 Ashley M. Gefen
Ashley M. Gefen- Phoenix Children’s Hospital, Department of Pediatrics, Division of Nephrology, Phoenix, AZ, United States
Nephrolithiasis (NL) is a common condition worldwide. The incidence of NL and nephrocalcinosis (NC) has been increasing, along with their associated morbidity and economic burden. The etiology of NL and NC is multifactorial and includes both environmental components and genetic components, with multiple studies showing high heritability. Causative gene variants have been detected in up to 32% of children with NL and NC. Children with NL and NC are genotypically heterogenous, but often phenotypically relatively homogenous, and there are subsequently little data on the predictors of genetic childhood NL and NC. Most genetic diseases associated with NL and NC are secondary to hypercalciuria, including those secondary to hypercalcemia, renal phosphate wasting, renal magnesium wasting, distal renal tubular acidosis (RTA), proximal tubulopathies, mixed or variable tubulopathies, Bartter syndrome, hyperaldosteronism and pseudohyperaldosteronism, and hyperparathyroidism and hypoparathyroidism. The remaining minority of genetic diseases associated with NL and NC are secondary to hyperoxaluria, cystinuria, hyperuricosuria, xanthinuria, other metabolic disorders, and multifactorial etiologies. Genome-wide association studies (GWAS) in adults have identified multiple polygenic traits associated with NL and NC, often involving genes that are involved in calcium, phosphorus, magnesium, and vitamin D homeostasis. Compared to adults, there is a relative paucity of studies in children with NL and NC. This review aims to focus on the genetic component of NL and NC in children.
1 Introduction
In the United States, nephrolithiasis (NL) is relatively common, affecting approximately 1 in 11 adults (Scales et al., 2012). A study of 12.7 million children in the United States noted a rate of NL of 54.1 cases per 100,000 person-years in 2016 (Ward et al., 2019). Multiple studies have noted that the incidence of NL and nephrocalcinosis (NC) has increased over the past decade, especially in adolescents and female children (Novak et al., 2009; Dwyer et al., 2012; Ward et al., 2019; Li et al., 2020). There is high morbidity associated with NL and NC, including an increased risk of chronic kidney disease (CKD) and kidney failure (Zhe and Hang, 2017). In addition, the economic burden associated with treatment is high with an annual expenditure of over $10 billion in the United States based on data from 2006 (Economic Impact of Urologic Disease, 2012). In adults, obesity, metabolic syndrome, hypertension, and diabetes have been associated with NL (Taylor et al., 2005; Lieske et al., 2006). The etiology of NL and NC is multifactorial and includes both environmental components and genetic components. Compared to adults, there is a relative paucity of studies in children with NL and NC. This review aims to focus on the genetic component of NL and NC in children.
2 Heritability of nephrolithiasis and nephrocalcinosis
Twin studies have illustrated 56% heritability for NL with a significantly higher frequency in female twins compared to male twins (Goldfarb et al., 2005; Goldfarb et al., 2018). Twin studies and large population studies have also shown heritability of serum and urine electrolytes, with that of serum calcium and 24-h urine calcium ranging from 33% to 37% and 44%–52%, respectively (Hunter et al., 2002; Moulin et al., 2017). Heritability of 80% has also been noted for serum 25-hydroxy-vitamin D levels (Wjst et al., 2007).
3 Genetic causes of nephrolithiasis and nephrocalcinosis in children
Causative gene variants have been detected in 11.5%–31.9% of children with NL and NC using high-throughput multiplex PCR with next-generation exon sequencing (Halbritter et al., 2015; Braun et al., 2016; Gefen et al., 2023). Causative variants have been detected in 29.4% of children with NL and NC using whole exome sequencing of 30 genes associated with monogenic NL and NC and in 32.5% of children with NL using whole exome sequencing of 38 genes associated with monogenic NL in a pediatric Chinese cohort.
Children with NL and NC are genotypically heterogenous, but often phenotypically relatively homogenous, and there are subsequently little data on the predictors of genetic childhood NL and NC. The discordance between phenotype and genotype is well illustrated by a study that showed that 23.9% and 7.3% of children with suspected Dent disease and primary hyperoxaluria (PH), respectively, based on their phenotype were found to have disease causing variants in unrelated genes (Cogal et al., 2021). Regarding identified risk factors for genetic NL in children, a Chinese study found the following: positive family history, consanguinity, younger age at onset, presence of concurrent NC, and CKD (Zhao et al., 2022). In contrast, an American study of children with NL and NC found that the only factor predictive of genetic disease was low serum bicarbonate (Gefen et al., 2023).
Given the high diagnostic yield of genetic testing in this population as well as the genetic heterogeneity, phenotypic homogeneity, and lack of well-established predictors of genetic NL and NC, performing targeted genetic testing broadly for children with NL and NC has been suggested. Genetic testing has become more readily available in recent years with high throughput exon sequencing with whole-exome sequencing or exon panels. Genetic testing may provide clinically meaningful information and affect long-term outcomes. According to multiple studies in children with kidney diseases including NL and NC, genetic testing has the power to alter clinical management and surveillance in >40% of cases (Halbritter et al., 2015; Amlie-Wolf et al., 2021; Jayasinghe et al., 2021).
3.1 Conditions with hypercalciuria
Most genetic diseases associated with NL and NC are secondary to hypercalciuria, including those secondary to hypercalcemia, renal phosphate wasting, renal magnesium wasting, distal renal tubular acidosis (RTA), proximal tubulopathies, mixed or variable tubulopathies, Bartter syndrome, hyperaldosteronism and pseudohyperaldosteronism, hyperparathyroidism and hypoparathyroidism, and other causes. Each of these categories will be expanded on in the following subsections.
3.1.1 Hypercalcemia and hypocalcemia
Table 1 contains a list of genetic causes of primary hypercalcemia and hypocalcemia with hypercalciuria and NL and/or NC. These conditions are due to variants in CASR and CYP24A1 and will be discussed in greater detail below.
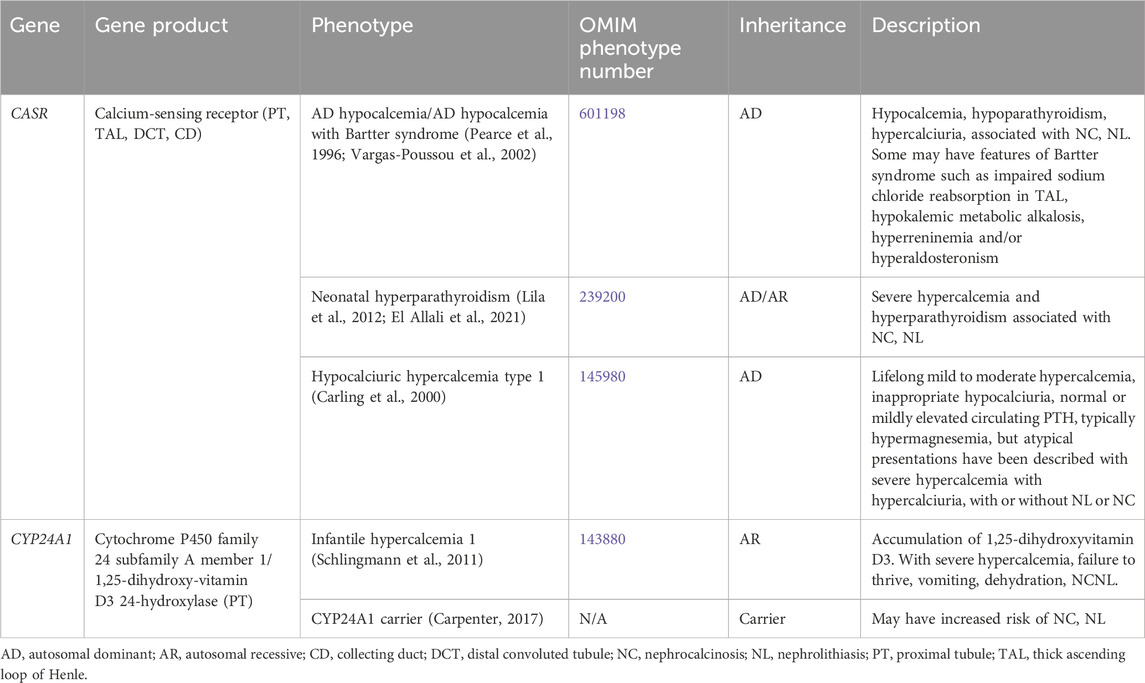
Table 1. Genetic causes of primary hypercalcemia and hypocalcemia with hypercalciuria, nephrolithiasis and/or nephrocalcinosis.
Supplementary Table S1 contains a list of genetic causes of secondary hypercalcemia and hypocalcemia with hypercalciuria and NL and/or NC. Conditions associated with secondary hypercalcemia include Williams-Beuren syndrome (7p11.23, autosomal dominant [AD] inheritance, OMIM phenotype number 194050), Oculoskeletodental syndrome (PIK32CA gene, autosomal recessive [AR] inheritance, OMIM phenotype number 618440) (Tiosano et al., 2019), Blue diaper syndrome (possibly PCSK1 gene, AR versus X-linked recessive [XLR] inheritance, OMIM phenotype number 211000) (Drummond et al., 1964; Distelmaier et al., 2024), Congenital surcease-isomaltase deficiency (SI gene, AR inheritance, OMIM phenotype number 222900) (Starnes and Welsh, 1970; Belmont et al., 2002), and Glucose/galactose malabsorption (SLC5A1 gene, AR inheritance, OMIM phenotype number 606824) (Soylu et al., 2008).
3.1.1.1 CASR gene
CASR encodes for the calcium-sensing receptor (CaSR), which is important in renal calcium homeostasis and expressed in the parathyroid gland and the kidney in the thick ascending loop of Henle (TAL) basolateral membrane (Figure 1), the PT, the distal convoluted tubule (DCT), and the CD (Brown and MacLeod, 2001). In the TAL, activation of CaSR by calcium inhibits NaCl reabsorption by inhibiting NKCC2 and ROMK (Gamba and Friedman, 2009). By inhibiting ROMK, the tubular lumen positive voltage is diminished, reducing the driving force for paracellular cation (including calcium) reabsorption (Gamba and Friedman, 2009).
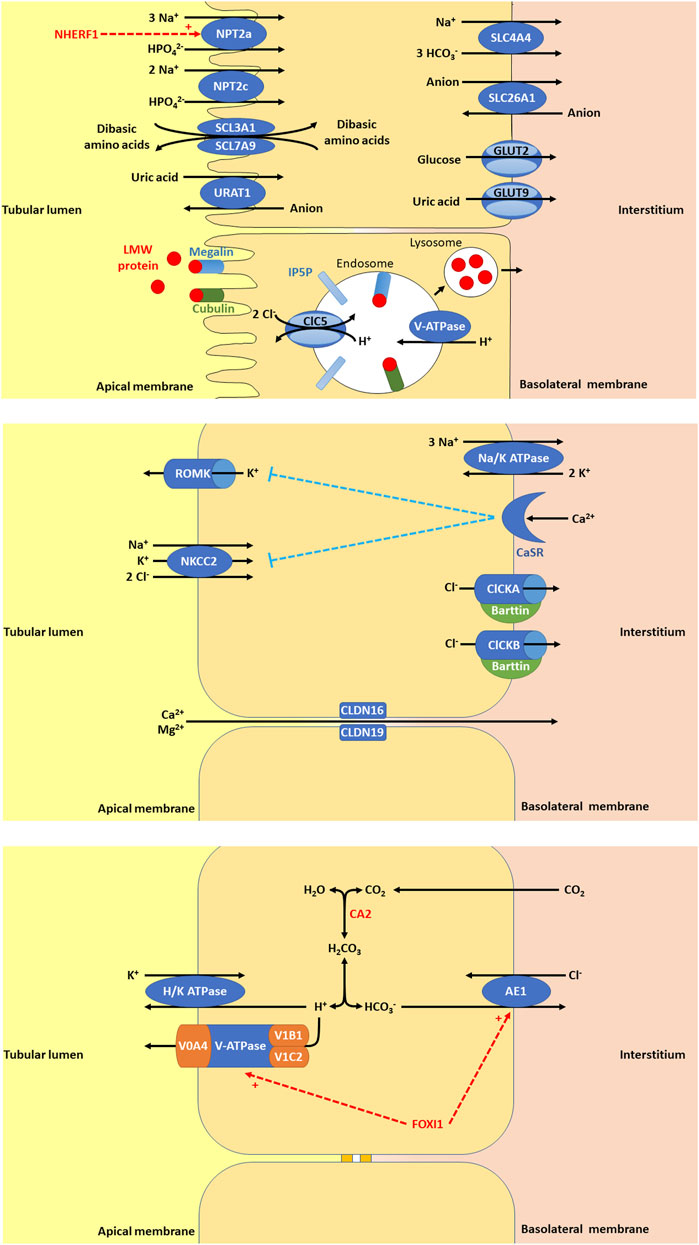
Figure 1. (A) In the proximal tubule (PT), there are a variety of apical membrane transporters, basolateral membrane transporters, and endosome transporters proteins that play important roles in PT function. Apical membrane transporters shown are sodium-dependent phosphate transport protein 2a (NPT2a, encoded by SLC34A1, whose activity is promoted by NHERF1, sodium/hydrogen exchange regulatory factor 1, encoded by SLC9A3R1), sodium-dependent phosphate transport protein 2c (NPT2c, encoded by SLC34A3), SLC3A1/SLC7A9 (encoded by SLC3A1 and SLC7A9), and urate-anion transporter (URAT1, encoded by SLC22A12). The basolateral membrane transporters shown are glucose transporter 2 (GLUT2, encoded for by SLC2A2), facilitated glucose transporter 9 (GLUT9)/voltage-driven urate transporter (encoded for by SLC2A9), solute carrier family 4 member 4 (SLC4A4, a sodium bicarbonate transporter encoded by SLC4A4), and solute carrier family 26 member 1 (SLC26A1, an electroneutral anion exchanger encoded by SLC26A1). Low molecular weight (LMW) protein in the tubular lumen binds megalin and cubulin and are endocytosed into endosomes, thought to be facilitated by inositol polyphosphate-5-phosphatase (IP5P), ended by OCRL, which is expressed in the glomerulus, the PT, the thick ascending loop of Henle (TAL), the distal convoluted tubule (DCT) and the collecting duct (CD). Endosome acidification is then mediated by V-ATPase and the chloride voltage-gated channel 5 (ClC5, encoded by CLCN5, expressed in the PT, alpha-intercalated cell (alpha-IC) and the TAL. Lysosomes form to allow the absorption of LMW protein and recycling of megalin and cubulin to the apical membrane. (B) In the TAL, there are a variety of apical membrane transporters, tight junction proteins, and basolateral membrane transporters, receptors, and proteins that play important roles in tubular function. Apical membrane transporters shown are kidney-specific Na-K-Cl symporter (NKCC2) and renal out-medullar potassium channel (ROMK). NKCC2 (encoded by SLC12A1) that is responsible for sodium, potassium, and chloride reabsorption from the tubular lumen. ROMK (encoded by KCNJ1) is responsible for excretion of potassium. The tight junction proteins shown are claudin 16 (CLDN16, encoded by CLDN16) and claudin 19 (CLDN19, encoded by CLDN19). CLDN16 and CLDN19 are necessary for paracellular reabsorption of calcium and magnesium. The basolateral membrane transporters and receptors shown include Na/K ATPase, chloride voltage-gated channel kidney A (ClCKA), chloride voltage-gated channel kidney B (ClCKB), Barttin, and calcium sensing receptor (CaSR). Na/K ATPase is responsible for generating the gradient for sodium entry across apical membranes via NKCC2 and subsequent exit of chloride across the basolateral membrane via ClCKA, ClCKB, and Barttin. ClCKA is encoded for by CLCNKA and ClCKB is encoded for by CLCNKB. These chloride channels allow chloride to exit the cell via the basolateral membrane. Barttin or chloride voltage-gated channel kidney (ClCK)-type accessory subunit beta, encoded for by BSND. ClCKA and ClCKB channels depend on the presence of the Barttin subunit for chloride transport. CaSR (encoded for by CASR), is expressed in the parathyroid gland and the kidney in the TAL, the PT, the DCT, and the CD. In the TAL, activation of CaSR by calcium inhibits NaCl reabsorption by inhibiting NKCC2 and ROMK. By inhibiting ROMK, the tubular lumen positive voltage is diminished, reducing the driving force for paracellular cation (including calcium) reabsorption. (C) In the alpha-IC of the CD, there are few key, apical membrane transporters, and basolateral membrane transporters that play important roles in tubular function. Carbonic anhydrase 2 (CA2) is an enzyme encoded by CA2 that is expressed in the CD (including the alpha-IC) and PT. CA2 catalyzes the combination of carbon dioxide and water to form carbonic acid (H2CO3), which then dissociates to protons (H+) and bicarbonate (HCO3−). The protons produced are excreted into the urine by exiting the cell through the apical membrane via H/K ATPase and V-ATPase, a proton transporter with V0 subunit A4 (V0A4, encoded by ATP6V0A4), V0 subunit B1 (V1B1, encoded by ATP6V1A1), and V1 subunit C2 (V1C2, encoded by ATP6V1C2). The bicarbonate produced is reabsorbed into the interstitium by exiting the cell thought the basolateral membrane via anion exchange protein 1 (AE1), a basolateral chloride bicarbonate counter transporter encoded by SLC4A1. Forkhead box I1 (FOXI1) encoded for by FOXI1 is a transcription factor that regulates the function of AE1 and V-ATPase.
AD hypocalcemia/AD hypocalcemia with Bartter syndrome (OMIM phenotype number 601198) is a condition resulting from activating variants in CASR, which increase the sensitivity of CaSR to extracellular calcium (Brown and MacLeod, 2001). Activation of CaSR in the parathyroid gland leads to inhibition of parathyroid hormone (PTH) release and subsequent hypoparathyroidism (Brown and MacLeod, 2001). Reduced PTH release leads to decreased calcium and phosphorus resorption in the bone (Brown and MacLeod, 2001). In the kidney, reduced PTH leads to decreased TAL and DCT reabsorption of calcium, increased reabsorption of phosphorus, and decreased production of 1,25-dihydroxyvitamin D3, leading to decreased absorption of calcium in the intestine (Brown and MacLeod, 2001). All of the above culminates in hypocalcemia, hypercalciuria, NC, and NL (Pearce et al., 1996). Some may have features of Bartter syndrome such as impaired sodium chloride reabsorption in the TAL, hypokalemic metabolic alkalosis, hyperreninemia and/or hyperaldosteronism (Vargas-Poussou et al., 2002). Treatment with minimal doses of calcium and vitamin D administration may be given to alleviate symptoms, but excess can exacerbate hypercalciuria, NC, and lead to kidney impairment (Pearce et al., 1996).
Neonatal hyperparathyroidism (OMIM phenotype number 239200) is an AD/AR condition resulting from inactivating variants in CASR that decrease the sensitivity of CaSR to extracellular calcium (Brown and MacLeod, 2001). Inactivation of CaSR in the parathyroid gland leads to stimulation of PTH release and subsequent hyperparathyroidism (Lila et al., 2012; El Allali et al., 2021). Increased PTH release leads to increased calcium and phosphorus resorption in the bone (Lila et al., 2012; El Allali et al., 2021). In the kidney, increased PTH leads to increased tubular reabsorption of calcium, decreased reabsorption of phosphorus, and increased production of 1,25-dihydroxyvitamin D3, leading to increased absorption of calcium in the intestine (Lila et al., 2012; El Allali et al., 2021). This culminates in severe hypercalcemia, NC, and NL (Lila et al., 2012; El Allali et al., 2021). Calcimimetic medications may be given treat this condition by increasing sensitivity of CaSR to extracellular calcium (El Allali et al., 2021).
Hypocalciuric hypercalcemia type 1 (OMIM phenotype number 145980) is an AD condition resulting from an inactivating variant in CASR, resulting in lifelong mild to moderate hypercalcemia, inappropriate hypocalciuria, and normal PTH to mild hyperparathyroidism (Carling et al., 2000). This condition may also have atypical presentations with severe hypercalcemia and hypercalciuria with or without NL or NC (Carling et al., 2000).
3.1.1.2 CYP24A1 gene
CYP24A1 encodes for cytochrome P450 family 24 subfamily A member 1 or 1,25-dihydroxyvitamin D3 24-hydroxylase, the enzyme primarily responsible for the catabolism of 1,25-dihydroxy- and 25-hydroxy-vitamin D, which is expressed in the PT (Schlingmann et al., 2011). Infantile hypercalcemia 1 (OMIM phenotype number 143880) is an AR condition resulting from an inactivating variant in CYP24A1, resulting in accumulation of 1,25-dihydroxyvitamin D3 (Schlingmann et al., 2011). This accumulation leads to severe hypercalcemia, failure to thrive, vomiting, dehydration, NC, and NL (Schlingmann et al., 2011). Data suggests that carriers (heterozygotes) of inactivating variants in CYP24A1 may have increased risk of NC and NL (Carpenter, 2017).
For treatment of Infantile hypercalcemia 1, generous hydration and a diet low in vitamin D and calcium are suggested (Schlingmann et al., 2011). CYP3A4 inducers (rifampin) or CYP27B1 modulators (fluconazole/ketoconazole/itraconazole) may help reduce 1,25-dihydroxyvitamin D3 levels and improve hypercalcemia and hypercalciuria (Sayers et al., 2015; Hawkes et al., 2017). Low doses of these medications are suggested, but the long-term safety is uncertain.
3.1.2 Renal phosphate wasting with hypercalciuria
Genetic causes of renal phosphate wasting with hypercalciuria and NL and/or NC are due to variants in SCL34A1, SLC34A3, and SLC9A3R1 (Table 2). These individuals typically require treatment with phosphate supplementation (Tieder et al., 1992).
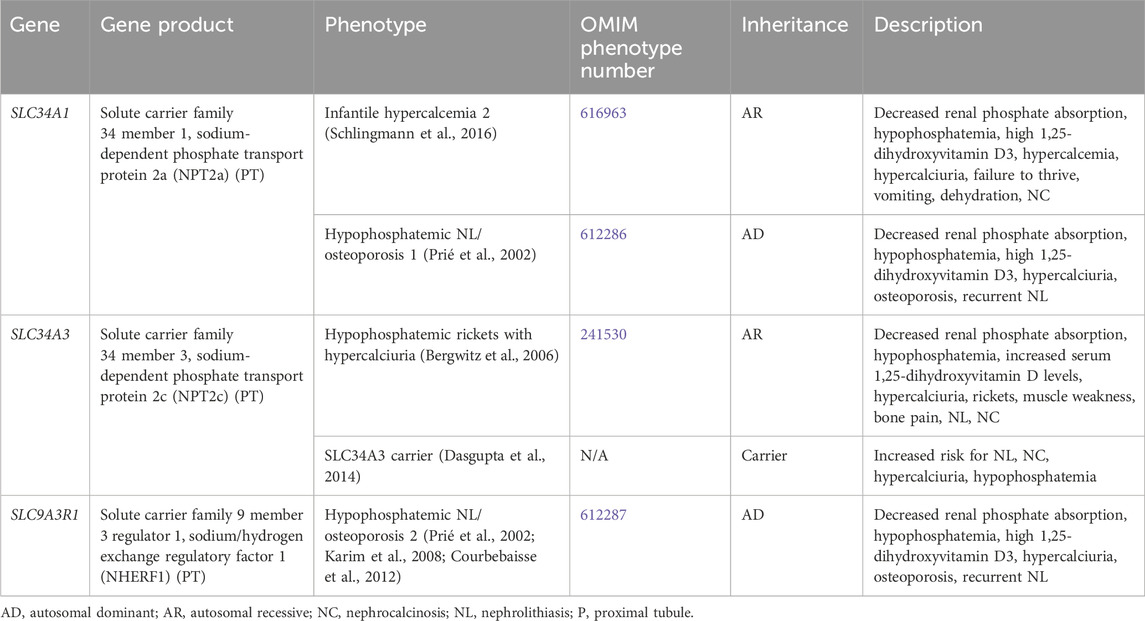
Table 2. Genetic causes of renal phosphate wasting with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.2.1 SLC34A1 gene
SLC34A1 encodes for the solute carrier family 34 member 1, the sodium-dependent phosphate transport protein 2a (NPT2a), which responsible for the majority of renal phosphate transport and is primarily expressed in the PT (Figure 1) (Schlingmann et al., 2016). Fanconi renotubular syndrome 2 (OMIM phenotype number 613388) is one of the conditions caused by homozygous variants in SLC34A1 but will not be discussed in this review as this condition has not been associated with NL or NC.
Infantile hypercalcemia 2 (OMIM phenotype number 616963) is an AR condition due to inactivating variants in SLC34A1, resulting in loss of NPT2a activity and subsequent renal phosphorus wasting with hypophosphatemia (Schlingmann et al., 2016). This hypophosphatemia results in increased production of 1,25-dihydroxyvitmain D3 with hypercalcemia, hypercalciuria, failure to thrive, vomiting, dehydration, and NC (Schlingmann et al., 2016). In addition to phosphate supplementation, generous hydration and a diet low in vitamin D and calcium are also suggested (Schlingmann et al., 2016). CYP3A4 inducers (rifampin) or CYP27B1 modulators (fluconazole/ketoconazole/itraconazole) may help reduce 1,25-dihydroxyvitamin D3 levels and improve hypercalcemia and hypercalciuria (Sayers et al., 2015; Hawkes et al., 2017). Low doses of these medications are suggested, but the long-term safety is uncertain.
Hypophosphatemic NL/osteoporosis 1 (OMIM phenotype number 612286) is an AD condition due to an inactivating variant in SLC34A1, resulting in loss of NPT2a activity and subsequent renal phosphorus wasting with hypophosphatemia (Prié et al., 2002). This hypophosphatemia results in increased production of 1,25-dihydroxyvitmain D3 with hypercalciuria, osteoporosis, and recurrent NL (Prié et al., 2002).
3.1.2.2 SLC34A3 gene
SLC34A3 encodes for the solute carrier family 34 member 3, the sodium-dependent phosphate transport protein 2c (NPT2c) which is primarily expressed in the PT (Figure 1) (Bergwitz et al., 2006). Hypophosphatemic rickets with hypercalciuria (OMIM phenotype number 241530) is an AR condition due to inactivating variants in SLC34A3, resulting in loss of NPT2c activity and subsequent renal phosphorus wasting with hypophosphatemia (Bergwitz et al., 2006). This hypophosphatemia results in increased production of 1,25-dihydroxyvitmain D3 with hypercalcemia, hypercalciuria, rickets, muscle weakness, bone pain, NL, and NC (Bergwitz et al., 2006). Data suggests that carriers (heterozygotes) of inactivating variants in SLC34A3 may have increased risk of NL, NC, hypercalciuria, and hypophosphatemia (Dasgupta et al., 2014).
3.1.2.3 SLC9A3R1 gene
SLC9A3R1 encodes for the solute carrier family 9 member 3 regulator 1/sodium/hydrogen exchange regulatory factor 1 (NHERF1), which is primarily expressed in the PT (Figure 1) (Karim et al., 2008; Courbebaisse et al., 2012). Hypophosphatemic NL/osteoporosis 2 (OMIM phenotype number 612287) is an AD condition due to an inactivating variant in SLC9A3R1, resulting in loss of NHERF1 activity with subsequent decrease in NPT2a expression, resulting in renal phosphorus wasting with hypophosphatemia (Courbebaisse et al., 2012). This subsequently results in increased production of 1,25-dihydroxyvitmain D3 with hypercalciuria, osteoporosis, and recurrent NL (Prié et al., 2002).
3.1.3 Renal magnesium wasting with hypercalciuria
Genetic causes of renal magnesium wasting with hypercalciuria and NL and/or NC are related to, CLDN16, CLDN19, and RRAGD, and are shown in Table 3. The conditions related to variants in these genes are discussed further below.
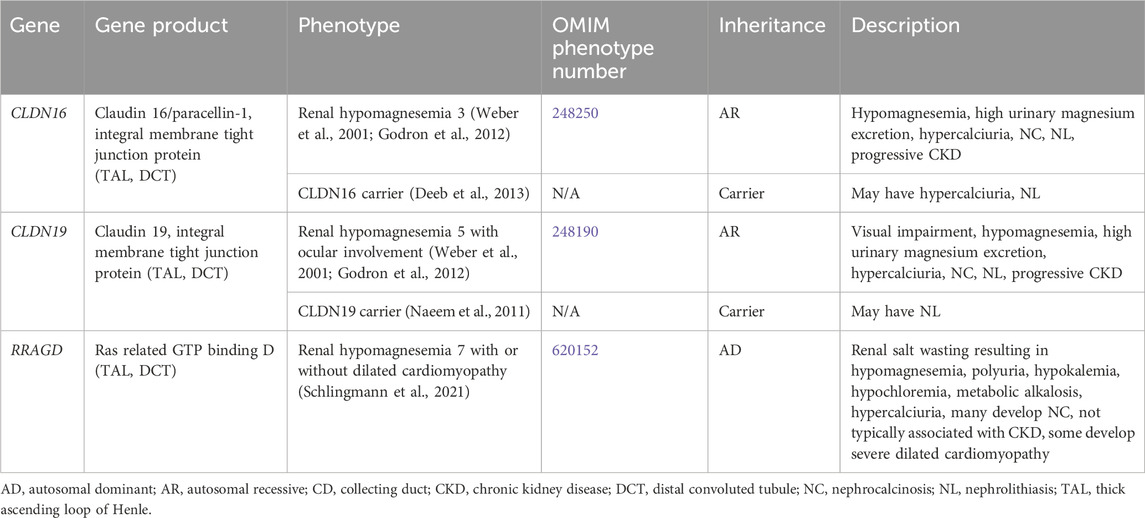
Table 3. Genetic causes of renal magnesium wasting with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.3.1 CLDN16 gene
CLDN16 encodes for Claudin 16 or paracellin-1, which is an integral membrane tight junction protein necessary for paracellular reabsorption of calcium and magnesium (Godron et al., 2012). CLDN16 is expressed in the TAL and DCT (Figure 1). Renal hypomagnesemia 3 (OMIM phenotype number 248250) is an AR condition due to inactivating variants in CLDN16, resulting reduced reabsorption of calcium (hypercalciuria)and magnesium (hypermagnesuria) with hypomagnesemia, NC, NL, and progressive CKD (Godron et al., 2012). Data suggests that carriers (heterozygotes) of inactivating variants in CLDN16 may have hypercalciuria and NL (Deeb et al., 2013).
Medical management of Renal hypomagnesemia 3 typically involves high-dose magnesium supplementation to replace renal losses, although this has not been shown to slow progression of CKD (Weber et al., 2001; Godron et al., 2012). Thiazide diuretics and indomethacin have been used in this condition but have not been shown to be successful in reducing hypercalciuria or hypermagnesuria or preventing progression of CKD (Weber et al., 2001; Godron et al., 2012). Definitive treatment can be achieved with kidney transplantation (Weber et al., 2001).
3.1.3.2 CLDN19 gene
CLDN19 encodes for Claudin 19, which is an integral membrane tight junction protein necessary for paracellular reabsorption of calcium and magnesium (Godron et al., 2012). CLDN19 is expressed in the TAL and DCT (Figure 1). Renal hypomagnesemia 5 with ocular involvement (OMIM phenotype number 248190) is an AR condition due to inactivating variants in CLDN19, resulting reduced reabsorption of calcium (hypercalciuria) and magnesium (hypermagnesuria)with hypomagnesemia, visual impairment, NC, NL, and progressive CKD (Godron et al., 2012). Data suggests that carriers (heterozygotes) of inactivating variants in CLDN16 may have NL (Naeem et al., 2011). Medical management of Renal hypomagnesemia 5 with ocular involvement and its efficacy are typically the same as that for Renal hypomagnesemia 3 caused by variants in CLDN16 (Weber et al., 2001; Godron et al., 2012).
3.1.3.3 RRAGD gene
RRAGD encodes for Ras related GTP binding D, which is expressed in the TAL and DCT (Schlingmann et al., 2021). Renal hypomagnesemia 7 with or without dilated cardiomyopathy (OMIM phenotype number 620152) is an AD condition due to an inactivating variant in RRAGD, resulting in activation of mTOR signaling, which leads to renal salt wasting resulting in hypomagnesemia, polyuria, hypokalemia, hypochloremia, metabolic alkalosis, hypercalciuria, and NC (Schlingmann et al., 2021). This condition is not typically associated with CKD, and some individuals develop severe dilated cardiomyopathy (Schlingmann et al., 2021). Optimal treatment strategy is unclear.
3.1.4 Distal renal tubular acidosis
Distal RTA results from the impaired ability to acidify the urine, which in turn leads to hyperchloremic metabolic acidosis (Karet, 2002). Distal RTA is associated and hypocitraturia with alkaline urine due to upregulation of citrate reabsorption in the PT, hypercalciuria and resultant NC and/or NL, and may be associated with hypokalemia (Karet, 2002; Lopez-Garcia et al., 2019). Treatment for distal RTA primarily consists of alkali therapy to correct metabolic acidosis, with potassium-containing medications such as potassium citrate being the preferred choice (Mohebbi and Wagner, 2018). Thiazide diuretics may be considered with severe hypercalciuria but may be complicated by polyuria and hypokalemia (Mohebbi and Wagner, 2018). Many individuals with chronic untreated or severe metabolic acidosis develop rickets or osteomalacia, although it appears as though most adults achieve normal adult height, especially those with better control of acidosis (Karet, 2002; Lopez-Garcia et al., 2019). CKD stages 2–4 have been reported in >80% of adults and in >30% of children with distal RTA with ESKD rarely occurring (Lopez-Garcia et al., 2019). In individuals with better control of acidosis, CKD incidence is lower (Lopez-Garcia et al., 2019). Table 4 shows genetic causes of distal RTA with hypercalciuria and NL and/or NC.
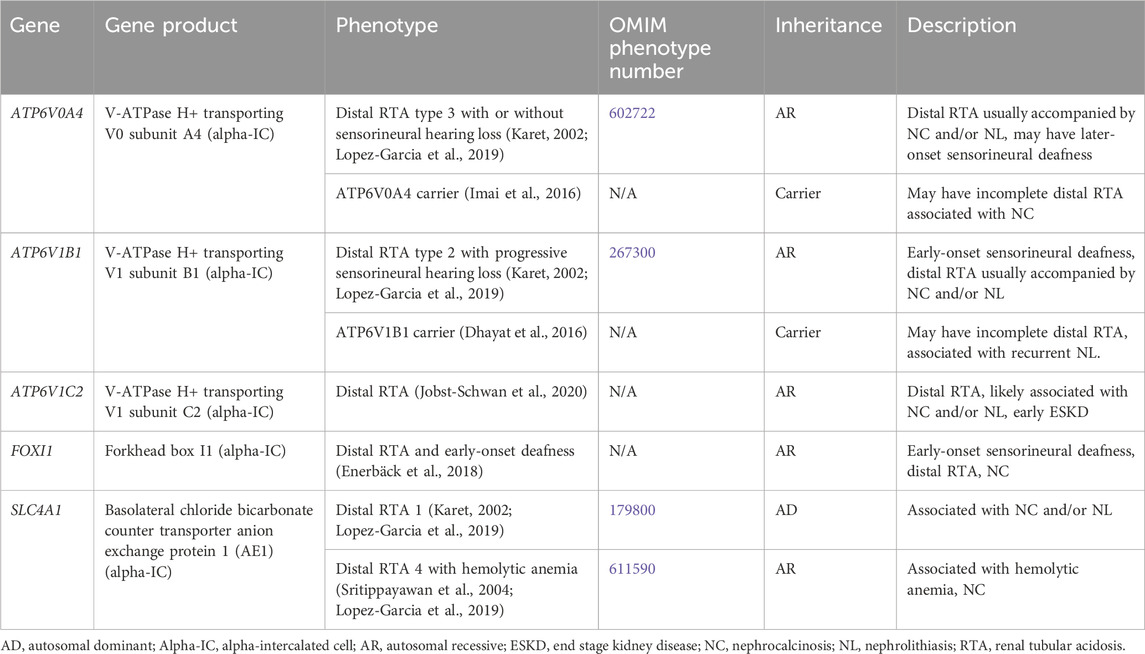
Table 4. Genetic causes of distal renal tubular acidosis with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.4.1 ATP6V0A4 gene
ATP6V0A4 encodes for V-ATPase H+ transporting V0 subunit A4, which excretes protons into the urine and is expressed in the alpha-IC of the CD (Figure 1) (Karet, 2002). Distal RTA type 3 with or without sensorineural hearing loss (OMIM phenotype number 602722) is an AR condition due to inactivating variants in ATP6V0A4, resulting in decreased urinary proton excretion, which leads to a distal RTA usually accompanied by NC and/or NL as well later-onset sensorineural deafness in >30% (Karet, 2002; Lopez-Garcia et al., 2019). Data suggests that carriers (heterozygotes) of inactivating variants in ATP6V0A4 may have an incomplete distal RTA associated with NC (Imai et al., 2016).
3.1.4.2 ATP6V1B1 gene
ATP6V1B1 encodes for V-ATPase H+ transporting V1 subunit B1, which excretes protons into the urine and is expressed in the alpha-IC of the CD (Figure 1) (Karet, 2002). Distal RTA type 2 with progressive sensorineural hearing loss (OMIM phenotype number 267300) is an AR condition due to inactivating variants in ATP6V1B1, resulting in decreased urinary proton excretion, which leads to a distal RTA usually accompanied by NC and/or NL as well early-onset sensorineural deafness in most (Lopez-Garcia et al., 2019). Data suggests that carriers (heterozygotes) of inactivating variants in ATP6VAB1 may have an incomplete distal RTA associated with recurrent NL (Dhayat et al., 2016).
3.1.4.3 ATP6V1C2 gene
ATP6V1C2 encodes for V-ATPase H+ transporting V1 subunit C2, which excretes protons into the urine and is expressed in the alpha-IC of the CD (Figure 1) (Jobst-Schwan et al., 2020). ATP6V1C2-associated distal RTA is an AR condition due to inactivating variants in ATP6V1C2, resulting in decreased urinary proton excretion, which leads to a distal RTA that is likely associated with NC and/or NL and with early ESKD (Jobst-Schwan et al., 2020).
3.1.4.4 SLC4A1 gene
SLC4A1 encodes for the basolateral chloride bicarbonate counter transporter anion exchange protein 1 (AE1), which reabsorbs bicarbonate from the urine into the circulation and is expressed in the alpha-IC of the CD (Figure 1) (Karet, 2002). Distal RTA 1 (OMIM phenotype number 179800) is an AD condition due to an inactivating variant in SLC4A1, resulting in decreased urinary bicarbonate reabsorption, which leads to a distal RTA usually accompanied by NC and NL (Karet, 2002; Lopez-Garcia et al., 2019). Distal RTA 4 with hemolytic anemia (OMIM phenotype number 611590) is an AR condition due to inactivating variants in SLC4A1, resulting in decreased urinary bicarbonate reabsorption, which leads to a distal RTA with hemolytic anemia and NC. (Sritippayawan et al., 2004; Lopez-Garcia et al., 2019).
3.1.4.5 FOXI1 gene
FOXI1 encodes for Forkhead box I1, which is a transcription factor that regulates the function of AE1, AE4, and V-ATPase subunits, and is expressed in the alpha-IC of the CD (Figure 1) (Enerbäck et al., 2018). Distal RTA and early-onset deafness is an AR condition due to inactivating variants in FOXI1, resulting in decreased urinary bicarbonate reabsorption, which leads to a distal RTA with early-onset sensorineural deafness and NC (Enerbäck et al., 2018).
3.1.5 Proximal tubulopathy
Proximal tubulopathy consists of dysfunction of the PT, which can lead to any combination of low molecular weight (LMW) proteinuria, aminoaciduria, glucosuria, urine bicarbonate wasting and RTA, hypercalciuria, hyperphosphaturia, high urinary potassium excretion, and uricosuria. NC and NL can occur with proximal tubulopathies, but less commonly so than with distal RTA. Another name for proximal tubulopathy is Fanconi renotubular syndrome. Treatment generally consists of supplementation to treat urinary solute losses.
Supplementary Table S2 contains a list of genetic causes of secondary proximal tubulopathy with hypercalciuria and NL and/or NC. Conditions that results in a secondary proximal tubulopathy include Hereditary fructose intolerance (ALDOB gene, AR inheritance (Higgins and Varney, 1966; Mass et al., 1966), OMIM phenotype number 229600), Wilson disease (ATP7B gene, AR inheritance, OMIM phenotype number 277900) (Di Stefano et al., 2012), Nephropathic cystinosis (CTNS gene, AR inheritance, OMIM phenotype number 219800) (Theodoropoulos et al., 1995), Tyrosinemia type 1 (FAH gene, AR inheritance, OMIM phenotype number 276700) (Forget et al., 1999), Congenital lactase deficiency (LCT gene, AR inheritance, OMIM phenotype number 223000) (Saarela et al., 1995), Mitochondrial DNA depletion syndrome 8A (encephalomyopathic type with renal tubulopathy) (RRM2B gene, AR inheritance, OMIM phenotype number 612075) (Finsterer and Scorza, 2017), Sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay (TRNT1 gene, AR inheritance, OMIM phenotype number 616084) (Wiseman et al., 2013), Arthrogryposis, renal dysfunction, and cholestasis 1 (VPS33B gene, AR inheritance, OMIM phenotype number 208085) (Holme et al., 2013), and Arthrogryposis, renal dysfunction, and cholestasis 2 (VIPAS39 gene, AR inheritance, OMIM phenotype number 613404) (Holme et al., 2013).
Primary inherited proximal tubulopathies associated with NC and/or NL are shown in Table 5 and will be discussed in greater detail below. Inactivating variants in EHHADH (encodes for enoyl-CoA hydratase and 3-hydroxyacyl CoA dehydrogenase) cause AD Fanconi renotubular syndrome 3 (OMIM phenotype number 615605), is associated with rickets, impaired growth, glucosuria, generalized aminoaciduria, phosphaturia, metabolic acidosis, LMW proteinuria, and hypercalciuria will not be discussed as there is no known association with NC or NL (Tolaymat et al., 1992; Klootwijk et al., 2014).
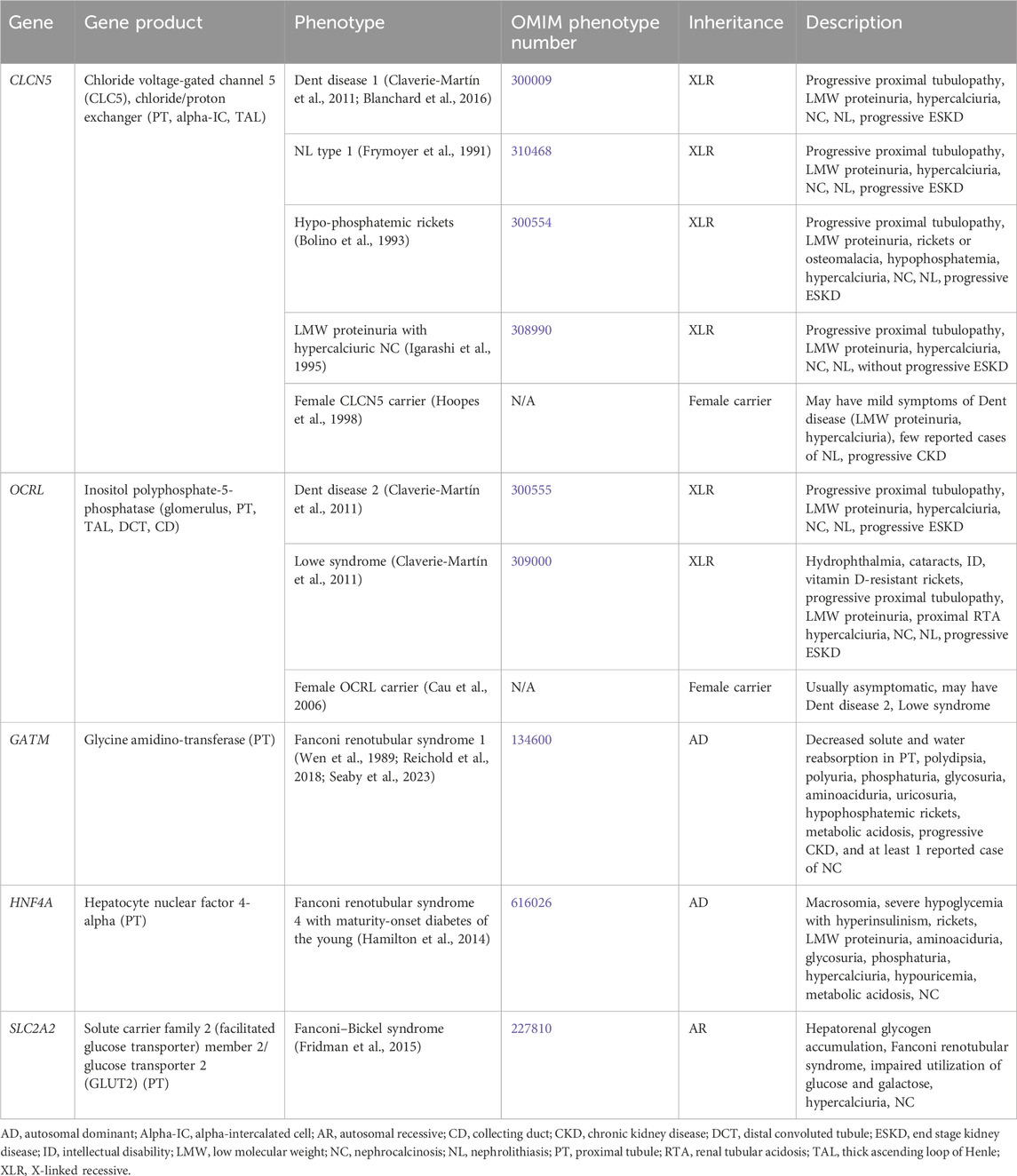
Table 5. Genetic causes of primary proximal tubulopathy with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.5.1 CLCN5 gene
CLCN5 encodes for chloride voltage-gated channel 5 (CLC5), a chloride/proton exchanger, and is expressed in the PT (Figure 1), alpha-IC, and TAL (Mansour-Hendili et al., 2015). Inactivating variants in CLCN5 appear to cause abnormal reabsorption of LMW proteins by disrupting PT vesicle acidification, impacting lysosome function and LMW protein processing (Nielsen et al., 2016). Inactivating variants in CLCN5 are associated with a spectrum of conditions (Claverie-Martín et al., 2011).
Dent disease 1 (OMIM phenotype number 300009) is an XLR condition resulting from inactivating variants in CLCN5. Hallmark features are progressive proximal renal tubulopathy with LMW proteinuria, hypercalciuria, NC, NL, and progressive ESKD (Claverie-Martín et al., 2011). Median age at ESKD onset has been reported as 40 years of age (Blanchard et al., 2016). Treatment is aimed at treating serum electrolyte disturbances (such as treating hypophosphatemia with phosphorus supplementation) and minimizing hypercalciuria, primarily with hydration and administration of thiazide diuretics. However, the administration thiazide diuretics to reduce hypercalciuria is controversial as there is no definite correlation between hypercalciuria and ESKD, and significant side effects have been reported including dehydration and hypokalemia (Blanchard et al., 2008). Kidney transplantation has been successful in these patients without recurrence of NL or NC (Scheinman, 1998). Due to the risk of worsening hypercalciuria with vitamin D supplementation, it should be reserved for patients who require it for treatment of rickets.
NL type 1 (OMIM phenotype number 310468) is an XLR condition resulting from inactivating variants in CLCN5. Hypophosphatemic rickets (OMIM phenotype number 300554) is an XLR condition resulting from inactivating variants in CLCN5. It is characterized by progressive proximal renal tubulopathy with LMW proteinuria, rickets or osteomalacia, hypophosphatemia, hypercalciuria, NC, NL, and progressive ESKD (Bolino et al., 1993). LMW proteinuria with hypercalciuric NC (OMIM phenotype number 308990) is an XLR condition resulting from inactivating variants in CLCN5. The presentation of LMW proteinuria with hypercalciuric NC resembles that of the conditions mentioned above, except that these individuals do not experience progressive ESKD (Igarashi et al., 1995). Although these three conditions have different OMIM phenotype numbers, it is unclear if they are truly separate conditions from Dent disease 1 as the spectrum of presenting symptoms are the same (Frymoyer et al., 1991). Data suggests that female carriers of inactivating variants in CLCN5 may have mild symptoms of Dent disease such as low-molecular-weight proteinuria, and hypercalciuria with few reported cases of NL and kidney insufficiency (Hoopes et al., 1998).
3.1.5.2 OCRL gene
OCRL encodes for inositol polyphosphate-5-phosphatase, which is thought to be important in endocytosis and cellular trafficking and is expressed throughout the body, including in the glomerulus, PT (Figure 1), TAL, DCT, and CD (Claverie-Martín et al., 2011). Inactivating variants in OCRL appear to cause abnormal protein trafficking required for PT solute reabsorption (Ooms et al., 2009). Inactivating variants in OCRL are associated with a spectrum of conditions (Claverie-Martín et al., 2011).
Dent disease 2 (OMIM phenotype number 300555) is an XLR condition resulting from inactivating variants in OCRL. Clinical features are shared with that of Dent disease 1 caused by variants in CLCN5 with progressive proximal renal tubulopathy with LMW proteinuria, hypercalciuria, NC, NL, and progressive ESKD (Claverie-Martín et al., 2011). Lowe syndrome (OMIM phenotype number 309000) is an XLR condition resulting from inactivating variants in OCRL. The presentation resembles that of Dent disease 2 but also involves other systemic symptoms of hydrophthalmia, cataracts, severely impaired intellectual development, vitamin D-resistant rickets, and proximal RTA (Claverie-Martín et al., 2011). Data suggests that female carriers of inactivating variants in ORCL are usually asymptomatic, but may have either Dent disease 2 or Lowe syndrome (Cau et al., 2006). Treatment of Dent disease 2 and Lowe syndrome is the same as for Dent disease 1, including treatment of electrolyte derangements such as acidosis.
3.1.5.3 GATM gene
GATM encodes for glycine amidinotransferase, which is expressed in the PT mitochondria (Reichold et al., 2018). An inactivating variant in GATM leads to Fanconi renotubular syndrome 1 (OMIM phenotype number 134600), an AD condition associated with decreased solute and water reabsorption in the PT with polydipsia, polyuria, phosphaturia, glycosuria, aminoaciduria, uricosuria, hypophosphatemic rickets, metabolic acidosis, progressive kidney insufficiency, and NC in 1 definitive case, and at least 1 case of AD Fanconi syndrome suspected to be secondary a variant in GATM, although genetic testing was not performed (Wen et al., 1989; Reichold et al., 2018; Seaby et al., 2023).
3.1.5.4 HNF4A gene
HNF4A encodes for hepatocyte nuclear factor 4-alpha, which in the kidney is expressed in the PT. An inactivating variant in HNF4A leads to Fanconi renotubular syndrome 4 with maturity-onset diabetes of the young (OMIM phenotype number 616026), an AD condition associated with macrosomia, severe hypoglycemia with hyperinsulinism, rickets, LMW proteinuria, aminoaciduria, glycosuria, phosphaturia, hypercalciuria, hypouricemia, metabolic acidosis, and NC (Hamilton et al., 2014).
3.1.5.5 SLC2A2 gene
SLC2A2 encodes for solute carrier family 2 (facilitated glucose transporter) member 2 or glucose transporter 2 (GLUT2), which mediates monosaccharide bidirectional transport in the liver, pancreas, intestines, and the renal PT (Figure 1) (Fridman et al., 2015). Inactivating variants in SLC2A2 lead to Fanconi–Bickel syndrome (OMIM phenotype number 227810), an AR condition associated with Fanconi renotubular syndrome, hepatorenal glycogen accumulation, impaired utilization of glucose and galactose, hypercalciuria, and NC (Fridman et al., 2015).
3.1.6 Mixed or variable tubulopathy
Genetic causes of mixed or variable tubulopathy with hypercalciuria and NL and/or NC are shown in Table 6. One such condition, Kearns-Sayre syndrome, is associated with various mitochondrial DNA (mtDNA) deletions and a variety of tubulopathies (AR inheritance, OMIM phenotype number 530000) (Finsterer and Scorza, 2017). Treatment for distal RTA primarily consists of alkali therapy to correct metabolic acidosis, and treatment of proximal tubulopathy primarily consists of replacement or urinary solute losses (Mohebbi and Wagner, 2018).
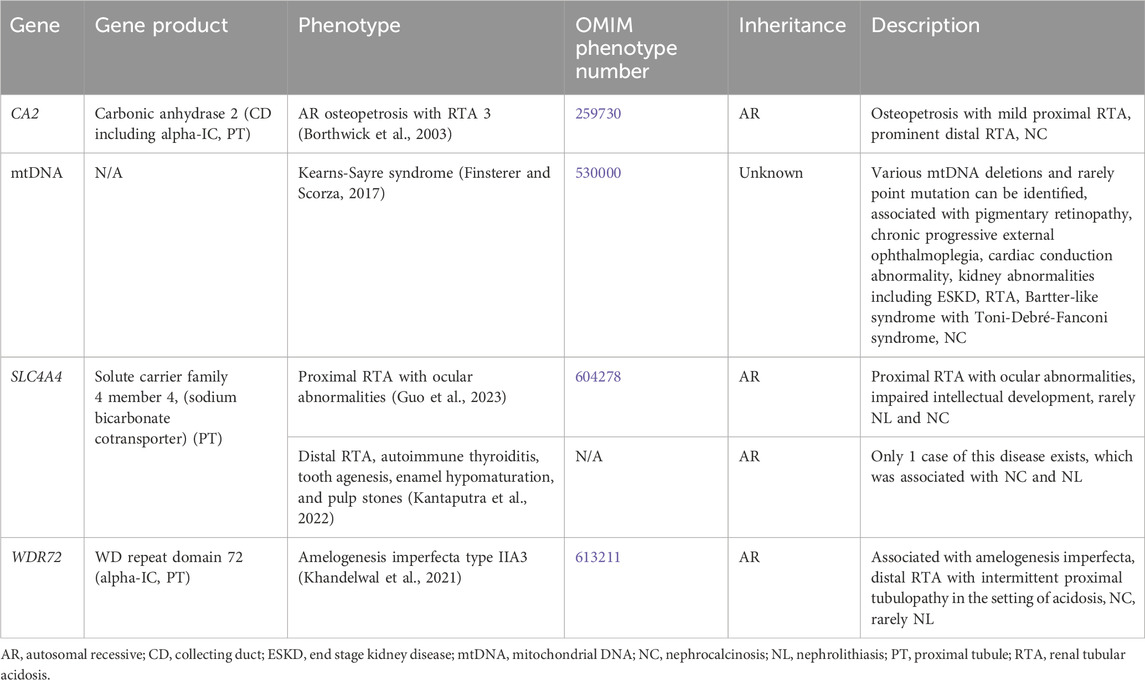
Table 6. Genetic causes of mixed or variable tubulopathy with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
3.1.6.1 CA2 gene
CA2 encodes for carbonic anhydrase 2 (CA2), which is expressed in the CD (including the alpha-IC) and PT (Figure 1). CA2 catalyzes the combination of carbon dioxide and water to form carbonic acid, which then dissociates to protons and bicarbonate (Whyte, 1993). Inactivating variants in CA2 lead to decreased proton and bicarbonate production in osteoclasts and the renal tubules and is associated AR Osteopetrosis with RTA 3 (OMIM phenotype number 259730). This condition is associated with both a proximal RTA and a distal RTA (Borthwick et al., 2003).
3.1.6.2 SLC4A4 gene
SLC4A4 encodes for solute carrier family 4 member 4, a sodium bicarbonate cotransporter that is important in the reabsorption of bicarbonate in the PT (Figure 1) (Guo et al., 2023). Inactivating variants in this gene are associated with two different conditions. Proximal RTA with ocular abnormalities (OMIM phenotype number 604278) is an AR condition associated with impaired intellectual development, and rarely NL and NC (Guo et al., 2023). Distal RTA, autoimmune thyroiditis, tooth agenesis, enamel hypomaturation, and pulp stones is an AR condition with only 1 case reported with the only case being associated with NC and NL (Kantaputra et al., 2022).
3.1.6.3 WDR72 gene
WDR72 encodes for WD repeat domain 72, implicated in the trafficking of V-ATPases and thought to play a role in sustained intracellular CaSR signaling through clathrin-mediated endocytosis (Khandelwal et al., 2021). WDR72 is expressed in the alpha-IC in the CD and PT (Khandelwal et al., 2021). Inactivating variants lead to the AR condition Amelogenesis imperfecta type IIA3 (OMIM phenotype number 613211) associated with a distal RTA and intermittent proximal tubulopathy in the setting of acidosis (Khandelwal et al., 2021). This condition is associated with NC and rarely NL.
3.1.7 Bartter syndrome
Bartter syndrome consists of a group of channelopathies that affect transporter proteins primarily in the TAL involved in sodium chloride reabsorption, which lead to urinary sodium losses, metabolic alkalosis, hypokalemia, hyperaldosteronism and/or hyperreninemia, and hypercalciuria due to loss of paracellular calcium reabsorption (Besouw et al., 2020). There are multiple forms of Bartter syndrome, all of which have been associated with NC (Table 7).
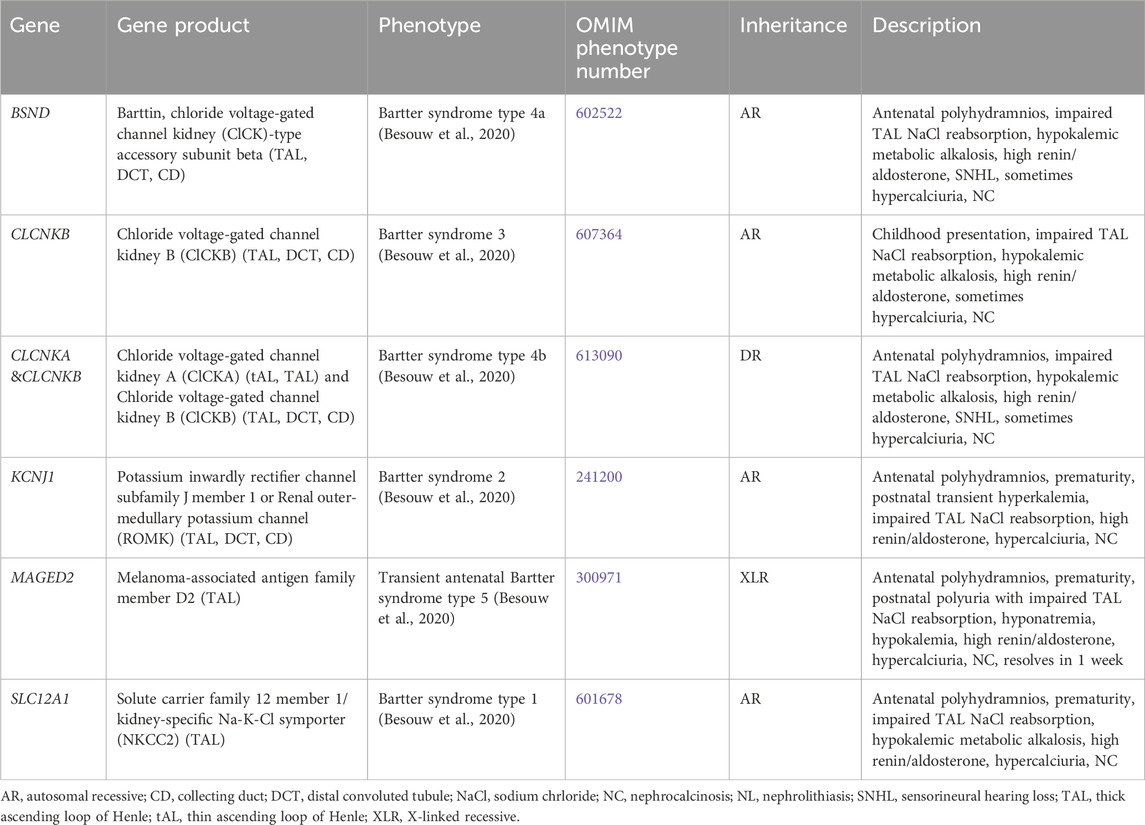
Table 7. Genetic causes of Bartter syndrome with hypercalciuria and nephrolithiasis and/or nephrocalcinosis.
Antenatal in presentation is present for Bartter syndrome type 1 (SLC12A1 gene, encodes for kidney-specific Na-K-Cl symporter [NKCC2], AR inheritance, OMIM phenotype number 601678) (Figure 1), Bartter syndrome type 2 (KCNJ1 gene, encodes for renal out-medullar potassium channel [ROMK], AR inheritance, OMIM phenotype number 241200) (Figure 1), Bartter syndrome type 4a (BSND gene, encodes for Barttin, AR inheritance, OMIM phenotype number 602522) (Figure 1), Bartter syndrome type 4b (CLCKA and CLCKB genes, encode for chloride voltage-gated channel kidney A [ClCKA] and chloride voltage-gated channel kidney B [ClCKB], digenic recessive [DR] inheritance, OMIM phenotype number 613090) (Figure 1), and Bartter syndrome type 5 (MAGED2 gene, XLR inheritance, OMIM phenotype number 300971) (Besouw et al., 2020). However, Bartter syndrome type 5 is transient and resolves after 1 week (Besouw et al., 2020). The only Bartter syndrome that presents in childhood is Bartter syndrome type 3 (CLCKB gene, encode for ClCKB, AR inheritance, OMIM phenotype number 607364) (Figure 1) (Besouw et al., 2020). Mainstays of treatment are hydration, sodium and potassium repletion, and nonsteroidal anti-inflammatory drugs (NSAIDs) that help to minimize urinary losses of electrolytes and water (Besouw et al., 2020).
3.1.8 Hyperaldosteronism and pseudohyperaldosteronism
Genetic causes of hyperaldosteronism and pseudohyperaldosteronism with hypercalciuria and NL and/or NC are shown in Supplementary Table S3. Studies have shown that individuals with hyperaldosteronism have a higher incidence of NL compared to the general population (Chang et al., 2022). Hyperaldosteronism has also been associated with NC, thought to be related to factors including increased sodium excretion and resultant hypercalciuria, hyperphosphaturia, hypocitraturia, and hypokalemia-associated ammonia-medicated nephropathy (Mittal et al., 2015). Genetic hyperaldosteronism conditions associated with NL and/or NC are Familial hyperaldosteronism type I/Glucocorticoid-remediable aldosteronism (CYP11B1 gene, AD inheritance, OMIM phenotype number 103900), Familial hyperaldosteronism type II (CLCN2 gene, AD inheritance, OMIM phenotype number 605635), Familial hyperaldosteronism type III (KCNJ5 gene, AD inheritance, OMIM phenotype number 613677), and Familial hyperaldosteronism type IV (CACNA1H gene, AD inheritance, OMIM phenotype number 617027) (Mittal et al., 2015; Chang et al., 2022). Pseudohyperaldosteronism type IIB/Gordon syndrome (WNK4 gene, AD inheritance, OMIM phenotype number 614491) is associated with hypercalciuria and NL (Mabillard and Sayer, 2019).
3.1.9 Hyperparathyroidism and hypoparathyroidism
Genetic causes of hyperparathyroidism and hypoparathyroidism with hypercalciuria and NL and/or NC are listed in Supplementary Table S4. Genetic causes of hyperparathyroidism with NL and/or NC are Familial primary hyperparathyroidism (CDC73 gene, AD inheritance, OMIM phenotype number 145000) and Hyperparathyroidism 4 (GCM2 gene, AD inheritance OMIM phenotype number 617343) (Lila et al., 2012; El Allali et al., 2021). Multiple endocrine neoplasia types I (131100), IIa (171400), and IV (610755) are also associated with hyperparathyroidism and NL and/or NC (Lila et al., 2012; El Allali et al., 2021). Hypoparathyroidism, sensorineural deafness, and renal dysplasia (GATA3 gene, AD inheritance OMIM phenotype number 146255) is associated with hypercalciuria and NC (Chenouard et al., 2013).
3.1.10 Other causes of hypercalciuria
Supplementary Table S5 lists other genetic causes of hypercalciuria and NL and/or NC. This includes Susceptibility to absorptive hypercalciuria/Familial idiopathic hypercalciuria (ADCY10 gene, AD inheritance, OMIM phenotype number 143870) (Reed et al., 2002), Hypophosphatasia (ALPL gene, AR and AD inheritance, OMIM phenotype numbers 241500, 241510, 146300) (Barvencik et al., 2011; Whyte et al., 2012; Rothenbuhler and Linglart, 2017), IMAGE syndrome (CDKN1C gene, AD inheritance, OMIM phenotype number 614732) (Balasubramanian et al., 2010), Beckwith-Wiedemann syndrome (CDKN1C, ICR1, KCNQ1OT1 genes, AD inheritance, OMIM phenotype number 130650) (Weksberg et al., 2010), Cystic fibrosis (CFTR gene, AR inheritance, OMIM phenotype number 219700) (Katz et al., 1988), Obstructive azoospermia with NL (CLDN2 gene, XLR inheritance, OMIM phenotype number 301060) (Askari et al., 2019), Amelogenesis imperfecta type IG/Enamel-renal syndrome (FAM20A gene, AR inheritance, OMIM phenotype number 204690) (Wang et al., 2013), Neurodevelopmental disorder with microcephaly, cataracts, and renal abnormalities (GEMIN4 gene, AR inheritance, OMIM phenotype number 617913) (Alazami et al., 2015), Somatic mosaic McCune-Albright syndrome (GNAS gene, mosaic inheritance, OMIM phenotype number 174800) (Kessel et al., 1992; Kirk et al., 1999), Mitochondrial DNA depletion syndrome 6 (MPV17 gene, AR inheritance, OMIM phenotype number 256810) (El-Hattab et al., 2018), Multiple congenital anomalies-hypotonia-seizures syndrome 3 (PIGT gene, AR inheritance, OMIM phenotype number 615398) (Kvarnung et al., 2013; Kohashi et al., 2018), SHORT syndrome (PIK3R1 gene, AD inheritance, OMIM phenotype number 269880) (Dyment et al., 2013), and Idiopathic hypercalciuria (VDR gene, AD inheritance) (Scott et al., 1999; Halbritter et al., 2015).
3.2 Conditions not primarily due to hypercalciuria
A minority of genetic diseases associated with NL and NC are not primarily due to hypercalciuria, including those secondary to hyperoxaluria, cystinuria, hyperuricosuria, xanthinuria, other metabolic disorders, and multifactorial etiologies. Each of these categories will be expanded on in detail in the following subsections.
3.2.1 Conditions with hyperoxaluria
Genetic causes of hyperoxaluria with NC and/or NL are discussed in greater detail below and are shown in Table 8 and Supplementary Table S6. General recommendations for children with hyperoxaluria include limiting foods high in oxalate, avoiding vitamin C supplementation, and adequate calcium intake (Copelovitch, 2012; Scoffone and Cracco, 2018).
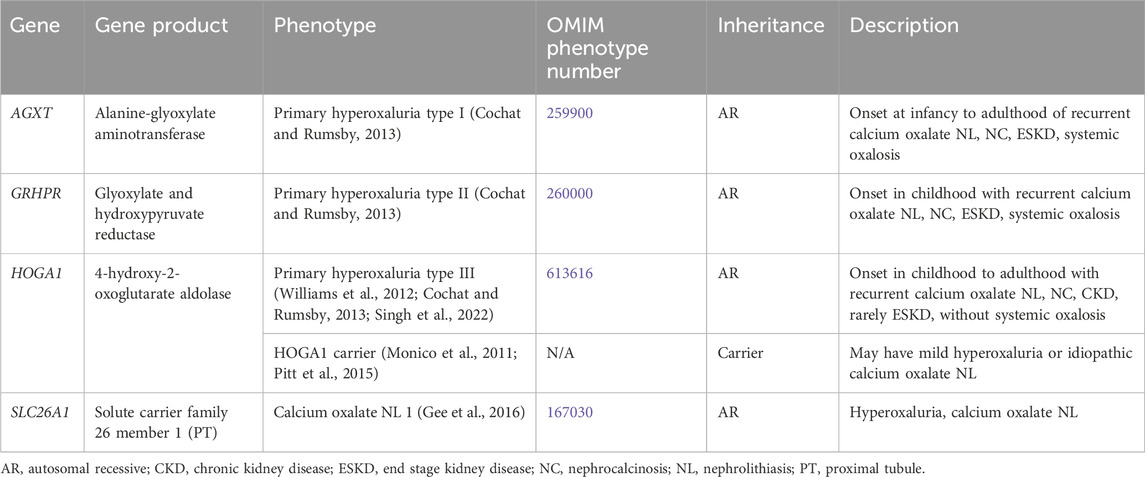
Table 8. Genetic causes of hyperoxaluria with nephrolithiasis and/or nephrocalcinosis.
3.2.1.1 AGXT gene
AGXT encodes for alanine-glyoxylate aminotransferase (AGT), which is a liver peroxisomal enzyme (Cochat and Rumsby, 2013). Inactivating variants in AGXT result in impaired metabolism of glyoxylate into glycine by AGT, leading to increased metabolism of glyoxylate by glyoxylate reductase/hydroxypyruvate reductase (GRHPR) to glycolate and by lactate dehydrogenase (LDH) into oxalate (Cochat and Rumsby, 2013). PH type I (PH1) (OMIM phenotype number 259900) accounts for approximately 80% of cases of PH (Cochat and Rumsby, 2013). This AR condition has onset in infancy to adulthood of recurrent calcium oxalate NL, NC, ESKD, and systemic oxalosis (widespread tissue deposition of calcium oxalate) (Cochat and Rumsby, 2013).
Treatments of PH1 that have been tested include substrate reduction therapy to target enzymes responsible for production of oxalate with RNA interference (RNAi) (targeting glycolate oxidase [GO] with Lumasiran, LDH with Nedosiran) and CRISPR (targeting GO, LDH) (Zabaleta et al., 2018; Martinez-Turrillas et al., 2022; Sas et al., 2022; Baum et al., 2023; Hayes et al., 2023), small molecules to prevent AGT mistargeting (Miyata et al., 2014), enhanced intestinal oxalate degradation using probiotics (Oxalobacter formigenes) or enzymes (oxalate decarboxylase) (Milliner et al., 2018; Lingeman et al., 2019; Quintero et al., 2020), and restoration of functional enzyme conformation with chaperone therapy (vitamin B6) (Fargue et al., 2013). Definitive treatment involves combined or sequential liver and kidney transplant (Loos et al., 2023).
3.2.1.2 GRHPR gene
GRHPR encodes for glyoxylate and hydroxypyruvate reductase (GRHPR), which an enzyme expressed throughout the body, including in the liver, specifically the hepatocyte cytosol and mitochondria (Cochat and Rumsby, 2013). Inactivating variants in GRHPR result in impaired metabolism of glyoxylate into glycolate by GRHRP, leading to increased metabolism of glyoxylate by AGT to glycine and by LDH into oxalate (Cochat and Rumsby, 2013). PH type II (PH2) (OMIM phenotype number 260000) is an AR condition that has onset in childhood with recurrent calcium oxalate NL, NC, ESKD, and systemic oxalosis (Cochat and Rumsby, 2013).
Treatments of PH2 that have been tested include substrate reduction therapy to target enzymes responsible production for oxalate with RNA interference (RNAi) (targeting LDH with Nedosiran) and CRISPR (targeting LDH) and enhanced intestinal oxalate degradation using probiotics (Oxalobacter formigenes) or enzymes (oxalate decarboxylase) (Milliner et al., 2018; Lingeman et al., 2019; Quintero et al., 2020; Martinez-Turrillas et al., 2022; Baum et al., 2023). Treatment with combined liver and kidney transplant has been successful in individuals with PH2 (Genena et al., 2023).
3.2.1.3 HOGA1 gene
HOGA1 encodes for 4-hydroxy-2-oxoglutarate aldolase (HOGA), which is a liver mitochondrial enzyme (Cochat and Rumsby, 2013). HOGA catalyzes metabolism from 4-hydroxy-2-oxoglutarate (HOG) to glyoxylate and pyruvate (Williams et al., 2012; Singh et al., 2022). The mechanism by which variants in HOGA1 result in PH is unclear (Williams et al., 2012; Singh et al., 2022). PH type III (PH3) (OMIM phenotype number 613616) accounts for approximately 10% of cases of PH (Williams et al., 2012; Singh et al., 2022). PH3 is an AR condition with onset in childhood to adulthood with recurrent calcium oxalate NL, NC, CKD, rarely ESKD, and without systemic oxalosis. Data suggests that carriers (heterozygotes) of inactivating variants in HOGA1 may have mild hyperoxaluria or idiopathic calcium oxalate NL (Monico et al., 2011; Pitt et al., 2015).
Treatments of PH3 that have been tested include substrate reduction therapy to target enzymes responsible production for oxalate with RNA interference (RNAi) (targeting LDH with Nedosiran) and CRISPR (targeting LDH) and enhanced intestinal oxalate degradation using probiotics (Oxalobacter formigenes) or enzymes (oxalate decarboxylase) (Milliner et al., 2018; Lingeman et al., 2019; Quintero et al., 2020; Martinez-Turrillas et al., 2022; Goldfarb et al., 2023).
3.2.1.4 SLC26A1 gene
SLC26A1 encodes for solute carrier family 26 member 1, an electroneutral anion exchanger (sulfate-oxalate, sulfate-bicarbonate, oxalate-bicarbonate) that is expressed in the PT (Figure 1) (Gee et al., 2016). Inactivating variants in SLC26A1 result Calcium oxalate NL 1 (OMIM phenotype number 167030), an AR condition with hyperoxaluria and calcium oxalate NL (Gee et al., 2016).
3.2.1.5 Other conditions with hyperoxaluria
Peroxisome biogenesis disorder A (Zellweger) and Peroxisome biogenesis disorder B (neonatal adrenoleukodystrophy [NALD] and infantile Refsum disease [IRD]) are characterized by deficient peroxisomal assembly with a generalized loss of peroxisomal functions (van Woerden et al., 2006; Alhazmi, 2018). Children with these disorders frequently have hyperoxaluria, hypothesized to be related to reduced glyoxylate conversion into glycine by AGT with increased oxalate production by LDH (van Woerden et al., 2006; Alhazmi, 2018).
3.2.1.5.1 Peroxisome biogenesis disorder A (Zellweger)
This is an AR condition associated with the absence of peroxisomes with severe neurologic dysfunction, craniofacial abnormalities, and liver dysfunction. It has been associated with hyperoxaluria with NL and NC with variants in PEX1 (Alhazmi, 2018), likely with variants in PEX5 (van Woerden et al., 2006), and possibly with variants in PEX3, PEX6, PEX 10, PEX 12, PEX13, PEX14, PEX16, PEX19, and PEX26 (Braverman et al., 2016; Alhazmi, 2018).
3.2.1.5.2 Peroxisome biogenesis disorder B (neonatal adrenoleukodystrophy [NALD] and infantile Refsum disease [IRD])
This is an AR condition generally associated with a milder phenotype than Zellweger. It has been associated with hyperoxaluria with NL and NC with variants in PEX1 and PEX3 (van Woerden et al., 2006; Maxit et al., 2017), likely with variants in PEX5 (van Woerden et al., 2006), and possibly with variants in PEX6, PEX7, PEX10, PEX11, PEX12, PEX13, PEX16, and PEX26 (Braverman et al., 2016; Alhazmi, 2018).
3.2.2 Conditions with cystinuria
Table 9 shows genetic causes of cystinuria with NL and/or NC, which consists of Cystinuria, and Hypotonia-cystinuria syndrome.
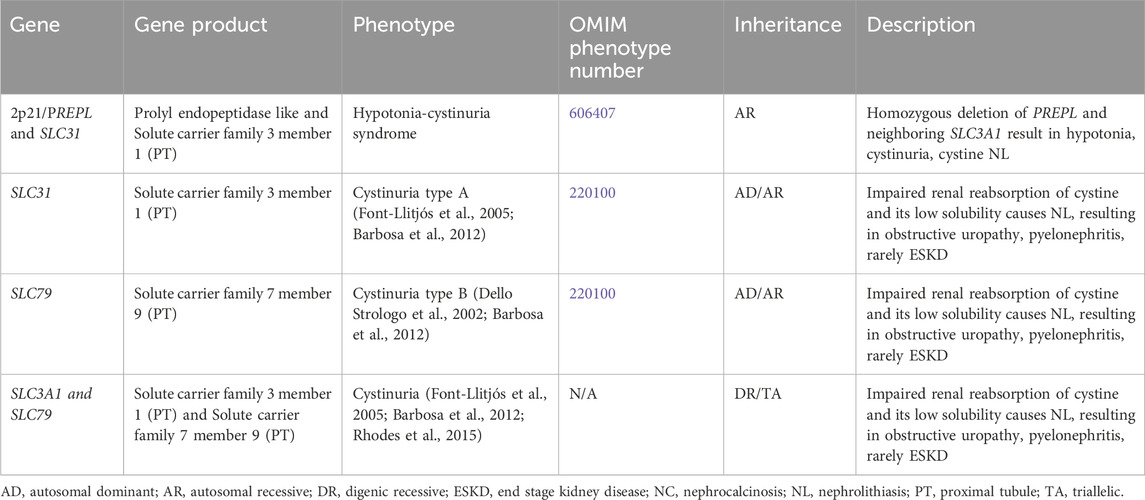
Table 9. Genetic causes of cystinuria with nephrolithiasis and/or nephrocalcinosis.
3.2.2.1 Cystinuria
This condition results in impaired renal reabsorption of cystine. Cystine’s low solubility causes the formation of NL, resulting in obstructive uropathy, pyelonephritis, and rarely ESKD (Barbosa et al., 2012). Cystinuria results from variants in SLC3A1 and SLC7A9, which encode for solute carrier family 3 member 1 and solute carrier family 7 member 9, respectively, both of which are expressed in the PT (Figure 1). This condition has been associated with one or two pathogenic variants in either gene (AD/AR, OMIM phenotype number 220100 for SLC3A1 (Font-Llitjós et al., 2005), OMIM phenotype number 220100 for SLC7A9 (Dello Strologo et al., 2002)), one pathogenic variant in each gene (DR) (Font-Llitjós et al., 2005), or two pathogenic variants in one gene with one pathogenic variant in the other gene (triallelic inheritance) (Rhodes et al., 2015).
Mainstays in therapy are urinary dilution with hyperhydration, decreased cystine excretion through low sodium diet and low protein (methionine) diet in adolescents and adults of <0.8 g protein per day, increased urinary cystine solubility by alkalinizing the urine to pH of 7.5 with potassium citrate, and increased urinary cystine solubility by conversion of cystine to cysteine (if cystine excretion is >3 mmol per day) with chelation agents such as D-penicillamine and tiopronin (Knoll et al., 2005). Tolvaptan has been shown to decrease urinary cystine concentrations by increasing diuresis (de Boer et al., 2012). Captopril and Bucillamine are other drugs thought to increasing urinary cystine solubility by conversion of cystine to cysteine, but the data is uncertain (Knoll et al., 2005; Moussa et al., 2020). L-cystine dimethyl ester (L-CDME) and L-cystine methyl ester (L-CME) are being studied as cystine crystal growth inhibitors and alpha-lipoic acid is being studied as a drug to increase urinary cystine solubility (Rimer et al., 2010; Zee et al., 2017).
3.2.2.2 Hypotonia-cystinuria syndrome
Chromosome 2p21 contains the following genes: PREPL and SLC3A1, which encode for prolyl endopeptidase like and solute carrier family 3 member 1, respectively (Jaeken et al., 2006). PREPL and SLC3A1 are expressed in the PT (Jaeken et al., 2006). Homozygous deletion of both PREPL and the neighboring gene SLC3A1 result in Hypotonia-cystinuria syndrome (AR inheritance, OMIM phenotype number 606407) with hypotonia, cystinuria, and cystine NL (Jaeken et al., 2006).
3.2.3 Purine metabolism disorders
Genetic causes of purine metabolism disorders with NL and/or NC are shown in Table 10. Purine metabolism is illustrated in Figure 2. The genes responsible for conditions with hyperuricosuria are HPRT1, PRPS1, SLC22A12, and SLC2A9. In addition, variation in the ZNF365 gene has been associated susceptibility to uric acid NL (complex inheritance, OMIM phenotype number 605990) (Gianfrancesco et al., 2003). Genetic causes of xanthinuria with NL and/or NC consist of Xanthinuria (XDH and MOCOS genes) and Molybdenum cofactor deficiency (MOCS1 and MOCS2 genes). Molybdenum cofactor deficiency C (GPHN gene, OMIM phenotype number 615501) has not been associated with reports of NL or NC and will not be discussed. Treatment of these conditions generally includes hyperhydration and low purine diet (Scoffone and Cracco, 2018).
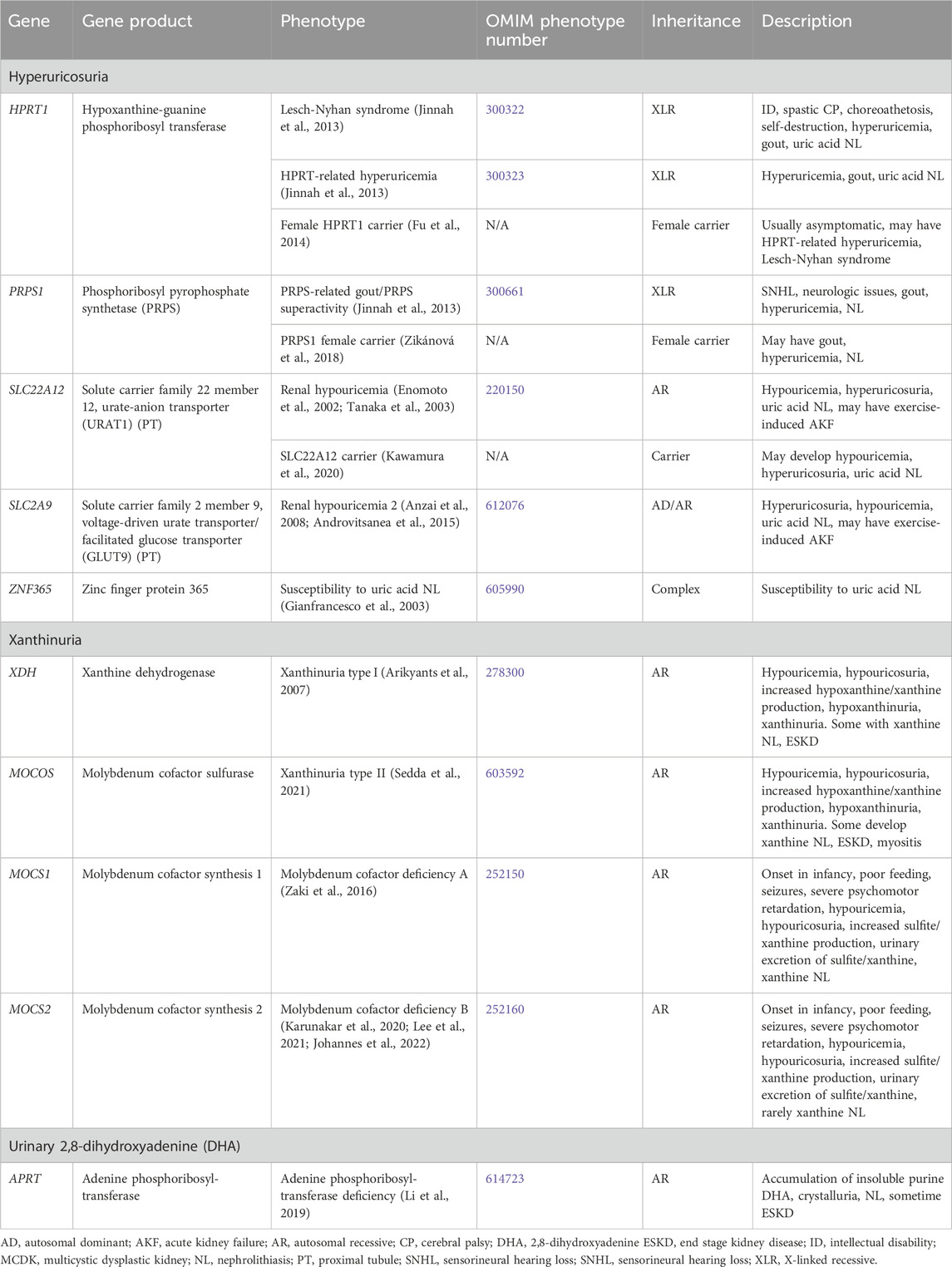
Table 10. Genetic causes of purine metabolism disorders associated with nephrolithiasis and/or nephrocalcinosis.
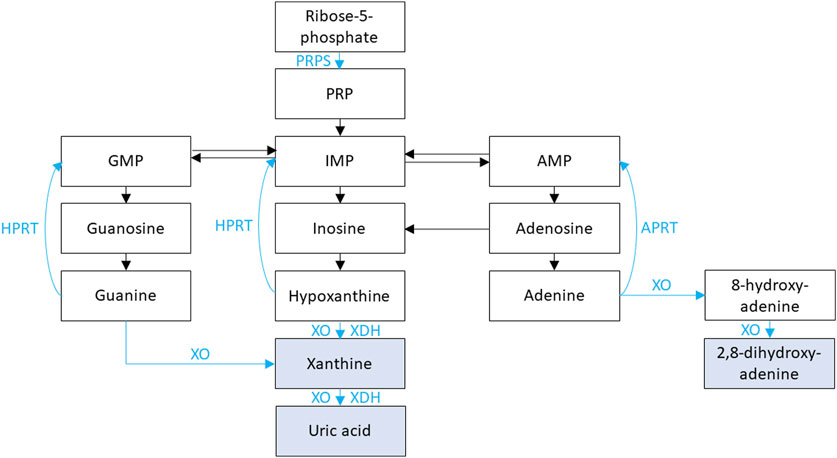
Figure 2. Purine metabolism is shown. Xanthine oxidase (XO) and xanthine dehydrogenase (XDH) facilitate the oxidation and reduction, respectively, of hypoxanthine to xanthine and of xanthine to uric acid. XO is also responsible for the oxidation of guanine to xanthine, the oxidation of adenine to 8-hydroxy-adenine, and the oxidation of 8-hydroxy-adenine to 2,8-dihydroxy-adenine. Phosphoribosyl pyrophosphate synthetase (PRPS) facilitates the conversion of adenosine triphosphate (ATP) and ribose-5-phosphate into phosphoribosyl pyrophosphate (PRP). PRP is subsequently converted to inosine monophosphate (IMP) then hypoxanthine. Hypoxanthine-guanine phosphoribosyl transferase (HPRT) is responsible for converting hypoxanthine IMP and guanine to guanosine monophosphate (GMP). Adenine phosphoribosyltransferase (APRT) is responsible for converting adenine to adenosine monophosphate (AMP).
3.2.3.1 Hyperuricosuria
3.2.3.1.1 HPRT1 gene
HPRT1 encodes for hypoxanthine-guanine phosphoribosyl transferase (HPRT), which plays an important role in purine nucleotide metabolism (Figure 2) (Jinnah et al., 2013). During normal purine metabolism, xanthine oxidase (XO) facilitates the oxidation of purines hypoxanthine and guanine to xanthine and oxidation of the xanthine to uric acid (Figure 2) (Jinnah et al., 2013). HPRT is responsible for converting the purine hypoxanthine to the purine nucleotide inosine monophosphate (IMP) and the purine guanine to the purine nucleotide guanosine monophosphate (GMP) (Jinnah et al., 2013). Conversion of purines to purine nucleotides by HPRT therefore results in decreased uric acid production. Therefore, inactivating variants in HPRT1 result in increased production of purines hypoxanthine and guanine and oxidation by XO to xanthine then uric acid with resultant hyperuricemia (Jinnah et al., 2013). Inactivating variants in HPRT1 are associated with a spectrum of conditions depending on degree of HPRT dysfunction.
Treatment for hyperuricemia in these conditions has included hyperhydration, xanthine oxidase inhibitors (allopurinol, febuxostat), recombinant urate oxidases (rasburicase), and urinary alkalinization (Torres et al., 2012; Madeo et al., 2019). Allopurinol is considered first line treatment, but caution must be used to avoid xanthine NL that can result from excessive doses (Torres et al., 2012; Madeo et al., 2019). Lesch-Nyhan syndrome (OMIM phenotype number 300322) is an XLR condition due to an inactivating variant in HPRT1, resulting in hyperuricemia with subsequent intellectual disability, involuntary movements, self-injurious behavior, gout, and uric acid NL (Madeo et al., 2019). HPRT-related hyperuricemia (OMIM phenotype number 300323) is an XLR condition due to an inactivating variant in HPRT1, resulting in hyperuricemia with subsequent gout and uric acid NL (Madeo et al., 2019). Data suggests that female carriers of inactivating variants in HPRT1 are usually asymptomatic but may have HPRT-related hyperuricemia or Lesch-Nyhan syndrome (Fu et al., 2014).
3.2.3.1.2 PRPS1 gene
PRPS1 encodes for phosphoribosyl pyrophosphate synthetase (PRPS), which plays an important role in purine nucleotide synthesis (Figure 2) (Jinnah et al., 2013). During normal purine metabolism, PRPS facilitates the conversion of adenosine triphosphate (ATP) and ribose-5-phosphate into phosphoribosyl pyrophosphate (PRP) (Jinnah et al., 2013). PRP is subsequently converted to the purine nucleotides IMP then hypoxanthine, and XO facilitates the oxidation of hypoxanthine to xanthine and conversion of xanthine to uric acid (Jinnah et al., 2013). Conversion of ribose-5-phosphate to PRP by PRPS therefore results in increased uric acid production. Therefore, activating variants in PRPS1 result in increased production PRPS, IMP, hypoxanthine, xanthine, and finally uric acid (Jinnah et al., 2013). Treatment for hyperuricemia in these conditions is like that of conditions associated with HPRT1 inactivating variants. PRPS-related gout/PRPS superactivity (OMIM phenotype number 300661) is an XLR condition due to an activating variant in PRPS1, resulting in hyperuricemia with sensorineural hearing loss, neurologic issues gout, and NL (Jinnah et al., 2013). Data suggests that female carriers of inactivating variants in PRPS1 may have gout, hyperuricemia, and NL (Zikánová et al., 2018).
3.2.3.1.3 SLC22A12 gene
SLC22A12 encodes for solute carrier family 22 member 12, which is a urate-anion transporter (URAT1) expressed in the PT responsible for luminal/apical uric acid reuptake and is important in the regulation of blood urate levels (Figure 1) (Enomoto et al., 2002). Renal hypouricemia (OMIM phenotype number 220150) is an AR condition due to inactivating variants in SLC22A1, which lead to decreased urate transport in the PT, resulting in hypouricemia, hyperuricosuria, and uric acid NL (Enomoto et al., 2002). This condition may be associated with severe complications such as exercise-induced acute kidney failure (Tanaka et al., 2003). Patients with this condition have been successfully treated with urinary alkalinization (Hisatome et al., 1993). Data suggests that carriers (heterozygotes) of inactivating variants in SLC22A12 may develop hypouricemia, hyperuricosuria, and uric acid NL (Kawamura et al., 2020).
3.2.3.1.4 SLC2A9 gene
SLC2A9 encodes for solute carrier family 2 member 9, which is a voltage-driven urate transporter and facilitated glucose transporter (GLUT9) expressed in the PT responsible for basolateral acid reuptake and is important in the regulation of blood urate levels (Figure 1) (Anzai et al., 2008). Renal hypouricemia 2 (OMIM phenotype number 612076) is an AD/AR condition due to inactivating variants in SLC2A9, resulting in impaired renal urate reabsorption with hyperuricosuria and subsequent hypouricemia and uric acid NL (Anzai et al., 2008). This condition may be associated with severe complications such as exercise-induced acute kidney failure (Androvitsanea et al., 2015). Patients with this condition have been successfully treated with urinary alkalinization (Hisatome et al., 1993).
3.2.3.2 Xanthinuria
3.2.3.2.1 XDH gene
XDH encodes for xanthine dehydrogenase (XDH) (Arikyants et al., 2007). During normal purine metabolism, XDH facilitates the conversion of hypoxanthine to xanthine and from xanthine to uric acid via reduction of NAD+ to NADH (Figure 2) (Arikyants et al., 2007). Xanthinuria type I (OMIM phenotype number 278300) is an AR condition due inactivating variants in XDH, resulting in hypouricemia with hypouricosuria, increased hypoxanthine production with hypoxanthinuria, and increased xanthine production with xanthinuria (Arikyants et al., 2007). Some individuals develop xanthine NL and/or ESKD (Arikyants et al., 2007). In addition to hyperhydration and low purine diet, inhibitors of xanthine crystallization have been tested successfully in vitro (Sedda et al., 2021).
3.2.3.2.2 MOCOS gene
MOCOS encodes for molybdenum cofactor sulfurase (MOCOS), which is required to activate XDH and aldehyde oxidase 1 (AOX1) (Sedda et al., 2021). During normal purine metabolism, XDH facilitates the conversion of hypoxanthine to xanthine and from xanthine to uric acid via reduction of NAD+ to NADH (Arikyants et al., 2007). The physiologic relevance of AOX1 is uncertain (Sedda et al., 2021). Xanthinuria type II (OMIM phenotype number 603592) is an AR condition due inactivating variants in MOCOS, resulting in hypouricemia with hypouricosuria, increased hypoxanthine production with hypoxanthinuria, and increased xanthine production with xanthinuria (Sedda et al., 2021). Some individuals develop xanthine NL, ESKD, and/or myositis (Sedda et al., 2021). Treatment is the same as for Xanthinuria type I (Sedda et al., 2021).
3.2.3.2.3 MOCS1 gene
MOCS1 encodes for molybdenum cofactor synthesis 1 (MOCS1), which is responsible for the conversion from guanosine triphosphate (GTP) to cyclic pyranopterin monophosphate (cPMP), the first step in the synthesis of molybdenum cofactor (MOCO) (Reiss and Hahnewald, 2011; Johannes et al., 2022). MOCO is required to activate XDH and sulfite oxidase (SUOX) (Reiss and Hahnewald, 2011). XDH facilitates the conversion of hypoxanthine to xanthine and from xanthine to uric acid via reduction of NAD+ to NADH and SUOX facilitates the oxidative degradation of sulfur-containing amino acids (Arikyants et al., 2007; Zaki et al., 2016).
Molybdenum cofactor deficiency A (OMIM phenotype number 252150) is an AR condition due inactivating variants in MOCS1, resulting in disease onset in infancy with poor feeding, intractable seizures, severe psychomotor retardation, hypouricemia with hypouricosuria, increased sulfite production with urinary excretion of sulfite, and increased xanthine production with xanthinuria and xanthine NL (Zaki et al., 2016; Johannes et al., 2022). Treatment with synthetic cPMP (Fisdenopterin) has been shown to be effective in this condition (Farrell et al., 2021).
3.2.3.2.4 MOCS2 gene
MOCS2 encodes for molybdenum cofactor synthesis 2, which is responsible for the conversion from cPMP to molybdopterin (MPT), the second step in the synthesis of molybdenum cofactor (MOCO) (Lee et al., 2021; Johannes et al., 2022). Molybdenum cofactor deficiency B (OMIM phenotype number 252160) is an AR condition due inactivating variants in MOCS2, resulting in disease onset in infancy with poor feeding, intractable seizures, severe psychomotor retardation, hypouricemia with hypouricosuria, increased sulfite production with urinary excretion of sulfite, and increased xanthine production with xanthinuria and rarely xanthine NL (Karunakar et al., 2020; Lee et al., 2021; Johannes et al., 2022). There is currently no effective therapy for this condition (Johannes et al., 2022).
3.2.3.3 Urinary 2,8-dihydroxyadenine (DHA)
3.2.3.3.1 APRT gene
APRT encodes for adenine phosphoribosyltransferase (APRT), which plays an important role in purine nucleotide metabolism (Figure 2) (Li et al., 2019). APRT is responsible for converting the purine nucleotide adenine to adenosine monophosphate (AMP), which subsequently is converted to the purine nucleoside adenosine, then to the nucleoside inosine (Li et al., 2019). Insosine is then converted to the purine hypoxanthine and XO facilitates the oxidation of hypoxanthine to xanthine and xanthine to uric acid (Figure 2) (Li et al., 2019; Johannes et al., 2022).
APRT deficiency (OMIM phenotype number 614723) is an AR condition due inactivating variants in APRT, resulting in inability of adenine to be converted to AMP (Li et al., 2019). XO then facilitates the oxidation of adenine to 8-hydroxyadenine then 2,8-hydroxyadenine (DHA), an insoluble purine that accumulates in the kidney with crystalluria, NL, and sometime ESKD (Li et al., 2019). Treatment of APRT deficiency involves low purine diet to limit DHA production and xanthine oxidase inhibitors (allopurinol and/or febuxostat) to reduce conversion of adenine to DHA (Li et al., 2019).
3.2.4 Other metabolic disorders
Supplementary Table S7 shows other genetic metabolic disorders with NL and/or NC. The conditions are 3-methylglutaconic aciduria type VIIB (CLPB gene, AR inheritance, OMIM phenotype number 616271) (Kanabus et al., 2015), Congenital disorder of glycosylation with defective fucosylation 1 (FUT8 gene, AR inheritance, OMIM phenotype number 618005) (Ng et al., 2018), Glycogen storage disease type 1A (G6PC gene, AR inheritance, OMIM phenotype number 232200) (Weinstein et al., 2001), Alkaptonuria (HGD gene, AR inheritance, OMIM phenotype number 203500) (Phornphutkul et al., 2002), 5-oxoprolinase deficiency (OPLAH gene, AD/AR inheritance, OMIM phenotype number 260005) (Larsson et al., 1981), Dicarboxylic aminoaciduria (SLC1A1 gene, AR inheritance, OMIM phenotype number 222730) (Bailey et al., 2011), Hyperglycinuria (SLC36A2 gene, AD inheritance, OMIM phenotype number 138500) (De Vries et al., 1957), and Hartnup disorder (SLC6A19 gene, AR inheritance OMIM phenotype number 234500) (Simoni et al., 2007).
3.2.5 Conditions with multifactorial etiologies
3.2.5.1 Disorders of inorganic pyrophosphate
Genetic disorders of inorganic pyrophosphate (PPi) with NL and/or NC are shown in Table 11. The genes responsible are ENPP1 and ABCC6.
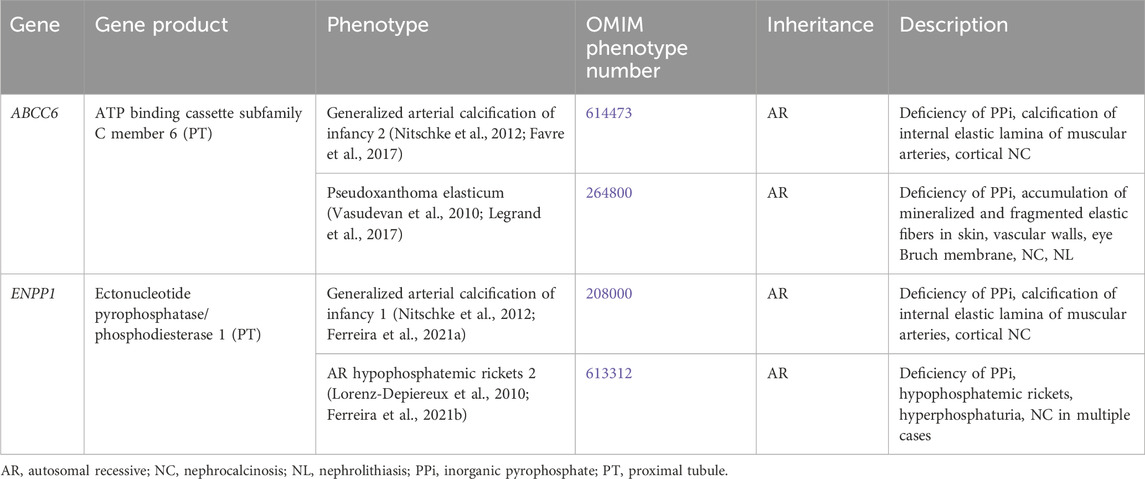
Table 11. Genetic disorders of inorganic pyrophosphate with nephrolithiasis and/or nephrocalcinosis.
3.2.5.1.1 ENPP1 gene
ENPP1 encodes for ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), which is responsible for generation of PPi and is expressed in the liver and the renal PT (Nitschke et al., 2012). Inactivating variants in ENPP1 are associated with a spectrum of conditions.
Generalized arterial calcification of infancy 1 (OMIM phenotype number 208000) is an AR condition due to inactivating variants in ENPP1, resulting in deficiency of PPi with calcification of the internal elastic lamina of muscular arteries and stenosis due to myointimal proliferation as well as cortical NC, possibly due to ischemia (Ferreira et al., 2021a). Bisphosphonate therapy was previously thought to improve survival, but based on recent studies, this may not be the case (Ferreira et al., 2021a).
AR hypophosphatemic rickets 2 (OMIM phenotype number 613312) is an AR condition due to inactivating variants in ENPP1, resulting in deficiency of PPi and subsequent hypophosphatemic rickets with hyperphosphaturia as well as NC in multiple cases (Lorenz-Depiereux et al., 2010; Ferreira et al., 2021b). Treatment with calcitriol and oral phosphate improve skeletal symptoms, although they do not appear to improve bone mineral density and are associated with NC (Ferreira et al., 2021a). Enzyme replacement therapy in mice has been associated with improved bone mineral density without NC or ESKD and is currently being studied in humans (Ferreira et al., 2021a).
3.2.5.1.2 ABCC6 gene
ABCC6 encodes for ATP binding cassette subfamily C member 6, which through an unclear mechanism is thought to play an important role in physiologic inhibition of arterial calcification through production of PPi. ABCC6 is expressed primarily in the liver and the renal PT (Favre et al., 2017). Inactivating variants in ABCC6 are associated with a spectrum of conditions.
Generalized arterial calcification of infancy 2 (OMIM phenotype number 614473) is an AR condition due to inactivating variants in ABCC6, resulting in deficiency of PPi with calcification of the internal elastic lamina of muscular arteries and stenosis due to myointimal proliferation as well as cortical NC, possibly due to ischemia (Nitschke et al., 2012; Ferreira et al., 2021a). Pseudoxanthoma elasticum (OMIM phenotype number 264800) is an AR condition due to inactivating variants in ABCC6, resulting in deficiency of PPi with accumulation of mineralized and fragmented elastic fibers in the skin, vascular walls, and Bruch membrane in the eye, as well as NC and NL (Vasudevan et al., 2010; Legrand et al., 2017). As with ENNP1 variants, bisphosphonate therapy may not be effective (Kawai et al., 2022).
3.2.5.2 Polycystic kidney disease
Genetic disorders with polycystic kidney disease (PKD) with NL and/or NC are shown in Supplementary Table S8, which consist of AR PKD (PKDH1 gene, OMIM phenotype number 263200) and AD PKD including AD PKD type 1 (PKD1 gene, OMIM phenotype number 173900), AD PKD type 2 (PKD2 gene, OMIM phenotype number 613095), AD PKD type 3 (GANAB gene, OMIM phenotype number 600666), AD PKD type 6 with or without polycystic liver disease (DNAJB11 gene, OMIM phenotype number 618061), and AD PKD type 7 (ALG5 gene, OMIM phenotype number 620056). AR PKD is associated with multifactorial NL, NC, and medullary sponge kidney (Adeva et al., 2006). AD PKD is associated with NL (usually uric acid or calcium oxalate), abnormal transport of ammonium, low urine pH, hypocitraturia, and sometimes a distal RTA (Torres et al., 1993).
3.2.5.3 Other disorders with multifactorial etiologies
Other genetic disorders with multifactorial etiologies of NL and/or NC are shown in Supplementary Table S9. The associated genes for these disorders are CLDN10 (Klar et al., 2017), EMC10 (Shao et al., 2021; Kaiyrzhanov et al., 2022), HSD11B2 (Lu et al., 2022), PAX2 (Bower et al., 2012), STRADA (Puffenberger et al., 2007; Bi et al., 2016; Nelson et al., 2018), and ZNF687 (Rendina et al., 2020).
3.3 Conditions with possible association
Supplementary Table S10 shows a list of genetic disorders possibly associated with NL and/or NC. Variants or deletions in the following genes or genetic locations are associated with 1–2 cases of NL and/or NC: 19q13.11 (Caubit et al., 2016), AGPAT2 (Haghighi et al., 2016), AMMECR1 (Andreoletti et al., 2017), ATIC (Ramond et al., 2020), ATP6V1E1 (Alazami et al., 2016), BSCL2 (Haghighi et al., 2016), CHST14 (Dundar et al., 2001), FGF23 (Chefetz et al., 2005), GAD1 (Neuray et al., 2020), GNB2 (Tan et al., 2022), IFIH1 (Buers et al., 2017), MTM1 (Herman et al., 1999), MYL9 (Kandler et al., 2020), ROR2 (Tufan et al., 2005), SLC45A1 (Srour et al., 2017), SRCAP (White et al., 2010), and TMEM67 (Lee et al., 2017).
Variants in the following genes are tested for using a number of commercially available gene panels, but upon review, no cases of NL and/or NC were confirmed: GALNT3 (Hyperphosphatemic familial tumoral calcinosis 1, AR inheritance, OMIM phenotype number 211900) and GNA11 (AD hypocalcemia 2, AD inheritance, OMIM phenotype number 615361). For the genetic condition Developmental and epileptic encephalopathy 41 (SLC1A2 gene, AD inheritance, OMIM phenotype number 617105), OMIM states there is an association with NC, but on review, no cases were able to be confirmed.
3.4 Genome-wide association studies
There have been several genome wide association studies (GWAS) of adult populations on multiple continents (North America, Europe, Asia) and in multiple countries (USA, Iceland, Netherlands, England, Japan) that have been summarized below (Thorleifsson et al., 2009; Gudbjartsson et al., 2010; Urabe et al., 2012; Oddsson et al., 2015; Howles et al., 2019; Ware et al., 2019; Curry et al., 2020). Although the results of these adult GWAS are important, there have not been any GWAS in children with NL or NC and it is unclear what the relevance of these findings are in the pediatric population.
Variants in TRPM6, which encodes for transient receptor potential melastatin 6 and mediates the renal and intestinal transport of magnesium, have been associated with urinary magnesium excretion (Ware et al., 2019). Variants in CYP24A1 associated with urinary calcium excretion, vitamin D metabolism, serum calcium levels, and recurrent nephrolithiasis (Howles et al., 2019; Ware et al., 2019). Loci in DGKD, DGKH, WDR72, GPIC1, and BCR were found to influence signaling of CaSR (Howles et al., 2019). DGKD and DGKH encode diacylglycerol kinase, which induce CaSR-mediated intracellular calcium release (Howles et al., 2019). GIPC1 encodes G-protein alpha-interacting protein C-terminus-interacting protein 1, which is thought to play a role in sustained intracellular CaSR signaling through clathrin-mediated endocytosis (Howles et al., 2019). BCR encodes breakpost cluster region, whose activation is induced by CaSR ligand binding (Howles et al., 2019).
Variants in CLDN14, ALPL, CASR, CLDN2, SLC34A1, AQP1, as well as DGKH have been significantly associated with NL (Thorleifsson et al., 2009; Urabe et al., 2012; Oddsson et al., 2015; Curry et al., 2020). CLDN14 regulates paracellular permeability in the kidney epithelial tight junctions (Thorleifsson et al., 2009). ALPL encodes an alkaline phosphatase that is widely expressed, including in the renal PT (Oddsson et al., 2015). CLDN2 encodes for a paracellular cation channel that mediates calcium reabsorption primarily in the PT (Curry et al., 2020). AQP1 encodes aquaporin 1, a water channel present in the PT, thin descending loop of Henle, and vasa recta responsible for urinary concentration and water reabsorption (Urabe et al., 2012). Variants in SLC34A1 and TRPV5 have been associated with recurrent NL and variants in UMOD have been associated with both CKD and a decreased risk of NL (Gudbjartsson et al., 2010; Oddsson et al., 2015). TRPV5 encodes an epithelial calcium channel at the apical membrane of the distal tubule that facilitates renal calcium transport, stimulated by PTH and 1,25-dihydroxy-vitamin D (Oddsson et al., 2015). UMOD encodes for uromodulin, the most abundant protein in the urine of mammals, and regulates endocytosis of TRPV5 (Gudbjartsson et al., 2010).
4 General prevention strategies
As a reminder, aside from specific interventions mentioned above, general dietary interventions for children with NL include proper hydration, low sodium diet, maintaining adequate calcium intake, adequate but not excessive protein intake, avoidance of sugar-sweetened beverages, and a diet rich in fruits and vegetables (Copelovitch, 2012; Ferraro et al., 2013; Escribano et al., 2014; Scoffone and Cracco, 2018). However, much of this is based on adult studies and more studies are necessary in children with NL and NC. Hydration, ideally with water, of 1.5–2 L/m2/day is recommended to help reduce lithogenic factor concentration (Copelovitch, 2012; Scoffone and Cracco, 2018). Studies in adults with hypercalciuria have suggested that a low sodium intake, normal calcium intake, and low protein intake reduce NL recurrence and urinary calcium excretion (Escribano et al., 2014). However, it is important that growing children receive adequate recommended daily intake of calcium and protein to grow and develop appropriately, and therefore it is recommended that children ingest adequate calcium and adequate but not excessive amounts of protein (Copelovitch, 2012; Scoffone and Cracco, 2018). In adults, sugar-sweetened beverage intake has been associated with increased risk of NL (Ferraro et al., 2013). A diet rich in fruits and vegetables is recommended as they are a rich source of potassium and citrate, generally considered to be inhibitors of NL formation (Copelovitch, 2012; Scoffone and Cracco, 2018).
5 Discussion
The etiology of NL and NC is complex and includes environmental as well as genetic factors. As genetic testing has become more advanced, efficient, and readily available, several polygenic traits and monogenic disorders have been implicated in NL and NC. The discovery of these genes and study of these genes has greatly expanded our knowledge of the renal tubules and their channels, transporters, and receptors. However, further studies are necessary, especially in children, to better be able to provide individualized and evidence-based treatments, including the use of precision-medicine approaches.
Author contributions
AG: Data curation, Writing–original draft, Writing–review and editing. JZ: Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
AG has received research support from Natera, Inc.
Conflict of interest
JZ is on the speaker bureau for Alnylam and Novo Nordisk.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1381174/full#supplementary-material
References
Adeva, M., El-Youssef, M., Rossetti, S., Kamath, P. S., Kubly, V., Consugar, M. B., et al. (2006). Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Med. Baltim. 85 (1), 1–21. doi:10.1097/01.md.0000200165.90373.9a
Alazami, A. M., Al-Qattan, S. M., Faqeih, E., Alhashem, A., Alshammari, M., Alzahrani, F., et al. (2016). Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 135 (5), 525–540. doi:10.1007/s00439-016-1660-z
Alazami, A. M., Patel, N., Shamseldin, H. E., Anazi, S., Al-Dosari, M. S., Alzahrani, F., et al. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 10 (2), 148–161. doi:10.1016/j.celrep.2014.12.015
Alhazmi, H. H. (2018). Renal oxalate stones in children with Zellweger spectrum disorders. Saudi J. Anaesth. 12, 332–334. doi:10.4103/sja.SJA_699_17
Amlie-Wolf, L., Baker, L., Hiddemen, O., Thomas, M., Burke, C., Gluck, C., et al. (2021). Novel genetic testing model: a collaboration between genetic counselors and nephrology. Am. J. Med. Genet. Part A 185 (4), 1142–1150. doi:10.1002/ajmg.a.62088
Andreoletti, G., Seaby, E. G., Dewing, J. M., O'Kelly, I., Lachlan, K., Gilbert, R. D., et al. (2017). AMMECR1: a single point mutation causes developmental delay, midface hypoplasia and elliptocytosis. J. Med. Genet. 54 (4), 269–277. doi:10.1136/jmedgenet-2016-104100
Androvitsanea, A., Stylianou, K., Maragkaki, E., Tzanakakis, M., Stratakis, S., Petrakis, I., et al. (2015). Vanishing urate, acute kidney injury episodes and a homozygous SLC2A9 mutation. Int. Urol. Nephrol. 47 (6), 1035–1036. doi:10.1007/s11255-015-1005-1
Anzai, N., Ichida, K., Jutabha, P., Kimura, T., Babu, E., Jin, C. J., et al. (2008). Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J. Biol. Chem. 283 (40), 26834–26838. doi:10.1074/jbc.C800156200
Arikyants, N., Sarkissian, A., Hesse, A., Eggermann, T., Leumann, E., and Steinmann, B. (2007). Xanthinuria type I: a rare cause of urolithiasis. Pediatr. Nephrol. 22 (2), 310–314. doi:10.1007/s00467-006-0267-3
Askari, M., Karamzadeh, R., Ansari-Pour, N., Karimi-Jafari, M. H., Almadani, N., Sadighi Gilani, M. A., et al. (2019). Identification of a missense variant in CLDN2 in obstructive azoospermia. J. Hum. Genet. 64 (10), 1023–1032. doi:10.1038/s10038-019-0642-0
Bailey, C. G., Ryan, R. M., Thoeng, A. D., Ng, C., King, K., Vanslambrouck, J. M., et al. (2011). Loss-of-function mutations in the glutamate transporter SLC1A1 cause human dicarboxylic aminoaciduria. J. Clin. Invest. 121 (1), 446–453. doi:10.1172/JCI44474
Balasubramanian, M., Sprigg, A., and Johnson, D. S. (2010). IMAGe syndrome: case report with a previously unreported feature and review of published literature. Am. J. Med. Genet. A 152a (12), 3138–3142. doi:10.1002/ajmg.a.33716
Barbosa, M., Lopes, A., Mota, C., Martins, E., Oliveira, J., Alves, S., et al. (2012). Clinical, biochemical and molecular characterization of cystinuria in a cohort of 12 patients. Clin. Genet. 81 (1), 47–55. doi:10.1111/j.1399-0004.2011.01638.x
Barvencik, F., Beil, F. T., Gebauer, M., Busse, B., Koehne, T., Seitz, S., et al. (2011). Skeletal mineralization defects in adult hypophosphatasia--a clinical and histological analysis. Osteoporos. Int. 22 (10), 2667–2675. doi:10.1007/s00198-011-1528-y
Baum, M. A., Langman, C., Cochat, P., Lieske, J. C., Moochhala, S. H., Hamamoto, S., et al. (2023). PHYOX2: a pivotal randomized study of nedosiran in primary hyperoxaluria type 1 or 2. Kidney Int. 103 (1), 207–217. doi:10.1016/j.kint.2022.07.025
Belmont, J. W., Reid, B., Taylor, W., Baker, S. S., Moore, W. H., Morriss, M. C., et al. (2002). Congenital sucrase-isomaltase deficiency presenting with failure to thrive, hypercalcemia, and nephrocalcinosis. BMC Pediatr. 2, 4. doi:10.1186/1471-2431-2-4
Bergwitz, C., Roslin, N. M., Tieder, M., Loredo-Osti, J. C., Bastepe, M., Abu-Zahra, H., et al. (2006). SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am. J. Hum. Genet. 78 (2), 179–192. doi:10.1086/499409
Besouw, M. T. P., Kleta, R., and Bockenhauer, D. (2020). Bartter and gitelman syndromes: questions of class. Pediatr. Nephrol. 35 (10), 1815–1824. doi:10.1007/s00467-019-04371-y
Bi, W., Glass, I. A., Muzny, D. M., Gibbs, R. A., Eng, C. M., Yang, Y., et al. (2016). Whole exome sequencing identifies the first STRADA point mutation in a patient with polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE). Am. J. Med. Genet. A 170 (8), 2181–2185. doi:10.1002/ajmg.a.37727
Blanchard, A., Curis, E., Guyon-Roger, T., Kahila, D., Treard, C., Baudouin, V., et al. (2016). Observations of a large Dent disease cohort. Kidney Int. 90 (2), 430–439. doi:10.1016/j.kint.2016.04.022
Blanchard, A., Vargas-Poussou, R., Peyrard, S., Mogenet, A., Baudouin, V., Boudailliez, B., et al. (2008). Effect of hydrochlorothiazide on urinary calcium excretion in dent disease: an uncontrolled trial. Am. J. Kidney Dis. 52 (6), 1084–1095. doi:10.1053/j.ajkd.2008.08.021
Bolino, A., Devoto, M., Enia, G., Zoccali, C., Weissenbach, J., and Romeo, G. (1993). Genetic mapping in the Xp11.2 region of a new form of X-linked hypophosphatemic rickets. Eur. J. Hum. Genet. 1 (4), 269–279. doi:10.1159/000472424
Borthwick, K. J., Kandemir, N., Topaloglu, R., Kornak, U., Bakkaloglu, A., Yordam, N., et al. (2003). A phenocopy of CAII deficiency: a novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J. Med. Genet. 40 (2), 115–121. doi:10.1136/jmg.40.2.115
Bower, M., Salomon, R., Allanson, J., Antignac, C., Benedicenti, F., Benetti, E., et al. (2012). Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum. Mutat. 33 (3), 457–466. doi:10.1002/humu.22020
Braun, D., Lawson, J., Gee, H., Halbritter, J., Shril, S., Tan, W., et al. (2016). Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin. J. Am. Soc. Nephrol. CJASN 11 (4), 664–672. doi:10.2215/CJN.07540715
Braverman, N. E., Raymond, G. V., Rizzo, W. B., Moser, A. B., Wilkinson, M. E., Stone, E. M., et al. (2016). Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 117 (3), 313–321. doi:10.1016/j.ymgme.2015.12.009
Brown, E. M., and MacLeod, R. J. (2001). Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 81 (1), 239–297. doi:10.1152/physrev.2001.81.1.239
Buers, I., Rice, G. I., Crow, Y. J., and Rutsch, F. (2017). MDA5-Associated neuroinflammation and the singleton-merten syndrome: two faces of the same type I interferonopathy spectrum. J. Interferon Cytokine Res. 37 (5), 214–219. doi:10.1089/jir.2017.0004
Carling, T., Szabo, E., Bai, M., Ridefelt, P., Westin, G., Gustavsson, P., et al. (2000). Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J. Clin. Endocrinol. metabolism 85 (5), 2042–2047. doi:10.1210/jcem.85.5.6477
Carpenter, T. (2017). CYP24A1 loss of function: clinical phenotype of monoallelic and biallelic mutations. J. steroid Biochem. Mol. Biol., 173. doi:10.1016/j.jsbmb.2017.01.006
Cau, M., Addis, M., Congiu, R., Meloni, C., Cao, A., Santaniello, S., et al. (2006). A locus for familial skewed X chromosome inactivation maps to chromosome Xq25 in a family with a female manifesting Lowe syndrome. J. Hum. Genet. 51 (11), 1030–1036. doi:10.1007/s10038-006-0049-6
Caubit, X., Gubellini, P., Andrieux, J., Roubertoux, P. L., Metwaly, M., Jacq, B., et al. (2016). TSHZ3 deletion causes an autism syndrome and defects in cortical projection neurons. Nat. Genet. 48 (11), 1359–1369. doi:10.1038/ng.3681
Chang, C. K., Chang, C. C., Wu, V. C., Geng, J. H., and Lee, H. Y. (2022). The relationship between renal stones and primary aldosteronism. Front. Endocrinol. (Lausanne) 13, 828839. doi:10.3389/fendo.2022.828839
Chefetz, I., Heller, R., Galli-Tsinopoulou, A., Richard, G., Wollnik, B., Indelman, M., et al. (2005). A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum. Genet. 118 (2), 261–266. doi:10.1007/s00439-005-0026-8
Chenouard, A., Isidor, B., Allain-Launay, E., Moreau, A., Le Bideau, M., and Roussey, G. (2013). Renal phenotypic variability in HDR syndrome: glomerular nephropathy as a novel finding. Eur. J. Pediatr. 172 (1), 107–110. doi:10.1007/s00431-012-1845-y
Claverie-Martín, F., Ramos-Trujillo, E., and García-Nieto, V. (2011). Dent's disease: clinical features and molecular basis. Pediatr. Nephrol. 26 (5), 693–704. doi:10.1007/s00467-010-1657-0
Cochat, P., and Rumsby, G. (2013). Primary hyperoxaluria. N. Engl. J. Med. 369 (7), 649–658. doi:10.1056/NEJMra1301564
Cogal, A., Arroyo, J., Shah, R., Reese, K., Walton, B., Reynolds, L., et al. (2021). Comprehensive genetic analysis reveals complexity of monogenic urinary stone disease. Kidney Int. Rep. 6 (11), 2862–2884. doi:10.1016/j.ekir.2021.08.033
Copelovitch, L. (2012). Urolithiasis in children: medical approach. Pediatr. Clin. North Am. 59 (4), 881–896. doi:10.1016/j.pcl.2012.05.009
Courbebaisse, M., Leroy, C., Bakouh, N., Salaün, C., Beck, L., Grandchamp, B., et al. (2012). A new human NHERF1 mutation decreases renal phosphate transporter NPT2a expression by a PTH-independent mechanism. PLoS One 7 (4), e34764. doi:10.1371/journal.pone.0034764
Curry, J., Saurette, M., Askari, M., Pei, L., Filla, M., Beggs, M., et al. (2020). Claudin-2 deficiency associates with hypercalciuria in mice and human kidney stone disease. J. Clin. investigation 130 (4), 1948–1960. doi:10.1172/JCI127750
Dasgupta, D., Wee, M. J., Reyes, M., Li, Y., Simm, P. J., Sharma, A., et al. (2014). Mutations in SLC34A3/NPT2c are associated with kidney stones and nephrocalcinosis. J. Am. Soc. Nephrol. 25 (10), 2366–2375. doi:10.1681/ASN.2013101085
de Boer, H., Roelofsen, A., and Janssens, P. M. (2012). Antidiuretic hormone antagonist to reduce cystine stone formation. Ann. Intern Med. 157, 459–460. doi:10.7326/0003-4819-157-6-201209180-00023
Deeb, A., Abood, S. A., Simon, J., Dastoor, H., Pearce, S. H., and Sayer, J. A. (2013). A novel CLDN16 mutation in a large family with familial hypomagnesaemia with hypercalciuria and nephrocalcinosis. BMC Res. Notes 6, 527. doi:10.1186/1756-0500-6-527
Dello Strologo, L., Pras, E., Pontesilli, C., Beccia, E., Ricci-Barbini, V., de Sanctis, L., et al. (2002). Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classification. J. Am. Soc. Nephrol. 13 (10), 2547–2553. doi:10.1097/01.asn.0000029586.17680.e5
De Vries, A., Kochwa, S., Lazebnik, J., Frank, M., and Djaldetti, M. (1957). Glycinuria, a hereditary disorder associated with nephrolithiasis. Am. J. Med. 23 (3), 408–415. doi:10.1016/0002-9343(57)90320-0
Dhayat, N. A., Schaller, A., Albano, G., Poindexter, J., Griffith, C., Pasch, A., et al. (2016). The vacuolar H+-ATPase B1 subunit polymorphism p.E161K associates with impaired urinary acidification in recurrent stone formers. J. Am. Soc. Nephrol. 27 (5), 1544–1554. doi:10.1681/ASN.2015040367
Di Stefano, V., Lionetti, E., Rotolo, N., La Rosa, M., and Leonardi, S. (2012). Hypercalciuria and nephrocalcinosis as early feature of Wilson disease onset: description of a pediatric case and literature review. Hepat. Mon. 12 (8), e6233. doi:10.5812/hepatmon.6233
Distelmaier, F., Herebian, D., Atasever, C., Beck-Woedl, S., Mayatepek, E., Strom, T. M., et al. (2024). Blue diaper syndrome and PCSK1 mutations. Pediatrics 141 (Suppl. ment_5), S501–S505. doi:10.1542/peds.2017-0548
Drummond, K. N., Michael, A. F., Ulstrom, R. A., and Good, R. A. (1964). The blue diaper syndrome: familial hypercalcemia with nephrocalcinosis and indicanuria: a new familial disease, with definition of the metabolic abnormality. Am. J. Med. 37 (6), 928–948. doi:10.1016/0002-9343(64)90134-2
Dundar, M., Kurtoglu, S., Elmas, B., Demiryilmaz, F., Candemir, Z., Ozkul, Y., et al. (2001). A case with adducted thumb and club foot syndrome. Clin. Dysmorphol. 10 (4), 291–293. doi:10.1097/00019605-200110000-00012
Dwyer, M., Krambeck, A., Bergstralh, E., Milliner, D., Lieske, J., and Rule, A. (2012). Temporal trends in incidence of kidney stones among children: a 25-year population based study. J. urology 188 (1), 247–252. doi:10.1016/j.juro.2012.03.021
Dyment, D. A., Smith, A. C., Alcantara, D., Schwartzentruber, J. A., Basel-Vanagaite, L., Curry, C. J., et al. (2013). Mutations in PIK3R1 cause SHORT syndrome. Am. J. Hum. Genet. 93 (1), 158–166. doi:10.1016/j.ajhg.2013.06.005
Economic Impact of Urologic Disease (2012). Urologic diseases in America US department of health and human services, public health service, national institutes of health, national institute of diabetes and digestive and kidney diseases. NIH publication No. 12-7865. Washington, DC: US Government Printing Office, 486–487.
El Allali, Y., Hermetet, C., Bacchetta, J., Amouroux, C., Rothenbuhler, A., Porquet-Bordes, V., et al. (2021). Presenting features and molecular genetics of primary hyperparathyroidism in the paediatric population. Eur. J. Endocrinol. 184 (2), 347–355. doi:10.1530/EJE-20-1119
El-Hattab, A. W., Wang, J., Dai, H., Almannai, M., Staufner, C., Alfadhel, M., et al. (2018). MPV17-related mitochondrial DNA maintenance defect: new cases and review of clinical, biochemical, and molecular aspects. Hum. Mutat. 39 (4), 461–470. doi:10.1002/humu.23387
Enerbäck, S., Nilsson, D., Edwards, N., Heglind, M., Alkanderi, S., Ashton, E., et al. (2018). Acidosis and deafness in patients with recessive mutations in FOXI1. J. Am. Soc. Nephrol. 29 (3), 1041–1048. doi:10.1681/ASN.2017080840
Enomoto, A., Kimura, H., Chairoungdua, A., Shigeta, Y., Jutabha, P., Cha, S. H., et al. (2002). Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 417 (6887), 447–452. doi:10.1038/nature742
Escribano, J., Balaguer, A., Roqué i Figuls, M., Feliu, A., and Ferre, N. (2014). Dietary interventions for preventing complications in idiopathic hypercalciuria. Cochrane Database Syst. Rev. 2014 (2), Cd006022. doi:10.1002/14651858.CD006022.pub4
Fargue, S., Rumsby, G., and Danpure, C. J. (2013). Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim. Biophys. Acta 1832 (10), 1776–1783. doi:10.1016/j.bbadis.2013.04.010
Farrell, S., Karp, J., Hager, R., Wang, Y., Adeniyi, O., Wang, J., et al. (2021). Regulatory news: nulibry (fosdenopterin) approved to reduce the risk of mortality in patients with molybdenum cofactor deficiency type A: FDA approval summary. J. Inherit. Metab. Dis. 44 (5), 1085–1087. doi:10.1002/jimd.12421
Favre, G., Laurain, A., Aranyi, T., Szeri, F., Fulop, K., Le Saux, O., et al. (2017). The ABCC6 transporter: a new player in biomineralization. Int. J. Mol. Sci. 18 (9), 1941. doi:10.3390/ijms18091941
Ferraro, P. M., Taylor, E. N., Gambaro, G., and Curhan, G. C. (2013). Soda and other beverages and the risk of kidney stones. Clin. J. Am. Soc. Nephrol. 8 (8), 1389–1395. doi:10.2215/CJN.11661112
Ferreira, C. R., Hackbarth, M. E., Ziegler, S. G., Pan, K. S., Roberts, M. S., Rosing, D. R., et al. (2021b). Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet. Med. 23 (2), 396–407. doi:10.1038/s41436-020-00983-0
Ferreira, C. R., Kavanagh, D., Oheim, R., Zimmerman, K., Stürznickel, J., Li, X., et al. (2021a). Response of the ENPP1-deficient skeletal phenotype to oral phosphate supplementation and/or enzyme replacement therapy: comparative studies in humans and mice. J. Bone Min. Res. 36 (5), 942–955. doi:10.1002/jbmr.4254
Finsterer, J., and Scorza, F. A. (2017). Renal manifestations of primary mitochondrial disorders. Biomed. Rep. 6 (5), 487–494. doi:10.3892/br.2017.892
Font-Llitjós, M., Jiménez-Vidal, M., Bisceglia, L., Di Perna, M., de Sanctis, L., Rousaud, F., et al. (2005). New insights into cystinuria: 40 new mutations, genotype-phenotype correlation, and digenic inheritance causing partial phenotype. J. Med. Genet. 42 (1), 58–68. doi:10.1136/jmg.2004.022244
Forget, S., Patriquin, H. B., Dubois, J., Lafortune, M., Merouani, A., Paradis, K., et al. (1999). The kidney in children with tyrosinemia: sonographic, CT and biochemical findings. Pediatr. Radiol. 29 (2), 104–108. doi:10.1007/s002470050551
Fridman, E., Zeharia, A., Markus-Eidlitz, T., and Haimi Cohen, Y. (2015). Phenotypic variability in patients with fanconi-bickel syndrome with identical mutations. JIMD Rep. 15, 95–104. doi:10.1007/8904_2014_303
Frymoyer, P. A., Scheinman, S. J., Dunham, P. B., Jones, D. B., Hueber, P., and Schroeder, E. T. (1991). X-linked recessive nephrolithiasis with renal failure. N. Engl. J. Med. 325 (10), 681–686. doi:10.1056/NEJM199109053251003
Fu, R., Ceballos-Picot, I., Torres, R. J., Larovere, L. E., Yamada, Y., Nguyen, K. V., et al. (2014). Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 137 (Pt 5), 1282–1303. doi:10.1093/brain/awt202
Gamba, G., and Friedman, P. A. (2009). Thick ascending limb: the Na(+):K (+):2Cl (-) co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. CaSR. Pflugers Arch. 458 (1), 61–76. doi:10.1007/s00424-008-0607-1
Gee, H. Y., Jun, I., Braun, D. A., Lawson, J. A., Halbritter, J., Shril, S., et al. (2016). Mutations in SLC26A1 cause nephrolithiasis. Am. J. Hum. Genet. 98 (6), 1228–1234. doi:10.1016/j.ajhg.2016.03.026
Gefen, A., Sethna, C., Cil, O., Perwad, F., Schoettler, M., Michael, M., et al. (2023). Genetic testing in children with nephrolithiasis and nephrocalcinosis. Pediatr. Nephrol. Berl. Ger. 38 (8), 2615–2622. doi:10.1007/s00467-023-05879-0
Genena, K. M., Sas, D. J., Milliner, D. S., and Lieske, J. C. (2023). Successful treatment of primary hyperoxaluria type 2 with a combined liver and kidney transplant. Kidney Int. Rep. 8, 1469–1472. doi:10.1016/j.ekir.2023.03.013
Gianfrancesco, F., Esposito, T., Ombra, M. N., Forabosco, P., Maninchedda, G., Fattorini, M., et al. (2003). Identification of a novel gene and a common variant associated with uric acid nephrolithiasis in a Sardinian genetic isolate. Am. J. Hum. Genet. 72 (6), 1479–1491. doi:10.1086/375628
Godron, A., Harambat, J., Boccio, V., Mensire, A., May, A., Rigothier, C., et al. (2012). Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin. J. Am. Soc. Nephrol. 7 (5), 801–809. doi:10.2215/CJN.12841211
Goldfarb, D., Avery, A., Beara-Lasic, L., Duncan, G., and Goldberg, J. (2018). A twin study of genetic influences on nephrolithiasis in women and men. Kidney Int. Rep. 4 (4), 535–540. doi:10.1016/j.ekir.2018.11.017
Goldfarb, D., Fischer, M., Keich, Y., and Goldberg, J. (2005). A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int. 67 (3), 1053–1061. doi:10.1111/j.1523-1755.2005.00170.x
Goldfarb, D. S., Lieske, J. C., Groothoff, J., Schalk, G., Russell, K., Yu, S., et al. (2023). Nedosiran in primary hyperoxaluria subtype 3: results from a phase I, single-dose study (PHYOX4). Urolithiasis 51 (1), 80. doi:10.1007/s00240-023-01453-3
Gudbjartsson, D., Holm, H., Indridason, O., Thorleifsson, G., Edvardsson, V., Sulem, P., et al. (2010). Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet. 6 (7), e1001039. doi:10.1371/journal.pgen.1001039
Guo, W., Ji, P., and Xie, Y. (2023). Genetic diagnosis and treatment of inherited renal tubular acidosis. Kidney Dis. (Basel) 9 (5), 371–383. doi:10.1159/000531556
Haghighi, A., Kavehmanesh, Z., Salehzadeh, F., Santos-Simarro, F., Van Maldergem, L., Cimbalistiene, L., et al. (2016). Congenital generalized lipodystrophy: identification of novel variants and expansion of clinical spectrum. Clin. Genet. 89 (4), 434–441. doi:10.1111/cge.12623
Halbritter, J., Baum, M., Hynes, A., Rice, S., Thwaites, D., Gucev, Z., et al. (2015). Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J. Am. Soc. Nephrol. JASN 26 (3), 543–551. doi:10.1681/ASN.2014040388
Hamilton, A. J., Bingham, C., McDonald, T. J., Cook, P. R., Caswell, R. C., Weedon, M. N., et al. (2014). The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a β cell phenotype. J. Med. Genet. 51 (3), 165–169. doi:10.1136/jmedgenet-2013-102066
Hawkes, C., Li, D., Hakonarson, H., Meyers, K., Thummel, K., and Levine, M. (2017). CYP3A4 induction by rifampin: an alternative pathway for vitamin D inactivation in patients with CYP24A1 mutations. J. Clin. Endocrinol. metabolism 102 (5), 1440–1446. doi:10.1210/jc.2016-4048
Hayes, W., Sas, D. J., Magen, D., Shasha-Lavsky, H., Michael, M., Sellier-Leclerc, A. L., et al. (2023). Efficacy and safety of lumasiran for infants and young children with primary hyperoxaluria type 1: 12-month analysis of the phase 3 ILLUMINATE-B trial. Pediatr. Nephrol. 38 (4), 1075–1086. doi:10.1007/s00467-022-05684-1
Herman, G. E., Finegold, M., Zhao, W., de Gouyon, B., and Metzenberg, A. (1999). Medical complications in long-term survivors with X-linked myotubular myopathy. J. Pediatr. 134 (2), 206–214. doi:10.1016/s0022-3476(99)70417-8
Higgins, R. B., and Varney, J. K. (1966). Dissolution of renal calculi in a case of hereditary fructose intolerance and renal tubular acidosis. J. Urol. 95 (3), 291–296. doi:10.1016/s0022-5347(17)63450-5
Hisatome, I., Tanaka, Y., Kotake, H., Kosaka, H., Hirata, N., Fujimoto, Y., et al. (1993). Renal hypouricemia due to enhanced tubular secretion of urate associated with urolithiasis: successful treatment of urolithiasis by alkalization of urine K+, Na(+)-citrate. Nephron 65 (4), 578–582. doi:10.1159/000187567
Holme, A., Hurcombe, J. A., Straatman-Iwanowska, A., Inward, C. I., Gissen, P., and Coward, R. J. (2013). Glomerular involvement in the arthrogryposis, renal dysfunction and cholestasis syndrome. Clin. Kidney J. 6 (2), 183–188. doi:10.1093/ckj/sfs182
Hoopes, R. R., Hueber, P. A., Reid, R. J., Braden, G. L., Goodyer, P. R., Melnyk, A. R., et al. (1998). CLCN5 chloride-channel mutations in six new North American families with X-linked nephrolithiasis. Kidney Int. 54 (3), 698–705. doi:10.1046/j.1523-1755.1998.00061.x
Howles, S. A., Wiberg, A., Goldsworthy, M., Bayliss, A. L., Gluck, A. K., Ng, M., et al. (2019). Genetic variants of calcium and vitamin D metabolism in kidney stone disease. Nat. Commun. 10 (1), 5175–5210. doi:10.1038/s41467-019-13145-x
Hunter, D., Lange, M., Snieder, H., MacGregor, A., Swaminathan, R., Thakker, R., et al. (2002). Genetic contribution to renal function and electrolyte balance: a twin study. Clin. Sci. Lond. Engl. 1979 103 (3), 259–265. doi:10.1042/cs1030259
Igarashi, T., Hayakawa, H., Shiraga, H., Kawato, H., Yan, K., Kawaguchi, H., et al. (1995). Hypercalciuria and nephrocalcinosis in patients with idiopathic low-molecular-weight proteinuria in Japan: is the disease identical to Dent's disease in United Kingdom? Nephron 69 (3), 242–247. doi:10.1159/000188464
Imai, E., Kaneko, S., Mori, T., Okado, T., Uchida, S., and Tsukamoto, Y. (2016). A novel heterozygous mutation in the ATP6V0A4 gene encoding the V-ATPase a4 subunit in an adult patient with incomplete distal renal tubular acidosis. Clin. Kidney J. 9, 424–428. doi:10.1093/ckj/sfw008
Jaeken, J., Martens, K., Francois, I., Eyskens, F., Lecointre, C., Derua, R., et al. (2006). Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am. J. Hum. Genet. 78 (1), 38–51. doi:10.1086/498852
Jayasinghe, K., Stark, Z., Kerr, P., Gaff, C., Martyn, M., Whitlam, J., et al. (2021). Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet. Med. official J. Am. Coll. Med. Genet. 23 (1), 183–191. doi:10.1038/s41436-020-00963-4
Jinnah, H. A., Sabina, R. L., and Van Den Berghe, G. (2013). Metabolic disorders of purine metabolism affecting the nervous system. Handb. Clin. Neurol. 113, 1827–1836. doi:10.1016/B978-0-444-59565-2.00052-6
Jobst-Schwan, T., Klämbt, V., Tarsio, M., Heneghan, J. F., Majmundar, A. J., Shril, S., et al. (2020). Whole exome sequencing identified ATP6V1C2 as a novel candidate gene for recessive distal renal tubular acidosis. Kidney Int. 97 (3), 567–579. doi:10.1016/j.kint.2019.09.026
Johannes, L., Fu, C. Y., and Schwarz, G. (2022). Molybdenum cofactor deficiency in humans. Molecules 27 (20), 6896. doi:10.3390/molecules27206896
Kaiyrzhanov, R., Rocca, C., Suri, M., Gulieva, S., Zaki, M. S., Henig, N. Z., et al. (2022). Biallelic loss of EMC10 leads to mild to severe intellectual disability. Ann. Clin. Transl. Neurol. 9 (7), 1080–1089. doi:10.1002/acn3.51602
Kanabus, M., Shahni, R., Saldanha, J. W., Murphy, E., Plagnol, V., Hoff, W. V., et al. (2015). Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation. J. Inherit. Metab. Dis. 38 (2), 211–219. doi:10.1007/s10545-015-9813-0
Kandler, J. L., Sklirou, E., Woerner, A., Walsh, L., Cox, E., and Xue, Y. (2020). Compound heterozygous loss of function variants in MYL9 in a child with megacystis-microcolon-intestinal hypoperistalsis syndrome. Mol. Genet. Genomic Med. 8 (11), e1516. doi:10.1002/mgg3.1516
Kantaputra, P., Guven, Y., Aksu, B., Kalayci, T., Doğan, C., Intachai, W., et al. (2022). Distal renal tubular acidosis, autoimmune thyroiditis, enamel hypomaturation, and tooth agenesis caused by homozygosity of a novel double-nucleotide substitution in SLC4A4. J. Am. Dent. Assoc. 153 (7), 668–676. doi:10.1016/j.adaj.2021.12.009
Karet, F. E. (2002). Inherited distal renal tubular acidosis. J. Am. Soc. Nephrol. 13 (8), 2178–2184. doi:10.1097/01.asn.0000023433.08833.88
Karim, Z., Gérard, B., Bakouh, N., Alili, R., Leroy, C., Beck, L., et al. (2008). NHERF1 mutations and responsiveness of renal parathyroid hormone. N. Engl. J. Med. 359 (11), 1128–1135. doi:10.1056/NEJMoa0802836
Karunakar, P., Krishnamurthy, S., Kasinathan, A., Hariharan, R., and Chidambaram, A. C. (2020). Renal stones in an infant with microcephaly and spastic quadriparesis: answers. Pediatr. Nephrol. 35 (6), 987–989. doi:10.1007/s00467-019-04449-7
Katz, S. M., Krueger, L. J., and Falkner, B. (1988). Microscopic nephrocalcinosis in cystic fibrosis. N. Engl. J. Med. 319 (5), 263–266. doi:10.1056/NEJM198808043190502
Kawai, K., Sato, Y., Kawakami, R., Sakamoto, A., Cornelissen, A., Mori, M., et al. (2022). Generalized arterial calcification of infancy (GACI): optimizing care with a multidisciplinary approach. J. Multidiscip. Healthc. 15, 1261–1276. doi:10.2147/JMDH.S251861
Kawamura, Y., Toyoda, Y., Ohnishi, T., Hisatomi, R., Higashino, T., Nakayama, A., et al. (2020). Identification of a dysfunctional splicing mutation in the SLC22A12/URAT1 gene causing renal hypouricaemia type 1: a report on two families. Rheumatol. Oxf. 59, 3988–3990. doi:10.1093/rheumatology/keaa461
Kessel, D., Hall, C. M., and Shaw, D. G. (1992). Two unusual cases of nephrocalcinosis in infancy. Pediatr. Radiol. 22 (6), 470–471. doi:10.1007/BF02013518
Khandelwal, P., Mahesh, V., Mathur, V. P., Raut, S., Geetha, T. S., Nair, S., et al. (2021). Phenotypic variability in distal acidification defects associated with WDR72 mutations. Pediatr. Nephrol. 36 (4), 881–887. doi:10.1007/s00467-020-04747-5
Kirk, J. M., Brain, C. E., Carson, D. J., Hyde, J. C., and Grant, D. B. (1999). Cushing's syndrome caused by nodular adrenal hyperplasia in children with McCune-Albright syndrome. J. Pediatr. 134 (6), 789–792. doi:10.1016/s0022-3476(99)70302-1
Klar, J., Piontek, J., Milatz, S., Tariq, M., Jameel, M., Breiderhoff, T., et al. (2017). Altered paracellular cation permeability due to a rare CLDN10B variant causes anhidrosis and kidney damage. PLoS Genet. 13 (7), e1006897. doi:10.1371/journal.pgen.1006897
Klootwijk, E. D., Reichold, M., Helip-Wooley, A., Tolaymat, A., Broeker, C., Robinette, S. L., et al. (2014). Mistargeting of peroxisomal EHHADH and inherited renal Fanconi's syndrome. N. Engl. J. Med. 370 (2), 129–138. doi:10.1056/NEJMoa1307581
Knoll, T., Zöllner, A., Wendt-Nordahl, G., Michel, M. S., and Alken, P. (2005). Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr. Nephrol. 20 (1), 19–24. doi:10.1007/s00467-004-1663-1
Kohashi, K., Ishiyama, A., Yuasa, S., Tanaka, T., Miya, K., Adachi, Y., et al. (2018). Epileptic apnea in a patient with inherited glycosylphosphatidylinositol anchor deficiency and PIGT mutations. Brain Dev. 40 (1), 53–57. doi:10.1016/j.braindev.2017.06.005
Kvarnung, M., Nilsson, D., Lindstrand, A., Korenke, G. C., Chiang, S. C., Blennow, E., et al. (2013). A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J. Med. Genet. 50 (8), 521–528. doi:10.1136/jmedgenet-2013-101654
Larsson, A., Mattsson, B., Wauters, E. A., van Gool, J. D., Duran, M., and Wadman, S. K. (1981). 5-oxoprolinuria due to hereditary 5-oxoprolinase deficiency in two brothers--a new inborn error of the gamma-glutamyl cycle. Acta Paediatr. Scand. 70 (3), 301–308. doi:10.1111/j.1651-2227.1981.tb16556.x
Lee, E. J., Dandamudi, R., Granadillo, J. L., Grange, D. K., and Kakajiwala, A. (2021). Rare cause of xanthinuria: a pediatric case of molybdenum cofactor deficiency B. Jpn. Soc. Nephrol. 10, 378–382. doi:10.1007/s13730-021-00572-3
Lee, S. H., Nam, T. S., Li, W., Kim, J. H., Yoon, W., Choi, Y. D., et al. (2017). Functional validation of novel MKS3/TMEM67 mutations in COACH syndrome. Sci. Rep. 7 (1), 10222. doi:10.1038/s41598-017-10652-z
Legrand, A., Cornez, L., Samkari, W., Mazzella, J. M., Venisse, A., Boccio, V., et al. (2017). Mutation spectrum in the ABCC6 gene and genotype-phenotype correlations in a French cohort with pseudoxanthoma elasticum. Genet. Med. 19 (8), 909–917. doi:10.1038/gim.2016.213
Li, J., Shingde, M., Nankivell, B. J., Tchan, M. C., Bose, B., Chapman, J. R., et al. (2019). Adenine phosphoribosyltransferase deficiency: a potentially reversible cause of CKD. Kidney Int. Rep. 4 (8), 1161–1170. doi:10.1016/j.ekir.2019.04.021
Li, Y., Bayne, D., Wiener, S., Ahn, J., Stoller, M., and Chi, T. (2020). Stone formation in patients less than 20 years of age is associated with higher rates of stone recurrence: results from the Registry for Stones of the Kidney and Ureter (ReSKU). J. Pediatr. urology 16 (3), e1–e373. doi:10.1016/j.jpurol.2020.03.014
Lieske, J., de la Vega, L., Gettman, M., Slezak, J., Bergstralh, E., Melton, L., et al. (2006). Diabetes mellitus and the risk of urinary tract stones: a population-based case-control study. Am. J. kidney Dis. official J. Natl. Kidney Found. 48 (6), 897–904. doi:10.1053/j.ajkd.2006.09.002
Lila, A. R., Sarathi, V., Jagtap, V., Bandgar, T., Menon, P. S., and Shah, N. S. (2012). Renal manifestations of primary hyperparathyroidism. Indian J. Endocrinol. Metab. 16 (2), 258–262. doi:10.4103/2230-8210.93745
Lingeman, J. E., Pareek, G., Easter, L., Pease, R., Grujic, D., Brettman, L., et al. (2019). ALLN-177, oral enzyme therapy for hyperoxaluria. Int. Urol. Nephrol. 51 (4), 601–608. doi:10.1007/s11255-019-02098-1
Loos, S., Kemper, M. J., Schmaeschke, K., Herden, U., Fischer, L., Hoppe, B., et al. (2023). Long-term outcome after combined or sequential liver and kidney transplantation in children with infantile and juvenile primary hyperoxaluria type 1. Front. Pediatr. 11, 1157215. doi:10.3389/fped.2023.1157215
Lopez-Garcia, S. C., Emma, F., Walsh, S. B., Fila, M., Hooman, N., Zaniew, M., et al. (2019). Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol. Dial. Transpl. 34 (6), 981–991. doi:10.1093/ndt/gfy409
Lorenz-Depiereux, B., Schnabel, D., Tiosano, D., Häusler, G., and Strom, T. M. (2010). Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am. J. Hum. Genet. 86 (2), 267–272. doi:10.1016/j.ajhg.2010.01.006
Lu, Y. T., Zhang, D., Zhang, Q. Y., Zhou, Z. M., Yang, K. Q., Zhou, X. L., et al. (2022). Apparent mineralocorticoid excess: comprehensive overview of molecular genetics. J. Transl. Med. 20 (1), 500. doi:10.1186/s12967-022-03698-9
Mabillard, H., and Sayer, J. A. (2019). The molecular genetics of Gordon syndrome. Genes (Basel). 10 (12), 986. doi:10.3390/genes10120986
Madeo, A., Di Rocco, M., Brassier, A., Bahi-Buisson, N., De Lonlay, P., and Ceballos-Picot, I. (2019). Clinical, biochemical and genetic characteristics of a cohort of 101 French and Italian patients with HPRT deficiency. Mol. Genet. Metab. 127 (2), 147–157. doi:10.1016/j.ymgme.2019.06.001
Mansour-Hendili, L., Blanchard, A., Le Pottier, N., Roncelin, I., Lourdel, S., Treard, C., et al. (2015). Mutation update of the CLCN5 gene responsible for dent disease 1. Hum. Mutat. 36 (8), 743–752. doi:10.1002/humu.22804
Martinez-Turrillas, R., Martin-Mallo, A., Rodriguez-Diaz, S., Zapata-Linares, N., Rodriguez-Marquez, P., San Martin-Uriz, P., et al. (2022). In vivo CRISPR-Cas9 inhibition of hepatic LDH as treatment of primary hyperoxaluria. Mol. Ther. Methods Clin. Dev. 25, 137–146. doi:10.1016/j.omtm.2022.03.006
Mass, R. E., Smith, W. R., and Walsh, J. R. (1966). The association of hereditary fructose intolerance and renal tubular acidosis. Am. J. Med. Sci. 251 (5), 516–523. doi:10.1097/00000441-196605000-00003
Maxit, C., Denzler, I., Marchione, D., Agosta, G., Koster, J., Wanders, R. J. A., et al. (2017). Novel PEX3 gene mutations resulting in a moderate zellweger spectrum disorder. JIMD Rep. 34, 71–75. doi:10.1007/8904_2016_10
Milliner, D., Hoppe, B., and Groothoff, J. (2018). A randomised Phase II/III study to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Urolithiasis 46 (4), 313–323. doi:10.1007/s00240-017-0998-6
Mittal, K., Anandpara, K., Dey, A. K., Sharma, R., Thakkar, H., Hira, P., et al. (2015). An association of chronic hyperaldosteronism with medullary nephrocalcinosis. Pol. J. Radiol. 80, 417–424. doi:10.12659/PJR.894674
Miyata, N., Steffen, J., Johnson, M. E., Fargue, S., Danpure, C. J., and Koehler, C. M. (2014). Pharmacologic rescue of an enzyme-trafficking defect in primary hyperoxaluria 1. Proc. Natl. Acad. Sci. U. S. A. 111 (40), 14406–14411. doi:10.1073/pnas.1408401111
Mohebbi, N., and Wagner, C. A. (2018). Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J. Nephrol. 31 (4), 511–522. doi:10.1007/s40620-017-0447-1
Monico, C. G., Rossetti, S., Belostotsky, R., Cogal, A. G., Herges, R. M., Seide, B. M., et al. (2011). Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin. J. Am. Soc. Nephrol. 6 (9), 2289–2295. doi:10.2215/CJN.02760311
Moulin, F., Ponte, B., Pruijm, M., Ackermann, D., Bouatou, Y., Guessous, I., et al. (2017). A population-based approach to assess the heritability and distribution of renal handling of electrolytes. Kidney Int. 92 (6), 1536–1543. doi:10.1016/j.kint.2017.06.020
Moussa, M., Papatsoris, A. G., Abou Chakra, M., and Moussa, Y. (2020). Update on cystine stones: current and future concepts in treatment. Intractable Rare Dis. Res. 9 (2), 71–78. doi:10.5582/irdr.2020.03006
Naeem, M., Hussain, S., and Akhtar, N. (2011). Mutation in the tight-junction gene claudin 19 (CLDN19) and familial hypomagnesemia, hypercalciuria, nephrocalcinosis (FHHNC) and severe ocular disease. Am. J. Nephrol. 34 (3), 241–248. doi:10.1159/000330854
Nelson, K., Jackman, C., Bell, J., Shih, C. S., Payne, K., Dlouhy, S., et al. (2018). Novel homozygous deletion in STRADA gene associated with polyhydramnios, megalencephaly, and epilepsy in 2 siblings: implications for diagnosis and treatment. J. Child. Neurol. 33 (14), 925–929. doi:10.1177/0883073818802724
Neuray, C., Maroofian, R., Scala, M., Sultan, T., Pai, G. S., Mojarrad, M., et al. (2020). Early-infantile onset epilepsy and developmental delay caused by bi-allelic GAD1 variants. Brain 143 (8), 2388–2397. doi:10.1093/brain/awaa178
Ng, B. G., Xu, G., Chandy, N., Steyermark, J., Shinde, D. N., Radtke, K., et al. (2018). Biallelic mutations in FUT8 cause a congenital disorder of glycosylation with defective fucosylation. Am. J. Hum. Genet. 102 (1), 188–195. doi:10.1016/j.ajhg.2017.12.009
Nielsen, R., Christensen, E. I., and Birn, H. (2016). Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. 89 (1), 58–67. doi:10.1016/j.kint.2015.11.007
Nitschke, Y., Baujat, G., Botschen, U., Wittkampf, T., du Moulin, M., Stella, J., et al. (2012). Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 90 (1), 25–39. doi:10.1016/j.ajhg.2011.11.020
Novak, T., Lakshmanan, Y., Trock, B., Gearhart, J., and Matlaga, B. (2009). Sex prevalence of pediatric kidney stone disease in the United States: an epidemiologic investigation. Urology 74 (1), 104–107. doi:10.1016/j.urology.2008.12.079
Oddsson, A., Sulem, P., Helgason, H., Edvardsson, V. O., Thorleifsson, G., Sveinbjörnsson, G., et al. (2015). Common and rare variants associated with kidney stones and biochemical traits. Nat. Commun. 6 (1), 7975–7979. doi:10.1038/ncomms8975
Ooms, L. M., Horan, K. A., Rahman, P., Seaton, G., Gurung, R., Kethesparan, D. S., et al. (2009). The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem. J. 419 (1), 29–49. doi:10.1042/BJ20081673
Pearce, S. H. S., Williamson, C., Kifor, O., Bai, M., Coulthard, M. G., Davies, M., et al. (1996). A familial syndrome of hypocalcemia with hypercalciuria due to mutations in the calcium-sensing receptor. N. Engl. J. Med. 335 (15), 1115–1122. doi:10.1056/NEJM199610103351505
Phornphutkul, C., Introne, W. J., Perry, M. B., Bernardini, I., Murphey, M. D., Fitzpatrick, D. L., et al. (2002). Natural history of alkaptonuria. N. Engl. J. Med. 347 (26), 2111–2121. doi:10.1056/NEJMoa021736
Pitt, J. J., Willis, F., Tzanakos, N., Belostotsky, R., and Frishberg, Y. (2015). 4-hydroxyglutamate is a biomarker for primary hyperoxaluria type 3. JIMD Rep. 15, 1–6. doi:10.1007/8904_2013_291
Prié, D., Huart, V., Bakouh, N., Planelles, G., Dellis, O., Gérard, B., et al. (2002). Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N. Engl. J. Med. 347 (13), 983–991. doi:10.1056/NEJMoa020028
Puffenberger, E. G., Strauss, K. A., Ramsey, K. E., Craig, D. W., Stephan, D. A., Robinson, D. L., et al. (2007). Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain 130 (Pt 7), 1929–1941. doi:10.1093/brain/awm100
Quintero, E., Bird, V. Y., Liu, H., Stevens, G., Ryan, A. S., Buzzerd, S., et al. (2020). A prospective, double-blind, randomized, placebo-controlled, crossover study using an orally administered oxalate decarboxylase (OxDC). Kidney360. 1 (11), 1284–1290. doi:10.34067/KID.0001522020
Ramond, F., Rio, M., Héron, B., Imbard, A., Marie, S., Billiemaz, K., et al. (2020). AICA-ribosiduria due to ATIC deficiency: delineation of the phenotype with three novel cases, and long-term update on the first case. J. Inherit. Metab. Dis. 43 (6), 1254–1264. doi:10.1002/jimd.12274
Reed, B. Y., Gitomer, W. L., Heller, H. J., Hsu, M. C., Lemke, M., Padalino, P., et al. (2002). Identification and characterization of a gene with base substitutions associated with the absorptive hypercalciuria phenotype and low spinal bone density. J. Clin. Endocrinol. Metab. 87 (4), 1476–1485. doi:10.1210/jcem.87.4.8300
Reichold, M., Klootwijk, E. D., Reinders, J., Otto, E. A., Milani, M., Broeker, C., et al. (2018). Glycine amidinotransferase (GATM), renal fanconi syndrome, and kidney failure. J. Am. Soc. Nephrol. 29 (7), 1849–1858. doi:10.1681/ASN.2017111179
Reiss, J., and Hahnewald, R. (2011). Molybdenum cofactor deficiency: mutations in GPHN, MOCS1, and MOCS2. Hum. Mutat. 32 (1), 10–18. doi:10.1002/humu.21390
Rendina, D., De Filippo, G., Merlotti, D., Di Stefano, M., Mingiano, C., Giaquinto, A., et al. (2020). Increased prevalence of nephrolithiasis and hyperoxaluria in paget disease of bone. J. Clin. Endocrinol. Metab. 105 (12), dgaa576. doi:10.1210/clinem/dgaa576
Rhodes, H. L., Yarram-Smith, L., Rice, S. J., Tabaksert, A., Edwards, N., Hartley, A., et al. (2015). Clinical and genetic analysis of patients with cystinuria in the United Kingdom. Clin. J. Am. Soc. Nephrol. 10 (7), 1235–1245. doi:10.2215/CJN.10981114
Rimer, J. D., An, Z., Zhu, Z., Lee, M. H., Goldfarb, D. S., Wesson, J. A., et al. (2010). Crystal growth inhibitors for the prevention of L-cystine kidney stones through molecular design. Science 330 (6002), 337–341. doi:10.1126/science.1191968
Rothenbuhler, A., and Linglart, A. (2017). Hypophosphatasia in children and adolescents: clinical features and treatment. Arch. Pediatr. 24 (5s2), 5s66–5s70. doi:10.1016/S0929-693X(18)30017-4
Saarela, T., Similä, S., and Koivisto, M. (1995). Hypercalcemia and nephrocalcinosis in patients with congenital lactase deficiency. J. Pediatr. 127 (6), 920–923. doi:10.1016/s0022-3476(95)70028-5
Sas, D. J., Magen, D., Hayes, W., Shasha-Lavsky, H., Michael, M., Schulte, I., et al. (2022). Phase 3 trial of lumasiran for primary hyperoxaluria type 1: a new RNAi therapeutic in infants and young children. Genet. Med. 24 (3), 654–662. doi:10.1016/j.gim.2021.10.024
Sayers, J., Hynes, A., Srivastava, S., Dowen, F., Quinton, R., Datta, H., et al. (2015). Successful treatment of hypercalcaemia associated with a CYP24A1 mutation with fluconazole. Clin. kidney J. 8 (4), 453–455. doi:10.1093/ckj/sfv028
Scales, C. D., Smith, A. C., Hanley, J. M., and Saigal, C. S.Urologic Diseases in America Project (2012). Prevalence of kidney stones in the United States. Eur. Urol. 62 (1), 160–165. doi:10.1016/j.eururo.2012.03.052
Scheinman, S. J. (1998). X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int. 53 (1), 3–17. doi:10.1046/j.1523-1755.1998.00718.x
Schlingmann, K., Kaufmann, M., Weber, S., Irwin, A., Goos, C., John, U., et al. (2011). Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N. Engl. J. Med. 365 (5), 410–421. doi:10.1056/NEJMoa1103864
Schlingmann, K. P., Jouret, F., Shen, K., Nigam, A., Arjona, F. J., Dafinger, C., et al. (2021). mTOR-activating mutations in RRAGD are causative for kidney tubulopathy and cardiomyopathy. J. Am. Soc. Nephrol. 32 (11), 2885–2899. doi:10.1681/ASN.2021030333
Schlingmann, K. P., Ruminska, J., Kaufmann, M., Dursun, I., Patti, M., Kranz, B., et al. (2016). Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J. Am. Soc. Nephrol. 27 (2), 604–614. doi:10.1681/ASN.2014101025
Scoffone, C. M., and Cracco, C. M. (2018). Pediatric calculi: cause, prevention and medical management. Curr. Opin. Urol. 28 (5), 428–432. doi:10.1097/MOU.0000000000000520
Scott, P., Ouimet, D., Valiquette, L., Guay, G., Proulx, Y., Trouvé, M. L., et al. (1999). Suggestive evidence for a susceptibility gene near the vitamin D receptor locus in idiopathic calcium stone formation. J. Am. Soc. Nephrol. 10 (5), 1007–1013. doi:10.1681/ASN.V1051007
Seaby, E. G., Turner, S., Bunyan, D. J., Seyed-Rezai, F., Essex, J., Gilbert, R. D., et al. (2023). A novel variant in GATM causes idiopathic renal Fanconi syndrome and predicts progression to end-stage kidney disease. Clin. Genet. 103 (2), 214–218. doi:10.1111/cge.14235
Sedda, D., Mackowiak, C., Pailloux, J., Culerier, E., Dudas, A., Rontani, P., et al. (2021). Deletion of mocos induces xanthinuria with obstructive nephropathy and major metabolic disorders in mice. Kidney360. 2 (11), 1793–1806. doi:10.34067/KID.0001732021
Shao, D. D., Straussberg, R., Ahmed, H., Khan, A., Tian, S., Hill, R. S., et al. (2021). A recurrent, homozygous EMC10 frameshift variant is associated with a syndrome of developmental delay with variable seizures and dysmorphic features. Genet. Med. 23 (6), 1158–1162. doi:10.1038/s41436-021-01097-x
Simoni, R. E., Gomes, L. N., Scalco, F. B., Oliveira, C. P., Aquino Neto, F. R., and de Oliveira, M. L. (2007). Uric acid changes in urine and plasma: an effective tool in screening for purine inborn errors of metabolism and other pathological conditions. J. Inherit. Metab. Dis. 30 (3), 295–309. doi:10.1007/s10545-007-0455-8
Singh, P., Viehman, J. K., Mehta, R. A., Cogal, A. G., Hasadsri, L., Oglesbee, D., et al. (2022). Clinical characterization of primary hyperoxaluria type 3 in comparison with types 1 and 2. Nephrol. Dial. Transpl. 37 (5), 869–875. doi:10.1093/ndt/gfab027
Soylu, Ö. B., Ecevit, Ç., Altınöz, S., Öztürk, A. A., Temizkan, A. K., Maeda, M., et al. (2008). Nephrocalcinosis in glucose-galactose malabsorption: nephrocalcinosis and proximal tubular dysfunction in a young infant with a novel mutation of SGLT1. Eur. J. Pediatr. 167 (12), 1395–1398. doi:10.1007/s00431-008-0681-6
Sritippayawan, S., Sumboonnanonda, A., Vasuvattakul, S., Keskanokwong, T., Sawasdee, N., Paemanee, A., et al. (2004). Novel compound heterozygous SLC4A1 mutations in Thai patients with autosomal recessive distal renal tubular acidosis. Am. J. Kidney Dis. 44 (1), 64–70. doi:10.1053/j.ajkd.2004.03.033
Srour, M., Shimokawa, N., Hamdan, F. F., Nassif, C., Poulin, C., Al Gazali, L., et al. (2017). Dysfunction of the cerebral glucose transporter SLC45A1 in individuals with intellectual disability and epilepsy. Am. J. Hum. Genet. 100 (5), 824–830. doi:10.1016/j.ajhg.2017.03.009
Starnes, C., and Welsh, J. (1970). Intestinal sucrase-isomaltase deficiency and renal calculi. N. Engl. J. Med. 282 (18), 1023–1024. doi:10.1056/NEJM197004302821809
Tan, N. B., Pagnamenta, A. T., Ferla, M. P., Gadian, J., Chung, B. H., Chan, M. C., et al. (2022). Recurrent de novo missense variants in GNB2 can cause syndromic intellectual disability. J. Med. Genet. 59 (5), 511–516. doi:10.1136/jmedgenet-2020-107462
Tanaka, M., Itoh, K., Matsushita, K., Wakita, N., Adachi, M., Nonoguchi, H., et al. (2003). Two male siblings with hereditary renal hypouricemia and exercise-induced ARF. Am. J. Kidney Dis. 42 (6), 1287–1292. doi:10.1053/j.ajkd.2003.08.032
Taylor, E., Mj, S., and Curhan, G. (2005). Obesity, weight gain, and the risk of kidney stones. JAMA 293 (4), 455–462. doi:10.1001/jama.293.4.455
Theodoropoulos, D. S., Shawker, T. H., Heinrichs, C., and Gahl, W. A. (1995). Medullary nephrocalcinosis in nephropathic cystinosis. Pediatr. Nephrol. 9 (4), 412–418. doi:10.1007/BF00866713
Thorleifsson, G., Holm, H., Edvardsson, V., Walters, G. B., Styrkarsdottir, U., Gudbjartsson, D. F., et al. (2009). Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat. Genet. 41 (8), 926–930. doi:10.1038/ng.404
Tieder, M., Arie, R., Bab, I., Maor, J., and Liberman, U. A. (1992). A new kindred with hereditary hypophosphatemic rickets with hypercalciuria: implications for correct diagnosis and treatment. Nephron 62 (2), 176–181. doi:10.1159/000187029
Tiosano, D., Baris, H. N., Chen, A., Hitzert, M. M., Schueler, M., Gulluni, F., et al. (2019). Mutations in PIK3C2A cause syndromic short stature, skeletal abnormalities, and cataracts associated with ciliary dysfunction. PLoS Genet. 15 (4), e1008088. doi:10.1371/journal.pgen.1008088
Tolaymat, A., Sakarcan, A., and Neiberger, R. (1992). Idiopathic Fanconi syndrome in a family. Part I. Clinical aspects. J. Am. Soc. Nephrol. 2 (8), 1310–1317. doi:10.1681/ASN.V281310
Torres, R. J., Puig, J. G., and Jinnah, H. A. (2012). Update on the phenotypic spectrum of Lesch-Nyhan disease and its attenuated variants. Curr. Rheumatol. Rep. 14 (2), 189–194. doi:10.1007/s11926-011-0231-5
Torres, V. E., Wilson, D. M., Hattery, R. R., and Segura, J. W. (1993). Renal stone disease in autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 22 (4), 513–519. doi:10.1016/s0272-6386(12)80922-x
Tufan, F., Cefle, K., Türkmen, S., Türkmen, A., Zorba, U., Dursun, M., et al. (2005). Clinical and molecular characterization of two adults with autosomal recessive Robinow syndrome. Am. J. Med. Genet. A 136 (2), 185–189. doi:10.1002/ajmg.a.30785
Urabe, Y., Tanikawa, C., Takahashi, A., Okada, Y., Morizono, T., Tsunoda, T., et al. (2012). A genome-wide association study of nephrolithiasis in the Japanese population identifies novel susceptible Loci at 5q35.3, 7p14.3, and 13q14.1. PLoS Genet. 8 (3), e1002541. doi:10.1371/journal.pgen.1002541
van Woerden, C. S., Groothoff, J. W., Wijburg, F. A., Duran, M., Wanders, R. J., Barth, P. G., et al. (2006). High incidence of hyperoxaluria in generalized peroxisomal disorders. Mol. Genet. Metab. 88 (4), 346–350. doi:10.1016/j.ymgme.2006.03.004
Vargas-Poussou, R., Huang, C., Hulin, P., Houillier, P., Jeunemaître, X., Paillard, M., et al. (2002). Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J. Am. Soc. Nephrol. JASN. 13 (9), 2259–2266. doi:10.1097/01.asn.0000025781.16723.68
Vasudevan, B., Shijith, K. P., Bahal, A., and Raghav, V. (2010). Pseudoxanthoma elasticum with renal cortical calcification. Med. J. Armed Forces India 66, 272–274. doi:10.1016/S0377-1237(10)80056-5
Wang, S. K., Aref, P., Hu, Y., Milkovich, R. N., Simmer, J. P., El-Khateeb, M., et al. (2013). FAM20A mutations can cause enamel-renal syndrome (ERS). PLoS Genet. 9 (2), e1003302. doi:10.1371/journal.pgen.1003302
Ward, J., Feinstein, L., Pierce, C., Lim, J., Abbott, K., Bavendam, T., et al. (2019). Pediatric urinary stone disease in the United States: the urologic diseases in America project. Urology 129, 180–187. doi:10.1016/j.urology.2019.04.012
Ware, E., Smith, J., Zhao, W., Ganesvoort, R., Curhan, G., Pollak, M., et al. (2019). Genome-wide association study of 24-hour urinary excretion of calcium, magnesium, and uric acid. Mayo Clin. Proc. Innovations, Qual. outcomes 3 (4), 448–460. doi:10.1016/j.mayocpiqo.2019.08.007
Weber, S., Schneider, L., Peters, M., Misselwitz, J., Rönnefarth, G., Böswald, M., et al. (2001). Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J. Am. Soc. Nephrol. 12 (9), 1872–1881. doi:10.1681/ASN.V1291872
Weinstein, D. A., Somers, M. J., and Wolfsdorf, J. I. (2001). Decreased urinary citrate excretion in type 1a glycogen storage disease. J. Pediatr. 138 (3), 378–382. doi:10.1067/mpd.2001.111322
Weksberg, R., Shuman, C., and Beckwith, J. B. (2010). Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18 (1), 8–14. doi:10.1038/ejhg.2009.106
Wen, S. F., Friedman, A. L., and Oberley, T. D. (1989). Two case studies from a family with primary Fanconi syndrome. Am. J. Kidney Dis. 13 (3), 240–246. doi:10.1016/s0272-6386(89)80059-9
White, S. M., Morgan, A., Da Costa, A., Lacombe, D., Knight, S. J., Houlston, R., et al. (2010). The phenotype of Floating-Harbor syndrome in 10 patients. Am. J. Med. Genet. A 152a (4), 821–829. doi:10.1002/ajmg.a.33294
Whyte, M. P. (1993). Carbonic anhydrase II deficiency. Clin. Orthop. Relat. Res. 294 (294), 52–63. doi:10.1097/00003086-199309000-00007
Whyte, M. P., Greenberg, C. R., Salman, N. J., Bober, M. B., McAlister, W. H., Wenkert, D., et al. (2012). Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 366 (10), 904–913. doi:10.1056/NEJMoa1106173
Williams, E. L., Bockenhauer, D., van't Hoff, W. G., Johri, N., Laing, C., Sinha, M. D., et al. (2012). The enzyme 4-hydroxy-2-oxoglutarate aldolase is deficient in primary hyperoxaluria type 3. Nephrol. Dial. Transpl. 27 (8), 3191–3195. doi:10.1093/ndt/gfs039
Wiseman, D. H., May, A., Jolles, S., Connor, P., Powell, C., Heeney, M. M., et al. (2013). A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood 122 (1), 112–123. doi:10.1182/blood-2012-08-439083
Wjst, M., Altmüller, J., Braig, C., Bahnweg, M., and André, E. (2007). A genome-wide linkage scan for 25-OH-D(3) and 1,25-(OH)2-D3 serum levels in asthma families. J. steroid Biochem. Mol. Biol. 103 (3-5), 799–802. doi:10.1016/j.jsbmb.2006.12.053
Zabaleta, N., Barberia, M., Martin-Higueras, C., Zapata-Linares, N., Betancor, I., Rodriguez, S., et al. (2018). CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat. Commun. 9 (1), 5454. doi:10.1038/s41467-018-07827-1
Zaki, M. S., Selim, L., El-Bassyouni, H. T., Issa, M. Y., Mahmoud, I., Ismail, S., et al. (2016). Molybdenum cofactor and isolated sulphite oxidase deficiencies: clinical and molecular spectrum among Egyptian patients. Eur. J. Paediatr. Neurol. 20 (5), 714–722. doi:10.1016/j.ejpn.2016.05.011
Zee, T., Bose, N., Zee, J., Beck, J. N., Yang, S., Parihar, J., et al. (2017). α-Lipoic acid treatment prevents cystine urolithiasis in a mouse model of cystinuria. Nat. Med. 23 (3), 288–290. doi:10.1038/nm.4280
Zhao, Y., Fang, X., He, L., Fan, Y., Li, Y., Xu, G., et al. (2022). Integration of exome sequencing and metabolic evaluation for the diagnosis of children with urolithiasis. World J. urology 40 (5), 2759–2765. doi:10.1007/s00345-020-03449-9
Zhe, M., and Hang, Z. (2017). Nephrolithiasis as a risk factor of chronic kidney disease: a meta-analysis of cohort studies with 4,770,691 participants. Urolithiasis 45 (5), 441–448. doi:10.1007/s00240-016-0938-x
Keywords: children, genetic kidney disease, kidney stones, nephrolithiasis, nephrocalcinosis
Citation: Gefen AM and Zaritsky JJ (2024) Review of childhood genetic nephrolithiasis and nephrocalcinosis. Front. Genet. 15:1381174. doi: 10.3389/fgene.2024.1381174
Received: 03 February 2024; Accepted: 04 March 2024;
Published: 28 March 2024.
Edited by:
Bohan Wang, Zhejiang University, ChinaReviewed by:
Elisa Cicerello, ULSS2 Marca Trevigiana, ItalySermin Saadeh, King Faisal Specialist Hospital and Research Centre, Saudi Arabia
Copyright © 2024 Gefen and Zaritsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashley M. Gefen, agefen@phoenixchildrens.com