 Xiaoming Zhao1
Xiaoming Zhao1 Zhonghua Yang
Zhonghua Yang- 1Department of Pediatric Orthopaedics, Shenyang Orthopaedic Hospital, Shenyang, China
- 2College of Police Dog Technology, Criminal Investigation Police University of China, Shenyang, China
- 3Department of Pediatric Surgery, Shengjing Hospital of China Medical University, Shenyang, China
- 4Key Laboratory of Health Ministry for Congenital Malformation, Shengjing Hospital of China Medical University, Shenyang, China
The most prevalent hip disease in neonates is developmental dysplasia of the hip (DDH). A timely and accurate diagnosis is required to provide the most effective treatment for pediatric patients with DDH. Heredity and gene variation have been the subject of increased attention and research worldwide as one of the factors contributing to the pathogenesis of DDH. Genome-wide association studies (GWAS), genome-wide linkage analyses (GWLA), and exome sequencing (ES) have identified variants in numerous genes and single-nucleotide polymorphisms (SNPs) as being associated with susceptibility to DDH in sporadic and DDH family patients. Furthermore, the DDH phenotype can be observed in animal models that exhibit susceptibility genes or loci, including variants in CX3CR1, KANSL1, and GDF5. The dentification of noncoding RNAs and de novo gene variants in patients with DDH-related syndrome has enhanced our understanding of the genes implicated in DDH. This article reviews the most recent molecular mechanisms and genetic factors that contribute to DDH.
Highlights
• Heredity and gene variation was one of the factors leading to the pathogenesis of DDH.
• Variants in coding and non-coding RNAs were related to the pathogenesis of DDH.
• The review on the genetic factors of DDH help to understand DDH-related genetic information contributing to the early screening and diagnosis.
Introduction
Developmental dysplasia of the hip (DDH) is a disorder characterized by an abnormal relationship between the femoral head and the acetabulum. Signs of DDH include luxation, subluxation, femoral head instability causing it to migrate in and out of the acetabulum, and multiple radiographic abnormalities reflecting inadequate acetabular formation. DDH is the most prevalent congenital defect in newborns, with an estimated incidence of 3–6/1,000 (Loder and Shafer, 2015). DDH alters hip biomechanics and overloads the articular cartilage, leading to early osteoarthritis. Early DDH detection allows for a simpler and more effective treatment. The Pavlik harness is usually the first-line treatment for infants younger than 6 months with DDH. If the DDH diagnosis is delayed, a pelvic or femoral osteotomy must be conducted within the next few years. However, this procedure can lead to lower limb discrepancy, gait instability, postural scoliosis, hip flexion contracture, gait abnormalities and ipsilateral permanent disabilities such as valgus knee (Yang et al., 2019). DDH is the leading cause of total hip replacement in younger populations, with an estimated incidence of 21%–29% (Vaquero-Picado et al., 2019). Therefore, early diagnosis is crucial for treating DDH to achieve a favorable clinical prognosis. Studies have demonstrated that the etiology of DDH is related to risk factors including maternal estrogen and progesterone imbalance, oligohydramnios, breech presentation during delivery, multiple pregnancy, clubfoot, female gender, ligament relaxation surgery, left hip joint, swaddling with lower extremities wrapped tightly together, macrosomia, and high altitude (Woodacre et al., 2016; Zhao et al., 2019). Of note, DDH is also considered to be a familial aggregation disease and typically affects multiple individuals within a family. The risk of developing DDH increased by 12 times in first-degree relatives of DDH patients (Gkiatas et al., 2019), while the risk of developing DDH in second-degree relatives is only 1.7 times higher than in the general population. According to the American Academy of Orthopedic Surgeons guidelines, breech position, family history, and symptoms of hip instability are risk factors that warrant ultrasound-based screening for DDH in infants (Schaeffer et al., 2018). Ultrasound-based screening has been implemented for infants with risk factors for DDH worldwide. DDH screening in pregnancy is exclusively dependent on ultrasound during the middle and third trimesters, as there is no evidence of hip dysplasia in fetuses aborted before 20 weeks of gestation (Dunn, 1976). This indicates that most changes in DDH are detected in the later stages of the intrauterine period. Therefore, the diagnosis and prediction of DDH during pregnancy are of significant importance, especially in the early stages of pregnancy. Given the familial susceptibility of DDH, understanding the genes underlying the pathogenesis of DDH and conducting screening for these genes during pregnancy will significantly enhance the early diagnosis and treatment of DDH.
DDH demonstrates a diverse array of signs and symptoms, ranging from mild ligament relaxation, subluxation, and complete dislocation of the femoral head to deformities characterized by various forms of systemic skeletal dysplasia or other systemic genetic abnormalities that may involve chromosomal, genetic, and epigenetic changes (Kenanidis et al., 2020). Recently, genetic studies have increasingly employed exome sequencing (ES), whole ES (WES), candidate gene association studies, genome-wide association studies (GWAS), and genome-wide linkage analyses (GWLA) due to their higher output and reliability. Advances in molecular analysis and sequencing technology have enabled researchers to conduct an in-depth study of familial inheritance in DDH. Several chromosomal loci that are closely associated with unilateral or bilateral hip dysplasia, such as CX3CR1, GDF5, and HOX, were identified through familial aggregation studies of multiple DDH patients (Feldman et al., 2010; Feldman et al., 2017; Kenanidis et al., 2020) (Table 1).

Table 1. Gene variants of developmental dysplasia of the hip (DDH) detected in family study.
Additionally, the pathogenic potential of family pedigree-screened variants of enhanced susceptibility, including TXNDC3, HOXB9, HSPG2, and ATP2B4, has been investigated in a disseminated population through a case-control study (Rouault et al., 2009; Qiao et al., 2017; Xu et al., 2021) (Table 2). Besides, since more and more reports show that DDH is one of the symptoms of the syndrome such as trisomy 21 (Bennet et al., 1982), 18q deletion syndrome (Yu et al., 2022), and Steel syndrome (Frigola et al., 2021), it is particularly necessary to summarize the results of genetic variants for these syndromes, which has demonstrated phenotypic variability and genetic heterogeneity (Yan et al., 2019) (Table 3). These evidences revealed that a wide range of genetic variants during embryonic development can lead to syndrome-associated DDH. In addition, it has recently been recognized that epigenetic modifications such as RNA methylation may also play an important role in the pathogenesis of DDH (Harsanyi et al., 2020). Moreover, due to the similarities between human and animal models (Doncheva et al., 2021), studies on the identification of DDH susceptibility gene variants are promising in animal models, including pigs, rats, mice, and rabbits. This research has the potential to provide novel insights into the pathogenesis of DDH through epigenetic analyses in vivo experiments (Kiapour et al., 2018; Ji et al., 2020; Wu et al., 2020; Liu et al., 2021). Therefore, an increasing number of DDH-related genes have been identified. In this paper, we systematically reviewed DDH-related genes and their functions to better understand DDH-related genetic data.

Table 2. Gene variants of developmental dysplasia of the hip (DDH) in different populations.

Table 3. Gene variants of developmental dysplasia of the hip (DDH) detected in syndrome.
Genes related to DDH pathogenesis
Asporin
Asporin (ASPN) gene is located on chromosome 9q22.31 and encodes extracellular matrix (ECM) protein that belongs to the small leucine-rich proteoglycan (SLRP) protein family. Members of the SLRP family are able to bind to other structural components of ECMs, such as collagen, as well as members of the transforming growth factor-beta (TGFβ) superfamily that temporarily reside in ECMs. A study of 370 Chinese patients with DDH showed that the protein encoded by ASPN in DDH patients had a higher 14-aspartic acid repeat sequence (ASPN D14) in the N-terminal region, indicating that ASPN D14 was positively correlated with DDH. The frequency of D13, a common allele encoding 13 aspartic acid repeats, was significantly reduced, suggesting that ASPN D13 may have a protective effect (Shi et al., 2011). In addition, copy number variation (CNV) analysis showed that copy number loss of ASPN in the 60 kb region of 9q22.31 was found in 9 of 64 patients with DDH (9/64), which was significantly higher than that in the control group (0/32). Given that deletion of copy number in ASPN is associated with severe DDH, all of these 9 patients required surgery (Sekimoto et al., 2017). Previous studies have shown that the ASPN D14 allele is a risk gene for knee osteoarthritis (OA) and the D13 allele is a protective genetic factor for OA, and recent studies have shown that the ASPN repeat polymorphism is associated with OA, lumbar disc degeneration (LDD), and DDH. Therefore, these results contribute to our understanding of ASPN on the risk of DDH genetics (Loughlin, 2011).
Bone morphogenetic proteins
Bone morphogenetic proteins (BMPs) are extracted from bone tissue, and more than 40 proteins of the BMP family have been identified. BMP2 has been widely studied for its important role in embryonic development and postnatal organ development, which can induce mesenchymal stem cells (MSCs) to osteogenesis and differentiation into chondrocytes (Bauge et al., 2014). In cell studies of DDH, proliferation of osteoblast cells was accompanied by upregulation of BMP2 expression (Xu J. et al., 2022). In a recent study, BMP2 was used for the treatment of DDH. Specifically, poly-ethylene glycol-lactic acid (PELA) electrospun fibrous scaffolds transplanted with BMP2 were used to repair the acetabular defect of the micropig. Samples taken after 24 weeks showed the appearance of large quantities of chondrocytes and the formation of part of new bone tissue, suggesting that the novel material with BMP2 component has the potential to treat DDH (Wu et al., 2020). However, in a population from Turkey (68 cases, 100 controls), no significant association between BMP2 (rs235768) polymorphism and DDH was found (Gumus et al., 2021).
BMP2-inducible kinase (BMP2K) is located on chromosome 4q21.21, belongs to the transforming growth factor-β superfamily. The protein encoded by BMP2K is a serine/threonine protein kinase with a nuclear localization signal, and its expression level is increased during BMP2-induced differentiation of mouse osteoblast cell lines and is critical for bone development and osteoblast differentiation. Thus, BMP2K variants may affect hip joint development (Zhao et al., 2017). WES was performed on three affected individuals and two unaffected individuals from four generations of the DDH family. Two new heterozygous mutations (c.1432_1440delCAGCAGCAG corresponding to p.Gln478_480del and c.1440_1441insCAG corresponding to p.Gln480ins) in exon 11 of BMP2K were found in three affected individuals and their unaffected grandmother, suggesting that the BMP2K variant may be a cause of DDH(31).
Genes of collagen
Collagen, the main component of connective tissue, is a marker of early cartilage degeneration and reflects the mechanical properties of the hip joint (Li et al., 2016). A case study of a 2-year-old girl with DDH reported that intraarticular cartilage tissue was found semi-free during surgery and assumed to originate from the femoral head. This accidental discovery prompted the investigators to conduct histopathological examination of the cartilage tissue. They found that cartilage tissue is composed of collagen fibers and chondroid tissue and expresses Collagen Type I (Chen et al., 2018). Collagen Type I is composed of Collagen Type I Alpha 1 Chain (COL1A1) and Collagen Type I Alpha 2 Chain (COL1A2) proteins encoded by the type I collagen gene. Autosomal dominant mutation in the type I collagen gene can cause osteogenesis imperfecta (Marini et al., 2017). COL1A1 is located on 17q21.3-q22.1 and encodes the a1 chain of type I collagen, which is also located in the 4 mb region of chromosome 17q21.32 associated with DDH reported by Feldman et al. (Feldman et al., 2010). Although a case-control study in a Caucasian population (239 cases and 239 controls) from western Brittany, France showed that 10 SNPs of COL1A1 have no association with DDH (Rouault et al., 2009), Chinese scholars detected three variations in the promoter region of COL1A1 by comparing 154 female DDH cases with 180 control cases, and believed that DDH of Chinese female is associated with a higher overall variation rate of COL1A1 gene (Zhao et al., 2013a). Bioinformatic analysis in a recent study conducted on 16 susceptibility genes of DDH showed that ASPN, TGFβ1, dickkopf WNT signaling pathway inhibitor 1 (DKK1), Interleukin 6 gene (IL-6), Teneurin transmembrane protein 3 gene (TENM3) and Growth differentiation factor 5 (GDF5) were significantly co-expressed with COL1A1 (Yang et al., 2022). As key regulatory genes influencing the formation of collagen fibers, COL1A1 and Collagen TypeIII Alpha1 Chain (COL3A1) were found to be downregulated by comparing the hip joint capsule of DDH and the control tissue using high-throughput sequencing, which was suspected to be one of the factors leading to joint capsule relaxation due to the loss of collagen fibers and fibroblasts (Yang et al., 2022). In addition, in a study of 12 patients with Arthrochalasia Ehlers-Danlos syndrome (aEDS), the majority (10/12) of patients had total or partial loss of COL1A1 or COL1A2 exon 6, five of whom underwent hip joint surgery. This finding suggested that COL1A1 exon loss may be associated with DDH formation (Ayoub et al., 2020). Fluoride intake was found to affect the degree of hip capsule relaxation. In rat animal models of DDH combined toxicity of fluoride, the incidence of DDH increased with increasing fluoride concentration, showing significantly downregulated COL1A1 and upregulated COL3A1 in hip capsule and fibroblast, accompanied by increased oxidative stress and apoptosis levels (Zhou et al., 2022).
Collagen Type II is mainly produced by chondrocytes and provides osmotic function and mechanical support to articular cartilage against compressive stress (Feng et al., 2017). Collagen Type II has been reported to be significantly upregulated in the acetabulum of rabbit DDH models (Zhang et al., 2015). The mRNA level of Collagen type II was significantly upregulated in acetabular cartilage of DDH models established in the swaddling position of Wistar rats (Ning et al., 2018). However, other studies compared Collagen expression in acetabular cartilage of patients with DDH and OA and found that Collagen Type II mRNA level was significantly decreased in DDH, indicating dysregulation of Collagen Type II in cartilage lesions (Feng et al., 2017). Collagen Type II Alpha 1 Chain (COL2A1) is located on chromosome 12, and the encoded protein is the most abundant structural protein in cartilage. Mutations in the alpha-1 chain are associated with the onset of rare familial OA (Palotie et al., 1989). Sequencing in two patients has reported that the heterozygous missense mutation of COL2A1 is associated with the Spondylometaphyseal dysplasia (SMD) corner fracture type (Machol et al., 2017). In addition, a heterozygous mutation of COL2A1 (c.2014G>T; p.(Gly672Cys)) was found by WES in a multigenerational family with progressive hip disease and mild SMD from the Namaqualand region of northwestern South Africa, which was also confirmed in 23 family members with DDH (Agenbag et al., 2020). Donatella Granchi et al. studied 50 healthy subjects and 143 patients undergoing total hip replacement for idiopathic OA or DDH-associated OA with PvuII restriction endonuclease and found that the COL2A1 allele was significantly correlated with DDH-associated OA. The probability of COL2A1 containing at least one P allele is increased by 3 times (95% confidence interval (CI), 1.120–8.730) (Agenbag et al., 2020). Since COL2A1 and vitamin D receptor (VDR) are adjacent genes at 12q13 that play a role in chondromorphism and bone mineralization, Granchi et al. reported that SNPs of COL2A1 and VDR genes were associated with DDH-associated OA in the Italian population (Granchi et al., 2002), but SNPs of COL2A1 and VDR genes did not appear to play a role in the association with non-syndromic DDH (Rubini et al., 2008).
Collagen type X was significantly higher in DDH than in the control group (Bo et al., 2012; Ning et al., 2018). A study compared the expression of Collagen in acetabular cartilage of patients with DDH and OA, and found that the mRNA level of Collagen Type II was significantly decreased in DDH, while that of Collagen Type X was significantly decreased. It is suggested that the imbalance of collagen may be related to DDH chondropathy (Feng et al., 2017). In addition, a domestic rabbit DDH model was established by inserting a meniscus into the hip joint to simulate limbus inersion after DDH closed reduction, and the acetabular index and Collagen Type X expression were significantly increased in this DDH model. These results suggest that limbus inversion can accelerate cartilage degeneration in DDH and lead to early OA (Liu et al., 2021).
Collagen type XI alpha 2 chain (COL11A2) is located on 6p21.32 and the encoded protein is indispensably involved in collagen formation and extracellular matrix construction in cartilage and bone. Collagen type XI and Collagen type II together constitute the main structure of articular cartilage, suggesting that COL11A1 may play a role in the development of DDH. A case-control candidate gene association study was conducted in 945 Han patients (350 patients with imaging diagnosis of DDH and 595 healthy controls). The expression of COL11A2 in DDH patients was significantly lower than that in the control group. In addition, the number of DDH patients with rs9277935TT genotype was significantly higher than that with GG genotype. The chondrogenic effect of COL11A2 was further verified in cell experiments, suggesting that chondrogenesis inhibited by COL11A2 rs9277935 variant may be a pathogenic effect of DDH (Xu et al., 2020).
Collagen type XXVII alpha 1 chain (COL27A1) is located on 9q32, encodes Collagen type XXVII alpha 1 chain and provides support for connective tissue structure, which is expressed in cartilage in the process of endochondral ossification (Mayo et al., 2009). Two consecutive infants of a closely related couple presented with severe abnormalities and were diagnosed with Steel syndrome (STLS), characterized by the characteristic features of dwarfism and dislocation of bilateral hip and radial heads. In both infants, two variant sites in COL27A1 (c.2G>A-p.Gly850Arg- and c.3249+1G>T) were identified by WES (Frigola et al., 2021). This is the first report of a COL27A1 variant in STLS. Due to the important role of the COL27A1 variant in STLS, some of the major skeletal features associated with Steel syndrome, such as shortened body length, scoliosis, and altered skull shape, were observed in COL27A1 knockout mice, although DDH was not present (Gonzaga-Jauregui et al., 2020), so the role of COL27A1 in human bone development deserves further study. The above evidence suggests that collagen is an important marker in DDH cartilage, suggesting disease development and prognosis of DDH.
C-X3-C motif chemokine receptor 1
C-X3-C motif chemokine receptor 1 (CX3CR1) is located on 3p21.3 and belongs to the G-protein-coupled transmembrane receptor superfamily. CX3CR1 is a co-receptor of the human immunodeficiency virus (HIV) and is overexpressed in the lymph nodes of HIV patients (Arenzana-Seisdedos and Parmentier, 2006)and plays an important role in cell adhesion, differentiation and proliferation of mesenchymal cells into chondrocytes or osteoblasts (Sun et al., 2017). Currently, the role of CX3CR1 in development is receiving increasing attention. For example, the CX3CR1 bone marrow lineage plays an important role in lymphatic endothelial remodeling during the development of the mammalian cardiovascular system (Cahill et al., 2021). Bioinformatic analysis of DDH susceptibility genes revealed that CX3CR1 and GDF5 modulate chondrogenesis through the classical Wnt signaling pathway (Yang et al., 2022). Feldman et al. has found the 2.61MB candidate region of chromosome 3 in 72 family members of four generations using GWLA, and then the CX3CR1 locus (rs3732378) variant in this candidate region was found in four members with severe symptoms using WES. Later, Sanger sequencing revealed that the variant was present in the DNA of all the 14 individuals with DDH symptoms (Feldman et al., 2013). Later, this team observed that in the CX3CR1 knockout mice had significantly increased acetabular diameter and joint space, and gait defects such as increased stance width and significantly shortened weight bearing stance time, consistent with human OA. These data provided suggestive evidence for the induction role of CX3CR1 gene in the pathogenesis of DDH (Feldman et al., 2017). In addition, several studies have reported that CX3CR1 (rs3732378) polymorphism is associated with DDH (Feldman et al., 2013; Basit et al., 2018; Gumus et al., 2021). Studies in the Turkish population showed that CX3CR1 (rs3732378) polymorphisms increased the risk of DDH by 12-fold in the covert and 75-fold in the overt group compared with the normal population (Gumus et al., 2021). Due to the increasing attention paid to the role of CX3CR1 in DDH, recent studies have evaluated the methylation status of CX3CR1 in DDH patients. However, the results showed that there was no significant difference in the methylation status of CX3CR1 gene between the 45 pairs of DDH patients and the control group, suggesting that DNA methylation of CX3CR1 is not involved in the pathogenesis of DDH (Nejadhosseinian et al., 2022).
Double von Willebrand factor A
Double von Willebrand factor A (DVWA), located on chromosome 3p24.3, is a novel gene identified by a Japanese GWAS. It was found to be specifically expressed in cartilage and binds to ß-tubulin to regulate chondrocyte differentiation (Farquharson et al., 1999; Meulenbelt et al., 2009). Polymorphism in DVWA (rs7639618 and rs11718863) has been reported to be strongly associated with the risk of OA in Japanese and Chinese patients (Miyamoto et al., 2008). The correlation between DVWA SNPs (rs7639618, rs9864422, and rs11718863) and DDH was analyzed in 368 DDH patients and 508 control patients, which showed that DVWA did not seem to be a risk factor for DDH etiology in Chinese Han population (Zhu et al., 2011).
Estrogen receptor 1
Estrogen receptor 1 (ESR1) is located on chromosome 6q25, is a sex steroid associated with the occurrence of osteoarthritis and osteoporosis, which plays a critical role in skeletal muscle homeostasis and motor function (Ikeda et al., 2019; Tang et al., 2021). ESR1 has been implicated in the pathogenesis and prognosis of DDH. The expression of ESR1 in the femoral head ligament and joint capsule of DDH patients was significantly higher than that in the control group, as demonstrated by immunohistochemical staining (Desteli et al., 2013). ESR1 consists of eight exons separated by seven introns. The mot widely studied polymorphic region is the Xba I-restricted fragment length polymorphism in intron 1. In the study of DDH, the Xba I genotype XX of ESR1 was found to be more common in patients with DDH than genotypes Xx and xx, indicating that genotype XX may be associated with increased risk of DDH (Kapoor et al., 2007). However, a Japanese report showed that SNPs in the Xba I site of ESR1 were not associated with the risk of DDH (Yamanaka et al., 2009). PvuII is also located in intron 1 of ESR1, and premenopausal women who are ESR1 PP homozygous have lower bone density than non-PP homozygous women (Rauhio et al., 2011). Although the ESR1 PvuII polymorphism is not associated with the risk of DDH formation, 90% of ESR1 PP patients had narrow joint space width and low mid-angle after surgery, suggesting that the ESR1 PvuII polymorphism is associated with poor prognosis of DDH (Yamanaka et al., 2009).
Growth differentiation factor 5
GDF5 is located on 20q11.22 and encodes bone morphogenetic proteins of the vertebrate-only TGF-β superfamily (Storm et al., 1994), which play a key role in bone and synovial joint formation and endochondral osteogenesis (Harsanyi et al., 2020). The GDF5-encoded protein is also a key component of the biological pathways involved in the prenatal and postnatal development of synovial joints (Chijimatsu and Saito, 2019). Many reports have shown that mutations in GDF5 are associated with abnormal bone development in humans (Genovesi et al., 2021; Rubin et al., 2021). At present, GDF5 is considered as a key pathogenic gene of DDH and has been studied more in DDH (Kiapour et al., 2018), however, the mechanism on GDF5 inducing to DDH still needs to be further studied. Short-footed mice with GDF5 inactivated mutation presented as shorter femur, smaller femoral head and neck, and smaller acetabulum. Cis-regulatory structure of Gdf5 locus in mice scanned and fine mapped with FVB-NJ strain confirmed many enhancers with DDH-associated risk variants in downstream of GDF5, and intervention of downstream genes can restore the above damaged key morphological features (Kiapour et al., 2018). Since GDF5 (SNP rs143383) is associated with susceptibility to OA, the relationship between GDF5 (SNP rs143383) and DDH was studied, which showed that GDF5 (rs143383) was significantly correlated with DDH. Especially in female DDH and severe DDH (Dai et al., 2008). Harsanyi et al. studied 118 cases of DDH in Slovakia, and the results showed that although GDF5 (SNP rs143383) was not the only factor contributing to the genetic component of DDH, the polymorphism of this site had an important influence on the pathogenesis of DDH (Harsanyi et al., 2021a). In addition, this team demonstrated that GDF5 (SNP rs143383) influenced the occurrence of DDH in 45 patients with DDH in Slovakia (Harsanyi et al., 2021b). In addition, case-control studies showed that methylation of the GDF5 promoter was significantly increased in cartilage samples from patients with DDH (Baghdadi et al., 2019). A GWLA analysis of 770 patients with DDH found that the heritable component of DDH was attributable to common genetic variations, accounting for 55%, and was evenly distributed across the autosomal and X chromosomes. This study also found a robust association between the GDF5 promoter (SNP rs143384) and DDH (Hatzikotoulas et al., 2018). A French study involving 239 pairs of Caucasian patients with DDH and controls found a significant association between GDF5 polymorphism (rs143384, rs143383) and DDH in Caucasians (Rouault et al., 2010). A study from China involving 192 DDH and 191 controls found an association between SNPs and DDH at two sites of GDF5 (rs224332 and rs224333) in Chinese women, suggesting that GDF5 may be a candidate gene for pahogenesis of DDH (Zhao et al., 2013b). As a number of studies have confirmed the genetic association between GDF5 and DDH, a functional knee cartilage structure for cartilage repair was generated using 3D bioprinted GDF5 scaffolds combined with bone marrow mesenchymal stem cells (BMSC), which were transplanted into rabbit knee joints to achieve long-term cartilage protection (Sun et al., 2019a). Recent studies have shown that GDF5 duplication loci can be associated with more than 20 different diseases. Muthuirulan et al. identified two of the most disease-causing variants, the GDF5 (rs4911178) variant was associated with DDH and the GDF5 (rs6060369) variant was associated with knee OA. Futher verification in GROW1 (where GFP5 rs4911178 resides in) enhancer null mouse has shown have clinically relevant DDH phenotypes and alterations to the orientation of the acetabulum (Muthuirulan et al., 2021).
Homeobox
The product of the homeobox (HOX) gene on chromosome 17 is a transcription factor associated with bone development and plays a role in embryonic limb development (Sauvegarde et al., 2016). Although a case-control study of a Caucasian population (239 cases and 239 controls) from western Brittany, France, showed no association between multiple SNPs of HOXB9 and DDH (Rouault et al., 2009), a subsequent study showed that DNA extraction from a multi-generation DDH family of 18 members revealed that a 4 Mb region abnormality on chromosome 17q21.32, which contains the entire HOXB homologous gene cluster, may be associated with DDH and was confirmed to be inherited in an autosomal dominant manner (Feldman et al., 2010). HOXB9 is expressed in human embryos at 5–9 weeks, which coincides with the formation of the original gluteal band, and is expressed in the lumbar spine, sacrum, and caudal regions of the embryo. (Chen and Behringer, 2022). HOXD9 regulates muscle cell differentiation and growth regulation as well as mesenchymal cell differentiation. A study of Chinese Han women with DDH found that HOXD9 (rs711819) was significantly correlated with DDH (Tian et al., 2012). Another study in the Han population showed that SNP of HOXD9 (rs2303486) may be associated with DDH formation in female patients (Hao et al., 2014).
Heparan sulfate proteoglycan 2 and ATPase plasma membrane Ca2+ transporting 4
Heparan sulfate proteoglycan 2 (HSPG2) is located on 1p36.12 and encodes a perlecan protein that crosslinks extracellular matrix components and cell surface molecules and is closely associated with musculoskeletal development in mice and humans (Lowe et al., 2014). ATPase plasma membrane Ca2+ transporting 4 (ATP2B4) is located on 1q32.1 and encodes the plasma membrane calcium transport ATPase 4 enzyme, which is involved in catalyzing ATP hydrolysis and Ca2 transport out of cells.ATP2B4 has been shown to maintain bone homeostasis in mice and humans by playing a regulatory role in undifferentiated and mature osteoclasts (Kim et al., 2012). Basit et al. excluded the currently known variation of DDH-related genes (ASPN,CX3CR1, DKK1, GDF5, HOXB9, HOXD9, PAPPA2,TGFβ1) in five members of a Saudi family (including three DDH patients) using WES and found common haplotypes of HSPG2 and ATP2B4 gene variants on chromosome 1 with incomplete penetrance. They predicted that HSPG2 regulates ATP2B4 expression through several transcription factors GATA1, MAZ, and RFX1 (Basit et al., 2017). Based on the above results, Sanger sequencing was performed in 250 sporadic cases of DDH in China and showed that SNPs of HSPG2(chr1:22201470), ATP2B4 (chr1:203682345), and prostaglandin F receptor (PTGFR) (chr1:79002214) were not associated with DDH in Chinese Han population (Xu X. et al., 2021). In addition, HSPG2 mutation has been reported in the first case of 18q deletion syndrome with DDH. This female child not only has the 18q deficiency syndrome such as developmental delay and cleft palate, but also has bilateral developmental hip dislocation, hypothyroidism, and recurrent fever. NGS testing of the child’s blood samples showed a 10 Mb deletion at 18q22.2 to q23 and two mutations of HSPG2, and no variation of DDH-related genes was found in the deletion region, suggesting that DDH may be related to HSPG2 mutation (Yu et al., 2022).
Interleukin 6 and transforming growth factor beta-1
IL-6 is located on 7p15.3, is an immunomodulatory cytokine that plays an important role in the pathogenesis of osteoporosis (Kany et al., 2019). Transforming growth factor beta-1 gene (TGFβ1) is located on 19q13.1–13.3, is also reported to be a proinflammatory cytokine and plays an important role in the pathogenesis of OA (Tachmazidou et al., 2019). IL6 and TGFβ1 have complex biological roles in bone metabolism, calcium and vitamin D levels and other physiological aspects (Eftekhari et al., 2018). As regulators of fibroblasts and perichondrium cells in tendon, the interaction between IL6 and TGFβ1 may affect the development of DDH. In Caucasian populations, the CC genotype located in the IL6 promoter region at site 572 (rs1800796) is associated with DDH and its secondary OA, and the “C allele carrier” in the TGFβ1 signaling sequence is also associated with DDH and its secondary OA (Kolundzic et al., 2011). However, another study of patients with DDH in Slovakia showed no association between IL6 (rs1800796) and DDH(73). Čengić et al. compared 68 cases of DDH secondary OA with 152 control cases and found that all three genotypes of TGFβ1 (29T>C) (rs1800470) and IL-6 572G>C (rs1800796) polymorphisms were present in the DDH secondary OA group. However, when family history polymorphism was evaluated, the homozygous mutation rates of TGFβ1T>C (rs1800470) and IL-6 572G>C (rs1800796) were found to be significantly higher in patients with positive family history than in patients with negative family history (Cengic et al., 2015). Ma et al. confirmed that the SNPs of TGFβ1 (rs1800470) and the SNPs of IL6 (rs1800796) were associated with DDH in a Han population (4206 DDH cases), but the SNP of IL6 (rs1800796) was weakly associated with DDH (Ma et al., 2017). Conversely, a study showed that SNPs of TGFβ1 (rs1800470) and IL6 (rs1800796) were not the main genetic factors causing DDH (Igrek et al., 2021). In addition, polymorphisms in the TGFβ1 gene were significantly associated with hip osteoarthritis in a Croatian population (Kolundzic et al., 2011).
LDL receptor related protein 1
LDL receptor related protein 1 (LRP1) is located on 12q13.3, encodes a member of the LDL receptor protein family and has high variability. Knockdown of LRP1 inhibited receptor activator of nuclear factor-κB (NFκB) ligand (RANKL) induced osteoclast formation and osteoclast differentiation and proliferation in macrophages (Kohara et al., 2020). A case report revealed an LRP1 variant associated with syndrome in a brother and sister characterized by cardiopulmonary dysfunction, paddle-shaped fingers and toes, and cloudy corneas. LRP1 knockout mice may also exhibit skeletal abnormalities during the embryonic period and abnormal limb morphology after birth (Mark et al., 2022). A study performed WES on 17 patients with DDH from eight families and targeted sequencing on 68 patients with sporadic DDH, and identified two LRP1 mutation sites in two families and seven sporadic cases. Missense variants of LRP1 knockout mice showed a DDH phenotype with acetabular and femoral head malformation in mice, which was milder in heterozygotes and more severe in homozygotes. In vitro experiments have confirmed that LRP1 deficiency leads to decreased autophagy levels and decreased chondrogenesis capacity (Yan et al., 2022).
Pappalysin 2
Pappalysin 2 (PAPPA2) is located on chromosome 1 (1q24), encodes PAPPA2, a 1,791-residue protein that is highly expressed in the human placenta during the first trimester. PAPPA2 was originally thought to be a placental-derived circulating protein and was later demonstrated to play a role in fetal embryo development, postnatal bone growth regulation, and reproductive function (Conover et al., 2011). PAPPA2 is also a protease of IGF-like growth factor binding protein 5 (IGFBP-5) and regulates insulin-like growth factors (IGFs) required for growth. PAPPA2 was found to be involved in hip joint development by IGF using the developing mouse hip joint. PAPPA2 specifically lyses IGFBP-5, resulting in the separation of the biological function of IGF1 from the complex containing IGFBP. In the acetabular and femoral head cartilage of PAPPA2 knockout mice, the chondroproliferation-related proteins Col1A1 and IGF1 were significantly decreased, while IGFBP-5 was significantly increased (Chen et al., 2019). At present, the function of PAPPA2 in fetal development has received more and more attention (Rosenkrantz et al., 2021; Haider et al., 2022), PAPPA2 was expressed in osteoblasts during fetal development and in differentiated chondrocytes during intrachondral bone formation (Jia et al., 2012), and PAPPA2 knockout mice showed reduced body length and femur shortening as adults (Conover et al., 2011). Another study found that the pelvic girdle and coccyx were shortened and the shape of the mandible and pelvic girdle were also altered according to geometric morphometric evaluation in PAPPA2 knockout mice. The influence of this variation on the size and shape of the pelvic girdle may indicate the correlation between PAPPA2 and DDH (Christians et al., 2013). In addition, a study using GWLA in a Chinese Han family of 24 individuals (5 DDH patients) found a significant association between PAPPA2 (rs726252) and DDH, and the PAPPA2 (rs726252) associated with DDH was verified in 310 DDH cases and 487 control cases (Christians et al., 2013). However, another study showed no significant difference of PAPPA2 (rs726252) in an extended Han population (697 patients with DDH and 707 patients) (Shi et al., 2014). Meanwhile, a study from Slovakia including 45 patients with DDH and 85 controls also showed that the PAPPA2 (rs726252) was not associated with DDH (Harsanyi et al., 2021b).
Substance P
Substance P (SP) is a highly conserved member of the tachykinin family and is widely expressed throughout the animal kingdom. As an 11-amino acid neuropeptide that preferentially activates the neurokinin-1 receptor (NK1R), it transmits damaging signals to secondary neurons in the spinal cord and brain stem via primary afferent fibers (Zieglgansberger, 2019). The contents of SP and its receptor NK-1R in the serum and synovium of patients with DDH and DDH-associated OA were significantly higher than those of the normal population. These results suggest that SP and NK-1R may be associated with hip dysfunction and chronic pain perception and may be involved in the progression from DDH to DDH-related OA (Wang et al., 2014). The expression of SP in the synovium was significantly upregulated in patients with severe DDH, but no such significant upregulation was observed in patients with moderate DDH. It was considered that the degree of articular cartilage wear would gradually increase, and the cushioning capacity of the cartilage would gradually disappear. During this process, gradually increased afferent stimulation of joint pain would cause the dorsal nerve root to release more SP (Li et al., 2019). Wang et al. showed that SP is significantly upregulated in the synovial membrane and synovial fluid of DDH, which is involved in the inflammatory process of arthritis by activating the NF-κB pathway (Wang et al., 2015).
Teneurin transmembrane protein 3 gene
The cytogenetic location of TENM3 is 4q34.3–q35.1, encoding teneurin transmembrane protein 3. TENM3 belongs to the tenogenic protein superfamily and plays an important role in cell aggregation and adhesion. Feldman et al. performed WES in patients with severe DDH in four generations of a family with intergenerational transmission and found that glutamine at position 2,665 of the TENM3 gene was changed into proline. Meanwhile, TENM3 knockout mice were found to have delayed development of the left acetabulum and left articular fossa, and matrix metallopeptidase 13 (MMP13), which inhibits differentiation, was overexpressed in the mouse bone marrow cells (Feldman et al., 2019). Recently, a study of 250 cases of DDH of Han nationality found that TENM3 (OMIM * 610083, chr4:183721398) was not associated with DDH (Xu et al., 2021).
Ubiquinol-cytochrome c reductase complex chaperone
Ubiquinol-cytochrome c reductase complex chaperone (UQCC) is located on 20q11.22, encodes a zinc-binding protein and is an important candidate gene involved in bone and cartilage development (Liu et al., 2010). Currently, UQCC is considered to be a susceptibility gene for DDH (Gkiatas et al., 2019). In a GWAS study of 386 patients with DDH and 558 healthy controls in the Chinese population, 12 SNPs of UQCC were found to be associated with DDH, and the UQCC (rs6060373) polymorphism significantly correlated with DDH was verified in 755 patients with DDH and 944 controls (Sun et al., 2015). However, a study from Turkey (68 DDHS and 100 controls) showed that UQCC rs6060373 was not associated with DDH (Gumus et al., 2021).
Vitamin D receptor
Vitamin D receptor (VDR), adjacent to the COL2A1 gene, is located on chromosome 12q13.11 and contains 11 exons with a length of approximately 75 kb. The product of VDR binds to vitamin binding D to initiate cellular signaling pathways that regulate and control calcium and phosphorus absorption, thereby affecting bone density. Studies have shown that the serum VD of patients with DDH is not different from that of the normal population, but the level of VDR is significantly lower than that of the normal population, indicating that reduced VDR may affect the prognosis of DDH (Topak et al., 2021). VDR polymorphism is associated with OA (Hassan et al., 2022). In the aforementioned study conducted by Donatella Granchi et al., the distribution of the BsmI polymorphism in VDR was also found to be different, with homozygous bb frequency being significantly higher in patients with DDH (Granchi et al., 2002). Previous study has suggested that homozygous t allele mutations in the TaqI region of VDR exon 9 were probably associated with higher acetabular index of DDH (Kapoor et al., 2007). However, a study from Japan on 64 DDH patients treated with rotational acetabular osteotomy found that rotational ApaI and Taq I gene polymorphism of VDR are not correlated with the formation and prognosis of DDH (Yamanaka et al., 2009). In addition, no significant correlation was found between four SNPs in the VDR gene (rs731236, rs1544410, rs7975232, and rs2228570) and DDH in a case-control analysis using 50 DDH patients and controls from the Saudi population (Jawadi et al., 2018).
Genes related to WNT
WNT is a large L-cysteine-rich family of glycoproteins ranging in size from 39 to 46 kD. The WNT pathway is involved in joint and chondrogenic embryonic development and joint formation (Cahill et al., 2021) and is associated with many stages of vertebrate limb development. Molecules in the WNT pathway are involved in proximal-distal growth, dorsoventral patterns, and the development and maintenance of cartilage, bone, muscle, and joint (Church and Francis-West, 2002). In the bioinformatic analysis of 16 DDH susceptibility genes, DKK1, FRZB, and WISP3 were members of the WNT pathway, suggesting that the WNT pathway may be involved in the pathogenesis of DDH (Yang et al., 2022). In the study of DDH, the proliferation of osteoblasts and chondrocytes was affected as interference with Wnt family member 1 (WNT1), which also affected the expression of GDF5 and WNT1 inducible signaling pathway protein 2 (WISP2), BMP2, and BMP4 (Xu et al., 2022). In the DDH rat model, the differentially expressed genes in the hip cartilage tissues of DDH and healthy control rats were analyzed by microarray. The results showed that WISP2 was significantly upregulated and the peroxisome proliferator activated receptor gamma (PPARG) signaling pathway played an important role in the development and degradation of cartilage in DDH by KEGG analysis. Acetabular cartilage degradation and apoptosis were significantly increased in DDH rats, accompanied by significantly upregulated WYP-2. Cellular experiments confirmed that WYP-2 negatively regulated the expression of PPARG in chondrocytes, suggesting that WYP-2 may promote the apoptosis of acetabular chondrocytes by regulating the expression of PPARG (Ji et al., 2020). The WNT1-inducible signaling pathway protein 3 gene (WISP3) plays an important role in chondrogenesis. It regulates the production of type 2 collagen through the IGF signaling pathway, as WISP3 is essential for physiological postnatal bone growth and cartilage homeostasis (Cui et al., 2007). Zhang et al. proved that five SNPs in WISP3 (rs69306665, rs1022313, rs1230345, rs17073268, and rs10456877) were associated with the formation of DDH. They also performed haplotype analyses showing that the GGCGG haplotype was associated with a higher risk of DDH, suggesting that WISP3 should be used as a reliable biomarker (Zhang et al., 2018). Wnt inhibitory factor 1 (WIF1), encoded by the WIF1 gene, is a lipid-binding protein that binds to the Wnt protein and consists of a unique and conserved WNT inhibitory factor (WIF) domain and five epidermal growth factor (EGF)-like domains. The WIF1 protein inhibits WNT signaling and is expressed primarily in the epiphysis and articular cartilage surface and promotes chondrogenesis (Zhang et al., 2022). The expression of WIF1 in the hip capsule of canine hip dysplasia was significantly lower than that in normal tissues (Todhunter et al., 2019). A comparative study of WIF1 expression in hip joint tissues of 586 Chinese Han patients with DDH and 987 healthy controls showed that WIF1 was significantly increased in DDH patients, and the expression of WIF1 (rs3782499) AA genotype patients was significantly higher than that of GG genotype patients. The results suggested that the polymorphism of WIF1 gene rs3782499 might be related to DDH formation (Sun et al., 2019b). Frizzled-Related Protein (FRZB) is located on 2q32.1, is an antagonist of the Wnt signaling pathway. FRZB is extremely important for chondroblast differentiation of stem cells, and loss of cartilage integrity was observed in FRZB -knockout mice (Lories et al., 2007). Xu et al. found a high expression of FRZB, which regulates integrin to affect cell adhesion pathways and cell diffusion in DDH, and the rs3768842 and rs2242040 genotypes of FRZB were significantly correlated with DDH (Xu R. et al., 2021).
TXNDC3
NME/NM23 family member 8 (TXNDC3) is located on 7p14.1, is associated with bone mineral density, chondrocytes, and OA (Yerges-Armstrong et al., 2014). A significant susceptibility of the TXNDC3 SNP with OA was confirmed in Caucasian and Chinese females (Shi et al., 2008; Machol et al., 2017). A study comparing 984 DDH patients with 2043 healthy controls from a Han population identified a TXNDC3 rs10250905 missense mutation in 15 members of seven families, confirming an association between the TXNDC3 rs10250905 and DDH (Qiao et al., 2017).
Gene variants in fewer studies and syndromes related to DDH
A case report reports an 8-month-old male child with DDH, microcephaly, mild motor retardation, left undescended testis, and patent ductus arteriosus. CNV of Affymetrix SNP array data showed a 437 kb microduplication on 22q11.21 involved in T-box transcription factor 1 (TBX1), catechol-O-methyltransferase (COMT), septin 5 (SEPT5), G protein subunit beta 1 like (GNB1L), thioredoxin reductase 2 (TXNRD2) and glycoprotein Ib plateletsubunit beta (GP1BB) (Weisfeld-Adams et al., 2012), which have not been reported to be associated with DDH. T-box genes, originally discovered in mice, are highly conserved in vertebrates and play a central role in posterior mesoderm formation and axial development (Schulte-Merker et al., 1994). Loss of Tbx1 results in a bone phenotype similar to cranial dysplasia (Funato et al., 2015). Tbx4 and Tbx5 are key regulators of interspecies limb growth and identification during embryonic development (Tickle, 2015). Currently, it has been reported that TBX4 rs3744438 was not correlated with DDH in Han population (505 children with DDH and 551 controls), while TBX4 (rs3744448) was significantly correlated with male DDH. Stratified analysis showed that TBX4 (rs3744448) was associated with hip dislocation, but not with subluxation or hip instability (Wang et al., 2010).
A study aiming at exploring new genes associated with DDH using GWAS in a Chinese Han cohort consisting of 386 DDH cases and 500 healthy controls has identified the minor allele rs61930502-A, and there is no gene located around rs61930502 either 20 kb upstream or 20 kb downstream. This intronic SNP tended to prevent DDH in a dominant manner and in addition, was also verified in another Chinese Han cohort consisting of 574 DDH patients and 569 healthy controls (Yan et al., 2019). Meanwhile, protein-protein interaction analysis also revealed that showed the most direct protein interacted with 409 DDH-associated proteins was heat shock 70 kDa protein 8 (HSPA8) in this study, indicating its possible role in.pathogenesis of DDH (Yan et al., 2019).
Zhang et al. performed GWLA on a four-generation Chinese family consisting of 24 members (5 DDH cases) and found the strongest linkage evidence on chromosomes 8q23-24, covering the 2.377 Mb SNP (rs724717-rs720132), which resided four genes: transcriptional repressor GATA binding 1 (TRPS1), eukaryotic translation initiation factor 3 subunit H (EIF3H), RAD21 cohesin complex component (RAD21) and rRNA-binding ribosome biosynthesis protein (UTP23). Variations in TRPS1 and/or TRPS2 may cause Trichorhinophalangeal Syndrome, which is characterized by fine, sparse, discolored, and slow-growing hair, malnourished nails, small breasts, short stature, short feet, short fingers and DDH (Maas et al., 1993). RAD21 is also involved in the pathogenesis of Trichorhinophalangeal Syndrome because of its nearby location of TRPS2 (Maas et al., 1993). EIF3H and UTP23 have not been reported to be associated with DDH. In addition, three consecutive markers (rs1919980, rs763853, and rs725124) showed on chromosome 12p12 was 0.642 Mb, in which the only known gene was SOX5 (Zhang et al., 2020).
Sanger sequencing of a DDH family from Saudi Arabia showed that no known variants in DDH genes (CX3CR1, ASPN, HOXB9, HOXD9, DKK1, GDF5, PAPPA2, TGFB1, and UFSP2). Instead, common homozygous regions on chromosomes 15q13.3 and 19p13.2 were identified and their locations were rs17228178-rs1534200 and rs466123-rs2112461, respectively. Notch receptor 2, chromodomain helicase DNA binding protein 7, DEAH-box helicase 36, pericentrin, and DNA polymerase epsilon, catalytic subunit were detected as genes or gene promoters in the above regions, but no DDH correlation was reported for these genes. In addition, the copy number of chr6p21.32 (chr6:33 053 906-33 069 893) was increased by approximately 15 kb in all affected individuals, and the sixth exon of HLA-DB1 was found in this region. The 6p21.32 (rs6905837) SNP is currently considered to be a novel gene associated with bone mineral density (Liu et al., 2020), but HLA-DB1 is not currently considered to be a pathogenic gene for DDH because partial gains in this region have also been found in non-diseased siblings of this family (Basit et al., 2018). Missense variant in KANSL1 (c.C767T; p.S256F) was identified as the pathogenic cause of DDH through ES, which was a novel variant co-inherited by all severely affected individuals in A 3-generation, 12-member family. Further study using CRISPR/Cas9 technology to construct KANSL1 mutant mouse has presented diminished acetabular chondrocytes and defects in acetabular cartilage (Xu et al., 2022)
Notably, DDH is increasingly being reported as one of the features of the syndrome, and additional gene variants were detected that may be associated with DDH besides the above-mentioned variants in HSPG2 (Yu et al., 2022), TBX1 (Weisfeld-Adams et al., 2012), COL1A1, COL1A2 (Ayoub et al., 2020), COL2A1 (Agenbag et al., 2020), and COL27A1 (Frigola et al., 2021) (Table 2; Table 3). Frameshift (c.1860_1861insT; p.Pro621fs) and missense variants (C2125 G > T; p.Val709Leu) of NTRK1 have been reported in patients from Jordan with congenital insensitivity to pain with anhidrosis who were complicated with DDH (3/7) (Masri et al., 2020). A heterozygous missense mutation in exon 6 of the alpha 1 subunit of the glycine receptor gene (GLRA1) (Arg271Gln) has been reported in two Japanese hyperekplexia patients, one of whom was diagnosed with hip dislocation (Kimura et al., 2006). The WDR73 homozygous missense mutation [NM_032856.3, c.287G > A (p. Arg96Lys)] was detected through WES in a patient with Galloway-Mowat syndrome. At 22 months of age, this patient exhibited microcephaly, infantile onset of central nervous system abnormalities, and the right hip demonstrated features of DDH with a shallow acetabular roof (Al-Rakan et al., 2018). Although DDH has been classified as one of the musculoskeletal manifestations of the Wiedemann-Steiner syndrome, it has only been described in two Wiedemann-Steiner syndrome patients with exons 2–10 deletion and de novo nonsense mutation, p.Gln 1978* of KMT2A (Ko et al., 2016). In two of the ten patients with intellectual disability, a de novo missense mutation c.509G>A (ENST00000418600); p.Arg170Gln (ENSP00000403636.2) of SYNGAP1 was confirmed through ES. This mutation was also associated with unilateral DDH (Parker et al., 2015). Mohammed et al. have reported the first case of a de novo heterozygous variant in FAR1 presenting as cataracts, spastic paraparesis, and speech delay (CSPSD) in the Middle Eastern region. This variant was identified through ES analysis, and it was also the second case of CSPSD with DDH (Almuqbil et al., 2022). Besides, c.808G > A(p.(G270S)) and c.942G > C(p.(W314C) of B3GALT6 were detected using Sanger sequencing in an Ehlers-Danlos syndrome (EDS) patient with widened right hip joint space (Han et al., 2022). Moreover, the patient with a partial trisomy 3q12-q23 de novo who could not be diagnosed as dup(3q) syndrome presented with mild mental retardation, postnatal growth retardation, facial dysmorphisms, and DDH(140). VPS33B (c.1157A > C (p.His386Pro)) were detected in a spainish patient of arthrogryposis, renal dysfunction, and cholestasis syndrome with bilateral DDH (Del Brío Castillo et al., 2019).
Non-coding RNAs
In the study of the correlation between the FRZB gene and DDH, upstream miRNAs regulating FRZB expression were found in the synovial fluid of patients with DDH. The expression of multiple miRNAs was significantly downregulated, among which miRNA-454 was the most significantly downregulated, and luciferase experiments confirmed that miRNA-454 could lead to the targeted inhibition of FRZB, suggesting that downregulated miR-454 upregulated the expression of FRZB and may play a role in promoting chondrogenesis in DDH joint tissues (Xu et al., 2021).
Cheng et al. compared the hip cartilage of DDH patients and control cases by whole transcriptome sequencing and identified 186 differential miRNA-targeted 175 DDH candidate genes from 1833 differential mRNAs, such as von Willebrand factor A domain containing 1, transmembrane protein 119 and signal peptide, CUB domain and EGF like domain containing 3. Gene ontology (GO) enrichment analysis detected 111 candidate terms for DDH such as skeletal system morphogenesis and skeletal phylogeny. KEGG pathway enrichment analysis identified 14 candidate DDH pathways, such as Hedgehog signaling pathway and Wnt signaling pathway (Cheng et al., 2021). The involvement of DDH-related miRNAs in the screening of DDH differential genes is conducive to further study of the pathogenesis of DDH.
Liu et al. identified 18 differentially-expressed miRNAs using the DDH rabbit model by high-throughput sequencing, and verified that the downregulation of miR-129-5p promoted the expression of its target gene GDF11, thus inducing the phosphorylation of SMAD3. This pathway can inhibit chondrocyte proliferation and cycling and reduce chondrocyte mineralization nodules, which may be associated with DDH-associated ossification of the acetabular roof (Liu et al., 2020). According to the above sequencing data, members of the same research group verified the low level of miR-1-3p in the acetabular roof cartilage in the rabbit DDH model, and verified the targeting relationship between miR-1-3p and SOX9 at the cellular level. With the silencing of miR-1-3p, SOX9 expression was upregulated. Genes associated with endochondral osteogenesis, such as RUNX2 and collagen X, were downregulated (Ding et al., 2021), and the reduction of mineralized nodules produced by cartilage suggested that miR-1-3p may be the cause of abnormal ossification in acetabular cartilage in DDH animals.
In addition, changes in long non-coding RNAs (lncRNAs) were also involved in the pathogenesis of DDH. lncRNA-H19 was downregulated in patients with DDH and in rat models. It has been found that lncRNA-H19 can inhibit DDH cartilage degeneration through competing endogenous RNAs (ceRNAs) mechanism competitive with miR-483e5p combined with Dusp5 in chondrocytes mimicking intermittent cyclic mechanical stress (ICMS) cell force (Wang et al., 2020). Experiments confirmed that lncRNA H19 played a positive role in inhibiting cartilage degeneration.
Discussion
Due to the diversity of clinical symptoms of DDH ranging from subtle characteristics of dysplastic hips, such as shallowness and underdevelopment of the acetabulum, to grossly deformed acetabulum, which may either be subluxated or dislocated, there is an absence of general official clinical consensus on the screening for DDH. Early diagnosis and management of DDH have been demonstrated to significantly improve treatment efficacy and prevent permanent disability successfully (Zhu et al., 2019). Ultrasound is considered the most reliable examination to screen for DDH in children under 3 years of age, but may not detect DDH in those with smaller lesions. In patients with mild hip instability detected by ultrasound at birth, 96% of the pathologic changes resolved spontaneously within the first 6 weeks after birth (Vaquero-Picado et al., 2019). Accurate imaging and clinical examination that are guided by genetic background knowledge will significantly facilitate the diagnosis and treatment process. Therefore, understanding the genetic basis of the pathogenesis of DDH and conducting early genetic screening may significantly achieve accurate diagnosis and early implementation of correct treatment for DDH and affect the prognosis of DDH.
At present, the genetic basis of DDH is widely acknowledged and proven by familial heredity, and significant progress has been made in the research on the molecular mechanism of DDH. A considerable number of DDH-related genes have been screened due to the extensive application of GWAS, GWLA, and WES. Despite the ongoing debate regarding the potential association of certain genes with DDH, it is unfeasible to disregard the genetic factors that influence DDH. Currently, among the two types of genetic modes of DDH being considered, type I DDH is a multifactorial genetic disease with variable phenotypes and multiple candidate genes including noncoding RNAs (Feldman et al., 2010). Interestingly, there are relatively more studies on DDH-related SNPs than CNVs in Type I DDH, although it has been reported that the DNA sequence diversity within the human genome is more influenced by CNVs than SNPs. Hence, CNVs can be utilized to study DDH and may contribute to the etiological understanding of the genetic basis of DDH in the future (Zamborsky et al., 2019). Notably, single gene variants detected in humans that have been mutated in animal models, such as CX3CR1, GFP5 (rs4911178 (G → A) locate in GROW1, a regulatory enhancer active in humans and mice), and KANSL1, have been demonstrated to have potential effects on the DDH phenotype (Feldman et al., 2017; Gumus et al., 2021; Xu et al., 2022). Additionally, SNPs of GDF5 and TGFβ1 have been demonstrated to be related to DDH in various races (Dai et al., 2008; Rouault et al., 2010; Zhao et al., 2013b; Hatzikotoulas et al., 2018; Sun et al., 2019a; Baghdadi et al., 2019; Harsanyi et al., 2021a; Harsanyi et al., 2021b; Muthuirulan et al., 2021). Nevertheless, it is important to acknowledge that the results were inconsistent across diverse populations. Despite the fact that gene variants such as ESR1, Hoxb9, IL6, PAPPA2, UQCC, and VDR were associated with DDH, other case-control studies did not report a significant difference (Granchi et al., 2002; Kapoor et al., 2007; Rouault et al., 2009; Yamanaka et al., 2009; Kolundzic et al., 2011; Jia et al., 2012; Christians et al., 2013; Hao et al., 2014; Jawadi et al., 2018; Feldman et al., 2019; Harsanyi et al., 2021b; Gumus et al., 2021).
The genetic pattern of type II DDH, which is autosomal inheritance, is less prevalent. Feldman et al. reported a four-generation family in Utah with a variable phenotype (Sun et al., 2017). The gene variants screened from familial genetic studies or detected in sporadic cases were probably pathogenic genes, some of which were further explored in case-control studies. The variant TXNDC3 rs10250905 was verified to be significantly associated with DDH both in 15 members of seven families and in case-control pairs consisting of 984 DDH children and 2043 healthy controls (Qiao et al., 2017). The variant CX3CR1 rs3732378 screened using WES in four DDH members of an American family has been demonstrated to increase the risk of DDH by 12 times in the covert group and 75 times in the overt group in the Turkish population (Feldman et al., 2013; Gumus et al., 2021) However, the results are not always consistent. The HSPG2 (c.3328G > T) and ATP2B4 (c.2264G > A) variants detected by WES in five members of a Saudi family were not associated with the susceptibility to DDH in the Chinese Han population (Basit et al., 2017; Xu et al., 2021). This result indicates that the genes inherited in DDH families may be different from those in sporadic cases and may play a prominent genetic role. These findings may have been influenced by the small number of cases, the heterogeneity of DDH in pathogenic genes, and racial disparities. These findings lay the foundation for an early and accurate diagnosis of DDH in the future. Therefore, we analyzed the co-expression, pathways, genetic interactions, physical interactions, shared protein domains of DDH related genes concluded in Tables 1 and 2 (Figures 1, 2), which has shown the potential reaction of them. Futhermore, enreiched pathways of these genes analyzed in String database were shown in Table 4, suggesting the potential role of COL2A1, COL11A2, and COL1A1 in the pathogensis of DHH. Meanwhile, it also suggested that DDH is probably caused by a combination of genes and can subsequently be affected by epigenetic, mechanical, or environmental factors (Zhang et al., 2020). In general, the genetic information from the DDH families and sporadic cases will enhance our understanding of DDH gene variants. Additional research with larger sample sizes could be highly valuable in terms of acquiring more information.
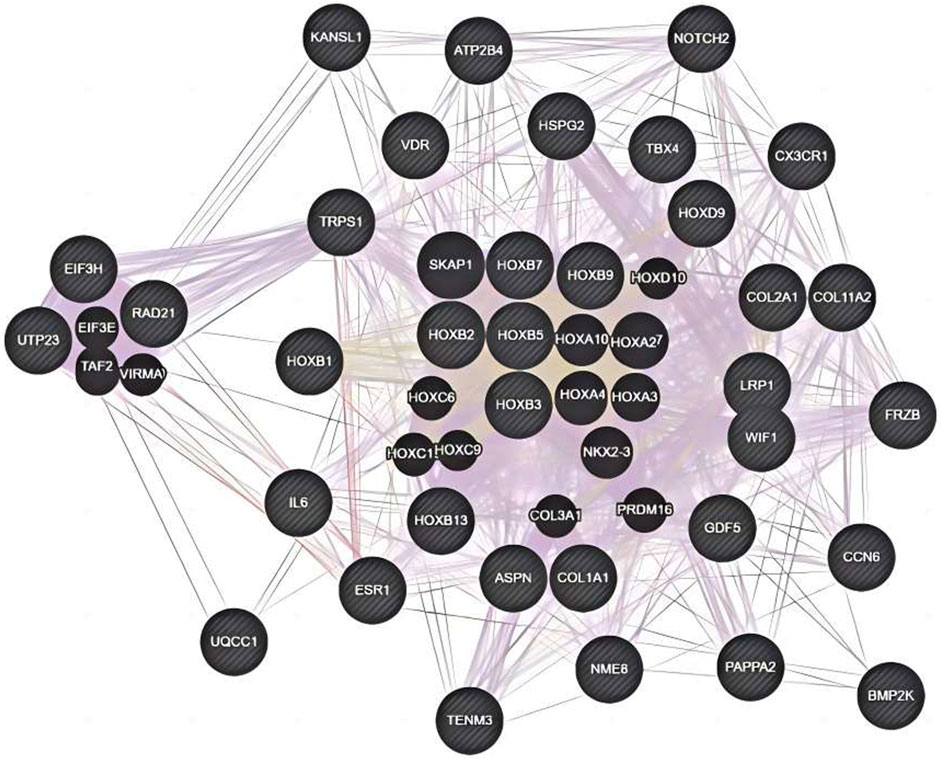
Figure 1. Co-expression, pathways, genetic interactions, physical interactions, shared protein domains for genes from Table 2 analyzed by Genemania software.
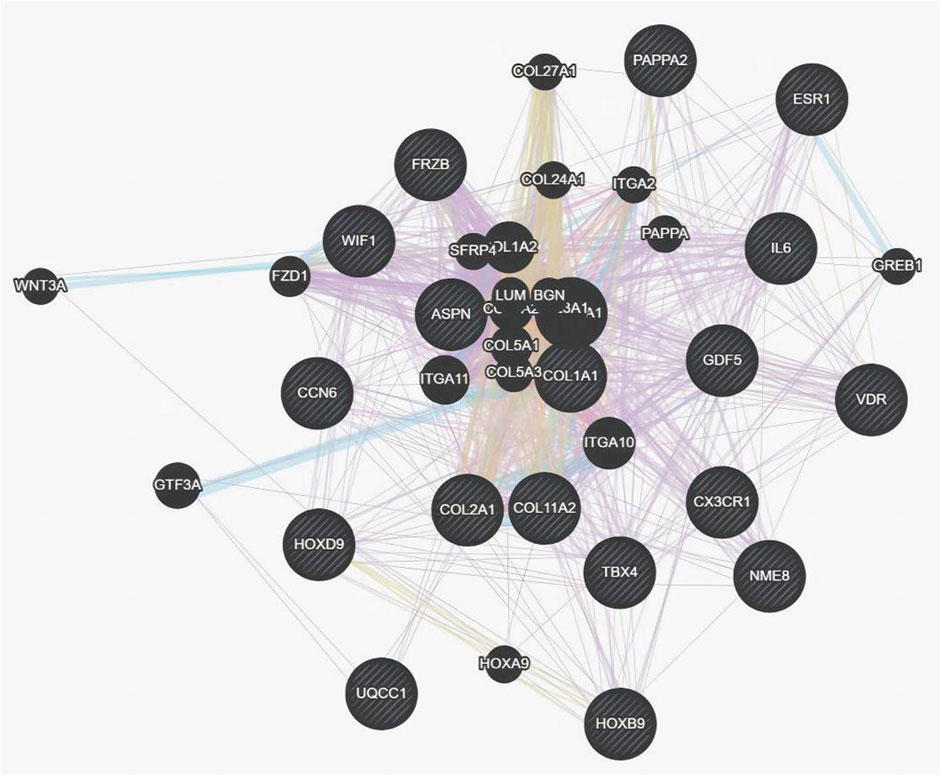
Figure 2. Co-expression, pathways, genetic interactions, physical interactions, shared protein domains for genes from Table 1 and Table 2 analyzed by Genemania software.
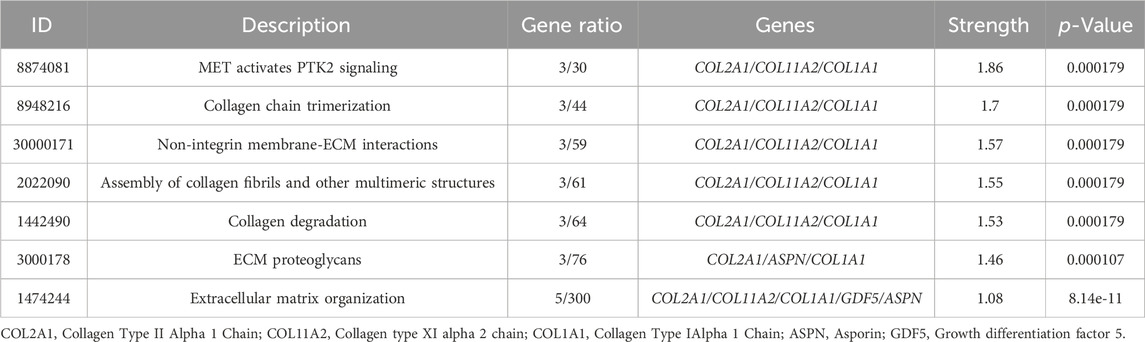
Table 4. Pathway enriched terms for gene variants in Tables 1 and 2.
Gene variants detected in patients with DDH-related syndrome also contributed to the increasing number of DDH susceptibility genes. In the 18q deletion syndrome, which includes bilateral DDH, HSPG2 variants were observed. Additionally, five members of the Saudi family were affected, despite the presence of distinct mutant loci (Basit et al., 2017; Yu et al., 2022). Ayoub et al. reported total or partial loss of COL1A1 exon 6 in patients with EDS type arthrochalasia (aEDS) in 2019 (Ayoub et al., 2020). Additionally, a higher overall variation rate of the COL1A1 gene in Chinese female DDH patients was reported 6 years previously (Zhao et al., 2013a) The application of NGS (such as WES and ES) to DDH-related syndromes has the potential to produce more precise results. This will enable the establishment of connections between DDH and other clinical presentations in syndromes as well as the pursuit of an understanding of the pathogenesis of DDH from the perspective of mutation-related genetics.Notably, although DDH has been suggested to be annual screened in trisomy 21 patient (van Gijzen et al., 2019), there is still blank in the genetic study of trisomy 21 children with DDH.
Although accumulating data has demonstrated that genetic components play a crucial role in the etiology of DDH, most studies examining the pathophysiologic mechanisms underlying the development of DDH are superficial. The genetic basis of DDH remains largely unknown, and researchers recommend that further studies on the molecular mechanism of DDH be carried out based on variant genes and loci.
The future of DDH treatment is early screening, diagnosis, and treatment, which can prevent or reduce the occurrence of DDH with appropriate genetic counseling. Without reliable genetic data, early treatment cannot be effectively implemented. Understanding DDH associated genetics will aid in early disease diagnosis, which is essential for proper disease management and prevention through genetic counseling.
Conclusion
There is a paucity of definitive data regarding the genetics of DDH. In this review, we highlighted the potentially crucial role of specific molecules in the pathogenesis of DDH, such as CX3CR1, KANSL1, GDF5, TGFβ1, HSPG2, and COL1A1, which merit further investigation. The data obtained by whole gene studies is beneficial for improving the accuracy of diagnosis when combined with clinical presentation and auxiliary detection. This leads to improved treatment efficiency and a reduced risk of movement impairment. However, it is important not to overlook the interindividual heterogeneity and the influence of environmental factors.
Author contributions
XZ: Writing–original draft. SL: Writing–review and editing. ZY: Writing–review and editing. YL: Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Basic Research project of the Education Department of Liaoning Province (grant number LJKMZ20221195) by ZY.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agenbag, G., Vorster, A., Julius, S., Ramesar, R., and Beighton, P. (2020). Namaqualand hip dysplasia in South Africa: the molecular determinant elucidated. S Afr. Med. J. 111 (1), 57–60. doi:10.7196/SAMJ.2020.v111i1.14561
Almuqbil, M., AbuMelha, A., and Albokhari, D. (2022). Milder presentation of autosomal dominant fatty acyl CoA reductase 1-related syndrome: report of the first Middle Eastern patient and review of the literature. Clin. Case Rep. 10 (10), e6307. doi:10.1002/ccr3.6307
Al-Rakan, M. A., Abothnain, M. D., Alrifai, M. T., and Alfadhel, M. (2018). Extending the ophthalmological phenotype of Galloway-Mowat syndrome with distinct retinal dysfunction: a report and review of ocular findings. BMC Ophthalmol. 18 (1), 147. doi:10.1186/s12886-018-0820-4
Arenzana-Seisdedos, F., and Parmentier, M. (2006). Genetics of resistance to HIV infection: role of co-receptors and co-receptor ligands. Semin. Immunol. 18 (6), 387–403. doi:10.1016/j.smim.2006.07.007
Ayoub, S., Ghali, N., Angwin, C., Baker, D., Baffini, S., Brady, A. F., et al. (2020). Clinical features, molecular results, and management of 12 individuals with the rare arthrochalasia Ehlers-Danlos syndrome. Am. J. Med. Genet. A 182 (5), 994–1007. doi:10.1002/ajmg.a.61523
Baghdadi, T., Nejadhosseinian, M., Shirkoohi, R., Mostafavi Tabatabaee, R., Tamehri, S. S., Saffari, M., et al. (2019). DNA hypermethylation of GDF5 in developmental dysplasia of the hip (DDH). Mol. Genet. Genomic Med. 7 (9), e887. doi:10.1002/mgg3.887
Basit, S., Albalawi, A. M., Alharby, E., and Khoshhal, K. I. (2017). Exome sequencing identified rare variants in genes HSPG2 and ATP2B4 in a family segregating developmental dysplasia of the hip. BMC Med. Genet. 18 (1), 34. doi:10.1186/s12881-017-0393-8
Basit, S., Alharby, E., Albalawi, A. M., and Khoshhal, K. I. (2018). Whole genome SNP genotyping in a family segregating developmental dysplasia of the hip detected runs of homozygosity on chromosomes 15q13.3 and 19p13.2. Congenit. Anom. (Kyoto) 58 (2), 56–61. doi:10.1111/cga.12235
Bauge, C., Girard, N., Lhuissier, E., Bazille, C., and Boumediene, K. (2014). Regulation and role of TGFβ signaling pathway in aging and osteoarthritis joints. Aging Dis. 5 (6), 394–405. doi:10.14336/AD.2014.0500394
Bennet, G. C., Rang, M., Roye, D. P., and Aprin, H. (1982). Dislocation of the hip in trisomy 21. J. Bone Jt. Surg. Br. 64 (3), 289–294. doi:10.1302/0301-620X.64B3.6212586
Bo, N., Peng, W., Xinghong, P., and Ma, R. (2012). Early cartilage degeneration in a rat experimental model of developmental dysplasia of the hip. Connect. Tissue Res. 53 (6), 513–520. doi:10.3109/03008207.2012.700346
Cahill, T. J., Sun, X., Ravaud, C., Villa Del Campo, C., Klaourakis, K., Lupu, I. E., et al. (2021). Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development 148 (3), dev194563. doi:10.1242/dev.194563
Cengic, T., Trkulja, V., Pavelic, S. K., Ratkaj, I., Markova-Car, E., Mikolaucic, M., et al. (2015). Association of TGFB1 29C/T and IL6 -572G/C polymorphisms with developmental hip dysplasia: a case-control study in adults with severe osteoarthritis. Int. Orthop. 39 (4), 793–798. doi:10.1007/s00264-015-2675-0
Chen, C. H., and Behringer, R. R. (2022). Transgenic human HOXB1-9 directs anterior-posterior axial skeleton pattern in Hoxb1-9 deficient mice. Differentiation 127, 1–11. doi:10.1016/j.diff.2022.07.002
Chen, J., Zhang, W. B., He, J. Z., Zhang, R., Cao, Y. Q., and Liu, X. (2018). Developmental dysplasia of the hip: a special pathology. Chin. J. Traumatol. 21 (4), 238–242. doi:10.1016/j.cjtee.2018.02.001
Chen, Y., Li, L., Wang, E., Zhang, L., and Zhao, Q. (2019). Abnormal expression of Pappa2 gene may indirectly affect mouse hip development through the IGF signaling pathway. Endocrine 65 (2), 440–450. doi:10.1007/s12020-019-01975-0
Cheng, B., Jia, Y., Wen, Y., Hou, W., Xu, K., Liang, C., et al. (2021). Integrative analysis of MicroRNA and mRNA sequencing data identifies novel candidate genes and pathways for developmental dysplasia of hip. Cartilage 13 (2_Suppl. l), 1618S–26S. doi:10.1177/1947603521990859
Chijimatsu, R., and Saito, T. (2019). Mechanisms of synovial joint and articular cartilage development. Cell Mol. Life Sci. 76 (20), 3939–3952. doi:10.1007/s00018-019-03191-5
Christians, J. K., de Zwaan, D. R., and Fung, S. H. (2013). Pregnancy associated plasma protein A2 (PAPP-A2) affects bone size and shape and contributes to natural variation in postnatal growth in mice. PLoS One 8 (2), e56260. doi:10.1371/journal.pone.0056260
Church, V. L., and Francis-West, P. (2002). Wnt signalling during limb development. Int. J. Dev. Biol. 46 (7), 927–936.
Conover, C. A., Boldt, H. B., Bale, L. K., Clifton, K. B., Grell, J. A., Mader, J. R., et al. (2011). Pregnancy-associated plasma protein-A2 (PAPP-A2): tissue expression and biological consequences of gene knockout in mice. Endocrinology 152 (7), 2837–2844. doi:10.1210/en.2011-0036
Cui, R. R., Huang, J., Yi, L., Xie, H., Zhou, H. D., Yuan, L. Q., et al. (2007). WISP3 suppresses insulin-like growth factor signaling in human chondrocytes. Mol. Cell Endocrinol. 279 (1-2), 1–8. doi:10.1016/j.mce.2007.08.007
Dai, J., Shi, D., Zhu, P., Qin, J., Ni, H., Xu, Y., et al. (2008). Association of a single nucleotide polymorphism in growth differentiate factor 5 with congenital dysplasia of the hip: a case-control study. Arthritis Res. Ther. 10 (5), R126. doi:10.1186/ar2540
Del Brío Castillo, R., Squires, J. E., and McKiernan, P. J. (2019). A novel mutation in VPS33B gene causing a milder ARC syndrome phenotype with prolonged survival. JIMD Rep. 47 (1), 4–8. doi:10.1002/jmd2.12027
Desteli, E. E., Piskin, A., Gulman, A. B., Kaymaz, F., Koksal, B., and Erdogan, M. (2013). Estrogen receptors in hip joint capsule and ligamentum capitis femoris of babies with developmental dysplasia of the hip. Acta Orthop. Traumatol. Turc 47 (3), 158–161. doi:10.3944/aott.2013.2772
Ding, R., Liu, X., Zhang, J., Yuan, J., Zheng, S., Cheng, X., et al. (2021). Downregulation of miR-1-3p expression inhibits the hypertrophy and mineralization of chondrocytes in DDH. J. Orthop. Surg. Res. 16 (1), 512. doi:10.1186/s13018-021-02666-1
Doncheva, N. T., Palasca, O., Yarani, R., Litman, T., Anthon, C., Groenen, M. A. M., et al. (2021). Human pathways in animal models: possibilities and limitations. Nucleic Acids Res. 49 (4), 1859–1871. doi:10.1093/nar/gkab012
Dunn, P. M. (1976). Perinatal observations on the etiology of congenital dislocation of the hip. Clin. Orthop. Relat. Res. 119, 11–22. doi:10.1097/00003086-197609000-00004
Eftekhari, H., Hosseini, S. R., Pourreza Baboli, H., Mafi Golchin, M., Heidari, L., Abedian, Z., et al. (2018). Association of interleukin-6 (rs1800796) but not transforming growth factor beta 1 (rs1800469) with serum calcium levels in osteoporotic patients. Gene 671, 21–27. doi:10.1016/j.gene.2018.05.118
Farquharson, C., Lester, D., Seawright, E., Jefferies, D., and Houston, B. (1999). Microtubules are potential regulators of growth-plate chondrocyte differentiation and hypertrophy. Bone 25 (4), 405–412. doi:10.1016/s8756-3282(99)00187-8
Feldman, G., Dalsey, C., Fertala, K., Azimi, D., Fortina, P., Devoto, M., et al. (2010). The Otto Aufranc Award: identification of a 4 Mb region on chromosome 17q21 linked to developmental dysplasia of the hip in one 18-member, multigeneration family. Clin. Orthop. Relat. Res. 468 (2), 337–344. doi:10.1007/s11999-009-1073-6
Feldman, G., Kappes, D., Mookerjee-Basu, J., Freeman, T., Fertala, A., and Parvizi, J. (2019). Novel mutation in Teneurin 3 found to co-segregate in all affecteds in a multi-generation family with developmental dysplasia of the hip. J. Orthop. Res. 37 (1), 171–180. doi:10.1002/jor.24148
Feldman, G., Offemaria, A., Sawan, H., Parvizi, J., and Freeman, T. A. (2017). A murine model for developmental dysplasia of the hip: ablation of CX3CR1 affects acetabular morphology and gait. J. Transl. Med. 15 (1), 233. doi:10.1186/s12967-017-1335-0
Feldman, G. J., Parvizi, J., Levenstien, M., Scott, K., Erickson, J. A., Fortina, P., et al. (2013). Developmental dysplasia of the hip: linkage mapping and whole exome sequencing identify a shared variant in CX3CR1 in all affected members of a large multigeneration family. J. Bone Min. Res. 28 (12), 2540–2549. doi:10.1002/jbmr.1999
Feng, W. J., Wang, H., Shen, C., Zhu, J. F., and Chen, X. D. (2017). Severe cartilage degeneration in patients with developmental dysplasia of the hip. IUBMB Life 69 (3), 179–187. doi:10.1002/iub.1606
Frigola, G., Del Rincon, O. G., Florian, V. B., Fita, A. V., Campos, B., Pauta, M., et al. (2021). Histopathology of recurrent Steel syndrome in fetuses caused by novel variants of COL27A1 gene. Virchows Arch. 479 (2), 413–418. doi:10.1007/s00428-020-02979-2
Funato, N., Nakamura, M., Richardson, J. A., Srivastava, D., and Yanagisawa, H. (2015). Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum. Mol. Genet. 24 (2), 424–435. doi:10.1093/hmg/ddu458
Gamerdinger, U., Bosse, K., Eggermann, T., Kalscheuer, V., Schwanitz, G., and Engels, H. (2006). First report of a partial trisomy 3q12-q23 de novo--FISH breakpoint determination and phenotypic characterization. Eur. J. Med. Genet. 49 (3), 225–234. doi:10.1016/j.ejmg.2005.07.002
Genovesi, M. L., Guadagnolo, D., Marchionni, E., Giovannetti, A., Traversa, A., Panzironi, N., et al. (2021). GDF5 mutation case report and a systematic review of molecular and clinical spectrum: expanding current knowledge on genotype-phenotype correlations. Bone 144, 115803. doi:10.1016/j.bone.2020.115803
Gkiatas, I., Boptsi, A., Tserga, D., Gelalis, I., Kosmas, D., and Pakos, E. (2019). Developmental dysplasia of the hip: a systematic literature review of the genes related with its occurrence. EFORT Open Rev. 4 (10), 595–601. doi:10.1302/2058-5241.4.190006
Gonzaga-Jauregui, C., Yesil, G., Nistala, H., Gezdirici, A., Bayram, Y., Nannuru, K. C., et al. (2020). Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide. Eur. J. Hum. Genet. 28 (9), 1243–1264. doi:10.1038/s41431-020-0632-x
Granchi, D., Stea, S., Sudanese, A., Toni, A., Baldini, N., and Giunti, A. (2002). Association of two gene polymorphisms with osteoarthritis secondary to hip dysplasia. Clin. Orthop. Relat. Res. 403 (403), 108–117. doi:10.1097/00003086-200210000-00018
Gumus, E., Temiz, E., Sarikaya, B., Yuksekdag, O., Sipahioglu, S., and Gonel, A. (2021). The association between BMP-2, UQCC1 and CX3CR1 polymorphisms and the risk of developmental dysplasia of the hip. Indian J. Orthop. 55 (1), 169–175. doi:10.1007/s43465-020-00235-y
Haider, S., Lackner, A. I., Dietrich, B., Kunihs, V., Haslinger, P., Meinhardt, G., et al. (2022). Transforming growth factor-β signaling governs the differentiation program of extravillous trophoblasts in the developing human placenta. Proc. Natl. Acad. Sci. U. S. A. 119 (28), e2120667119. doi:10.1073/pnas.2120667119
Han, S., Xu, X., Wen, J., Wang, J., Xiao, S., Pan, L., et al. (2022). New genetic mutations in a Chinese child with Ehlers-Danlos syndrome-like spondyloepimetaphyseal dysplasia: a case report. Front. Pediatr. 10, 1073748. doi:10.3389/fped.2022.1073748
Hao, Z., Dai, J., Shi, D., Xu, Z., Chen, D., Zhao, B., et al. (2014). Association of a single nucleotide polymorphism in HOXB9 with developmental dysplasia of the hip: a case-control study. J. Orthop. Res. 32 (2), 179–182. doi:10.1002/jor.22507
Harsanyi, S., Zamborsky, R., Krajciova, L., Bohmer, D., Kokavec, M., and Danisovic, L. (2021a). Association analysis of GDF5 and contributing factors in developmental dysplasia of the hip in infants. Ortop. Traumatol. Rehabil. 23 (5), 335–339. doi:10.5604/01.3001.0015.4348
Harsanyi, S., Zamborsky, R., Krajciova, L., Kokavec, M., and Danisovic, L. (2020). Developmental dysplasia of the hip: a review of etiopathogenesis, risk factors, and genetic aspects. Med. Kaunas. 56 (4), 153. doi:10.3390/medicina56040153
Harsanyi, S., Zamborsky, R., Krajciova, L., Kokavec, M., and Danisovic, L. (2021b). Genetic study of IL6, GDF5 and PAPPA2 in association with developmental dysplasia of the hip. Genes (Basel) 12 (7), 986. doi:10.3390/genes12070986
Hassan, M. H., Elsadek, A. A. M., Mahmoud, M. A., and Elsadek, B. E. M. (2022). Vitamin D receptor gene polymorphisms and risk of knee osteoarthritis: possible correlations with TNF-α, macrophage migration inhibitory factor, and 25-hydroxycholecalciferol status. Biochem. Genet. 60 (2), 611–628. doi:10.1007/s10528-021-10116-0
Hatzikotoulas, K., Roposch, A., Consortium, DDHCC, Shah, K. M., Clark, M. J., Bratherton, S., et al. (2018). Genome-wide association study of developmental dysplasia of the hip identifies an association with GDF5. Commun. Biol. 1, 56. doi:10.1038/s42003-018-0052-4
Igrek, S., Onay, T., Akgulle, A. H., Polat, M., Guney, A. I., and Muratli, H. H. (2021). The association of interleukin-6 (IL-6) -572G/C and transforming growth factor beta 1 (TGFB1) 29C/T single nucleotide polymorphisms (SNPs) with developmental dysplasia of the hip: a case control study. Acta Chir. Orthop. Traumatol. Cech 88 (5), 339–343. doi:10.55095/achot2021/050
Ikeda, K., Horie-Inoue, K., and Inoue, S. (2019). Functions of estrogen and estrogen receptor signaling on skeletal muscle. J. Steroid Biochem. Mol. Biol. 191, 105375. doi:10.1016/j.jsbmb.2019.105375
Jawadi, A. H., Wakeel, A., Tamimi, W., Nasr, A., Iqbal, Z., Mashhour, A., et al. (2018). Association analysis between four vitamin D receptor gene polymorphisms and developmental dysplasia of the hip. J. Genet. 97 (4), 925–930. doi:10.1007/s12041-018-0984-y
Ji, X., Liu, T., Zhao, S., Li, J., Li, L., and Wang, E. (2020). WISP-2, an upregulated gene in hip cartilage from the DDH model rats, induces chondrocyte apoptosis through PPARγ in vitro. FASEB J. 34 (4), 4904–4917. doi:10.1096/fj.201901915R
Jia, J., Li, L., Zhao, Q., Zhang, L., Ru, J., Liu, X., et al. (2012). Association of a single nucleotide polymorphism in pregnancy-associated plasma protein-A2 with developmental dysplasia of the hip: a case-control study. Osteoarthr. Cartil. 20 (1), 60–63. doi:10.1016/j.joca.2011.10.004
Kany, S., Vollrath, J. T., and Relja, B. (2019). Cytokines in inflammatory disease. Int. J. Mol. Sci. 20 (23), 6008. doi:10.3390/ijms20236008
Kapoor, B., Dunlop, C., Wynn-Jones, C., Fryer, A. A., Strange, R. C., and Maffulli, N. (2007). Vitamin D and oestrogen receptor polymorphisms in developmental dysplasia of the hip and primary protrusio acetabuli--a preliminary study. J. Negat. Results Biomed. 6, 7. doi:10.1186/1477-5751-6-7
Kenanidis, E., Gkekas, N. K., Karasmani, A., Anagnostis, P., Christofilopoulos, P., and Tsiridis, E. (2020). Genetic predisposition to developmental dysplasia of the hip. J. Arthroplasty 35 (1), 291–300. doi:10.1016/j.arth.2019.08.031
Kiapour, A. M., Cao, J., Young, M., and Capellini, T. D. (2018). The role of Gdf5 regulatory regions in development of hip morphology. PLoS One 13 (11), e0202785. doi:10.1371/journal.pone.0202785
Kim, H. J., Prasad, V., Hyung, S. W., Lee, Z. H., Lee, S. W., Bhargava, A., et al. (2012). Plasma membrane calcium ATPase regulates bone mass by fine-tuning osteoclast differentiation and survival. J. Cell Biol. 199 (7), 1145–1158. doi:10.1083/jcb.201204067
Kimura, M., Taketani, T., Horie, A., Isumi, H., Sejima, H., and Yamaguchi, S. (2006). Two Japanese families with hyperekplexia who have a Arg271Gln mutation in the glycine receptor alpha 1 subunit gene. Brain Dev. 28 (4), 228–231. doi:10.1016/j.braindev.2005.08.007
Ko, J. M., Yoo, Y., Seo, J., Cho, J. S., Choi, M., Chae, J. H., et al. (2016). Wiedemann-steiner syndrome with 2 novel KMT2A mutations: variable severity in psychomotor development and musculoskeletal manifestation. J. Child Neurology 32 (2), 237–242. doi:10.1177/0883073816674095
Kohara, Y., Haraguchi, R., Kitazawa, R., and Kitazawa, S. (2020). Knockdown of Lrp1 in RAW264 cells inhibits osteoclast differentiation and osteoclast-osteoblast interactions in vitro. Biochem. Biophys. Res. Commun. 523 (4), 961–965. doi:10.1016/j.bbrc.2020.01.065
Kolundzic, R., Trkulja, V., Mikolaucic, M., Kolundzic, M. J., Pavelic, S. K., and Pavelic, K. (2011). Association of interleukin-6 and transforming growth factor-β1 gene polymorphisms with developmental hip dysplasia and severe adult hip osteoarthritis: a preliminary study. Cytokine 54 (2), 125–128. doi:10.1016/j.cyto.2011.02.004
Li, D., Wang, H., He, J. Y., Wang, C. L., Feng, W. J., Shen, C., et al. (2019). Inflammatory and fibrosis infiltration in synovium associated with the progression in developmental dysplasia of the hip. Mol. Med. Rep. 19 (4), 2808–2816. doi:10.3892/mmr.2019.9910
Li, J., Hua, X., Jones, A. C., Williams, S., Jin, Z., Fisher, J., et al. (2016). The influence of the representation of collagen fibre organisation on the cartilage contact mechanics of the hip joint. J. Biomech. 49 (9), 1679–1685. doi:10.1016/j.jbiomech.2016.03.050
Liu, J., Zhou, W., Chen, Y., and Li, L. (2021). Acetabular development and fate of inverted limbus in rabbits: experimental observation from an animal model. J. Orthop. Res. 39 (12), 2595–2603. doi:10.1002/jor.25005
Liu, L., Zhao, M., Xie, Z. G., Liu, J., Peng, H. P., Pei, Y. F., et al. (2020a). Twelve new genomic loci associated with bone mineral density. Front. Endocrinol. (Lausanne) 11, 243. doi:10.3389/fendo.2020.00243
Liu, X., Deng, X., Ding, R., Cheng, X., and Jia, J. (2020b). Chondrocyte suppression is mediated by miR-129-5p via GDF11/SMAD3 signaling in developmental dysplasia of the hip. J. Orthop. Res. 38 (12), 2559–2572. doi:10.1002/jor.24713
Liu, Y., Zan, L., Zhao, S., Huang, H., Li, Y., Tang, Z., et al. (2010). Molecular cloning, expression and characterization of bovine UQCC and its association with body measurement traits. Mol. Cells 30 (5), 393–401. doi:10.1007/s10059-010-0129-5
Loder, R. T., and Shafer, C. (2015). The demographics of developmental hip dysplasia in the Midwestern United States (Indiana). J. Child. Orthop. 9 (1), 93–98. doi:10.1007/s11832-015-0636-1
Lories, R. J., Peeters, J., Bakker, A., Tylzanowski, P., Derese, I., Schrooten, J., et al. (2007). Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 56 (12), 4095–4103. doi:10.1002/art.23137
Loughlin, J. (2011). Knee osteoarthritis, lumbar-disc degeneration and developmental dysplasia of the hip--an emerging genetic overlap. Arthritis Res. Ther. 13 (2), 108. doi:10.1186/ar3291
Lowe, D. A., Lepori-Bui, N., Fomin, P. V., Sloofman, L. G., Zhou, X., Farach-Carson, M. C., et al. (2014). Deficiency in perlecan/HSPG2 during bone development enhances osteogenesis and decreases quality of adult bone in mice. Calcif. Tissue Int. 95 (1), 29–38. doi:10.1007/s00223-014-9859-2
Ma, W., Zha, Z., Chen, K., Chen, H., Wu, Y., Ma, J., et al. (2017). Genetic association study of common variants in TGFB1 and IL-6 with developmental dysplasia of the hip in Han Chinese population. Sci. Rep. 7 (1), 10287. doi:10.1038/s41598-017-11185-1
Maas, S., Shaw, A., Bikker, H., Hennekam, R. C. M., et al. (1993). “Trichorhinophalangeal syndrome,” in GeneReviews((R)). M. P. Adam, D. B. Everman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and L. J. H. Bean (Seattle (WA).
Machol, K., Jain, M., Almannai, M., Orand, T., Lu, J. T., Tran, A., et al. (2017). Corner fracture type spondylometaphyseal dysplasia: overlap with type II collagenopathies. Am. J. Med. Genet. A 173 (3), 733–739. doi:10.1002/ajmg.a.38059
Marini, J. C., Forlino, A., Bachinger, H. P., Bishop, N. J., Byers, P. H., Paepe, A., et al. (2017). Osteogenesis imperfecta. Nat. Rev. Dis. Prim. 3, 17052. doi:10.1038/nrdp.2017.52
Mark, P. R., Murray, S. A., Yang, T., Eby, A., Lai, A., Lu, D., et al. (2022). Autosomal recessive LRP1-related syndrome featuring cardiopulmonary dysfunction, bone dysmorphology, and corneal clouding. Cold Spring Harb. Mol. Case Stud. 8 (6), a006169. doi:10.1101/mcs.a006169
Masri, A., Shboul, M., Khasawneh, A., Jadallah, R., Almustafa, A., Escande-Beillard, N., et al. (2020). Congenital insensitivity to pain with anhidrosis syndrome: a series from Jordan. Clin. Neurol. Neurosurg. 189, 105636. doi:10.1016/j.clineuro.2019.105636
Mayo, J. L., Holden, D. N., Barrow, J. R., and Bridgewater, L. C. (2009). The transcription factor Lc-Maf participates in Col27a1 regulation during chondrocyte maturation. Exp. Cell Res. 315 (13), 2293–2300. doi:10.1016/j.yexcr.2009.04.020
Meulenbelt, I., Chapman, K., Dieguez-Gonzalez, R., Shi, D., Tsezou, A., Dai, J., et al. (2009). Large replication study and meta-analyses of DVWA as an osteoarthritis susceptibility locus in European and Asian populations. Hum. Mol. Genet. 18 (8), 1518–1523. doi:10.1093/hmg/ddp053
Miyamoto, Y., Shi, D., Nakajima, M., Ozaki, K., Sudo, A., Kotani, A., et al. (2008). Common variants in DVWA on chromosome 3p24.3 are associated with susceptibility to knee osteoarthritis. Nat. Genet. 40 (8), 994–998. doi:10.1038/ng.176
Muthuirulan, P., Zhao, D., Young, M., Richard, D., Liu, Z., Emami, A., et al. (2021). Joint disease-specificity at the regulatory base-pair level. Nat. Commun. 12 (1), 4161. doi:10.1038/s41467-021-24345-9
Nejadhosseinian, M., Haerian, H., Shirkoohi, R., Karami, J., and Mortazavi, S. M. J. (2022). Evaluation of CX3CR1 gene DNA methylation in developmental dysplasia of the hip (DDH). J. Orthop. Surg. Res. 17 (1), 436. doi:10.1186/s13018-022-03324-w
Ning, B., Jin, R., Wan, L., and Wang, D. (2018). Cellular and molecular changes to chondrocytes in an in vitro model of developmental dysplasia of the hip-an experimental model of DDH with swaddling position. Mol. Med. Rep. 18 (4), 3873–3881. doi:10.3892/mmr.2018.9384
Palotie, A., Vaisanen, P., Ott, J., Ryhanen, L., Elima, K., Vikkula, M., et al. (1989). Predisposition to familial osteoarthrosis linked to type II collagen gene. Lancet. 1 (8644), 924–927. doi:10.1016/s0140-6736(89)92507-5
Parker, M. J., Fryer, A. E., Shears, D. J., Lachlan, K. L., McKee, S. A., Magee, A. C., et al. (2015). De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am. J. Med. Genet. A 167A (10), 2231–2237. doi:10.1002/ajmg.a.37189
Qiao, L., Yan, W., Dai, J., Yao, Y., Chen, D., Xu, Z., et al. (2017). A novel missense variant in TXNDC3 is associated with developmental dysplasia of the hip in Han Chinese population. Int. J. Clin. Exp. Pathol. 10 (10), 10483–10488.
Rauhio, A., Uusi-Rasi, K., Kunnas, T., Nikkari, S. T., Kannus, P., and Sievanen, H. (2011). Estrogen receptor-1 genotype is associated with bone structure in premenopausal obese women. Maturitas 68 (4), 362–367. doi:10.1016/j.maturitas.2010.12.006
Rosenkrantz, J. L., Gaffney, J. E., Roberts, V. H. J., Carbone, L., and Chavez, S. L. (2021). Transcriptomic analysis of primate placentas and novel rhesus trophoblast cell lines informs investigations of human placentation. BMC Biol. 19 (1), 127. doi:10.1186/s12915-021-01056-7
Rouault, K., Scotet, V., Autret, S., Gaucher, F., Dubrana, F., Tanguy, D., et al. (2009). Do HOXB9 and COL1A1 genes play a role in congenital dislocation of the hip? Study in a Caucasian population. Osteoarthr. Cartil. 17 (8), 1099–1105. doi:10.1016/j.joca.2008.12.012
Rouault, K., Scotet, V., Autret, S., Gaucher, F., Dubrana, F., Tanguy, D., et al. (2010). Evidence of association between GDF5 polymorphisms and congenital dislocation of the hip in a Caucasian population. Osteoarthr. Cartil. 18 (9), 1144–1149. doi:10.1016/j.joca.2010.05.018
Rubin, S., Agrawal, A., Stegmaier, J., Krief, S., Felsenthal, N., Svorai, J., et al. (2021). Application of 3D MAPs pipeline identifies the morphological sequence chondrocytes undergo and the regulatory role of GDF5 in this process. Nat. Commun. 12 (1), 5363. doi:10.1038/s41467-021-25714-0
Rubini, M., Cavallaro, A., Calzolari, E., Bighetti, G., and Sollazzo, V. (2008). Exclusion of COL2A1 and VDR as developmental dysplasia of the hip genes. Clin. Orthop. Relat. Res. 466 (4), 878–883. doi:10.1007/s11999-008-0120-z
Sauvegarde, C., Paul, D., Bridoux, L., Jouneau, A., Degrelle, S., Hue, I., et al. (2016). Dynamic pattern of HOXB9 protein localization during oocyte maturation and early embryonic development in mammals. PLoS One 11 (10), e0165898. doi:10.1371/journal.pone.0165898
Schaeffer, E. K., Study Group, I., and Mulpuri, K. (2018). Developmental dysplasia of the hip: addressing evidence gaps with a multicentre prospective international study. Med. J. Aust. 208 (8), 359–364. doi:10.5694/mja18.00154
Schulte-Merker, S., van Eeden, F. J., Halpern, M. E., Kimmel, C. B., and Nusslein-Volhard, C. (1994). No tail (ntl) is the zebrafish homologue of the mouse T (Brachyury) gene. Development 120 (4), 1009–1015. doi:10.1242/dev.120.4.1009
Sekimoto, T., Ishii, M., Emi, M., Kurogi, S., Funamoto, T., Yonezawa, Y., et al. (2017). Copy number loss in the region of the ASPN gene in patients with acetabular dysplasia: ASPN CNV in acetabular dysplasia. Bone Jt. Res. 6 (7), 439–445. doi:10.1302/2046-3758.67.BJR-2016-0094.R1
Shi, D., Dai, J., Zhu, P., Qin, J., Zhu, L., Zhu, H., et al. (2011). Association of the D repeat polymorphism in the ASPN gene with developmental dysplasia of the hip: a case-control study in Han Chinese. Arthritis Res. Ther. 13 (1), R27. doi:10.1186/ar3252
Shi, D., Nakamura, T., Nakajima, M., Dai, J., Qin, J., Ni, H., et al. (2008). Association of single-nucleotide polymorphisms in RHOB and TXNDC3 with knee osteoarthritis susceptibility: two case-control studies in East Asian populations and a meta-analysis. Arthritis Res. Ther. 10 (3), R54. doi:10.1186/ar2423
Shi, D., Sun, W., Xu, X., Hao, Z., Dai, J., Xu, Z., et al. (2014). A replication study for the association of rs726252 in PAPPA2 with developmental dysplasia of the hip in Chinese Han population. Biomed. Res. Int. 2014, 979520. doi:10.1155/2014/979520
Storm, E. E., Huynh, T. V., Copeland, N. G., Jenkins, N. A., Kingsley, D. M., and Lee, S. J. (1994). Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature 368 (6472), 639–643. doi:10.1038/368639a0
Sun, Y., Wang, C., Hao, Z., Dai, J., Chen, D., Xu, Z., et al. (2015). A common variant of ubiquinol-cytochrome c reductase complex is associated with DDH. PLoS One 10 (4), e0120212. doi:10.1371/journal.pone.0120212
Sun, Y., Wang, F., Sun, X., Wang, X., Zhang, L., and Li, Y. (2017). CX3CR1 regulates osteoarthrosis chondrocyte proliferation and apoptosis via Wnt/β-catenin signaling. Biomed. Pharmacother. 96, 1317–1323. doi:10.1016/j.biopha.2017.11.080
Sun, Y., You, Y., Dai, K., Zhang, J., Yan, M., and Zhang, Y. (2019b). Genetic variant of WIF1 gene is functionally associated with developmental dysplasia of the hip in Han Chinese population. Sci. Rep. 9 (1), 285. doi:10.1038/s41598-018-36532-8
Sun, Y., You, Y., Jiang, W., Zhai, Z., and Dai, K. (2019a). 3D-bioprinting a genetically inspired cartilage scaffold with GDF5-conjugated BMSC-laden hydrogel and polymer for cartilage repair. Theranostics 9 (23), 6949–6961. doi:10.7150/thno.38061
Tachmazidou, I., Hatzikotoulas, K., Southam, L., Esparza-Gordillo, J., Haberland, V., Zheng, J., et al. (2019). Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 51 (2), 230–236. doi:10.1038/s41588-018-0327-1
Tang, J., Liu, T., Wen, X., Zhou, Z., Yan, J., Gao, J., et al. (2021). Estrogen-related receptors: novel potential regulators of osteoarthritis pathogenesis. Mol. Med. 27 (1), 5. doi:10.1186/s10020-021-00270-x
Tian, W., Zhao, L., Wang, J., Suo, P., Wang, J., Cheng, L., et al. (2012). Association analysis between HOXD9 genes and the development of developmental dysplasia of the hip in Chinese female Han population. BMC Musculoskelet. Disord. 13, 59. doi:10.1186/1471-2474-13-59
Tickle, C. (2015). How the embryo makes a limb: determination, polarity and identity. J. Anat. 227 (4), 418–430. doi:10.1111/joa.12361
Todhunter, R. J., Garrison, S. J., Jordan, J., Hunter, L., Castelhano, M. G., Ash, K., et al. (2019). Gene expression in hip soft tissues in incipient canine hip dysplasia and osteoarthritis. J. Orthop. Res. 37 (2), 313–324. doi:10.1002/jor.24178
Topak, D., Seyithanoglu, M., Dogar, F., Karadeniz, A. A., Tanriverdi, B., Ozan, F., et al. (2021). Are vitamin D and vitamin D receptor levels different in children with developmental dysplasia of the hip? J. Orthop. Surg. Res. 16 (1), 24. doi:10.1186/s13018-020-02162-y
van Gijzen, A. F. M., Rouers, E. D. M., van Douveren, FQMP, Dieleman, J., Hendriks, J. G. E., Halbertsma, F. J. J., et al. (2019). Developmental dysplasia of the hip in children with Down syndrome: comparison of clinical and radiological examinations in a local cohort. Eur. J. Pediatr. 178 (4), 559–564. doi:10.1007/s00431-019-03322-x
Vaquero-Picado, A., Gonzalez-Moran, G., Garay, E. G., and Moraleda, L. (2019). Developmental dysplasia of the hip: update of management. EFORT Open Rev. 4 (9), 548–556. doi:10.1302/2058-5241.4.180019
Wang, C. L., Zuo, B., Li, D., Zhu, J. F., Xiao, F., Zhang, X. L., et al. (2020). The long noncoding RNA H19 attenuates force-driven cartilage degeneration via miR-483-5p/Dusp5. Biochem. Biophys. Res. Commun. 529 (2), 210–217. doi:10.1016/j.bbrc.2020.05.180
Wang, H., Zhang, X., He, J. Y., Zheng, X. F., Li, D., Li, Z., et al. (2015). Increasing expression of substance P and calcitonin gene-related peptide in synovial tissue and fluid contribute to the progress of arthritis in developmental dysplasia of the hip. Arthritis Res. Ther. 17 (1), 4. doi:10.1186/s13075-014-0513-1
Wang, H., Zheng, X. F., Zhang, X., Li, Z., Shen, C., Zhu, J. F., et al. (2014). Increasing substance P levels in serum and synovial tissues from patients with developmental dysplasia of the hip (DDH). BMC Musculoskelet. Disord. 15, 92. doi:10.1186/1471-2474-15-92
Wang, K., Shi, D., Zhu, P., Dai, J., Zhu, L., Zhu, H., et al. (2010). Association of a single nucleotide polymorphism in Tbx4 with developmental dysplasia of the hip: a case-control study. Osteoarthr. Cartil. 18 (12), 1592–1595. doi:10.1016/j.joca.2010.09.008
Weisfeld-Adams, J. D., Edelmann, L., Gadi, I. K., and Mehta, L. (2012). Phenotypic heterogeneity in a family with a small atypical microduplication of chromosome 22q11.2 involving TBX1. Eur. J. Med. Genet. 55 (12), 732–736. doi:10.1016/j.ejmg.2012.08.011
Woodacre, T., Ball, T., and Cox, P. (2016). Epidemiology of developmental dysplasia of the hip within the UK: refining the risk factors. J. Child. Orthop. 10 (6), 633–642. doi:10.1007/s11832-016-0798-5
Wu, R., Gao, G., and Xu, Y. (2020). Electrospun fibers immobilized with BMP-2 mediated by polydopamine combined with autogenous tendon to repair developmental dysplasia of the hip in a porcine model. Int. J. Nanomedicine 15, 6563–6577. doi:10.2147/IJN.S259028
Xu, J., Ye, W., Li, H., and Xu, L. (2022a). WNT1 expression influences the development of dysplasia of the hip via regulating RBPMS2/NOG-BMP2/4-GDF5- WISP2 pathway. Nucleosides Nucleotides Nucleic Acids 41 (8), 765–777. doi:10.1080/15257770.2022.2081337
Xu, R., Jiang, X., Lu, J., Wang, K., Sun, Y., and Zhang, Y. (2020). Genetic variant of COL11A2 gene is functionally associated with developmental dysplasia of the hip in Chinese Han population. Aging (Albany NY) 12 (9), 7694–7703. doi:10.18632/aging.103040
Xu, R., Zhang, F., Lu, J., Wang, K., Pan, P., Sun, Y., et al. (2021b). Secreted frizzled-related protein 3 was genetically and functionally associated with developmental dysplasia of the hip. Aging (Albany NY) 13 (8), 11281–11295. doi:10.18632/aging.202815
Xu, X., Bi, X., Wang, J., Gui, R., Li, T., Li, L., et al. (2022b). Identification of KANSL1 as a novel pathogenic gene for developmental dysplasia of the hip. J. Mol. Med. Berlin, Ger. 100 (8), 1159–1168. doi:10.1007/s00109-022-02220-4
Xu, X., Wang, B., Chen, Y., Zhou, W., and Li, L. (2021a). Replicative verification of susceptibility genes previously identified from families with segregating developmental dysplasia of the hip. Ital. J. Pediatr. 47 (1), 140. doi:10.1186/s13052-021-01087-4
Yamanaka, M., Ishijima, M., Tokita, A., Sakamoto, Y., Kaneko, H., Maezawa, K., et al. (2009). Association of oestrogen receptor gene polymorphism with the long-term results of rotational acetabular osteotomy. Int. Orthop. 33 (4), 1155–1164. doi:10.1007/s00264-009-0730-4
Yan, W., Hao, Z., Tang, S., Dai, J., Zheng, L., Yu, P., et al. (2019). A genome-wide association study identifies new genes associated with developmental dysplasia of the hip. Clin. Genet. 95 (3), 345–355. doi:10.1111/cge.13483
Yan, W., Zheng, L., Xu, X., Hao, Z., Zhang, Y., Lu, J., et al. (2022). Heterozygous LRP1 deficiency causes developmental dysplasia of the hip by impairing triradiate chondrocytes differentiation due to inhibition of autophagy. Proc. Natl. Acad. Sci. U. S. A. 119 (37), e2203557119. doi:10.1073/pnas.2203557119
Yang, S., Zusman, N., Lieberman, E., and Goldstein, R. Y. (2019). Developmental dysplasia of the hip. Pediatrics 143 (1), e20181147. doi:10.1542/peds.2018-1147
Yang, W., Jin, G., Qian, K., Zhang, C., Zhi, W., Yang, D., et al. (2022). Comprehensive bioinformatics analysis of susceptibility genes for developmental dysplasia of the hip. Intractable Rare Dis. Res. 11 (2), 70–80. doi:10.5582/irdr.2022.01043
Yerges-Armstrong, L. M., Yau, M. S., Liu, Y., Krishnan, S., Renner, J. B., Eaton, C. B., et al. (2014). Association analysis of BMD-associated SNPs with knee osteoarthritis. J. Bone Min. Res. 29 (6), 1373–1379. doi:10.1002/jbmr.2160
Yu, S., Wang, C., Lei, K., Leng, X., Zhang, L., Tian, F., et al. (2022). Case report: genetic analysis of a child with 18q deletion syndrome and developmental dysplasia of the hip. BMC Med. Genomics 15 (1), 199. doi:10.1186/s12920-022-01345-2
Zamborsky, R., Kokavec, M., Harsanyi, S., Attia, D., and Danisovic, L. (2019). Developmental dysplasia of hip: perspectives in genetic screening. Med. Sci. (Basel). 7 (4), 59. doi:10.3390/medsci7040059
Zhang, C. H., Gao, Y., Hung, H. H., Zhuo, Z., Grodzinsky, A. J., and Lassar, A. B. (2022). Creb5 coordinates synovial joint formation with the genesis of articular cartilage. Nat. Commun. 13 (1), 7295. doi:10.1038/s41467-022-35010-0
Zhang, J., Yan, M., Zhang, Y., Yang, H., and Sun, Y. (2018). Association analysis on polymorphisms in WISP3 gene and developmental dysplasia of the hip in Han Chinese population: a case-control study. Gene 664, 192–195. doi:10.1016/j.gene.2018.04.020
Zhang, L., Xu, X., Chen, Y., Li, L., Zhang, L., and Li, Q. (2020a). Mapping of developmental dysplasia of the hip to two novel regions at 8q23-q24 and 12p12. Exp. Ther. Med. 19 (4), 2799–2803. doi:10.3892/etm.2020.8513
Zhang, S., Doudoulakis, K. J., Khurwal, A., and Sarraf, K. M. (2020b). Developmental dysplasia of the hip. Br. J. Hosp. Med. (Lond) 81 (7), 1–8. doi:10.12968/hmed.2020.0223
Zhang, X., Meng, Q., Ma, R., Chen, G., Cheng, L., and Shen, J. (2015). Early acetabular cartilage degeneration in a rabbit model of developmental dysplasia of the hip. Int. J. Clin. Exp. Med. 8 (8), 14505–14512.
Zhao, L., Ma, Q., Feng, X., Fan, L., Jiao, Q., Wang, S., et al. (2019). Screening for developmental dysplasia of the hip in infants in tibet identifies increased prevalence associated with altitude. Med. Sci. Monit. 25, 5771–5775. doi:10.12659/MSM.916456
Zhao, L., Pan, H., Wang, J., Cheng, Z., Cheng, L., Wang, B., et al. (2013b). Two single nucleotide polymorphisms in the GDF5 gene are associated with development dysplasia of the hip in Chinese female population. Sci. China Life Sci. 56 (11), 1063–1065. doi:10.1007/s11427-013-4514-0
Zhao, L., Tian, W., Pan, H., Zhu, X., Wang, J., Cheng, Z., et al. (2013a). Variations of the COL1A1 gene promoter and the relation to developmental dysplasia of the hip. Genet. Test. Mol. Biomarkers 17 (11), 840–843. doi:10.1089/gtmb.2013.0179
Zhao, L., Zhou, Z., Wang, S., Jiao, Q., Wu, J., Ma, F., et al. (2017). A recurrent mutation in bone morphogenetic proteins-2-inducible kinase gene is associated with developmental dysplasia of the hip. Exp. Ther. Med. 13 (5), 1773–1778. doi:10.3892/etm.2017.4191
Zhou, W., Luo, W., Liu, D., Canavese, F., Li, L., and Zhao, Q. (2022). Fluoride increases the susceptibility of developmental dysplasia of the hip via increasing capsular laxity triggered by cell apoptosis and oxidative stress in vivo and in vitro. Ecotoxicol. Environ. Saf. 234, 113408. doi:10.1016/j.ecoenv.2022.113408
Zhu, L., Shi, D., Dai, J., Qin, J., Fan, J., Wang, Z., et al. (2011). Lack of evidence for association between DVWA gene polymorphisms and developmental dysplasia of the hip in Chinese Han population. Rheumatol. Int. 31 (7), 883–887. doi:10.1007/s00296-010-1410-9
Zhu, L. Q., Su, G. H., Dai, J., Zhang, W. Y., Yin, C. H., Zhang, F. Y., et al. (2019). Whole genome sequencing of pairwise human subjects reveals DNA mutations specific to developmental dysplasia of the hip. Genomics 111 (3), 320–326. doi:10.1016/j.ygeno.2018.02.006
Keywords: developmental dysplasia of the hip, susceptibility gene, single-nucleotide polymorphisms, CX3CR1, GDF5
Citation: Zhao X, Liu S, Yang Z and Li Y (2024) Molecular mechanisms and genetic factors contributing to the developmental dysplasia of the hip. Front. Genet. 15:1413500. doi: 10.3389/fgene.2024.1413500
Received: 07 April 2024; Accepted: 16 July 2024;
Published: 02 August 2024.
Edited by:
Roy Morello, University of Arkansas for Medical Sciences, United StatesReviewed by:
Ottmar Distl, University of Veterinary Medicine Hannover, GermanyAndre Travessa, Centro Hospitalar Lisboa Norte (CHLN), Portugal
Copyright © 2024 Zhao, Liu, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Li, bGl5b25nQHNqLWhvc3BpdGFsLm9yZw==; Zhonghua Yang, eWFuZ3poQHNqLWhvc3B0aWFsLm9yZw==