 Guojian Ma1,2
Guojian Ma1,2 Jingya Xu
Jingya Xu- 1Breeding Department, Wuhan COFCO Meat Co., Ltd., Wuhan, Hubei, China
- 2COFCO Nutrition and Health Research Institute, Beijing, China
Backfat thickness (BFT) and feed conversion ratio (FCR) are important commercial traits in the pig industry. With the increasing demand for human health and meat production, identifying functional genomic regions and genes associated with these commercial traits is critical for enhancing production efficiency. In this research, we conducted a genome-wide association study (GWAS) on a Landrace population comprising 4,295 individuals with chip data for BFT and FCR. Our analysis revealed a total of 118 genome-wide significant signals located on chromosomes SSC1, SSC2, SSC7, SSC12, and SSC13, respectively. Furthermore, we identified 10 potential regions associated with the two traits and annotated the genes within these regions. In addition, enrichment analysis was also performed. Notably, candidate genes such as SHANK2, KCNQ1, and ABL1 were found to be associated with BFT, whereas NAP1L4, LSP1, and PPFIA1 genes were related to the FCR. Our findings provide valuable insights into the genetic architecture of these two traits and offer guidance for future pig breeding efforts.
Introduction
The increasing demand for human health and food nutrition has become a challenge due to the rapid growth of global population. Consequently, enhancing the production efficiency of livestock products has become vital for the livestock industry and sustainable development (Mehrabi et al., 2020). Swine is one of the most important economic livestock in the world, providing a diverse range of products to meet human needs. The rapid development of breeding methods, such as genomic selection, has effectively reduced the genetic interval between pig generations and significantly improved the performance of commercial pig breeds by increasing prediction accuracy (Knol et al., 2016). As consumers’ demand for healthier meat products increases, pigs have been bred for lower fat content and higher lean meat. Previous research uncovered that daily energy intake is related to whole-body fat composition in male pigs (Liu et al., 2021), and leaner pigs tend to exhibit higher feed efficiency. Therefore, understanding the genetic architecture of these commercial traits is essential.
Feed conversion ratio (FCR) and backfat thickness (BFT) are primary commercial phenotypes in the pig industry and have been extensively analyzed by numerous researchers. Candidate genes, such as phospholipase A2 group IB (PLA2G1B), have been reported to be associated with feed efficiency by influencing lipid catabolism (Fu L. et al., 2020; Hollie and Hui, 2011). Additionally, the members of the insulin-like growth factor family, such as IGF1 and IGF2, have been found to affect the growth rate and feed conversion efficiency (Zhu et al., 2014). Backfat thickness is another important trait in pig production as it impacts lean meat yield and the popularity of pork meat (Hoa et al., 2021). Many loci on SSC1, SSC5, SSC6, SSC7, and SSC12, as well as candidate genes such as MC4R, IGF2, and LEPR, were found to be related to backfat thickness (Gozalo-Marcilla et al., 2021; Fu M. et al., 2023).
Over the past 15 years, genome-wide association studies (GWASs) have been employed to investigate the linkage between genomic markers and records of various traits (Abdellaoui et al., 2023). This approach has facilitated the identification of numerous quantitative trait nucleotides (QTNs) and candidate genes associated with FCR and BFT (Li W. et al., 2022; Delpuech et al., 2021; Ding et al., 2022; Miao et al., 2023), which provided deep insights into these commercial traits and improved the quality of meat production. To date, about 55,688 quantitative trait loci (QTLs) have been released by pig QTLdb (Hu et al., 2005). However, due to the complexity of these quantitative traits, many QTLs remain unknown.
Using a Landrace population with genomic chip data, a total of 4,295 individuals with two important commercial traits, including BFT at 100 kg and FCR, were analyzed in this study. Related variants with annotated candidate genes within candidate regions were detected using a mixed-effects linear model in a genome-wide association study. Furthermore, linkage disequilibrium (LD) block analysis with candidate regions and enrichment analysis of candidate genes were also performed. The main objectives of this research were to identify the associated genomic regions and candidate genes of BFT and FCR within our population. In addition, we also used a multi-omics swine database (Fu Y. et al., 2020) to prioritize the candidate genes in order to provide an understanding of the majority of candidate genes.
Results
Summary of phenotype and genotype data
In this research, we used a Landrace population consisting of 4,295 individuals with chip-level genotype data derived from a functional SNP lipid chip. Summary statistics, including the sample size, mean of phenotype values, standard deviation of phenotypes, and coefficient of variation, are provided in Table 1. Additionally, phenotype distribution plots are shown in Supplementary Figure 1. The mean values for BFT (100 kg) and FCR in our population are 10.22 and 2.3 with standard deviations of 1.99 and 0.22 and the coefficients of variation are 0.2 and 0.09, respectively. According to Table 1 and Supplementary Figure 1, both traits can be used for further analysis. Quality control was performed on the genomic data. After imputation and filtering, 100,235 SNPs on autosomes remained for the association study, excluding those with a minor allele frequency (MAF) less than 0.01. The marker density on each chromosome is shown in Figure 1A. LD decay analysis was also conducted using PopLDdecay (Zhang et al., 2019), and the results are shown in Figure 1B.

Table 1. Summary statistics of phenotypes.
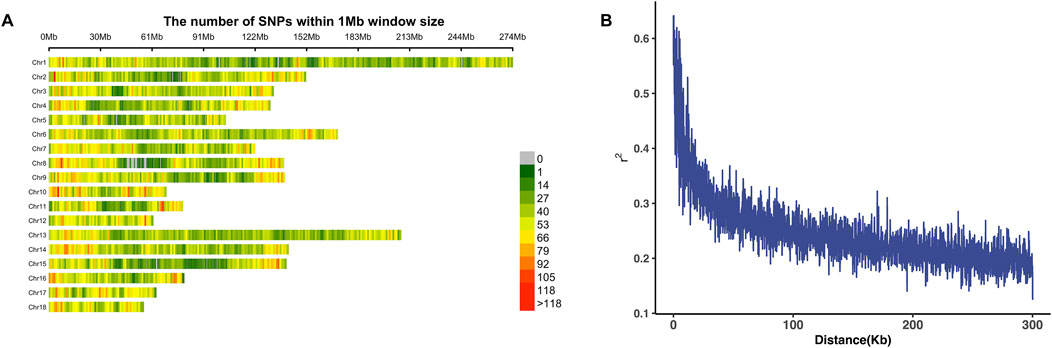
Figure 1. Density plot of genome variants and LD decay plot. (A) Genotype density plot of whole genome variants. The SNPs were counted using a 1-Mb window size, and the legend shows different colors representing the number of SNPs. (B) LD decay plot of the Landrace population.
Genome-wide association studies
Genome-wide association studies were performed through a mixed-effects linear model in the rMVP package (Yin et al., 2021). Sex, farms, and the first three principle components were included as fixed effects, whereas the additive genetic effects were considered the random effects variable. A Bonferroni cutoff of 0.05/N was used as a significant threshold, where N represents the number of SNPs. The details of quality control and genotype data are described in Methods. A total of 69 significant SNPs distributed on SSC1, SSC2, SSC7, and SSC12 were identified to be associated with backfat thickness. The details of these SNPs are shown in Supplementary Table 1. Manhattan and quantile–quantile (QQ) plots for BFT (100 kg) are shown in Figure 2, with a lambda value of 0.93 indicating minimal population inflation in GWAS. Based on the LD decay results, a distance of 300 kb was determined to define the candidate regions around each significant signal, and the candidate regions are shown in Table 2. Regions with overlapping areas were merged into one region, and a total of 244 genes were annotated within these candidate regions. After annotation, the genes were prioritized by a multi-omics database called ISwine (Fu Y. et al., 2020). Further details are provided in Supplementary Table 3.
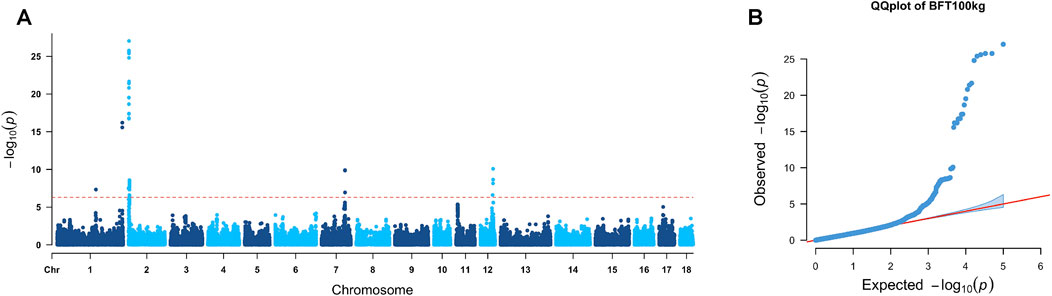
Figure 2. GWAS results of BFT (100 kg). (A) In the Manhattan plot of backfat thickness, the red line represents the Bonferroni cutoff, which was 0.05/N, and N represents the number of variants used in the analysis. (B) QQ plot for the GWAS of backfat thickness. The x-axis represents the expected
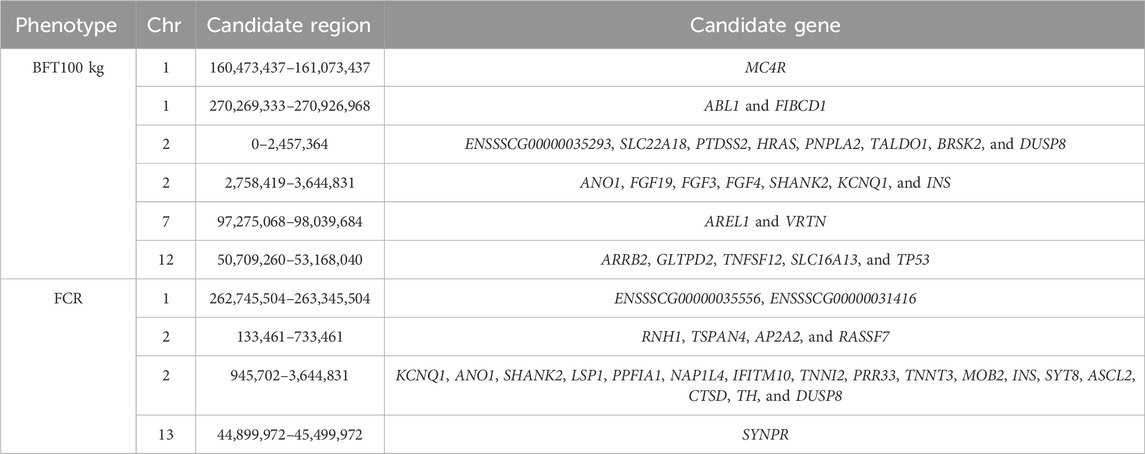
Table 2. Summary of the identified candidate regions and genes of phenotypes.
A total of 49 significant markers located on SSC1, SSC2, and SSC13, respectively, were found to be related to the FCR. Manhattan and QQ plots of FCR GWAS results are presented in Figure 3, where the lambda value was 0.96 for FCR GWAS results. The candidate regions and genes for the FCR are provided in Table 2, and the details of gene annotation in the candidate regions are displayed in Supplementary Table 4.
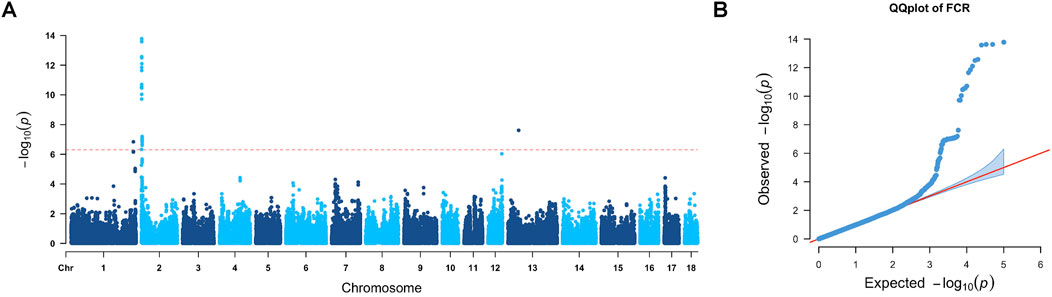
Figure 3. GWAS results of the FCR. (A) In the Manhattan plot of the feed conversion ratio, the red line represents the Bonferroni cutoff, which was 0.05/N, and N represents the number of variants used in the analysis. (B) QQ plot for the GWAS of the FCR. The x-axis represents the expected
Gene ontology annotation analysis and enrichment results
The candidate genes were annotated with Gene Ontology (GO) and the Kyoto Encyclopedia for Genes and Genomes (KEGG) database using IAnimal (Fu Y. et al., 2023). The details for the gene annotation results of BFT are shown in Supplementary Figure 2 and Supplementary Table 5. A total of 161 GO terms were significantly enriched, comprising 94 biological processes (BPs), 21 cellular components (CCs), and 46 molecular functions (MFs). Notably, several significant GO terms were associated with backfat thickness, such as positive regulation of insulin secretion (P = 0.019), intermembrane lipid transfer (P = 0.028), and lipid transfer activity (P = 0.041). Significant pathways, such as the MAPK signaling pathway (P =
Enrichment results for the FCR are shown in Supplementary Figure 3 and Supplementary Table 6. A total of 37 KEGG pathways were significantly enriched (P < 0.05). We also found 132 significant GO terms, including 94 BPs, 12 CCs, and 26 MFs. The positive regulation of insulin secretion, involved in the cellular response to glucose stimuli, is associated with nutrient absorption and energy metabolism, which were significantly enriched (P = 0.0057), and anoctamin 1 (ANO1) was involved in this process. Furthermore, some digestion-related pathways, such as protein digestion and absorption, were detected with suggestive P-values. Potassium voltage-gated channel subfamily Q member 1 (KCNQ1) was involved in this pathway and has been reported to be related to pig feed efficiency (Xiang et al., 2024).
Linkage disequilibrium block analysis
The LD blocks around the peak signals were analyzed and plotted using LDBlockShow (Dong et al., 2020). These plots are shown in Figure 4, Supplementary Figure 4, and Supplementary Figure 5. Figure 4 highlights the most significant regions of GWAS results for the two traits. Multiple LD blocks were observed around the top signals, with an overlapping region between BFT and FCR results, spanning from 2.76 to 3.64 Mb on SSC2, indicating that this region may have an influence on both FCR and BFT.
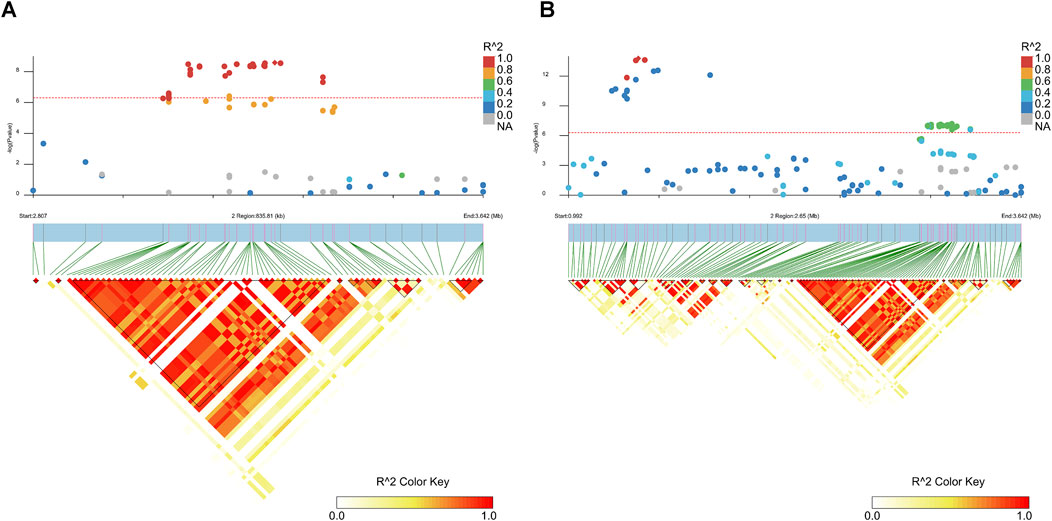
Figure 4. Linkage disequilibrium plot for SNPs within the most significant candidate regions. (A) Linkage disequilibrium plot for SNPs within Chr2: 2,758,419–3,644,831, which is the candidate region for BTF (100 kg). The red line represents the Bonferroni cutoff, which was 0.05/N, where N represents the number of variants used in the analysis. The legend on the right side represents the different R2 values of SNPs around the regions of peak SNP detection. (B) Linkage disequilibrium plot for SNPs within Chr2: 945,702–3,644,831, which is the candidate region for BTF (100 kg); the red line represents the Bonferroni cutoff, which was 0.05/N, where N represents the number of variants used in the analysis. The legend on the right side represents the different R2 values of SNPs around the regions of peak SNP detection.
Discussion
In our research, the GWAS approach was used to identify related genomic regions and candidate genes for backfat thickness and feed conversion ratio; a total of 4,295 Landraces were involved and analyzed. Our results showed 10 candidate regions on the genome for two commercial traits, and among the identified candidate genes, we found many reported genes that should be related to these two traits.
There were 46 overlapping signals between the GWAS results for BFT and FCR, suggesting that these variants may have influences on both traits. Some genes in these candidate regions were found to be related to both backfat thickness and feed conversion ratio. For example, as a member of the SHANK protein family, SHANK2 (SH3 and multiple ankyrin repeat domains 2) was reported to be associated with childhood obesity (Comuzzie et al., 2012). It was strongly highlighted as a candidate gene for backfat thickness in an association study based on imputed whole-genome data from a multi-breed population (Li J. et al., 2022). Interestingly, SHANK2 was found to be associated with average daily gain and the meat-to-fat ratio in pooled F2-designed pigs (Falker-Gieske et al., 2019). Since SHANK2 was also found as a candidate gene for the FCR, it may play an important role in both backfat thickness and feed conversion ratio.
The MAPK signaling pathway was significantly enriched in the KEGG analysis results for backfat thickness (P =
MC4R is a major gene influencing fatness in pigs. It is also involved in the regulation of feeding behavior and body weight in mice and humans. A missense mutation in this gene leads to increased fat accumulation in pigs (Ovilo et al., 2006). MC4R has also been shown to affect growth, feed intake, and backfat thickness in pigs, according to previous studies by Gozalo-Marcilla et al. (2021), Lee et al. (2020), Galve et al. (2012), and Piórkowska et al. (2010). We also found that AREL1 and VRTN on SSC7, which have previously been associated with body length, teat number, and intramuscular fat content (Hong et al., 2021; Park et al., 2023; Hirose et al., 2013; Yang et al., 2016), are also associated with meat production traits, thus influencing backfat.
Pigs are known for their outstanding olfactory abilities, which are attributed to their abundant functional olfactory receptors (Groenen et al., 2012). Odors affect pig reproduction and also have an influence on early food preferences (Brunjes et al., 2016). In the region of 262,745,504–263,345,504 on SSC1, we detected ENSSSCG00000035556 and ENSSSCG00000031416 to be associated with the FCR. These genes were enriched in an MF term of olfactory receptor activity (P = 0.015). In addition to these two genes, nine other genes (ENSSSCG00000036003, ENSSSCG00000032805, ENSSSCG00000032825, ENSSSCG00000027589, ENSSSCG00000037454, ENSSSCG00000027732, ENSSSCG00000031516, ENSSSCG00000026287, and ENSSSCG00000035439) were also enriched in this term and may be related. However, based on the prioritization results, ENSSSCG00000035556 and ENSSSCG00000031416 had higher scores, suggesting that these two genes may have a more significant impact on the FCR. The KEGG pathway analysis also identified significant enrichment in an olfactory-related pathway, namely, olfactory transduction (P = 0.046). ANO1 (anoctamin 1) was part of this pathway, along with 11 other genes enriched for olfactory receptor activity, as mentioned earlier. Olfactory receptors can perceive odor, transform biochemical signaling events into electrical impulses, and send them to the brain (Ma, 2007). This pathway was also found to be significantly enriched and related to residual feed intake (Do et al., 2014). Therefore, ANO1 may affect the FCR through olfactory-related pathways. ANO1 also affects fat deposition in pigs (Shi et al., 2022).
Feed efficiency can be largely influenced by energy metabolism and digestion. KCNQ1 (potassium voltage-gated channel subfamily Q member 1) was the most significantly related gene among these candidate genes. It plays an important role in fetal development in mice and humans, and it was reported to be associated with development. KCNQ1 is an imprinted gene expressed paternally in pigs (Wu et al., 2020) that influences nutrient absorption by regulating gastric acid secretion, as well as salt and glucose homeostasis (Sun and MacKinnon, 2020). It is also found to be involved in a related KEGG pathway that facilitates the digestion and absorption of proteins. In a recent multi-omics study, KCNQ1, along with SYT8, TNNI2, ASCL2, MOB2, DUSP8, TH, PRR33, TNNT3, IFITM10, CTSD, and INS, was identified as a candidate gene for the FCR in Large White boars (Xiang et al., 2024). These genes were also observed in our results. Interestingly, KCNQ1 was investigated in an association study as one of the candidate genes in the backfat thickness of pigs (Lee et al., 2018). The LSP1 gene has been reported to be associated with weight loss after dry-curing of hams (Faggion et al., 2024). Additionally, it can stimulate myogenic factors and influence skeletal muscle development in pigs (Albuquerque et al., 2021). Since skeletal muscle plays a key role in energy storage and consumption and is closely linked to energy metabolism, LSP1 may impact muscle development, thereby affecting pig growth and FCR. TSPAN4 and AP2A2 were also reported to be associated with growth in pigs with LSP1 (Faggion et al., 2024). In addition to LSP1, PPF1A1 was found to be differently expressed in the top three canonical pathways (Liu et al., 2015). In an epigenome-wide skeletal muscle study, NAP1L4 was detected to have CpG positions hypermethylated within its promoters (Ponsuksili et al., 2019). RASSF7 is a candidate gene for the FCR and a member of the N-terminal Ras association domain family. Studies have shown that knocking down RASSF7 restricts cell growth (Recino et al., 2010), and a deficiency in lysine negatively impacts the expression of RASSF7 (Wang et al., 2017). A mitochondrial protein, RNH1, was found to be associated with angiogenesis in porcine corpus luteum (Likszo et al., 2021). Synaptoporin (SYNPR) is one of the tetratransmembrane transport vesicle proteins, which is distributed in the digestive system (Liu et al., 2022). It has been found to be genome-wide associated with autoimmune hepatitis (AIH) in humans (Li Y. et al., 2022). We identified SYNPR as one of the significant candidate genes on SSC13. Therefore, it may affect feed efficiency through digestion.
In conclusion, we performed a GWAS based on a Landrace population to investigate two commercial traits. Our analysis identified 118 significant signals, from which 10 candidate regions were selected. Candidate genes within these regions were annotated and further analyzed using GO and KEGG pathways. Among the identified candidate genes, MC4R, SLC22A13, and INS were associated with backfat thickness, whereas ENSSSCG00000035556, SHANK2, KCNQ1, and LSP1 were related to the feed conversion ratio. Overall, our research provides deeper insights into the genetic basis of these traits and could inform future pig breeding efforts.
Materials and methods
Collection of Landrace population and phenotypes
A total of 4,295 Landrace individuals were used in this research, collected from three different great-grandparent farms of COFCO Joycome Foods Co., Ltd. All the pigs were raised under uniform feeding and management standards during the measurement period. Original records, including daily feed intake and weight for each pig, were automatically collected using the Pig Performance Testing System (Nedap, Groenlo, Netherlands). Outliers in these records were removed. The start and end dates of the test, along with the initial and final weights, weight gain, and total feed consumed during the test, were recorded. The FCR was then calculated as the total feed intake divided by the weight gain. The backfat thickness was measured using living B-ultrasonography at the end of the test, and the measured traits were then adjusted to a body weight of 100 kg. Details and distribution of phenotypes are shown in Table 1 and Supplementary Figure 1.
Genotyping, imputation, and quality control
Genomic DNA was extracted from ear tissue samples using the Tecan Freedom EVO NGS Workstation and the MagPure Tissue DNA KF Kit (MD5112-02), with a concentration of ≥40 ng/µL and a quantity of ≥1 µg. After that, the samples were genotyped using the Porcine 80K functional SNP genotyping chip by Wuhan Yingzi Gene Co., Ltd., using target capture sequencing technology. A total of 187,255 variants were included in the original genotype data. Quality control was performed using PLINK 1.90 (Purcell et al., 2007). Markers with a call rate <90% and MAF < 0.01 and variants on the sex chromosomes were excluded. This resulted in the retention of 100,240 variants, with a total genotyping rate of 0.99. Beagle 5.4 (Browning et al., 2018) was applied to impute genotype data. After imputation, quality control was performed again to remove markers with MAF < 0.01. A total of 100,235 SNPs and 4,295 individuals were left for further analysis at last.
Genome-wide association study
GWAS analysis was performed by fitting a mixed-effects linear model using the following equation in the rMVP (Yin et al., 2021) package:
where
Identification of candidate regions, genes, and enrichment analysis
LD decay analysis was performed to detect the size of the candidate region using PopLDdecay (Zhang et al., 2019). A window size of 300 kb was determined based on the LD decay results. Significant SNPs were then annotated to nearby genes within a 300 kb upstream or downstream range and prioritized using the multi-omics swine knowledgebase ISwine (Fu Y. et al., 2020). The candidate regions were uploaded in the section “Search by Region” on ISwine to obtain a candidate gene list. Then, we used “Prioritize” in the tool section to prioritize the candidate genes for each commercial trait and downloaded the results. Enrichment analyses, including GO and KEGG pathway analyses, were performed using enrichment tools from the IAnimal database (Fu Y. et al., 2023). The LD blocks around the top QTN plot were generated by LDBlockShow (Dong et al., 2020).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://figshare.com/, https://figshare.com/articles/dataset/genotype_data_of_Landrace_GWAS/27861177.
Ethics statement
The animal studies were approved by Breeding Department, Wuhan COFCO Meat Co., Ltd. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
GM: conceptualization, writing–review and editing, formal analysis, and resources. XT: formal analysis, data curation, and writing–review and editing. YY: writing–review and editing. TZ: data curation, formal analysis, and writing–review and editing. JW: data curation, formal analysis, and writing–review and editing. XC: data curation, formal analysis, and writing–review and editing. JX: conceptualization, investigation, methodology, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by STI2030-Major Projects [2023ZD0404707].
Acknowledgments
The authors would like to thank Shanxia Yang, Zijia Liu, and Jingya Zhang for the suggestions and help in this study.
Conflict of interest
Authors GM, XT, YY, TZ, JW, XC, and JX were employed by Wuhan COFCO Meat Co., Ltd.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1505197/full#supplementary-material
SUPPLEMENTARY TABLE 1 | Details of GWAS results for BFT.
SUPPLEMENTARY TABLE 2 | Details of GWAS results for FCR.
SUPPLEMENTARY TABLE 3 | Details of annotated genes for BFT.
SUPPLEMENTARY TABLE 4 | Details of annotated genes for FCR.
SUPPLEMENTARY TABLE 5 | Details of GO and KEGG results for BFT.
SUPPLEMENTARY TABLE 6 | Details of GO and KEGG results for FCR.
References
Abdellaoui, A., Yengo, L., Verweij, K. J., and Visscher, P. M. (2023). 15 years of GWAS discovery: realizing the promise. Am. J. Hum. Genet. 110, 179–194. doi:10.1016/j.ajhg.2022.12.011
Albuquerque, A., Óvilo, C., Núñez, Y., Benítez, R., López-Garcia, A., García, F., et al. (2021). Transcriptomic profiling of skeletal muscle reveals candidate genes influencing muscle growth and associated lipid composition in Portuguese local pig breeds. Animals 11, 1423. doi:10.3390/ani11051423
Aouadi, M., Laurent, K., Prot, M., Le Marchand-Brustel, Y., Binétruy, B., and Bost, F. (2006). Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes 55, 281–289. doi:10.2337/diabetes.55.02.06.db05-0963
Blaj, I., Tetens, J., Preuß, S., Bennewitz, J., and Thaller, G. (2018). Genome-wide association studies and meta-analysis uncovers new candidate genes for growth and carcass traits in pigs. PloS one 13, e0205576. doi:10.1371/journal.pone.0205576
Browning, B. L., Zhou, Y., and Browning, S. R. (2018). A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348. doi:10.1016/j.ajhg.2018.07.015
Brunjes, P. C., Feldman, S., and Osterberg, S. K. (2016). The pig olfactory brain: a primer. Chem. senses 41, 415–425. doi:10.1093/chemse/bjw016
Comuzzie, A. G., Cole, S. A., Laston, S. L., Voruganti, V. S., Haack, K., Gibbs, R. A., et al. (2012). Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PloS one 7, e51954. doi:10.1371/journal.pone.0051954
Delpuech, E., Aliakbari, A., Labrune, Y., Fève, K., Billon, Y., Gilbert, H., et al. (2021). Identification of genomic regions affecting production traits in pigs divergently selected for feed efficiency. Genet. Sel. Evol. 53, 49. doi:10.1186/s12711-021-00642-1
Ding, R., Zhuang, Z., Qiu, Y., Ruan, D., Wu, J., Ye, J., et al. (2022). Identify known and novel candidate genes associated with backfat thickness in Duroc pigs by large-scale genome-wide association analysis. J. Animal Sci. 100, skac012. doi:10.1093/jas/skac012
Do, D. N., Strathe, A. B., Ostersen, T., Pant, S. D., and Kadarmideen, H. N. (2014). Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front. Genet. 5, 307. doi:10.3389/fgene.2014.00307
Dong, S.-S., He, W.-M., Ji, J.-J., Zhang, C., Guo, Y., and Yang, T.-L. (2020). LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Briefings Bioinforma. 22, bbaa227. doi:10.1093/bib/bbaa227
Faggion, S., Bonfatti, V., and Carnier, P. (2024). Genome-wide association study for weight loss at the end of dry-curing of hams produced from purebred heavy pigs. Animals 14, 1983. doi:10.3390/ani14131983
Falker-Gieske, C., Blaj, I., Preuß, S., Bennewitz, J., Thaller, G., and Tetens, J. (2019). GWAS for meat and carcass traits using imputed sequence level genotypes in pooled F2-designs in pigs. G3 Genes, Genomes, Genet. 9, 2823–2834. doi:10.1534/g3.119.400452
Fu, L., Jiang, Y., Wang, C., Mei, M., Zhou, Z., Jiang, Y., et al. (2020a). A genome-wide association study on feed efficiency related traits in Landrace pigs. Front. Genet. 11, 692. doi:10.3389/fgene.2020.00692
Fu, M., Zhou, X., Liu, Z., Wang, T., and Liu, B. (2023a). Genome-wide association study reveals a genomic region on SSC7 simultaneously associated with backfat thickness, skin thickness and carcass length in a Large White× Tongcheng advanced generation intercross resource population. Anim. Genet. 54, 216–219. doi:10.1111/age.13285
Fu, Y., Liu, H., Dou, J., Wang, Y., Liao, Y., Huang, X., et al. (2023b). IAnimal: a cross-species omics knowledgebase for animals. Nucleic Acids Res. 51, D1312–D1324. doi:10.1093/nar/gkac936
Fu, Y., Xu, J., Tang, Z., Wang, L., Yin, D., Fan, Y., et al. (2020b). A gene prioritization method based on a swine multi-omics knowledgebase and a deep learning model. Commun. Biol. 3, 502. doi:10.1038/s42003-020-01233-4
Galve, A., Burgos, C., Silió, L., Varona, L., Rodríguez, C., Ovilo, C., et al. (2012). The effects of leptin receptor (LEPR) and melanocortin-4 receptor (MC4R) polymorphisms on fat content, fat distribution and fat composition in a Duroc× Landrace/Large White cross. Livest. Sci. 145, 145–152. doi:10.1016/j.livsci.2012.01.010
Gozalo-Marcilla, M., Buntjer, J., Johnsson, M., Batista, L., Diez, F., Werner, C. R., et al. (2021). Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genet. Sel. Evol. 53, 76. doi:10.1186/s12711-021-00671-w
Groenen, M. A., Archibald, A. L., Uenishi, H., Tuggle, C. K., Takeuchi, Y., Rothschild, M. F., et al. (2012). Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398. doi:10.1038/nature11622
Hirose, K., Mikawa, S., Okumura, N., Noguchi, G., Fukawa, K., Kanaya, N., et al. (2013). Association of swine vertnin (VRTN) gene with production traits in D uroc pigs improved using a closed nucleus breeding system. Animal Sci. J. 84, 213–221. doi:10.1111/j.1740-0929.2012.01066.x
Hoa, V. B., Seo, H. W., Seong, P. N., Cho, S. H., Kang, S. M., Kim, Y. S., et al. (2021). Back-fat thickness as a primary index reflecting the yield and overall acceptance of pork meat. Animal Sci. J. 92, e13515. doi:10.1111/asj.13515
Hollie, N. I., and Hui, D. Y. (2011). Group 1B phospholipase A₂ deficiency protects against diet-induced hyperlipidemia in mice. J. lipid Res. 52, 2005–2011. doi:10.1194/jlr.M019463
Hong, Y., Ye, J., Dong, L., Li, Y., Yan, L., Cai, G., et al. (2021). Genome-wide association study for body length, body height, and total teat number in large white pigs. Front. Genet. 12, 650370. doi:10.3389/fgene.2021.650370
Hu, Z.-L., Dracheva, S., Jang, W., Maglott, D., Bastiaansen, J., Rothschild, M. F., et al. (2005). A QTL resource and comparison tool for pigs: PigQTLDB. Mamm. Genome 16, 792–800. doi:10.1007/s00335-005-0060-9
Knol, E. F., Nielsen, B., and Knap, P. W. (2016). Genomic selection in commercial pig breeding. Anim. Front. 6, 15–22. doi:10.2527/af.2016-0003
Lee, J., Kim, Y., Cho, E., Cho, K., Sa, S., Kim, Y., et al. (2020). Genomic analysis using Bayesian methods under different genotyping platforms in Korean Duroc pigs. Animals 10, 752. doi:10.3390/ani10050752
Lee, J.-B., Kang, H.-C., Kim, E.-H., Kim, Y.-J., Yoo, C.-K., Choi, T.-J., et al. (2018). Genome-wide association study identifies positional candidate genes affecting back fat thickness trait in pigs. Korean J. Agric. Sci. 45, 707–713. doi:10.7744/kjoas.20180055
Li, D., Huang, M., Zhuang, Z., Ding, R., Gu, T., Hong, L., et al. (2021). Genomic analyses revealed the genetic difference and potential selection genes of growth traits in two duroc lines. Front. Veterinary Sci. 8, 725367. doi:10.3389/fvets.2021.725367
Li, J., Wu, J., Jian, Y., Zhuang, Z., Qiu, Y., Huang, R., et al. (2022b). Genome-wide association studies revealed significant QTLs and candidate genes associated with backfat and loin muscle area in pigs using imputation-based whole genome sequencing data. Animals 12, 2911. doi:10.3390/ani12212911
Li, W., Wang, Z., Luo, S., Wu, J., Zhou, L., and Liu, J. (2022a). Genome-wide association analysis and genetic parameters for feed efficiency and related traits in Yorkshire and Duroc pigs. Animals 12, 1902. doi:10.3390/ani12151902
Li, Y., Sun, Y., Liu, Y., Wang, B., Li, J., Wang, H., et al. (2022c). Genome-wide meta-analysis identifies susceptibility loci for autoimmune hepatitis type 1. Hepatology 76, 564–575. doi:10.1002/hep.32417
Likszo, P., Skarzynski, D. J., and Moza Jalali, B. (2021). Changes in porcine corpus luteum proteome associated with development, maintenance, regression, and rescue during estrous cycle and early pregnancy. Int. J. Mol. Sci. 22, 11740. doi:10.3390/ijms222111740
Liu, F., Brewster, C. J., Gilmour, S. L., Henman, D. J., Smits, R. J., Luxford, B. G., et al. (2021). Relationship between energy intake and growth performance and body composition in pigs selected for low backfat thickness. J. Animal Sci. 99, skab342. doi:10.1093/jas/skab342
Liu, L., Yao, X., Wang, Y., Hu, R., Fan, C., Gong, H., et al. (2022). Physins in digestive system neoplasms. Adv. Clin. Chem. 111, 157–176. doi:10.1016/bs.acc.2022.08.002
Liu, X., Du, Y., Trakooljul, N., Brand, B., Muráni, E., Krischek, C., et al. (2015). Muscle transcriptional profile based on muscle fiber, mitochondrial respiratory activity, and metabolic enzymes. Int. J. Biol. Sci. 11, 1348–1362. doi:10.7150/ijbs.13132
Ma, H., Zhang, S., Zhang, K., Zhan, H., Peng, X., Xie, S., et al. (2019). Identifying selection signatures for backfat thickness in Yorkshire pigs highlights new regions affecting fat metabolism. Genes 10, 254. doi:10.3390/genes10040254
Ma, M. (2007). Encoding olfactory signals via multiple chemosensory systems. Crit. Rev. Biochem. Mol. Biol. 42, 463–480. doi:10.1080/10409230701693359
Mehrabi, Z., Gill, M., Wijk, M. v., Herrero, M., and Ramankutty, N. (2020). Livestock policy for sustainable development. Nat. Food 1, 160–165. doi:10.1038/s43016-020-0042-9
Miao, Y., Zhao, Y., Wan, S., Mei, Q., Wang, H., Fu, C., et al. (2023). Integrated analysis of genome-wide association studies and 3D epigenomic characteristics reveal the BMP2 gene regulating loin muscle depth in Yorkshire pigs. PLoS Genet. 19, e1010820. doi:10.1371/journal.pgen.1010820
Nosková, A., Mehrotra, A., Kadri, N., Neuenschwander, S., Hofer, A., and Pausch, H. (2022). Detecting QTL for two lowly correlated traits using multi-trait and meta-analysis approaches in Swiss Large White pigs. Proc. 12th World Congr. Genet. Appl. Livest. Prod. (WCGALP) Tech. species orientated innovations animal Breed. contribution Genet. solving Soc. challenges, 1630–1633.
Ovilo, C., Fernández, A., Rodríguez, M., Nieto, M., and Silió, L. (2006). Association of MC4R gene variants with growth, fatness, carcass composition and meat and fat quality traits in heavy pigs. Meat Sci. 73, 42–47. doi:10.1016/j.meatsci.2005.10.016
Park, J. (2024). Genome-wide association study to reveal new candidate genes using single-step approaches for productive traits of Yorkshire pig in Korea. Anim. Biosci. 37, 451–460. doi:10.5713/ab.23.0255
Park, J., Do, K. T., Park, K. D., and Lee, H. K. (2023). Genome-wide association study using a single-step approach for teat number in Duroc, Landrace and Yorkshire pigs in Korea. Anim. Genet. 54, 743–751. doi:10.1111/age.13357
Piórkowska, K., Tyra, M., Rogoz, M., Ropka-Molik, K., Oczkowicz, M., and Różycki, M. (2010). Association of the melanocortin-4 receptor (MC4R) with feed intake, growth, fatness and carcass composition in pigs raised in Poland. Meat Sci. 85, 297–301. doi:10.1016/j.meatsci.2010.01.017
Ponsuksili, S., Trakooljul, N., Basavaraj, S., Hadlich, F., Murani, E., and Wimmers, K. (2019). Epigenome-wide skeletal muscle DNA methylation profiles at the background of distinct metabolic types and ryanodine receptor variation in pigs. BMC Genomics 20, 492. doi:10.1186/s12864-019-5880-1
Pulit, S. L., Stoneman, C., Morris, A. P., Wood, A. R., Glastonbury, C. A., Tyrrell, J., et al. (2019). Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 28, 166–174. doi:10.1093/hmg/ddy327
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi:10.1086/519795
Recino, A., Sherwood, V., Flaxman, A., Cooper, W. N., Latif, F., Ward, A., et al. (2010). Human RASSF7 regulates the microtubule cytoskeleton and is required for spindle formation, Aurora B activation and chromosomal congression during mitosis. Biochem. J. 430, 207–213. doi:10.1042/BJ20100883
Saltiel, A. R., and Kahn, C. R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806. doi:10.1038/414799a
Schumann, T., König, J., von Loeffelholz, C., Vatner, D. F., Zhang, D., Perry, R. J., et al. (2021). Deletion of the diabetes candidate gene Slc16a13 in mice attenuates diet-induced ectopic lipid accumulation and insulin resistance. Commun. Biol. 4, 826. doi:10.1038/s42003-021-02279-8
Shi, L., Wang, L., Fang, L., Li, M., Tian, J., Wang, L., et al. (2022). Integrating genome-wide association studies and population genomics analysis reveals the genetic architecture of growth and backfat traits in pigs. Front. Genet. 13, 1078696. doi:10.3389/fgene.2022.1078696
Sun, J., and MacKinnon, R. (2020). Structural basis of human KCNQ1 modulation and gating. Cell 180, 340–347. doi:10.1016/j.cell.2019.12.003
VanRaden, P. M. (2008). Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423. doi:10.3168/jds.2007-0980
Wang, H., Wang, X., Yan, D., Sun, H., Chen, Q., Li, M., et al. (2022). Genome-wide association study identifying genetic variants associated with carcass backfat thickness, lean percentage and fat percentage in a four-way crossbred pig population using SLAF-seq technology. BMC Genomics 23, 594. doi:10.1186/s12864-022-08827-8
Wang, T., Feugang, J. M., Crenshaw, M. A., Regmi, N., Blanton, J. R., and Liao, S. F. (2017). A systems biology approach using transcriptomic data reveals genes and pathways in porcine skeletal muscle affected by dietary lysine. Int. J. Mol. Sci. 18, 885. doi:10.3390/ijms18040885
Wang, X., Sun, B., Jiang, Q., Wu, R., Cai, M., Yao, Y., et al. (2018). mRNA m6A plays opposite role in regulating UCP2 and PNPLA2 protein expression in adipocytes. Int. J. Obes. 42, 1912–1924. doi:10.1038/s41366-018-0027-z
Wu, Y.-Q., Zhao, H., Li, Y.-J., Khederzadeh, S., Wei, H.-J., Zhou, Z.-Y., et al. (2020). Genome-wide identification of imprinted genes in pigs and their different imprinting status compared with other mammals. Zoological Res. 41, 721–725. doi:10.24272/j.issn.2095-8137.2020.072
Xiang, Y., Sun, J., Ma, G., Dai, X., Meng, Y., Fu, C., et al. (2024). Integrating multi-omics data to identify key functional variants affecting feed efficiency in large white boars. Genes 15, 980. doi:10.3390/genes15080980
Yamamoto, T., Iizuka, Y., Izumi-Yamamoto, K., Shirota, M., Mori, N., Tahara, Y., et al. (2024). Overexpression of Slc22a18 facilitates fat accumulation in mice. Biochem. Biophysical Res. Commun. 712, 149922. doi:10.1016/j.bbrc.2024.149922
Yang, J., Huang, L., Yang, M., Fan, Y., Li, L., Fang, S., et al. (2016). Possible introgression of the VRTN mutation increasing vertebral number, carcass length and teat number from Chinese pigs into European pigs. Sci. Rep. 6, 19240. doi:10.1038/srep19240
Yin, L., Zhang, H., Tang, Z., Xu, J., Yin, D., Zhang, Z., et al. (2021). rMVP: a memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genomics Proteomics Bioinforma. 19, 619–628. doi:10.1016/j.gpb.2020.10.007
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M., and Yang, T. L. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. doi:10.1093/bioinformatics/bty875
Zhang, D., Wu, W., Huang, X., Xu, K., Zheng, C., and Zhang, J. (2021). Comparative analysis of gene expression profiles in differentiated subcutaneous adipocytes between Jiaxing Black and Large White pigs. BMC genomics 22, 61–13. doi:10.1186/s12864-020-07361-9
Keywords: pigs, feed efficiency, backfat thickness, GWAS, candidate genes
Citation: Ma G, Tan X, Yan Y, Zhang T, Wang J, Chen X and Xu J (2025) A genome-wide association study identified candidate regions and genes for commercial traits in a Landrace population. Front. Genet. 15:1505197. doi: 10.3389/fgene.2024.1505197
Received: 02 October 2024; Accepted: 09 December 2024;
Published: 06 January 2025.
Edited by:
Xiao-Lin Wu, Council on Dairy Cattle Breeding, United StatesCopyright © 2025 Ma, Tan, Yan, Zhang, Wang, Chen and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingya Xu, eHVqaW5neWFAY29mY28uY29t