 Carla Semedo1†
Carla Semedo1† Bruno António Cardoso
Bruno António Cardoso- 1Universidade Católica Portuguesa, Centro de Investigação Interdisciplinar em Saúde, Sintra, Portugal
- 2Universidade Católica Portuguesa, Faculdade de Medicina, Sintra, Portugal
- 3Department of Hematology, Hospital da Luz Lisboa, Lisboa, Portugal
Hematopoiesis is a complex and tightly regulated process that drives the formation of mature blood cells from a single hematopoietic stem cell. This complex process occurs within the bone marrow, which, once disrupted or deregulated, subverts normal hematopoietic development, allowing leukemic cells to emerge, proliferate, and thrive. Notably, several cellular populations and paracrine factors within the bone marrow fuel leukemia expansion and progression. This review presents an overview of the main microenvironmental components that promote myeloid leukemia progression, discussing the emerging therapeutical strategies that target both leukemic cells and the supportive bone marrow microenvironment – targeting both the seed and the soil.
1 Hematopoiesis
Hematopoiesis is the lifelong, complex, and hierarchical mechanism that orchestrates the differentiation of all blood cells. From a single cell, hematopoiesis produces white blood cells, red blood cells, and platelets, which are responsible for immune responses, oxygen delivery, and stopping bleeding in case of blood vessel damage, respectively (1).
This process relies on hematopoietic stem cells (HSCs), which possess the unique capacity for self-renewal and differentiation into two primary lineages: lymphoid and myeloid. From HSCs, common lymphoid progenitors generate B and T lymphocytes and dendritic and natural killer cells. Moreover, common myeloid progenitors initiate the production of erythrocytes, megakaryocytes, macrophages, granulocytes, and other cell types from this lineage (2–4).
HSCs progressively lose their self-renewal capability and become committed to specific lineages upon differentiation (5, 6). This model is considered a tree-like hierarchy. However, single-cell sequencing technologies have brought numerous advantages, including insights into cell heterogeneity, developmental processes, identification of rare cell types and subpopulations, and tracking cell fate decisions. According to these studies, it is suggested that multipotent HSCs gradually become lineage committed through a transitional process, where the progenitor cells are in intermediate states before their full differentiation. Therefore, current views perceive HSC differentiation as a continuum (7–9).
HSC differentiation occurs in specialized microenvironments (or niches) located within the bone marrow (BM) that are intricately regulated and sustained by a combination of intrinsic factors, like transcription factors and epigenetic regulators, extrinsic factors (2, 5), and also non-hematopoietic cells (10).
Extrinsic factors like cytokines, chemokines, and growth factors secreted by BM cellular components also regulate hematopoiesis. Among the most pivotal for HSC regulation are the stromal cell-derived factor 1 (SDF-1, also known as CXCL12), which binds to the CXC-chemokine receptor 4 (CXCR4), and stem cell factor (SCF) that interacts with the c-Kit receptor (CD117) (1, 5). Additionally, interleukin-3 (IL-3), interleukin-5 (IL-5), interleukin-6 (IL-6), thrombopoietin (TPO), Fms-like tyrosine kinase 3 ligand (FLT3L), and vascular endothelial growth factor (VEGF) are other paracrine molecules essential for hematopoiesis (6).
2 Leukemia
Leukemia is a BM disorder characterized by the abnormal proliferation of mutant HSCs and progenitor cells, disrupting regular hematopoietic differentiation. While this malignant disorder comprises two primary categories—myeloid and lymphoid— this review will mainly focus on myeloid malignancies: Acute Myeloid Leukemia (AML), Chronic Myeloid Leukemia (CML), and BCR-ABL1 negative Myeloproliferative Neoplasms (MPN).
Extensive research over the years has identified several mutations occurring within the developing HSCs that result in AML (11, 12). The mutations implicated in AML can be divided into two main groups. The first group, class I mutations, provide cells with a proliferative advantage, while class II mutations predominantly interfere with hematopoietic differentiation and subsequent apoptosis (13).
Mutations in the FLT3 and KIT genes are canonical examples of class I mutations in AML. These genes encode tyrosine kinase receptors, whose mutation initiates internal signaling pathways, leading to uncontrolled growth and expansion of HSCs, contributing to leukemogenesis (14, 15). On the other hand, class II mutations encompass examples such as those found in the CEBPA and RUNX1 genes, which encode transcription factors crucial for hematopoietic differentiation (16, 17).
Nonetheless, AML extends beyond the class I/II mutations framework, encompassing a spectrum of additional genetic alterations that involve pivotal epigenetic regulators and housekeeping genes (11, 13). The DNMT3A gene encodes a methyltransferase crucial for HSC differentiation. Mutations in DNMT3A are early events in leukemogenesis, leading to diminished enzymatic activity (18). Similarly, mutations in TET2, occurring at the onset of leukemogenesis, result in DNA hypermethylation. This aberrant methylation alters the expression of genes critical to HSC function, thereby deregulating hematopoiesis (19). Housekeeping genes are also crucial in AML. The NPM1 gene encodes a chaperone protein involved in cellular homeostasis. Mutations in NPM1 disrupt its subcellular localization, affecting the stabilization and localization of essential proteins, such as p14ARF, a regulator of the p53 pathway (20). Moreover, the IDH1 and IDH2 genes, which encode enzymes essential for metabolic processes, are also mutated in AML. These mutations lead to the production of 2-Hydroxyglutarate (2-HG), an oncometabolite that inhibits DNA demethylation enzymes (21).
The BCR-ABL1 fusion oncogene characterizes CML, and the resulting fusion protein exhibits constitutive kinase activity. Such constitutive activation sustains the fueling of downstream signaling pathways that are associated with cell growth and proliferation (JAK-STAT, PI3K-Akt, and MEK-ERK), contributing to the leukemic transformation of HSCs and disease progression (22, 23).
Essential Thrombocytosis (ET), Polycythemia vera (PV), and Primary myelofibrosis (PMF) constitute a distinct subset of myeloid malignancies known as BCR-ABL1 negative myeloproliferative neoplasms (MPN), distinguished by specific genetic alterations in the JAK2, MPL, and CALR genes. The JAK2V617F mutation, characterized by the substitution of valine with phenylalanine at codon 617, results in the continuous phosphorylation of JAK2 kinase, constitutively activating the JAK-STAT pathway and disrupting HSC regulation. Similarly, mutations in the MPL gene, which encodes a thrombopoietin receptor critical for megakaryopoiesis, also lead to autonomous activation of the JAK-STAT pathway, enabling abnormal HSC proliferation. Moreover, the CALR gene encodes calreticulin that regulates protein folding and calcium signaling, and mutations in this gene modulate receptor signaling activation, ultimately driving aberrant proliferation and survival in developing HSCs (24).
3 The bone marrow microenvironment
The BM is a remarkably complex tissue, housing a variety of cell types, both hematopoietic and non-hematopoietic, that support HSC differentiation and expansion (25–27). Recent advances in cutting-edge imaging techniques, such as confocal and intravital microscopy, more complex and physiological animal models, and RNA sequencing studies, led to identifying several cell types that promote HSC development and their localization within specific BM niches. However, this increased level of resolution has also led to conflicting data across different studies. These discrepancies arise from variations in techniques, animal models, and the types of bones analyzed, making the precise localization of HSCs within the BM a subject of ongoing debate and controversy (3, 28). Nonetheless, despite these current controversies and conflictual data, in this review, we decided to discuss cellular components of the BM microenvironment, focusing on two particular regions: the endosteal niche, which is adjacent to the bone endosteum, and the perivascular niche, located within the BM central region (6, 29).
The endosteal niche comprises mainly osteoblasts (OBs) and osteoclasts (OCs). OBs play a pivotal role by providing essential paracrine signals, such as SDF-1, SCF, and Osteopontin (OPN), that are crucial for HSC function, homing, self-renewal, and quiescence (30–34). In myeloid neoplasia, OBs demonstrate a tumor-suppressor role, as reduced numbers in patient samples correlate with disease progression (35). Conversely, restoring OB frequency in mouse models decreases leukemic burden and extends survival (36). Additionally, myeloid leukemia cells have been found to influence OB differentiation, promoting their proliferation and expansion even with chemotherapy exposure (36–40). The OCs are responsible for bone reabsorption (41) and contribute to HSC regulation by physically creating endosteal niches (42) and degrading paracrine factors implicated in HSC mobilization (43). However, studies investigating the role of OCs in modulating myeloid leukemia progression are scarce.
The perivascular niche is a highly dynamic microenvironment characterized by several cell types like adipocytes, Endothelial cells (ECs), Sympathetic neural cells (SNCs) and Mesenchymal Stem cells (MSCs).
Adipocytes, originating from MSCs, are reservoirs for HSCs and progenitor cells (44) and can support hematopoiesis by secreting SCF (45–47). In leukemia, adipocytes are critical in the dysregulation of cellular energetics by providing fatty acids for cell metabolism (48), sustaining cell survival and migration, and conferring protection against chemotherapy cytotoxicity (49–52). Interestingly, obesity, associated with increased adipose tissue, has been shown to correlate with poorer outcomes (53).
The BM vasculature forms a network of arterioles and sinusoids that deliver essential nutrients and oxygen (54). The ECs are the key components, reside at the interface between blood vessels and the BM, and express distinctive surface markers like CD31, MECA-32, VE-Cadherin, VCAM-1, and VEGFR-2 (55, 56). Moreover, ECs regulate HSC homeostasis by secreting SDF-1 and SCF and expressing Notch ligands (57, 58). In myeloid leukemias, blasts migrate towards ECs, establishing direct interactions through cell adhesion molecules and indirect effects via paracrine factors, also providing protection against chemotherapy-induced cytotoxicity (59–65).
The central nervous system (CNS) also regulates hematopoiesis (66), and central to this regulation are the SNCs (29, 67). By releasing catecholamines, sympathetic neuronal fibers innervate OBs and MSCs and modulate SDF-1 and SCF secretion, thereby modulating HSC homeostasis. Additionally, non-myelinating Schwann cells govern HSC quiescence through transforming growth factor – β (TGF-β) signaling (68–70). In leukemic pre-clinical models, reduced SNC activity resulted in remodeled BM, which, in turn, affected MSCs and sympathetic neurons, leading to leukemic blast proliferation and expansion (39, 71).
MSCs differentiate into several cell types like OBs, adipocytes, and stromal cells (72). These cells exhibit a wide range of surface markers like Nestin, Leptin receptor (LepR), CD51, CD140a, and Sca-1, reflecting their functional diversity regarding self-renewal, multipotency, and distribution (73, 74). Moreover, MSCs are the main source of SCF and SDF-1 within the BM (57, 75), particularly the MSC Nestin+ LepR+ cells (76, 77). In leukemia, MCSs facilitate the homing and retention of AML blasts and, due to their increased Notch and NF-κB signaling, also stimulate the proliferation, survival, and chemoresistance of leukemic blasts (78–82). Similarly, in CML, SDF-1 expression levels also increase the proliferation and chemoresistance of leukemic blasts (83). In MPN, MSCs emerge as crucial drivers of fibrosis, and several molecular players have been identified: the alarmin complex S100A8/S100A9 and the TGFβ, JAK2/STAT3, and NFκB signaling pathways (84, 85).
4 Targeting the bone marrow microenvironment
Leukemia patients frequently resist treatment, often due to the emergence of genetic mutations that alter the drug targets (86). Nonetheless, BM components are also crucial modulators of leukemia pathophysiology, and recognizing its importance in protecting leukemic cells from therapeutic interventions stirred the development of alternative strategies that disrupt this symbiosis. Thus, the concept of targeting both the ‘seed’ (leukemic cells) and the ‘soil’ (the supportive BM microenvironment) has emerged. It suggests that effective treatment requires eliminating leukemic cells and their supportive microenvironment and has gained traction due to compelling evidence from (pre)-clinical studies demonstrating that a combined treatment strategy yields superior efficacy (6, 87–91). Recently, we discussed four distinct strategies: adhesion molecules, angiogenesis, hypoxia, and the SDF-1/CXCR4 axis (6). Here, we explore the latest advancements and broaden our discussion to other topics (Figure 1 and Table 1).
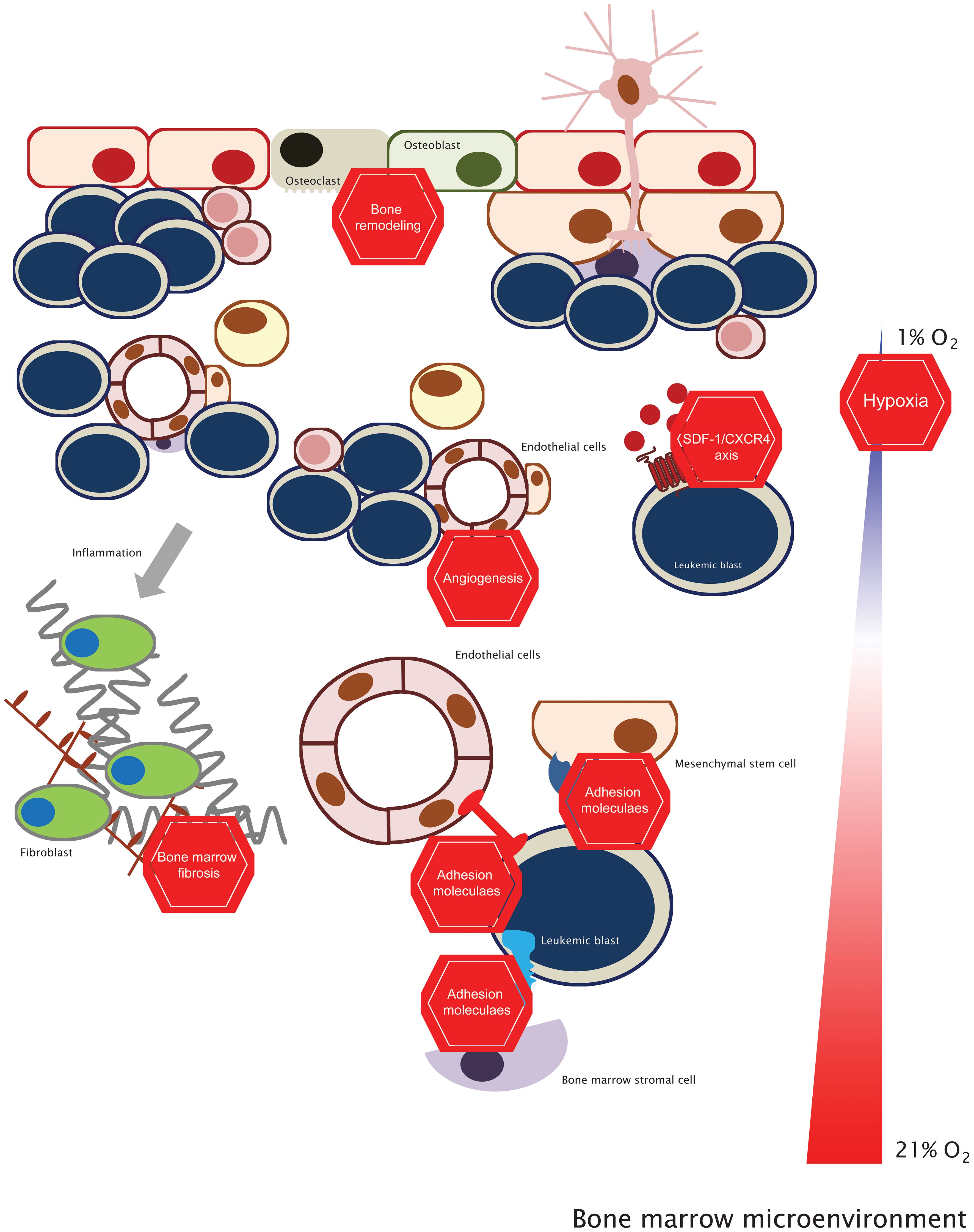
Figure 1 Targeting the bone marrow microenvironment. This concept stems from the fact that targeting leukemic cells (with standard chemotherapy or targeted therapy) and inhibiting the BM microenvironmental factors will be more effective in the clinical setting than just targeting leukemic cells. See the text for further details.
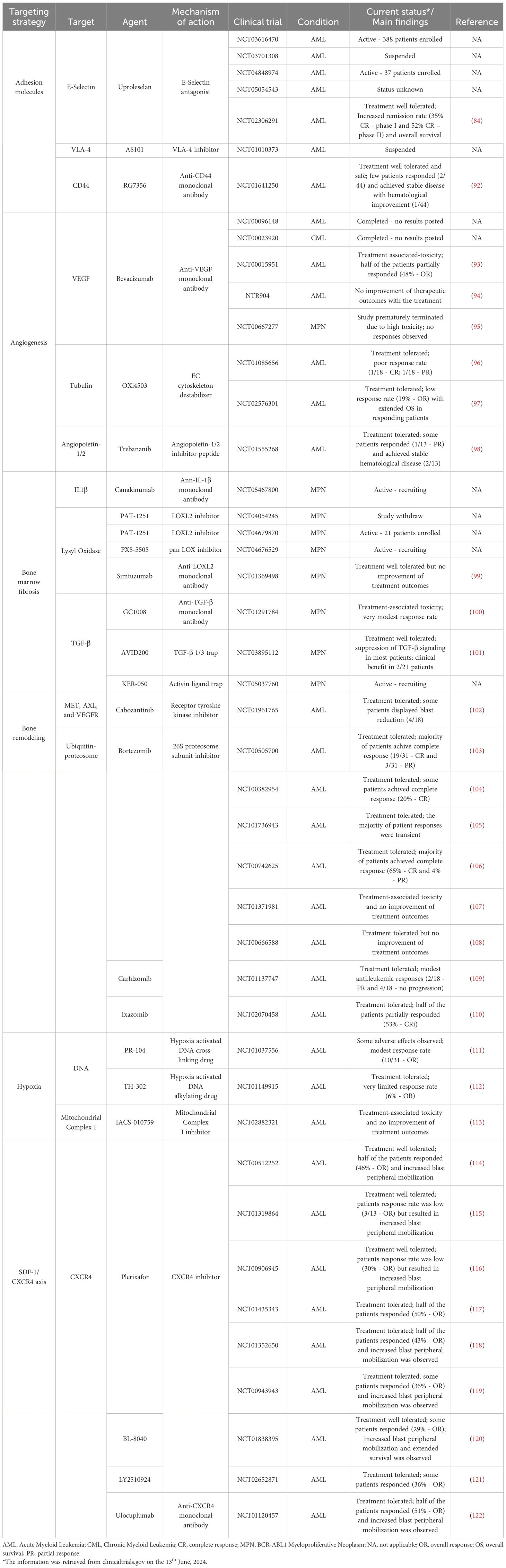
Table 1 Summary of the therapeutic strategies targeting the bone marrow microenvironment.
4.1 Adhesion molecules
The physical interaction between leukemia cells and their microenvironment is crucial for their sustained expansion, and several adhesion molecules are under investigation. The E-Selectin receptor is an essential regulator of HSC function and mediates the chemoresistance of AML blasts (123). Uproleselan, an E-Selectin inhibitor, enhanced chemotherapy efficacy and reduced leukemia burden in an AML pre-clinical model (60). Currently, it is undergoing evaluation in several clinical trials (phase I/II/III) to assess its efficacy in combination with chemotherapy (NCT03616470, NCT03701308, NCT04848974, and NCT05054543). Notably, preliminary data from a phase I/II trial (NCT02306291) showed impressive patient responses, with high remission rates and reduced mortality observed by combining both strategies (124). VLA-4 is implicated in AML proliferation and chemoresistance (80, 92), and its inhibition in mouse models increased chemotherapeutic effects and extended mouse survival (93–95). Targeting VLA-4 is currently being evaluated in a phase II clinical trial (NCT01010373). The CD44 receptor regulates the BM homing of leukemic blasts (125, 126). In AML and CML pre-clinical models, CD44 inhibition with antibodies reduced leukemia burden (96, 97, 127). RG7356, an anti-CD44 monoclonal antibody, was evaluated in a phase I clinical trial (NCT01641250) and showed encouraging results on safety and tolerability (98).
4.2 Angiogenesis
The vasculature is another BM component whose targeting is an appealing therapeutic strategy in leukemia. One notable target, VEGF, is a crucial mediator of angiogenesis and is implicated in AML chemoresistance (62). Bevacizumab, an anti-VEGF antibody, was approved to treat solid cancers, but its efficacy in myeloid neoplasia has been very limited (128–130). Another compound, Combretastatin-A1-diphosphate (OXi4503), disrupts ECs microtubules, hindering the vascular architecture (131). In a pre-clinical AML model, OXi4503 disrupted BM vasculature, decrease tumor burden, and extend mouse survival (132). However, despite its safety and tolerability in clinical trials (NCT01085656, NCT02576301), it resulted in modest response rates when combined with standard chemotherapy (133, 134). The interaction between Angiopoietin and its receptor Tie is also pivotal in regulating AML physiology (99). Trebananib (AMG386), an Angiopoietin inhibitor, underwent evaluation in a phase I clinical trial (NCT01555268), but the outcomes were disappointing, with minimal patient response (100).
4.3 Bone marrow fibrosis
The development of BM fibrosis is the hallmark of PMF, the most aggressive condition in MPN (135). Fibrosis is characterized by the BM deposition of reticulin and collagen fibers, and pro-inflammatory cytokines, lysyl oxidase (LOX), and TGF-β signaling have been shown to modulate this process (101, 102, 136, 137).
IL-1β is one such pro-inflammatory cytokine (102, 137), and Canakinumab (anti-IL1β antibody) is currently being evaluated in a phase II clinical trial (NCT05467800). LOX is an extracellular enzyme that catalyzes the collagen-elastin cross-link, promoting fibrosis (136) and is upregulated in PMF patients (103, 104). Importantly, several clinical trials evaluated LOX inhibition without any known results (NCT04054245, NCT04679870, and NCT04676529). Nonetheless, Simtuzumab, an anti-LOX antibody, was assessed in a phase II clinical trial (NCT01369498) with minimal improvement of BM fibrosis (105). Regarding the TGF-β signaling, GC-1008, an anti-TGF-β antibody, demonstrated a modest reduction in spleen size reduction and anemia recovery (NCT01291784) (106). However, the AVID200, a potent and selective TGFβ 1/3 trap, suppressed TGF-β signaling and resolved BM fibrosis in a pre-clinical model (107). In the clinical setting (NCT03895112), AVID200 was tolerated and suppressed TGF-β signaling (108). The KER-050 compound is another TGF-β inhibitor currently being evaluated in clinical trials (NCT05037760).
4.4 Bone remodeling
Osteolytic lesions are common in cancer patients, resulting from dysregulated bone remodeling due to OB/OC dynamic imbalance. Cabozantinib, a receptor tyrosine kinase inhibitor, exhibits bone remodeling activity by inhibiting OC activity and bone resorption (138). In a phase I clinical trial (NCT01961765), Cabozantinib was well tolerated and demonstrated suppressive signaling activity in leukemic blasts (109).
The ubiquitin-proteasome network is an attractive target due to its importance in bone metabolism. Proteasome inhibitors, like Bortezomib, were tested in AML due to their clinical impact in Multiple Myeloma (MM) (110). Bortezomib, when combined with chemotherapy, was well tolerated (NCT00505700, NCT00382954) (139, 140) but failed to elicit sustained responses and delay disease progression in older (NCT01736943, NCT00742625) (141, 142) and pediatric patients (NCT01371981, NCT00666588) (111, 143). Other proteasome inhibitors like Carfilzomib and Ixazomib demonstrated bone-modulating capabilities by regulating OB/OC cellular function (112). In clinical trials, Carfilzomib demonstrated tolerability and induced modest anti-leukemic activity (NCT01137747) (113), but remarkably, Ixazomib treatment in combination with chemotherapy, induced responses in half of the patients (NCT02070458) (144).
4.5 Hypoxia
The BM is highly hypoxic, contributing to chemoresistance in leukemic cells by upregulating Hypoxia-inducible factor 1 (HIF-1) (145). Hypoxia-activated drugs are unique compounds that remain inactive under normoxic conditions but are activated in low oxygen conditions (hypoxia) and induce cytotoxicity by interfering with DNA synthesis (146). In AML, hypoxia-induced drugs (PR-104, TH-302, and IACS-010759) demonstrated robust efficacy in pre-clinical models by reducing tumor burden and extending survival (114, 147, 148). Unfortunately, phase I clinical trials revealed limited efficacy for PR-104 and TH-302 (NCT01037556, NCT01149915) (115, 116), while IACS-010759, besides limited efficacy, also resulted in increased toxicity in the patients (NCT02882321) (117).
4.6 SDF-1/CXCR4 axis
This signaling axis is essential in the BM homing of leukemic cells, rendering it a desirable target for therapeutic intervention and several compounds have been developed to neutralize it, including Plerixafor [Mozobil – approved for clinical use – (118)], BL-8040, LY2510924, and Ulocuplumab. In myeloid neoplasia, therapeutic blockade of the SDF-1/CXCR4 axis led to leukemic cell peripheral mobilization and increased sensitivity to chemotherapy in pre-clinical models (119–122), spurring the clinical investigation of this pathway. Plerixafor treatment in AML patients demonstrated safety and tolerability (NCT00512252, NCT01319864) (149, 150), and its combination with chemotherapy (NCT00906945 and NCT01435343) (151, 152), hypomethylating agents (NCT01352650) (153) and signaling inhibitors (NCT00943943) (154) yielded promising results regarding leukemic blast reduction and peripheral mobilization. Other strategies, like BL-8040 and LY2510924 (CXCR4 inhibitors) and Ulocuplumab (Anti-CXCR4 antibody), have also undergone clinical evaluation and demonstrated favorable safety profiles, tolerability, reduced leukemic burden, and increased peripheral blast mobilization (NCT01838395, NCT02652871, and NCT01120457) (155–157).
5 Conclusions
Several seminal discoveries unveiled the significant roles of the BM; it is also a crucial supportive microenvironment for the proliferation and survival of leukemic cells (6). The interactions between the leukemic cells and the BM microenvironment are complex, and this symbiosis facilitates the expansion, thriving, and evasion of chemotherapeutic cytotoxic effects by the leukemic blasts. The therapeutic approach in myeloid neoplasia, particularly AML, is evolving, with increasing consideration given to the interactions between leukemic cells and the BM.
Here, we discussed several BM targets currently under evaluation in clinical trials (Table 1), with promising results combined with standard therapy. Such therapeutic strategies include adhesion molecules, BM fibrosis, and the SDF-1/CXCR4 axis. These clinical studies should be reinforced and expanded to more extensive clinical trials and other myeloid malignancies like CML and MPN.
In sharp contrast, targeting other BM components, like vasculature and bone remodeling, yielded disappointing results with very dismal patient responses and associated toxicity. These results underscore the importance of carefully evaluating these targeting strategies, the molecular targets, and even the drug design.
Nonetheless, it is expected that shortly, some of the most promising targets will receive approval from regulatory agencies like the FDA and EMA, thus integrating into the arsenal available to clinicians. Such integration will enhance the outcomes and prognosis for patients with leukemia, particularly in the AML context.
Finally, it is crucial to continue expanding the therapeutic options in myeloid neoplasia by identifying novel BM microenvironmental components and elucidating their significance in leukemic cell expansion.
Author contributions
CS: Writing – original draft, Writing – review & editing. RC: Writing – original draft, Writing – review & editing. AA: Writing – review & editing. BC: Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Centro de Investigação Interdisciplinar em Saúde (CIIS) and Faculdade de Medicina da Universidade Católica Portuguesa (FM-UCP) internal funding, and Fundação para a Ciência e a Tecnologia (FCT) within the project UIDP/04279/2020.
Acknowledgments
We want to thank our funding agencies and the organizations that have provided financial support for this research. We apologize to all the authors whose work, although important to the field, was not included in this review due to space limitations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Olson OC, Kang YA, Passegué E. Normal hematopoiesis is a balancing act of self-renewal and regeneration. Cold Spring Harb Perspect Med. (2020) 10. doi: 10.1101/cshperspect.a035519
2. O’Reilly E, Zeinabad HA, Szegezdi E. Hematopoietic versus leukemic stem cell quiescence: Challenges and therapeutic opportunities. Blood Rev. (2021) 50. doi: 10.1016/j.blre.2021.100850
3. Comazzetto S, Shen B, Morrison SJ. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev Cell. (2021) 56:1848–60. doi: 10.1016/j.devcel.2021.05.018
4. Groarke EM, Young NS. Aging and hematopoiesis. Clinics Geriatric Med. (2019) 35:285–93. doi: 10.1016/j.cger.2019.03.001
5. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. (2019) 20:303–20. doi: 10.1038/s41580-019-0103-9
6. Cardoso BA. The bone marrow niche – the tumor microenvironment that ensures leukemia progression. Adv Exp Med Biol. (2020) 1219:259–293. doi: 10.1007/978-3-030-34025-4_14
7. Watcham S, Kucinski I, Gottgens B. New insights into hematopoietic differentiation landscapes from single-cell RNA sequencing. Blood. (2019) 133:1415–26. doi: 10.1182/blood-2018-08-835355
8. Ye F, Huang W, Guo G. Studying hematopoiesis using single-cell technologies. J Hematol Oncol. (2017) 10:27. doi: 10.1186/s13045-017-0401-7
9. Zhang P, Li X, Pan C, Zheng X, Hu B, Xie R, et al. Single-cell RNA sequencing to track novel perspectives in HSC heterogeneity. Stem Cell Res Ther. (2022) 13:39. doi: 10.1186/s13287-022-02718-1
10. Hoggatt J, Pelus LM. Hematopoiesis. In: Brenner’s encyclopedia of genetics, 2nd ed. Academic Press, Elsevier (2013). doi: 10.1016/B978-0-12-374984-0.00686-0
11. Lagunas-Rangel FA, Chávez-Valencia V, Gómez-Guijosa MÁ, Cortes-Penagos C. Acute myeloid leukemia—genetic alterations and their clinical prognosis. Int J Hematology-Oncol Stem Cell Res. (2017) 11:328–39.
12. Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol. (2011) 4. doi: 10.1186/1756-8722-4-36
13. Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. (2002) 3:179–98. doi: 10.1146/annurev.genom.3.032802.115046
14. Bystrom R, Levis MJ. An update on FLT3 in acute myeloid leukemia: pathophysiology and therapeutic landscape. Curr Oncol Rep. (2023) 25:369–78. doi: 10.1007/s11912-023-01389-2
15. Katagiri S, Chi S, Minami Y, Fukushima K, Shibayama H, Hosono N, et al. Mutated KIT tyrosine kinase as a novel molecular target in acute myeloid leukemia. Int J Mol Sci. (2022) 23:4694. doi: 10.3390/ijms23094694
16. Yokota A, Huo L, Lan F, Wu J, Huang G. The clinical, molecular, and mechanistic basis of RUNX1 mutations identified in hematological Malignancies. Mol Cells. (2020) 43:145–52. doi: 10.14348/molcells.2019.0252
17. Prada-Arismendy J, Arroyave JC, Röthlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. (2017) 31:63–76. doi: 10.1016/j.blre.2016.08.005
18. The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
19. Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. (2015) 29:910–22. doi: 10.1101/gad.260174.115
20. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. (2014) 28:311–20. doi: 10.1038/leu.2013.338
21. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
22. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. (2020) 34:966–84. doi: 10.1038/s41375-020-0776-2
23. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am J Hematol. (2020) 95:691–709. doi: 10.1002/ajh.25792
24. Tefferi A, Pardanani A. Myeloproliferative neoplasms: A contemporary review. JAMA Oncol. (2015) 1:97–105. doi: 10.1001/jamaoncol.2015.89
25. Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. A hypothesis. Blood Cells. (1978) 4:7–25.
26. Rossi DJ, Seita J, Czechowicz A, Bhattacharya D, Bryder D, Weissman IL. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle. (2007) 6:2371–6. doi: 10.4161/cc.6.19.4759
27. Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. (1993) 81:2844–53. doi: 10.1182/blood.V81.11.2844.bloodjournal81112844
28. Kokkaliaris KD. Dissecting the spatial bone marrow microenvironment of hematopoietic stem cells. Curr Opin Oncol. (2020) 32:154–61. doi: 10.1097/CCO.0000000000000605
29. Seita J, Weissman IL. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip Rev: Syst Biol Med. (2010) 2:640–53. doi: 10.1002/wsbm.86
30. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. (2003) 425:841–6. doi: 10.1038/nature02040
31. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. (2003) 425:836–41. doi: 10.1038/nature02041
32. Ponomaryov T, Peled A, Petit I, Taichman RS, Habler L, Sandbank J, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest. (2000) 106:1331–9. doi: 10.1172/JCI10329
33. Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grünewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. (2005) 201:1781–91. doi: 10.1084/jem.20041992
34. Cordeiro-Spinetti E, Taichman RS, Balduino A. The bone marrow endosteal niche: how far from the surface? J Cell Biochem. (2015) 116:6–11. doi: 10.1002/jcb.24952
35. Krevvata M, Silva BC, Manavalan JS, Galan-Diez M, Kode A, Matthews BG, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood. (2014) 124:2834–46. doi: 10.1182/blood-2013-07-517219
36. Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood. (2012) 119:540–50. doi: 10.1182/blood-2011-04-348151
37. Palani HK, Ganesan S, Balasundaram N, Venkatraman A, Korula A, Abraham A, et al. Ablation of Wnt signaling in bone marrow stromal cells overcomes microenvironment-mediated drug resistance in acute myeloid leukemia. Sci Rep. (2024) 14:8404. doi: 10.1038/s41598-024-58860-8
38. Tomasoni C, Arsuffi C, Donsante S, Corsi A, Riminucci M, Biondi A, et al. AML alters bone marrow stromal cell osteogenic commitment via Notch signaling. Front Immunol. (2023) 14. doi: 10.3389/fimmu.2023.1320497
39. Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes Malignancy in an altered hematopoietic stem cell Niche. Cell Stem Cell. (2014) 15:365–75. doi: 10.1016/j.stem.2014.06.020
40. Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. (2013) 13:285–99. doi: 10.1016/j.stem.2013.06.009
41. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. (2003) 423:337–42. doi: 10.1038/nature01658
42. Lymperi S, Ersek A, Ferraro F, Dazzi F, Horwood NJ. Inhibition of osteoclast function reduces hematopoietic stem cell numbers in vivo. Blood. (2011) 117:1540–9. doi: 10.1182/blood-2010-05-282855
43. Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. (2006) 12:657–64. doi: 10.1038/nm1417
44. Han J, Koh YJ, Moon HR, Ryoo HG, Cho CH, Kim I, et al. Adipose tissue is an extramedullary reservoir for functional hematopoietic stem and progenitor cells. Blood. (2010) 115:957–64. doi: 10.1182/blood-2009-05-219923
45. Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. (2017) 19:891–903. doi: 10.1038/ncb3570
46. Zhu RJ, Wu MQ, Li ZJ, Zhang Y, Liu KY. Hematopoietic recovery following chemotherapy is improved by BADGE-induced inhibition of adipogenesis. Int J Hematol. (2013) 97:58–72. doi: 10.1007/s12185-012-1233-4
47. Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. (2009) 460:259–63. doi: 10.1038/nature08099
48. Herroon MK, Rajagurubandara E, Hardaway AL, Powell K, Turchick A, Feldmann D, et al. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget. (2013) 4:2108–23. doi: 10.18632/oncotarget.v4i11
49. Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. (2017) 77:1453–64. doi: 10.1158/0008-5472.CAN-16-1645
50. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
51. Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. (2017) 129:1320–32. doi: 10.1182/blood-2016-08-734798
52. Sabbah R, Saadi S, Shahar-Gabay T, Gerassy S, Yehudai-Resheff S, Zuckerman T. Abnormal adipogenic signaling in the bone marrow mesenchymal stem cells contributes to supportive microenvironment for leukemia development. Cell Communicat Signaling. (2023) 21:277. doi: 10.1186/s12964-023-01231-z
53. Meloni G, Proia A, Capria S, Romano A, Trapé G, Trisolini S, et al. Obesity and autologous stem cell transplantation in acute myeloid leukemia. Bone Marrow Transplant. (2001) 28:365–7. doi: 10.1038/sj.bmt.1703145
54. Nombela-Arrieta C, Pivarnik G, Winkel B, Canty KJ, Harley B, Mahoney JE, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. (2013) 15:533–43. doi: 10.1038/ncb2730
55. Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. (2012) 18:1651–7. doi: 10.1038/nm.2969
56. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. (2010) 10:138–46. doi: 10.1038/nrc2791
57. Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. (2012) 481:457–62. doi: 10.1038/nature10783
58. Greenbaum A, Hsu YMS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. (2013) 495:227–30. doi: 10.1038/nature11926
59. Erbani J, Tay J, Barbier V, Levesque JP, Winkler IG. Acute myeloid leukemia chemo-resistance is mediated by E-selectin receptor CD162 in bone marrow niches. Front Cell Dev Biol. (2020) 8. doi: 10.3389/fcell.2020.00668
60. Barbier V, Erbani J, Fiveash C, Davies JM, Tay J, Tallack MR, et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat Commun. (2020) 11:2042. doi: 10.1038/s41467-020-15817-5
61. Pezeshkian B, Donnelly C, Tamburo K, Geddes T, Madlambayan GJ. Leukemia mediated endothelial cell activation modulates leukemia cell susceptibility to chemotherapy through a positive feedback loop mechanism. PloS One. (2013) 8:e60823. doi: 10.1371/journal.pone.0060823
62. Poulos MG, Gars EJ, Gutkin MC, Kloss CC, Ginsberg M, Scandura JM, et al. Activation of the vascular niche supports leukemic progression and resistance to chemotherapy. Exp Hematol. (2014) 42:976–86.e3. doi: 10.1016/j.exphem.2014.08.003
63. Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc Natl Acad Sci. (2002) 99:4349–54. doi: 10.1073/pnas.072586399
64. Zhang J, Ye J, Ma D, Liu N, Wu H, Yu S, et al. Cross-talk between leukemic and endothelial cells promotes angiogenesis by VEGF activation of the Notch/Dll4 pathway. Carcinogenesis. (2013) 34:667–77. doi: 10.1093/carcin/bgs386
65. Becker PS. Dependence of acute myeloid leukemia on adhesion within the bone marrow microenvironment. Sci World J. (2012) 2012:1–4. doi: 10.1100/2012/856467
66. Cosentino M, Marino F, Maestroni GJM. Sympathoadrenergic modulation of hematopoiesis: a review of available evidence and of therapeutic perspectives. Front Cell Neurosci. (2015) 9. doi: 10.3389/fncel.2015.00302
67. Méndez-, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. (2008) 452:442–7. doi: 10.1038/nature06685
68. Kalinkovich A, Spiegel A, Shivtiel S, Kollet O, Jordaney N, Piacibello W, et al. Blood-forming stem cells are nervous: Direct and indirect regulation of immature human CD34+ cells by the nervous system. Brain Behav Immun. (2009) 23:1059–65. doi: 10.1016/j.bbi.2009.03.008
69. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. (2011) 147:1146–58. doi: 10.1016/j.cell.2011.09.053
70. Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. (2006) 124:407–21. doi: 10.1016/j.cell.2005.10.041
71. Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. (2014) 512:78–81. doi: 10.1038/nature13383
72. Méndez-, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. (2010) 466:829–34. doi: 10.1038/nature09262
73. Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, et al. PDGFRα and CD51 mark human Nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J Exp Med. (2013) 210:1351–67. doi: 10.1084/jem.20122252
74. Houlihan DD, Mabuchi Y, Morikawa S, Niibe K, Araki D, Suzuki S, et al. Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-α. Nat Protoc. (2012) 7:2103–11. doi: 10.1038/nprot.2012.125
75. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. (2013) 495:231–5. doi: 10.1038/nature11885
76. Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. (2010) 33:387–99. doi: 10.1016/j.immuni.2010.08.017
77. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. (2006) 25:977–88. doi: 10.1016/j.immuni.2006.10.016
78. Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. (2004) 64:2817–24. doi: 10.1158/0008-5472.CAN-03-3693
79. Kamga PT, Bassi G, Cassaro A, Midolo M, Di Trapani M, Gatti A, et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget. (2016) 7:21713–27. doi: 10.18632/oncotarget.v7i16
80. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. (2014) 123:2691–702. doi: 10.1182/blood-2013-06-511527
81. Forte D, García-Fernández M, Sánchez-Aguilera A, Stavropoulou V, Fielding C, Martín-Pérez D, et al. Bone marrow mesenchymal stem cells support acute myeloid leukemia bioenergetics and enhance antioxidant defense and escape from chemotherapy. Cell Metab. (2020) 32:829–843.e9. doi: 10.1016/j.cmet.2020.09.001
82. Viñado AC, Calvo IA, Cenzano I, Olaverri D, Cocera M, San Martin-Uriz P, et al. The bone marrow niche regulates redox and energy balance in MLL::AF9 leukemia stem cells. Leukemia. (2022) 36:1969–79. doi: 10.1038/s41375-022-01601-5
83. Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz Á, et al. Mesenchymal niche-specific expression of cxcl12 controls quiescence of treatment-resistant leukemia stem cells. Cell Stem Cell. (2019) 24:769–784.e6. doi: 10.1016/j.stem.2019.02.018
84. Leimkühler NB, Gleitz HFE, Ronghui L, Snoeren IAM, Fuchs SNR, Nagai JS, et al. Heterogeneous bone-marrow stromal progenitors drive myelofibrosis via a druggable alarmin axis. Cell Stem Cell. (2021) 28:637–652.e8. doi: 10.1016/j.stem.2020.11.004
85. Vukotić M, Kapor S, Dragojević T, Đikić D, Mitrović Ajtić O, Diklić M, et al. Inhibition of proinflammatory signaling impairs fibrosis of bone marrow mesenchymal stromal cells in myeloproliferative neoplasms. Exp Mol Med. (2022) 54:273–84. doi: 10.1038/s12276-022-00742-y
86. Stelmach P, Trumpp A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica. (2023) 108:353–66. doi: 10.3324/haematol.2022.280800
87. Agarwal P, Bhatia R. Influence of bone marrow microenvironment on leukemic stem cells. Advances in Cancer Research (2015) 127:227–52. doi: 10.1016/bs.acr.2015.04.007
88. Shafat MS, Gnaneswaran B, Bowles KM, Rushworth SA. The bone marrow microenvironment – Home of the leukemic blasts. Blood Rev. (2017) 31:277–86. doi: 10.1016/j.blre.2017.03.004
89. Kuek V, Hughes AM, Kotecha RS, Cheung LC. Therapeutic targeting of the leukaemia microenvironment. Int J Mol Sci. (2021) 22:6888. doi: 10.3390/ijms22136888
90. Sendker S, Waack K, Reinhardt D. Far from health: the bone marrow microenvironment in AML, A leukemia supportive shelter. Children. (2021) 8:371. doi: 10.3390/children8050371
91. Skelding KA, Barry DL, Theron DZ, Lincz LF. Bone marrow microenvironment as a source of new drug targets for the treatment of acute myeloid leukaemia. Int J Mol Sci. (2022) 24:563. doi: 10.3390/ijms24010563
92. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. (2003) 9:1158–65. doi: 10.1038/nm909
93. Hsieh YT, Jiang E, Pham J, Kim HN, Abdel-Azim H, Khazal S, et al. VLA4 blockade in acute myeloid leukemia. Blood. (2013) 122:3944. doi: 10.1182/blood.V122.21.3944.3944
94. Matsunaga T, Fukai F, Miura S, Nakane Y, Owaki T, Kodama H, et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia. (2008) 22:353–60. doi: 10.1038/sj.leu.2405017
95. Layani-Bazar A, Skornick I, Berrebi A, Pauker MH, Noy E, Silberman A, et al. Redox modulation of adjacent thiols in vla-4 by as101 converts myeloid leukemia cells from a drug-resistant to drug-sensitive state. Cancer Res. (2014) 74:3092–103. doi: 10.1158/0008-5472.CAN-13-2159
96. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. (2006) 12:1167–74. doi: 10.1038/nm1483
97. da Cruz L, McConkey F, Feng N, Pereira D, Hahn S, Rubinstein D, et al. Anti-CD44 antibody, ARH460–16-2, binds to human AML CD34+CD38- cancer stem cells and demonstrates anti-tumor activity in an AML xenograft model. Cancer Res. (2008) 68:3976.
98. Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget. (2016) 7:32532–42. doi: 10.18632/oncotarget.v7i22
99. Reikvam H, Hatfield KJ, Lassalle P, Olsnes Kittang A, Ersvær E, Bruserud Ø. Targeting the angiopoietin (Ang)/Tie-2 pathway in the crosstalk between acute myeloid leukaemia and endothelial cells: Studies of Tie-2 blocking antibodies, exogenous Ang-2 and inhibition of constitutive agonistic Ang-1 release. Expert Opin Investigational Drugs. (2010) 19:169–83. doi: 10.1517/13543780903485659
100. Wang ES, Fetterly G, Brady W, Tan W, Greene J, Gaudy A, et al. Clinical and biologic effects of the angiopoietin 1/2 neutralizing peptibody, trebananib (AMG 386), in acute myeloid leukemia patients. Blood. (2013) 122:2701. doi: 10.1182/blood.V122.21.2701.2701
101. Piasecki A., Leiva O., Ravid K.. Lysyl oxidase inhibition in primary myelofibrosis: A renewed strategy. Arch Stem Cell Ther. (2020) 1:23–7. doi: 10.46439/stemcell
102. Rai S, Grockowiak E, Hansen N, Luque Paz D, Stoll CB, Hao-Shen H, et al. Inhibition of interleukin-1β reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat Commun. (2022) 13:5346. doi: 10.1038/s41467-022-32927-4
103. Abbonante V, Chitalia V, Rosti V, Leiva O, Matsuura S, Balduini A, et al. Upregulation of lysyl oxidase and adhesion to collagen of human megakaryocytes and platelets in primary myelofibrosis. Blood. (2017) 130:829–31. doi: 10.1182/blood-2017-04-777417
104. Tadmor T, Bejar J, Attias D, Mischenko E, Sabo E, Neufeld G, et al. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am J Hematol. (2013) 88:355–8. doi: 10.1002/ajh.23409
105. Verstovsek S, Savona MR, Mesa RA, Dong H, Maltzman JD, Sharma S, et al. A phase 2 study of simtuzumab in patients with primary, post-polycythaemia vera or post-essential thrombocythaemia myelofibrosis. Br J Haematol. (2017) 176:939–49. doi: 10.1111/bjh.14501
106. Mascarenhas J, Li T, Sandy L, Newsom C, Petersen B, Godbold J, et al. Anti-transforming growth factor-β therapy in patients with myelofibrosis. Leuk Lymphoma. (2014) 55:450–2. doi: 10.3109/10428194.2013.805329
107. Varricchio L, Iancu-Rubin C, Upadhyaya B, Zingariello M, Martelli F, Verachi P, et al. TGF-β1 protein trap AVID200 beneficially affects hematopoiesis and bone marrow fibrosis in myelofibrosis. JCI Insight. (2021) 6:e145651. doi: 10.1172/jci.insight.145651
108. Mascarenhas J, Migliaccio AR, Kosiorek H, Bhave R, Palmer J, Kuykendall A, et al. A phase ib trial of AVID200, a TGFβ 1/3 trap, in patients with myelofibrosis. Clin Cancer Res. (2023) 29:3622–32. doi: 10.1158/1078-0432.CCR-23-0276
109. Fathi AT, Blonquist TM, Hernandez D, Amrein PC, Ballen KK, McMasters M, et al. Cabozantinib is well tolerated in acute myeloid leukemia and effectively inhibits the resistance-conferring FLT3/tyrosine kinase domain/F691 mutation. Cancer. (2018) 124:306–14. doi: 10.1002/cncr.31038
110. Cengiz Seval G, Beksac M. The safety of bortezomib for the treatment of multiple myeloma. Expert Opin Drug Saf. (2018) 17:953–62. doi: 10.1080/14740338.2018.1513487
111. Horton TM, Perentesis JP, Gamis AS, Alonzo TA, Gerbing RB, Ballard J, et al. A Phase 2 study of bortezomib combined with either idarubicin/cytarabine or cytarabine/etoposide in children with relapsed, refractory or secondary acute myeloid leukemia: A report from the Children’s Oncology Group. Pediatr Blood Cancer. (2014) 61:1754–60. doi: 10.1002/pbc.v61.10
112. Accardi F, Toscani D, Bolzoni M, Dalla Palma B, Aversa F, Giuliani N. Mechanism of action of bortezomib and the new proteasome inhibitors on myeloma cells and the bone microenvironment: Impact on myeloma-induced alterations of bone remodeling. BioMed Res Int. (2015) 2015. doi: 10.1155/2015/172458
113. Wartman LD, Fiala MA, Fletcher T, Hawkins ER, Cashen A, DiPersio JF, et al. A phase I study of carfilzomib for relapsed or refractory acute myeloid and acute lymphoblastic leukemia. Leuk Lymphoma. (2016) 57:728–30. doi: 10.3109/10428194.2015.1076930
114. Liu F, Kalpage HA, Wang D, Edwards H, Hüttemann M, Ma J, et al. Cotargeting of mitochondrial complex I and bcl-2 shows antileukemic activity against acute myeloid leukemia cells reliant on oxidative phosphorylation. Cancers (Basel). (2020) 12:2400. doi: 10.3390/cancers12092400
115. Konopleva M, Thall PF, Yi CA, Borthakur G, Coveler A, Bueso-Ramos C, et al. Phase I/II study of the hypoxia-activated prodrug PR104 in refractory/relapsed acute myeloid leukemia and acute lymphoblastic leukemia. Haematologica. (2015) 100:927–34. doi: 10.3324/haematol.2014.118455
116. Badar T, Handisides DR, Benito JM, Richie MA, Borthakur G, Jabbour E, et al. Phase I study of evofosfamide, an investigational hypoxia-activated prodrug, in patients with advanced leukemia. Am J Hematol. (2016) 91:800–5. doi: 10.1002/ajh.24415
117. Yap TA, Daver N, Mahendra M, Zhang J, Kamiya-Matsuoka C, Meric-Bernstam F, et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: phase I trials. Nat Med. (2023) 29:115–26. doi: 10.1038/s41591-022-02103-8
118. De Clercq E. Mozobil® (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir Chem Chemother. (2019) 27:204020661982938. doi: 10.1177/2040206619829382
119. Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. (2009) 113:6206–14. doi: 10.1182/blood-2008–06-162123
120. Zeng Z, Xi Shi Y, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. (2009) 113:6215–24. doi: 10.1182/blood-2008–05-158311
121. Abraham M, Klein S, Bulvik B, Wald H, Weiss ID, Olam D, et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16–1 expression. Leukemia. (2017) 31:2336–46. doi: 10.1038/leu.2017.82
122. Cho BS, Zeng Z, Mu H, Wang Z, Konoplev S, McQueen T, et al. Antileukemia activity of the novel peptidic CXCR4 antagonist LY2510924 as monotherapy and in combination with chemotherapy. Blood. (2015) 126:222–32. doi: 10.1182/blood-2015-02-628677
123. Chien S, Haq SU, Pawlus M, Moon RT, Estey EH, Appelbaum FR, et al. Adhesion of acute myeloid leukemia blasts to E-selectin in the vascular niche enhances their survival by mechanisms such as wnt activation. Blood. (2013) 122:61. doi: 10.1182/blood.V122.21.61.61
124. DeAngelo DJ, Jonas BA, Liesveld JL, Bixby DL, Advani AS, Marlton P, et al. Phase 1/2 study of uproleselan added to chemotherapy in patients with relapsed or refractory acute myeloid leukemia. Blood. (2022) 139:1135–46. doi: 10.1182/blood.2021010721
125. Krause DS, Lazarides K, Von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat Med. (2006) 12:1175–80. doi: 10.1038/nm1489
126. Allouche M, Charrad RS, Bettaieb A, Greenland C, Grignon C, Smadja- Joffe F. Ligation of the CD44 adhesion molecule inhibits drug-induced apoptosis in human myeloid leukemia cells. Blood. (2000) 96:1187–90. doi: 10.1182/blood.V96.3.1187
127. Hellqvist E, Holm F, Mason CN, Runza V, Weigand S, Sadarangani A, et al. CD44 monoclonal antibody-enhanced clearance of chronic myeloid leukemia stem cells from the Malignant niche. Blood. (2013) 122:858. doi: 10.1182/blood.V122.21.858.858
128. Karp JE, Gojo I, Pili R, Gocke CD, Greer J, Guo C, et al. Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemias: Therapy with sequential 1-β -D-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin Cancer Res. (2004) 10:3577–85. doi: 10.1158/1078-0432.CCR-03-0627
129. Ossenkoppele GJ, Stussi G, Maertens J, Van Montfort K, Biemond BJ, Breems D, et al. Addition of bevacizumab to chemotherapy in acute myeloid leukemia at older age: A randomized phase 2 trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood. (2012) 120:4706–11. doi: 10.1182/blood-2012-04-420596
130. Mesa RA, Silver RT, Verstovsek S, Mascarenhas J, Kessler CM, Rondelli D, et al. Single agent bevacizumab for myelofibrosis: Results of the myeloproliferative disorders research consortium trial. Haematologica. (2013) 98:1421–3. doi: 10.3324/haematol.2012.083337
131. Hua J, Sheng Y, Pinney KG, Garner CM, Kane RR, Prezioso JA, et al. Oxi4503, a novel vascular targeting agent: Effects on blood flow and antitumor activity in comparison to combretastatin A-4 phosphate. Anticancer Res. (2003) 23:1433–40.
132. Benezra M, Phillips E, Tilki D, Ding BS, Butler J, Dobrenkov K, et al. Serial monitoring of human systemic and xenograft models of leukemia using a novel vascular disrupting agent. Leukemia. (2012) 26:1771–8. doi: 10.1038/leu.2012.48
133. Stockton SS, Pettiford L, Cline C, Chaplin D, Hsu JW, Wingard JR, et al. The vascular disrupting agent OXi4503 in relapsed and refractory AML and MDS. Blood. (2015) 126:4936. doi: 10.1182/blood.V126.23.4936.4936
134. Uckun FM, Cogle CR, Lin TL, Qazi S, Trieu VN, Schiller G, et al. A phase 1B clinical study of combretastatin a1 diphosphate (OXI4503) and cytarabine (ARA-C) in combination (OXA) for patients with relapsed or refractory acute myeloid leukemia. Cancers (Basel). (2020) 12:74. doi: 10.3390/cancers12010074
136. Zahr AA, Salama ME, Carreau N, Tremblay D, Verstovsek S, Mesa R, et al. Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica. (2016) 101:660–71. doi: 10.3324/haematol.2015.141283
137. Rahman MFU, Yang Y, Le BT, Dutta A, Posyniak J, Faughnan P, et al. Interleukin-1 contributes to clonal expansion and progression of bone marrow fibrosis in JAK2V617F-induced myeloproliferative neoplasm. Nat Commun. (2022) 13:5347. doi: 10.1038/s41467-022-32928-3
138. Fioramonti M, Santini D, Iuliani M, Ribelli G, Manca P, Papapietro N, et al. Cabozantinib targets bone microenvironment modulating human osteoclast and osteoblast functions. Oncotarget. (2017) 8:20113–121. doi: 10.18632/oncotarget.v8i12
139. Attar EC, DeAngelo DJ, Supko JG, D’Amato F, Zahrieh D, Sirulnik A, et al. Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin Cancer Res. (2008) 14:1446–54. doi: 10.1158/1078-0432.CCR-07-4626
140. Howard DS, Liesveld J, Phillips GL, Hayslip J, Weiss H, Jordan CT, et al. A phase I study using bortezomib with weekly idarubicin for treatment of elderly patients with acute myeloid leukemia. Leuk Res. (2013) 37:1502–8. doi: 10.1016/j.leukres.2013.09.003
141. Tomlinson BK, Tuscano JM, Abedi M, Welborn J, Arora M, O’Donnell RT, et al. A phase II study of bortezomib in combination with pegylated liposomal doxorubicin for acute myeloid leukemia. Am J Hematol. (2019) 94:E291–E294. doi: 10.1002/ajh.25605
142. Attar EC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, et al. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J Clin Oncol. (2013) 31:923–9. doi: 10.1200/JCO.2012.45.2177
143. Aplenc R, Meshinchi S, Sung L, Alonzo T, Choi J, Fisher B, et al. Bortezomib with standard chemotherapy for children with acute myeloid leukemia does not improve treatment outcomes: a report from the Children’s Oncology Group. Haematologica. (2020) 105:1879–86. doi: 10.3324/haematol.2019.220962
144. Advani AS, Cooper B, Visconte V, Elson P, Chan R, Carew J, et al. A phase I/II trial of MEC (Mitoxantrone, etoposide, cytarabine) in combination with ixazomib for relapsed refractory acute myeloid leukemia. Clin Cancer Res. (2019) 25:4231–7. doi: 10.1158/1078-0432.CCR-18-3886
145. Frolova O, Samudio I, Benito J, Jacamo R, Kornblau SM, Markovic A, et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther. (2012) 13:858–70. doi: 10.4161/cbt.20838
146. Mistry IN, Thomas M, Calder EDD, Conway SJ, Hammond EM. Clinical advances of hypoxia-activated prodrugs in combination with radiation therapy. Int J Radiat Oncol Biol Phys. (2017) 98:1183–96. doi: 10.1016/j.ijrobp.2017.03.024
147. Benito J, Shi Y, Szymanska B, Carol H, Boehm I, Lu H, et al. Pronounced hypoxia in models of murine and human leukemia: High efficacy of hypoxia-activated prodrug PR-104. PloS One. (2011) 6:e23108. doi: 10.1371/journal.pone.0023108
148. Portwood S, Lal D, Hsu YC, Vargas R, Johnson MK, Wetzler M, et al. Activity of the hypoxia-activated prodrug, TH-302, in preclinical human Acute myeloid leukemia models. Clin Cancer Res. (2013) 19:6506–19. doi: 10.1158/1078-0432.CCR-13-0674
149. Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. (2012) 119:3917–24. doi: 10.1182/blood-2011-10-383406
150. Cooper TM, Sison EAR, Baker SD, Li L, Ahmed A, Trippett T, et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium study (POE 10-03). Pediatr Blood Cancer. (2017) 64. doi: 10.1002/pbc.26414
151. Uy GL, Rettig MP, Stone RM, Konopleva MY, Andreeff M, McFarland K, et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. (2017) 7:e542–2. doi: 10.1038/bcj.2017.21
152. Martínez-Cuadrón D, Boluda B, Martínez P, Bergua J, Rodríguez-Veiga R, Esteve J, et al. A phase I–II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann Hematol. (2018) 97:763–72. doi: 10.1007/s00277-018-3229-5
153. Roboz GJ, Ritchie EK, Dault Y, Lam L, Marshall DC, Cruz NM, et al. Phase I trial of plerixafor combined with decitabine in newly diagnosed older patients with acute myeloid leukemia. Haematologica. (2018) 103:1308–16. doi: 10.3324/haematol.2017.183418
154. Borthakur G, Zeng Z, Cortes JE, Chen H, Huang X, Konopleva M, et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD -mutated acute myelogenous leukemia patients. Am J Hematol. (2020) 95:1296–303. doi: 10.1002/ajh.25943
155. Borthakur G, Ofran Y, Tallman MS, Foran J, Uy GL, DiPersio JF, et al. BL-8040 CXCR4 antagonist is safe and demonstrates antileukemic activity in combination with cytarabine for the treatment of relapsed/refractory acute myelogenous leukemia: An open-label safety and efficacy phase 2a study. Cancer. (2021) 127:1246–59. doi: 10.1002/cncr.33338
156. Boddu P, Borthakur G, Koneru M, Huang X, Naqvi K, Wierda W, et al. Initial report of a phase I study of LY2510924, idarubicin, and cytarabine in relapsed/refractory acute myeloid leukemia. Front Oncol. (2018) 8. doi: 10.3389/fonc.2018.00369
157. Becker PS, Foran JM, Altman JK, Yacoub A, Castro JE, Sabbatini P, et al. Targeting the CXCR4 pathway: safety, tolerability and clinical activity of ulocuplumab (BMS-936564), an anti-CXCR4 antibody, in relapsed/refractory acute myeloid leukemia. Blood. (2014) 124:386. doi: 10.1182/blood.V124.21.386.386
Keywords: hematopoiesis, leukemia, myeloid neoplasia, bone marrow microenvironment, bone marrow targeting
Citation: Semedo C, Caroço R, Almeida A and Cardoso BA (2024) Targeting the bone marrow niche, moving towards leukemia eradication. Front. Hematol. 3:1429916. doi: 10.3389/frhem.2024.1429916
Received: 08 May 2024; Accepted: 01 July 2024;
Published: 22 July 2024.
Edited by:
Valentina Giudice, University of Salerno, ItalyReviewed by:
Francesco La Rocca, Madonna delle Grazie Hospital, ItalyPia Sommerkamp, European Molecular Biology Laboratory Heidelberg, Germany
Copyright © 2024 Semedo, Caroço, Almeida and Cardoso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno António Cardoso, bacardoso@ucp.pt
†These authors have contributed equally to this work and share first authorship