 Boxuan Li1†
Boxuan Li1† Ping Sun
Ping Sun- 1Center of Prenatal Diagnosis, Department of Obstetrics and Gynecology, Qilu Hospital of Shandong University, Jinan, China
- 2Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Qilu Hospital of Shandong University, Jinan, China
- 3Suzhou Research Institute of Shandong University, Suzhou, China
Background: Intellectual disability (ID) is defined by cognitive and social adaptation defects. Variants in the SYNGAP1 gene, which encodes the brain-specific cytoplasmic protein SYNGAP1, are commonly associated with ID. The aim of this study was to identify novel SYNGAP1 gene variants in Chinese individuals with ID and evaluate the pathogenicity of the detected variants.
Methods: Whole exome sequencing (WES) was performed on 113 patients diagnosed with ID. In the study, two de novo variants in SYNGAP1 were identified. Sanger sequencing was used to confirm these variants. Minigene assays were used to verify whether the de novo intronic variant in SYNGAP1 influenced the normal splicing of mRNA.
Results: Two de novo heterozygous pathogenic variants in SYNGAP1, c.333del and c.664-2A>G, were identified in two ID patients separately. The c.333del variant has been reported previously as a de novo finding in a child with ID, while the c.664-2A>G variant was novel de novo intronic variant, which has not been reported in the literature. Functional studies showed that c.664-2A>G could cause aberrant splicing, resulting in exon 7 skipping and a 16bp deletion within exon 7.
Conclusion: We identified two de novo pathogenic heterozygous variants in SYNGAP1 in two patients with ID, among which the c.664-2A>G variant was a novel de novo pathogenic variant. Our findings further enrich the variant spectrum of the SYNGAP1 gene and provide a research basis for the genetic diagnosis of ID.
Introduction
Intellectual disability (ID) is characterized by cognitive and social adaptation defects, which typically occur before the age of 18 (Chelly et al., 2006). ID is the most common severe disability in children, affecting approximately 1%–3% of the population (Goldenberg and Saugier-Veber, 2010). ID is classified as either a syndromic or non-syndromic (NSID) form. The majority of ID patients have the NSID form of the disease, which is mainly characterized by the lack of relevant morphological, radiological, and metabolic features (Hane et al., 1996). ID patients often need lifelong rehabilitation support treatment, causing substantial psychological and economic burdens for families and society. Determining the genetic and molecular basis of ID remains a significant challenge in neuroscience.
The SYNGAP1 gene encodes the brain-specific RAS GTPase-activating protein SYNGAP1, which is an important component of the N-methyl-D-aspartate receptor (NMDA) complex and plays a pivotal role in neuronal synaptic development, structure, and plasticity (Clement et al., 2012). De novo variants of SYNGAP1 are a common cause of NSID, autism spectrum disorders (ASD), and epilepsy (Berryer et al., 2013; Mignot et al., 2016). De novo nonsense variants in SYNGAP1 cause haploinsufficiency, resulting in a neurodevelopmental disorder known as intellectual developmental disorder (OMIM #612621), with phenotypes including ID, motor disorders, and epilepsy. The effects of these variants demonstrate the importance of SYNGAP1 in developing the nervous system and brain (Agarwal et al., 2019). Currently, approximately 0.7%–1% of ID cases are caused by SYNGAP1 variants (Mignot et al., 2016).
In this study, we identified two de novo heterozygous pathogenic variants in the SYNGAP1, c. 333del and c.664-2A>G, in two patients with ID, among which the c.664-2A>G variant was a novel de novo pathogenic variant. The patients exhibited generalized developmental delay, motor retardation, hypotonia, and severe language impairment. To assess the impact of c.664-2A>G variant on splicing, we performed a minigene splicing assay. We found that the c.664-2A>G variant causes aberrant splicing, which would result in impaired function of the SYNGAP1 protein and consequently contribute to the occurrence of ID.
Methods and materials
Subjects
Two female patients from unrelated families were diagnosed with intellectual developmental disorder from a clinical cohort of 113 cases with ID from between January 2019 and January 2023 at Qilu Hospital of Shandong University. All the patients were diagnosed by experienced experts of the hospital according to the DSM-5 criteria. Among these patients with ID, 25 cases were combined with epilepsy and 49 cases were combined with other structural anomalies. The age of the children ranged from 12 months to 18 years, with a median age of 8 years. The etiology of these patients was unknown. All of these patients underwent karyotyping and chromosome microarray (CMA) analysis, and these results were inconclusive, following these samples were processed for WES. We collected peripheral blood and clinical information from their families. The families accepted the inheritance consultation and signed the informed consent form before the genetic test. This study was authorized by the Ethics Committee of Qilu Hospital of Shandong University.
DNA extraction and whole exome sequencing (WES)
The genomic DNA for sequencing was obtained from peripheral blood. The extraction steps were conducted according to the instructions of the DNA extraction kit (Tiangen Biotech). WES was performed on the DNA from the affected individual and sequenced on NovaSeq 6000 platforms (Illumina) with 150 bp paired-end reads. Reads data were aligned with the GRCh37/hg19 human reference sequence. The single-nucleotide variants (SNVs) and other variants were called with the Genome Analysis Toolkit (GATK). The variants were annotated using Annovar software. During the annotation, several public databases such as Clinvar, gnomAD, PubMed, HGMD, dbNSFP, etc., were used. Variants with allele frequencies higher than 1% in any public databases (ExAC Browser and gnomAD) were excluded. De novo variants were analyzed from sequencing data by DeNovoGear software (Ramu et al., 2013). The candidate variants were confirmed in the patients with ID by Sanger sequencing.
Minigene assay
The SYNGAP1 c.664-2A>G variant is located at the splice-acceptor site of exon 7. We obtained the SYNGAP1 fragment [intron6 (192bp)-Exon7 (99bp)-intron7 (547bp)] with restriction sites (KpnI and BamHI) from human genomic DNA by nested PCR amplification and then cloned it into a pcMINI plasmid using nucleic acid endonuclease and DNA ligase. The pcMINI vector contain ExonA-IntronA-multiple cloning site-IntronB-ExonB (Bioeagle Biotech Company). Exon A and Exon B simulate exon 6 and exon 8, respectively. The pcMINI-SYNGAP1-MUT (c.664-2A>G) plasmids were produced using a QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent) with pcMINI-SYNGAP1-WT (wild-type) plasmid as the template. Both WT and mutant plasmids contained the whole sequence of exon 7 and a portion of the upstream and downstream intron sequences. The recombinant plasmids were transiently transfected into HEK293T and HeLa cells according to the transfection reagent instructions. After the transfected cells were cultured for 48 h, total RNA was extracted with Trizol (TaKaRa), and cDNA was acquired with HifairTM 1st Strand cDNA Synthesis SuperMix (TEASEN). The RT-PCR products was analyzed by electrophoresis on 2% agarose gels containing ethidium bromide and visualized by exposure to ultraviolet light. Each DNA band was purified by DNA Gel Exctration Kit (SIMGEN). Direct sequencing of purified RT-PCR products was performed with the Big Dye Terminator Cycler Sequencing Ready Reaction Kit (Applied Biosystems) on the ABI3730xl Genetic Analyzer (Applied Biosystems). Primers used for minigene assay of SYNGAP1 were as follows: SYNGAP1-F1: 5′-AACTCCTGGGCTCAAGTGAC-3′; SYNGAP1-R1: 5′-TGGGTAAAGCTTGGCCAGAT-3′; SYNGAP1-F2:5′-AGCACTTTGGGAGGCTGAAT-3′; SYNGAP1-R2: 5′-GAGGTTGCAGTGAGCCAAGA-3′; MINI-SYNGAP1-KpnI-F: 5′-GGTAGGTACCCTGGGGAGGGCCAAAGGACA-3′; MINI-SYNGAP1-BamHI-R: 5′-TAGTGGATCCGAGAATAGCTGACAGAACTG-3′; SYNGAP1-c.664-2A>G-F: 5′-TCCACACTCCTTTCTGGGTAACAACTTCATC-3′; SYNGAP1-c.664-2A>G-R: 5′-GATGAAGTTGTTACCCAGAAAGGAGTGTGGA -3′.
Results
De novo heterozygous variants were identified
Two de novo heterozygous pathogenic variants in SYNGAP1, NM_006772.3: c. 333del and c.664-2A>G, were identified in two ID patients separately, as detailed in Table 1. The c.333del variant has been reported previously as a de novo finding in a child with ID (Carvill et al., 2013). The c.333del variant is predicted to cause loss of normal protein function either through protein truncation or nonsense-mediated mRNA decay. Conversely, the c.664-2A>G variant was a novel de novo intronic variant identified in a child with ID, which has not been reported in the literature and databases (ClinVar, DVD, PubMed, HGMD, etc.) (Figure 1).
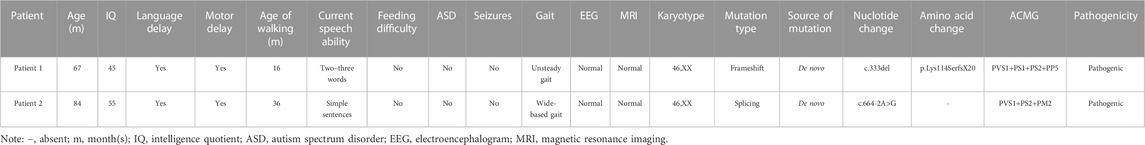
TABLE 1. Variations of SYNGAP1 genes identified in two ID patients and their clinical characteristics.
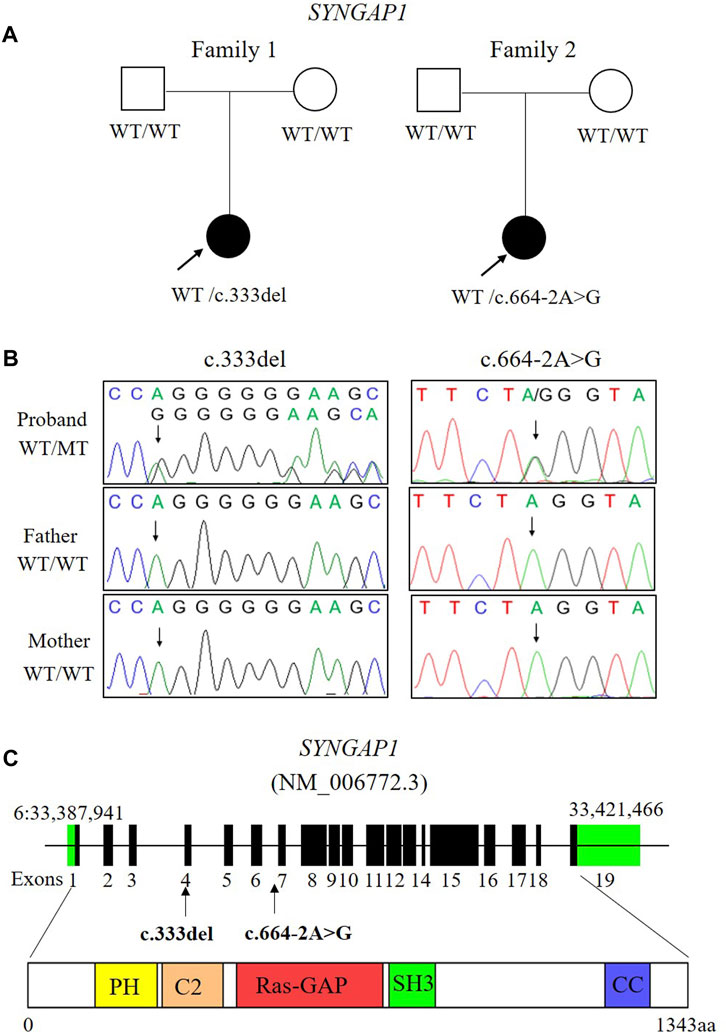
FIGURE 1. Two de novo variants of SYNGAP1 were identified in two patients with ID. (A) Families pedigree and genotype are shown. The probands with ID underwent WES. Filled symbols represent affected individuals. (B) Sanger sequencing chromatograms of the SYNGAP1 variants in these families. (C) Localization of the SYNGAP1: c. 333del and c.664-2A>G variant found in the study. The amino acid (aa) positions are referenced to RefSeq number NM_006772.3 (isoform-1: 1343 aa). Various predicted SYNGAP1 domains are showed: PH, pleckstrin homology domain (amino acid positions 150–251), C2 domain (amino acid positions 263–362), Ras-GAP (amino acid positions 392–729), SH3 (amino acid positions 785–815), coiled coil (CC; amino acid positions 1189–1262).
Clinical characteristics of the NSID affected individual
Both patients in their respective families, carrying de novo variants in SYNGAP1, exhibit delays in intellectual and motor developmental. These two patients had impaired speaking ability and were speech disabled, along with symptoms such as hypotonia, muscle flaccidity, and a wide-based/unsteady gait. Notably, they didn’t exhibit feeding difficulty, autism spectrum disorder (ASD), epilepsy, or microcephaly. We conducted a comprehensive assessment for ASD on these two patients, which included psychological tests, clinical examinations, and consideration of their family medical history. We utilized standardized assessment tools such as the Autism Diagnostic Observation Schedule (ADOS) for ASD evaluation and found no symptoms of ASD in these patients. Additionally, we conducted initial psychological tests on both patients to assess cognitive functioning and identify any coexisting conditions, and the results indicate that both of these patients don't exhibit symptoms of autism. Both patients had normal electroencephalograms (EEGs) and normal karyotypes (Table 1). There is no family history related to developmental disorders. The two patients are girls and are the only children in their respective families. One patient is 5 years old and the other is 7 years old, and their parents are healthy. They have experienced an overall delay in developmental milestones. For example, the patient which carrying the c.664-2A>G variant was independently sitting and walking later than children of the same age. She didn't achieve independent walking until the age of 3 years, and her gait was extensive and unstable. At the age of 7, her intelligence quotient (IQ) was measured at 55 on the Tanaka-Binet IQ Scale V. Furthermore, she exhibits delayed language development and only uses short and simple sentences with limited vocabulary.
Expression of SYNGAP1 mRNA in transfected cells with recombinant plasmids
The c.333del variant has been reported previously as a de novo finding, so we did not perform functional experiments on it. We performed in vitro experiments on the novel de novo splicing variant in SYNGAP gene. To investigate the influence of the c.664-2A>G variant on splicing, we conducted a minigene splicing assay. The pcMINI-SYNGAP1-WT and pcMINI-SYNGAP1-MUT (c.664-2A>G) plasmids were transiently transfected into 293T and HeLa cells (Figure 2A). The total RNA was extracted and reverse-transcribed into cDNA after transfection over 48 h. The cDNA was amplified by PCR and analyzed by agarose gel electrophoresis. Agarose gel electrophoresis showed that the mutant-type (MUT) had two bands, all bands were smaller than the WT band, and their migrations were relatively faster (Figure 2B). DNA sequencing results showed that the WT minigene (pcMINI-SYNGAP1-WT) transcribed normal mRNA composed of exon 7 (Figure 2C, band a), while the c.664-2A>G mutant minigene caused abnormal splicing, resulting in exon 7 skipping (Figures 2C, D, band b) and a 16 bp deletion within exon 7 (Figures 2C, D, band c), which reveals that this variant may be a crucial mechanism for the pathogenesis of ID. Exon 7 skipping could result in the loss of 33 amino acids (c.664_762del p.Val222_Lys254del), and the deletion of 16bp in exon 7 could result in a frameshift of amino acids at position 222 and a premature stop codon (c.664_679del p.Val222Glufs*24). The aberrant splicing is predicted to abolish the pleckstrin homology (PH, pos. 150–251aa) and C-terminal domains, suggesting that it would prevent the SYNGAP1 protein from performing its normal functions. SYNGAP1 c.664-2A>G may damage cognitive and social adaptation development by impairing maturation of dendritic spine synapses in neurons (Figure 3).
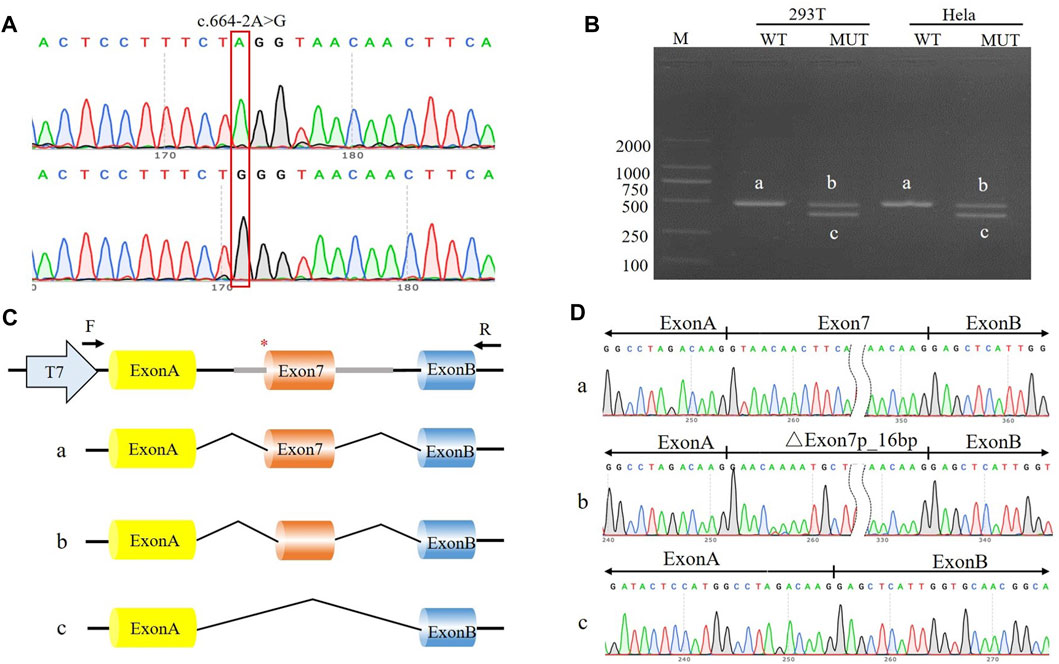
FIGURE 2. The effect of the c.664-2A>G variant on splicing was assessed through a minigene assay. (A) Construction of the pcMINI-SYNGAP1-WT/MUT vector, which contain exon 7 and flanking intronic sequences of WT or mutant type (c.664-2A>G) of the SYNGAP1 gene. (B) Minigene assay performed in 293T and Hela cells transfected with the pcMINI-SYNGAP1-WT/MUT vector. The PCR products were isolated by gel electrophoresis. The SYNGAP1 splicing products of wild-type (band a) and variant type (band b and c) are shown. (C, D) Schematic diagram of minigene construction and sanger sequencing of PCR products.
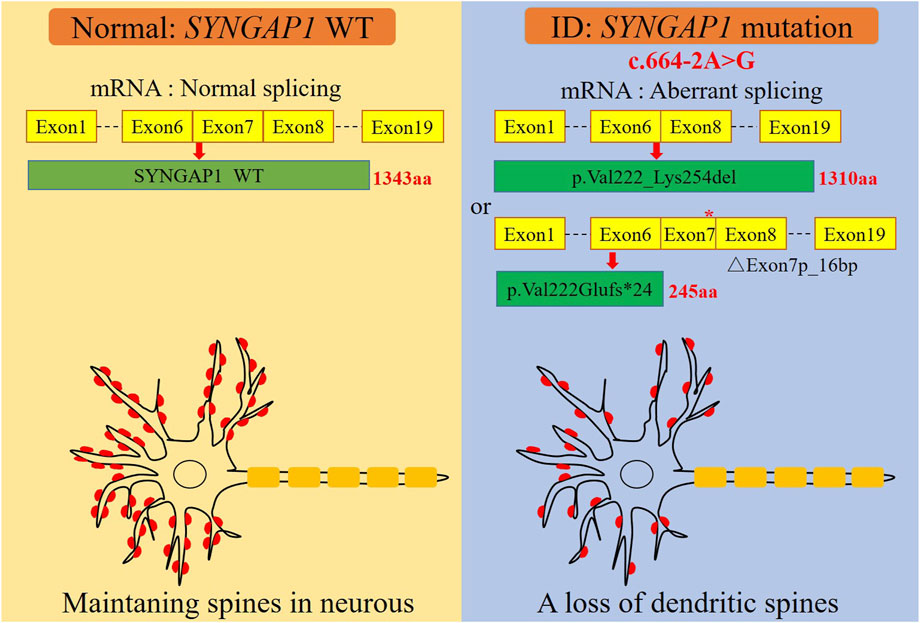
FIGURE 3. A graphical summary of the mechanism of dendritic spine loss caused by the SYNGAP1 c.664-2A>G variant. The SYNGAP1 c.664-2A>G variant causing aberrant splicing and dendritic spine loss.
Discussion
In this study, we identified two de novo heterozygous pathogenic variants in SYNGAP1 among 113 patients with ID. The c.333del variant has been previously reported as a de novo finding in a child with ID (Carvill et al., 2013), while the other splicing variant c.664-2A>G has not been reported in any literature or databases. The c.333del variant is predicted to cause loss of normal protein function either through protein truncation or nonsense-mediated mRNA decay. The phenotypes of the ID patient, which carrying the c.664-2A>G variant, are similar to the previously published truncating variant in SYNGAP1. Although the intronic variants may have more deleterious effects than the exonic variants, they are underexplored (Kallel-Bouattour et al., 2017). To further evaluate the deleterious effects of the intronic variant c.664-2A>G, we conducted a minigene assay to investigate its impact on mRNA splicing. The minigene experiment results showed that the intronic variant c.664-2A>G causes aberrant splicing of SYNGAP1. The c.664-2A>G variant would result in exon 7 skipping and partial exon 7 deletion, which would abolish critical functional domains and impair the function of the SYNGAP1 protein.
There is a potential acceptor site located 16 bp upstream of exon 7 in SYNGAP1. After c.664-2A>G variant, this site is activated for splicing. The c.664-2A>G variant disrupts the original acceptor site, potentially leading to the recognition of the alternative splicing site that causes a 16 bp deletion on upstream of exon 7. Alternatively, the disruption of the original acceptor site might result in direct skipping of exon 7 during splicing, resulting in the overall deletion of exon 7, much like how a gene in a database might have multiple normal transcripts.
SYNGAP1 is an important gene that is necessary for neuronal development, and its dysfunction is associated with ID (Jeyabalan and Clement, 2016). SYNGAP1 is located on human chromosome 6, contains 19 exons, and generates approximately a 6 kb transcript, which encodes a brain-specific synaptic Ras GTP-ase activating protein. The impairment of SYNGAP1 function may make patients with ID susceptible to seizures by increasing the recruitment of AMPA receptors at postsynaptic glutamatergic synapses, which leads to increased transmission of excitatory synapses (Hamdan et al., 2009). In large-scale studies, almost all SYNGAP1 variants associated with NSID, ASD, and epilepsy are loss-of-function and lead to SYNGAP1 haploinsufficiency, resulting in intellectual developmental disorder (Hamdan et al., 2011; Berryer et al., 2013; Carvill et al., 2013; Fieremans et al., 2016).In our study, as well as previous observations, suggest that the SYNGAP1 c.664-2A>G variant would cause ID mainly through a mechanism of haploinsufficiency.
NSID poses a challenge for clinicians because of the absence of specific clinical features to guide them toward an etiological diagnosis. The identification of novel variants in known pathogenic genes or novel ID genes suggests that molecular diagnostic approaches are becoming increasingly significant in unraveling the underlying causes of this condition. In the present study, the two patients presented with comprehensive developmental delays, particularly motor milestones and language development, and exhibited behavioral disorders. We identified pathogenic variants in SYNGAP1 in both of these patients by WES. Based on clinical and genetic features, the patients were diagnosed with intellectual developmental disorder. De novo SYNGAP1 variants were initially reported to cause ID, accounting for approximately 0.62% of all the patients in the Deciphering Developmental Disorders (DDD) study (Hamdan et al., 2011; Wright et al., 2015). Six patients with SYNGAP1 variants exhibited moderate to-severe ID due to severe language impairment (Hamdan et al., 2011). Studies involving rodent models with the deletion of the SYNGAP1 allele showed abnormal formation and maturation of dendritic spines in neurons, altered excitatory-inhibitory (E/I) balance, and changed the critical period of development, suggesting that heterozygous variants also have the potential to disrupt brain function in humans and lead to ID through the mechanism of haploinsufficiency (Rumbaugh et al., 2006; Guo et al., 2009; Muhia et al., 2010).
In conclusion, we identified two de novo pathogenic heterozygous variants in SYNGAP1, c. 333del and c.664-2A>G, among which the c.664-2A>G variant was a novel de novo pathogenic variant. Based on previous findings from others and our research, nonsense variants in SYNGAP1 remain the most common variant type leading to ID. This study further enriched the variant landscape of SYNGAP1 in ID and provided a basis for the clinical diagnosis and genetic counseling of ID.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: https://ngdc.cncb.ac.cn/search/?dbId=hra&q=HRA005421.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Qilu Hospital, Shandong University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
BL: Data curation, Formal Analysis, Validation, Writing–original draft. YW: Data curation, Formal Analysis, Writing–original draft. DH: Funding acquisition, Methodology, Writing–review and editing. ZS: Data curation, Investigation. LZ: Investigation, Methodology. NL: Investigation, Methodology. RY: Project administration, Resources. PS: Conceptualization, Project administration, Resources, Supervision, Writing–review and editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (82001509) and Natural Science Foundation of Jiangsu Province (BK20200233).
Acknowledgments
We thank the families and clinical staff for participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agarwal, M., Johnston, M. V., and Stafstrom, C. E. (2019). SYNGAP1 mutations: clinical, genetic, and pathophysiological features. Int. J. Dev. Neurosci. 78, 65–76. doi:10.1016/j.ijdevneu.2019.08.003
Berryer, M. H., Hamdan, F. F., Klitten, L. L., Moller, R. S., Carmant, L., Schwartzentruber, J., et al. (2013). Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 34 (2), 385–394. doi:10.1002/humu.22248
Carvill, G. L., Heavin, S. B., Yendle, S. C., McMahon, J. M., O'Roak, B. J., Cook, J., et al. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45 (7), 825–830. doi:10.1038/ng.2646
Chelly, J., Khelfaoui, M., Francis, F., Cherif, B., and Bienvenu, T. (2006). Genetics and pathophysiology of mental retardation. Eur. J. Hum. Genet. 14 (6), 701–713. doi:10.1038/sj.ejhg.5201595
Chen, T., Chen, X., Zhang, S., Zhu, J., Tang, B., Wang, A., et al. (2021). The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinforma. 19 (4), 578–583. doi:10.1016/j.gpb.2021.08.001
Clement, J. P., Aceti, M., Creson, T. K., Ozkan, E. D., Shi, Y., Reish, N. J., et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151 (4), 709–723. doi:10.1016/j.cell.2012.08.045
Fieremans, N., Van Esch, H., Holvoet, M., Van Goethem, G., Devriendt, K., Rosello, M., et al. (2016). Identification of intellectual disability genes in female patients with a skewed X-inactivation pattern. Hum. Mutat. 37 (8), 804–811. doi:10.1002/humu.23012
Goldenberg, A., and Saugier-Veber, P. (2010). Genetics of mental retardation. Pathol. Biol. Paris. 58 (5), 331–342. doi:10.1016/j.patbio.2009.09.013
Guo, X., Hamilton, P. J., Reish, N. J., Sweatt, J. D., Miller, C. A., and Rumbaugh, G. (2009). Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology 34 (7), 1659–1672. doi:10.1038/npp.2008.223
Hamdan, F. F., Daoud, H., Piton, A., Gauthier, J., Dobrzeniecka, S., Krebs, M. O., et al. (2011). De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol. Psychiatry 69 (9), 898–901. doi:10.1016/j.biopsych.2010.11.015
Hamdan, F. F., Gauthier, J., Spiegelman, D., Noreau, A., Yang, Y., Pellerin, S., et al. (2009). Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 360 (6), 599–605. doi:10.1056/NEJMoa0805392
Hane, B., Schroer, R. J., Arena, J. F., Lubs, H. A., Schwartz, C. E., and Stevenson, R. E. (1996). Nonsyndromic X-linked mental retardation: review and mapping of MRX29 to Xp21. Clin. Genet. 50 (4), 176–183. doi:10.1111/j.1399-0004.1996.tb02622.x
Jeyabalan, N., and Clement, J. P. (2016). SYNGAP1: mind the Gap. Front. Cell Neurosci. 10, 32. doi:10.3389/fncel.2016.00032
Kallel-Bouattour, R., Belguith-Maalej, S., Zouari-Bradai, E., Mnif, M., Abid, M., and Hadj Kacem, H. (2017). Intronic variants of SLC26A4 gene enhance splicing efficiency in hybrid minigene assay. Gene 620, 10–14. doi:10.1016/j.gene.2017.03.043
CNCB-NGDC Members and Partners (2023). Database resources of the national genomics data center, China national center for bioinformation in 2023. Nucleic Acids Res. 51 (D1), D18–D28. doi:10.1093/nar/gkac1073
Mignot, C., von Stulpnagel, C., Nava, C., Ville, D., Sanlaville, D., Lesca, G., et al. (2016). Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J. Med. Genet. 53 (8), 511–522. doi:10.1136/jmedgenet-2015-103451
Muhia, M., Yee, B. K., Feldon, J., Markopoulos, F., and Knuesel, I. (2010). Disruption of hippocampus-regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. Eur. J. Neurosci. 31 (3), 529–543. doi:10.1111/j.1460-9568.2010.07079.x
Ramu, A., Noordam, M. J., Schwartz, R. S., Wuster, A., Hurles, M. E., Cartwright, R. A., et al. (2013). DeNovoGear: de novo indel and point mutation discovery and phasing. Nat. Methods 10 (10), 985–987. doi:10.1038/nmeth.2611
Rumbaugh, G., Adams, J. P., Kim, J. H., and Huganir, R. L. (2006). SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc. Natl. Acad. Sci. U. S. A. 103 (12), 4344–4351. doi:10.1073/pnas.0600084103
Keywords: SYNGAP1, whole exome sequencing (WES), intellectual disability, minigene, variant
Citation: Li B, Wang Y, Hou D, Song Z, Zhang L, Li N, Yang R and Sun P (2023) Identification and functional characterization of de novo variant in the SYNGAP1 gene causing intellectual disability. Front. Genet. 14:1270175. doi: 10.3389/fgene.2023.1270175
Received: 03 August 2023; Accepted: 09 October 2023;
Published: 19 October 2023.
Edited by:
Hua Li, Shanghai Jiao Tong University, ChinaReviewed by:
Abhijeet Botre, KEM Hospital Research Centre, IndiaYoichi Araki, Johns Hopkins University, United States
Copyright © 2023 Li, Wang, Hou, Song, Zhang, Li, Yang and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Sun, c3A3NzIyM0AxNjMuY29t
†These authors have contributed equally to this work and share first authorship