 Swe Swe Hlaing
Swe Swe Hlaing Christine Jane Kurian
Christine Jane Kurian Jennie Tan2
Jennie Tan2 Eric Behling
Eric Behling
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Hematol. , 19 December 2022
Sec. Immunobiology and Immunotherapy
Volume 1 - 2022 | https://doi.org/10.3389/frhem.2022.1039821
Hemophagocytic lymphohistiocytosis (HLH) is a highly detrimental syndrome that can progress to multiorgan failure, necessitating the resources of an intensive care unit, with a mortality rate as high as 40%. Secondary HLH is usually triggered by infection, most often from a viral infection or malignancy. Management of HLH in adults is challenging as treatment algorithms targeting hyperinflammation are based on pediatric protocols, such as HLH-94 and HLH-2004. To our knowledge, there are only a few reported cases of HLH secondary to ehrlichiosis infection and none in elderly patients with multiple comorbidities. Here, we present a unique case of HLH secondary to ehrlichiosis infection in an 82-year-old female successfully treated with antibiotics and steroids.
Hemophagocytic lymphohistiocytosis (HLH) is a highly detrimental syndrome that can progress to multiorgan failure, necessitating the resources of an intensive care unit, with a mortality rate as high as 40% (1).
Familial/primary HLH is common in children and caused by genetic mutations. Secondary HLH is caused by a trigger such as infection [typically viral, such as Epstein–Barr virus, human immunodeficiency virus, and cytomegalovirus, but also bacterial, parasitic, and fungal organisms (2)], malignancy [commonly lymphoma (3)], autoimmune conditions (4) or something else, such as organ transplantation, surgery, or hemodialysis (4). Both primary and secondary HLH are hyperferritinemic, hyperinflammatory syndromes with a common terminal pathway, but they have different pathogenic roots. HLH is an aberrant immune response driven by T cells and associated with potentially fatal cytokine storms (5).
An 82-year-old-female with a history of coronary artery disease, hypertension, congestive heart failure, and hyperlipidemia presented with 2 months of myalgia, fatigue, and waxing and waning mentation. On admission, she had a fever of 103°F and was hypoxic, with 86% oxygen saturation on room air, and placed on 2 liters of nasal cannula. A chest CT, as per the pulmonary embolism (PE) protocol, was performed, and it did not show any blood clot or source of infection such as pneumonia. Because infection was suspected, with presenting symptoms including fever and altered mental status, an abdominal CT was performed to rule out any source of infection. Borderline splenomegaly (12.1 cm) was observed, and no other source of infection was noted. A head CT was performed, given the altered mental status, and it did not show any acute pathology. The patient’s admission laboratory values are described in detail in Table 1. Her white blood cell (WBC) count was 2,200 cells and the platelet count was 30,000/μl. Her baseline cell counts were normal. Subsequent laboratory results showed a lactate dehydrogenase (LDH) level of 1700 U/l, ferritin level of 76,000 ng/ml, aspartate transaminase (AST) level of 238 U/l, alanine transaminase (ALT) level of 7676 U/l, triglyceride level of 207 mg/dl, and a fibrinogen level of 197 mg/dl, and a prothrombin time (PT) of 13.7 sec, a partial thromboplastin time (PTT) of 99.5 sec and an international normalized ratio (INR) of 1.2.
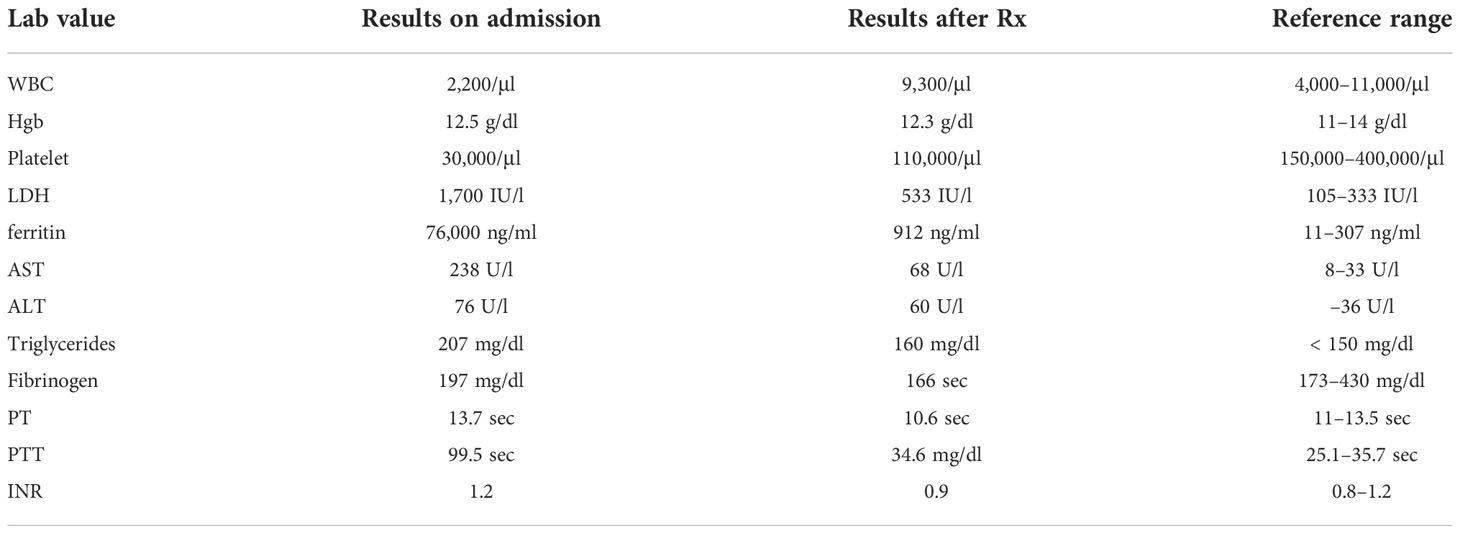
Table 1 Laboratory values on admission and post treatment (Rx) with doxycycline and dexamethasone along with reference ranges.
A peripheral smear did not reveal any schistocytes; hence, concerns about thrombotic thrombocytopenic purpura (TTP) and disseminated intravascular coagulation (DIC) were low. Pancultures, hepatitis serologies, and other viral infection serologies, including Epstein–Barr virus (EBV), cytomegalovirus (CMV), and Lyme, Borrelia, and Rocky Mountain spotted fever infections, were also checked. Given the significant fever, pancytopenia, and elevated levels of ferritin, HLH was highly suspected; the HScore was 181 points, which was significant, with 70%–80% probability of hemophagocytic syndrome. The patient started empiric steroid therapy with 10 mg/m2 of dexamethasone daily. Although her peripheral smear did not show signs of HLH, such as hemophagocytosis, subsequent bone marrow biopsy revealed hemophagocytic cells (Figure 1). Later, her infectious work-up came back positive for ehrlichiosis (via a PCR assay). She was immediately placed on a 10-day course of doxycycline therapy. She improved significantly and her use of dexamethasone tapered off. Her soluble IL-2 receptor assay level, sent on admission, resulted later in the hospital stay and was 13439 pg/ml (reference value 241–848 pg/ml).
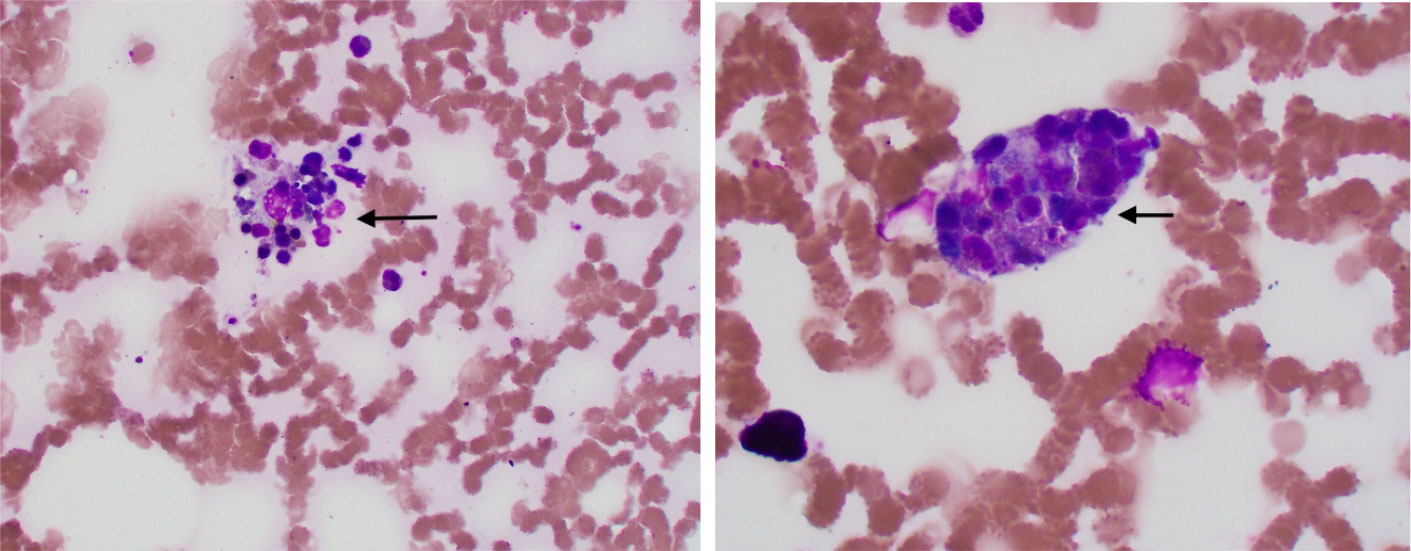
Figure 1 Black arrow showing hemophagocytic cells in bone marrow biopsy.
Patients with HLH usually present with a triad of fever, bicytopenia, and splenomegaly. Symptoms range from mild fatigue, purpura, rash, bleeding diathesis, diarrhea, and arthralgia to severe sepsis-like syndromes. These symptoms together with a rapid clinical deterioration raise the possibility of HLH, even in the presence of proper antibiotic therapy and/or the absence of any infectious focus.
The diagnosis of HLH is challenging because no single clinical or laboratory parameter has the sensitivity and specificity required for unambiguous HLH diagnosis. In adults, diagnosis is based on HLH-2004 diagnosis criteria in conjunction with clinical judgment and the patient’s history. An HLH-94 clinical trial proposed a standardized set of five diagnostic criteria for HLH (6). These were revised for the HLH-2004 (7), and it was established that individuals need to meet five or more of the eight diagnostic criteria presented in Table 2.
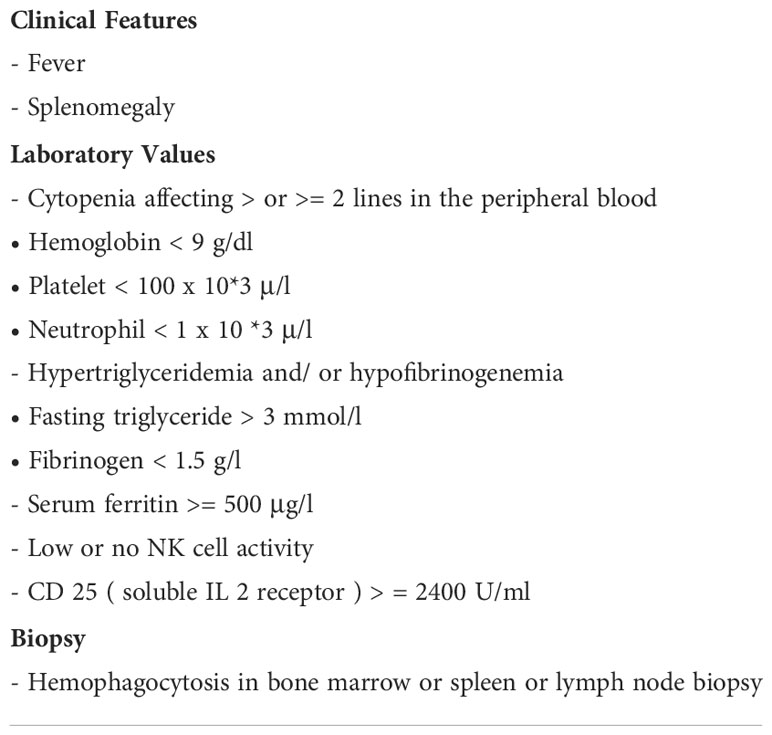
Table 2 HLH-2004 diagnostic criteria.
In some instances, HLH may be strongly considered and HLH-directed treatment may be initiated, even though five criteria are not fulfilled (8). This HLH-2004 criteria, developed for children, has not been fully verified for adults and is still based on professional opinions. Our patient had seven out of the eight HLH-2004 diagnostic criteria (NK cell activity was not tested because she had already met the criteria).
The HLH probability calculator (HScore) is a web-based online calculator developed retrospectively in adult patients that may be a helpful diagnostic tool (9) (Table 3). Our patient had a HScore of 181 points.
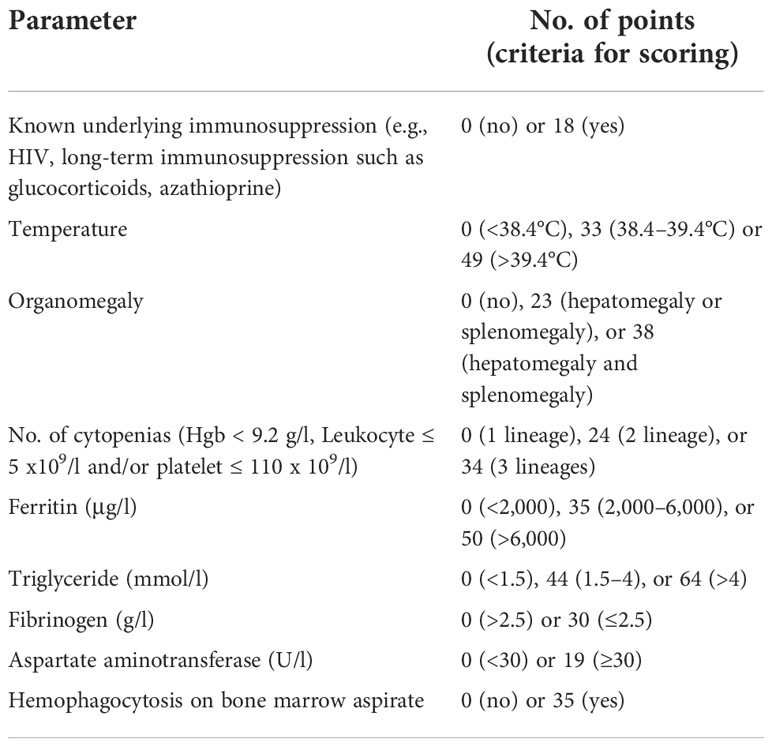
Table 3 HScore for diagnosis of HLH.
As for treatment, the HLH-94 protocol relates to corticosteroids, typically dexamethasone, cyclosporine A, intrathecal therapy (methotrexate if there are progressive neurological symptoms or if an abnormal CSF has not improved) and etoposide destroying activated T cells and suppress inflammatory cytokine production (10). However, elderly patients may have many chronic comorbidities, making them more vulnerable to end-organ damage caused by a cytokine storm in HLH and HLH-94 chemotherapy. A reduced etoposide frequency (twice weekly to once weekly) or dose reduction (150 mg/m2 to 50–100 mg/m2) may be considered.
Given that viral infections are the most frequent trigger for HLH, the prognosis improved greatly when treated promptly using HLH-94 protocols, with variable intensity and length of treatment depending on the severity of HLH. The pathogenesis of secondary HLH from an underlying viral infection is because of the inability of the immune system to adequately control the hyperstimulatory effect of the trigger. The acquired immune dysfunction leads to a severe life-threatening condition. Some experts recommend the addition of rituximab to HLH-directed therapy as it will be effective in clearing the reservoir of the virus in EBV-triggered HLH (11).
In HLH induced by intracellular infections, such as tuberculosis, leishmaniasis, or rickettsia disease (ehrlichiosis), patients usually do not need HLH-94-like treatment. Patients with infections from pathogens that affect the monocyte–macrophage system may experience HLH; however, immunosuppression as provided by the HLH-94 regimen should be avoided because these bacteria typically respond to particular antibiotic therapies.
Tetracyclines or chloramphenicol are used to treat rickettsioses, whereas amphotericin B is used to treat leishmanias (12) and quadruple antibiotic therapy is used to treat tuberculosis (13).
Although it is not common, patients may have refractory or relapsing HLH. Salvage treatment may be required using combined chemotherapy and consolidation with allogeneic stem cell transplant. The treatment decision needs to be individualized based on the most likely triggering cause.
Anakinra is a very quick-acting medication used in individuals who are in a serious condition. Interleukin-1 (IL-1)-mediated innate immune system dysfunction is a key factor in disease etiology, and anakinra inhibits this key mediator of hyperinflammation. To diagnose and treat the underlying condition, anakinra is required in the acute phase of HLH. It is not a therapeutic medication, but rather dampens the pathogenic, hyperinflammatory response. By slowing down the disease’s course and helping the patient with HLH survive the critical period, it makes it possible to treat the underlying condition. Anakinra has a good safety profile compared with other HLH therapies (14).
Utilizing short-term corticosteroids with or without intravenous immunoglobulin (IVIG) is another conservative strategy. Cell counts and ferritin levels are examined to see how well the treatment is working.
Ehrlichiosis is a tick-borne infection, and it usually manifests as fever, hypotension, confusion, acute renal failure, and coagulopathy. The principal vector of Ehrlichia chaffeensis is the lone star tick. Its incubation period is 1–2 weeks and a rash is reported in only 36% of cases (15). A wide range of laboratory abnormalities is noted in ehrlichiosis including leucopenia, thrombocytopenia, transaminitis, elevated LDH levels, and anemia (16). The complications of ehrlichiosis are not limited to seizure, renal failure, respiratory failure, sepsis, and HLH. The diagnosis is usually obtained using PCR testing. Given a high level of clinical suspicion, it should be treated at once with antimicrobial therapy, such as doxycycline.
In this clinical scenario, the patient’s initial presentations were vague. Both HLH and ehrlichiosis infection can cause bicytopenia and transaminitis. The treatment decision was challenging as she was an elderly frail lady with multiple comorbidities. Given the high level of suspicion of HLH, she was started on dexamethasone treatment initially while the infection work-up was pending. Fortunately, she responded well to doxycycline and her HLH was also resolved. She was given 10 mg/m2 of doxycycline in weeks 1 and 2, 5 mg/m2 in weeks 3 and 4, 2.5 mg/m2 in weeks 5 and 6, and 1.25 mg/m2 in week 7, after which it was stopped.
We chose to treat the patient with dexamethasone over IVIG as she had underlying heart failure. Anakinra was not chosen as our patient was an elderly frail lady with multiple comorbidities and she was also improving with dexamethasone and doxycycline.
The following table is the reported cases of HLH precipitated by ehrlichiosis in the literature along with the treatment provided and their outcomes (Table 4). All these patients presented with fever and pancytopenia of different ranges and they were diagnosed with ehrlichiosis.

Table 4 Reported cases of HLH precipitated by ehrlichiosis in the literature.
Interestingly, the immunosenescence effect of NK cells diminishes with age. As a result, viral infections are said to become more common as people age. It is brought on by multiple factors, including NK cell-mediated cytolysis and elimination (23). Aging is associated with a loss in NK cell population, phenotype, and functions, which contributes to poor immunomodulation, raises the risk of infection, and precipitates life-threatening conditions.
A reasonable index of suspicion for HLH should be present in any patients with fever, pancytopenia, high ferritin levels, and low fibrinogen levels. As long as five of the eight diagnostic criteria are met, a positive bone biopsy is not necessary to justify empiric treatment. Current guidelines recommend the use of steroids, IVIG, and etoposide. Early recognition of underlying triggers must be addressed promptly to reduce the risk of mortality. Although the drugs used in pediatric HLH have shown efficacy in treating adult HLH, research involving novel agents is ongoing.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Karlsson T. Secondary haemophagocytic lymphohistiocytosis: Experience from the uppsala university hospital. Upsala J Med Sci (2015) 120(4):257–62. doi: 10.3109/03009734.2015.1064500
2. Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis (2007) 7(12):814–22. doi: 10.1016/S1473-3099(07)70290-6
3. Lehmberg K, Nichols KE, Henter JI, Girschikofsky M, Greenwood T, Jordan M, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica (2015) 100(8):997–1004. doi: 10.3324/haematol.2015.123562
4. Carter SJ, Tattersall RS, Ramanan AV. Macrophage activation syndrome in adults: recent advances in pathophysiology, diagnosis and treatment. Rheumatol (Oxford England) (2019) 58(1):5–17. doi: 10.1093/rheumatology/key006
5. Schulert GS, Canna SW. Convergent pathways of the hyperferritinemic syndromes. Int Immunol (2018) 30(5):195–203. doi: 10.1093/intimm/dxy012
6. Henter JI, Elinder G, Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. the FHL study group of the histiocyte society. Semin Oncol (1991) 18(1):29–33.
7. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer (2007) 48(2):124–31. doi: 10.1002/pbc.21039
8. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: Primary forms and predisposing conditions. Curr Opin Immunol (2017) 49:20–6. doi: 10.1016/j.coi.2017.08.004
9. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol (Hoboken NJ) (2014) 66(9):2613–20. doi: 10.1002/art.38690
10. Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: Consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol In Pract (2018) 6(5):1508–17. doi: 10.1016/j.jaip.2018.05.031
11. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J haematology (2013) 162(3):376–82. doi: 10.1111/bjh.12386
12. Gagnaire MH, Galambrun C, Stéphan JL. Hemophagocytic syndrome: A misleading complication of visceral leishmaniasis in children–a series of 12 cases. Pediatrics (2000) 106(4):E58. doi: 10.1542/peds.106.4.e58
13. Falzon D, Schünemann HJ, Harausz E, González-Angulo L, Lienhardt C, Jaramillo E, et al. World health organization treatment guidelines for drug-resistant tuberculosis, 2016 update. Eur Respir J (2017) 49(3):1602308. doi: 10.1183/13993003.02308-2016
14. . Available at: https://www.england.nhs.uk/wp-content/uploads/2021/10/1924-Clinical-commissioning-policy-anakinra-for-haemophagocytic-lymphohistiocytosis-.pdf.
15. Fishbein DB, Dawson JE, Robinson LE. Human ehrlichiosis in the united states, 1985 to 1990. Ann Internal Med (1994) 120(9):736–43. doi: 10.7326/0003-4819-120-9-199405010-00003
16. Bakken JS, Aguero-Rosenfeld ME, Tilden RL, Wormser GP, Horowitz HW, Raffalli JT, et al. Serial measurements of hematologic counts during the active phase of human granulocytic ehrlichiosis. Clin Infect Dis an Off Pub. Infect Dis Soc America (2001) 32(6):862–70. doi: 10.1086/319350
17. Otrock ZK, Gonzalez MD, Eby CS. Ehrlichia-induced hemophagocytic lymphohistiocytosis: A case series and review of literature. Blood cells molecules Dis (2015) 55(3):191–3. doi: 10.1016/j.bcmd.2015.06.009
18. Pandey R, Kochar R, Kemp S, Rotaru D, Shah SV. Ehrlichiosis presenting with toxic shock-like syndrome and secondary hemophagocytic lymphohistiocytosis. J Ark Med Soc (2013) 109(13):280–2.
19. Kumar N, Goyal J, Goel A, Shakoory B, Chatham W. Macrophage activation syndrome secondary to human monocytic ehrlichiosis. Indian J Hematol Blood Transfus. (2014) 30(Suppl 1):145–7. doi: 10.1007/s12288-013-0299-3
20. Kaplan RM, Swat SA, Singer BD. Human monocytic ehrlichiosis complicated by hemophagocytic lymphohistiocytosis and multi-organ dysfunction syndrome. Diagn Microbiol Infect Dis (2016) 86(3):327–8. doi: 10.1016/j.diagmicrobio.2016.08.007
21. Badireddi S, Joshi M. Hemophagocytic lymphohistiocytosis: An unreported complication of ehrlichiosis in adults. Chest (2012) 142(4):415A. doi: 10.1378/chest.1390907
22. Provenzano JD, Kamerzell TJ, Reda E, Hawkinson DJ, Sharpe MR. Diagnosis and treatment of hemophagocytic lymphohistiocytosis in an adult patient with ehrlichiosis. Case Rep Inten Med (2015) 2(2):44. doi: 10.5430/crim.v2n2p44
Keywords: HLH, ehrlichiosis, secondary HLH, immune hyperactivation, steroid
Citation: Hlaing SS, Kurian CJ, Tan J, Behling E and Hussein AKA (2022) Case Report: A unique case of secondary hemophagocytic lymphohistiocytosis from ehrlichiosis infection. Front. Hematol. 1:1039821. doi: 10.3389/frhem.2022.1039821
Received: 08 September 2022; Accepted: 22 November 2022;
Published: 19 December 2022.
Edited by:
Emmanouil Nikolousis, European University Cyprus, CyprusReviewed by:
Alexandros Kanellopoulos, The University of Sheffield, United KingdomCopyright © 2022 Hlaing, Kurian, Tan, Behling and Hussein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swe Swe Hlaing, c3dlc3dlLmhsYWluZ0Bjcm96ZXIub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.