 Yunxia Qi1
Yunxia Qi1 Xiaolong He2Biao Wang2
Xiaolong He2Biao Wang2 Chaoyun Yang1Lai Da2Bin Liu3Wenguang Zhang4
Chaoyun Yang1Lai Da2Bin Liu3Wenguang Zhang4 Shaoyin Fu2*Yongbin Liu2,5*
Shaoyin Fu2*Yongbin Liu2,5*- 1College of Animal Sciences, Xichang University, Xichang, China
- 2Animal Husbandry Institute, Inner Mongolia Academy of Agricultural and Animal Husbandry Sciences, Hohhot, China
- 3Inner Mongolia BIONEW Technology Co., LTD., Hohhot, China
- 4College of Life Sciences, Inner Mongolia Agricultural University, Hohhot, China
- 5State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock (R2BGL), College of Life Sciences, Inner Mongolia University, Hohhot, China
Tail type of sheep, which may be affected by many genes with a complex mechanism, is an important economic trait concerned by both raiser and consumers. Here, we employed two sheep breeds with extreme phenotypes - Mongolian sheep (short-fat-tailed) and Bamei Mutton sheep (long-thin-tailed) to analyze the genetic differences at the genomic level and find candidate genes associated with tail phenotype. The results of population structure analysis showed that the LD decay rate of Mongolian sheep was greater than that of Bamai Mutton sheep. When K = 2, the two populations were obviously separated with a certain degree of mixing. From 49 sheep individuals, 20,270,930 and 2,479,474 SNPs and Indels were identified, respectively. Selection signals were detected based on FST, π-Ratio, and XP-EHH. These three methods identified 85 candidate genes, of which PDGFD, GLIS1, AR, and FGF9 were reported to be associated with tail fat deposition, while VRTN associated with tail length in sheep tail phenotype; the others were novel genes that may play important roles in sheep tail phenotype formation. Gene annotation revealed that these candidate genes mainly participate in pathways associated with fat deposition or lipid metabolism. This study provided insight into sheep tail type development and a guide for molecular breeding.
1 Introduction
Sheep (Ovis aries), as a major source of meat, milk, fiber and leather for mankind, has been domesticated since Mesolithic period, ∼11,000 years ago (Chessa et al., 2009; Alberto et al., 2018; Deng et al., 2020). During this long procedure sheep has evolved diverse phenotypes such as coat color, horn, tail type, etc., under natural and artificial selection (Kijas et al., 2009). Of these phenotypes, tail type is an important economic trait concerned by both producers and consumers. According to its length and fat deposition, sheep tail can be divided into five major types: long-fat tail, short-fat tail, long-thin tail, short-thin tail, and fat-rumped tail. It is widely believed that the wild ancestors of sheep were thin-tailed, while the fat-tailed sheep breeds emerged as an adaptive response to harsh and challenging environmental conditions (such as climate fluctuation, drought, and food scarcity) (Atti et al., 2004; Pourlis, 2011; Moradi et al., 2012; Kalds et al., 2021). Fat-tailed sheep could deposit up to 20% of their carcass weight as fat in the tail (Yousefi et al., 2012). The large amount of tail fat serves as an energy source for sheep, and also provided people valuable edible fat in the era of material scarcity. Nowadays, however, with the increasing incidences of obesity and cardiovascular disease, people prefer a diet low in fat and high in protein. On the other hand, with the popularization of intensive and semi-intensive feeding management, fat-tail of sheep has lost its original advantages and brought inconvenience to production management, such as inconvenience for mating (Kridli and Said, 1999) and locomotion (Orihuela and Ungerfeld, 2019). In addition, a large amount of fat deposition in tails may reduce feed conversion rate and even affect the carcass quality (Safdarian et al., 2008; Yousefi et al., 2012). Yousefi et al. found that the thin-tailed breed accumulated more intramuscular fat in longissimus dorsi muscle and had lower shear force and better eating quality, tenderness, and drip loss than the fat-tailed breed (Yousefi et al., 2012). In practice, tail docking (O’Donovan et al., 1973; Shelton et al., 1991; Bicer et al., 1992; Moharrery, 2007; Wang et al., 2018) and cross-breeding (Kashan et al., 2005; Khaldari et al., 2008; Abdullah et al., 2010) are usually taken to reduce tail size and length. It was reported that tail docking may improve lambs’ growth, slaughter performance and mutton quality (Atti and Mahouachi, 2011; Marai et al., 1987; Bicer et al., 1992; Abouheif et al., 1993; Bing et al., 2006). However, tail docking is stressful and risky, and has been banned in several countries to improve animal welfare (Eck et al., 2019). Cross-breeding takes time and efforts, and the results are usually unsatisfactory. Currently, how to breed short-thin-tailed sheep through molecular breeding methods has become a focus of sheep breeders, and the key to solve this problem is to identify genes related to tail phenotype of sheep. There were some research on tail phenotype and several promising genes such as PDGFD, BMP2 and TBXT, etc., associated with tail phenotype had been suggested (Yuan et al., 2017; Zhi et al., 2018; Dong et al., 2020; Mastrangelo et al., 2019; Pan et al., 2019; Moradi et al., 2012), but most of the studies focused either on fat deposition or tail length, and the results are usually inconsistent. The genetic mechanics underlying tail phenotype still remain unclear.
Mongolian sheep, short-fat-tailed (Figure 1A), is the most widely distributed and abundant sheep breed in China. It is mainly distributed in Inner Mongolia Autonomous Region, northeast, north, and northwest of China. Mongolian sheep is an ancient indigenous breed formed by natural and artificial selection for a long time, and is favored by local herdsman and consumers because of its rough feeding resistance, cold resistance, drought resistance, and high-quality meat. Bamei Mutton sheep, which is long-thin-tailed (Figures 1B, C), is the first dual-purpose breed that was bred in China by crossing local fine-mixed sheep as the maternal line with German Merino sheep as the paternal line. It contains 6.25% bloodline of Mongolian sheep. Bamei Mutton sheep is mainly distributed in Bayannur City of Inner Mongolia Autonomous Region, China. It is characterized by resistance to rough forage, strong stress resistance, good adaptability, rapid weight gain in lamb fattening, and early sexual maturity. In the present study, we performed whole genome resequencing of the two breeds with extreme tail phenotypes to investigate selection signatures and candidate genes associated with tail phenotype (fat vs. thin and long vs. short) based on three statistical tests, including fixation index (FST), π-Ratio, and cross-population extended haplotype homozygosity test (XP-EHH). The candidate genes identified in our study provided the basis for understanding the molecular mechanism of tail phenotype in sheep.

Figure 1. Tail phenotype of sheep. (A) Short-fat-tailed Mongolian sheep. (B) Long-thin-tailed Bamei Mutton sheep after docking. (C) long-thin-tailed lamb of Bamei Mutton sheep before docking.
2 Materials and methods
2.1 Sample collection, DNA extraction, and sequencing
A total of 28 Mongolian sheep (MG) and 21 Bamei Mutton sheep (BM) were selected from Inner Mongolia Autonomous Region, China. The sheep were raised and managed under the same condition. All individuals were typical of the breeds and unrelated according to pedigree records or owner’s information. Blood samples were collected and returned to laboratory on dry ice. Genomic DNA was extracted from the blood samples following the standard phenol-chloroform extraction procedure. DNA samples that passed the test (D260 nm/D280 nm = 1.7–1.9) were randomly interrupted into fragments of 500 bp in length. Paired-end sequencing libraries were constructed according to the manufacturer’s instructions (Illumina Inc., San Diego, CA, USA) and sequenced on the Illumina HiSeq Xten Sequencer (Illumina Inc.) with PE150 module.
2.2 Alignments and variant identification
After filtering out low quality reads, the 150-bp paired-end clean reads were mapped onto the sheep reference genome Oar v.4.0 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000298735.2) with BWA-MEM using the default parameters (Li and Durbin, 2009). After alignments, SNP calling was performed using SAMtools and Genome Analysis Toolkit (GATK, v.3.8) (Nekrutenko and Taylor, 2012). All SNPs were filtered using the ‘Variant Filtration’ module of GATK with the standard parameters as below: Window 4; Variants with QD (quality depth) < 4.0; FS (Phred-scaled p-value using Fisher’s exact test to detect strand bias) > 60.0; MQ < 40.0; -G_filter “GQ < 20”.
The implementation of SAMtools mpileup (v.1.8) (Li, 2011) was run in a multi-sample mode to calculate genotype likelihoods from the aligned reads for all samples simultaneously. The parameters -E and -t were used to recalculate (and apply) base alignment quality and produce per-sample genotype annotations, respectively. Then, the estimated genotype likelihoods were converted into genotypes using BCFtools call using the -v and -m flags to output variable sites only, and permitted sites to have more than two alternative alleles, respectively.
Based on the annotation file of the sheep reference genome Oar v.4.0, a transcript FASTA file for database was built using the retrieve_seq_from_fasta.pl module of ANNOVAR, and then the functional annotation for each SNP was performed using the table_annovar.pl module of ANNOVAR (Wang et al., 2010).
2.3 Population structure analysis
SNPs were pruned the in high levels of pair-wise LD using PLINK v.1.9 (Purcell et al., 2007) with the parameter (−-indep-pair-wise 50 5 0.2) to perform principal component analysis (PCA) and ADMIXTURE analysis. PCA of whole-genome SNPs for all 49 individuals was conducted using the GCTA v.1.24.2 (Yang et al., 2011). Furthermore, population structure analysis was carried out using the ADMIXTURE v1.3 (Alexander and Lange, 2011) with kinship (K) ranged from 2 to 5. The unrooted Neighbor-joining (NJ) tree was constructed with TASSEL using the matrix of pairwise genetic distances and visualized with iTOL (https://itol.embl.de/). The LD decay for each group was measured using PopLDdecay (Zhang et al., 2019) with default parameters.
2.4 Genome-wide selective sweep test
To identify the selective sweep regions, we performed genome-wide scans of selection signals using three metrics: allele frequency based methods FST (Weir and Hill, 2002), π-Ratio (Danecek et al., 2011), and haplotype-based method XP-EHH (Sabeti et al., 2007a).
The SNPs were filtered with parameters (--maf 0.05 -max-missing 0.90) using PLINK v.1.9 (Purcell et al., 2007). The FST was calculated using VCFtools (Danecek et al., 2011) with parameter “--weir-fst-pop group1 --weir-fst-pop group2 --fst-window-size 50000 --fst-window-step 20000 --maf 0.05 --max-missing 0.90”. Then the FST values were normalized (ZFST) using the Ztransformation method (Rubin et al., 2010). The genetic diversity (π-Ratio) was calculated using VCFtools with parameters “--keep gropu1/gropu2 --window-pi 50000 --window-pi-step 20000 --maf 0.05 --max-missing 0.90” and python scripts. The overlap of the top 5% windows in each method was considered as candidate signatures of selection.
The XP-EHH was performed for every SNP using the default settings by selscan v.1.1 (Szpiech and Hernandez, 2014), and genotypes were phased using Beagle (Browning and Browning, 2007) with default parameters. The genome-wide raw XP-EHH statistics were standardized to a distribution with zero mean and unit variance. SNPs in the top 0.1% are taken as significant SNPs. Significant regions are identified by combining SNPs of significant XP-EHH scores that are less than 200 kb apart. If two SNPs both have significant XP-EHH scores and were less than 200 kb apart, then the two SNPs formed a region.
In the π-Ratio and XP-EHH tests, the BM sheep were used as the target population, and the MG sheep as the reference population.
2.5 Gene ontology enrichment and KEGG pathway analyses
According to genome annotation, a gene was assumed to be under positive selection if it overlapped with a selection signal. To obtain an in-depth view of the biological significance of the candidate genes, online Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were conducted by retrieving O. aries in self-built database (No.AH96240) on AnnotationHub website (https://annotationhub.bioconductor.org/). Protein-Protein Interaction (PPI) analysis was performed using STRING database (https://cn.string-db.org/).
3 Results
3.1 Overview of sequencing quality
After sequencing and data quality control, more than 100 million clean reads were obtained in the MG and BM groups, respectively. The number of clean bases in the BM group was found to be more than 4G greater than that in the MG group. The mapping rate is greater than 98% in both groups with an average depth of 7.31 × (Table 1; Supplementary Table S1).
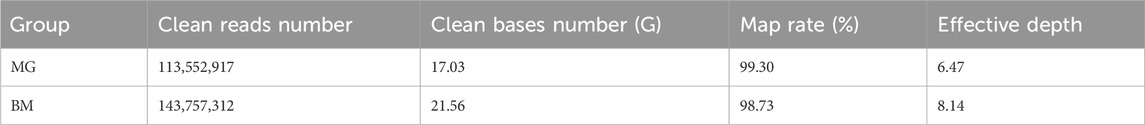
Table 1. Overview of sequencing statistics.
3.2 SNP identification
In total, 29,926,218 and 23,122,081 SNPs, 3,755,604 and 5,005,409 Indels were identified in MG and BM sheep, respectively, among which, 20,270,930 SNPs and 2,479,474 Indels are common in MG and BM sheep (Figure 2). Functional annotation of the polymorphic sites showed that the vast majority of SNPs and Indels were present either in intergenic regions (62.2% and 60.8%, respectively) or in intronic regions (34.8% and 36.3%, respectively). Exons contained 0.60% of the total variation (Table 2). These results indicate that the variants on the MG and BM genomes differ.
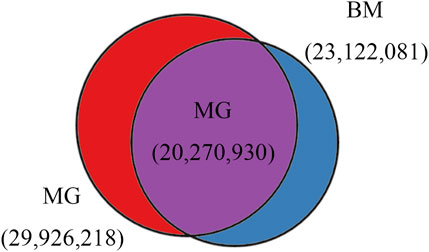
Figure 2. SNPs identified in BM and MG sheep.
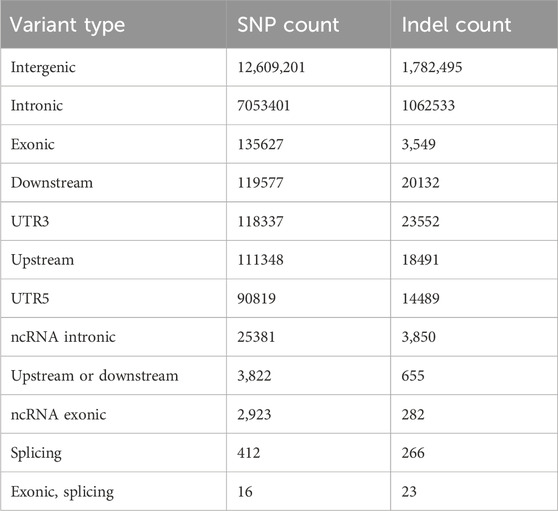
Table 2. Annotation of SNPs/Indels.
3.3 Population genetic structure
Following the identification of the SNPs, PCA, phylogenetic relationship analysis, and population genetic structure analysis were conducted for all the individuals. The PCA results showed that the BM and MG sheep were clearly separated (Figure 3A), and the NJ tree also produced similar results, with BM and MG sheep divided into 2 clades (Figure 3B), which indicated that there is a certain degree of genetic distance between BM and MG sheep. The results of population genetic structure (Figure 3C) showed that for the BM group, the consistency within groups was better when K = 2 and K = 4, while for the MG, it was better when K = 2 and K = 3, indicating that the consistency of the individuals within the groups was better, and that differences existed between the groups. Furthermore, when K = 2, BM and MG sheep were obviously divided into two subgroups, but with a certain degree of mixing, which was consistent with the fact that BM sheep contains a certain amount of ancestry of MG sheep. When K = 3, no new subgroups appeared in the experimental population. The aforementioned results demonstrate that there are distinctions between BM and GM sheep, which may be attributed to tail phenotype variation. In order to further explore the genetic diversity, linkage disequilibrium, in terms of the correlation coefficient (r2), was calculated for BM and MG sheep populations. As shown in Figure 3D, the faster LD decay was observed in the MG population, which indicates that MG sheep had higher genetic diversity, and BM sheep had higher degree of domestication and greater intensity of selection.
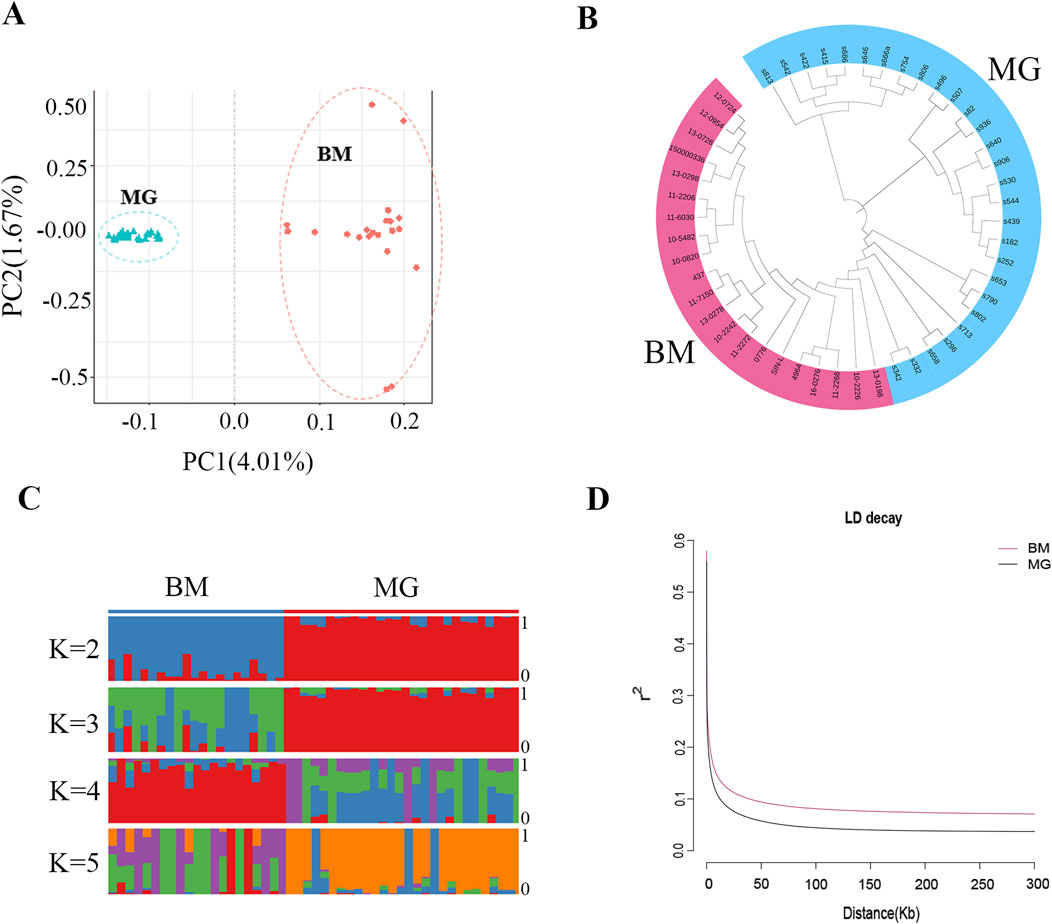
Figure 3. Population genetic structures of BM and MG sheep. (A) Principal component analysis (PCA) of 49 sheep individuals. (B) Neighbor-joining (NJ) tree constructed from SNPs data of the two populations. (C) Model-based clustering of sheep individuals using ADMIXTURE with K = 2–5. (D) Correlation coefficients (r2) were calculated for the MG and BM sheep over 50-kb windows.
3.4 Detection of selective sweeps
Due to the genetic separation between MG and BM sheep, selective sweep analysis using FST, π-Ratio and XP-EHH were performed to investigate selection signals in BM sheep, which would facilitate the identification of target genes. The results demonstrated that the majority of SNPs exhibited moderate genetic variance within the population (Figure 4A, FST < 0.15). Additionally, some SNPs exhibited high genetic variance and high genetic variability on chromosomes 13, 16, and 17, suggesting that these SNPs may be mutations specific to the BM and MG populations. A further 544,123 SNPs were identified under the conditions of FST ≥ 0.2 and π-Ratio ≤0.397 (Figure 4B). The combined analysis of FST and π-Ratio revealed that 1884 genes (representing the top 10% of genes) were identified by log2 (Pi_BM/GM)_ZFST, while 294 genes were identified by XP_EHH (Figure 4C). Eventually, a total of 85 overlapping genes were identified as candidate genes by log2 (Pi_BM/GM)_ZFST and XP_EHH (Figure 4D; Supplementary Table S2). Of these candidate genes, some were known to be related to sheep tail phenotype, such as fat deposition associated genes PDGFD, GLIS1, AR, FGF9, and vertebral number variation associated gene VRTN; some were novel genes that may have relationship with sheep tail phenotype formation.
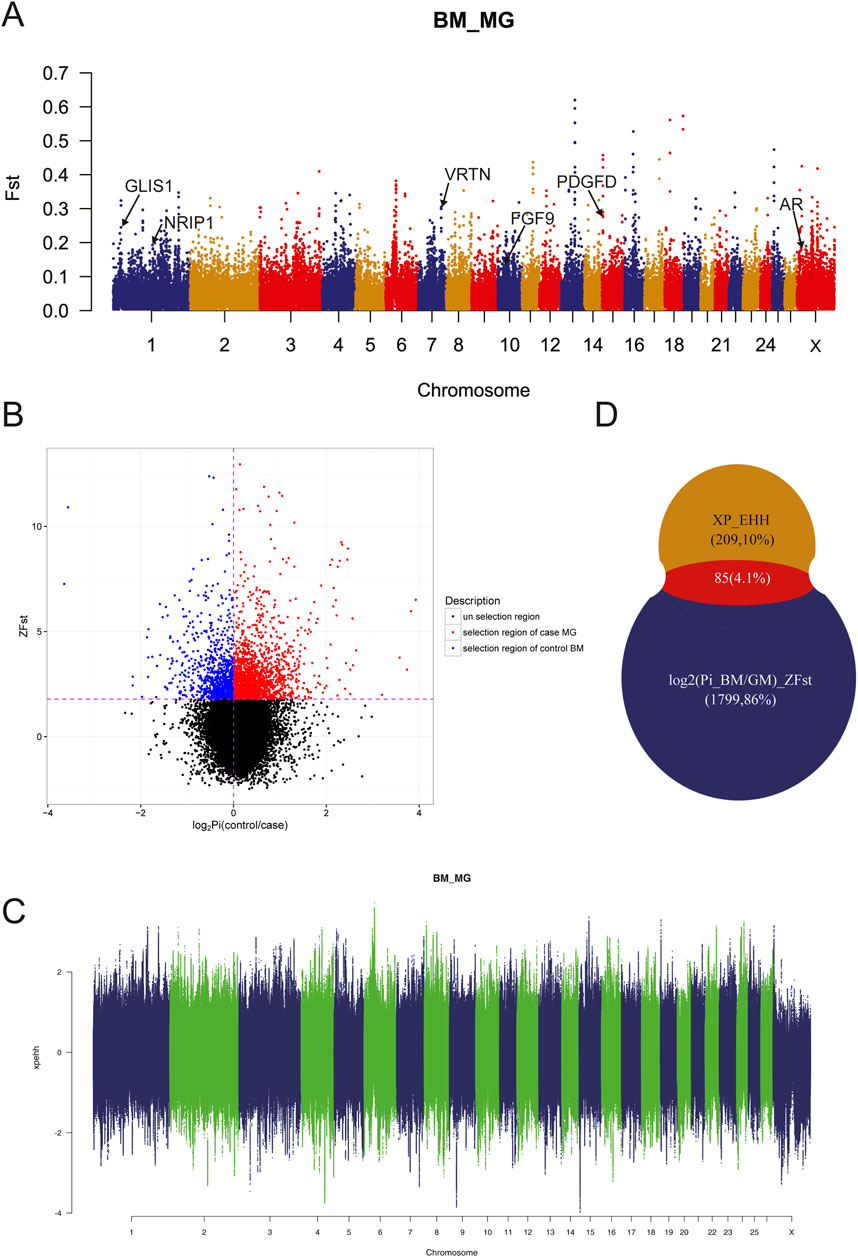
Figure 4. Genomic Selection analyses. (A) Selective signals detected by FST. (B) Selective signals detected by log2 (Pi_BM/GM)_ZFST. (C) Selective signals detected by XP_EHH. (D) Overlapping genes identified by log2 (Pi_BM/GM)_ZFST and XP_EHH.
3.5 Function annotation of the selected genes
Functional association of the 85 candidate genes was further investigated by GO and KEGG analysis. For GO analysis, the biological processes mainly focused on ‘cellular response to organic substance’, ‘cellular response to lipid’, ‘cellular response to organic cyclic compound’, ‘negative regulation of cell development’, ‘beta stimulus regulation of DTPase activity’, and so on (Figure 5A) (p < 0.05). In terms of KEGG, several signaling pathways related to lipid metabolism were significantly enriched, including ‘phospholipase D signaling pathway’, ‘glycerophospholipid metabolism’, ‘glycerolipid metabolism’, and ‘fatty acid elongation’ (Figure 5B). In addition, some significant pathways were also enriched, such as ‘MAPK pathway’, ‘P13K Akt signaling pathway’, ‘Calcium signaling pathway’ and ‘ras signaling pathway’. The interaction of KEGG pathways and the relationship between genes and pathways was show in Figure 5B. Furthermore, these candidate genes showed close functional association (Figure 6).
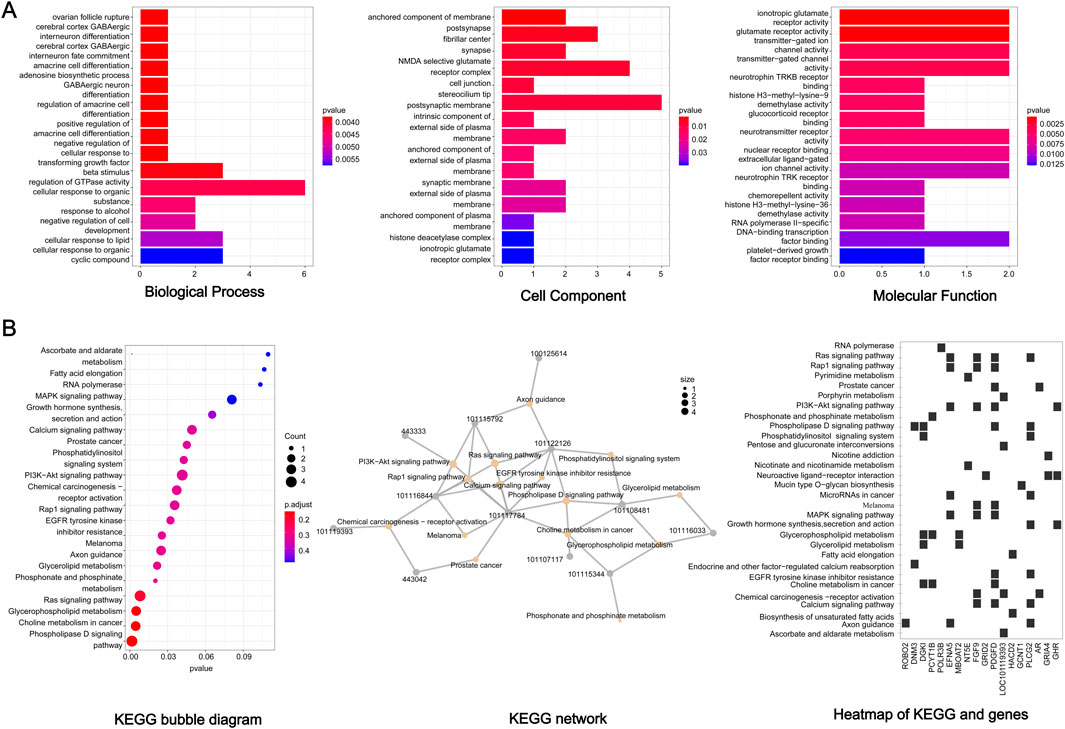
Figure 5. GO and KEGG analysis of the 85 candidate genes. (A) GO analysis. (B) KEGG analysis.
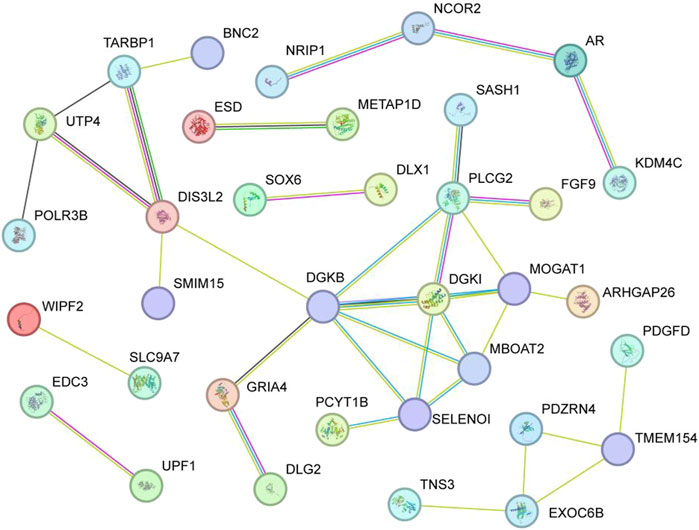
Figure 6. Protein-Protein Interaction (PPI) of the 85 candidate genes.
4 Discussion
In the past decade, the improvement of sheep tail phenotype has gradually developed from utilization of traditional hybridization or tail-docking to molecular breeding technology. To date, several methods are employed to detect the selective sweeps in various livestock genomes. In the present study, we used three metrics, allele frequency-based methods FST, π-Ratio, and haplotype-based method XP-EHH, to identify genome-wide selective sweep regions. The power of each test was different, and any set of candidate genes may contain some false positives (Nielsen et al., 2007). FST measures the genetic differentiation between populations (Holsinger and Weir, 2009); π-Ratio identifies the differences in nucleotide divergence between populations (Sun et al., 2020); XP-EHH detects ongoing or nearly fixed selective sweeps by comparing haplotypes between the two populations (Sabeti et al., 2007b). Combining multiple tests can improve the power of detecting selection signatures (Zeng et al., 2007), making the results more reliable. We considered the overlapping genes derived from three methods as candidate genes and eventually identified 85 candidate genes. Among these genes, PDGFD (Dong et al., 2020; Zhu et al., 2021; Pan et al., 2019; Mastrangelo et al., 2019; Zhao et al., 2020; Li et al., 2020a; li et al., 2020b; Wang et al., 2022; Wei et al., 2015; Luo et al., 2021), GLIS1 (Luo et al., 2021), NRIP1 (RIP140) (Xu et al., 2017), AR (Moradi et al., 2022), FGF9 (Moioli et al., 2015), and VRTN (Mastrangelo et al., 2019; Zhu et al., 2021; Moioli et al., 2015) were formerly reported to be involved in regulation of tail fat deposition or tail length.
Many studies have recently highlighted the platelet-derived growth factor D (PDGFD) gene as a new sheep tail phenotype pattern maker (Wei et al., 2015; Pan et al., 2019; Dong et al., 2020; Li Q. et al., 2020; Li X. et al., 2020; Luo et al., 2021; Mastrangelo et al., 2019). Dong et al. found that the expression of the PDGFD gene is higher in fat-tailed breeds than in thin-tailed breeds, and a similar result was observed in obese mice and human after analyzing a public transcriptomic data (Dong et al., 2020). Overexpression of PDGFD in ovine preadipocytes could promote adipogenic differentiation, and the expression levels of two adipogenesis marker genes (PPARc and LPL) increased after PDGFD overexpression (Li Q. et al., 2020). Furthermore, oil red O staining showed that the number of lipid drops was higher in the PDGFD-overexpressing group than in the control group (Li Q. et al., 2020). These studies indicated that PDGFD gene plays a positive regulation role in the fat deposition process of sheep tail. Interestingly, there were also different discoveries about the expression profile of the PDGFD gene. Li X. et al. (2020) identified four PDGFD transcripts (I, II, III, and IV), and transcript I was the most differentially expressed transcripts between the thin-tailed and the fat-tailed/fat-rumped sheep breeds. Notably, PDGFD expression (at the mRNA and protein levels) was consistently negatively correlated with fat deposition in sheep tails (Li X. et al., 2020). The highest PDGFD gene expression level was observed in the thin-tailed Chinese Merino sheep, followed sequentially by the small fat-tailed Han sheep, the large fat-tailed Han sheep, and the fat-rumped Altay sheep (Li X. et al., 2020). The authors inclined to the idea of involving the PDGFRb signaling (a receptor of PDGFD) in inhibiting the differentiation of white adipocytes by regulating the expression of two key transcriptional regulators of adipogenesis (PPARc2 and C/EBPa) (Olson and Soriano, 2011; Kalds et al., 2021). In the present study we found that PDGFD gene was strongly selected by Bamei Mutton sheep, combined with previous researches, indicating that PDGFD was involved in regulating the fat deposition process of sheep tail, but how to regulate this process still needs more in-depth research.
GLIS1 is a zinc finger protein that acts as both an activator and repressor of transcription (Kim et al., 2002). The temporal and spatial expression of GLIS1 is consistent with mesoderm differentiation during mouse embryonic development (Nakashima et al., 2002). Later, Tosic et al. discovered that GLIS1was highly expressed in bipotent muscle satellite cells. But when overexpressed, increased occupancy of GLIS1 is observed at the promoters of adipogenic genes Adipoq, Cebpa and Ucp1, and drives brown adipogenesis both in vitro and in vivo, indicating that GLIS1 was a novel pro-adipogenic transcription factor (Tosic et al., 2018). Most recently, GLIS1was detected as a candidate gene of selective signature of sheep tail phenotype (Luo et al., 2021). A non-synonymous point mutation (g.27807636G > T) was found within GLIS1 in two fat-tailed Chinese indigenous sheep breeds (Mongolian sheep and Small Tail Han sheep) compared with two thin-tailed dairy sheep (DairyMeade and East Friesian), and resulted in a Pro to Thr substitution (Luo et al., 2021). In our study, GLIS1 was also strongly selected in Bamei Mutton sheep compared with Mongolian sheep. Taken together, GLIS1, as a pro-adipogenic factor, may plays a key role in mesodermal cell differentiation during fetal development in fat-tailed sheep to initiate differentiation of pre-adipocytes and fat accumulation (Luo et al., 2021).
Nuclear receptor interacting protein one gene (NRIP1, also known as RIP140), encodes a nuclear protein also known as receptor-interacting protein 140 (RIP140). RIP140 is widely expressed and plays an important role in regulating lipid and glucose metabolism (Leonardsson et al., 2004; Ho et al., 2011; Hochberg et al., 2015). RIP140 interacts with multiple adipocyte-specific genes, such as uncoupling protein 1 (UCP1), mitochondrial fatty acid transporter carnitine palmitoyl transferase 1 (CPT1) and lipid droplet protein cell death-inducing DFFA-like effector A (CIDEA). The expression of these genes is characteristic of brown adipose tissue (Nautiyal et al., 2013). Previous studies in adipocyte cell models also revealed that RIP140 functions as a corepressor of catabolic pathways, including fatty acid oxidation, oxidative phosphorylation, glycolysis and tricarboxylic acid cycle (Christian et al., 2005; Powelka et al., 2006). Moreover, Xu et al. conducted a genome-wide association study using phenotypes and genotypes of two breeds of contrasting tail types (Small-tailed and Large-tailed Han sheep breeds) to identify functional genes and variants associated with fat deposition, and revealed that RIP140 was a strong candidate for fat deposition in the tails of sheep (Xu et al., 2017), which is consistent with our results.
It has been proved that androgen receptor (Ar) gene gets participate in lipid binding (Huang et al., 2009), and has a negative function in fat deposition in both mice and human beings (Rubinow et al., 2015; Kim et al., 2019). Adipose tissue macrophages express the androgen receptor (AR) and regulate adipose tissue remodeling. Thus, testosterone signaling in macrophages could alter the paracrine function of these cells and thereby contribute to the metabolic effects of androgens in men (Rubinow et al., 2015). In order to determine whether the loss of AR signaling in hematopoietic cells results in greater fat accumulation, Rubinow et al. performed a metabolic phenotyping study in male mice. C57BL/6J male mice (ages 12–14 weeks) underwent bone marrow transplant from either wild-type (WT) or AR knockout (ARKO) donors (n = 11–13 per group). Mice were fed a high-fat diet (60% fat) for 16 weeks. At baseline, 8 and 16 weeks, glucose and insulin tolerance tests were performed, and body composition was analyzed with fat-water imaging by MRI. No differences in body weight were observed between mice transplanted with WT bone marrow [WT (WTbm)] or ARKO bone marrow [WT (ARKObm)] prior to initiation of the high-fat diet. After 8 weeks of high-fat feeding, WT (ARKObm) mice exhibited significantly more visceral and total fat mass than WT (WTbm) animals. Resultant data indicate that AR signaling in hematopoietic cells influences body fat distribution in male mice, and the absence of hematopoietic AR plays a permissive role in visceral fat accumulation. These findings demonstrate a metabolic role for AR signaling in marrow-derived cells and suggest a novel mechanism by which androgen deficiency in men might promote increased adiposity (Rubinow et al., 2015). Kim et al. also discovered that blocking AR can decreases the expression of CPTI (one of long-chain fatty acid (LCFA) transport proteins) in the skeletal muscle, which reduces fat metabolism. Thus, reducing sex hormones or suppressing the sensitivity of AR can inhibit energy efficiency and fat metabolism by suppressing CPTI (Kim et al., 2019). However, the effect of AR gene on fat deposition in sheep is rarely reported. In our study, AR was identified as a promising candidate gene of sheep tail phenotype, and the same results were found in two Iranian thin- and fat-tailed sheep Breeds (Moradi et al., 2022). Combined with the above studies, we speculate that AR may promote energy efficiency and fat metabolism of sheep, thereby inhibit fat deposition in tail.
Fibroblast growth factor 9 (FGF9) is a protein-coding gene that plays an important role in the regulation of embryonic development, cell proliferation, cell differentiation, and cell migration (Moioli et al., 2015). FGF9 expressed not only in white adipose tissue (WAT) of human, but also in brown adipose tissue (BAT) while exposed to cold, regulating the development of adipose tissue (Mejhert et al., 2010; Grefhorst et al., 2015). In mammals, WAT stores fat and BAT dissipates fat to produce heat. FGF9 was selected as a candidate gene associated with tail phenotype in both our and a recent study (Moioli et al., 2015), indicating that FGF9 may plays a role in fat deposition and metabolism of sheep, but the mechanism underlying it is unclear.
In addition to fat deposition, the number of caudal vertebrae also affects tail phenotype by affecting the length of sheep tail. It has been reported that Vertnin (VRTN) gene is a key candidate gene associated with the variation of vertebral number in sheep and pigs (Duan et al., 2018; Zhang et al., 2017). As a transcriptional inhibitor, VRTN independently regulates the expression of BMP2 gene in the dorsoventral axis through combination with its regulatory sequence, thereby enabling normal development of embryos along the dorsoventral axis (Shao et al., 2017). In a study of genome-wide scan of selection signatures of sheep tail phenotype, Mastrangelo et al. identified VRTN as a key candidate gene; and the tail of the five fat-tailed sheep breeds where this signature was detected was definitively longer than the tail of the thin-tailed breeds (Mastrangelo et al., 2019). The same results were also found in Moioli et al., Zhu et al., and our study. In the current study, the tail length of Bamei Mutton sheep is longer than that of Mongolian sheep, with the caudal vertebrae number is 20–30 and 10–14, respectively. These results indicated that VRTN may be an important gene involved in regulating the development of sheep caudal vertebrae, and affect the tail phenotype of sheep by affecting the tail length.
Except for above mentioned genes, the rest of the candidate genes may also play important roles in regulating sheep tail phenotype, although there was not enough evidence at presence based on publicly available information. Therefore, further studies and experiments are needed to confirm the soles and mechanisms of these genes in formation of sheep tail phenotype. In conclusion, our results provide a strong foundation for studying the regulation of tail phenotype in sheep and do offer hope that the causal mutations and the mode of inheritance of this trait will soon be discovered by further experimentation.
Data availability statement
The data presented in the study are deposited in the SRA repository, accession number PRJNA1192386 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1192386).
Ethics statement
The animal studies were approved by the Animal Ethics Committee of Inner Mongolia Academy of Agricultural and Animal Husbandry Sciences. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
YQ: Funding acquisition, Writing–original draft, Writing–review and editing, Methodology, Visualization. XH: Methodology, Resources, Writing–review and editing. BW: Data curation, Methodology, Writing–review and editing. CY: Visualization, Writing–review and editing, Data curation. LD: Resources, Writing–review and editing. BL: Data curation, Methodology, Validation, Visualization, Writing–review and editing. WZ: Conceptualization, Writing–review and editing. SF: Funding acquisition, Methodology, Writing–review and editing. YL: Conceptualization, Funding acquisition, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Modern Agricultural Industry Technology System of China (CARS-38), the Doctoral Start-up Fund Project of Xichang University (YBZ202406), the Inner Mongolia Autonomous Region Open Competition Projects (2022JBGS0024), and the Inner Mongolia Autonomous Region Science and Technology Plan (2023YFHH0114).
Conflict of interest
Author BL was employed by Inner Mongolia BIONEW Technology Co., LTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1509177/full#supplementary-material
References
Abdullah, A. Y., Kridli, R. T., Shaker, M. M., and Obeidat, M. D. (2010). Investigation of growth and carcass characteristics of pure and crossbred Awassi lambs. Small Ruminant Res. 94, 167–175. doi:10.1016/j.smallrumres.2010.08.005
Abouheif, M. A., Kraidees, M. S., and Shatat, R. A. (1993). Performance and carcass traits of docked and intact fat-tailed Najdi lambs. Asian-Australasian J. Animal Sci. 6 (1), 135–138. doi:10.5713/ajas.1993.135
Alberto, F. J., Boyer, F., Orozco-terWengel, P., Streeter, I., Servin, B., de Villemereuil, P., et al. (2018). Convergent genomic signatures of domestication in sheep and goats. Nat. Commun. 9 (1), 813. Published 2018 Mar 6. doi:10.1038/s41467-018-03206-y
Alexander, D. H., and Lange, K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinforma. 12, 246. Published 2011 Jun 18. doi:10.1186/1471-2105-12-246
Atti, N., Bocquier, F., and Khaldi, G. (2004). Performance of the fat-tailed Barbarine sheep in its environment: adaptive capacity to alter-nation of underfeeding and re-feeding periods. A review. Animal Res. 53, 165–176. doi:10.1051/animres:2004012
Atti, N., and Mahouachi, M. (2011). The effects of diet, slaughter weight and docking on growth, carcass composition and meat quality of fat-tailed Barbarine lambs. A review. A Rev. Trop. Anim. Health Prod. 43 (7), 1371–1378. doi:10.1007/s11250-011-9865-6
Bicer, O., Pekel, E., and Guney, O. (1992). Effects of docking on growth performance and carcass characteristics of fat-tailed Awassi ram lambs. Small Ruminant Res. 8, 353–357. doi:10.1016/0921-4488(92)90216-q
Bing, I. M., Aygn, T., kdal, G., and Yılmaz, A. (2006). The effects of docking on fattening performance and carcass characteristics in fat-tailed Norduz male lambs. Small Ruminant Res. 64 (1), 101–106. doi:10.1016/j.smallrumres.2005.04.011
Browning, S. R., and Browning, B. L. (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81 (5), 1084–1097. doi:10.1086/521987
Chessa, B., Pereira, F., Arnaud, F., Amorim, A., Goyache, F., Mainland, I., et al. (2009). Revealing the history of sheep domestication using retrovirus integrations. Science 324 (5926), 532–536. doi:10.1126/science.1170587
Christian, M., Kiskinis, E., Debevec, D., Leonardsson, G., White, R., and Parker, M. G. (2005). RIP140-targeted repression of gene expression in adipocytes. Mol. Cell Biol. 25 (21), 9383–9391. doi:10.1128/MCB.25.21.9383-9391.2005
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
Deng, J., Xie, X. L., Wang, D. F., Zhao, C., Lv, F. H., Li, X., et al. (2020). Paternal origins and migratory episodes of domestic sheep. Curr. Biol. 30 (20), 4085–4095.e6. doi:10.1016/j.cub.2020.07.077
Dong, K., Yang, M., Han, J., Ma, Q., Han, J., Song, Z., et al. (2020). Genomic analysis of worldwide sheep breeds reveals PDGFD as a major target of fat-tail selection in sheep. BMC Genomics 21 (1), 800. doi:10.1186/s12864-020-07210-9
Duan, Y., Zhang, H., Zhang, Z., Gao, J., Yang, J., Wu, Z., et al. (2018). VRTN is required for the development of thoracic vertebrae in mammals. Int. J. Biol. Sci. 14 (6), 667–681. Published 2018 Apr 30. doi:10.7150/ijbs.23815
Eck, K., Kunz, E., Mendel, C., Luhken, G., and Medugorac, I. (2019). Morphometric measurements in lambs as a basis for future mapping studies. Small Ruminant Res. 181, 57–64. doi:10.1016/j.smallrumres.2019.04.007
Grefhorst, A., van den Beukel, J. C., van Houten, E. L., Steenbergen, J., Visser, J. A., and Themmen, A. P. (2015). Estrogens increase expression of bone morphogenetic protein 8b in brown adipose tissue of mice. Biol. Sex. Differ. 6, 7. Published 2015 Apr 3. doi:10.1186/s13293-015-0025-y
Ho, P. C., Chuang, Y. S., Hung, C. H., and Wei, L. N. (2011). Cytoplasmic receptor-interacting protein 140 (RIP140) interacts with perilipin to regulate lipolysis. Cell Signal 23 (8), 1396–1403. doi:10.1016/j.cellsig.2011.03.023
Hochberg, I., Tran, Q. T., Barkan, A. L., Saltiel, A. R., Chandler, W. F., and Bridges, D. (2015). Gene expression signature in adipose tissue of acromegaly patients. PLoS One 10 (6), e0129359. Published 2015 Jun 18. doi:10.1371/journal.pone.0129359
Holsinger, K. E., and Weir, B. S. (2009). Genetics in geographically structured populations: defining, estimating and interpreting F(ST). Nat. Rev. Genet. 10 (9), 639–650. doi:10.1038/nrg2611
Huang, W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4 (1), 44–57. doi:10.1038/nprot.2008.211
Kalds, P., Luo, Q., Sun, K., Zhou, S., Chen, Y., and Wang, X. (2021). Trends towards revealing the genetic architecture of sheep tail patterning: promising genes and investigatory pathways. Anim. Genet. 52 (6), 799–812. doi:10.1111/age.13133
Kashan, N. E. J., Azar, G. H. M., Afzalzadeh, A., and Salehi, A. (2005). Growth performance and carcass quality of fattening lambs from fat-tailed and tailed sheep breeds. Small Ruminant Res. 60, 267–271. doi:10.1016/j.smallrumres.2005.01.001
Khaldari, M., Kashan, N. E., Afzalzadeh, A., and Salehi, A. (2008). Growth and carcass characteristics of crossbred progeny from lean-tailed and fat-tailed sheep breeds. South Afr. J. Animal Sci. 37, 51–56. doi:10.4314/sajas.v37i1.4026
Kijas, J. W., Townley, D., Dalrymple, B. P., Heaton, M. P., Maddox, J. F., McGrath, A., et al. (2009). A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS One 4 (3), e4668. doi:10.1371/journal.pone.0004668
Kim, J., Park, J., Kim, N., Park, H. Y., and Lim, K. (2019). Inhibition of androgen receptor can decrease fat metabolism by decreasing carnitine palmitoyl transferase I levels in skeletal muscles of trained mice. Nutr. Metab. (Lond). 16, 82. Published 2019 Nov 27. doi:10.1186/s12986-019-0406-z
Kim, Y. S., Lewandoski, M., Perantoni, A. O., Kurebayashi, S., Nakanishi, G., and Jetten, A. M. (2002). Identification of Glis1, a novel Gli-related, Kruppel-like zinc finger protein containing transactivation and repressor functions. J. Biol. Chem. 277 (34), 30901–30913. doi:10.1074/jbc.M203563200
Kridli, R., and Said, S. (1999). Libido testing and the effect of exposing sexually naïve Awassi rams to estrous ewes on sexual performance. Small Rumin. Res. 32 (2), 149–152. doi:10.1016/s0921-4488(98)00168-0
Leonardsson, G., Steel, J. H., Christian, M., Pocock, V., Milligan, S., Bell, J., et al. (2004). Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc. Natl. Acad. Sci. U. S. A. 101 (22), 8437–8442. doi:10.1073/pnas.0401013101
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27 (21), 2987–2993. doi:10.1093/bioinformatics/btr509
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, Q., Lu, Z., Jin, M., Fei, X., Quan, K., Liu, Y., et al. (2020a). Verification and analysis of sheep tail type-associated PDGF-D gene polymorphisms. Anim. (Basel) 10 (1), 89. Published 2020 Jan 6. doi:10.3390/ani10010089
Li, X., Yang, J., Shen, M., Xie, X. L., Liu, G. J., Xu, Y. X., et al. (2020b). Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 11 (1), 2815. Published 2020 Jun 4. doi:10.1038/s41467-020-16485-1
Luo, R., Zhang, X., Wang, L., Zhang, L., Li, G., and Zheng, Z. (2021). GLIS1, a potential candidate gene affect fat deposition in sheep tail. Mol. Biol. Rep. 48 (5), 4925–4931. doi:10.1007/s11033-021-06468-w
Marai, I. F. M., Nowar, M. S., Bahgat, L. B., and Owen, J. B. (1987). Effect of docking and shearing on growth and carcass traits of fat-tailed Ossimi sheep. J. Agric. Sci. 109 (3), 513–518. doi:10.1017/S0021859600081727
Mastrangelo, S., Moioli, B., Ahbara, A., Latairish, S., Portolano, B., Pilla, F., et al. (2019). Genome-wide scan of fat-tail sheep identifies signals of selection for fat deposition and adaptation. Animal Prod. Sci. 59, 835–848. doi:10.1071/an17753
Mejhert, N., Galitzky, J., Pettersson, A. T., Bambace, C., Blomqvist, L., Bouloumié, A., et al. (2010). Mapping of the fibroblast growth factors in human white adipose tissue. J. Clin. Endocrinol. Metab. 95 (5), 2451–2457. doi:10.1210/jc.2009-2049
Moharrery, A. (2007). Effect of docking and energy of diet on carcass fat characteristics in fat-tailed Badghisian sheep. Small Ruminant Res. 69, 208–216. doi:10.1016/j.smallrumres.2005.12.021
Moioli, B., Pilla, F., and Ciani, E. (2015). Signatures of selection identify loci associated with fat tail in sheep. J. Anim. Sci. 93 (10), 4660–4669. doi:10.2527/jas.2015-9389
Moradi, M. H., Nejati-Javaremi, A., Moradi-Shahrbabak, M., Dodds, K. G., Brauning, R., and McEwan, J. C. (2022). Hitchhiking mapping of candidate regions associated with fat deposition in Iranian thin and fat tail sheep breeds suggests new insights into molecular aspects of fat tail selection. Anim. (Basel) 12 (11), 1423. Published 2022 May 31. doi:10.3390/ani12111423
Moradi, M. H., Nejati-Javaremi, A., Moradi-Shahrbabak, M., Dodds, K. G., and McEwan, J. C. (2012). Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genet. 13, 10. Published 2012 Feb 26. doi:10.1186/1471-2156-13-10
Nakashima, M., Tanese, N., Ito, M., Auerbach, W., Bai, C., Furukawa, T., et al. (2002). A novel gene, GliH1, with homology to the Gli zinc finger domain not required for mouse development. Mech. Dev. 119 (1), 21–34. doi:10.1016/s0925-4773(02)00291-5
Nautiyal, J., Christian, M., and Parker, M. G. (2013). Distinct functions for RIP140 in development, inflammation, and metabolism. Trends Endocrinol. Metab. 24 (9), 451–459. doi:10.1016/j.tem.2013.05.001
Nekrutenko, A., and Taylor, J. (2012). Next-generation sequencing data interpretation: enhancing reproducibility and accessibility. Nat. Rev. Genet. 13 (9), 667–672. doi:10.1038/nrg3305
Nielsen, R., Hellmann, I., Hubisz, M., Bustamante, C., and Clark, A. G. (2007). Recent and ongoing selection in the human genome. Nat. Rev. Genet. 8 (11), 857–868. doi:10.1038/nrg2187
O’Donovan, P. B., Ghadaki, M. B., Behesti, R. D., Saleh, B. A., and Rollinson, D. H. L. (1973). Performance and carcass composition of docked and control fat-tailed Kellakui lambs. Animal Sci. 16, 67–76. doi:10.1017/s0003356100034887
Olson, L. E., and Soriano, P. (2011). PDGFRβ signaling regulates mural cell plasticity and inhibits fat development. Dev. Cell 20 (6), 815–826. doi:10.1016/j.devcel.2011.04.019
Orihuela, A., and Ungerfeld, R. (2019). Tail docking in sheep (Ovis aries): a review on the arguments for and against the procedure,advantages/disadvantages, methods, and new evidence to revisit the topic. Livest. Sci. 230, 103837. doi:10.1016/j.livsci.2019.103837
Pan, Z., Li, S., Liu, Q., Wang, Z., Zhou, Z., Di, R., et al. (2019). Rapid evolution of a retro-transposable hotspot of ovine genome underlies the alteration of BMP2 expression and development of fat tails. BMC Genomics 20 (1), 261. Published 2019 Apr 2. doi:10.1186/s12864-019-5620-6
Pourlis, A. F. (2011). A review of morphological characteristics relating to the production and reproduction of fat-tailed sheep breeds. Trop. Anim. Health Prod. 43 (7), 1267–1287. doi:10.1007/s11250-011-9853-x
Powelka, A. M., Seth, A., Virbasius, J. V., Kiskinis, E., Nicoloro, S. M., Guilherme, A., et al. (2006). Suppression of oxidative metabolism and mitochondrial biogenesis by the transcriptional corepressor RIP140 in mouse adipocytes. J. Clin. Invest 116 (1), 125–136. doi:10.1172/JCI26040
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Rubin, C. J., Zody, M. C., Eriksson, J., Meadows, J. R. S., Sherwood, E., Webster, M. T., et al. (2010). Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464 (7288), 587–591. doi:10.1038/nature08832
Rubinow, K. B., Wang, S., den Hartigh, L. J., Subramanian, S., Morton, G. J., Buaas, F. W., et al. (2015). Hematopoietic androgen receptor deficiency promotes visceral fat deposition in male mice without impairing glucose homeostasis. Andrology 3 (4), 787–796. doi:10.1111/andr.12055
Sabeti, P. C., Varilly, P., Fry, B., Lohmueller, J., Hostetter, E., Cotsapas, C., et al. (2007a). Genome-wide detection and characterization of positive selection in human populations. Nature 449 (7164), 913–918. doi:10.1038/nature06250
Sabeti, P. C., Varilly, P., Fry, B., Lohmueller, J., Hostetter, E., Cotsapas, C., et al. (2007b). Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918. doi:10.1038/nature06250
Safdarian, M., Zamiri, M. J., Hashemi, M., and Noorolahi, H. (2008). Relationships of fat-tail dimensions with fat-tail weight and carcass characteristics at different slaughter weights of Torki-Ghashghaii sheep. Meat Sci. 80 (3), 686–689. doi:10.1016/j.meatsci.2008.03.007
Shao, M., Wang, M., Liu, Y. Y., Ge, Y. W., Zhang, Y. J., and Shi, D. L. (2017). Vegetally localised Vrtn functions as a novel repressor to modulate bmp2b transcription during dorsoventral patterning in zebrafish. Development 144 (18), 3361–3374. doi:10.1242/dev.152553
Shelton, M., Willingham, T., Thompson, P., and Roberts, E. M. (1991). Influence of docking and castration on growth and carcass traits of fat-tail Karakul, Rambouillet and crossbred lambs. Small Ruminant Res. 4, 235–243. doi:10.1016/0921-4488(91)90147-i
Sun, T., Huang, G. Y., Wang, Z. H., Teng, S. H., Cao, Y. H., Sun, J. L., et al. (2020). Selection signatures of Fuzhong Buffalo based on whole-genome sequences. BMC Genomics 21 (1), 674. Published 2020 Sep 29. doi:10.1186/s12864-020-07095-8
Szpiech, Z. A., and Hernandez, R. D. (2014). selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 31 (10), 2824–2827. doi:10.1093/molbev/msu211
Tosic, M., Allen, A., Willmann, D., Lepper, C., Kim, J., Duteil, D., et al. (2018). Lsd1 regulates skeletal muscle regeneration and directs the fate of satellite cells. Nat. Commun. 9 (1), 366. Published 2018 Jan 25. doi:10.1038/s41467-017-02740-5
Wang, F., Shao, J., He, S., Guo, Y., Pan, X., Wang, Y., et al. (2022). Allele-specific expression and splicing provide insight into the phenotypic differences between thin- and fat-tailed sheep breeds. J. Genet. Genomics 49 (6), 583–586. doi:10.1016/j.jgg.2021.12.008
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, Y. Q., Zhong, R. Z., Fang, Y., and Zhou, D. W. (2018). Influence of tail docking on carcass characteristics, meat quality and fatty acid composition of fat-tail lambs. Small Ruminant Res. 162, 17–21. doi:10.1016/j.smallrumres.2017.09.005
Wei, C., Wang, H., Liu, G., Wu, M., Cao, J., Liu, Z., et al. (2015). Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genomics 16 (1), 194. Published 2015 Mar 17. doi:10.1186/s12864-015-1384-9
Weir, B. S., and Hill, W. G. (2002). Estimating F-statistics. Annu. Rev. Genet. 36, 721–750. doi:10.1146/annurev.genet.36.050802.093940
Xu, S. S., Ren, X., Yang, G. L., Xie, X. L., Zhao, Y. X., Zhang, M., et al. (2017). Genome-wide association analysis identifies the genetic basis of fat deposition in the tails of sheep (Ovis aries). Anim. Genet. 48 (5), 560–569. doi:10.1111/age.12572
Yang, J., Lee, S. H., Goddard, M. E., and Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88 (1), 76–82. doi:10.1016/j.ajhg.2010.11.011
Yousefi, A. R., Kohram, H., Zare Shahneh, A., Nik-khah, A., and Campbell, A. W. (2012). Comparison of the meat quality and fatty acid composition of traditional fat-tailed (Chall) and tailed (Zel) Iranian sheep breeds. Meat Sci. 92 (4), 417–422. doi:10.1016/j.meatsci.2012.05.004
Yuan, Z., Liu, E., Liu, Z., Kijas, J. W., Zhu, C., Hu, S., et al. (2017). Selection signature analysis reveals genes associated with tail type in Chinese indigenous sheep. Anim. Genet. 48 (1), 55–66. doi:10.1111/age.12477
Zeng, K., Shi, S., and Wu, C. I. (2007). Compound tests for the detection of hitchhiking under positive selection. Mol. Biol. Evol. 24 (8), 1898–1908. doi:10.1093/molbev/msm119
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M., and Yang, T. L. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35 (10), 1786–1788. doi:10.1093/bioinformatics/bty875
Zhang, Z., Sun, Y., Du, W., He, S., Liu, M., and Tian, C. (2017). Effects of vertebral number variations on carcass traits and genotyping of Vertnin candidate gene in Kazakh sheep. Asian-Australas J. Anim. Sci. 30 (9), 1234–1238. doi:10.5713/ajas.16.0959
Zhao, F., Deng, T., Shi, L., Wang, W., Zhang, Q., Du, L., et al. (2020). Genomic scan for selection signature reveals fat deposition in Chinese indigenous sheep with extreme tail types. Anim. (Basel) 10 (5), 773. Published 2020 Apr 29. doi:10.3390/ani10050773
Zhi, D., Da, L., Liu, M., Cheng, C., Zhang, Y., Wang, X., et al. (2018). Whole genome sequencing of hulunbuir short-tailed sheep for identifying candidate genes related to the short-tail phenotype. G3 (Bethesda) 8 (2), 377–383. doi:10.1534/g3.117.300307
Keywords: sheep, tail type, selection signature analysis, FST, π-ratio, XP-EHH
Citation: Qi Y, He X, Wang B, Yang C, Da L, Liu B, Zhang W, Fu S and Liu Y (2024) Selection signature analysis reveals genes associated with tail phenotype in sheep. Front. Genet. 15:1509177. doi: 10.3389/fgene.2024.1509177
Received: 10 October 2024; Accepted: 19 November 2024;
Published: 11 December 2024.
Edited by:
Fei Hao, Northumbria University, United KingdomReviewed by:
Yufang Liu, Hebei University of Engineering, ChinaRan Li, Northwest A&F University, China
Copyright © 2024 Qi, He, Wang, Yang, Da, Liu, Zhang, Fu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongbin Liu, eWJsaXVAaW11LmVkdS5jbg==; Shaoyin Fu, ZnVzaGFvMTIzNEAxMjYuY29t