 Joanna Walczak-Sztulpa1†Anna Wawrocka1†Cenna Doornbos2Ronald van Beek2,3Anna Sowińska-Seidler1Aleksander Jamsheer1,4Ewelina Bukowska-Olech1Anna Latos-Bieleńska1Ryszard Grenda5Ernie M. H. F. Bongers2Miriam Schmidts6,7Ewa Obersztyn8Maciej R. Krawczyński1,4‡Machteld M. Oud2,3*‡
Joanna Walczak-Sztulpa1†Anna Wawrocka1†Cenna Doornbos2Ronald van Beek2,3Anna Sowińska-Seidler1Aleksander Jamsheer1,4Ewelina Bukowska-Olech1Anna Latos-Bieleńska1Ryszard Grenda5Ernie M. H. F. Bongers2Miriam Schmidts6,7Ewa Obersztyn8Maciej R. Krawczyński1,4‡Machteld M. Oud2,3*‡- 1Department of Medical Genetics, Poznan University of Medical Sciences, Poznan, Poland
- 2Department of Human Genetics, Radboud University Medical Center, Nijmegen, Netherlands
- 3Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, Netherlands
- 4Centers for Medical Genetics, Poznan, Poland
- 5Department of Nephrology, Kidney Transplantation and Hypertension, The Children’s Memorial Health Institute, Warsaw, Poland
- 6Center for Pediatrics and Adolescent Medicine, University Hospital Freiburg, Freiburg University Faculty of Medicine, Freiburg, Germany
- 7CIBSS—Centre for Integrative Biological Signalling Studies, Freiburg University, Freiburg, Germany
- 8Department of Medical Genetics, Institute of Mother and Child, Warsaw, Poland
Ciliopathies are rare congenital disorders, caused by defects in the cilium, that cover a broad clinical spectrum. A subgroup of ciliopathies showing significant phenotypic overlap are known as skeletal ciliopathies and include Jeune asphyxiating thoracic dysplasia (JATD), Mainzer-Saldino syndrome (MZSDS), cranioectodermal dysplasia (CED), and short-rib polydactyly (SRP). Ciliopathies are heterogeneous disorders with >187 associated genes, of which some genes are described to cause more than one ciliopathy phenotype. Both the clinical and molecular overlap make accurate diagnosing of these disorders challenging. We describe two unrelated Polish patients presenting with a skeletal ciliopathy who share the same compound heterozygous variants in IFT140 (NM_014,714.4) r.2765_2768del; p.(Tyr923Leufs*28) and exon 27–30 duplication; p.(Tyr1152_Thr1394dup). Apart from overlapping clinical symptoms the patients also show phenotypic differences; patient 1 showed more resemblance to a Mainzer-Saldino syndrome (MZSDS) phenotype, while patient 2 was more similar to the phenotype of cranioectodermal dysplasia (CED). In addition, functional testing in patient-derived fibroblasts revealed a distinct cilium phenotyps for each patient, and strikingly, the cilium phenotype of CED-like patient 2 resembled that of known CED patients. Besides two variants in IFT140, in depth exome analysis of ciliopathy associated genes revealed a likely-pathogenic heterozygous variant in INTU for patient 2 that possibly affects the same IFT-A complex to which IFT140 belongs and thereby could add to the phenotype of patient 2. Taken together, by combining genetic data, functional test results, and clinical findings we were able to accurately diagnose patient 1 with “IFT140-related ciliopathy with MZSDS-like features” and patient 2 with “IFT140-related ciliopathy with CED-like features”. This study emphasizes that identical variants in one ciliopathy associated gene can lead to a variable ciliopathy phenotype and that an in depth and integrated analysis of clinical, molecular and functional data is necessary to accurately diagnose ciliopathy patients.
1 Introduction
Ciliopathies are a group of rare disorders that show clinical and genetic heterogeneity. The majority of ciliopathies are recessively inherited, however, few autosomal dominant forms (e.g. polycystic kidney disease, ADPKD) and X-linked inheritance patterns (e.g. retinitis pigmentosa, RP) have been reported (Tsang and Sharma, 2018; Ma, 2021). To date 35 ciliopathies and >187 associated disease genes have been described in literature (Reiter and Leroux, 2017). Those ciliopathies presenting with significant skeletal abnormalities are collectively called skeletal ciliopathies, i.e. Jeune asphyxiating thoracic dystrophy (JATD), short-rib polydactyly syndrome (SRPS), Mainzer-Saldino syndrome (MZSDS) and cranioectodermal dysplasia (CED). The skeletal ciliopathy cluster of CED (also called Sensenbrenner syndrome, MIM#218330, MIM#613610, MIM#614099, MIM#614378), MZSDS (MIM#266920, MIM#615630) and JATD (MIM#611263, MIM#613091, MIM#613819, MIM#614376, MIM%208,500) share significant genetic and phenotypic overlap. All three syndromes are characterized by skeletal features; short stature, rhizomelic limb shortening, and narrowing of the thorax to a variable degree where JATD shows the most severe form. A defining feature of JATD and MZSDS is pelvic abnormalities, which distinguishes the two syndromes from CED. While ectodermal abnormalities (i.e. nail, teeth and hair abnormalities) and dysmorphic craniofacial features such as frontal bossing and craniosynostosis are typical for CED and moderately common for MZSDS. The combination of renal and retinal abnormalities is typical for MZSDS, but can also occur in CED and to a much lesser extent in JATD. The major cause of skeletal ciliopathies are defects in genes that encode components of the intraflagellar transport complex A (IFT-A) and complex B (IFT-B). IFT is an evolutionarily conserved bidirectional trafficking machinery that is driven by kinesin- and dynein motor complexes and is essential for proper cilia function and structure. The IFT-B complex is composed of 16 proteins and is responsible for the anterograde transport, while the much smaller IFT-A complex is associated with retrograde transport. The IFT-A complex can be divided into a three core proteins; IFT122, IFT140 and WDR19, and three peripheral proteins; IFT43, WDR35, TTC21B. Dysfunctional IFT140 results in an accumulation of IFT proteins at the ciliary tip and in cilium shortening (Piperno et al., 1998; Miller et al., 2013). Cilia play an important role in diverse signaling pathways including Hedgehog (Hh),Wingless (Wnt), platelet-derived growth factor receptors (PDGFR), mammalian target of rapamycin (mTOR), G-protein coupled receptors (GPCR), Hippo and Notch. These pathways are crucial for normal embryonic development and for tissue homeostasis after birth (Wheway et al., 2019).
Variants in IFT140 are implicated in the pathogenesis of CED and MZSDS, but also in JATD, Bardet-Biedl syndrome (BBS), Optiz trigonocephaly C syndrome (OTCS), and isolated retinitis pigmentosa (RP) displaying the complexity of ciliopathies (Cole and Snell, 2009; Schmidts et al., 2013; Bifari et al., 2016; Schaefer et al., 2016; Pena-Padilla et al., 2017). Here, we present a clinical, molecular and functional study of two unrelated patients with identical compound heterozygous variants in IFT140 and show that the observed clinical differences were supported by distinctive cilium phenotypes of the two patients. Moreover, we suggest an alternative description of MZSDS to better resemble the variability seen between patients within this cohort, i.e. “IFT140-related ciliopathy with MZSDS- or CED-like features”.
2 Materials and Methods
2.1 Ethical Considerations
The study was conducted to the ethical tenets of the Declaration of Helsinki and in agreement with the “Code of Conduct for responsible use of patient-derived material”. This study was approved by the Bioethics Committee at Poznan University of Medical Sciences in compliance with The Good Clinical Practice (GCP) and Polish law. Written informed consent was obtained from the patients and their parents.
2.2 Collection of Samples
Genomic DNA from both patients and their parents was extracted from whole blood and used for exome sequencing (ES) using a standard method. Skin-derived fibroblasts from both patients were obtained and used for immunofluorescent imaging.
2.3 Exome Sequencing
Genomic DNA from the affected patients and the unaffected parents of patient 2 were used for ES . Prior to sequencing, samples were prepared with the Twist library preparation kit (TWIST Bioscience, San Francisco, CA, United States) followed by sequencing on an Illumina Novaseq sequencer (Illumina, San Diego, CA, United States). Reads were aligned to the human genome assembly GRCh37(HG19) using the Burrows-Wheeler aligner version 0.7.13. Variant calling was performed for SNV with GATK HaplotypeCaller version 3.4–46, and for CNV with ExomeDepth version 1.1.12.
2.4 IFT140 Transcript Analysis
RNA was isolated from skin-derived cultured fibroblasts using a standard method. Samples were prepared using the Illumina Stranded mRNA Prep kit according to the manufacturer’s instructions. A total of 18 samples were pooled, equimolar, and sequenced on a SP flowcell using the Illumina NovaSeq6000 (Illumina, San Diego, CA, United States). Demultiplexing and sample analysis with DRAGEN RNA Pipeline 3.7.5 was performed on the Illumina BaseSpace platform.
2.5 Cilium Phenotyping Using the Automated ALPACA Tool
The cilium phenotype for both patients was determined using a standardized immunofluorescent assay in skin-derived fibroblasts as described by Doornbos et al. (Doornbos et al., 2021). In brief, the fibroblasts were cultured under standard cell culture conditions on glass coverslips. Cilia formation was stimulated by replacement of the culture medium containing 20% fetal calf serum (FCS) to medium containing 0.2% FCS for 48 h prior to staining. Upon fixation, permeabilization and blocking, the cells were stained with antibodies to visualize different cilium parameters, i.e. ciliogenesis, cilium length, and retrograde IFT. The ciliogenesis parameter represents the percentage of ciliated cells and is visualized by acetylated-α-tubulin + ARL13B + PCNT. The cilium length was measured using the ALPACA tool, described in detail by Doornbos et al., based on the length of the combined acetylated-α-tubulin and ARL13B signal. The ALPACA tool was also used to measure the surface area of IFT88 and an increased surface area of IFT88 indicates defective retrograde transport. The cilium length and the IFT88 measurement data were combined and plotted into a previously generated reference graph containing data from six control fibroblast lines and ten skeletal ciliopathy patient fibroblast lines (Doornbos et al., 2021).
3 Clinical Report
Detailed clinical characteristics of two unrelated male patients, showing clinical overlap but diagnosed with a different ciliopathy based on differences in the clinical phenotype, presented in Table 1 and Figures 1A–J.
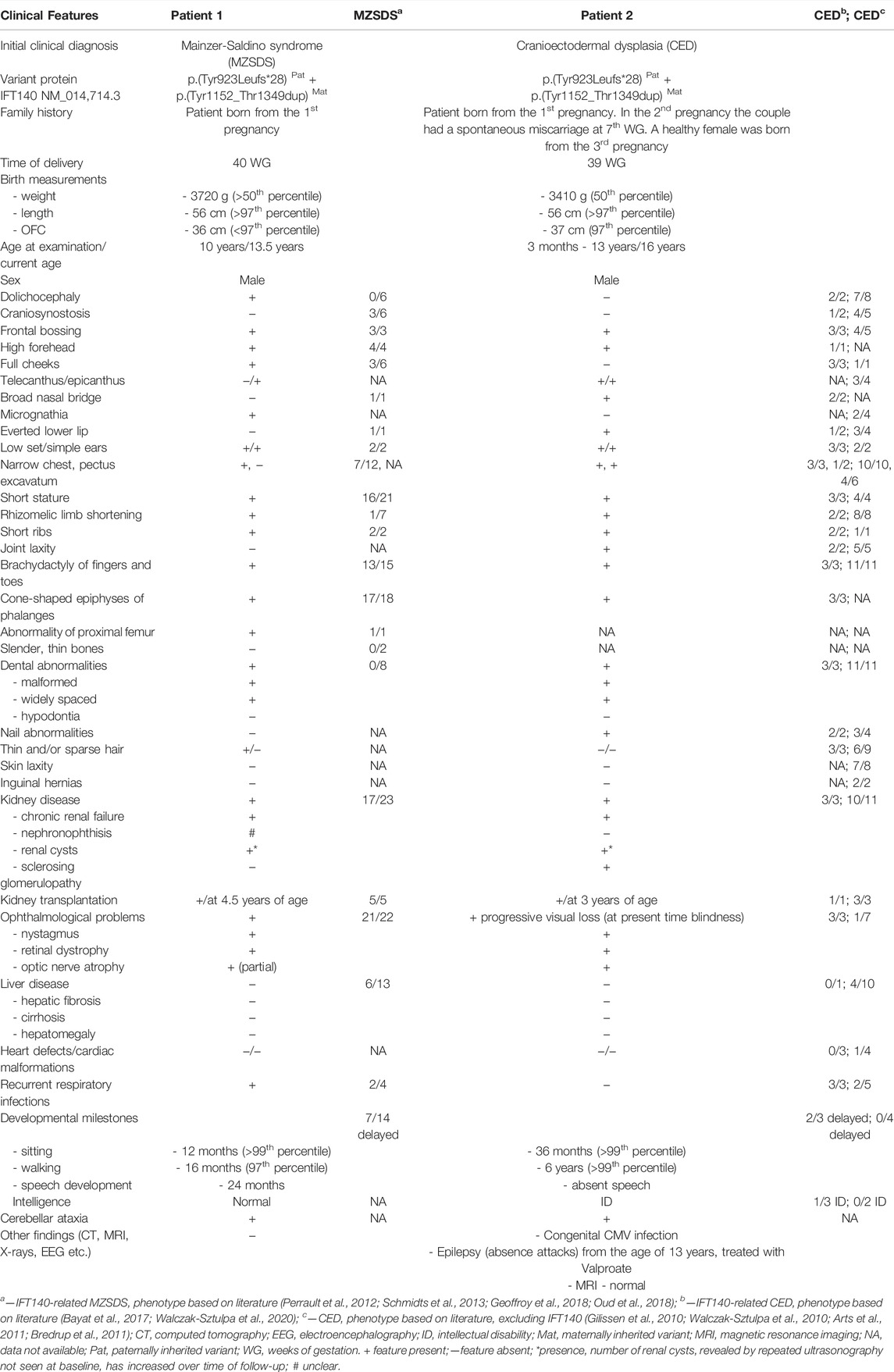
TABLE 1. Clinical features of patients with compound heterozygous variants in IFT140 described in this study and in literature.
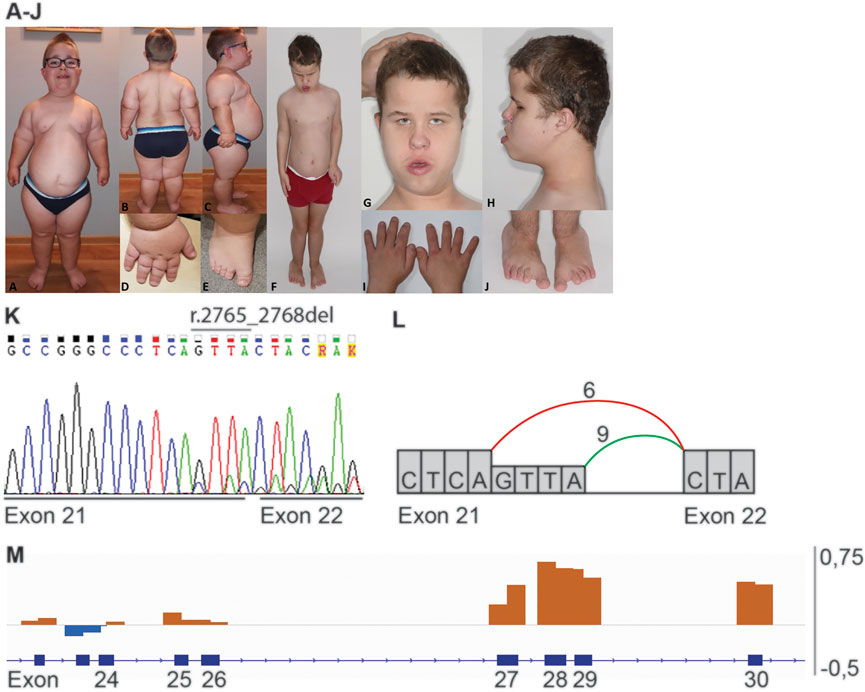
FIGURE 1. Phenotype and genotype of patients 1 and 2. Proximal limb shortening, narrow thorax, frontal bossing, high forehead, low set and simple ears is present in both patients (A,F,G,H). (A–E) Clinical features of patient 1: dolichocephalic head shape, full cheeks, micrognathia (A) obesity and hyperlordosis (B,C), brachydactyly and sandal gap (D,E). (F–J) Clinical features of patient 2: pectus excavatum (F), epicanthal folds and telecanthus, broad nasal bridge, everted lower lip (G), brachydactyly and sandal gap (I,J). (K) Sanger sequence of heterozygous variant r.2765_2768del in IFT140 (NM_014714.3: c.2767_2768+2del, p.(Tyr923Leufs*28)), representative for patient 1 and 2. (L) Schematic representation of the splicing effect caused by IFT140 r.2765_2768del. The coloured lines indicate the splicing between exons, in green the wildtype splicing between exon 21 and 22 and in red the aberrant splicing seen in patients 1 and 2. (M) Schematic overview of the heterozygous exon 27–30 duplication (p. (Tyr1152_Thr1394dup)) detected in patients 1 and 2. Orange bars indicate an increase in coverage and blue bars indicate a decrease in coverage.
In summary, patient 1, a 10-year old boy was referred to a clinical geneticist due to severe retinal degradation, facial dysmorphisms, bone dysplasia, skeletal abnormalities and chronic renal failure (after kidney transplantation at 4.5 years of age). Patient 2, a 16-year old boy, presented with facial dysmorphisms, global developmental delay, epilepsy (from 14 years old), abnormal body proportions, skeletal abnormalities, ESRD and congenital CMV infection. The skeletal abnormalities in both patients include narrowing of the chest, short stature, rhizomelic limb shortening, brachydactyly, and cone-shaped epiphyses of phalanges. Besides the significant clinical overlap between patients 1 and 2 there are also typical phenotypic differences, including the presence of dolichocephaly, micrognathia and thin hair in patient 1 but not in patient 2, and, the presence of a broad nasal bridge, everted lower lip, nail abnormalities and joint laxity only in patient 2. In addition, there were striking differences in the development of both patients. Patient 1 showed normal psychomotor development and intelligence, while patient 2 showed developmental delay with intellectual disability and epilepsy. Even though both patients developed early-onset end-stage renal disease (ESRD) the development of the disease was different. Patient 1 developed ESRD at the age of 3 years, while patient 2 was diagnosed with rapid end-stage renal failure due to diffuse glomerulosclerosis and fibrosis at the age of 9 months. Moreover, patient 2 presented with a bilateral vesicoureteral reflux (VUR) grade II.
4 Results
4.1 ES Revealed Identical Causative Variants in IFT140
A targeted analysis was performed selecting exonic and intronic position -20 to +8 variants in 170 ciliopathy associated genes (Radboudumc ciliopathy gene panel version DG3.00 (Radboudumc)) with a frequency <1% in dbSNP151, <5% in in-house db (containing data from >22.000 exomes), and <1% in gnomAD (version 2.1.1). This resulted in 16 variants for patient 1 and 19 variants for patient 2 (Supplementary Tables S1 and S2). CNV analysis was performed by selecting for segments containing any of the 170 ciliopathy associated genes and a frequency <1% in the in-house db. This resulted in two duplications for patient 1 and one duplication for patient 2 (Supplementary Table S3). Subsequently, the combined variants from SNV and CNV were filtered for a recessive inheritance model and a matching phenotype, resulting in a single gene, IFT140, in both patients. Both probands carry the same variants in IFT140 Chr16 (GRCh37):g.1575886_1575889del; NM_014,714.4: r.2765_2768del; p.(Tyr923Leufs*28) and exon 27–30 duplication; p.(Tyr1152_Thr1349dup) (Figure 1K-M). Segregation analysis in both families fit with a recessive inheritance pattern. The deletion variant in IFT140 r.2765_2768 has previously been reported as a likely pathogenic variant in ClinVar (VCV000863072.3) in a patient with retinal dystrophy, as well as, a patient with MZSDS. The duplication variant in IFT140 NC_000016.9:g.1568118(NM_014714.4):c (4182 + 99)_(3558)dup is absent from ClinVar, but has been detected in two Polish skeletal ciliopathy patients and in eight families reported by Geoffroy (Geoffroy et al., 2018; Walczak-Sztulpa et al., 2020). These include six families (seven patients) diagnosed with MZSDS, one patient with JATD and one affected individual with features of CED.
A larger duplication, including the variant detected in patients 1 and 2, has been reported once in a patient with a skeletal ciliopathy (VCV000523177.1). In addition to the variants in IFT140, a heterozygous variant c.1354G > A; p.Ala452Thr in INTU (NM_015693.3) was detected in ES data from patient 2. The minor allele frequency of INTU variant c.1354G > A is 0.246% in GnomAD v2.1. 1 (accessed on 19 May 2022). The detected variant was previously described as likely pathogenic in a patient with nephronophthisis and growth retardation by Toriyama et al. (Toriyama et al., 2016). Furthermore, the molecular data from patient 2 was analyzed for rare variants (same filters applied as described above) in genes associated with intellectual disability (ID) (Radboudumc ID gene panel version DG3.00 (Radboudumc, 2021)) and analyzed for ID-associated CNVs. The resulting variant list was filtered for both autosomal recessive and autosomal dominant inheritance models and subsequently checked for phenotypic overlap. This approach did not lead to candidate variants that could explain the ID phenotype seen in patient 2.
4.2 Aberrant Splicing of IFT140 Transcript
RNA isolated from skin-derived fibroblasts from patients 1 and 2 was used for IFT140 transcript analysis. Compared to the reference fibroblast sample both patient samples showed aberrant splicing at the splice donor site of exon 21 where ES revealed a 4bp deletion (Figure 1L). Approximately half of the reads originated from position r.2764 indicating a 4bp deletion r.2765_2768del and would result in a frameshift.
4.3 Different Cilium Phenotype Between Patients 1 and 2
Skin-derived fibroblasts from patients 1 and 2 were used to determine the cilium phenotype based on three parameters; ciliogenesis, cilium length, and retrograde IFT (Supplementary Figure S1). The results were compared to the “healthy cilium phenotype” and that of two distinct skeletal ciliopathy cohorts, ATD and CED, described by Doornbos et al. (Table 2) (Doornbos et al., 2021). Fibroblasts from both patients showed normal ciliogenesis >90% and a significantly increased IFT88 measurement of 0.99 ± 0.08µm2 for patient 1 and 0.79 ± 0.08µm2 for patient 2. While the cilium length for patient 1 was, although slightly increased, within normal range (3.71 ± 0.07 µm), the cilia of patient 2 were significantly shorter (3.04 ± 0.15 µm) (Figure 2). Based on these cilium length and IFT88 measurements, the cilium phenotype of patient 2 resembles that seen in CED patients whereas, the results of patient 1 are clearly abnormal, the cilium phenotype does not cluster with ATD nor CED.
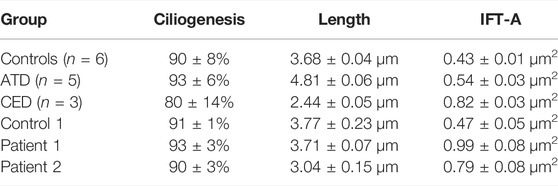
TABLE 2. Cilium phenotypes. The cilium phenotypes of three clusters; control, Jeune asphyxiating thoracic dysplasia and cranioectodermal dysplasia published by Doornbos et al. (Doornbos et al., 2021). Followed by the measurements from this study, a control line, patient 1 and patient 2. The cilium phenotypes are represented by the ciliogenesis, cilium length and IFT-A (the IFT88 measurement along the ciliary axoneme).
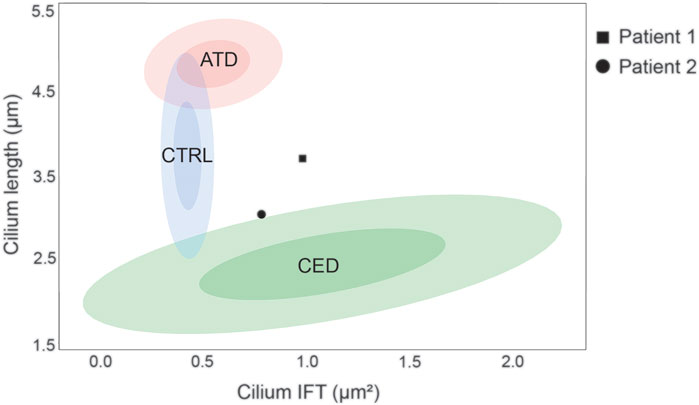
FIGURE 2. Ciliopathy cilium phenotype clusters. The cilium phenotype clusters are based on two cilium parameters; cilium length (Y-axis) and IFT88 measurement (X-axis) published in Doornbos et al. (Doornbos et al., 2021). The confidence intervals (CI) of 0.5 and 0.9 are indicated per identifiable group, i.e. the control, ATD, and CED cohorts. The cilium phenotype of patient 1 showed a normal cilium length (3.71 ± 0.07 µm) and an increased IFT88 measurement (0.99 ± 0.08µm2), therefore it does not fit in any cluster. Patient 2 showed a decreased cilium length (3.04 ± 0.15 µm) and an increased IFT88 measurement (0.79 ± 0.08µm2), therefore it is positioned on the border of the CED cluster.
5 Discussion
The two patients described in this report share identical compound heterozygous variants in IFT140. Despite the large phenotypic overlap, the observed clinical differences between patients 1 and 2 led to the suspicion of two different diagnoses. Functional experiments were requested to further investigate the clinical variability, and indeed, the different cilium phenotypes of each patient emphasized the observed clinical variability. The cilium phenotyping data placed patient 2 (decreased cilium length and increased IFT88 measurement) on the border of the established CED cluster, whereas patient 1 (normal cilium length and increased IFT88 measurement) did not fit in any previously defined cluster. Additional experiments are required to further elucidate the exact underlying mechanism that caused this difference in cilium phenotype.
One explanation for the cilium phenotypic difference could be the effect of the likely pathogenic variant found in INTU in the ES of patient 2, which was not present in patient 1. Interestingly, INTU has been described to interact with the IFT-A protein complex (Toriyama et al., 2016). Protein interaction studies showed that the ciliogenesis and planar polarity effector (CPLANE) complex proteins, consisting of INTU, FUZ, and WDPCP, interact with all six components of the retrograde IFT complex. The absence of CPLANE inhibits peripheral IFT proteins (IFT43, WDR35, and TTC21B) to localize to the basal body of the cilium and therefore do not assemble onto the IFT core proteins (IFT122, IFT140, WDR19). These data show that there is a connection between INTU and IFT-A components. Therefore, we can speculate that the detected INTU variant adds to the burden of the IFT140 variants on proper functioning of the IFT-A complex. Recent literature has shown that mutations in INTU are causative for a short-rib polydactyly syndrome phenotype (Toriyama et al., 2016; Bruel et al., 2018). The INTU variant c.1354G > A that was detected in patient 2 has previously been described to be causative in a 13-year old female with nephronophthisis, ESRD at 10 years of age and growth retardation. The authors suggest that the variant may be hypomorphic since the affected residue is poorly conserved and the patient has a milder phenotype compared to other INTU patients. It is possible that the likely pathogenic INTU variant contributes to the more severe phenotype seen in patient 2 presented in this study, however, further functional studies are required to provide more evidence.
The heterogeneity of ciliopathies is well known. With over 187 associated genes and a pleiotropy of ciliopathy genes for which one gene can cause different phenotypes (Reiter and Leroux, 2017). One example is WDR19 which is associated with JATD, CED, NPHP, RP, and Senior Løken syndrome (Bredrup et al., 2011; Coussa et al., 2013). In this report, we present two patients with identical compound heterozygous variants and a variable clinical phenotype. A similar case to what we found with the INTU variant, was presented by Maglic et al. who described two families with variants in TMEM231 (Maglic et al., 2016). In these two families, of whom the fathers are identical twins, both had children affected with a ciliopathy caused by compound heterozygous variants in TMEM231. The mothers carried two different missense variants in TMEM231 and the fathers carried the same variant. Family 1 had four children with Joubert syndrome (MIM#614970) of which three of the children had an identical phenotype whereas the fourth child was more severely affected. They suggest that an additional variant in the ciliopathy gene BBS10, only present in the fourth child, could explain the more severe phenotype. Moreover, the child in the second family presented with Meckel-Gruber syndrome (MIM#615397). Although the compound heterozygous variants between the two families are not identical, it is intriguing to find two different diagnoses in these two closely related families.
We cannot exclude that the developmental delay, ID and epilepsy are a result of the congenital CMV infection that patient 2 had. Recent literature showed that younger children (age 0–2) have an increased risk of developing epilepsy upon experiencing a congenital CMV infection (Lin et al., 2021).
While JATD and CED have cardinal features distinguishing one from the other, this is less clear for MZSDS as we showed in this study. Both presented patients, as well as, published patients with causative variants in IFT140 display a variable phenotype sharing features with both JATD and CED. This may indicate that the classic description of the skeletal ciliopathy cluster, CED, MZSDS and JATD, does not adequately cover the observed variability within each cohort. Instead “IFT140-related ciliopathy with MZSDS- and/or CED-like features” could be considered to better represent the phenotype of the patient. In our study, the CED-like features such as the ectodermal- and craniofacial dysmorphisms were more prominent in patient 2 compared to patient 1, and together with the suggestive cilium phenotype results we would consider two different diagnoses for each patient. Patient 1 was diagnosed with “IFT140-related ciliopathy with MZSDS-like features” and patient 2 with “IFT140-related ciliopathy with CED-like features”. The intra- and interfamilial clinical variability in patients with identical casual variants has been reported in ciliopathy patients. It is of note that the presence of modifying alleles, environmental factors, and other mechanisms such as epigenetics may play a role in the phenotypic variability. However, it is difficult to precisely define the impact of these factors on the clinical manifestation (Mitchison and Valente, 2017; Walczak-Sztulpa et al., 2021).
The definition of a diagnosis is not set in stone and calls for careful consideration of different facets of the disease. The initial clinical examination, often the guideline for ES analysis, will determine the course of the care path followed by the patient. In our experience the clinical symptoms are leading in defining the differential diagnosis for a patient, but we strongly believe that the addition and integration of molecular genetics data and functional work to the clinical phenotype is crucial to come to an accurate diagnosis for each patient. We showed that the observed clinical differences between two skeletal ciliopathy patients carrying identical causative variants were supported by different cilium phenotypes. Moreover, we suggest to describe the presented phenotypes as “IFT140-related ciliopathies with MZSDS- or CED-like features” to better represent a variable disease cohort. We advocate the necessity of combining clinical, molecular, and functional data to accurately diagnose ciliopathy patients.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Bioethics Committee at Poznan University of Medical Sciences, Poznan, Poland. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
MO, JW-S, and AW designed the experiments. RvB, CD, and MS performed the functional experiments. EO, AS-S, AJ, and EB-O contributed to the molecular testing. EO, MRK, AL-B, MS, EB, and RG performed clinical examinations. JW-S, MO, and AW wrote the manuscript. All authors read and approved the manuscript.
Funding
MS acknowledges funding from the European Research Council (ERC) (ERC starting grant No. 716344), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation): Project-ID 431984000–SFB 1453 and under Germany’s Excellence Strategy (CIBSS—EXC-2189—project ID 390939984).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the patients, parents, and family members for their participation in this study. We thank R. Derks for practical work and L.E.L.M Vissers and R. Roepman for their support of this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.931822/full#supplementary-material
References
Arts, H. H., Bongers, E. M. H. F., Mans, D. A., van Beersum, S. E. C., Oud, M. M., Bolat, E., et al. (2011). C14ORF179 Encoding IFT43 Is Mutated in Sensenbrenner Syndrome. J. Med. Genet. 48 (6), 390–395. doi:10.1136/jmg.2011.088864
Bayat, A., Kerr, B., Douzgou, S., and Study, D. D. D. (2017). The Evolving Craniofacial Phenotype of a Patient with Sensenbrenner Syndrome Caused by IFT140 Compound Heterozygous Mutations. Clin. Dysmorphol. 26 (4), 247–251. doi:10.1097/MCD.0000000000000169
Bifari, I. N., Elkhamary, S. M., Bolz, H. J., and Khan, A. O. (2016). The Ophthalmic Phenotype ofIFT140-Related Ciliopathy Ranges from Isolated to Syndromic Congenital Retinal Dystrophy. Br. J. Ophthalmol. 100 (6), 829–833. doi:10.1136/bjophthalmol-2015-307555
Bredrup, C., Saunier, S., Oud, M. M., Fiskerstrand, T., Hoischen, A., Brackman, D., et al. (2011). Ciliopathies with Skeletal Anomalies and Renal Insufficiency Due to Mutations in the IFT-A Gene WDR19. Am. J. Hum. Genet. 89 (5), 634–643. doi:10.1016/j.ajhg.2011.10.001
Bruel, A.-L., Levy, J., Elenga, N., Defo, A., Favre, A., Lucron, H., et al. (2018). INTU -related Oral-Facial-Digital Syndrome Type VI: A Confirmatory Report. Clin. Genet. 93 (6), 1205–1209. doi:10.1111/cge.13238
Cole, D. G., and Snell, W. J. (2009). SnapShot: Intraflagellar Transport. Cell 137 (4), 784. doi:10.1016/j.cell.2009.04.053
Coussa, R. G., Otto, E. A., Gee, H. Y., Arthurs, P., Ren, H., Lopez, I., et al. (2013). WDR19 : An Ancient, Retrograde, Intraflagellar Ciliary Protein Is Mutated in Autosomal Recessive Retinitis Pigmentosa and in Senior‐Loken Syndrome. Clin. Genet. 84 (2), 150–159. doi:10.1111/cge.12196
Doornbos, C., van Beek, R., Bongers, E. M. H. F., Lugtenberg, D., Klaren, P. H. M., Vissers, L. E. L. M., et al. (2021). Cell-based Assay for Ciliopathy Patients to Improve Accurate Diagnosis Using ALPACA. Eur. J. Hum. Genet. 29 (11), 1677–1689. doi:10.1038/s41431-021-00907-9
Geoffroy, V., Stoetzel, C., Scheidecker, S., Schaefer, E., Perrault, I., Bär, S., et al. (2018). Whole-genome Sequencing in Patients with Ciliopathies Uncovers a Novel Recurrent Tandem Duplication in IFT140. Hum. Mutat. 39 (7), 983–992. doi:10.1002/humu.23539
Gilissen, C., Arts, H. H., Hoischen, A., Spruijt, L., Mans, D. A., Arts, P., et al. (2010). Exome Sequencing Identifies WDR35 Variants Involved in Sensenbrenner Syndrome. Am. J. Hum. Genet. 87 (3), 418–423. doi:10.1016/j.ajhg.2010.08.004
Lin, C.-H., Chou, I.-C., Lee, I.-C., and Hong, S.-Y. (2021). Cytomegalovirus Infection in Infancy May Increase the Risk of Subsequent Epilepsy and Autism Spectrum Disorder in Childhood. Children 8 (11), 1040. doi:10.3390/children8111040
Ma, M. (2021). Cilia and Polycystic Kidney Disease. Seminars Cell & Dev. Biol. 110, 139–148. doi:10.1016/j.semcdb.2020.05.003
Maglic, D., Stephen, J., Malicdan, M. C. V., Guo, J., Fischer, R., Konzman, D., et al. (2016). TMEM231Gene Conversion Associated with Joubert and Meckel-Gruber Syndromes in the Same Family. Hum. Mutat. 37 (11), 1144–1148. doi:10.1002/humu.23054
Miller, K. A., Ah-Cann, C. J., Welfare, M. F., Tan, T. Y., Pope, K., Caruana, G., et al. (2013). Cauli: A Mouse Strain with an Ift140 Mutation that Results in a Skeletal Ciliopathy Modelling Jeune Syndrome. PLoS Genet. 9 (8), e1003746. doi:10.1371/journal.pgen.1003746
Mitchison, H. M., and Valente, E. M. (2017). Motile and Non-motile Cilia in Human Pathology: from Function to Phenotypes. J. Pathol. 241 (2), 294–309. doi:10.1002/path.4843
Oud, M. M., Latour, B. L., Bakey, Z., Letteboer, S. J., Lugtenberg, D., Wu, K. M., et al. (2018). Cellular Ciliary Phenotyping Indicates Pathogenicity of Novel Variants in IFT140 and Confirms a Mainzer-Saldino Syndrome Diagnosis. Cilia 7, 1. doi:10.1186/s13630-018-0055-2
Peña-Padilla, C., Marshall, C. R., Walker, S., Scherer, S. W., Tavares-Macías, G., Razo-Jiménez, G., et al. (2017). Compound Heterozygous Mutations in theIFT140gene Cause Opitz Trigonocephaly C Syndrome in a Patient with Typical Features of a Ciliopathy. Clin. Genet. 91 (4), 640–646. doi:10.1111/cge.12924
Perrault, I., Saunier, S., Hanein, S., Filhol, E., Bizet, A. A., Collins, F., et al. (2012). Mainzer-Saldino Syndrome Is a Ciliopathy Caused by IFT140 Mutations. Am. J. Hum. Genet. 90 (5), 864–870. doi:10.1016/j.ajhg.2012.03.006
Piperno, G., Siuda, E., Henderson, S., Segil, M., Vaananen, H., and Sassaroli, M. (1998). Distinct Mutants of Retrograde Intraflagellar Transport (IFT) Share Similar Morphological and Molecular Defects. J. Cell Biol. 143 (6), 1591–1601. doi:10.1083/jcb.143.6.1591
Radboudumc. 2021. Radboudumc Gene Panels. [Online]. Available: https://www.radboudumc.nl/en/afdelingen/genetica/about-us/genomediagnostics/services/gene-panels [Accessed 2021].
Reiter, J. F., and Leroux, M. R. (2017). Genes and Molecular Pathways Underpinning Ciliopathies. Nat. Rev. Mol. Cell Biol. 18 (9), 533–547. doi:10.1038/nrm.2017.60
Schaefer, E., Stoetzel, C., Scheidecker, S., Geoffroy, V., Prasad, M. K., Redin, C., et al. (2016). Identification of a Novel Mutation Confirms the Implication of IFT172 (BBS20) in Bardet-Biedl Syndrome. J. Hum. Genet. 61 (5), 447–450. doi:10.1038/jhg.2015.162
Schmidts, M., Frank, V., Eisenberger, T., Al Turki, S., Bizet, A. A., Antony, D., et al. (2013). Combined NGS Approaches Identify Mutations in the Intraflagellar Transport Gene IFT140 in Skeletal Ciliopathies with Early Progressive Kidney Disease. Hum. Mutat. 34 (5), 714–724. doi:10.1002/humu.22294
Toriyama, M., Lee, C., Lee, C., Taylor, S. P., Duran, I., Cohn, D. H., et al. (2016). The Ciliopathy-Associated CPLANE Proteins Direct Basal Body Recruitment of Intraflagellar Transport Machinery. Nat. Genet. 48 (6), 648–656. doi:10.1038/ng.3558
Tsang, S. H., and Sharma, T. (2018). X-Linked Retinitis Pigmentosa. Adv. Exp. Med. Biol. 1085, 31–35. doi:10.1007/978-3-319-95046-4_8
Walczak-Sztulpa, J., Eggenschwiler, J., Osborn, D., Brown, D. A., Emma, F., Klingenberg, C., et al. (2010). Cranioectodermal Dysplasia, Sensenbrenner Syndrome, Is a Ciliopathy Caused by Mutations in the IFT122 Gene. Am. J. Hum. Genet. 86 (6), 949–956. doi:10.1016/j.ajhg.2010.04.012
Walczak-Sztulpa, J., Posmyk, R., Bukowska-Olech, E. M., Wawrocka, A., Jamsheer, A., Oud, M. M., et al. (2020). Compound Heterozygous IFT140 Variants in Two Polish Families with Sensenbrenner Syndrome and Early Onset End-Stage Renal Disease. Orphanet J. Rare Dis. 15 (1), 36. doi:10.1186/s13023-020-1303-2
Walczak‐Sztulpa, J., Wawrocka, A., Stańczyk, M., Pesz, K., Dudarewicz, L., Chrul, S., et al. (2021). Interfamilial Clinical Variability in Four Polish Families with Cranioectodermal Dysplasia and Identical Compound Heterozygous Variants in WDR35. Am. J. Med. Genet. 185 (4), 1195–1203. doi:10.1002/ajmg.a.62067
Keywords: IFT140, skeletal ciliopathy, cilium phenotype, MZSDS-like features, CED-like features
Citation: Walczak-Sztulpa J, Wawrocka A, Doornbos C, van Beek R, Sowińska-Seidler A, Jamsheer A, Bukowska-Olech E, Latos-Bieleńska A, Grenda R, Bongers EMHF, Schmidts M, Obersztyn E, Krawczyński MR and Oud MM (2022) Identical IFT140 Variants Cause Variable Skeletal Ciliopathy Phenotypes—Challenges for the Accurate Diagnosis. Front. Genet. 13:931822. doi: 10.3389/fgene.2022.931822
Received: 29 April 2022; Accepted: 15 June 2022;
Published: 07 July 2022.
Edited by:
Steven LC Pei, Yale University, United StatesReviewed by:
Joshi Stephen, Baylor College of Medicine, United StatesAlanna Strong, Children’s Hospital of Philadelphia, United States
Peter Harris, Mayo Clinic, United States
Copyright © 2022 Walczak-Sztulpa, Wawrocka, Doornbos, van Beek, Sowińska-Seidler, Jamsheer, Bukowska-Olech, Latos-Bieleńska, Grenda, Bongers, Schmidts, Obersztyn, Krawczyński and Oud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Machteld M. Oud, Machteld.Oud@radboudumc.nl
†These authors have contributed equally and share first authorship
‡These authors have contributed equally and share last authorship