
Gene dosage can also play a key role in heterotic expression. Studies of the expression levels of nuclear and mitochondrial genes in maize aneuploids showed consistent effects on quantitative traits (Lee et al., 1996; Auger et al., 2001). Regarding this point, the comparison between what happens in diploids and in polyploids, in particular newly synthesized autopolyploids, can be very interesting. Polyploidy, as a general effect, leads to an increase in gene expression levels in proportion to the gene dosage conferred by the level of ploidy, as has been demonstrated for many genes in corn euploid series (monoploid, diploid, triploid and tetraploid) (Osborn et al., 2003). In diploids, the consequences of gene dosage were analyzed for many developmental regulatory genes, such as those that control plant architecture (tbl in maize), fruit size (fw2.2 in tomato) and flowering time (FLC in Arabidopsis and Brassica). Overall, the results achieved suggest that gene dosages play a key role in controlling quantitative traits. Dosage effects were observed in heterozygous genotypes that exhibit intermediate levels of gene expression and phenotypic expression compared to homozygous genotypes characterized by null or very low expression and high expression. Therefore, in the case of genes affected by the number of copies, polyploidy can increase the potential variation in their expression levels and, consequently, the phenotypic expression of the corresponding characters. The three possible genotypes at the diploid level contrast with nine possible genotypes at the tetraploid level. Additionally, the phenotypic effects increase from three in the diploids to five in the tetraploids (the marked genotypes to which each of the diploid genomes contributes two alleles (A/a and A’/a’) could be fixed in autogamous species with tetraploid disomic heredity). This phenomenon could also not widen the range of phenotypic variation with respect to diploids, although in polyploids, it is possible that the highest and the lowest gene doses give rise to even more extreme antagonistic phenotypes, but what counts is the creation of more intermediate phenotypic classes some of which could have a selective advantage. In theory, both autopolyploids and allopolyploids can benefit from this effect, although the intermediate genotypic classes would be stabilized in polyploid disomic heredity and not in of polysomic inheritance, only in the presence of an autogamous reproductive system. A clear dosage effect was also found by studying the expression profiles of thousands of genes in maize endosperm. Data showed that the level of gene expression in the endosperm triploid tissue is mainly dependent on the dosage since, from a quantitative point of view, transcript levels reflect the genomic contribution of the parents, showing a maternal versus paternal ratio of 2:1. However, about 8% of the genes analyzed showed significant deviations, showing non-additive expression. In hybrids, the expression levels of these genes were very similar to those observed in one of the two parental lines. Furthermore, genes with expression levels similar to those of the mother were clearly superior to genes with expression levels similar to paternal ones (Guo et al., 2003). If heterosis in polyploids is truly conditioned by gene dosage effects at a number of loci, these data could also be related to what happens in diploids since many of the loci for quantitative characters show an absence of dominance or semi-dominance effects (Tanksley, 1993). This means that the heterozygous genotype for a specific QTL shows a phenotype different from that of the two homozygotes and closer to that one or the other homozygote, although it has been shown that most of the time the heterozygote has an intermediate value between the two homozygotes. The modulated gene expression variation due to dosage effects in combination with the creation of allelic variants is considered responsible for the expression of new and more advantageous characters in polyploids (Osborn et al., 2003).
Experimental evidence suggests that, in addition to the genetic basis for heterosis, epigenetic and other non-genetic processes may also play an important role. Epigenetics is defined as the study of heritable changes in gene expression that do not depend on an organism’s genotype but rather on changes to the chromatin structure and/or the post-transcriptional process. In particular, DNA methylation-, histone modification-, and small RNA (sRNA)-related pathways are the main mechanisms that lead plants to translate their genotype in distinct directions (Rehman et al., 2021). However, the majority of current research on potential connections between epigenetics and heterosis is based on statistical correlation models without a detailed explanation of the mechanisms at play. Since it would take some effort to accurately determine how epigenetics contributes to heterosis, this offers an intriguing area of study for plant breeding science.
To understand the epigenetic impact of the alterations in gene expression levels in F1 hybrids and their parental lines, global DNA methylation analysis and sRNA profiling are frequently used. The relationship between DNA methylation and heterotic effects in F1 hybrids has been confirmed in a number of studies, observing higher overall levels of DNA methylation in F1 hybrids than in their parents (Chen, 2013; Greaves et al., 2015). In model Arabidopsis, the role of epigenetic control in the expression of heterosis was demonstrated by using epiRILs with altered levels and distributions of DNA methylation along the genome: in F1 hybrids, these epigenetic modifications alter biological processes and phenotypic features such as blooming, fruiting, yield, energy, metabolism, and biomass, and light and hormone signalling (Zhang et al., 2016). Additionally, one study showed that a great majority of cytosine methylation sites in maize parental lines had changed in their hybrids, confirming the possible relevance of methylation-pattern remodelling for heterosis in major crops (Hochholdinger and Hoecker, 2007). Additional investigations on several other major crop plants further supported this hypothesis, and a strong correlation between cytosine methylation patterns and genetic expression changes among both rice hybrids and their inbred parental lines was also observed (He et al., 2010). The methylation of DNA can be directly driven by sRNA molecules that participate in the RNA-directed DNA methylation (RdDM) pathway, post-transcriptional silencing, and transcriptional silencing. To silence transcriptional genes, 24 nucleotide sRNAs target adjacent regions for DNA methylation (Melnyk et al., 2011). Several reports supported the hypothesis that these multi-functional sRNAs might also be involved in heterosis manifestation, since their accumulation in hybrids of model species was significantly lower than that in parents, correlating to decreased methylation patterns (Groszmann et al., 2011). This correlation was confirmed in many independent studies carried out on wheat, maize and rice, in which sRNA accumulation analysis showed significant variation in sRNA amounts between hybrids and their respective parental lines (Chen, 2010; Chodavarapu et al., 2012; Kenan-Eichler et al., 2011). However, the specific methylated regions and the mechanisms influencing hybrid performance remain unclear and have become a stimulating topic to be explored in horticulture research.
In addition, studying the relationship between histone modification and heterosis is particularly challenging because of its complexity. To date, the well-known genome of the model Arabidopsis has been the focus of the most prominent investigations of the potential impact of histone modification on heterosis. By observing Arabidopsis F1 hybrids’ circadian clock and associated genes, it was discovered that the transcription of these genes changed in association with histone modifications. Plants that successfully maintain synchronization between their internal circadian cycle and their environment are more robust than those that do not (Kim et al., 2017). Further studies on crop plants, especially the cereal models maize and rice, have also been performed to confirm the relationship between histone modification and heterosis in addition to the evidence from Arabidopsis investigations. For example, maize F1 hybrids showed significant expression variations in histone mark patterns compared to their parental inbred lines, providing a starting point for the investigation of specific histone modifications regulating crop hybrid performance (Jahnke et al., 2010). In rice, using high-throughput ChIP-Seq, the marks H3K4me3, H3K9ac, and H3K27me3 were analysed in the ‘japonica’ and ‘indica’ subspecies and their F1 hybrids, confirming differential enrichment of H3K4me3 and H3K27me3 between the hybrids and their parents (He et al., 2010).
The expression and transcriptional regulation of most genes are controlled by a complex network of regulatory factors, such as transcription factors. In general, the control mechanism can be positive or negative depending on whether the product of the regulatory gene is able to promote or suppress the expression of one or more structural genes. In particular, the modulation is deeply influenced by regulatory factors, which are real transcription factors that exert their effect directly on the activity of RNA polymerase or other regulatory proteins by recognizing specific DNA sequences. When a regulatory factor is able to promote the binding of generic transcription factors and that of RNA polymerase with the specific region of the promoter, intensifying the level of transcription, it is called a transcription activation factor. Otherwise, if the intervention of the regulatory factor induces a transcription silencing, it is called a transcription repression factor. In plants, there are numerous genes encoding transcription factors, that are required for the initiation of transcription by one of the RNA polymerases or regulatory transcription factors, involved in transcription intensification or repression. The number of regulatory factors that act in diploids is high, although lower than that of regulatory factors that act in polyploids. The complexity of regulatory factors is certainly greater in heterozygotes than in homozygotes. This complexity increases, for example, three times for the regulatory factors corresponding to dimeric proteins (for example, A1A1 or A2A2 in the inbred lines versus A1A1, A1A2 and A2A2 in hybrids). The action and interaction of regulatory factors, each of which is encoded by a distinct gene and acts on the level of expression of fundamental genes in a hierarchical manner, are different in homozygotes compared with heterozygotes. The functioning of regulatory networks in hybrids depends on how the factors encoded by the individual genomes interact between them: it is likely that this activity in the hybrid genome is quite modified compared to that in the single parental genomes and that the changes are more marked in hybrids produced by crossing genetically divergent inbred lines. The typical homozygous condition of inbred lines can simplify the operation of these networks due to the presence of alleles identical at many loci. In hybrids the functioning of regulatory networks should be less prone to alterations when inbred lines are genetically similar. However, the overall effects on gene expression are difficult to predict since some regulatory factors act as promoters and others as inhibitors of gene expression. Today, it is believed that interactions between regulatory factors play an important role in the vigour increase associated with heterozygosity. Both diploids and polyploids have specific mechanisms for maintaining high levels of allelic variability at individual loci: in both cases, in fact, the overall vigour of the plants is positively correlated with heterozygosity or with the diversity of the genome. Some authors hypothesize that the expression model found in hybrids is not additive because of the interaction of regulatory factors encoded by different alleles inherited from single parental genomes, which results in new levels of expression of those genes under their control in the heterozygous condition (Hammerle and Ferrus, 2003; Auger et al., 2005).
Based on the information acquired so far, it is believed that the combination of divergent genomes of the same species (Zea mays) or of different species of the same genus (Drosophila) may determine alterations in regulatory genes with consequent modulations of the expression of fundamental genes. Changes in gene expression may be due not only to allele dosage effects (Lee et al., 1996; Auger et al., 2001) and/or allele interactions (Osborn et al., 2003) of regulatory genes, but also to effects related to specific trans-acting elements that are able to act on the expression of most of the genes of a given genome (Guo et al., 1996; Birchler et al., 2001), as demonstrated by studies conducted in aneuploids (Birchler and Newton, 1981; Matzke et al., 2003). It can be hypothesized that the reduction in gene expression usually observed in monosomic and trisomic lines represents a detrimental factor for the vigour responsible for the phenotypic alterations associated with aneuploidy. Most of the regulatory genes show action strongly conditioned by allelic dosage effects, while their target genes usually show normal dominance/recessive relationships between the possible allelic variants (Birchler et al., 2003). A possible explanation for the dichotomy between dominance relationships and dosage effects is provided by analyses conducted on yeast genes whose expression is conditioned by the number of copies (Papp et al., 2003). The genes that at the haploid level exert insufficient action for the development of cells encode for polypeptides that participate in the formation of functional molecular complexes under diploid conditions. In plants it is known that genes with regulatory action very often preside over the synthesis of polypeptides that form of protein complexes composed of by multiple subunits. Based on several experimental observations, it can be assumed that even in multicellular organisms regulatory genes exert action dependent on their dosage, and the constitutive genes that control metabolic functions are less conditioned by dosage effects (Birchler et al., 2001). Consequently, the manifestation of quantitative traits could be controlled to a large extent by a plurality of loci whose action is dependent on the dosage. According to this hypothesis, heterosis is determined by the efficiency of specific alleles of regulatory genes, which in turn control the expression of the genes at the QTL most directly responsible for the expression of quantitative characters (Birchler et al., 2003). One aspect that has recently been considered concerns the inactivation of maternal and/or paternal alleles during gamete formation (genomic imprinting). The molecular mechanism that controls this phenomenon in maize is believed to be responsible for the quantitative deviations of some gene transcripts in hybrids with respect to those in the parental lines (Guo et al., 2003). Variations in the allelic expression of autosomal genes, which are not attributable to genomic imprinting, have recently been documented in mouse hybrids (Cowles et al., 2002). These variations in transcript levels, considered to be real regulatory mechanisms, have been attributed to polymorphisms found in intergenic regions, mostly differences in non-coding DNA sequences, while epigenetic phenomena have not been observed. At present, it is believed that the nucleotide differences present in the regulatory region of a gene may condition the expression pattern and be the cause of the variation at the phenotypic level (Knight, 2004). In conclusion, considering the overexpression of many fundamental genes, such as housekeeping genes, as hypothesized by Birchler et al. (Birchler et al., 2003), heterosis could be traced back to the action of regulatory genes or to the alteration of regulatory networks (Song and Messing, 2003).
In addition to regulatory factors, there are numerous physiological factors that also contribute to the heterosis phenomenon (Birchler et al., 2003; Chen, 2013; Fujimoto et al., 2018). Among these, hormone balance has been invoked to explain heterosis. Plant hormones are the master regulators of plant architecture and function. In particular, there is substantial evidence demonstrating that gibberellins (Gas) are the most prominent factors related to heterotic phenotypes that manifest themselves in shoot growth, regulation of stem elongation, flowering development, and leaf senescence. This association has been deeply revised for many crops (Zanewich and Rood, 2020); however, at present, the research is still focused on model species, and little information is available for horticultural crops.
Numerous investigations of F1 hybrids, in which genes putatively involved in heterosis were modulated, revealed altered phytohormone-related pathways (Shen et al., 2017), as observed in the cases of commercial crops such as rice (He et al., 2010), maize (He et al., 2012), and Brassica (Shen et al., 2017). The influence of the phytohormone GA on hybrid vigour has been largely investigated in maize, in which it was shown that hybrids have a higher GA content than their inbred parents. For maize, Zanewich and Rood (1988) discovered in their studies on shoot growth that hybrids had higher levels of endogenous Gas than their parental inbred lines, confirming an influence of phytohormones on heterosis. Additionally, the authors reported that a lack of endogenous GA prevented the inbred lines from growing normally, which could result in inbreeding depression. This direct correlation between GA levels and heterosis was also observed in other agronomically important species, such as sorghum, even if the effects were not as pronounced as those in maize. In addition, analogous observations, in terms of a more rapid shoot growth rate in hybrids than in inbred lines due to a higher content and altered metabolism of GA, have been reported in other plant species (e.g., poplar) (Stuber, 1997). Additionally, in wheat hybrids, higher GA4 levels were observed, demonstrating that the increased elongation of the uppermost internode contributed mostly to the heterosis in plant height (Zhang, 2007).
The expression regulation of plant hormone-related genes and their putative involvement in developmental processes was investigated in Brassicaceae (Kong et al., 2020). Up- and downregulated DEGs associated with ABA, auxin, ethylene and GA in commercial Chinese cabbage hybrids were identified. Previous research revealed that reduced expression of genes related to defence-responsive plant hormones may contribute to heterosis by lowering the energy cost for defence and allocating resources to plant growth (Gonzalez-Bayon et al., 2019). In particular, the ABA-responsive gene ABF4 (BraA03g038820.3C), upregulated in the hybrid, was identified to be expressed in guard cells of Arabidopsis (Na and Metzger, 2017), while it was noted that the increase in PYL8 (BraA02g014770.3C) expression mediates responses such as stomatal closure that result in poor transpiration during drought conditions (Lim et al., 2013). The upregulation of GASA6 (BraA02g022450.3C), GA3OX1 (BraA06g011600.3C), GAST1 (BraA01g040480.3C) and GA2OX8 (BraA02g011510.3C) was also observed in cabbage hybrids relative to their parents. It was found that changes in the leaf cells due to hormonal variations at early stages of development may lead to a continuing increase in leaf size in F1 hybrids of cabbage, and this property could be important in driving the increased biomass of the hybrid compared with those of the parents (Saeki et al., 2016).
Finally, changing the expression peaks of circadian clock genes may also have an impact on other biological pathways, such as those that regulate the expression of phytohormone signalling genes, which are altered in plant allopolyploids and hybrids (Chen, 2013).
Over time, a sizable amount of literature on heterosis has been developed, the majority of which is difficult to exhaustively portray and comprehend. Since the beginning of the 20th century, when the first hypothesis for the heterosis phenomenon was formulated, this topic has been a constant in international research, although the focus has been, for a long time, on its implications in agricultural practices rather than on elucidating its scientific basis. Scientists could not get beyond the enunciation of genetic theories, dominance and over-dominance in the first place, simply trying to describe hypothetical processes rather than explaining their biological and molecular fundamentals without knowing the mechanism of action and interaction of the genes.
Although the understanding of heterosis applies mainly to model cereal crops, like such as rice and maize, with the introduction of new molecular approaches based on genome-wide techniques, it has been possible to expand this knowledge to the horticultural species. There are several reasons why exploiting heterosis in vegetable crops is important since their productivity and quality can be increased via exploitation of this phenomenon, which also strengthen plant tolerance to biological and environmental challenges. Vegetable crop plants are critical for human health and their yield improvement results are crucial. Heterosis has played and will continue to play a part in meeting this demand: new F1 varieties have been developed on the basis their heterotic vigour, and several vegetables currently have a significant market share held by F1 types. Other types of F1 hybrids, such as interspecific and intraspecific hybrids obtained by crossing parental lines within the same genus, are frequently utilized in crop breeding. In contrast to intra-subspecies crossings and given the greater genetic distance of their parents, interspecific or inter-subspecies hybrids may exhibit higher heterosis. Interspecific hybrids have more vegetative biomass, but the features of the two original species are usually different enough that it is challenging to establish criteria for heterosis estimation. However, there may be significant opportunities for greater yield potential through interspecific or inter-subspecies crosses.
The efficacy of molecular breeding has been demonstrated, and through marker-assisted selection strategies, it has been used to exploit resistance genes for reducing disease susceptibility in crops, for instance, or for identifying the suitable parentals carrying genomic and phenotypic traits of interest, yield, for breeding new progenies. There is now a wealth of knowledge regarding the diallelic breeding method for combining agronomic characteristics and yield-related QTLs. As high-throughput genotyping approaches are being developed, more loci associated with phenotypic traits will probably be discovered, which could encourage the use of MAS for F1 hybrid breeding, including in horticultural species.
Integrated transcriptome, metabolome, proteome, and epigenome (omics) analyses were carried out that provided new information in numerous vegetable crops, even if the analysed stages and tissues have inevitably impacted the obtainable transcriptomic profiles. However, these multi-omics analyses have provided insight into heterosis and facilitates the exploitation of heterosis not only in breeding of major model crops, but also in the new frontiers in horticulture research. In fact, the characterisation with the other omics techniques could be crucial in helping uncover useful biomarkers of heterosis, as the proteome and metabolome could better reflect the plant phenotype than its transcriptome. Furthermore, the level of tissue and stage variation in the horticulture crops’ epigenomes is still largely unknown, but there is some indication that epigenetics play a consistent role in heterosis, and for this reason, future research should be focused on comprehending this molecular aspect of heterosis. Currently, it is thought that heterosis is the direct consequence of the interaction between genomes that results in intricate changes at the genetic, epigenetic, biochemical and regulatory network levels. The association between the genetic hypotheses and the molecular processes leading to heterosis will likely be clarified in the coming years as a result of a knowledge of the connections between various multi- and trans-disciplinary investigations. Thus, current developments in novel genetic and genomic technologies are expected to increase our understanding of the intricate relationship between the structural organization of the genome, its gene expression, and the resulting phenotype.
Conceptualization, ML and GB. Investigation and resources, SF, FS, FP and AV. Data curation, SF and FS. Writing—original draft preparation, FS, FP, and AV. Writing—review and editing, SF, FP and AV. Visualization, SF, FP, AV and GB. Supervision, ML and GB. Project administration, G.B. Funding Acquisition G.B. All authors contributed to the article and approved the submitted version.
The study was carried out within the Agritech National Research Center and received funding from the European Union Next-Generation EU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) – MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4—D.D. 1032 17/06/2022, CN00000022). In particular, our study represents a research paper within the Task 4.1.1. (Spoke 4).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) GB, AV, FP declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adams K. L., Cronn R., Percifield R., Wendel J. F. (2003). Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. U.S.A. 100, 4649–4654. doi: 10.1073/pnas.0630618100
Aguilar M., Prieto P. (2021). Telomeres and subtelomeres dynamics in the context of early chromosome interactions during meiosis and their implications in plant breeding. Front. Plant Sci. 12, 672489. doi: 10.3389/fpls.2021.672489
Am E.-A., El-Hadi A., M Fathy H., Abdein M. (2014). Heterosis, Heritability and Combining Abilities for some Earliness Traits in Squash (Cucurbita pepo, L.). Alexandria Sci. Exchange Journal: Int. Q. J. Sci. Agric. Environments 35, 203–214. doi: 10.21608/asejaiqjsae.2014.2612
Auger D. L., Gray A. D., Ream T. S., Kato A., Coe E. H. Jr., Birchler J. A. (2005). Nonadditive gene expression in diploid and triploid hybrids of maize. Genetics 169, 389–397. doi: 10.1534/genetics.104.032987
Auger D. L., Newton K. J., Birchler J. A. (2001). Nuclear gene dosage effects upon the expression of maize mitochondrial genes. Genetics 157, 1711–1721. doi: 10.1093/genetics/157.4.1711
Baby J., Thomas T., Dennis Thomas T. (2023). “Molecular markers for harnessing heterosis,” in Molecular marker techniques. Ed. Kumar N. (Singapore: Springer). doi: 10.1007/978-981-99-1612-2_1
Basunanda P., Radoev M., Ecke W., Friedt W., Becker H. C., Snowdon R. J. (2010). Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). Theor. Appl. Genet. 120, 271–281. doi: 10.1007/s00122-009-1133-z
Behr M., Sergeant K., Leclercq C. C., Planchon S., Guignard C., Lenouvel A., et al. (2018). Insights into the molecular regulation of monolignol-derived product biosynthesis in the growing hemp hypocotyl. BMC Plant Biol. 18, 1. doi: 10.1186/s12870-017-1213-1
Bhakta M. S., Jones V. A., Vallejos C. E. (2015). Punctuated distribution of recombination hotspots and demarcation of pericentromeric regions in Phaseolus vulgaris L. PloS One 10, e0116822. doi: 10.1371/journal.pone.0116822
Birchler J. A., Auger D. L., Riddle N. C. (2003). In search of the molecular basis of heterosis. Plant Cell 15, 2236–2239. doi: 10.1105/tpc.151030
Birchler J. A., Bhadra U., Bhadra M. P., Auger D. L. (2001). Dosage-dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev. Biol. 234, 275–288. doi: 10.1006/dbio.2001.0262
Birchler J. A., Newton K. J. (1981). Modulation of protein levels in chromosomal dosage series of maize: the biochemical basis of aneuploid syndromes. Genetics 99, 247–266. doi: 10.1093/genetics/99.2.247
Birchler J. A., Yao H., Chudalayandi S., Vaiman D., Veitia R. A. (2010). Heterosis. Plant Cell 22, 2105–2112. doi: 10.1105/tpc.110.076133
Birdseye D., De Boer L. A., Bai H., Zhou P., Shen Z. X., Schmelz E. A., et al. (2021). Plant height heterosis is quantitatively associated with expression levels of plastid ribosomal proteins. Proc. Natl. Acad. Sci. USA 118, e2109332118. doi: 10.1073/pnas.2109332118j1of7
Bruce A. B. (1910). The mendelian theory of heredity and the augmentation of vigor. Science 32, 627–628. doi: 10.1126/science.32.827.627-a
Brunner S., Fengler K., Morgante M., Tingey S., Rafalski A. (2005). Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 17, 343–360. doi: 10.1105/tpc.104.025627
Burton J. W., Brownie C. (2006). Heterosis and inbreeding depression in two soybean single crosses. Crop Sci. 46, 2643–2648. doi: 10.2135/cropsci2006.03.0156
Buso J. A., Boiteux L. S., Peloquin S. J. (2003). Tuber yield and quality of 4x-2x (FDR) potato progenies derived from the wild diploid species solanum berthaultii and Solanum tarijense. Plant Breed 122, 229–232. doi: 10.1046/j.1439-0523.2000.00037.x
Bustamante F. O., Aliyeva-Schnorr L., Fuchs J., Beier S., Houben A. (2017). Correlating the genetic and physical map of barley chromosome 3H revealed limitations of the FISH-based mapping of nearby single-copy probes caused by the dynamic structure of metaphase chromosomes. Cytogenet. Genome Res. 152, 90–96. doi: 10.1159/000478631
Chandel R., Sadashiva A. T., Ravishankar K. V., Das A., Rout B. M., Singh S. (2021). Genetic combining, heterosis analysis for horticultural traits in tomato (Solanum lycopersicum L.) using ToLCV-resistant lines and molecular validation of Ty genes. Plant Genet. Resour. 19, 512–521. doi: 10.1017/S1479262121000630
Chen Z. (2013). Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 14, 471–482. doi: 10.1038/nrg3503
Chen H. M., Chen L. T., Patel K., Li Y. H., Baulcombe D. C., Wu S. H. (2010). 22-nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. U S A. 107 (34), 15269–15274. doi: 10.1073/pnas.1001738107
Cheng S. H., Zhuang J. Y., Fan Y. Y., Du J. H., Cao L. Y. (2007). Progress in research and development on hybrid rice: a super-domesticate in China. Ann. Bot. 100, 959–966. doi: 10.1093/aob/mcm121
Choi K., Henderson I. R. (2015). Meiotic recombination hotspots - a comparative view. Plant J. 83, 52–61. doi: 10.1111/tpj.12870
Choi K., Zhao X., Kelly K. A., Venn O., Higgins J. D., Yelina N. E., et al. (2013). Arabidopsis meiotic crossover hot spots overlap with H2A.Z nucleosomes at gene promoters. Nat. Genet. 45, 1327–1336. doi: 10.1038/ng.2766
Chodavarapu R. K., Feng S., Ding B., Simon S. A., Lopez D., Jia Y., et al. (2012). Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. U S A. 109 (30), 12040–12045. doi: 10.1073/pnas.1209297109
Civardi L., Xia Y., Edwards K. J., Schnable P. S., Nikolau B. J. (1994). The relationship between genetic and physical distances in the cloned a1-sh2 interval of the Zea mays L. genome. Proc. Natl. Acad. Sci. U.S.A. 91, 8268–8272. doi: 10.1073/pnas.91.17.8268
Colombo N., Galmarini C. R. (2017). The use of genetic, manual and chemical methods to control pollination in vegetable hybrid seed production: a review. Plant Breed. 136, 287–299. doi: 10.1111/pbr.12473
Cowles C. R., Hirschhorn J. N., Altshuler D., Lander E. S. (2002). Detection of regulatory variation in mouse genes. Nat. Genet. 32, 432–437. doi: 10.1038/ng992
Cros D., Denis M., Bouvet J. M., Sanchez L. (2015). Long-term genomic selection for heterosis without dominance in multiplicative traits: case study of bunch production in oil palm. BMC Genomics 16, 651. doi: 10.1186/s12864-015-1866-9
Crow J. F. (1948). Alternative hypotheses of hybrid vigor. Genetics 33, 477–487. doi: 10.1093/genetics/33.5.477
Crow J. F. (1999). Dominance and overdominance. Genet. Exploitation Heterosis Crops, 49–58. doi: 10.2134/1999.geneticsandexploitation.c5
da Rosa S. S., Ribeiro N. D., Jost E., et al. (2010). Potential for increasing the zinc content in common bean using genetic improvement. Euphytica 175, 207–213. doi: 10.1007/s10681-010-0163-6
Dafna A., Halperin I., Oren E., Isaacson T., Tzuri G., Meir A., et al. (2021). Underground heterosis for yield improvement in melon. J. Exp. Bot. 72, 6205–6218. doi: 10.1093/jxb/erab219
Dan Z., Liu P., Huang W., Zhou W., Yao G., Hu J., et al. (2014). Balance between a higher degree of heterosis and increased reproductive isolation: a strategic design for breeding inter-subspecific hybrid rice. PloS One 9, e93122. doi: 10.1371/journal.pone.0093122
Diaz A., Martin-Hernandez A. M., Dolcet-Sanjuan R., Garces-Claver A., Alvarez J. M., Garcia-Mas J., et al. (2017). Quantitative trait loci analysis of melon (Cucumis melo L.) domestication-related traits. Theor. Appl. Genet. 130, 1837–1856. doi: 10.1007/s00122-017-2928-y
Dobzhansky T. (1952). Nature and origin of heterosis. Heterosis: a record of researches directed toward explaining and utilizing the vigor of hybrids. Nat. origin heterosis 218–223.
Dooner H. K., Martinez-Ferez I. M. (1997). Germinal excisions of the maize transposon activator do not stimulate meiotic recombination or homology-dependent repair at the bz locus. Genetics 147, 1923–1932. doi: 10.1093/genetics/147.4.1923
Duvick D. N. (2005). GENETIC PROGRESS IN YIELD OF UNITED STATES MAIZE (Zea mays L.). Maydica 50, 193–202.
Faria M. V., Zaluski W. L., Rosa J., Rossi E. S., Resende J. T. V., Kobori R. F., et al. (2019). Genetic divergence among inbred onion lines and correlation with heterosis and combining ability. Genet. Mol. Res. 18. doi: 10.4238/gmr18316
Fernandez-Silva I., Moreno E., Eduardo I., Arus P., Alvarez J. M., Monforte A. J. (2009). On the genetic control of heterosis for fruit shape in melon (Cucumis melo L.). J. Hered 100, 229–235. doi: 10.1093/jhered/esn075
Fievet J. B., Nidelet T., Dillmann C., De Vienne D. (2018). Heterosis is a systemic property emerging from non-linear genotype-phenotype relationships: evidence from in vitro genetics and computer simulations. Front. Genet. 9. doi: 10.3389/fgene.2018.00159
Figueiredo A. S., Resende J. T., Faria M. V., Da-Silva P. R., Fagundes B. S., Morales R. G. (2016). Prediction of industrial tomato hybrids from agronomic traits and ISSR molecular markers. Genet. Mol. Res. 15, 1–13. doi: 10.4238/gmr.15027981
Fortuny A. P., Bueno R. A., Pereira Da Costa J. H., Zanor M. I., Rodriguez G. R. (2021). Tomato fruit quality traits and metabolite content are affected by reciprocal crosses and heterosis. J. Exp. Bot. 72, 5407–5425. doi: 10.1093/jxb/erab222
Fu H., Dooner H. K. (2002). Intraspecific violation of genetic colinearity and its implications in maize. Proc. Natl. Acad. Sci. U.S.A. 99, 9573–9578. doi: 10.1073/pnas.132259199
Fu D. H., Xiao M. L., Hayward A., Fu Y., Liu G., Jiang G. J., et al. (2014). Utilization of crop heterosis: a review. Euphytica 197, 161–173. doi: 10.1007/s10681-014-1103-7
Fujimoto R., Uezono K., Ishikura S., Osabe K., Peacock W. J., Dennis E. S. (2018). Recent research on the mechanism of heterosis is important for crop and vegetable breeding systems. Breed Sci. 68 (2), 145–158. doi: 10.1270/jsbbs.17155
Gardner C. O., Lonnquist J. H. (1959). Linkage and the degree of dominance of genes controlling quantitative characters in maize 1. Agron. J. 51, 524–528. doi: 10.2134/agronj1959.00021962005100090005x
Geleta L. F., Labuschagne M. T., Viljoen C. D. (2004). Relationship between heterosis and genetic distance based on morphological traits and AFLP markers in pepper. Plant Breed. 123, 467–473. doi: 10.1111/j.1439-0523.2004.01017.x
Gill B. S., Gill K. S., Friebe B., Endo T. R. (1997). Expanding genetic maps: Reevaluation of the relationship between chiasmata and crossovers. In: Henriques-Gil N., Parker J. S., Puertas M. J. (eds) Chromosomes Today (Dordrecht: Springer). doi: 10.1007/978-94-009-1537-4_17
Gitonga V. W., Stolker R., Koning-Boucoiran C. F., Aelaei M., Visser R. G., Maliepaard C., et al. (2016). Inheritance and QTL analysis of the determinants of flower color in tetraploid cut roses. Mol. Breed 36, 143. doi: 10.1007/s11032-016-0565-9
Gonçalves-Vidigal M. C., Silvério L., Elias H. T., Vidigal Filho P. S., Kvitschal M. V., Retuci V. S., et al. (2008). Combining ability and heterosis in common bean cultivars. Pesquisa Agropecuaria Bras. 43 (9), 1143–1150. doi: 10.1590/S0100-204X2008000900007
Gonda I., Ashrafi H., Lyon D. A., Strickler S. R., Hulse-Kemp A. M., Ma Q., et al. (2019). Sequencing-based bin map construction of a tomato mapping population, facilitating high-resolution quantitative trait loci detection. Plant Genome 12, 180010. doi: 10.3835/plantgenome2018.02.0010
Gonzalez-Bayon R., Shen Y., Groszmann M., Zhu A., Wang A., Allu A. D., et al. (2019). Senescence and defense pathways contribute to heterosis. Plant Physiol. 180 (1), 240–252. doi: 10.1104/pp.18.01205
Greaves I. K., Gonzalez-Bayon R., Wang Li, Zhu A., Liu P.-C., Groszmann M., et al. (2015). Epigenetic changes in hybrids. Plant Physiol. 168, 1197–1205. doi: 10.1104/pp.15.00231
Groszmann M., Greaves I. K., Albertyn Z. I., Scofield G. N., Peacock W. J., Dennis E. S. (2011). Changes in 24-nt siRNA levels in arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc. Natl. Acad. Sci. U S A. 108 (6), 2617–2622. doi: 10.1073/pnas.1019217108
Guo M., Davis D., Birchler J. A. (1996). Dosage effects on gene expression in a maize ploidy series. Genetics 142, 1349–1355. doi: 10.1093/genetics/142.4.1349
Guo M., Rupe M. A., Danilevskaya O. N., Yang X., Hu Z. (2003). Genome-wide mRNA profiling reveals heterochronic allelic variation and a new imprinted gene in hybrid maize endosperm. Plant J. 36, 30–44. doi: 10.1046/j.1365-313x.2003.01852.x
Guo M., Rupe M. A., Zinselmeier C., Habben J., Bowen B. A., Smith O. S. (2004). Allelic variation of gene expression in maize hybrids. Plant Cell 16, 1707–1716. doi: 10.1105/tpc.022087
Guo T., Yang N., Tong H., Pan Q., Yang X., Tang J., et al. (2014). Genetic basis of grain yield heterosis in an "immortalized F(2)" maize population. Theor. Appl. Genet. 127, 2149–2158. doi: 10.1007/s00122-014-2368-x
Gur A., Semel Y., Cahaner A., Zamit D. (2004). Real Time QTL of complex phenotypes in tomato interspecific introgression lines. Trends Plant Sci. 9. doi: 10.1016/j.tplants.2004.01.003
Gur A., Zamir D. (2015). Mendelizing all components of a pyramid of three yield QTL in tomato. Front. Plant Sci. 6. doi: 10.3389/fpls.2015.01096
Hallauer A. R., Carena M. J., Miranda Filho J. B. (2010). Heterosis. Quantitative Genet. maize Breed. doi: 10.1007/978-1-4419-0766-0
Hammerle B., Ferrus A. (2003). Expression of enhancers is altered in Drosophila melanogaster hybrids. Evol. Dev. 5, 221–230. doi: 10.1046/j.1525-142x.2003.03030.x
Hanifei M., Gholizadeh A., Khodadadi M., Mehravi S., Hanifeh M., Edwards D., et al. (2022). Dissection of genetic effects, heterosis, and inbreeding depression for phytochemical traits in coriander. Plants 11, 2959. doi: 10.3390/plants11212959
Havey M. J. (1998). Molecular analyses and heterosis in the vegetables: can we breed them like maize? Concepts Breed. Heterosis Crop Plants 25. doi: 10.2135/cssaspecpub25.c9
Hayes H. K., Jones D. F. (1916). First generation crosses in cucumbers. Report of the Connecticut Agricultural Experiment Station, Connecticut Agricultural Experiment Station, United States, pp. 319–322.
He G., Chen B., Wang X., Li X., Li J., He H., et al. (2013). Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol. 14 (6), R57. doi: 10.1186/gb-2013-14-6-r57
He G., Zhu X., Elling A. A., Chen L., Wang X., Guo L., et al. (2010). Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 22 (1), 17–33. doi: 10.1105/tpc.109.072041
Hochholdinger F., Hoecker N. (2007). Towards the molecular basis of heterosis. Trends Plant Sci. 12, 427–432. doi: 10.1016/j.tplants.2007.08.005
Hu Y., Xiong J., Shalby N., Zhuo C., Jia Y., Yang Q. Y., et al. (2022). Comparison of dynamic 3D chromatin architecture uncovers heterosis for leaf size in Brassica napus. J. Adv. Res. 42, 289–301. doi: 10.1016/j.jare.2022.01.001
Huang X., Yang S., Gong J., et al. (2015). Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun. 6, 6258. doi: 10.1038/ncomms7258
Huang X., Han B. (2014). Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 65, 531–551. doi: 10.1146/annurev-arplant-050213-035715
Huang W., Mackay T. F. (2016). The genetic architecture of quantitative traits cannot be inferred from variance component analysis. PloS Genet. 12, e1006421. doi: 10.1371/journal.pgen.1006421
Huang X., Yang S., Gong J., Zhao Q., Feng Q., Zhan Q., et al. (2016). Genomic architecture of heterosis for yield traits in rice. Nature 537, 629–633. doi: 10.1038/nature19760
Jagosz B. (2011). The relationship between heterosis and genetic distances based on RAPD and AFLP markers in carrot. Plant Breed. 130, 574–579. doi: 10.1111/j.1439-0523.2011.01877.x
Jahnke S., Sarholz B., Thiemann A., Kühr V., Gutiérrez-Marcos J. F., Geiger H. H., et al. (2010). Heterosis in early seed development: a comparative study of F1 embryo and endosperm tissues 6 days after fertilization. Theor. Appl. Genet. 120 (2), 389–400. doi: 10.1007/s00122-009-1207-y
Jeong S. Y., Ahmed N. U., Jung H. J., Kim H. T., Park J. I., Nou I. S. (2017). Discovery of candidate genes for heterosis breeding in Brassica oleracea L. Acta Physiologiae Plantarum 39, 1–12. doi: 10.1007/s11738-017-2474-x
Jones D. F. (1917). Dominance of linked factors as a means of accounting for heterosis. Genetics 2, 466–479. doi: 10.1093/genetics/2.5.466
Jones H. A., Clarke A. E. (1943). Inheritance of male-sterility in the onion and the production of hybrid seed. Proc. America Soc. Horticult. Sci. 43, 189–194.
Kaushik P. (2019). Line x Tester Analysis for Morphological and Fruit Biochemical Traits in Eggplant (Solanum melongena L.) Using Wild Relatives as Testers. Agronomy-Basel 9, 185. doi: 10.3390/agronomy9040185
Kaushik P., Plazas M., Prohens J., Vilanova S., Gramazio P. (2018). Diallel genetic analysis for multiple traits in eggplant and assessment of genetic distances for predicting hybrids performance. PloS One 13, e0199943. doi: 10.1371/journal.pone.0199943
Kawamura K., Kawanabe T., Shimizu M., Nagano A. J., Saeki N., Okazaki K., et al. (2016). Genetic distance of inbred lines of Chinese cabbage and its relationship to heterosis. Plant Gene 5, 1–7. doi: 10.1016/j.plgene.2015.10.003
Kenan-Eichler M., Leshkowitz D., Tal L., Noor E., Melamed-Bessudo C., Feldman M., et al. (2011). Wheat hybridization and polyploidization results in deregulation of small RNAs. Genetics 188 (2), 263–272. doi: 10.1534/genetics.111.128348
Kim J. A., Kim H. S., Choi S. H., Jang J. Y., Jeong M. J., Lee S. I. (2017). The importance of the circadian clock in regulating plant metabolism. Int. J. Mol. Sci. 18 (12), 2680. doi: 10.3390/ijms18122680
Knight J. C. (2004). Allele-specific gene expression uncovered. Trends Genet. 20, 113–116. doi: 10.1016/j.tig.2004.01.001
Kong X., Chen L., Wei T., Zhou H., Bai C., Yan X., et al. (2020). Transcriptome analysis of biological pathways associated with heterosis in Chinese cabbage. Genomics 112, 4732–4741. doi: 10.1016/j.ygeno.2020.08.011
Krieger U., Lippman Z. B., Zamir D. (2010). The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 42, 459–463. doi: 10.1038/ng.550
Kumar A., Sharma V., Jain B. T., Kaushik P. (2020). Heterosis breeding in eggplant (Solanum melongena L.): gains and provocations. Plants (Basel) 9, 403. doi: 10.3390/plants9030403
Kumar S., Singh P. K. (2004). Mechanisms for hybrid development in vegetables. J. New Seeds 6, 381–407. doi: 10.1300/J153v06n04_05
Lai J., Li R., Xu X., et al. (2010). Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42, 1027–1030. doi: 10.1038/ng.684
Lee E. A., Darrah L. L., Coe E. H. (1996). Dosage effects on morphological and quantitative traits in maize aneuploids. Genome 39, 898–908. doi: 10.1139/g96-113
Lehner B. (2011). Molecular mechanisms of epistasis within and between genes. Trends Genet. 27, 323–331. doi: 10.1016/j.tig.2011.05.007
Leonardi A., Damerval C., Hebert Y., Gallais A., De Vienne D. (1991). Association of protein amount polymorphism (PAP) among maize lines with performances of their hybrids. Theor. Appl. Genet. 82, 552–560. doi: 10.1007/BF00226790
Li J., Chitwood J., Menda N., Mueller L., Hutton S. F. (2018). Linkage between the I-3 gene for resistance to Fusarium wilt race 3 and increased sensitivity to bacterial spot in tomato. Theor. Appl. Genet. 131, 145–155. doi: 10.1007/s00122-017-2991-4
Li D., Huang Z., Song S., Xin Y., Mao D., Lv Q., et al. (2016). Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc. Natl. Acad. Sci. U.S.A. 113, E6026–E6035. doi: 10.1073/pnas.1610115113
Li D., Lu X., Zhu Y., Pan J., Zhou S., Zhang X., et al. (2022). The multi-omics basis of potato heterosis. J. Integr. Plant Biol. 64, 671–687. doi: 10.1111/jipb.13211
Li X., Lv H. H., Zhang B., Fang Z. Y., Yang L. M., Zhuang M., et al. (2023). Dissection of two QTL clusters underlying yield-related heterosis in the cabbage founder parent 01-20. Hortic. Plant J. 9, 77–88. doi: 10.1016/j.hpj.2022.05.002
Li M.-Y., Hou X.-L., Wang F., Tan G.-F., Xu Z.-S., Xiong A.-S. (2018). Advances in the research of celery, an important apiaceae vegetable crop. Crit. Rev. Biotechnol. 38, 172–183. doi: 10.1080/07388551.2017.1312275
Lian Q., Fu Q., Xu Y., Hu Z., Zheng J., Zhang A., et al. (2021). QTLs and candidate genes analyses for fruit size under domestication and differentiation in melon (Cucumis melo L.) based on high resolution maps. BMC Plant Biol. 21, 126. doi: 10.1186/s12870-021-02904-y
Lichten M., Goldman A. S. (1995). Meiotic recombination hotspots. Annu. Rev. Genet. 29, 423–444. doi: 10.1146/annurev.ge.29.120195.002231
Lim C. W., Baek W., Han S. W., Lee S. C. (2013). Arabidopsis PYL8 plays an important role for ABA signaling and drought stress responses. Plant Pathol. J. 29 (4), 471–476. doi: 10.5423/PPJ.NT.07.2013.0071
Lin Z., Qin P., Zhang X., Fu C., Deng H., Fu X., et al. (2020). Divergent selection and genetic introgression shape the genome landscape of heterosis in hybrid rice. Proc. Natl. Acad. Sci. U S A. 117 (9), 4623–4631. doi: 10.1073/pnas.1919086117
Lippman Z. B., Zamir D. (2007). Heterosis: revisiting the magic. Trends Genet. 23. doi: 10.1016/j.tig.2006.12.006
Liu T., Duan W., Chen Z., Yuan J., Xiao D., Hou X., et al. (2020b). Enhanced photosynthetic activity in pak choi hybrids is associated with increased grana thylakoids in chloroplasts. Plant J. 103, 2211–2224. doi: 10.1111/tpj.14893
Liu C., Liu X., Wang X. A., Han Y., Meng H., Cheng Z. (2022). Heterosis prediction system based on non-additive genomic prediction models in cucumber (Cucumis sativus L.). Scientia Hortic. 293, 110677. doi: 10.1016/j.scienta.2021.110677
Liu H., Wang Q., Chen M., Ding Y., Yang X., Liu J., et al. (2020a). Genome-wide identification and analysis of heterotic loci in three maize hybrids. Plant Biotechnol. J. 18, 185–194. doi: 10.1111/pbi.13186
Luan F. S., Sheng Y. Y., Wang Y. H., Staub J. E. (2010). Performance of melon hybrids derived from parents of diverse geographic Origins. Euphytica 173, 1–16. doi: 10.1007/s10681-009-0110-6
Manna F., Martin G., Lenormand T. (2011). Fitness landscapes: an alternative theory for the dominance of mutation. Genetics 189, 923–937. doi: 10.1534/genetics.111.132944
Matzke M. A., Mette M. F., Kanno T., Matzke A. J. (2003). Does the intrinsic instability of aneuploid genomes have a causal role in cancer? Trends Genet. 19, 253–256. doi: 10.1016/s0168-9525(03)00057-x
Mehraj H., Akter A., Miyaji N., Miyazaki J., Shea D. J., Fujimoto R., et al. (2020). Genetics of clubroot and fusarium wilt disease resistance in brassica vegetables: the application of marker assisted breeding for disease resistance. Plants (Basel) 9, 726. doi: 10.3390/plants9060726
Melchinger A. E. (1999). Genetic diversity and heterosis. Genet. Exploitation Heterosis Crops, 99–118. doi: 10.2134/1999.geneticsandexploitation.c10
Melchinger A. E., Gumber R. K. (1998). Overview of heterosis and heterotic groups in agronomic crops. Concepts Breed. Heterosis Crop Plants, 29–44. doi: 10.2135/cssaspecpub25.c3
Melchinger A. E., Utz H. F., Schön C. C. (2000). From Mendel to Fisher: the power and limits of QTL mapping for quantitative traits. Vortr Pflanzenzucht 48, 132–142.
Melnyk C. W., Molnar A., Bassett A., Baulcombe D. C. (2011). Mobile 24 nt small RNAs direct transcriptional gene silencing in the root meristems of arabidopsis thaliana. Curr. Biol. 21 (19), 1678–1683. doi: 10.1016/j.cub.2011.08.065
Miyaji N., Fujimoto R. (2018). Chapter Eight - Hybrid Vigor: Importance of Epigenetic Processes and Consequences for Breeding. Editor(s): Mirouze M., Bucher E., Gallusci P., Adv. Botanical Res. 88, 247–275. doi: 10.1016/bs.abr.2018.10.001
Moll R. H., Lonnquist J. H., Fortuno J. V., Johnson E. C. (1965). The relationship of heterosis and genetic divergence in maize. Genetics 52, 139–144. doi: 10.1093/genetics/52.1.139
Morgante M., Salamini F. (2003). From plant genomics to breeding practice. Curr. Opin. Biotechnol. 14, 214–219. doi: 10.1016/s0958-1669(03)00028-4
Moustafa Y. M. M., Yui S., Uemura M. (2006). Chilling tolerance and field performance of an F1 cooking tomato cultivar, nitaki-koma, relative to its parents. Breed. Sci. 56, 269–276. doi: 10.1270/jsbbs.56.269
Na J. K., Metzger J. D. (2017). Guard-cell-specific expression of arabidopsis ABF4 improves drought tolerance of tomato and tobacco. Mol. Breed. 37, 154. doi: 10.1007/s11032-017-0758-x
Natalini A., Acciarri N., Cardi T. (2021). Breeding for nutritional and organoleptic quality in vegetable crops: The case of tomato and cauliflower. Agriculture 11, 606. doi: 10.3390/agriculture11070606
Obiadalla-Ali H. A., Mohamed N. E., Khaled A. G. (2015). Inbreeding, outbreeding and RAPD markers studies of faba bean (Vicia faba L.) crop. J. Adv. Res. 6, 859–868. doi: 10.1016/j.jare.2014.07.004
Osborn T. C., Pires J. C., Birchler J. A., Auger D. L., Chen Z. J., Lee H. S., et al. (2003). Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19, 141–147. doi: 10.1016/s0168-9525(03)00015-5
Pan Y., Wang Z., Zhan W., Deng L. (2018). Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 34, 1473–1480. doi: 10.1093/bioinformatics/btx822
Pandey S. K., Dasgupta T., Rathore A., Vemula A. (2018). Relationship of parental genetic distance with heterosis and specific combining ability in sesame (Sesamum indicum L.) based on phenotypic and molecular marker analysis. Biochem. Genet. 56, 188–209. doi: 10.1007/s10528-017-9837-2
Papp B., Pal C., Hurst L. D. (2003). Dosage sensitivity and the evolution of gene families in yeast. Nature 424, 194–197. doi: 10.1038/nature01771
Patella A., Scariolo F., Palumbo F., Barcaccia G. (2019). Genetic structure of cultivated varieties of radicchio (Cichorium intybus l.): A comparison between F1 hybrids and synthetics. Plants (Basel) 8 (7), 213. doi: 10.3390/plants8070213
Paterson A. H., Lander E. S., Hewitt J. D., Peterson S., Lincoln S. E., Tanksley S. D. (1988). Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 335, 721–726. doi: 10.1038/335721a0
Pearson O. H. (1933). Study of the life history of brassica oleracea. Botanical Gazette 94, 534–550. doi: 10.1086/334326
Phillips P. C. (2008). Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9, 855–867. doi: 10.1038/nrg2452
Pickett A. A., Galwey N. W. (1997). A further evaluation of hybrid wheat. Plant varieties seeds 10, 15–32.
Pott D. M., Durán-Soria S., Osorio S., et al. (2021). Combining metabolomic and transcriptomic approaches to assess and improve crop quality traits. CABI Agric. Biosci. 2, 1. doi: 10.1186/s43170-020-00021-8
Rafalski A., Morgante M. (2004). Corn and humans: recombination and linkage disequilibrium in two genomes of similar size. Trends Genet. 20, 103–111. doi: 10.1016/j.tig.2003.12.002
Rahman M. M., Sarker U., Swapan M. A., Raihan M. S., Oba S., Alamri S., et al. (2022). Combining ability analysis and marker-based prediction of heterosis in yield reveal prominent heterotic combinations from diallel population of rice. Agronomy 12. doi: 10.3390/agronomy12081797
Rehman A. U., Dang T., Qamar S., Ilyas A., Fatema R., Kafle M., et al. (2021). Revisiting plant heterosis–from field scale to molecules. Genes 12, 1688. doi: 10.3390/genes12111688
Rajendran S., Bae J. H., Park M. W., Oh J. H., Jeong H. W., Lee Y. K., et al. (2022). Tomato yield effects of reciprocal hybridization of solanum lycopersicum cultivars M82 and micro-tom. Plant Breed. Biotechnol. 10, 37–48. doi: 10.9787/pbb.2022.10.1.37
Reif J. C., Melchinger A. E., Xia X. C., Warburton M. L., Hoisington D. A., Vasal S. K., et al. (2003). Genetic distance based on simple sequence repeats and heterosis in tropical maize populations. Crop Sci. 43, 1275–1282. doi: 10.2135/cropsci2003.1275
Rodríguez-Burruezo A., Prohens J., Nuez F. (2008). Performance of hybrids between local varieties of eggplant (Solanum melongena) and its relation to the mean of parents and to morphological and genetic distances among parents. Eur. J. Hortic. Sci. 73, 76.
Romagnoli S., Maddaloni M., Livini C., Motto M. (1990). Relationship between gene expression and hybrid vigor in primary root tips of young maize (Zea mays L.) plantlets. Theor. Appl. Genet. 80, 769–775. doi: 10.1007/BF00224190
Saeki N., Kawanabe T., Ying H., Shimizu M., Kojima M., Abe H., et al. (2016). Molecular and cellular characteristics of hybrid vigour in a commercial hybrid of Chinese cabbage. BMC Plant Biol. 16, 45. doi: 10.1186/s12870-016-0734-3
Santamaria P., Signore A. (2021). How has the consistency of the Common catalogue of varieties of vegetable species changed in the last ten years? Scientia Hortic. 277, 109805. doi: 10.1016/j.scienta.2020.109805
Scariolo F., Palumbo F., Barcaccia G. (2022). Molecular characterization and genetic structure evaluation of breeding populations of fennel (Foeniculum vulgare mill.). Agronomy 12, 542. doi: 10.3390/agronomy12030542
Scariolo F., Palumbo F., Farinati S., Barcaccia G. (2023). Pipeline to design inbred lines and F1 hybrids of leaf chicory (Radicchio) using male sterility and genotyping-by-Sequencing. Plants (Basel) 12 (6), 1242. doi: 10.3390/plants12061242
Schnable P. S., Wise R. P. (1998). The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 3. doi: 10.1016/S1360-1385(98)01235-7
Semel Y., Nissenbaum J., Menda N., Zinder M., Krieger U., Issman N., et al. (2006). Overdominant quantitative trait loci for yield and fitness in tomato. Proc. Natl. Acad. Sci. U.S.A. 103, 12981–12986. doi: 10.1073/pnas.0604635103
Shapturenko M. N., Tarutina L. A., Mishin L. A., Kilchevsky A. V., Khotyleva L. V. (2014). DNA divergence as a criterion of a sweet pepper (Capsicum annuum L.) selection for heterosis. Russian J. Genet. 50, 123–130. doi: 10.1134/S1022795414020148
Shapturenko M., Tarutina L., Pechkovskaya T., Mishin L., Kilchevsky A., Khotyleva L. (2013). “Hybrid performance in sweet pepper relative to parental diversity detected by RAPD and ISSR markers,” in Breakthroughs in the genetics and breeding of capsicum and eggplant; Proceedings of the XV EUCARPIA meeting, 2013. EUCARPIA Meeting on Genetics and Breeding of Capsicum and Eggplant, 15th Torino 2-4 September 2013 Italy
Sharma M., Singh Y., Singh S. K., Dhangrah V. K. (2016). Exploitation of gynoecious lines in cucumber (Cucumis sativus L.) for heterosis breeding. Int. J. Bio-resource Stress Manage. 7, 184–190. doi: 10.23910/ijbsm/2016.7.2.1557
Sharma A., Yadav R., Sheoran R., Kaushik D., Mohanta T. K., Sharma K., et al. (2023). Estimation of heterosis and the combining ability effect for yield and its attributes in field pea (Pisum sativum l.) using PCA and GGE biplots. Horticulturae 9, 256. doi: 10.3390/horticulturae9020256
Shen G., Hu W., Zhang B., Xing Y. (2015). The regulatory network mediated by circadian clock genes is related to heterosis in rice. J. Integr. Plant Biol. 57, 300–312. doi: 10.1111/jipb.12240
Shen Y., Sun S., Hua S., Shen E., Ye C. Y., Cai D., et al. (2017). Analysis of transcriptional and epigenetic changes in hybrid vigor of allopolyploid brassica napus uncovers key roles for small RNAs. Plant J. 91 (5), 874–893. doi: 10.1111/tpj.13605
Shiraki S., Fujiwara K., Kamiya Y., Akter M. A., Dennis E. S., Fujimoto R., et al. (2023). Studies on the molecular basis of heterosis in arabidopsis thaliana and vegetable crops. Horticulturae 9. doi: 10.3390/horticulturae9030366
Shu H. Y., Zhou H., Mu H. L., Wu S. H., Jiang Y. L., Yang Z., et al. (2021). Integrated Analysis of mRNA and Non-coding RNA Transcriptome in Pepper (Capsicum chinense) Hybrid at Seedling and Flowering Stages. Front. Genet. 12. doi: 10.3389/fgene.2021.685788
Shull G. H. (1908). The composition of a field of maize. J. Heredity 4, 296–301. doi: 10.1093/jhered/os-4.1.296
Si W., Yuan Y., Huang J., Zhang X., Zhang Y., Zhang Y., et al. (2015). Widely distributed hot and cold spots in meiotic recombination as shown by the sequencing of rice F2 plants. New Phytol. 206, 1491–1502. doi: 10.1111/nph.13319
Silva Dias J. (2014). Guiding strategies for breeding vegetable cultivars. Agric. Sci. 5, 9–32. doi: 10.4236/as.2014.51002
Singh B., Singh P., Singh B. (2018). Heterosis for economic traits in single cross-hybrids of radish (Raphanus sativus L.). Vegetable Sci. 45, 45–49.
Song R., Messing J. (2003). Gene expression of a gene family in maize based on noncollinear haplotypes. Proc. Natl. Acad. Sci. U.S.A. 100, 9055–9060. doi: 10.1073/pnas.1032999100
Song X., Ni Z., Yao Y., Zhang Y., Sun Q. (2009). Identification of differentially expressed proteins between hybrid and parents in wheat (Triticum aestivum L.) seedling leaves. Theor. Appl. Genet. 118, 213–225. doi: 10.1007/s00122-008-0890-4
Soyk S., Lemmon Z. H., Oved M., Fisher J., Liberatore K. L., Park S. J., et al. (2017). Bypassing negative epistasis on yield in tomato imposed by a domestication gene. Cell 169, 1142–1155.e12. doi: 10.1016/j.cell.2017.04.032
Spaldon S., Hussain S., Jabeen N., Lay P. (2015). Heterosis studies for earliness, fruit yield and yield attributing traits in chilli (Capsicum Annum L.). Bioscan 10, 813–818.
Stapley J., Feulner P. G. D., Johnston S. E., Santure A. W., Smadja C. M. (2017). Variation in recombination frequency and distribution across eukaryotes: patterns and processes. Philos. Trans. R Soc. Lond B Biol. Sci. 372, 20160455. doi: 10.1098/rstb.2016.0455
Steiner W. W., Smith G. R. (2005). Natural meiotic recombination hot spots in the Schizosaccharomyces pombe genome successfully predicted from the simple sequence motif M26. Mol. Cell Biol. 25, 9054–9062. doi: 10.1128/MCB.25.20.9054-9062.2005
Stitzer M. C., Ross-Ibarra J. (2018). Maize domestication and gene interaction. New Phytol. 220, 395–408. doi: 10.1111/nph.15350
Stuber C. W., Lincoln S. E., Wolff D. W., Helentjaris T., Lander E. S. (1992). Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics 132, 823–839. doi: 10.1093/genetics/132.3.823
Stuber C. (1997). “The biology and physiology of heterosis,” in In: A book of abstracts. the genetics and exploitation of heterosis in crops: An international symposium, 108–109.
Sun S., Zhou Y., Chen J., et al. (2018). Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 50, 1289–1295. doi: 10.1038/s41588-018-0182-0
Talbert P. B., Henikoff S. (2010). Centromeres convert but don't cross. PloS Biol. 8 (3), e1000326. doi: 10.1371/journal.pbio.1000326
Tamta S., Singh J. P. (2017). Heterosis in tomato for growth and yield traits. Int. J. Vegetable Sci. 24, 169–179. doi: 10.1080/19315260.2017.1407857
Tanksley S. D. (1993). Mapping polygenes. Annu. Rev. Genet. 27, 205–233. doi: 10.1146/annurev.ge.27.120193.001225
Thouet J., Quinet M., Ormenese S., Kinet J. M., Perilleux C. (2008). Revisiting the involvement of SELF-PRUNING in the sympodial growth of tomato. Plant Physiol. 148, 61–64. doi: 10.1104/pp.108.124164
Tian F., Bradbury P. J., Brown P. J., Hung H., Sun Q., Flint-Garcia S., et al. (2011). Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 43, 159–162. doi: 10.1038/ng.746
Timmermans S., Berg M. (1997). Standardization in action: Achieving local universality through medical protocols. Soc. Stud. Sci. 27, 273–305. doi: 10.1177/030631297027002003
Tonosaki K., Fujimoto R., Dennis E. S., Raboy V., Osabe K. (2023). Corrigendum: Will epigenetics be a key player in crop breeding? Front. Plant Sci. 14. doi: 10.3389/fpls.2023.1157933
Turner S. D., Maurizio P. L., Valdar W., Yandell B. S., Simon P. W. (2018). Dissecting the genetic architecture of shoot growth in carrot (Daucus carota L.) using a diallel mating design. G3 (Bethesda) 8, 411–426. doi: 10.1534/g3.117.300235
Verma V. K., Kalia P. (2017). Combining ability analysis and its relationship with gene action and heterosis in early maturity cauliflower. Proc. Natl. Acad. Sciences India Section B: Biol. Sci. 87, 877–884. doi: 10.1007/s40011-015-0664-0
von Maydell D., Stache A. M., El Menuawy A., et al. (2021). On heterosis, inbreeding depression and general combining ability in annual caraway (Carum carvi). Euphytica 217, 163. doi: 10.1007/s10681-021-02893-x
Wang M. L., Khera P., Pandey M. K., Wang H., Qiao L., Feng S., et al. (2015). Genetic mapping of QTLs controlling fatty acids provided insights into the genetic control of fatty acid synthesis pathway in peanut (Arachis hypogaea L.). PloS One 10, e0119454. doi: 10.1371/journal.pone.0119454
Wang C., Tang S., Zhan Q., Hou Q., Zhao Y., Zhao Q., et al. (2019). Dissecting a heterotic gene through GradedPool-Seq mapping informs a rice-improvement strategy. Nat. Commun. 10, 2982. doi: 10.1038/s41467-019-11017-y
Wang Q., Yan T., Long Z., Huang L. Y., Zhu Y., Xu Y., et al. (2021). Prediction of heterosis in the recent rapeseed (Brassica napus) polyploid by pairing parental nucleotide sequences. PloS Genet. 17, e1009879. doi: 10.1371/journal.pgen.1009879
Ward R. A., Kim K.-S., Diers B. W. (2017). Yield drag associated with the soybean aphid resistance gene rag2 from PI 200538. Crop Sci. 57, 3035–3042. doi: 10.2135/cropsci2017.04.0236
Wei W. H., Hemani G., Haley C. S. (2014). Detecting epistasis in human complex traits. Nat. Rev. Genet. 15, 722–733. doi: 10.1038/nrg3747
Weinreich D. M., Lan Y., Wylie C. S., Heckendorn R. B. (2013). Should evolutionary geneticists worry about higher-order epistasis? Curr. Opin. Genet. Dev. 23, 700–707. doi: 10.1016/j.gde.2013.10.007
Wolko J., Dobrzycka A., Bocianowski J., Bartkowiak-Broda I. (2019). Estimation of heterosis for yield-related traits for single cross and three-way cross hybrids of oilseed rape (Brassica napus L.). Euphytica 215, 156. doi: 10.1007/s10681-019-2482-6
Wright S. I., Agrawal N., Bureau T. E. (2003). Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 13, 1897–1903. doi: 10.1101/gr.1281503
Xiao Y., Jiang S., Cheng Q., et al. (2021). The genetic mechanism of heterosis utilization in maize improvement. Genome Biol. 22, 148. doi: 10.1186/s13059-021-02370-7
Xiao J., Li J., Yuan L., Tanksley S. D. (1995). Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics 140, 745–754. doi: 10.1093/genetics/140.2.745
Yang J., Mezmouk S., Baumgarten A., Buckler E. S., Guill K. E., Mcmullen M. D., et al. (2017). Incomplete dominance of deleterious alleles contributes substantially to trait variation and heterosis in maize. PloS Genet. 13, e1007019. doi: 10.1371/journal.pgen.1007019
Yi Q., Liu Y., Hou X., Zhang X., Li H., Zhang J., et al. (2019). Genetic dissection of yield-related traits and mid-parent heterosis for those traits in maize (Zea mays L.). BMC Plant Biol. 19, 392. doi: 10.1186/s12870-019-2009-2
Yu D., Gu X., Zhang S., Dong S., Miao H., Gebretsadik K., et al. (2021). Molecular basis of heterosis and related breeding strategies reveal its importance in vegetable breeding. Hortic. Res. 8, 120. doi: 10.1038/s41438-021-00552-9
Zanewich K. P., Rood S. B. (2020). Gibberellins and heterosis in crops and trees: An integrative review and preliminary study with brassica. Plants 9, 139. doi: 10.3390/plants9020139
Zanewich K. P., Rood S. B. (1988). Gibberellins: A phytohormonal basis for heterosis in maize. In: Science (JSTOR). 241 (4870), 1216–1218. Available at: http://www.jstor.org/stable/1702725 (Accessed 25 Oct. 2023).
Zhang W. L., Wang X., Song L., Xie K., Xu Y., Bao X., et al. (2020). Cloning and sequence analysis of CsMYB108 gene in cucumber (Cucumis sativa L.). Mol. 18, 4555–4561.
Zhang C., Yang Z., Tang D., Zhu Y., Wang P., Li D., et al. (2021). Genome design of hybrid potato. Cell 184, 3873–3883.e12. doi: 10.1016/j.cell.2021.06.006
Zhang H.-Y., He H., Chen L.-B., Li L., Liang M.-Z., Wang X.-F., et al. (2008). A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol. Plant 1, 720–731. doi: 10.1093/mp/ssn022
Zhang Y., Ni Z., Yao Y., et al. (2007). Gibberellins and heterosis of plant height in wheat (Triticum aestivum l.). BMC Genet. 8, 40. doi: 10.1186/1471-2156-8-40
Zhang Q., Wang D., Lang Z., He L., Yang L., Zeng L., et al. (2016). Methylation interactions in arabidopsis hybrids require RNA-directed DNA methylation and are influenced by genetic variation. Proc. Natl. Acad. Sci. U S A. 113 (29), E4248–E4256. doi: 10.1073/pnas.1607851113
Zhao G., Lian Q., Zhang Z., Fu Q., He Y., Ma S., et al. (2019). A comprehensive genome variation map of melon identifies multiple domestication events and loci influencing agronomic traits. Nat. Genet. 51, 1607–1615. doi: 10.1038/s41588-019-0522-8
Keywords: heterosis, hybrid vigour, horticultural crop breeding, genetics, genomics, QTLs
Citation: Farinati S, Scariolo F, Palumbo F, Vannozzi A, Barcaccia G and Lucchin M (2023) Heterosis in horticultural crop breeding: combining old theoretical bases with modern genomic views. Front. Hortic. 2:1250875. doi: 10.3389/fhort.2023.1250875
Received: 30 June 2023; Accepted: 19 October 2023;
Published: 06 November 2023.
Edited by:
Maria Raffaella Ercolano, University of Naples Federico II, ItalyReviewed by:
Reeta Bhatia, Indian Council of Agricultural Research, IndiaCopyright © 2023 Farinati, Scariolo, Palumbo, Vannozzi, Barcaccia and Lucchin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianni Barcaccia, Z2lhbm5pLmJhcmNhY2NpYUB1bmlwZC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.