
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 17 February 2025
Sec. Applied Genetic Epidemiology
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1502839
 Ziang Meng1†Chumeng Zhang2†Shuai Liu3Wen Li4Yue Wang4Qingyi Zhang4
Ziang Meng1†Chumeng Zhang2†Shuai Liu3Wen Li4Yue Wang4Qingyi Zhang4 Bichen Peng4Weiyi Ye4Yue Jiang4
Bichen Peng4Weiyi Ye4Yue Jiang4 Yingchao Song4
Yingchao Song4 Miao Guo5*
Miao Guo5* Xiao Chang4*
Xiao Chang4* Lei Shao1*
Lei Shao1*Introduction: COVID-19 severity has been linked to immune factors, with excessive immune responses like cytokine storms contributing to mortality. However, the genetic basis of these immune responses is not well understood. This study aimed to explore the genetic connection between COVID-19 severity and blood cell traits, given their close relationship with immunity.
Materials and methods: GWAS summary statistics for COVID-19 and blood cell counts were analyzed using Linkage Disequilibrium Score Regression (LDSC) to estimate genetic correlations and heritabilities. For traits with significant correlations, a Multi-Trait GWAS Analysis (MTAG) was performed to identify pleiotropic loci shared between COVID-19 and blood cell counts.
Results: Our MTAG analysis identified four pleiotropic loci associated with COVID-19 severity, five loci linked to hospitalized cases, and one locus related to general patients. Among these, two novel loci were identified in the high-risk population, with rs55779981 located near RAVER1 and rs73009538 near CARM1. In hospitalized patients, two previously unrecognized loci were detected, namely, rs115545251 near GFI1 and rs3181049 near RAVER1, while in general patients, rs11065822 near CUX2 emerged as a newly identified locus. We also identified potential target genes, including those involved in inflammation signaling (CARM1), endothelial dysfunction (INTS12), and antiviral immune response (RAVER1), which may require further investigation.
Conclusion: Our study offers insights into the genetic overlap between COVID-19 and immune factors, suggesting potential directions for future research and clinical exploration.
COVID-19 is an extreme and clinically homogeneous disease phenotype (Gao et al., 2021; Oran and Topol, 2020). Infection with the coronavirus triggers the onset of the disease through a series of immune responses (Jackson et al., 2022). According to data from the World Health Organization (WHO), over 760 million cases and 6.9 million deaths have been recorded globally since December 2019, although the actual numbers are believed to be higher. The pandemic has not only disrupted global economic and trade development but has also imposed a significant socioeconomic burden on individuals, families, and societies worldwide. While most infected individuals experience mild to moderate respiratory illness and recover without specialized treatment, older adults or those with pre-existing conditions such as cardiovascular disease, diabetes, chronic respiratory diseases, or cancer are at a higher risk of severe outcomes or hospitalization. Notably, COVID-19 can affect individuals of any age and may lead to severe illness or death (Gao et al., 2021).
Epidemiological studies have identified severe risk factors associated with COVID-19, such as hypertension, coronary heart disease (Espiritu et al., 2024), type II diabetes (Hopkins et al., 2024), and thrombosis (Lindström et al., 2019). Furthermore, emerging evidence suggests that genetic factors play a significant role in determining susceptibility to COVID-19 (van der Made et al., 2020). To uncover the genetic basis of COVID-19, various genome-wide association studies (GWAS) have been conducted, with the largest study identifying 49 significant loci involving 24,202 severe cases (Pairo-Castineira et al., 2023). Recently, the development of GWAS multi-trait analysis (MTAG) provides a powerful approach to explore the genetic overlap and common etiologies between complex traits. By jointly analyzing multiple genetically correlated traits, MTAG can identify pleiotropic genetic variants that influence related phenotypes, revealing shared biological pathways and genetic interactions among various conditions.
Many studies have highlighted changes in blood measurements of COVID-19 patients (Zhou et al., 2021). COVID-19 is closely associated with various hematological abnormalities, including lymphopenia, elevated inflammatory markers such as C-reactive protein (CRP) and ferritin levels, as well as abnormalities in the coagulation pathway. These hematological changes provide important clues for understanding the mechanisms of the virus and its effects on the body. Notably, lymphopenia, although the underlying mechanisms remain unclear, has increasing evidence suggesting that lymphocytes play a crucial role in the pathophysiology of viral infections (Hegde, 2020; Song et al., 2024). Immune factors are closely related to blood cell counts, particularly white blood cell counts, indicating a strong relationship between the virus and the immune system, with immune factors playing significant roles in viral infections and disease progression (Jackson et al., 2022). Additionally, endothelial dysfunction is associated with COVID-19, and angiogenic T cells (Tangs) play a key role in endothelial repair (Liu et al., 2024). The emergence of a hypercoagulable state is also linked to COVID-19 (Valencia et al., 2024). Overall, this series of hematological risk factors interact to influence the course of COVID-19, with each factor potentially having a unique genetic background.
This study aims to quantify the genetic correlation between COVID-19 and related hematological traits. By conducting a comprehensive multi-trait genome-wide association study (MTAG), we identified common pleiotropic loci that influence both COVID-19 susceptibility and blood cell counts, providing deeper insights into the shared genetic architecture of COVID-19 and immune-related blood traits. These findings may help identify potential genetic markers that contribute to the clinical manifestations of COVID-19, thereby informing the development of targeted treatments.
The COVID-19 GWAS data used in this study are from the COVID-19 Host Genetics Initiative (HGI), specifically from the round seven release, using European population data. The study includes data on three different COVID-19 phenotypes: very severe respiratory confirmed COVID-19 vs. population (COVID-19-a2) with a total sample size of 1,072,442 and 13,769 cases; hospitalized COVID-19 vs. population (COVID-19-b2) with a total sample size of 2,062,805 and 32,519 cases; and general COVID-19 vs. population (COVID-19-c2) with a total sample size of 2,475,240 and 122,616 cases. The release date for this data is 8 April 2022. The study encompasses contributions from numerous research partners, including but not limited to Estonian Biobank (EstBB), UK Biobank (UKBB), FinnGen, Generation Scotland, deCODE, and the Million Veterans Program (MVP). Each dataset has been filtered and standardized to ensure consistency and reliability in the analysis. Specific details and descriptions of the datasets are provided in Supplementary Table S1.
For the study on hematological traits, data was sourced from the trans-ethnic and ancestry-specific blood-cell genetics study published in Cell. This comprehensive study investigated blood-cell genetics across 746,667 individuals from five global populations (Chen et al., 2020). For our research, we focused on the European subset, including data on lymphocyte count (524923 Europeans), basophil count (474,001 Europeans), eosinophil count (474237 Europeans), neutrophil count (519,288 Europeans), and monocyte count (521,594 Europeans). Specific details of the datasets are provided in Supplementary Table S2.
The cross-ancestry genetic correlation (Rg) between each pair of COVID-19 phenotypes and blood-cell traits was evaluated using LD Score Regression (LDSC) and GWAS summary statistics. LDSC is an established method that identifies genetic correlations between complex traits and diseases, providing etiological insights and helping to prioritize likely causal relationships. The primary challenges in estimating genetic correlation from GWAS data with traditional methods include the unavailability of individual genotype data and widespread sample overlap among meta-analyses. LDSC overcomes these challenges by using a technique called cross-trait LD Score regression, which requires only GWAS summary statistics and is not biased by sample overlap (Bulik-Sullivan et al., 2015). The formula used in LDSC is as follows:
where
Research has demonstrated that certain functional categories within the genome disproportionately contribute to the heritability of complex diseases (Trynka et al., 2013). Stratified LD Score Regression (s-LDSC) is a method developed to analyze these contributions by partitioning heritability from GWAS summary statistics while accounting for linkage disequilibrium (LD). This approach is computationally efficient and capable of handling large sample sizes, making it suitable for genome-wide studies involving extensive datasets. It leverages genome-wide information without relying solely on SNPs reaching genome-wide significance, allowing for a more comprehensive analysis. s-LDSC enables the identification of cell type-specific elements and other functional genomic regions that significantly contribute to the polygenic architecture of complex traits and diseases, providing insights into their biological mechanisms and helping prioritize genomic regions for further functional characterization (Finucane et al., 2015).
This study utilized annotation data constructed from six chromatin marks (DHS, H3K27ac, H3K36me3, H3K4me1, H3K4me3, and H3K9ac) across 88 cell types or tissues from the Roadmap project to partition the SNP heritability of various traits. For each chromatin mark, the cell type-specific annotations were further divided into nine categories: adipose, central nervous system, digestive system, cardiovascular, musculoskeletal and connective tissue, immune and blood, liver, pancreas, and others (Finucane et al., 2018). The annotation-specific enrichment values for each trait were converted into a color scale and visualized through hierarchical clustering.
To complement the genome-wide genetic correlation estimated by LDSC, which aggregates information across all variants in the genome, we employed ρ-HESS to quantify the local genetic correlation between pairs of traits (Finucane et al., 2018). ρ-HESS is designed to measure the correlation due to genetic variation at small regions in the genome, providing a more granular understanding of the genetic architecture. This technique only requires GWAS summary data and does not assume any specific distribution of causal variant effect sizes, while accounting for linkage disequilibrium (LD) and overlapping GWAS samples. The analysis using ρ-HESS involves several steps: first, it computes the eigenvalues of LD matrices and the squared projections of GWAS effect size vectors onto the eigenvectors of LD matrices for each trait. Next, it estimates the local SNP-heritability of each trait using these projections. Finally, ρ-HESS uses the local SNP-heritability estimates to calculate local genetic covariance estimates and their standard errors. In our study, all significant trait pairs identified in the global genetic correlation analysis (LDSC, P < 0.01) underwent local genetic correlation analysis using ρ-HESS. This allowed us to investigate which specific local genomic regions contributed to the global genetic correlation. A Bonferroni-corrected p-value of less than 0.05/n was considered statistically significant (n = Hypothesis testing quantity).
Multi-Trait Analysis of GWAS (MTAG) is a method designed for the joint analysis of summary statistics from genome-wide association studies (GWAS) of different traits, possibly including overlapping samples. MTAG enhances the power to detect loci by analyzing GWAS summary statistics from related traits together, accounting for sample overlap and incomplete genetic correlation. It begins by filtering variants to remove non-common SNPs, duplicated SNPs, or SNPs with strand ambiguity (Turley et al., 2018). MTAG then estimates pairwise genetic correlations between traits using LD Score Regression (LDSC) and utilizes these estimates to calibrate the variance-covariance matrix of the random effect component (Bulik-Sullivan et al., 2015). Following this, MTAG performs a random-effect meta-analysis to produce SNP-level summary statistics. In our application, we prioritized nominally significant pleiotropic SNPs that achieved genome-wide significance (P < 1.67 × 10⁻⁸) in the multi-trait analysis and suggestive significance (P < 1 × 10−3) in the original single-trait GWAS. This threshold accounts for the number of independent tests conducted based on the traditional GWAS threshold of (P < 5 × 10⁻⁸), ensuring that the significance level is adjusted to minimize the risk of false positives. This method not only boosts the discovery of associated loci but also improves the interpretability of genetic associations through more informative bioinformatics analyses, thereby providing deeper insights into the genetic architecture of complex traits. In this study, our new loci refer to genetic loci that have not been reported in the relevant subtypes.
Colocalization (Giambartolomei et al., 2014) and gene-based analysis are critical for understanding the molecular basis of associations identified in genetic studies. This methodology integrates multiple association datasets, such as those from GWAS and gene expression studies, to assess whether two association signals are consistent with a shared causal variant. In our study, we applied this approach to investigate the relationship between COVID-19 and blood-cell traits. We used the Coloc tool to perform Bayesian colocalization analysis, aiming to identify loci where association signals for COVID-19 and blood-cell traits colocalize. This analysis uses default priors and considers a colocalization significant when the posterior probability of a shared causal variant (PP.H4) is greater than 0.8.
To identify genes significantly associated with complex traits, we utilized S-PrediXcan with multiple eQTL datasets from GTEx v8 (a total of 49 tissues). The expression weights used were derived from GTEx v8 multi-tissue expression data, and were estimated and provided by Junghyun Jung from the Mancuso lab. In the S-PrediXcan analysis (Barbeira et al., 2018), we used gene expression and genetic variation (SNP) data from a small reference set of individuals to infer the expression of cis-acting genetic components in a larger set of phenotyped individuals based on their SNP genotype data. The predicted expression is presented in the form of a linear model, with weights determined by the correlation between SNPs and gene expression in the training data while accounting for linkage disequilibrium among SNPs. We associated gene expression with traits by conducting a transcriptome-wide association study (TWAS) to identify significant expression-trait associations. Based on Bonferroni correction, the genome-wide significance threshold for TWAS was set at P-TWAS <1 × 10−6 focusing on loci established to be associated with COVID-19 risk that also showed relevance in the TWAS analysis.
We investigated the genetic correlation between COVID-19 and blood cell counts (Figure 1). Interestingly, while the correlation is weak among general patients (Rg = −0.03, p = 0.36), it becomes significantly stronger in severe patients (Rg = −0.07, p = 0.04) and hospitalized patients (Rg = −0.11, p = 2 × 10−4), suggesting a possible relationship with the severity of the disease. Importantly, our findings indicate a significant negative correlation between lymphocyte counts and COVID-19. Beyond lymphocytes, no significant correlations were found between COVID-19 and white blood cell counts (Supplementary Table S3). Most viral infections are associated with an increase in lymphocyte counts. Interestingly, several epidemiological studies have shown that COVID-19 infection leads to a decrease in lymphocyte levels (Chen et al., 2023; Niu et al., 2022; Xiong et al., 2020). Notably, this finding aligns with the results we obtained from our genetic analyses.
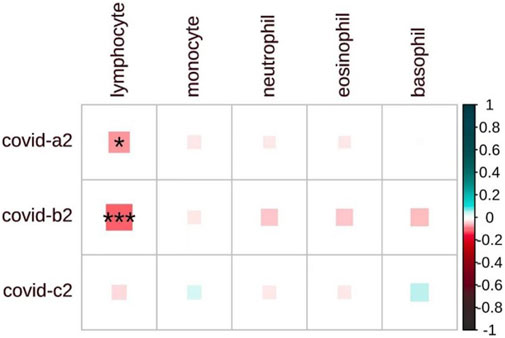
Figure 1. The heatmap displays the pairwise genome-wide genetic correlations between COVID-19 and blood cell counts. COVID-19-a2 for severe cases, COVID-19-b2 for hospitalized cases, and COVID-19-c2 for general cases. The matrix shows the strength of correlation (size of each square), with significant associations marked by an asterisk (P < 0.05). The shade of each square denotes a positive correlation (blue) or a negative correlation (red), with the depth of the shade reflecting the magnitude of the correlation.
For pulmonary COVID-19 and its subtypes, we further divided the heritability of SNP according to six chromatin markers and nine cell types. We found that each subtype of COVID-19 patients showed a similar pattern in the six chromatin markers of lymphocyte count (Supplementary Figure S1). It is worth noting that the respiratory system tissues or cell types, such as lungs, significantly enrich the chromatin markers H3K27Ac, H3K4me3 and H3K4me1 of COVID-19 or its subtypes. We also observed significant genetic enrichment of COVID-19 and its subtypes in musculoskeletal/connective tissue, digestive system, nervous system and other tissue or cell types. Interestingly, we also observed significant genetic enrichment of T cells, NK cells, and monocytes related to immune factors in the blood system, consistent with previous research findings (Ma et al., 2021; Ma et al., 2022).
Building on these significant findings, we conducted a comprehensive whole genome scan to identify specific genomic regions that may influence the shared heritability of various genetically related traits (Supplementary Table S4). During our statistical analysis, we adjusted for multiple testing to ensure the accuracy and reliability of our results. Notably, we discovered a significant correlation between COVID-19 and lymphocyte counts, particularly in the specific genomic region 19p13.2. This finding is particularly noteworthy given that previous studies have indicated multiple GWAS signals in the 19p13.2 region associated with COVID-19 (Figure 2). This research not only reinforces the importance of the 19p13.2 locus in the genetic architecture of COVID-19 but also provides new insights and a deeper understanding of the genetic mechanisms underlying this disease (Ferreira et al., 2022).
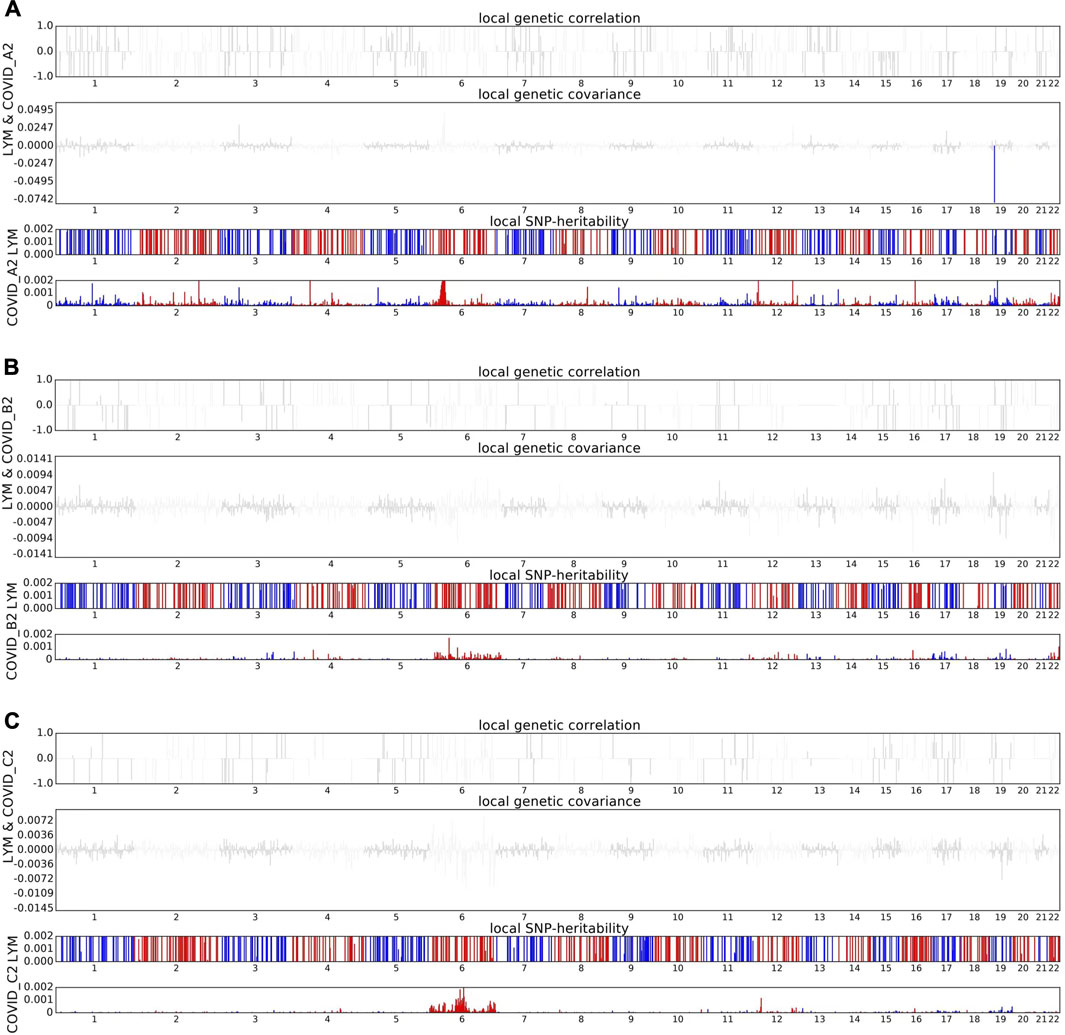
Figure 2. Local genetic correlation between COVID-19 and lymphocyte count. (A) The Manhattan plot shows the estimates of local genetic correlation, genetic covariance, and SNP heritability between severe COVID-19 and lymphocyte count. (B) The Manhattan plot shows the estimates of local genetic correlation, genetic covariance, and SNP heritability between hospitalized COVID-19 and lymphocyte count. (C) The Manhattan plot shows the estimates of local genetic correlation, genetic covariance, and SNP heritability between general COVID-19 and lymphocyte count. Red bars represent loci showing significant local genetic correlation after multiple testing adjustment (P < 0.05/sample size).
The evidence obtained from genetic correlation analysis has prompted us to further investigate the potential shared pleiotropic loci between COVID-19 and lymphocyte count. We employed Multi-Trait Analysis of GWAS (MTAG) to perform a multi-trait analysis of COVID-19 and its subtypes in relation to lymphocyte count, with special attention to SNPs achieving genome-wide significance (P < 1.66 × 10−8). To ensure the reliability of our results, we conducted comprehensive quality control (QC) analyses. The inflation lambda values for the original GWAS datasets were 1.055, 1.067, and 1.078 for severe COVID-19 (COVID-19-a2), hospitalized COVID-19 (COVID-19-b2), and general COVID-19 (COVID-19-c2), respectively, and 1.140 for lymphocyte count. The slightly higher lambda value for lymphocyte count may be attributed to its highly polygenic nature and the large sample size of the dataset, which can increase the observed inflation coefficient. In comparison, the MTAG results showed lower lambda values of 1.048 (COVID-19-a2), 1.041 (COVID-19-b2), and 1.023 (COVID-19-c2), indicating no significant inflation. Manhattan and QQ plots for these analyses are provided in Supplementary Figures S2, S3, confirming the robustness and consistency of our findings. These QC results further validate the reliability of our MTAG analysis and its ability to identify pleiotropic loci shared between COVID-19 and lymphocyte count.
In severe cases, we identified four loci, of which two are not reported previously. Notably, a signal at rs73009538 was discovered in the CARM1 gene, which is implicated in regulating gene transcription, growth, and RNA splicing, among other biological processes (Lai et al., 2021). We also identified a locus (rs55779981) within the RAVER1 gene region. Subsequently, among the four loci detected between hospitalized COVID-19 patients and lymphocyte count, two are not reported previously, specifically rs115545251 (1q22.1) and rs3181049 (19p13.3). The locus rs115545251 near the GFI1 gene. It is worth noting that another locus associated with hospitalized patients (rs3181049) and a locus identified in severe patients (rs55779981) are both located in the RAVER1 gene region. Interestingly, previous animal studies have suggested that other loci within the RAVER1 region, such as rs74956615 (19q13.2), increase susceptibility to coronavirus through complex mechanisms (Fink-Baldauf et al., 2022). In other words, these three loci, including two newly identified and one known locus, are densely located within the RAVER1 region yet remain independent of each other. For the general patient population, our LDSC analysis did not confirm a significant genetic correlation between COVID-19 and lymphocyte count. Despite the lack of significant genetic correlation, we conducted an MTAG analysis between hospitalized COVID-19 and lymphocyte counts, which revealed a previously unreported nominally significant loci (rs11065822) in the general population.
In this study, we employed the Coloc method to assess the Bayesian colocalization between COVID-19 and lymphocyte counts for each pleiotropic locus identified in the Multi-Trait Analysis of GWAS (MTAG). We identified three regions with PP.H4 values greater than 0.9, suggesting a high level of colocalization between COVID-19 and lymphocyte counts at these loci. Of the three colocalized loci, 19p13.2 was also identified in previous local genetic correlation analyses (Table1), reinforcing the consistency of our findings. This alignment not only bolsters the credibility of our genome-wide association results but also sheds new light on the genetic factors contributing to COVID-19 susceptibility, highlighting the potential for shared genetic pathways in immune response. To further explore the 19p13.2 (rs73009538) region in detail, we utilized the Locuscompare tool to visualize the association signals within this region. This analysis further confirmed the colocalization of genetic signals for COVID-19 susceptibility and lymphocyte counts at 19q13.2 (Figure 3) (Pruim et al., 2010).
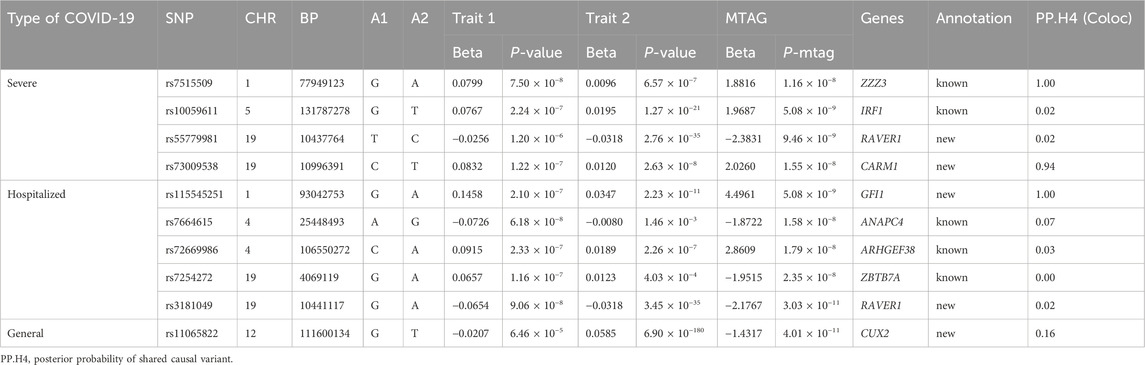
Table 1. Multi-trait meta-analysis between COVID-19 and lymphocyte.

Figure 3. The regional plots of the 19p13.2 locus associated with COVID-19 using Locuscompare. New locus rs73009538. Purple diamond indicates the lead SNP, and circles represent the other SNPs in the region, with coloring from the linkage disequilibrium (r2, based on the 1000 Genomes Project Europeans) between each SNP and the lead SNP.
We conducted a Transcriptome-Wide Association Study (TWAS) to investigate gene expression profiles associated with COVID-19 susceptibility and related phenotypes. TWAS integrates genotype and expression data, allowing for the identification of genes significantly linked to specific traits, thereby offering insights into gene regulation and disease association. Additionally, we have included Manhattan plots and QQ plots for the TWAS analyses, which can be accessed on Figshare (DOI: 10.6084/m9.figshare.28005845). In most tissues, the TWAS inflation factor (λ > 1.2) indicates moderate inflation, likely driven by the large sample size of the COVID-19 dataset. Similar levels of inflation have been observed in Alzheimer’s disease studies (Wingo et al., 2021), reflecting the increased statistical power to detect polygenic associations in large-scale datasets. These findings suggest that the observed inflation is primarily attributable to true signals rather than systematic bias.
The findings were largely consistent with earlier studies (Pathak et al., 2020) (Supplementary Tables S5–S7). In severe case A2, the eQTL rs10059611 was associated with ACSL6 in brain tissue, LncRNA in leukocytes and the stomach, microRNA in lung, and RAD50 in the uterus, pancreas, and skeletal muscle. These associations suggest that rs10059611 might have pleiotropic effects across multiple tissues, influencing diverse biological pathways relevant to severe COVID-19 outcomes. In hospitalized case B2, the eQTL rs7664615 was associated with PI4K2B in the pituitary, a gene involved in early T cell activation and immune regulation. In the thyroid, it was linked to SLC34A2, which plays a role in phosphate transport and cellular metabolism, and in the cerebellum, it was connected to ZCCHC4, a zinc finger protein involved in RNA processing and regulation. Additionally, rs72669986, located in the ARHGEF38 gene, was associated with GSTCD in the breast, esophagus, skin, and tibial nerve; SLC34A2 in the thyroid; and INTS12 in leukocytes, fibroblasts, prostate, and thyroid.
Furthermore, the TWAS analysis identified loci that provide additional insights into discoveries from GWAS studies. In severe case A2, the locus rs2236645 on chromosome 21 was associated with ATP5PO in the lung, brain, and skin, as well as MRPS6 in the esophagus and left atrium. Both genes are involved in mitochondrial function, a pathway that may play a critical role during early COVID-19 infection, as the virus relies on enhanced mitochondrial metabolism to support replication. In hospitalized case B2, rs3785632 was linked to SNHG26, a long non-coding RNA (lncRNA), and rs717624 was associated with NCOR1 in the testis, which regulates transcriptional activity. Additionally, rs3910266 was connected to SNX19 in the brain, a gene potentially involved in intracellular trafficking and cellular response mechanisms.
This study is one of the first comprehensive investigations into the genetic underpinnings of severe COVID-19, specifically focusing on the interplay between inflammation-induced lung damage and genetic susceptibility. It is evident that severe cases are largely driven by inflammation-mediated by immunological factors, with the progression and severity of the disease influenced by both environmental and genetic factors. Our findings highlight the crucial role of genetic determinants in shaping susceptibility, disease progression, and mortality in COVID-19, underscoring the need for further research to better understand these mechanisms and inform the development of targeted therapeutic interventions.
Recently, a Genome-Wide Association Study (GWAS) for COVID-19, launched by the GenOMICC, ISARIC4C, and SCOURGE consortia, collected genetic data from 24,202 severe COVID-19 patients across diverse ancestries. This study identified 49 genome-wide significant associations, of which 16 were newly reported (Pairo-Castineira et al., 2023). Beyond the known features of COVID-19 immune susceptibility, we found that several established COVID-19 genetic loci also impact the inflammatory response, a key risk factor for the progression and prognosis of COVID-19.
Based on the aforementioned evidence, we investigated the shared genetic architecture between different COVID-19 subtypes and other leukocytes. We observed a widespread negative correlation between lymphocyte counts and COVID-19, with a stronger correlation and more pronounced reduction in lymphocyte counts observed in more severe cases, such as critical and hospitalized patients. This phenomenon can be attributed to the direct viral infection of lymphocytes, particularly T cells and B cells, resulting in cellular death and a reduction in lymphocyte numbers (Rouse and Horohov, 1986). Concurrently, the immune system initiates a robust inflammatory response, which may escalate into a cytokine storm. The massive release of inflammatory mediators can induce lymphocyte apoptosis (programmed cell death), further diminishing lymphocyte numbers (Delogu et al., 2008; Peppelenbosch and van Deventer, 2004). Additionally, COVID-19 infection may cause immune suppression (Abbasi, 2021; Mehta et al., 2020), especially in severe cases. The increase in suppressive cytokines, such as IL-10 and TGF-β, may inhibit lymphocyte proliferation and function, further reducing their numbers. In summary, lymphopenia induced by COVID-19 infection is a complex process potentially involving direct viral effects, immune response, and immune suppression, with these pathophysiological responses being more pronounced in severe cases compared to milder ones. This suggests that lymphocyte count may partially reflect disease severity and prognosis. This finding can complement widely used imaging techniques, aiding clinicians in more effectively assessing patient conditions and guiding the design of clinical treatment plans. By monitoring lymphocyte counts, we can more accurately track disease progression and adjust therapeutic strategies as needed, ultimately improving overall patient outcomes.
Given the strong association between viral infection and lymphocyte count, we conducted deeper analyses to explore the shared genetic mechanisms between these phenotypes. We identified significant local genetic associations in the 19p13.2 region among severely ill patients, supported by previous studies (Ferreira et al., 2022). To deepen our analysis, we employed the MTAG approach to identify shared genetic loci between COVID-19 subtypes and lymphocyte count. By enhancing statistical power, we successfully identified 14 pleiotropic loci associated with COVID-19 and lymphocyte count, five of which were newly discovered and previously unreported in this subtype.
In severe cases, four nominally significant loci were identified, two of which are not reported previously. rs73009538 in the CARM1 region associated with type I interferon, and the deficiency of type I IFN response is one of the key factors in severe COVID-19 (Hadjadj et al., 2020). CARM1 is crucial for lung epithelial cell function (O'Brien et al., 2010), and its deficiency can lead to respiratory developmental issues, possibly linked to its role in gene transcription and cell growth. Additionally, CARM1 acts as a transcriptional coactivator for NF-kappaB, significantly influencing inflammatory responses (Srour et al., 2022). Notably, research from the Getx study found that rs73009538 serves as an eQTL for SMARCA4, a key component of the SWI/SNF complex that regulates immune responses and gene expression (Rhoads et al., 2018), particularly ACE2, the primary receptor for COVID-19 (Zoufaly et al., 2020). Mutations in SMARCA4 may disrupt mSWI/SNF complex function, resulting in lower ACE2 levels and potentially reducing cellular susceptibility to viral infection (Wei et al., 2023). RAVER1 is a key factor in inducing antiviral responses, capable of promoting the host’s immune reaction to viruses and enhancing antiviral capabilities (Chen et al., 2013). Meanwhile, RAVER1 is involved in mediating inflammation-related cell death processes, thereby regulating apoptosis or other pathways of cell death (Malireddi et al., 2023; Zhang et al., 2024). A deficiency in RAVER1 may lead to increased susceptibility of the host to COVID-19, potentially resulting in a more severe progression of the disease following infection. Interestingly, rs10059611 is an eQTL of LINC02863. While lincRNAs usually do not encode proteins, some small ORFs within them can be translated, though the mechanisms are unclear (Matsumoto et al., 2017). Additionally, lincRNAs can regulate protein-coding gene expression by interacting with RNA, DNA, or proteins (Atianand et al., 2017; Kong et al., 2024), influencing immune cell development (Brazão et al., 2016) and inflammation suppression (Atianand et al., 2016; Yue et al., 2020).
We identified five nominally significant loci associated with hospitalized COVID-19 cases, including two previously unreported loci: rs115545251 (located at 1q22.1), rs3181049 (located at 19q13.3). The rs115545251 locus, located near the GFI1 gene, is linked to hospitalized COVID-19 cases, with colocalization analysis confirming its association with lymphocyte count. The GFI1 gene, which encodes a zinc finger protein, promotes the proliferation of lymphocytes and granulocytes and functions as a transcriptional repressor involved in various biological activities. Mutations in the GFI1 gene, which is crucial for lymphocyte proliferation and regulation (Guo et al., 2021; Möröy and Khandanpour, 2011; 2019),can lead to lymphopenia and enhanced Th2 inflammatory responses (Sarkar et al., 2021; Zhu et al., 2006), potentially resulting in immune dysregulation and severe COVID-19 progression, highlighting the importance of rs115545251 in this context. The locus identified in hospitalized patients (rs3181049) and the locus found in severe patients (rs55779981) are both located in the RAVER1 gene region, further supporting our hypothesis: The defects in the RAVER1 gene not only reduce antiviral capability but also increase susceptibility to the disease, thereby raising the incidence of severe cases.
Additionally, our transcriptome-wide association study identified the rs72669986 locus in the ARHGEF38 region as a potential regulator of INTS12 expression, which is predominantly found in epithelial and cells and plays a critical role in cellular activity by regulating protein synthesis pathways (Kheirallah et al., 2017; Obeidat et al., 2013). INTS12 may influence cellular signaling pathways essential for the proliferation, differentiation, and survival of cells, thereby impacting COVID-19 susceptibility and disease severity while interacting with various genes related to lung function within complex gene networks (Obeidat et al., 2013). These findings shed light on the molecular mechanisms governing lung function and suggest potential targets for future interventions and treatments for related diseases. The eQTL rs7664615, located on ANAPC4 in GWAS. In the pituitary, it projects to the PI4K2B gene, whose encoded protein is involved in early T cell activation; in the thyroid, it projects to SLC34A2, which codes for a protein that regulates transmembrane protein transport; and in the cerebellum, it projects to ZCCHC4. COVID-19 can directly or indirectly affect the thyroid, leading to thyroid-related diseases, possibly secondary to the hypothalamic-pituitary-thyroid (HPT) axis, which may be related to the virus’s impact on the immune system (Lui et al., 2024). Additionally, thyroid hormones play a critical role as regulators of immune activity at the cellular level (Jaeger et al., 2021), contributing to lymphocyte homeostasis (De Vito et al., 2011). The zinc finger protein encoded by ZCCHC4 is involved in the process of viral clearance and plays an important role in regulating both innate and adaptive immune responses (Fu and Blackshear, 2017).
While our study provides valuable insights into the shared genetic architecture between COVID-19 and immune-related traits, several limitations should be noted. First, the lack of integration with single-cell RNA sequencing (scRNA-seq) data limits the resolution of our findings, particularly in identifying specific immune cell subtypes and pathways. Future research could leverage methods like scPagwas (Ma et al., 2023) to explore these aspects further. Additionally, functional validation of the identified loci was beyond the scope of this study, leaving the mechanistic roles of these loci to be explored in future experiments. Another key limitation is the use of binary and quantitative traits in our MTAG analysis. While this method may present some challenges in terms of accuracy and interpretability, it has been widely used in similar studies and remains a reasonable approach under the current constraints (Chang et al., 2024; Guo et al., 2020; Jiang et al., 2024). Lastly, the study primarily focuses on European ancestry populations, and further research is needed to assess the generalizability of these findings across diverse ancestries.
In this study, we conducted an in-depth multi-phenotype analysis of various COVID-19 subtypes and lymphocyte counts. By jointly analyzing these traits and enhancing statistical power, we identified five not reported previously genetic loci, shedding light on the pleiotropic genetic architecture between them. While some loci may exhibit vertical pleiotropy due to the causal relationship between COVID-19 and lymphocyte counts, others may display horizontal pleiotropy, directly influencing both traits. Further functional studies are needed to investigate these loci and their underlying mechanisms, contributing to a deeper understanding of COVID-19 susceptibility.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
ZM: Writing–review and editing, Data curation, Formal Analysis, Methodology, Visualization. CZ: Writing–original draft, Data curation, Formal Analysis, Investigation. SL: Investigation, Writing–original draft, Conceptualization. WL: Data curation, Writing–original draft. YW: Writing–original draft, Data curation. QZ: Methodology, Writing–original draft. BP: Validation, Writing–original draft. WY: Validation, Writing–original draft. YJ: Validation, Writing–review and editing. YS: Validation, Writing–review and editing. MG: Supervision, Writing–review and editing. XC: Writing–review and editing, Funding acquisition, Conceptualization, Supervision. LS: Supervision, Writing–review and editing, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Special Funds of Taishan Scholar Project, China (tsqn202211224), the National Natural Science Foundation of China (32270661), the Excellent Youth Science Fund Project (Overseas) of Shandong, China (2023HWYQ-082), and the Shandong Province Higher Education Institution Youth Innovation and Technology Support Program (2023KJ179). Special Funds of Taishan Scholar Project, China (tsqn202211224). The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1502839/full#supplementary-material
Abbasi, J. (2021). Researchers tie severe immunosuppression to chronic COVID-19 and virus variants. Jama 325 (20), 2033–2035. doi:10.1001/jama.2021.7212
Atianand, M. K., Caffrey, D. R., and Fitzgerald, K. A. (2017). Immunobiology of long noncoding RNAs. Annu. Rev. Immunol. 35, 177–198. doi:10.1146/annurev-immunol-041015-055459
Atianand, M. K., Hu, W., Satpathy, A. T., Shen, Y., Ricci, E. P., Alvarez-Dominguez, J. R., et al. (2016). A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell 165 (7), 1672–1685. doi:10.1016/j.cell.2016.05.075
Barbeira, A. N., Dickinson, S. P., Bonazzola, R., Zheng, J., Wheeler, H. E., Torres, J. M., et al. (2018). Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9 (1), 1825. doi:10.1038/s41467-018-03621-1
Brazão, T. F., Johnson, J. S., Müller, J., Heger, A., Ponting, C. P., and Tybulewicz, V. L. (2016). Long noncoding RNAs in B-cell development and activation. Blood 128 (7), e10–e19. doi:10.1182/blood-2015-11-680843
Bulik-Sullivan, B., Finucane, H. K., Anttila, V., Gusev, A., Day, F. R., Loh, P. R., et al. (2015). An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47 (11), 1236–1241. doi:10.1038/ng.3406
Chang, X., Zhi, L., Jiang, Y., Yu, L., Li, L., Song, Y., et al. (2024). Multi-trait genetic analysis of asthma and eosinophils uncovers novel loci in east asians. doi:10.21203/rs.3.rs-5425540/v1
Chen, G., Zhao, X., Chen, X., and Liu, C. (2023). Early decrease in blood lymphocyte count is associated with poor prognosis in COVID-19 patients: a retrospective cohort study. BMC Pulm. Med. 23 (1), 453. doi:10.1186/s12890-023-02767-z
Chen, H., Li, Y., Zhang, J., Ran, Y., Wei, J., Yang, Y., et al. (2013). RAVER1 is a coactivator of MDA5-mediated cellular antiviral response. J. Mol. Cell Biol. 5 (2), 111–119. doi:10.1093/jmcb/mjt006
Chen, M. H., Raffield, L. M., Mousas, A., Sakaue, S., Huffman, J. E., Moscati, A., et al. (2020). Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182 (5), 1198–1213.e14. doi:10.1016/j.cell.2020.06.045
Delogu, G., Famularo, G., Tellan, G., Marandola, M., Antonucci, A., Signore, M., et al. (2008). Lymphocyte apoptosis, caspase activation and inflammatory response in septic shock. Infection 36 (5), 485–487. doi:10.1007/s15010-008-7070-y
De Vito, P., Incerpi, S., Pedersen, J. Z., Luly, P., Davis, F. B., and Davis, P. J. (2011). Thyroid hormones as modulators of immune activities at the cellular level. Thyroid 21 (8), 879–890. doi:10.1089/thy.2010.0429
Espiritu, A. I., Pilapil, J. C. A., Aherrera, J. A. M., Sy, M. C. C., Anlacan, V. M. M., Villanueva, E. Q. I., et al. (2024). Outcomes of patients with COVID-19 and coronary artery disease and heart failure: findings from the Philippine CORONA study. BMC Res. Notes 17 (1), 14. doi:10.1186/s13104-023-06677-5
Ferreira, L. C., Gomes, C. E. M., Rodrigues-Neto, J. F., and Jeronimo, S. M. B. (2022). Genome-wide association studies of COVID-19: connecting the dots. Infect. Genet. Evol. 106, 105379. doi:10.1016/j.meegid.2022.105379
Fink-Baldauf, I. M., Stuart, W. D., Brewington, J. J., Guo, M., and Maeda, Y. (2022). CRISPRi links COVID-19 GWAS loci to LZTFL1 and RAVER1. EBioMedicine 75, 103806. doi:10.1016/j.ebiom.2021.103806
Finucane, H. K., Bulik-Sullivan, B., Gusev, A., Trynka, G., Reshef, Y., Loh, P. R., et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47 (11), 1228–1235. doi:10.1038/ng.3404
Finucane, H. K., Reshef, Y. A., Anttila, V., Slowikowski, K., Gusev, A., Byrnes, A., et al. (2018). Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50 (4), 621–629. doi:10.1038/s41588-018-0081-4
Fu, M., and Blackshear, P. J. (2017). RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat. Rev. Immunol. 17 (2), 130–143. doi:10.1038/nri.2016.129
Gao, M., Piernas, C., Astbury, N. M., Hippisley-Cox, J., O'Rahilly, S., Aveyard, P., et al. (2021). Associations between body-mass index and COVID-19 severity in 6·9 million people in England: a prospective, community-based, cohort study. Lancet Diabetes Endocrinol. 9 (6), 350–359. doi:10.1016/s2213-8587(21)00089-9
Giambartolomei, C., Vukcevic, D., Schadt, E. E., Franke, L., Hingorani, A. D., Wallace, C., et al. (2014). Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10 (5), e1004383. doi:10.1371/journal.pgen.1004383
Guo, Y., Rist, P. M., Daghlas, I., Giulianini, F., Kurth, T., Chasman, D. I., et al. (2020). A genome-wide cross-phenotype meta-analysis of the association of blood pressure with migraine. Nat. Commun. 11 (1), 3368. doi:10.1038/s41467-020-17002-0
Guo, Z., Zhang, Z., Prajapati, M., and Li, Y. (2021). Lymphopenia caused by virus infections and the mechanisms beyond. Viruses 13 (9), 1876. doi:10.3390/v13091876
Hadjadj, J., Yatim, N., Barnabei, L., Corneau, A., Boussier, J., Smith, N., et al. (2020). Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369 (6504), 718–724. doi:10.1126/science.abc6027
Hegde, S. (2020). Many paths to COVID-19 lymphocyte dysfunction. Nat. Rev. Immunol. 20 (7), 408. doi:10.1038/s41577-020-0361-y
Hopkins, R., Young, K. G., Thomas, N. J., Godwin, J., Raja, D., Mateen, B. A., et al. (2024). Risk factor associations for severe COVID-19, influenza and pneumonia in people with diabetes to inform future pandemic preparations: UK population-based cohort study. BMJ Open 14 (1), e078135. doi:10.1136/bmjopen-2023-078135
Jackson, C. B., Farzan, M., Chen, B., and Choe, H. (2022). Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 23 (1), 3–20. doi:10.1038/s41580-021-00418-x
Jaeger, M., Sloot, Y. J. E., Horst, R. T., Chu, X., Koenen, H., Koeken, V., et al. (2021). Thyrotrophin and thyroxine support immune homeostasis in humans. Immunology 163 (2), 155–168. doi:10.1111/imm.13306
Jiang, Y., Song, Y., Li, Y., Tong, Y., Ding, H., Li, L., et al. (2024). Multi-trait genetic analysis identifies novel pleiotropic loci for stroke and hematological traits or risk factors. Fundam. Res. doi:10.1016/j.fmre.2024.05.004
Kheirallah, A. K., de Moor, C. H., Faiz, A., Sayers, I., and Hall, I. P. (2017). Lung function associated gene Integrator Complex subunit 12 regulates protein synthesis pathways. BMC Genomics 18 (1), 248. doi:10.1186/s12864-017-3628-3
Kong, X., Wang, Q., Wang, X., Yang, K., Nie, S., Li, Y., et al. (2024). LINC01002 functions as a ceRNA to regulate FRMD8 by sponging miR-4324 for the development of COVID-19. Virol. J. 21 (1), 109. doi:10.1186/s12985-024-02382-2
Lai, Y., Li, X., Li, T., Nyunoya, T., Chen, K., Kitsios, G. D., et al. (2021). Endotoxin stabilizes protein arginine methyltransferase 4 (PRMT4) protein triggering death of lung epithelia. Cell Death Dis. 12 (9), 828. doi:10.1038/s41419-021-04115-7
Lindström, S., Wang, L., Smith, E. N., Gordon, W., van Hylckama Vlieg, A., de Andrade, M., et al. (2019). Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood 134 (19), 1645–1657. doi:10.1182/blood.2019000435
Liu, X., Hua, L., Chu, J., Zhou, W., Jiang, F., Wang, L., et al. (2024). Endothelial dysfunction and disease severity in COVID-19: insights from circulating Tang cell counts as a potential biomarker. Int. Immunopharmacol. 130, 111788. doi:10.1016/j.intimp.2024.111788
Lui, D. T. W., Lee, C. H., Woo, Y. C., Hung, I. F. N., and Lam, K. S. L. (2024). Thyroid dysfunction in COVID-19. Nat. Rev. Endocrinol. 20 (6), 336–348. doi:10.1038/s41574-023-00946-w
Ma, Y., Deng, C., Zhou, Y., Zhang, Y., Qiu, F., Jiang, D., et al. (2023). Polygenic regression uncovers trait-relevant cellular contexts through pathway activation transformation of single-cell RNA sequencing data. Cell Genom 3 (9), 100383. doi:10.1016/j.xgen.2023.100383
Ma, Y., Huang, Y., Zhao, S., Yao, Y., Zhang, Y., Qu, J., et al. (2021). Integrative genomics analysis reveals a 21q22.11 locus contributing risk to COVID-19. Hum. Mol. Genet. 30 (13), 1247–1258. doi:10.1093/hmg/ddab125
Ma, Y., Qiu, F., Deng, C., Li, J., Huang, Y., Wu, Z., et al. (2022). Integrating single-cell sequencing data with GWAS summary statistics reveals CD16+monocytes and memory CD8+T cells involved in severe COVID-19. Genome Med. 14 (1), 16. doi:10.1186/s13073-022-01021-1
Malireddi, R. K. S., Bynigeri, R. R., Mall, R., Nadendla, E. K., Connelly, J. P., Pruett-Miller, S. M., et al. (2023). Whole-genome CRISPR screen identifies RAVER1 as a key regulator of RIPK1-mediated inflammatory cell death, PANoptosis. iScience 26 (6), 106938. doi:10.1016/j.isci.2023.106938
Matsumoto, A., Pasut, A., Matsumoto, M., Yamashita, R., Fung, J., Monteleone, E., et al. (2017). mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 541 (7636), 228–232. doi:10.1038/nature21034
Mehta, P., McAuley, D. F., Brown, M., Sanchez, E., Tattersall, R. S., Manson, J. J., et al. (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395 (10229), 1033–1034. doi:10.1016/s0140-6736(20)30628-0
Möröy, T., and Khandanpour, C. (2011). Growth factor independence 1 (Gfi1) as a regulator of lymphocyte development and activation. Semin. Immunol. 23 (5), 368–378. doi:10.1016/j.smim.2011.08.006
Möröy, T., and Khandanpour, C. (2019). Role of GFI1 in epigenetic regulation of MDS and AML pathogenesis: mechanisms and therapeutic implications. Front. Oncol. 9, 824. doi:10.3389/fonc.2019.00824
Niu, J., Sareli, C., Mayer, D., Visbal, A., and Sareli, A. (2022). Lymphopenia as a predictor for adverse clinical outcomes in hospitalized patients with COVID-19: a single center retrospective study of 4485 cases. J. Clin. Med. 11 (3), 700. doi:10.3390/jcm11030700
Obeidat, M., Miller, S., Probert, K., Billington, C. K., Henry, A. P., Hodge, E., et al. (2013). GSTCD and INTS12 regulation and expression in the human lung. PLoS One 8 (9), e74630. doi:10.1371/journal.pone.0074630
O'Brien, K. B., Alberich-Jordà, M., Yadav, N., Kocher, O., Diruscio, A., Ebralidze, A., et al. (2010). CARM1 is required for proper control of proliferation and differentiation of pulmonary epithelial cells. Development 137 (13), 2147–2156. doi:10.1242/dev.037150
Oran, D. P., and Topol, E. J. (2020). Prevalence of asymptomatic SARS-CoV-2 infection: a narrative review. Ann. Intern Med. 173 (5), 362–367. doi:10.7326/m20-3012
Pairo-Castineira, E., Rawlik, K., Bretherick, A. D., Qi, T., Wu, Y., Nassiri, I., et al. (2023). GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature 617 (7962), 764–768. doi:10.1038/s41586-023-06034-3
Pathak, G. A., Singh, K., Miller-Fleming, T. W., Wendt, F. R., Ehsan, N., Hou, K., et al. (2020). Integrative analyses identify susceptibility genes underlying COVID-19 hospitalization. medRxiv. doi:10.1101/2020.12.07.20245308
Peppelenbosch, M. P., and van Deventer, S. J. (2004). T cell apoptosis and inflammatory bowel disease. Gut 53 (11), 1556–1558. doi:10.1136/gut.2004.040824
Pruim, R. J., Welch, R. P., Sanna, S., Teslovich, T. M., Chines, P. S., Gliedt, T. P., et al. (2010). LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26 (18), 2336–2337. doi:10.1093/bioinformatics/btq419
Rhoads, T. W., Burhans, M. S., Chen, V. B., Hutchins, P. D., Rush, M. J. P., Clark, J. P., et al. (2018). Caloric restriction engages hepatic RNA processing mechanisms in rhesus monkeys. Cell Metab. 27 (3), 677–688. doi:10.1016/j.cmet.2018.01.014
Rouse, B. T., and Horohov, D. W. (1986). Immunosuppression in viral infections. Rev. Infect. Dis. 8 (6), 850–873. doi:10.1093/clinids/8.6.850
Sarkar, M. H., Yagi, R., Endo, Y., Koyama-Nasu, R., Wang, Y., Hasegawa, I., et al. (2021). IFNγ suppresses the expression of GFI1 and thereby inhibits Th2 cell proliferation. PLoS One 16 (11), e0260204. doi:10.1371/journal.pone.0260204
Song, M., Liu, X., Shen, W., Wang, Z., Wu, J., Jiang, J., et al. (2024). IFN-γ decreases PD-1 in T lymphocytes from convalescent COVID-19 patients via the AKT/GSK3β signaling pathway. Sci. Rep. 14 (1), 5038. doi:10.1038/s41598-024-55191-6
Srour, N., Khan, S., and Richard, S. (2022). The influence of arginine methylation in immunity and inflammation. J. Inflamm. Res. 15, 2939–2958. doi:10.2147/jir.S364190
Trynka, G., Sandor, C., Han, B., Xu, H., Stranger, B. E., Liu, X. S., et al. (2013). Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 45 (2), 124–130. doi:10.1038/ng.2504
Turley, P., Walters, R. K., Maghzian, O., Okbay, A., Lee, J. J., Fontana, M. A., et al. (2018). Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50 (2), 229–237. doi:10.1038/s41588-017-0009-4
Valencia, I., Lumpuy-Castillo, J., Magalhaes, G., Sánchez-Ferrer, C. F., Lorenzo, Ó., and Peiró, C. (2024). Mechanisms of endothelial activation, hypercoagulation and thrombosis in COVID-19: a link with diabetes mellitus. Cardiovasc Diabetol. 23 (1), 75. doi:10.1186/s12933-023-02097-8
van der Made, C. I., Simons, A., Schuurs-Hoeijmakers, J., van den Heuvel, G., Mantere, T., Kersten, S., et al. (2020). Presence of genetic variants among young men with severe COVID-19. Jama 324 (7), 663–673. doi:10.1001/jama.2020.13719
Wei, J., Patil, A., Collings, C. K., Alfajaro, M. M., Liang, Y., Cai, W. L., et al. (2023). Pharmacological disruption of mSWI/SNF complex activity restricts SARS-CoV-2 infection. Nat. Genet. 55 (3), 471–483. doi:10.1038/s41588-023-01307-z
Wingo, A. P., Liu, Y., Gerasimov, E. S., Gockley, J., Logsdon, B. A., Duong, D. M., et al. (2021). Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer's disease pathogenesis. Nat. Genet. 53 (2), 143–146. doi:10.1038/s41588-020-00773-z
Xiong, S., Liu, L., Lin, F., Shi, J., Han, L., Liu, H., et al. (2020). Clinical characteristics of 116 hospitalized patients with COVID-19 in Wuhan, China: a single-centered, retrospective, observational study. BMC Infect. Dis. 20 (1), 787. doi:10.1186/s12879-020-05452-2
Yue, L., Yin, X., Hao, F., Dong, J., Ren, X., Xu, O., et al. (2020). Long noncoding RNA Linc00632 inhibits interleukin-13-induced inflammatory cytokine and mucus production in nasal epithelial cells. J. Innate Immun. 12 (1), 116–128. doi:10.1159/000500420
Zhang, B., Orning, P., Lehman, J. W., Dinis, A., Torres-Ulloa, L., Elling, R., et al. (2024). Raver1 links Ripk1 RNA splicing to caspase-8-mediated pyroptotic cell death, inflammation, and pathogen resistance. bioRxiv. doi:10.1101/2024.11.27.625707
Zhou, J., Sun, Y., Huang, W., and Ye, K. (2021). Altered blood cell traits underlie a major genetic locus of severe COVID-19. J. Gerontol. A Biol. Sci. Med. Sci. 76 (8), e147–e154. doi:10.1093/gerona/glab035
Zhu, J., Yamane, H., Cote-Sierra, J., Guo, L., and Paul, W. E. (2006). GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 16 (1), 3–10. doi:10.1038/sj.cr.7310002
Keywords: COVID-19, lymphocyte, immune response, genome-wide cross-trait analysis, transcriptome-wide association studies
Citation: Meng Z, Zhang C, Liu S, Li W, Wang Y, Zhang Q, Peng B, Ye W, Jiang Y, Song Y, Guo M, Chang X and Shao L (2025) Exploring genetic loci linked to COVID-19 severity and immune response through multi-trait GWAS analyses. Front. Genet. 16:1502839. doi: 10.3389/fgene.2025.1502839
Received: 27 September 2024; Accepted: 27 January 2025;
Published: 17 February 2025.
Edited by:
Goo Jun, University of Texas Health Science Center at Houston, United StatesReviewed by:
Yunlong Ma, University of Pennsylvania, United StatesCopyright © 2025 Meng, Zhang, Liu, Li, Wang, Zhang, Peng, Ye, Jiang, Song, Guo, Chang and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miao Guo, c2R0YWdtQDE2My5jb20=; Xiao Chang, Y2hhbmd4aWFvQHNkZm11LmVkdS5jbg==; Lei Shao, c2hhb2xlaTAzMjhAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.