 Xingyue Gu
Xingyue Gu Yijie Ding2,3*
Yijie Ding2,3*- 1State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, China
- 2Yangtze Delta Region Institute (Quzhou), University of Electronic Science and Technology of China, Quzhou, Zhejiang, China
- 3Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China
- 4Beidahuang Industry Group General Hospital, Harbin, China
There is a great deal of importance to SNARE proteins, and their absence from function can lead to a variety of diseases. The SNARE protein is known as a membrane fusion protein, and it is crucial for mediating vesicle fusion. The identification of SNARE proteins must therefore be conducted with an accurate method. Through extensive experiments, we have developed a model based on graph-regularized k-local hyperplane distance nearest neighbor model (GHKNN) binary classification. In this, the model uses the physicochemical property extraction method to extract protein sequence features and the SMOTE method to upsample protein sequence features. The combination achieves the most accurate performance for identifying all protein sequences. Finally, we compare the model based on GHKNN binary classification with other classifiers and measure them using four different metrics: SN, SP, ACC, and MCC. In experiments, the model performs significantly better than other classifiers.
1 Introduction
SNAREs mediate most of the intracellular membrane fusion events, and mammalian cells have over 30 members of the SNARE family, each found in a different subcellular compartment (Jahn and Scheller, 2006; van Dijk et al., 2008). SNAREs may encode aspects of membrane transport specificity, but the mechanisms by which they achieve specificity remain controversial (Ferro-Novick and Jahn, 1994; Rothman, 1994). Studies have shown that SNAREs are targets of CNT proteases, thus establishing the importance of SNARE proteins for synaptic neurotransmission (Schiavo et al., 1992; Blasi et al., 1993; Schiavo et al., 1993; Yamasaki et al., 1994a; Yamasaki et al., 1994b; Schiavo et al., 1995). Therefore, the accurate identification of SNARE proteins is particularly necessary and important. Up to now, experimenters have used a number of methods to identify SNARE proteins from a biological perspective. Traditional biological experiments have the disadvantage of long lead times and high costs. Machine learning and data mining have ushered in a new era of protein prediction (Xiong et al., 2012; Cao et al., 2014; Wei et al., 2014; Chen et al., 2016; Ding et al., 2016; Wei et al., 2016; Zou et al., 2016; Zeng et al., 2017a; Zhang et al., 2017; Xiong et al., 2018a; Bu et al., 2018; Liao et al., 2018; Wei et al., 2018; Cao et al., 2019; Chao et al., 2019; Chen et al., 2019; Le and Nguyen, 2019; Liu et al., 2019; Małysiak-Mrozek et al., 2019; Meng et al., 2019; Ghulam et al., 2020; Guo et al., 2021; Qian et al., 2022; Tiwari et al., 2022).
In this paper, a classification model based on the GKHNN algorithm is adopted to accurately identify SNARE proteins. Three datasets are used: a cross-training dataset, an independent-validation dataset, and the all-dataset containing all samples. To obtain 188-dimensional sample attribute features, the physicochemical property extraction method was used in this study to extract the sample features from three datasets. The sample sets of the three datasets were very unbalanced. In order to minimize the interference of the unbalanced datasets on the binary classification accuracy, this experiment uses the SMOTE upsampling method to make the positive and negative samples balanced. The GKHNN classifier model performs binary classification on each of the three datasets while comparing the classification results with the 2DCNN algorithm. Meanwhile, the binary classification experimental results are compared with other four classifiers on the complete dataset to verify the high accuracy of this model in classifying SNARE proteins; different feature extraction methods are used to compare the experimental results obtained using the 188D feature extraction method selected in this experimental model to verify the effectiveness of this experimental model feature extraction method; two other protein datasets in this field are selected for classification, and the generalizability of this experimental model was verified by comparing with previous experiments. This study uses four measures, namely, accuracy (ACC), sensitivity (SN), specificity (SP), and the Mathews correlation coefficient (MCC), to measure the degree of accuracy of the algorithmic model classification.
The structure and content of this paper are as follows: Section 1 describes the importance of identifying SNARE proteins as well as the structure and distribution of this paper. Section 2 describes the construction of the experimental dataset, preprocessing, and number of samples, as well as the specific experimental procedure of this experiment. Also, the physical and chemical property extraction method, the SMOTE dataset balancing method, and the classifier algorithm GKHNN are described in detail. Section 3 describes the comparison of experimental results when the specific parameters of this experimental model are taken at different values, the comparison of experimental results between this experimental model and other classifiers, the comparison of experimental results using the 188D feature extraction method used in this experiment and the four other common feature extraction methods, and the comparison of experimental results when this experimental model is applied to other datasets. The discussion of the current work is given in Section 4.
2 Materials and methods
2.1 Data retrieval and pretreatment
2.1.1 Dataset
Datasets were collected from the UniProt database (Consortium, 2015), which is one of the most comprehensive database resources for protein sequences. First, all proteins annotated with the keyword “snare” were collected from the UniProt database. It is worth noting that the proteins collected were all reviewed (extracted from the literature and assessed by the administrator for calculation and analysis). Subsequently, more than 30% of the redundant sequences were removed by the BLAST database (Altschul et al., 1997). After this process, only 245 SNARE proteins remained, and the number of proteins was not sufficient to build an accurate deep learning model. Therefore, we used a truncation level of 100% in the cross-training dataset to build a significant model. In the independent dataset, we still used a 30% similarity level to assess the classification performance of the model. This is critical for testing the model (Le and Nguyen, 2019).
In order to build a classification model with high classification accuracy, the dataset has a crucial role to play. The negative dataset collected should be similar to the positive dataset in terms of structure and function of the proteins. After considering the structure and function of the positive protein sequence, SNARE, the vesicle transporter protein, which is a general protein that includes the SNARE protein, was chosen as the positive dataset for this experiment. Thus, the problem was transformed into a binary classification problem for SNARE proteins and vesicular transporter proteins (vesicular transport proteins are referred to as non-SNARE proteins). We removed redundant datasets and those with greater than 30% similarity between the two datasets. Finally, we divided the data into cross-training, independent-validation, and the all-dataset. The details of the three datasets used in this study are shown in Table 1.

TABLE 1. Number of raw SNARE and non-SNARE proteins in the cross-training dataset, independent-validation dataset, and all-dataset.
2.1.2 Experimental procedure
The specific process for this experiment is as follows:
(1) After obtaining the data shown in Table 1, the cross-training dataset, the independent-validation dataset, and the negative and positive samples in the all-dataset were extracted using the physicochemical property extraction method to obtain 188-dimensional sample attribute features, respectively. In the cross-training dataset, there are 644*188-dimensional SNARE protein attributes and 2,234*188-dimensional non-SNARE protein attributes. In the independent-validation dataset, there are 38*188-dimensional SNARE proteins and 349*188-dimensional non-SNARE proteins. In the all-dataset, there are 682*188 dimensional SNARE proteins and 2,583*188 dimensional non-SNARE proteins.
(2) Due to the high imbalance in the number of positive and negative samples in the dataset, the cross-training dataset, the independent-validation dataset, and the all-dataset were upsampled separately using SMOTE. The number of positive and negative samples for the cross-training dataset was 2,200 and 2,234, respectively; the number of positive and negative samples for the independent-validation dataset was 350 and 349; and the number of negative and positive samples for the all-dataset was 2,550 and 2,583, respectively. This resulted in a balance of negative and positive samples. The specific numbers of negative and positive samples in the three datasets are shown in Table 2.
(3) Positive and negative samples from the independent-validation dataset, the cross-training dataset, and the all-dataset were classified using the GKHNN classifier and measured using four metrics based on specificity (spec), sensitivity (recall), Matthews correlation coefficient (mcc), and accuracy (acc). Specific results on the accuracy of the classification model are given in detail in Section 3.

TABLE 2. Number of SNARE and non-SNARE proteins in the cross-training dataset, the independent-validation dataset, and the all-dataset after SMOTE equilibration.
The exact procedure of the experiment is shown in Figure 1.
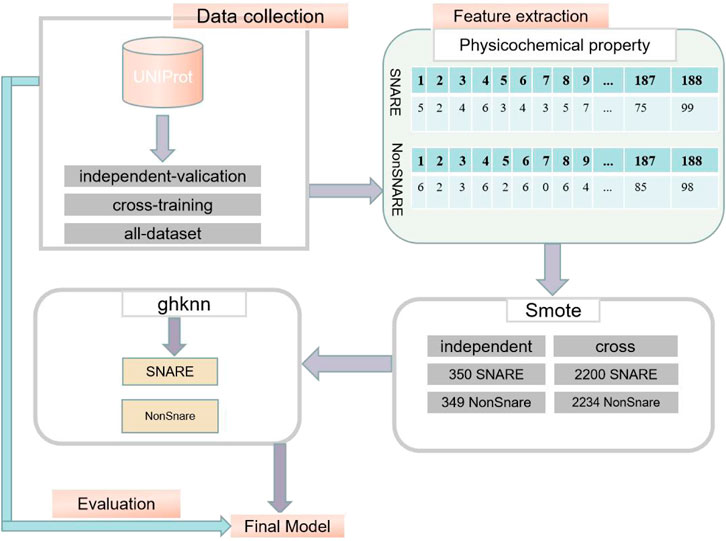
FIGURE 1. Experimental procedure diagram.
2.2 Physicochemical property extraction method
To extract sample features, we used the physicochemical property extraction method. The composition and position of protein molecules as well as its physicochemical characteristics have been used by previous researchers to extract protein features (Dubchak et al., 1995; Shen et al., 2017; Wang et al., 2017; Yu et al., 2017; Xiong et al., 2018b; Qiao et al., 2018; Zhang et al., 2018; Shen et al., 2019; Zou et al., 2019; Liu et al., 2020). Cai et al. (2003) used a physicochemical property feature extraction method to extract protein features, where the composition, distribution, and physical properties of amino acids were included. In one category, there are 20 amino acid features denoted as F1...F20, calculated using the following Eq. 1:
Here, the 20 features are represented by F, L denotes the length of the protein sequence, and
The other category is the physicochemical properties represented by 168 features, which are extracted from eight physicochemical properties of the protein, including polarity, secondary structure, polarizability, normalized van der Waals volume, and hydrophobicity. Each property has 21 features, for e.g., the 21 features of polarity are represented by F21–F41; hydrophobicity features are represented by F42–F52, calculated using the following Eq. 2:
According to the surface tension, the 20 amino acids are divided into three groups (Cai et al., 2003). Therefore,
where
Here, the numerator
This results in 20 features of amino acid composition and 21 features represented by each of the eight physicochemical properties of the protein, for a total of 168 features. This gives a total of 188 protein features (Gao and Li, 2020).
2.3 Sampling method
2.3.1 SMOTE upsampling technique
The negative and positive samples in the cross-training dataset, the independent-validation datasets and all-datasets are: 2,234, 349, and 644; 38,682, and 2,583, respectively. The positive and negative samples are quite unbalanced. To calculate the k nearest neighbors of an instance x in each of the few classes, the Euclidean distance between the instance x and the other instances in that class is calculated using SMOTE algorithm. The sampling rate N is determined by the imbalance rate. Several instances are chosen from the k nearest neighbors of x (Gao and Li, 2020). Finally, the following Eq. 5 is used to build an instance
2.4 Graph-regularized k-local hyperplane distance nearest neighbor model
Although HKNN algorithm incorporates a local hyperplane classification algorithm for better classification prediction performance, it is still in the original input space in terms of feature measurement. Its objective function is the distance from the sample x to the cth hyperplane:
where H in Eq. 6 denotes the hyperplane; k is the nearest neighbor, which refers to the nearest sample threshold of the test sample x; V denotes the matrix consisting of k nearest neighbor sample vectors of the test sample x; and N is the center of mass of the k nearest samples to the sample point x.
The GHKNN introduces kernel learning techniques and graph regularization terms to improve the generalization ability of the model and the association between samples. If the dimensionality of the original feature space is low, the model is not able to find a reasonable classification hyperplane. Therefore, by mapping the features in the original space to a suitable high-dimensional feature space and solving (6) using the kernel technique, the model is not able to find a reasonable classification hyperplane as the dimensionality of the original feature space is low, so it needs to be spatially projected to a high-dimensional space to find a reasonable classification hyperplane. Let x be mapped to f (let
where
In order to solve the parameters of the model, let the derivative of the left-hand side of the equation is 0 and
where
where
The following equation calculates the distance between the cth hyperplane and the test sample x. Here,
Finally, when assigning the test sample x to class c, the following results are obtained (Sun et al., 2021; Ding et al., 2022):
3 Results
3.1 Model assessment
This experiment used several commonly used evaluation metrics to measure the accuracy of model classification, including sensitivity (SN) (Chou, 2001; Chou, 2011; Lai et al., 2017; Liu et al., 2017; Ding et al., 2022), specificity (SP), accuracy (ACC), and Matthews correlation coefficient (MCC) (Matthews, 1975; Yu et al., 2015; Zeng et al., 2017b; Wei et al., 2017; Jia et al., 2018; Zhang et al., 2019a; Zhang et al., 2019b; Shan et al., 2019; Zeng et al., 2019; Hong et al., 2020). The four evaluation indicators are given in the following four formulas:
A false-positive, a true-negative, a false-negative, and a true-positive are, respectively, referred to as FP, TN, FN, and TP (Hou et al., 2020).
3.2 Parameter adjustment
The GKHNN classifier has three variable parameters:
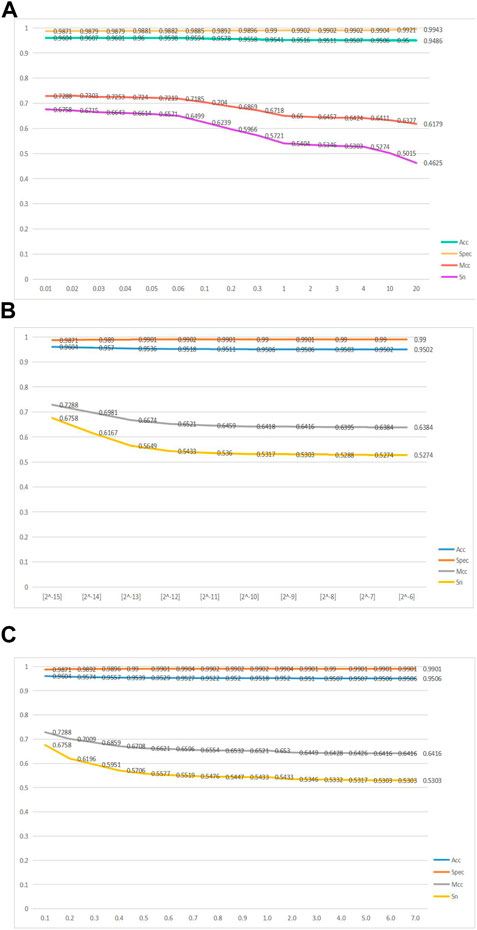
FIGURE 2. Line graphs of the four metrics, namely, acc, spec, mcc, and sn, as the variable parameters
Figure 2A shows a line graph of the GKHNN classifier bifurcating the positive and negative sample sets as the parameter
Figure 2B shows a line graph of the four metrics, acc, spec, mcc, and sn, continuously changing as the parameter
Figure 2C shows a line graph of how the four metrics, acc, spec, mcc, and sn, change as
Ultimately, based on the aforementioned line graphs, it was concluded that GKHNN classification performed best when the values of 0.01, [2^-15], and 0.1 were set for

TABLE 3. Values of parameters
3.3 Comparison with other methods
Table 4 shows the comparison of the classification results of GKHNN with 2DCNN on the all-dataset, the independent-validation dataset (Iv dataset), and the cross-training dataset (Ct dataset). As shown in the table, on the Ct dataset, the classification result values of this experimental model are higher than those of 2DCNN; on the Iv dataset, only the experimental result value of Sn is slightly lower than that of 2DCNN, and the other three metrics are higher than those of 2DCNN. Therefore, it seems that the present experimental model outperforms 2DCNN in terms of binary classification on these datasets in a comprehensive manner.
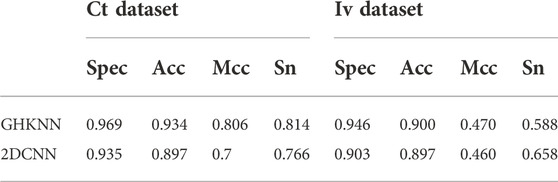
TABLE 4. Values of Sn, Acc, Spec, and Mcc of the GHKNN and 2DCNN classifiers on the Iv dataset and Ct dataset.
Figure 3 shows the comparison of the classification effect of the GHKNN classifier with four other classifiers: random forest (RF), support vector machine (SVM), k-local hyperplane distance nearest neighbor (HKNN), and k nearest neighbor (KNN), on the three datasets. As can be seen from the graph, the Acc, Spec, and Mcc values of GHKNN are higher than those of the other four classifiers, and only the Sn value is slightly lower.
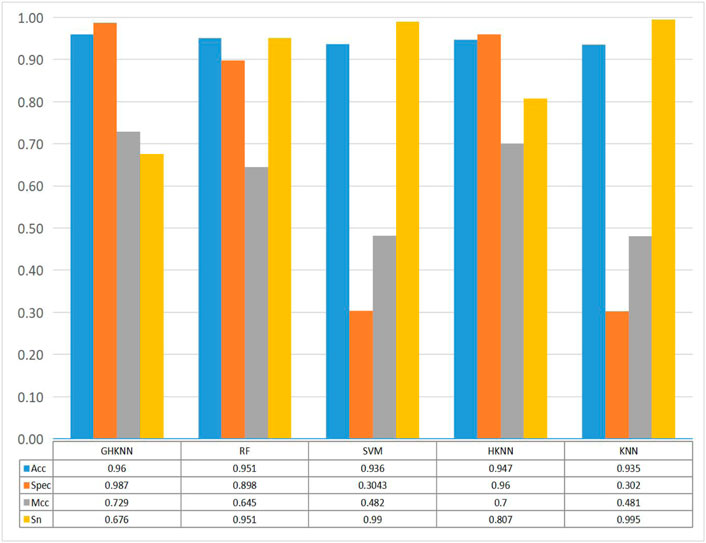
FIGURE 3. Histograms of the four metrics, namely, sn, acc, spec and mcc, obtained from the classification of the dataset by the five classifiers: GHKNN, RF, SVM, HKNN, and KNN.
Collectively, it seems that the classification effect of the GHKNN classifier exceeds that of the other classifiers, and thus, this experimental model has the best classification effect on the three datasets.
3.4 Comparison with other feature extraction methods
Table 5 shows the experimental comparison results of the 188D feature extraction method used in this experiment with four other common feature extraction methods with representative values: AAC, CTDC, GAAC, and CKSAAGP. From the table, it can be seen that the values of Acc, spec, mcc, and sn of the 188D feature extraction method used in this experiment are much higher than those of the other four extraction methods
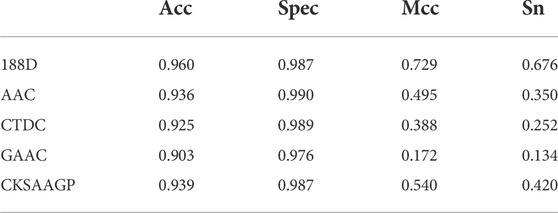
TABLE 5. Comparison of Sn, Acc, Spec, and Mcc values obtained by using GHKNN-based classification on this experimental dataset using five extraction feature methods: 188D, AAC, CTDC, GAAC, and CKSAAGP.
3.5 Comparison with previous classification results on other datasets
Table 6 shows the results of this experimental model on the G protein-coupled receptor (GPCR) dataset compared with those of Liao et al. (2016) and on the vesicular transport protein (vesi) dataset compared with those of Le and Nguyen (2019). As can be seen from the table, on the GPCR dataset, the values of the present experimental model is lower than the predicted value of Liao only on spec, which is about 0.04, and outperforms Liao on all three measures of acc, mcc, and sens; on the vesi dataset, the values of the present experimental model is lower than those of Nguyen on the index of sens, and the other three measures are higher than those of Nguyen. Collectively, it seems that the present experimental model outperforms the previous experimental results in classification on both GPCR and vesi datasets.
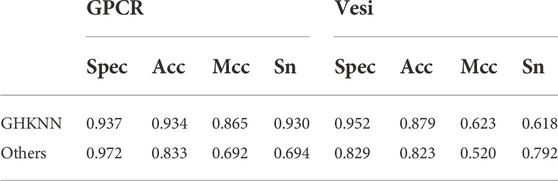
TABLE 6. Classification results using this experimental model on the GPCR and vesi datasets and the comparison with the previous experimental results.
4 Discussion
This study shows that the GKHNN-based binary classifier has good classification results for proteins. Compared to several other classifiers, the four metrics, spec, recall, mcc, and acc, reached high values on the three datasets of this study, which have some application value. In the study of the binary classification problem of protein sequences, both in the field of machine learning and deep learning, the classification accuracy of protein sequences is highly variable due to the individual performance of each protein sequence, resulting in significant differences in the classification results using different classifiers. As a result, we are committed to finding more general classification models with wider applicability in the future.
Data availability statement
Publicly available datasets were analyzed in this study. These data can be found at: https://github.com/ gugu131300/GHKNN_code.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The work was supported by the National Natural Science Foundation of China (No. 62172076) and the Special Science Foundation of Quzhou (2021D004).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.935717/full#supplementary-material
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25 (17), 3389–3402. doi:10.1093/nar/25.17.3389
Blasi, J., Chapman, E. R., Link, E., Binz, T., YamaSaki, S., De Camilli, P., et al. (1993). Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature 365 (6442), 160–163. doi:10.1038/365160a0
Bu, H., Hao, J., Guan, J., and Zhou, S. (2018). Predicting enhancers from multiple cell lines and tissues across different developmental stages based on SVM method. Curr. Bioinform. 13 (6), 655–660. doi:10.2174/1574893613666180726163429
Cai, C., Han, L. Y., Ji, Z. L., Chen, X., and Chen, Y. Z. (2003). SVM-prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 31 (13), 3692–3697. doi:10.1093/nar/gkg600
Cao, R., Wang, Z., Wang, Y., and Cheng, J. (2014). Smoq: A tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinforma. 15 (1), 120–128. doi:10.1186/1471-2105-15-120
Cao, Y., Wang, S., Guo, Z., Huang, T., and Wen, S. (2019). Synchronization of memristive neural networks with leakage delay and parameters mismatch via event-triggered control. Neural Netw. 119, 178–189. doi:10.1016/j.neunet.2019.08.011
Chao, L., Wei, L., and Zou, Q. (2019). SecProMTB: Support vector machine-based classifier for secretory proteins using imbalanced data sets applied to Mycobacterium tuberculosis. Proteomics 19, e1900007. doi:10.1002/pmic.201900007
Chen, W., Ding, H., Feng, P., Lin, H., and Chou, K. C. (2016). iACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 7 (13), 16895–16909. doi:10.18632/oncotarget.7815
Chen, W., Lv, H., Nie, F., and Lin, H. (2019). i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 35 (16), 2796–2800. doi:10.1093/bioinformatics/btz015
Chou, K.-C. (2011). Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 273 (1), 236–247. doi:10.1016/j.jtbi.2010.12.024
Chou, K.-C. (2001). Using subsite coupling to predict signal peptides. Protein Eng. 14 (2), 75–79. doi:10.1093/protein/14.2.75
Consortium, U. (2015). UniProt: A hub for protein information. Nucleic Acids Res. 43 (D1), D204–D212. doi:10.1093/nar/gku989
Ding, Y., Tang, J., and Guo, F. (2016). Identification of protein–protein interactions via a novel matrix-based sequence representation model with amino acid contact information. Int. J. Mol. Sci. 17 (10), 1623. doi:10.3390/ijms17101623
Ding, Y., Yang, C., Tang, J., and Guo, F. (2022). Identification of protein-nucleotide binding residues via graph regularized k-local hyperplane distance nearest neighbor model. Appl. Intell. (Dordr). 52 (6), 6598–6612. doi:10.1007/s10489-021-02737-0
Dubchak, I., MuchnIk, I., Holbrook, S. R., and Kim, S. H. (1995). Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U. S. A. 92 (19), 8700–8704. doi:10.1073/pnas.92.19.8700
Ferro-Novick, S., and Jahn, R. (1994). Vesicle fusion from yeast to man. Nature 370 (6486), 191–193. doi:10.1038/370191a0
Gao, X., and Li, G. (2020). A KNN model based on manhattan distance to identify the SNARE proteins. IEEE Access 8, 112922–112931. doi:10.1109/access.2020.3003086
Ghulam, A., Lei, X., Guo, M., and Bian, C. (2020). Comprehensive analysis of features and annotations of pathway databases. Curr. Bioinform. 15 (8), 803–820. doi:10.2174/1574893615999200413123352
Guo, X. Y., et al. (2021). An efficient multiple kernel support vector regression model for assessing dry weight of hemodialysis patients. Curr. Bioinform. 16 (2), 284–293. doi:10.2174/15748936mta3hmzqt1
Hong, Z., Zeng, X., Wei, L., and Liu, X. (2020). Identifying enhancer–promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics 36 (4), 1037–1043. doi:10.1093/bioinformatics/btz694
Hou, R., Wang, L., and Wu, Y.-J. (2020). Predicting atp-binding cassette transporters using the random forest method. Front. Genet. 11, 156. doi:10.3389/fgene.2020.00156
Jahn, R., and Scheller, R. H. (2006). SNAREs—Engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7 (9), 631–643. doi:10.1038/nrm2002
Jia, C., Zuo, Y., and Zou, Q. (2018). O-GlcNAcPRED-II: An integrated classification algorithm for identifying O-GlcNAcylation sites based on fuzzy undersampling and a K-means PCA oversampling technique. Bioinformatics 34 (12), 2029–2036. doi:10.1093/bioinformatics/bty039
Lai, H.-Y., Chen, X. X., Chen, W., Tang, H., and Lin, H. (2017). Sequence-based predictive modeling to identify cancerlectins. Oncotarget 8 (17), 28169–28175. doi:10.18632/oncotarget.15963
Le, N. Q. K., and Nguyen, V.-N. (2019). SNARE-CNN: A 2D convolutional neural network architecture to identify SNARE proteins from high-throughput sequencing data. PeerJ. Comput. Sci. 5, e177. doi:10.7717/peerj-cs.177
Le, N. Q. K., Yapp, E. K. Y., NagasuNdaramN., , Chua, M. C. H., and Yeh, H. Y. (2019). Computational identification of vesicular transport proteins from sequences using deep gated recurrent units architecture. Comput. Struct. Biotechnol. J. 17, 1245–1254. doi:10.1016/j.csbj.2019.09.005
Liao, Z., Ju, Y., and Zou, Q. (2016). Prediction of G Protein-Coupled receptors with SVM-prot features and random forest. Scientifica, 8309253.
Liao, Z., Li, D., Wang, X., Li, L., and Zou, Q. (2018). Cancer diagnosis through IsomiR expression with machine learning method. Curr. Bioinform. 13 (1), 57–63. doi:10.2174/1574893611666160609081155
Liu, B., Chen, S., Yan, K., and Weng, F. (2019). iRO-PsekGCC: identify DNA replication origins based on pseudo k-tuple GC composition. Front. Genet. 10, 842. doi:10.3389/fgene.2019.00842
Liu, B., Jiang, S., and Zou, Q. (2020). HITS-PR-HHblits: Protein remote homology detection by combining PageRank and hyperlink-induced topic search. Briefings Bioinforma. 21 (1), 298–308.
Liu, B., Yang, F., and Chou, K.-C. (2017). 2L-piRNA: A two-layer ensemble classifier for identifying piwi-interacting RNAs and their function. Mol. Ther. Nucleic Acids 7, 267–277. doi:10.1016/j.omtn.2017.04.008
Małysiak-Mrozek, B., Baron, T., and Mrozek, D. (2019). Spark-IDPP: High-throughput and scalable prediction of intrinsically disordered protein regions with spark clusters on the cloud. Clust. Comput. 22 (2), 487–508. doi:10.1007/s10586-018-2857-9
Matthews, B. W. (1975). Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 405 (2), 442–451. doi:10.1016/0005-2795(75)90109-9
Meng, C., Jin, S., Wang, L., Guo, F., and Zou, Q. (2019). AOPs-SVM: A sequence-based classifier of antioxidant proteins using a support vector machine. Front. Bioeng. Biotechnol. 7, 224. doi:10.3389/fbioe.2019.00224
Qian, Y. Q., Meng, H., Lu, W., Liao, Z., Ding, Y., and Wu, H. (2022). Identification of DNA-binding proteins via hypergraph based laplacian support vector machine. Curr. Bioinform. 17 (1), 108–117. doi:10.2174/1574893616666210806091922
Qiao, Y., Xiong, Y., Gao, H., Zhu, X., and Chen, P. (2018). Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinforma. 19 (1), 14–16. doi:10.1186/s12859-018-2009-5
Rothman, J. E. (1994). Mechanisms of intracellular protein transport. Nature 372 (6501), 55–63. doi:10.1038/372055a0
Schiavo, G. G., BenFenatiF., , Poulain, B., RossettO, O., Polverino de Laureto, P., DasGupta, B. R., et al. (1992). Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359 (6398), 832–835. doi:10.1038/359832a0
Schiavo, G., Santucci, A., Dasgupta, B. R., Mehta, P. P., Jontes, J., BenFenatiF., , et al. (1993). Botulinum neurotoxins serotypes A and E cleave SNAP-25 at distinct COOH-terminal peptide bonds. FEBS Lett. 335 (1), 99–103. doi:10.1016/0014-5793(93)80448-4
Schiavo, G., Shone, C. C., Bennett, M. K., Scheller, R. H., and MonteCuCCo, C. (1995). Botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J. Biol. Chem. 270 (18), 10566–10570. doi:10.1074/jbc.270.18.10566
Shan, X., Wang, X., Chu, Y., Zhang, Y., Xiong, Y., et al. (2019). Prediction of CYP450 enzyme–substrate selectivity based on the network-based label space division method. J. Chem. Inf. Model. 59 (11), 4577–4586. doi:10.1021/acs.jcim.9b00749
Shen, C., Ding, Y., Tang, J., Song, J., and Guo, F. (2017). Identification of DNA–protein binding sites through multi-scale local average blocks on sequence information. Molecules 22 (12), 2079. doi:10.3390/molecules22122079
Shen, Y., Tang, J., and Guo, F. (2019). Identification of protein subcellular localization via integrating evolutionary and physicochemical information into Chou’s general PseAAC. J. Theor. Biol. 462, 230–239. doi:10.1016/j.jtbi.2018.11.012
Sun, M., et al. (2021). Membrane protein identification via multi-view graph regularized k-local hyperplane distance nearest neighbor model. IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE.
Tiwari, P., Dehdashti, S., Obeid, A. K., Marttinen, P., and Bruza, P. (2022). Kernel method based on non-linear coherent states in quantum feature space. J. Phys. A Math. Theor. 55 (35), 355301. doi:10.1088/1751-8121/ac818e
van Dijk, A. D., Bosch, D., ter Braak, C. J. F., van der Krol, A. R., and van Ham, R. C. H. J. (2008). Predicting sub-Golgi localization of type II membrane proteins. Bioinformatics 24 (16), 1779–1786. doi:10.1093/bioinformatics/btn309
Wang, Y., Ding, Y., Guo, F., Wei, L., and Tang, J. (2017). Improved detection of DNA-binding proteins via compression technology on PSSM information. PloS one 12 (9), e0185587. doi:10.1371/journal.pone.0185587
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and promising identification of human MicroRNAs by incorporating a high-quality negative set. IEEE/ACM Trans. Comput. Biol. Bioinform. 11 (1), 192–201. doi:10.1109/TCBB.2013.146
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017). Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi:10.1016/j.artmed.2017.03.001
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018). ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34 (23), 4007–4016. doi:10.1093/bioinformatics/bty451
Wei, L., Zou, Q., Liao, M., Lu, H., and Zhao, Y. (2016). A novel machine learning method for cytokine-receptor interaction prediction. Comb. Chem. High. Throughput Screen. 19 (2), 144–152. doi:10.2174/1386207319666151110122621
Xiong, Y., Liu, J., Zhang, W., and Zeng, T. (2012). Prediction of heme binding residues from protein sequences with integrative sequence profiles. Proteome Sci. 10 (1), S20. doi:10.1186/1477-5956-10-S1-S20
Xiong, Y., Wang, Q., Yang, J., Zhu, X., and Wei, D. Q. (2018). PredT4SE-Stack: Prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 9, 2571. doi:10.3389/fmicb.2018.02571
Xiong, Y., Wang, Q., Yang, J., Zhu, X., and Wei, D. Q. (2018). PredT4SE-stack: Prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 9, 2571. doi:10.3389/fmicb.2018.02571
Yamasaki, S., Baumeister, A., Binz, T., Blasi, J., Link, E., CornilleF., , et al. (1994). Cleavage of members of the synaptobrevin/VAMP family by types D and F botulinal neurotoxins and tetanus toxin. J. Biol. Chem. 269 (17), 12764–12772. doi:10.1016/s0021-9258(18)99941-2
Yamasaki, S., Binz, T., Hayashi, T., Szabo, E., YamasakiN., , EklundM., , et al. (1994). Botulinum neurotoxin type G proteolyses the Ala81-Ala82 bond of rat synaptobrevin 2. Biochem. Biophys. Res. Commun. 200 (2), 829–835. doi:10.1006/bbrc.1994.1526
Yu, L., Huang, J., Ma, Z., Zhang, J., Zou, Y., and Gao, L. (2015). Inferring drug-disease associations based on known protein complexes. BMC Med. Genomics 8 (2), S2–S13. doi:10.1186/1755-8794-8-S2-S2
Yu, L., Zhao, J., and Gao, L. (2017). Drug repositioning based on triangularly balanced structure for tissue-specific diseases in incomplete interactome. Artif. Intell. Med. 77, 53–63. doi:10.1016/j.artmed.2017.03.009
Zeng, X., Ding, N., Rodriguez-Paton, A., and Zou, Q. (2017). Probability-based collaborative filtering model for predicting gene–disease associations. BMC Med. Genomics 10 (5), 76–53. doi:10.1186/s12920-017-0313-y
Zeng, X., Lin, W., Guo, M., and Zou, Q. (2017). A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 13 (6), e1005420. doi:10.1371/journal.pcbi.1005420
Zeng, X., Lin, Y., He, Y., Lu, L., Min, X., and Rodriguez-Paton, A. (2019). Deep collaborative filtering for prediction of disease genes. IEEE/ACM Trans. Comput. Biol. Bioinform. 17 (5), 1639–1647. doi:10.1109/TCBB.2019.2907536
Zhang, W., Li, Z., Guo, W., Yang, W., and Huang, F. (2019). A fast linear neighborhood similarity-based network link inference method to predict microRNA-disease associations. IEEE/ACM Trans. Comput. Biol. Bioinform. 18 (2), 405–415. doi:10.1109/TCBB.2019.2931546
Zhang, W., Liu, T. H., He, Y., Pan, H. Q., Yin, X. P., et al. (2019). Sflln: A sparse feature learning ensemble method with linear neighborhood regularization for predicting drug–drug interactions. Biol. Psychiatry 497, 189–201. doi:10.1016/j.biopsych.2018.06.019
Zhang, W., Qu, Q., Zhang, Y., and Wang, W. (2018). The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 273, 526–534. doi:10.1016/j.neucom.2017.07.065
Zhang, X., Zou, Q., Rodriguez-Paton, A., and Zeng, X. (2017). Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 16 (1), 283–291. doi:10.1109/TCBB.2017.2776280
Zhao, S. L., (2020). MK-FSVM-SVDD: A multiple kernel-based fuzzy SVM model for predicting DNA-binding proteins via support vector data description. 70.
Zhu, Q., Fan, Y., and Pan, X. (2021). Fusing multiple biological networks to effectively predict miRNA-disease associations. Curr. Bioinform. 16 (3), 371–384. doi:10.2174/1574893615999200715165335
Zou, Q., et al. (2017). Scalable data mining algorithms in computational biology and biomedicine. Hindawi.
Zou, Q., Li, J., Song, L., Zeng, X., and Wang, G. (2016). Similarity computation strategies in the microRNA-disease network: A survey. Brief. Funct. Genomics 15 (1), 55–64. doi:10.1093/bfgp/elv024
Zou, Q., Wang, Z., Guan, X., Liu, B., Wu, Y., and Lin, Z. (2013). An approach for identifying cytokines based on a novel ensemble classifier. Biomed. Res. Int. 2013, 686090. doi:10.1155/2013/686090
Zou, Q., Xing, P., Wei, L., and Liu, B. (2019). Gene2vec: Gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. Rna 25 (2), 205–218. doi:10.1261/rna.069112.118
Keywords: SNARE proteins, GHKNN, SMOTE, identify protein sequences, physicochemical property extraction method
Citation: Gu X, Ding Y, Xiao P and He T (2022) A GHKNN model based on the physicochemical property extraction method to identify SNARE proteins. Front. Genet. 13:935717. doi: 10.3389/fgene.2022.935717
Received: 16 August 2022; Accepted: 02 November 2022;
Published: 23 November 2022.
Edited by:
Zhibin Lv, Sichuan university, ChinaReviewed by:
Ke Han, Harbin University of Commerce, ChinaCong Shen, Tianjin University of Technology, China
Copyright © 2022 Gu, Ding, Xiao and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengfeng Xiao, eGlhb3BmQHNldS5lZHUuY24=; Tao He, MTkxMjA1MzkzMDBAMTYzLmNvbQ==; Yijie Ding, d3V4aV9keWpAMTYzLmNvbQ==