 Dinu Antony1,2Elif Gulec Yilmaz3†Alper Gezdirici3†Lennart Slagter1Zeineb Bakey1,2
Dinu Antony1,2Elif Gulec Yilmaz3†Alper Gezdirici3†Lennart Slagter1Zeineb Bakey1,2 Helen Bornaun4Ibrahim Cansaran Tanidir5
Helen Bornaun4Ibrahim Cansaran Tanidir5 Tran Van Dinh6Han G. Brunner1,7
Tran Van Dinh6Han G. Brunner1,7 Peter Walentek8,9Sebastian J. Arnold9,10
Peter Walentek8,9Sebastian J. Arnold9,10 Rolf Backofen6,9
Rolf Backofen6,9 Miriam Schmidts1,2,9*
Miriam Schmidts1,2,9*- 1Genome Research Division, Human Genetics Department, Radboud University Medical Center and Radboud Institute for Molecular Life Sciences, Nijmegen, Netherlands
- 2Center for Pediatrics and Adolescent Medicine, University Hospital Freiburg, Faculty of Medicine, Freiburg, Germany
- 3Department of Medical Genetics, University of Health Sciences, Istanbul Kanuni Sultan Suleyman Training and Research Hospital, Istanbul, Turkey
- 4Department of Pediatric Cardiology, University of Health Sciences, Istanbul Kanuni Sultan Suleyman Training and Research Hospital, Istanbul, Turkey
- 5Department of Pediatric Cardiology, Basaksehir Cam and Sakura City Hospital, Istanbul, Turkey
- 6Bioinformatics Group, Department of Computer Science, University of Freiburg, Freiburg, Germany
- 7Maastricht University Medical Center and GROW School of Oncology and Development, Maastricht University, Maastricht, Netherlands
- 8Renal Division, Department of Medicine, University Hospital Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 9CIBSS- Centre for Integrative Biological Signalling Studies, University of Freiburg, Freiburg, Germany
- 10Institute of Experimental and Clinical Pharmacology and Toxicology, Faculty of Medicine, University of Freiburg, Freiburg, Germany
Laterality defects are defined by the perturbed left–right arrangement of organs in the body, occurring in a syndromal or isolated fashion. In humans, primary ciliary dyskinesia (PCD) is a frequent underlying condition of defective left–right patterning, where ciliary motility defects also result in reduced airway clearance, frequent respiratory infections, and infertility. Non-motile cilia dysfunction and dysfunction of non-ciliary genes can also result in disturbances of the left–right body axis. Despite long-lasting genetic research, identification of gene mutations responsible for left–right patterning has remained surprisingly low. Here, we used whole-exome sequencing with Copy Number Variation (CNV) analysis to delineate the underlying molecular cause in 35 mainly consanguineous families with laterality defects. We identified causative gene variants in 14 families with a majority of mutations detected in genes previously associated with PCD, including two small homozygous CNVs. None of the patients were previously clinically diagnosed with PCD, underlining the importance of genetic diagnostics for PCD diagnosis and adequate clinical management. Identified variants in non-PCD-associated genes included variants in PKD1L1 and PIFO, suggesting that dysfunction of these genes results in laterality defects in humans. Furthermore, we detected candidate variants in GJA1 and ACVR2B possibly associated with situs inversus. The low mutation detection rate of this study, in line with other previously published studies, points toward the possibility of non-coding genetic variants, putative genetic mosaicism, epigenetic, or environmental effects promoting laterality defects.
Introduction
Laterality defects in humans occur with a frequency of about 1:10,000 (Lin et al., 2014). Situs inversus totalis presents with a complete mirror image of normal organ asymmetry throughout the body. In contrast, in situs inversus thoracalis or abdominalis (heterotaxy), only a subset of organs is affected. While situs inversus usually does not impair organ function, this is different in left or right isomerism or situs ambiguous, accounting for up to 5% of all laterality defect cases. Heterotaxy is commonly associated with complex heart defects such as transposition of the great arteries can significantly affect the quality of life and life expectancy (Lin et al., 2014). Furthermore, polysplenia can be observed with left isomerism, while right isomerism is associated with asplenia and increased risk for serious infections (Fliegauf et al., 2007) (Figure 1)
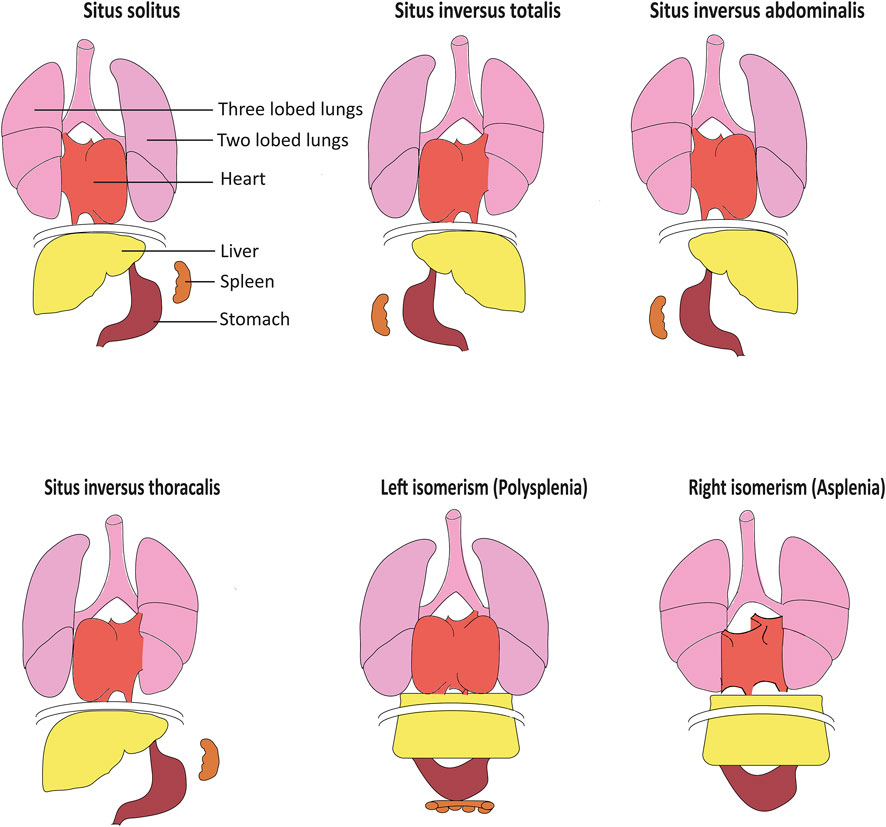
FIGURE 1. Organ arrangement patterns in humans. While situs solitus represents the most frequent left–right organ arrangement, situs inversus represents the corresponding mirror image. In situs inversus thoracalis and situs inversus abdominalis, left–right patterning is inversed solely above or below the diaphragm. Left isomerism often results in polysplenia while accordingly, right isomerism can cause asplenia. Isomerism often also causes severe structural heart defects including defective arrangement of the great blood vessels, adapted from Fliegauf et al. (2007).
Left–right body asymmetry in vertebrates is determined very early in embryonic development, such as embryonic day 7.5 in mice (Marszalek et al., 1999) and 6–12 somite stage in zebrafish (Kawakami et al., 2005). In mammals, the so-called embryonic node is a transiently present structure which exhibits motile 9 + 0 monocilia and non-motile 9 + 0 monocilia (Figures 2A,B). Both types of cilia are considered essential for left–right patterning as functional defects of both motile and non-motile cilia can result in laterality defects in vertebrates (Pennekamp et al., 2015). Dysfunction of genes encoding for proteins essential for the ciliary motility apparatus such as dynein arm components results in left–right patterning defects in humans (Leigh et al., 2019) and mice (Tan et al., 2007), most often combined with recurrent respiratory tract infections due to impaired mucociliary clearance and often infertility (Fliegauf et al., 2007). This combination of laterality defects, bronchiectasis, and sinusitis is also referred to as Kartagener syndrome, observed in approximately 50% of all primary ciliary dyskinesia (PCD) cases (Knowles et al., 2016) (Supplementary Table S1). Of note, mutations in genes encoding for proteins associated with the central pair microtubules and radial spokes result in PCD but do not cause situs abnormalities due to the 9 + 0 ciliary ultrastructure in the node compared to a 9 + 2 ultrastructure of motile airway cilia (Knowles et al., 2016). This includes mutations in RSPH4A, HYDIN, MCIDAS, RSPH9, RPGR, CCDC65, RSPH1, CCDC164, and STK36 (Edelbusch et al., 2017). Further, mutations in CCNO (Wallmeier et al., 2014) and MCIDAS (Boon et al., 2014) cause mucociliary clearance disorders without laterality disturbances. Likewise, mutations in genes encoding for ciliary proteins unrelated to motility in humans and mice can also result in a disturbed left–right body axis, including PKD2 (Pennekamp et al., 2002; Bataille et al., 2011), INVERSIN (Simons et al., 2005), NPHP3 (Olbrich et al., 2003), and PKD1L1 (Field et al., 2011; Vetrini et al., 2016). These mutations are usually combined with additional developmental defects such as renal-hepatic dysplasia, polycystic kidney disease, nephronophthisis, chondrodysplasias, brain malformation, or retinal degeneration (Fliegauf et al., 2007; Deng et al., 2015) (Supplementary Table S1).
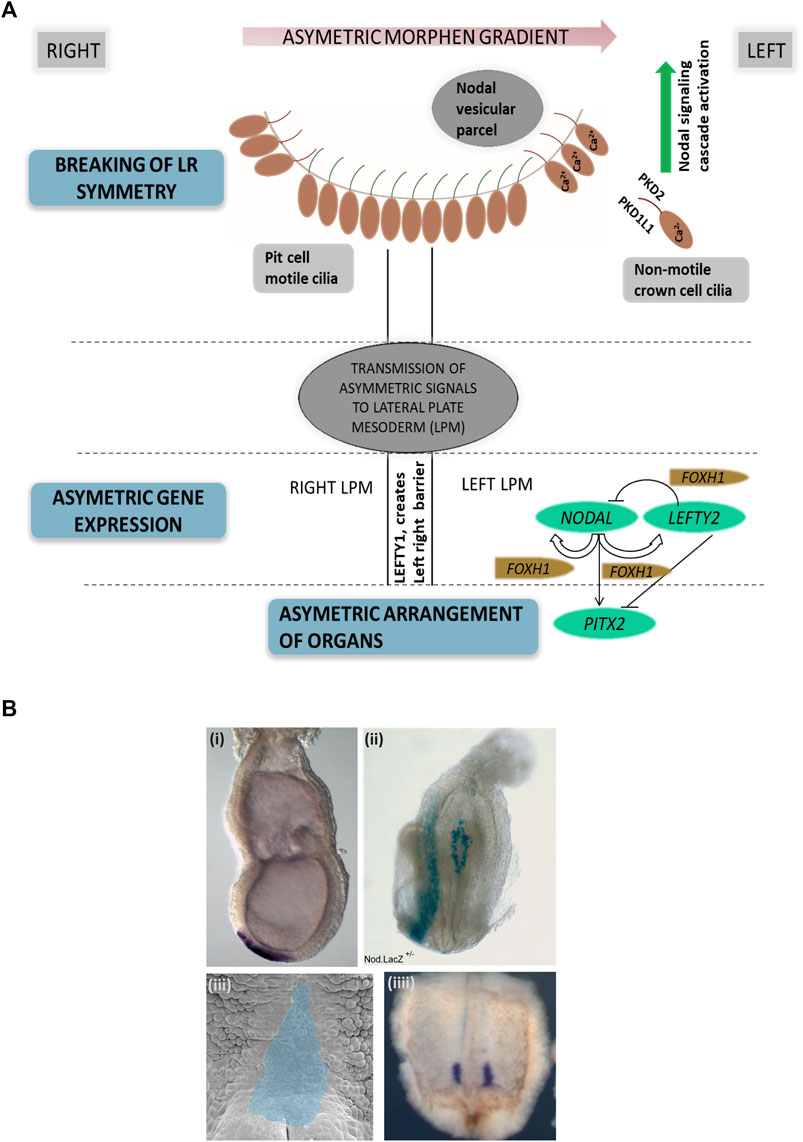
FIGURE 2. Left–right asymmetry in mammals. (A) Simplified representation of left–right patterning initiation in mammals. Breaking of left–right asymmetry is initiated by motile cilia in the embryonic node generating a leftward fluid flow followed by signal propagation by non-motile cilia. Here, PKD2 and PKD1L1 channels at the ciliary membrane enable calcium currents, resulting in elevated Ca2+ concentrations on the left side of the node. Node asymmetry is then propgated to the lateral plate mesoderm, which in turn causes NODAL expression in the left lateral plate mesoderm, followed by PITX2 expression. LEFTY2 acts as a feedback inhibitor restricting the range of nodal signaling. LEFTY1 helps in maintaining the midline barrier. (B) Examples of left–right organizing centers in vertebrates. (i) In situ hybridization visualizing the embryonic node (Shh, purple), e7.5 mouse embryo; (ii) Nodal.LacZ reporter expression (blue) marking the embryonic node and the lateral left plate mesoderm in an E8.5 mouse embryo; (iii) Xenopus embryo stage 17 archenteron roof in ventral perspective (pseudocolored in blue) where cilia-mediated leftward flow initiates left–right asymmetry; (iiii) Xenopus embryo stage 17 with nodal expression marked in purple (in situ hybridization).
Normal motile cilia function is crucial for generating a leftward flow within the embryonic node in mammals (Nonaka et al., 1998), initiating cell signaling pathways driving the establishment of body laterality. The “two-cilia hypothesis” proposes that a leftward flow generated by motile node pit cell cilia is sensed by non-motile node crown cell cilia which causes calcium influx determining the left side of the embryonic node (McGrath et al., 2003). A second model proposes that the leftward nodal flow results in a morphogen gradient, including vesicular particles being transported toward the left side of the node where they release their content, creating a morphogen gradient (Tanaka et al., 2005) (Figure 2A). The definition of the left side of the embryonic node subsequently leads to the activation of the TGFβ-superfamily components, namely, NODAL and LEFTY2. The nodal antagonist LEFTY2 prevents the activation of NODAL on the right side of the lateral plate mesoderm (Juan and Hamada, 2001; Shiratori and Hamada, 2006). On the left side, NODAL consecutively induces PITX2 expression on the left side of the lateral plate mesoderm, enabling asymmetric heart, lung, and spleen development (Norris, 2012). Midline LEFTY1 expression confines NODAL, LEFTY2, and PITX2 expression to the left side as LEFTY1 null mice display bilateral expression of NODAL, LEFTY2, and PITX2 (Meno et al., 1998; Capdevila et al., 2000), (Grimes and Burdine, 2017) (Figure 2A). It is therefore not surprising that nodal dysfunction in mammals causes laterality defects (Mohapatra et al., 2009).
In addition to nodal signaling, Bmp, Notch, Hedgehog, and Fgf signaling also play important roles for left–right establishment (Sempou and Khokha, 2019). In humans, mutations in ZIC3 (Gebbia et al., 1997), the TGF-beta signaling molecule GDF1 (Karkera et al., 2007) or ACVR2B (Kosaki et al., 1999; Ma et al., 2012), and the metalloprotease MMP21 (Guimier et al., 2015) have been associated with left–right patterning defects. However, past genetic studies investigating laterality defects in humans have had a surprisingly low yield of causative mutations, leaving up to 90% of cases genetically unexplained (Table 1). As progress has since been made with the identification of novel laterality defect genes and some of the previous studies did not use exome sequencing but targeted Sanger sequencing or gene panel sequencing, we performed an exome-based study in a cohort of 37 Turkish individuals with laterality defects to determine the rate of mutations in known disease genes and to identify novel disease-causing alleles.
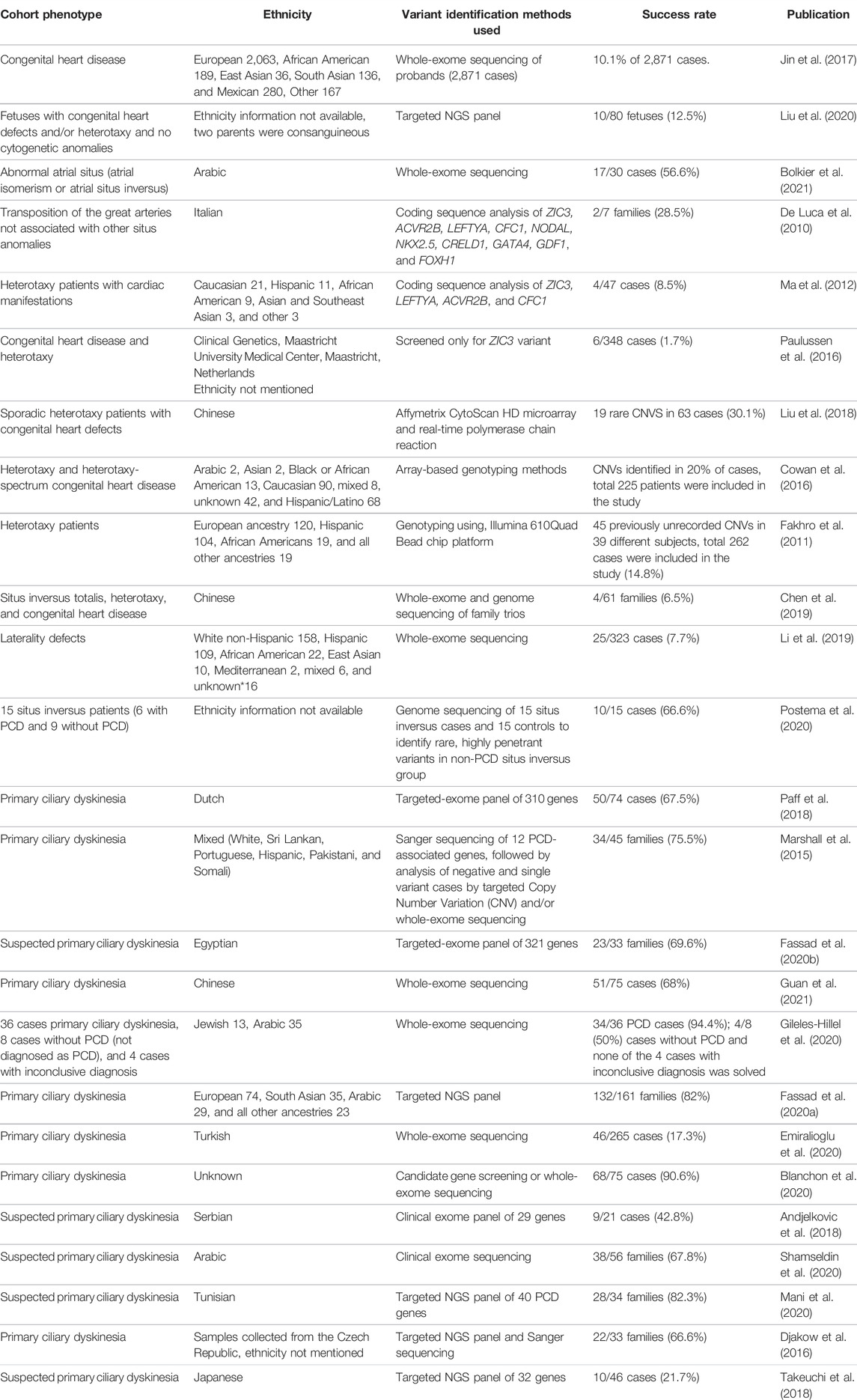
TABLE 1. Summary of previously published studies describing genetic screening in cohorts with laterality defects. Search terms included “laterality defect,” “heterotaxy,” “situs inversus,” “primary ciliary dyskinesia,” and “genetic” or “mutation.” Success rates were stated as presented in the manuscripts or calculated from the results presented in the publications.
Materials and Methods
DNA Samples
Inclusion criteria were the presence of situs inversus totalis, thoracalis or abdominalis, or left or right isomerism with or without the presence of structural heart defects. The majority of cases were children younger than 10 years, presenting to the local genetics clinic after a referral from local pediatricians or pediatric cardiologists, while the remaining cases were fetal cases diagnosed by prenatal ultrasound. None of the included children was reported by the parents to present with daily wet cough or chronic rhinitis. Informed consent was obtained from all the participants or their legal guardians. Ethical approval was obtained from the local ethics committee and samples were processed at Radboudumc in Nijmegen under the diagnostic innovation program to establish a genetic diagnosis (CMO-2006/048). Genomic DNA extraction was performed from EDTA blood samples using the standard salting out method (Miller et al., 1988) or commercially available kits such as Qiagen genomic DNA extraction kit (Germantown, MD, United States). The concentration of DNA was determined using the Nanodrop (Thermo Fisher Scientific, Waltham, MA, United States) or by Qubit 2.0 (Life Technologies, Carlsbad, CA, United States).
Whole-Exome Sequencing
Exome sequencing was performed as previously described (Loges et al., 2018). In brief, 2–5 micrograms of DNA from index cases were subjected to whole-exome sequencing (WES) at Novogene, Hong Kong. Exome capture was performed using the Agilent SureSelect Human All Exon V5 Kit; sequencing was performed using an Illumina HiSeq 2500 machine. Paired-end sequencing was performed resulting in sequences of 150 bases from each end of the fragments. UCSC hg19 was used as a reference genome. VarScan version 2.2.5 and MuTec and GATK Somatic Indel Detector were used to detect single-nucleotide variants (SNV) and InDels. Data were then filtered in-house using a minor allele frequency (MAF) < 1% in public control databases including dbSNP, ExAc, and gnomAD, and remaining variants were first filtered for known disease-causing genes with an emphasis on diseases compatible with the patient phenotype (laterality disorder) and genes causing laterality defects in animal models. We then filtered genes encoding for proteins present in the cilia proteome and subsequently used a list of genes expressed highly in the mouse embryonic node (S. J. Arnold, unpublished data) for further gene prioritization. A complete list of genes used for filtering is shown in Supplementary Table S1. Open exome analysis was subsequently performed for cases where no likely causative allele could be detected. We prioritized homozygous variants in known consanguineous families; however, compound heterozygosity or dominant inheritance was likewise considered. Additionally, visual BAM file inspection was performed for homozygous CNVs in genes previously associated with laterality defects, and exome data files were further analyzed for the presence of CNVs using ExomeDepth (Plagnol et al., 2012).
PCR and Sanger Sequencing
To confirm deletions identified by CNV analysis, respective exons (DNAI2 exons 8–13 and CCDC40 exons 1–3) were amplified using the one taq master mix following the manufacturer’s instructions (New England Biolabs, Massachusetts, United States). Primers were designed using the primer blast tool, and sequences of the primers are available on request.
Results
We performed genetic diagnostics using exome sequencing in 37 individuals, from 35 unrelated Turkish families, presenting with laterality defects to local genetics clinics. The included cases neither had a clinical history of PCD nor had an official diagnosis of PCD; however, airway ciliary motility assessment or nasal nitric oxide (NO) measurement was not performed prior to genetic investigations. Exome sequencing and CNV analysis identified likely causative variants in 14 families and candidate variants of unknown significance in 3 families, with the majority of mutations found in genes encoding for ciliary proteins of which in turn most encode for components of the ciliary motility apparatus such as dynein arms (DNAH5, DNAI1, DNAI2, DNAH9, and DNAH6), dynein arm-docking complex components (CCDC114), dynein assembly complex components (DNAAF3) or 96 nm ruler components (CCDC39 and CCDC40), and MNS1, a protein interacting with CCDC114 in vitro (Table 2). The vast majority of identified variants were found in a homozygous state which reflects the mainly autosomal recessive inheritance pattern seen in ciliopathies and the consanguineous nature of our subject population. In accordance with previous findings in mucociliary clearance disorders, we identified mainly nonsense, frameshift, or splice site variants (Fassad et al., 2020a). Of all causative variants identified, 8 variants have not been previously reported as disease-causing. In three additional families, we identified potential candidate disease variants in GJA1 in SI-18, in DNAH6 in SI-15, and in CCDC39 and ACVR2B in SI-47 (Table 2).
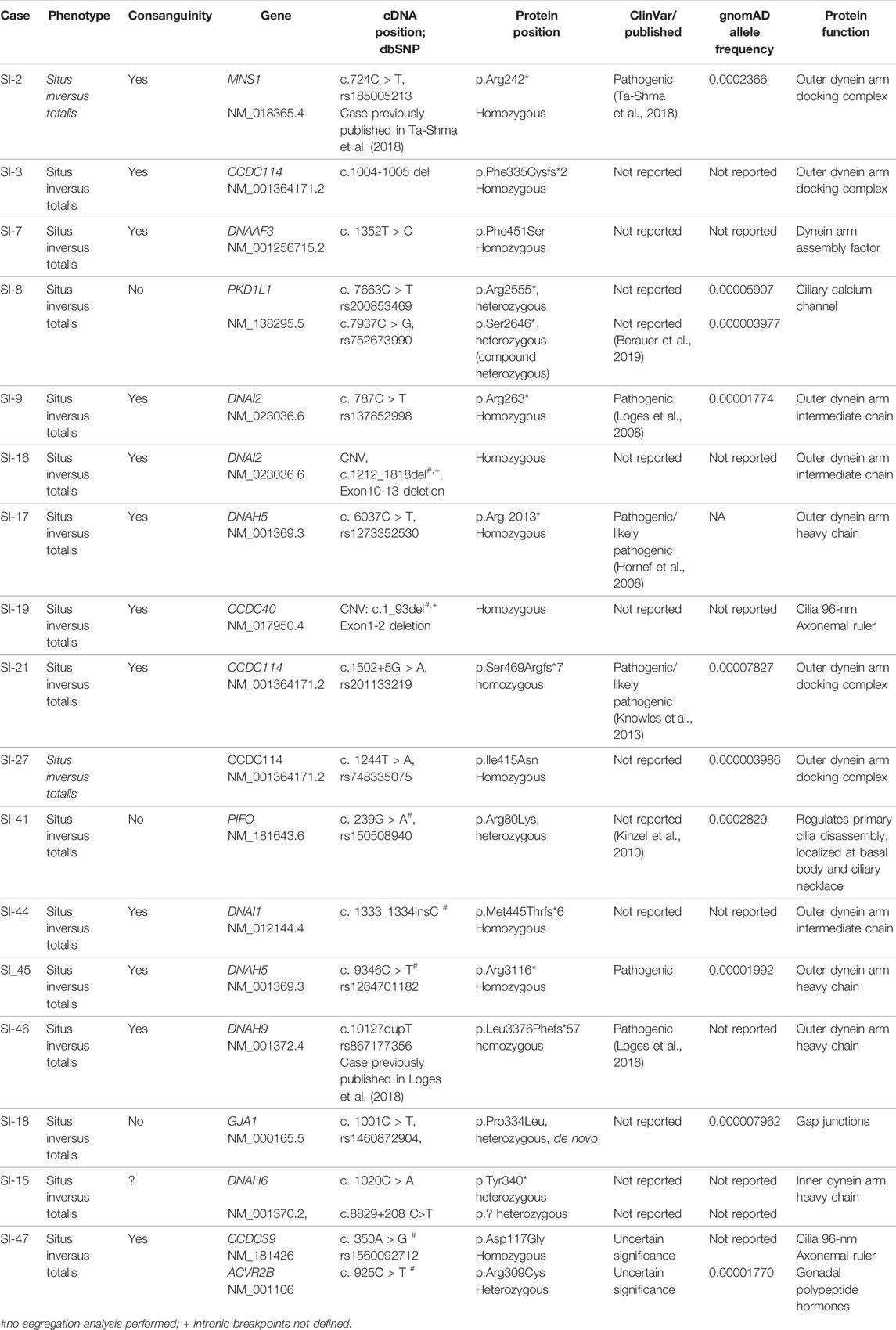
TABLE 2. Genetic variants identified by exome sequencing in 35 laterality defect families.
Variants Identified in Genes Encoding for Ciliary Motility Component-Related Genes
In total, 12 out of 15 likely disease-causing variants were identified in genes encoding for ciliary motility related proteins associated with axonemal dynein motor, including two nonsense DNAH5 variants in SI-17 and SI-45: p.Arg 2013* previously published by Hornef et al. (2006) and p.Arg3116* not previously reported as disease-causing. In SI-46, we identified a homozygous frameshift variant in DNAH9, p.Leu3376Phefs*57 (case and mutation meanwhile reported by Loges et al. (2018).
We further found three families to carry variants in genes encoding for dynein intermediate chains: DNAI1 c.1333_1334insC, p.Met445Thrfs*6 not previously reported as disease-causing or in gnomAD for SI-44 and DNAI2 p.Arg263* previously identified by Loges et al. in SI-9 (Loges et al., 2008) and a homozygous CNV encompassing DNAI2 exons 10–13 (c.1212_1818del) in SI-16 not previously reported and confirmed by PCR (Supplementary Figures S1, S2). Our analysis further revealed 3 homozygous variants in the outer dynein arm-docking complex (ODA-DC) component CCDC114: in SI-3, we identified c.1004-1005del, p.Phe335Cysfs*2 not previously reported, while in SI-21, we detected the previously reported splicing variant, c.1502+5G > A (Knowles et al., 2013) and a not previously reported homozygous missense, p.Ile415Asn in SI-27. Knowles et al. found that the splice site change in intron 12 results in a frameshift and introduces a stop codon (p.Ser469Argfs*7). The homozygous missense change p.Ile415Asn in SI-27 is not reported in ClinVar, predicted deleterious by SIFT, probably damaging (0.997) by PolyPhen and polymorphism by mutation taster. The mutant residue is bigger, and this will change the 3D structure and might lead to bumps. The different hydrophobicity of the wild-type and mutant residues could cause a loss of protein–protein interactions (mutation prediction program used: HOPE; https://www3.cmbi.umcn.nl/hope/). Segregation analysis revealed the mother to be a heterozygous carrier; however, the variant was not detected in the father. This could be due to a heterozygous not detected deletion on the other allele, non-paternity, de novo occurrence of the mutation in the child or maternal uniparental disomy. In SI-2, we identified a homozygous nonsense change (p.Arg242*) in MNS1 (case and variant reported in Ta-Shma et al., 2018 (Ta-Shma et al., 2018). Interestingly, this variant was identified in several unrelated individuals with laterality defects and perturbed male fertility but no respiratory symptoms (Ta-Shma et al., 2018). MNS1 interacts with CCDC114, suggesting likewise a role for outer dynein arm docking (Ta-Shma et al., 2018). In SI-7, we identified a not previously reported homozygous missense variant, p.Phe451Ser in DNAAF3. The variant is probably damaged (0.998) by PolyPhen prediction and polymorphism using mutation taster analysis. The wild-type and mutant amino acids differ in size with the mutant residue being smaller, and this might influence the protein structure and protein–protein interactions. Furthermore, due to differences in hydrophobicity of the wild-type and mutant residue, hydrophobic interactions, either in the core of the protein or on the surface, could be lost (https://www3.cmbi.umcn.nl/hope/).
CNV analysis revealed a novel homozygous deletion encompassing c.1_93 of CCDC40 in SI-19 conformed by PCR Supplementary Figures S3, S4. We could not identify the intronic breakpoints of the CNVs identified in this study.
Variants Identified in Other Ciliary Genes
We identified mutations in two genes encoding for ciliary proteins not related to ciliary motility, PIFO and PKD1L1. PIFO p.Arg80Lys was identified heterozygously in SI-41, previously reported by Kinzel et al. in two cases in a heterozygous state (Kinzel et al., 2010). One previously reported case represented a deceased neonate with thoracic–abdominal situs inversus, cystic kidneys, and liver fibrosis, and a second case suffered from isolated double-outlet right ventricle (Kinzel et al., 2010). PIFO has been previously identified as a gene expressed in the node by Tamplin et al. using microarray analysis in Foxa2 mutant mouse embryos (Tamplin et al., 2008). Parental DNA was not available to confirm de novo occurrence of the variant in our case. PKD1L1 p.Arg2555* and p.Ser2646* were identified as compound heterozygosity in SI-8, both not reported in ClinVar to date. Biallelic variants in PKD1L1 have been previously reported in individuals with laterality defects and congenital heart malformations (Vetrini et al., 2016; Postema et al., 2020; Bolkier et al., 2021; Correa et al., 2021). The variant p.Ser2646* identified in our study was also reported by Berauer et al. in a case with polysplenia, biliary atresia, abdominal heterotaxy, congenital heart disease, and vascular anomalies (Berauer et al., 2019). PKD1L1 seems to form a ciliary protein complex with PKD2 in primary crown cell cilia in the node, and it thought to play a role in flow sensing (Field et al., 2011).
Candidate Variants Identified
Candidate variants were identified in three families (SI-18, SI-15 and SI-47). SI-18 carries a heterozygous de novo missense variant in GJA1 (p.Pro334Leu). The mutant residue is bigger, and this might lead to bumps. The mutation is located within a stretch of residues annotated in UniProt as a special protein–protein interaction region. The wild-type residue is a proline. Prolines are known to be very rigid and therefore induce a special backbone conformation which might be required at this position. The mutation can disturb this special conformation (https://www3.cmbi.umcn.nl/hope/). This variant is not reported in ClinVar and has a frequency of 0.000007962 in gnomad. The variant is predicted to be benign (0.020) by PolyPhen, disease-causing by mutation taster (score 98), and tolerated by SIFT prediction. The amino acid position is conserved in mammals including mice, chicken, and Xenopus while zebrafish carries an alanine instead here.
In SI-15, we identified a heterozygous stop variant, c.1020C > A, p.Tyr340* and a heterozygous deep-intronic substitution, c.8229+208C>T in DNAH6. While the Alamut program predicted only minimal splice site probabilities for the mutant allele compared to the wild-type sequence, ESE finder predicted a slightly changed pattern of ESE binding (Supplementary Figures S5, S6).
In SI-47, we identified variants in two genes, CCDC39 and ACVR2B: a homozygous missense variant in CCDC39 (p.Asp117Gly), not reported in gnomAD but in ClinVar as a variant of uncertain significance (VUS). The variant is predicted as polymorphism by mutation taster and PolyPhen benign with a score of 0.351. The variant is located at the first coiled–coil domain (16–122 amino acids; UniProt). Additionally, we identified a heterozygous variant in ACVR2B, p.Arg309Cys, predicted damaging by SIFT, probably damaging (1.000) by PolyPhen, and disease-causing by mutation taster, likewise reported as VUS in ClinVar. The variant is located in the protein kinase-binding domain (190–480 amino acids). Segregation analysis could not be performed to establish if the variant is de novo. Several previous studies have reported heterozygous ACVR2B variants in individuals with heterotaxy (Kosaki et al., 1999; Ma et al., 2012; Li et al., 2019). ACVR2B functions in the NODAL/TGF-beta signaling pathway (Li et al., 2019). A summary of all genetic findings is shown in Table 2 and Figures 3, 4.
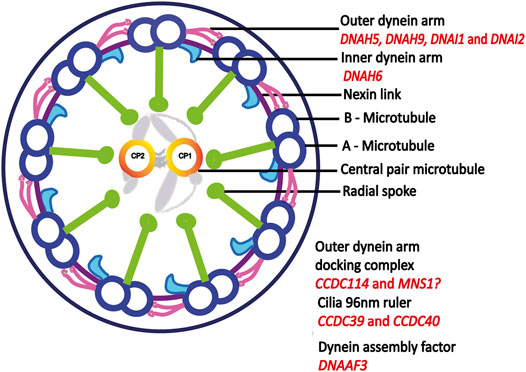
FIGURE 3. Motile cilia ultrastructure. Motile cilia are microtubule based structures, dynein arms powers the sliding motion of microtubules which in turn generates ciliary movement. Outer dynein arm-docking complex helps in the attachment of dynein arms to A-microtubule. Dynein assembly factor is important for the assembly of both outer and inner dynein arms. Mutations in central pair and radial spoke genes does not cause laterality defect, adapted from (Antony et al., 2021). Proteins encoded by genes identified in this study are indicated in red.
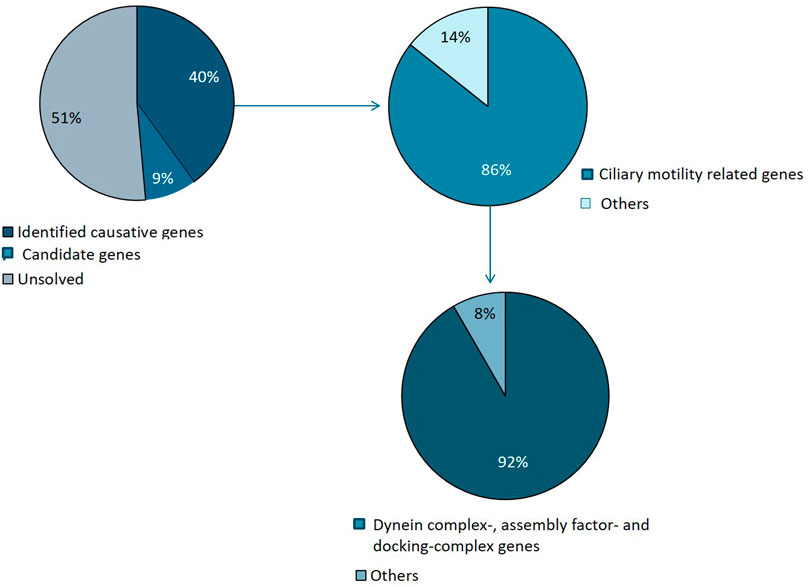
FIGURE 4. Summary of genetic variants identified. Causative mutations were identified in 14 families and candidate variants in 3 families. The majority of identified variants were in ciliary genes, mainly encoding motility apparatus components, of which the majority represents dynein complex related factors.
Discussion
Over the last 2 decades, genetic analyses of individuals with laterality defects have greatly advanced our understanding of establishment of left–right asymmetry. Eight variants reported here have not been previously reported as disease-causing, two of which are CNVs. This provides useful information for human genetic diagnostics and underlines the importance of CNV analysis.
Given the fact that none of the cases included in this study had a formal diagnosis of PCD nor was suspected with PCD clinically, we identified pathogenic or likely pathogenic variants in a rather large number of genes known to cause PCD when dysfunctional, including DNAH5, DNAH9, CCDC114, DNAI1, DNAI2, CCDC39, and CCDC40. DNAH5 encodes an outer dynein arm γ—heavy chain distributed panaxonemally in respiratory cilia, and biallelic loss of function mutations are a frequent cause of PCD (Hornef et al., 2006). DNAH9 encodes an outer dynein arm (ODA) heavy chain only present in one of 2 ODA subtypes, subtype 2. ODA2 localization is restricted to the distal half of the cilium and accordingly, and DNAH9 dysfunction results in a very subtle respiratory phenotype with perturbed distal ciliary movements (Loges et al., 2018). CCDC114 mutation causes the absence of the outer dynein arm in affected individuals, and majority of the cilia are static with few showing some stiff movements (Knowles et al., 2013; Onoufriadis et al., 2013). DNAAF3 encodes a cytoplasmic dynein preassembly factor. Dynein preassembly factors play a crucial role for the assembly of both outer and inner dynein arms and dysfunction results in cilia lacking dynein arms, causing ciliary immotility (Mitchison et al., 2012). DNAI1 and DNAI2 encode outer dynein arm intermediate chains and dysfunction of these genes results in the absence of outer dynein arms causing immotile cilia or cilia with minimum residual motility (Guichard et al., 2001; Loges et al., 2008). CCDC40 encodes a 96-nm ruler protein, and CCDC40 loss of function results in a disorganized microtubule arrangement and radial spokes and nexin link defects and absence of inner dynein arms. The respiratory cilia of the individuals carrying biallelic CCDC40 loss of function variants show defective ciliary beat regulation and ciliary movements with a reduced amplitude (Becker-Heck et al., 2011). CCDC39 likewise encodes a 96-nm ruler protein in motile cilia and dysfunction results in disorganized microtubules, defects in radial spokes, inner dynein arms, and nexin links, similar to what is observed with dysfunction of CCDC40 (Merveille et al., 2011). Our findings confirm genetic PCD studies showing that DNAH5 and CCDC40 mutations represent common underlying causes (Fassad et al., 2020a).
We further identified candidate variants in DNAH6 in SI-15. In humans, DNAH6 encodes a heavy chain of the inner dynein arm (Hom et al., 2011). Li et al. suggested that DNAH6 mutations play a role in laterality defects (Li et al., 2016) when they reported an individual with heterotaxy carrying a heterozygous DNAH6 mutation and a heterozygous DNAI1 mutation, suggesting digenic inheritance/genetic interaction. Moreover, 5/6 patients in their study with heterozygous DNAH6 mutations also harbored heterozygous DNAH5 mutations. The subthreshold double-morpholino knockdown of DNAH6/DNAH5 and DNAH6/DNAI1 in zebrafish resulted in likewise heterotaxy. Re-analyzing the exome data for potentially causative heterozygous mutations in DNAI1 and DNAH5 in SI-15 did not yield any new causative variants. Compound heterozygous mutations in DNAH6 are also reported in patients with sperm flagella defects (Li et al., 2018; Tu et al., 2019).
The PIFO heterozygous variant we identified was previously reported to cause laterality defects in a single individual of Northern European descent (Kinzel et al., 2010). PIFO plays an important role in primary cilia disassembly through AuraA activation and it is localized at the basal body (Kinzel et al., 2010). The p.Arf80Lys variant seemed to inhibit AurA activation. Furthermore, overexpression caused cilia disassembly defects (Kinzel et al., 2010). In contrast to the previously described case, no liver or kidney cysts have been observed in our case to date; however, later development in life cannot be excluded and appropriate clinical monitoring has been initiated. Our study confirms that the identified PIFO variant is a rare cause of left–right pattern defects in humans, and it seems likely that future studies will reveal more disease-causing PIFO alleles.
PKD1L1 is a subunit of the heterodimeric TRP channel thought to regulate calcium currents (Delling et al., 2013) and is important for confining the NODAL expression to the left side during the left–right axis determination (Grimes et al., 2016). The PKD1L1 p.Arg2555* and p.Ser2646* variants we identified in this study are located at the extracellular domains before the trans-membrane domains 8 and 10 respectively (Rodriguez et al., 2019; Correa et al., 2021). These two compound heterozygous changes identified in this study could affect the function of trans-membrane domains 8–10, extracellular domains between them, and an intracellular coiled–coil domain, which interacts with PKD2. The PKD1L1 variants are similarly identified in other laterality defect cases at N-terminal extracellular domains preceding immunoglobulin-like PKD, REJ (receptor egg jelly), GPR (G-protein-coupled receptor proteolytic site), trans-membrane, and extracellular domains in the middle of the trans-membrane domains and intracellular C-terminal coiled–coil domain (Berauer et al., 2019; Rodriguez et al., 2019; Correa et al., 2021).
The association of GJA1 with laterality defects or congenital heart disease remains unsure. GJA1 encodes connexion 43 and is important for the formation of gap junction and inter cellular channels (Britz-Cunningham et al., 1995). Gap junctions could play an important role in the transfer of signals from the node to the lateral plate mesoderm in vertebrates (Saund et al., 2012). Britz-Cunningham et al. reported potentially disease-causing missense variants in GJA1 in 6 patients with complex cardiac malformations and laterality defects (Britz-Cunningham et al., 1995). However, two later studies were not able to replicate these findings (Casey and Ballabio, 1995; Splitt et al., 1995). In 2001, Dasgupta et al. suggested that the inability to replicate the findings of the initial study could be caused by a PCR amplification bias toward the wild-type allele. Dasgupta et al. therefore used denaturing gradient gel electrophoresis (DGGE), followed by separate sequencing of two alleles and detected two heterozygous missense variants and 2 synonymous variants in eight individuals with hypoplastic left heart syndrome and one individual with atrioventricular canal defect. However, the four variants identified were identical to the pseudogene sequence at the nucleotide level (97% homology) (Dasgupta et al., 2001). Mutations in GJA1 have further been identified in cases with autosomal recessive and dominant oculodentodigital dysplasia, where cardiac anomalies are rare (OMIM: 257850, 164200). Clinically, our cases did not exhibit features of oculodentodigital dysplasia, where about 50% of all cases represent de novo alleles.
Huang et al. could not confirm that GJA1 dysfunction causes heart defects or left–right patterning defects in mice nor did they identify GJA1 disease-causing variants in a cohort of 300 patients with congenital heart disease (Britz-Cunningham et al., 1995; Dasgupta et al., 2001; Huang et al., 2011). However, in a sudden infant death case, a de novo mutation in GJA1 was detected and the patient tissue also demonstrated a mosaic expression of connexin43 (GJA1) (Van Norstrand et al., 2012). To our knowledge, no human cases with a laterality defect and a dominant GJA1 de novo variant have been described in the literature. The novel GJA1 variant we identified (p.Pro334Leu) was located in the UBQLN4 (ubiquilin 4)-binding region. The interaction of GJA1 with this ubiquitin-like and ubiquitin-associated domain-containing protein is important for its homeostasis and function (Su et al., 2010). Putatively, the mutation could influence the GJA1–UBQLN4 interaction.
The variant of unknown significance in ACVR2B we identified in SI-47 represents the only variant we identified in a gene encoding for a nodal signaling pathway component. To date, less than ten cases with ACVR2B dominant variants have been reported (Kosaki et al., 1999; Ma et al., 2012; Li et al., 2019). Among those, the same variant (p.Arg40His) has been identified in four cases. Generally, mutations in ACRV2B seem to represent a rare cause of laterality defects (Ma et al., 2012). As we also identified a VUS in CCDC39 in SI-47, the role of the ACRV2B variant we identified remains unclear.
To date, only few studies investigating cohorts of cases with laterality defects have been published and those have generally exhibited low mutation detection rates (Table 1), including a large study identifying pathogenic variants in just 25/323 cases (Li et al., 2019). Among non-PCD cases, mutations in ZIC3 and NODAL appear to be the most common genetic causes (Li et al., 2019). In our cohort, the rate of detected likely disease-causing variants was somewhat higher (17 of 35 families (49%), potentially reflecting the large proportion of not previously clinically identified PCD cases and a high rate of consanguineous families in our series. The patients in our study were neither selected for the presence of additional symptoms indicating PCD nor tested for motile cilia dysfunction, but still we identified the majority of causative mutations in genes associated with the ciliary motility apparatus. This is in line with previous studies where 42% of congenital heart disease patients with heterotaxy defects that had a genetic cause were found to carry mutations in mucociliary clearance disorder genes (Nakhleh et al., 2012) as well as with a mouse recessive forward genetics screen for congenital heart defects (Li et al., 2015). Furthermore, mutation detection rates are higher among human PCD cases than isolated laterality defect cases (Table 1). Overall, dysfunction of proteins associated with ciliary motility seems to be responsible for the majority of human laterality defects where a monogenetic cause can be identified with the currently used genetic diagnostics methods (Table 1). Confirming previous studies, we find that mutations in genes encoding for dynein motor complex components make up a large proportion of identified genetic variants in genes associated with ciliary motility. None of the cases in our study carrying mutations in genes associated with PCD had previously been diagnosed with PCD underlines the importance of genetics for PCD diagnostics, especially when specialized PCD diagnostics are unavailable locally, as the diagnosis of PCD allows appropriate clinical management which can significantly alter the disease course (Storm van’s Gravesande and Omran, 2005; Davis et al., 2019).
Conclusion
Our study provides a comprehensive list of laterality defect candidate genes based on human and animal studies. Future studies are needed to further validate candidate genes in regulating laterality patterning. In light of our findings, motile ciliary genes could be prioritized in laterality defect genetic screening studies. The overall low mutation detection rate in our laterality defect cohort (49%) and previous reports (Table 1) suggests the existence of non-coding monogenetic variants, genetic mosaicism within embryonic tissues or presence of epigenetic or environmental and non-genetic factors playing a significant role(s) in defective left-right patterning.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Innovative diagnostic program, Radboud UMC Nijmegen, Netherlands. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
DA, SJA, PW, RB, ZB, RB, LS, TVD, and MS performed experiments and analyzed data. EYG, AG, ICT and HB performed clinical evaluations. DA and MS wrote the manuscript. HGB critically revised the manuscript. All the authors read and approved the manuscript. MS supervised the study.
Funding
This study was funded by the European Research Council (ERC) (ERC starting grant to MS, grant No. 716344), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)–Project-ID 431984000–SFB 1453) to MS, SJA, and PW and Radboud University Medical Center and Radboud University (Hypatia Research grant and Excellence Fellowship to MS). SJA acknowledges funding from the German Research Foundation (DFG) (AR 732/3-1, project grant AR 732/2-1). This study was supported by the Deutsche Forschungsgemeinschaft (DFG) under the Emmy Noether Programme (grant WA3365/2-1) to PW and under Germany’s Excellence Strategy (CIBSS — EXC-2189 — project ID 390939984) to PW, SJA and MS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all participating families and referring clinicians for their participation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.861236/full#supplementary-material
References
Andjelkovic, M., Minic, P., Vreca, M., Stojiljkovic, M., Skakic, A., Sovtic, A., et al. (2018). Genomic Profiling Supports the Diagnosis of Primary Ciliary Dyskinesia and Reveals Novel Candidate Genes and Genetic Variants. PLoS One 13, e0205422. doi:10.1371/journal.pone.0205422
Antony, D., Brunner, H. G., and Schmidts, M. (2021). Ciliary Dyneins and Dynein Related Ciliopathies. Cells 10 (8), 1885. doi:10.3390/cells10081885
Bataille, S., Demoulin, N., Devuyst, O., Audrézet, M.-P., Dahan, K., Godin, M., et al. (2011). Association of PKD2 (Polycystin 2) Mutations with Left-Right Laterality Defects. Am. J. Kidney Dis. 58, 456–460. doi:10.1053/j.ajkd.2011.05.015
Becker-Heck, A., Zohn, I. E., Okabe, N., Pollock, A., Lenhart, K., B, Sullivan-Brown, J., et al. (2011). The Coiled-Coil Domain Containing Protein CCDC40 Is Essential For Motile Cilia Function And Left-Right Axis Formation. Nature Genet. 43, 79–84.
Berauer, J. P., Mezina, A. I., Okou, D. T., Sabo, A., Muzny, D. M., Gibbs, R. A., et al. (2019). Identification of Polycystic Kidney Disease 1 Like 1 Gene Variants in Children with Biliary Atresia Splenic Malformation Syndrome. Hepatology 70, 899–910. doi:10.1002/hep.30515
Blanchon, S., Legendre, M., Bottier, M., Tamalet, A., Montantin, G., Collot, N., et al. (2020). Deep Phenotyping, Including Quantitative Ciliary Beating Parameters, and Extensive Genotyping in Primary Ciliary Dyskinesia. J. Med. Genet. 57, 237–244. doi:10.1136/jmedgenet-2019-106424
Bolkier, Y., Barel, O., Marek-Yagel, D., Atias-Varon, D., Kagan, M., Vardi, A., et al. (2021). Whole-Exome Sequencing Reveals a Monogenic Cause in 56% of Individuals with Laterality Disorders and Associated Congenital Heart Defects. J. Med. Genet. doi:10.1136/jmedgenet-2021-107775
Boon, M., Wallmeier, J., Ma, L., Loges, N. T., Jaspers, M., Olbrich, H., et al. (2014). MCIDAS Mutations Result In A Mucociliary Clearance Disorder With Reduced Generation Of Multiple Motile Cilia. Nat Commun. 5, 4418. doi:10.1038/ncomms5418
Britz-Cunningham, S. H., Shah, M. M., Zuppan, C. W., and Fletcher, W. H. (1995). Mutations of the Connexin43 Gap-Junction Gene in Patients with Heart Malformations and Defects of Laterality. N. Engl. J. Med. 332, 1323–1330. doi:10.1056/nejm199505183322002
Capdevila, J., Vogan, K. J., Tabin, C. J., and Izpisúa Belmonte, J. C. (2000). Mechanisms of Left-Right Determination in Vertebrates. Cell 101, 9–21. doi:10.1016/s0092-8674(00)80619-4
Casey, B., and Ballabio, A. (1995). Connexin43 Mutations in Sporadic and Familial Defects of Laterality. N. Engl. J. Med. 333, 941–942. doi:10.1056/NEJM199510053331415
Chen, W., Zhang, Y., Yang, S., Shi, Z., Zeng, W., Lu, Z., et al. (2019). Bi-Allelic Mutations in NUP205 and NUP210 Are Associated with Abnormal Cardiac Left-Right Patterning. Circ. Genom Precis Med. 12, e002492. doi:10.1161/CIRCGEN.119.002492
Correa, A. R. E., Endrakanti, M., Naini, K., Kabra, M., and Gupta, N. (2021). Hydrops Fetalis in PKD1L1 ‐Related Heterotaxy: Report of Two Foetuses and Expanding the Phenotypic and Molecular Spectrum. Ann. Hum. Genet. 85, 138–145. doi:10.1111/ahg.12417
Cowan, J. R., Tariq, M., Shaw, C., Rao, M., Belmont, J. W., Lalani, S. R., et al. (2016). Copy Number Variation as a Genetic Basis for Heterotaxy and Heterotaxy-Spectrum Congenital Heart Defects. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371 (1710), 20150406. doi:10.1098/rstb.2015.0406
Dasgupta, C., Martinez, A.-M., Zuppan, C. W., Shah, M. M., Bailey, L. L., and Fletcher, W. H. (2001). Identification of Connexin43 (α1) gap junction Gene Mutations in Patients with Hypoplastic Left Heart Syndrome by Denaturing Gradient Gel Electrophoresis (DGGE). Mutat. Research/Fundamental Mol. Mech. Mutagenesis 479, 173–186. doi:10.1016/s0027-5107(01)00160-9
Davis, S. D., Rosenfeld, M., Lee, H.-S., Ferkol, T. W., Sagel, S. D., Dell, S. D., et al. (2019). Primary Ciliary Dyskinesia: Longitudinal Study of Lung Disease by Ultrastructure Defect and Genotype. Am. J. Respir. Crit. Care Med. 199, 190–198. doi:10.1164/rccm.201803-0548oc
De Luca, A., Sarkozy, A., Consoli, F., Ferese, R., Guida, V., Dentici, M. L., et al. (2010). Familial Transposition of the Great Arteries Caused by Multiple Mutations in Laterality Genes. Heart 96, 673–677. doi:10.1136/hrt.2009.181685
Delling, M., Decaen, P. G., Doerner, J. F., Febvay, S., and Clapham, D. E. (2013). Primary Cilia Are Specialized Calcium Signalling Organelles. Nature 504, 311–314. doi:10.1038/nature12833
Deng, H., Xia, H., and Deng, S. (2015). Genetic Basis of Human Left-Right Asymmetry Disorders. Expert Rev. Mol. Med. 16, e19. doi:10.1017/erm.2014.22
Djakow, J., Kramná, L., Dušátková, L., Uhlík, J., Pursiheimo, J.-P., Svobodová, T., et al. (2016). An Effective Combination of Sanger and Next Generation Sequencing in Diagnostics of Primary Ciliary Dyskinesia. Pediatr. Pulmonol. 51, 498–509. doi:10.1002/ppul.23261
Edelbusch, C., Cindrić, S., Dougherty, G. W., Loges, N. T., Olbrich, H., Rivlin, J., et al. (2017). Mutation of Serine/Threonine Protein Kinase 36 (STK36) Causes Primary Ciliary Dyskinesia with a Central Pair Defect. Hum. Mutat. 38, 964–969. doi:10.1002/humu.23261
Emiralioglu, N., Taskiran, E. Z., Kosukcu, C., Bilgic, E., Atilla, P., Kaya, B., et al. (2020). Genotype and Phenotype Evaluation of Patients with Primary Ciliary Dyskinesia: First Results from Turkey. Pediatr. Pulmonol 55, 383–393. doi:10.1002/ppul.24583
Fakhro, K. A., Choi, M., Ware, S. M., Belmont, J. W., Towbin, J. A., Lifton, R. P., et al. (2011). Rare Copy Number Variations In Congenital Heart Disease Patients Identify Unique Genes In Left-Right Patterning. PNAS 108, 2915–2920. doi:10.1073/pnas.1019645108
Fassad, M. R., Patel, M. P., Shoemark, A., Cullup, T., Hayward, J., Dixon, M., et al. (2020a). Clinical Utility of NGS Diagnosis and Disease Stratification in a Multiethnic Primary Ciliary Dyskinesia Cohort. J. Med. Genet. 57, 322–330. doi:10.1136/jmedgenet-2019-106501
Fassad, M. R., Shoman, W. I., Morsy, H., Patel, M. P., Radwan, N., Jenkins, L., et al. (2020b). Clinical and Genetic Spectrum in 33 Egyptian Families with Suspected Primary Ciliary Dyskinesia. Clin. Genet. 97, 509–515. doi:10.1111/cge.13661
Field, S., Riley, K.-L., Grimes, D. T., Hilton, H., Simon, M., Powles-Glover, N., et al. (2011). Pkd1l1 Establishes Left-Right Asymmetry and Physically Interacts with Pkd2. Development 138, 1131–1142. doi:10.1242/dev.058149
Fliegauf, M., Benzing, T., and Omran, H. (2007). When Cilia Go Bad: Cilia Defects and Ciliopathies. Nat. Rev. Mol. Cel Biol 8, 880–893. doi:10.1038/nrm2278
Gebbia, M., Ferrero, G. B., Pilia, G., Bassi, M. T., Aylsworth, A. S., Penman-Splitt, M., et al. (1997). X-Linked Situs Abnormalities Result from Mutations in ZIC3. Nat. Genet. 17, 305–308. doi:10.1038/ng1197-305
Gileles-Hillel, A., Mor-Shaked, H., Shoseyov, D., Reiter, J., Tsabari, R., Hevroni, A., et al. (2020). Whole-Exome Sequencing Accuracy in the Diagnosis of Primary Ciliary Dyskinesia. ERJ Open Res. 6 (4), 00213–02020. doi:10.1183/23120541.00213-2020
Grimes, D. T., and Burdine, R. D. (2017). Left-Right Patterning: Breaking Symmetry to Asymmetric Morphogenesis. Trends Genet. 33, 616–628. doi:10.1016/j.tig.2017.06.004
Grimes, D. T., Keynton, J. L., Buenavista, M. T., Jin, X., Patel, S. H., Kyosuke, S., et al. (2016). Genetic Analysis Reveals a Hierarchy of Interactions between Polycystin-Encoding Genes and Genes Controlling Cilia Function during Left-Right Determination. Plos Genet. 12, e1006070. doi:10.1371/journal.pgen.1006070
Guan, Y., Yang, H., Yao, X., Xu, H., Liu, H., Tang, X., et al. (2021). Clinical and Genetic Spectrum of Children with Primary Ciliary Dyskinesia in China. Chest 159, 1768–1781. doi:10.1016/j.chest.2021.02.006
Guichard, C., Harricane, M.-C., Lafitte, J.-J., Godard, P., Zaegel, M., Tack, V., et al. (2001). Axonemal Dynein Intermediate-Chain Gene (DNAI1) Mutations Result In Situs Inversus And Primary Ciliary Dyskinesia (Kartagener Syndrome). Am. J. Hum. Genet. 68, 1030–1035.
Guimier, A., Gabriel, G. C., Bajolle, F., Tsang, M., Liu, H., Noll, A., et al. (2015). MMP21 Is Mutated in Human Heterotaxy and Is Required for normal Left-Right Asymmetry in Vertebrates. Nat. Genet. 47, 1260–1263. doi:10.1038/ng.3376
Hom, E. F. Y., Witman, G. B., Harris, E. H., Dutcher, S. K., Kamiya, R., Mitchell, D. R., et al. (2011). A Unified Taxonomy for Ciliary Dyneins. Cytoskeleton 68, 555–565. doi:10.1002/cm.20533
Hornef, N., Olbrich, H., Horvath, J., Zariwala, M. A., Fliegauf, M., Loges, N. T., et al. (2006). DNAH5Mutations Are a Common Cause of Primary Ciliary Dyskinesia with Outer Dynein Arm Defects. Am. J. Respir. Crit. Care Med. 174, 120–126. doi:10.1164/rccm.200601-084oc
Huang, G.-Y., Xie, L.-J., Linask, K. L., Zhang, C., Zhao, X.-Q., Yang, Y. i., et al. (2011). Evaluating the Role of Connexin43 in Congenital Heart Disease: Screening for Mutations in Patients with Outflow Tract Anomalies and the Analysis of Knock-In Mouse Models. J. Cardiovasc. Dis. Res. 2, 206–212. doi:10.4103/0975-3583.89804
Jin, S. C., Homsy, J., Zaidi, S., Lu, Q., Morton, S., Depalma, S. R., et al. (2017). Contribution of Rare Inherited and De Novo Variants in 2,871 Congenital Heart Disease Probands. Nat. Genet. 49, 1593–1601. doi:10.1038/ng.3970
Juan, H., and Hamada, H. (2001). Roles of Nodal-Lefty Regulatory Loops in Embryonic Patterning of Vertebrates. Genes Cells 6, 923–930. doi:10.1046/j.1365-2443.2001.00481.x
Karkera, J. D., Lee, J. S., Roessler, E., Banerjee-Basu, S., Ouspenskaia, M. V., Mez, J., et al. (2007). Loss-of-Function Mutations in Growth Differentiation Factor-1 (GDF1) Are Associated with Congenital Heart Defects in Humans. Am. J. Hum. Genet. 81, 987–994. doi:10.1086/522890
Kawakami, Y., Raya, Á., Raya, R. M., Rodríguez-Esteban, C., and Belmonte, J. C. I. (2005). Retinoic Acid Signalling Links Left-Right Asymmetric Patterning and Bilaterally Symmetric Somitogenesis in the Zebrafish Embryo. Nature 435, 165–171. doi:10.1038/nature03512
Kinzel, D., Boldt, K., Davis, E. E., Burtscher, I., Trümbach, D., Diplas, B., et al. (2010). Pitchfork Regulates Primary Cilia Disassembly and Left-Right Asymmetry. Develop. Cel 19, 66–77. doi:10.1016/j.devcel.2010.06.005
Knowles, M. R., Leigh, M. W., Ostrowski, L. E., Huang, L., Carson, J. L., Hazucha, M. J., et al. (2013). Exome Sequencing Identifies Mutations in CCDC114 as a Cause of Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 92, 99–106. doi:10.1016/j.ajhg.2012.11.003
Knowles, M. R., Zariwala, M., and Leigh, M. (2016). Primary Ciliary Dyskinesia. Clin. Chest Med. 37, 449–461. doi:10.1016/j.ccm.2016.04.008
Kosaki, R., Gebbia, M., Kosaki, K., Lewin, M., Bowers, P., Towbin, J. A., et al. (1999). Left-Right Axis Malformations Associated with Mutations inACVR2B, the Gene for Human Activin Receptor Type IIB. Am. J. Med. Genet. 82, 70–76. doi:10.1002/(sici)1096-8628(19990101)82:1<70::aid-ajmg14>3.0.co;2-y
Leigh, M. W., Horani, A., Kinghorn, B., O’Connor, M. G., Zariwala, M. A., and Knowles, M. R. (2019). Primary Ciliary Dyskinesia (PCD): A Genetic Disorder of Motile Cilia. Transl Sci. Rare Dis. 4, 51–75. doi:10.3233/trd-190036
Li, A. H., Hanchard, N. A., Azamian, M., D’Alessandro, L. C. A., Coban-Akdemir, Z., Lopez, K. N., et al. (2019). Genetic Architecture of Laterality Defects Revealed by Whole Exome Sequencing. Eur. J. Hum. Genet. 27, 563–573. doi:10.1038/s41431-018-0307-z
Li, L., Sha, Y. W., Xu, X., Mei, L. B., Qiu, P. P., Ji, Z. Y., et al. (2018). DNAH6 Is a Novel Candidate Gene Associated with Sperm Head Anomaly. Andrologia 50 (4), e12953. doi:10.1111/and.12953
Li, Y., Klena, N. T., Gabriel, G. C., Liu, X., Kim, A. J., Lemke, K., et al. (2015). Global Genetic Analysis in Mice Unveils central Role for Cilia in Congenital Heart Disease. Nature 521, 520–524. doi:10.1038/nature14269
Li, Y., Yagi, H., Onuoha, E. O., Damerla, R. R., Francis, R., Furutani, Y., et al. (2016). DNAH6 and its Interactions with PCD Genes in Heterotaxy and Primary Ciliary Dyskinesia. Plos Genet. 12, e1005821. doi:10.1371/journal.pgen.1005821
Lin, A. E., Krikov, S., Riehle-Colarusso, T., Frías, J. L., Belmont, J., Anderka, M., et al. (2014). Laterality Defects in the National Birth Defects Prevention Study (1998-2007): Birth Prevalence and Descriptive Epidemiology. Am. J. Med. Genet. 164, 2581–2591. doi:10.1002/ajmg.a.36695
Liu, C., Cao, R., Xu, Y., Li, T., Li, F., Chen, S., et al. (2018). Rare Copy Number Variants Analysis Identifies Novel Candidate Genes in Heterotaxy Syndrome Patients With Congenital Heart Defects. Genome Med. 10 (1), 40. doi:10.1186/s13073-018-0549-y
Liu, H., Giguet‐Valard, A. G., Simonet, T., Szenker‐Ravi, E., Lambert, L., Vincent‐Delorme, C., et al. (2020). Next‐Generation Sequencing in a Series of 80 Fetuses with Complex Cardiac Malformations And/or Heterotaxy. Hum. Mutat. 41, 2167–2178. doi:10.1002/humu.24132
Loges, N. T., Antony, D., Maver, A., Deardorff, M. A., Güleç, E. Y., Gezdirici, A., et al. (2018). Recessive DNAH9 Loss-Of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects. Am. J. Hum. Genet. 103, 995–1008. doi:10.1016/j.ajhg.2018.10.020
Loges, N. T., Olbrich, H., Fenske, L., Mussaffi, H., Horvath, J., Fliegauf, M., et al. (2008). DNAI2 Mutations Cause Primary Ciliary Dyskinesia with Defects in the Outer Dynein Arm. Am. J. Hum. Genet. 83, 547–558. doi:10.1016/j.ajhg.2008.10.001
Ma, L., Selamet Tierney, E. S., Lee, T., Lanzano, P., and Chung, W. K. (2012). Mutations in ZIC3 and ACVR2B Are a Common Cause of Heterotaxy and Associated Cardiovascular Anomalies. Cardiol. Young 22, 194–201. doi:10.1017/s1047951111001181
Mani, R., Belkacem, S., Soua, Z., Chantot, S., Montantin, G., Tissier, S., et al. (2020). Primary Ciliary Dyskinesia Gene Contribution in Tunisia: Identification of a Major Mediterranean Allele. Hum. Mutat. 41, 115–121. doi:10.1002/humu.23905
Marshall, C. R., Scherer, S. W., Zariwala, M. A., Lau, L., Paton, T. A., Stockley, T., et al. (2015). Whole-Exome Sequencing and Targeted Copy Number Analysis in Primary Ciliary Dyskinesia. G3 (Bethesda) 5, 1775–1781. doi:10.1534/g3.115.019851
Marszalek, J. R., Ruiz-Lozano, P., Roberts, E., Chien, K. R., and Goldstein, L. S. B. (1999). Situs Inversus and Embryonic Ciliary Morphogenesis Defects in Mouse Mutants Lacking the KIF3A Subunit of Kinesin-II. Proc. Natl. Acad. Sci. 96, 5043–5048. doi:10.1073/pnas.96.9.5043
McGrath, J., Somlo, S., Makova, S., Tian, X., and Brueckner, M. (2003). Two Populations of Node Monocilia Initiate Left-Right Asymmetry in the Mouse. Cell 114, 61–73. doi:10.1016/s0092-8674(03)00511-7
Meno, C., Shimono, A., Saijoh, Y., Yashiro, K., Mochida, K., Ohishi, S., et al. (1998). lefty-1 Is Required for Left-Right Determination as a Regulator of Lefty-2 and Nodal. Cell 94, 287–297. doi:10.1016/s0092-8674(00)81472-5
Merveille, A.-C., Davis, E. E., Becker-Heck, A., Legendre, M., Amirav, I., Bataille, G., et al. (2011). CCDC39 Is Required for Assembly of Inner Dynein Arms and the Dynein Regulatory Complex and for normal Ciliary Motility in Humans and Dogs. Nat. Genet. 43, 72–78. doi:10.1038/ng.726
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A Simple Salting Out Procedure for Extracting DNA from Human Nucleated Cells. Nucl. Acids Res. 16, 1215. doi:10.1093/nar/16.3.1215
Mitchison, H. M., Schmidts, M., Loges, N. T., Freshour, J., Dritsoula, A., Hirst, R. A., et al. (2012). Mutations In Axonemal Dynein Assembly Factor DNAAF3 Cause Primary Ciliary Dyskinesia. Nature Genet. 44, 381–389.
Mohapatra, B., Casey, B., Li, H., Ho-Dawson, T., Smith, L., Fernbach, S. D., et al. (2009). Identification and Functional Characterization of NODAL Rare Variants in Heterotaxy and Isolated Cardiovascular Malformations. Hum. Mol. Genet. 18, 861–871. doi:10.1093/hmg/ddn411
Nakhleh, N., Francis, R., Giese, R. A., Tian, X., Li, Y., Zariwala, M. A., et al. (2012). High Prevalence of Respiratory Ciliary Dysfunction in Congenital Heart Disease Patients with Heterotaxy. Circulation 125, 2232–2242. doi:10.1161/circulationaha.111.079780
Nonaka, S., Tanaka, Y., Okada, Y., Takeda, S., Harada, A., Kanai, Y., et al. (1998). Randomization of Left-Right Asymmetry Due to Loss of Nodal Cilia Generating Leftward Flow of Extraembryonic Fluid in Mice Lacking KIF3B Motor Protein. Cell 95, 829–837. doi:10.1016/s0092-8674(00)81705-5
Norris, D. P. (2012). Cilia, Calcium and the Basis of Left-Right Asymmetry. BMC Biol. 10, 102. doi:10.1186/1741-7007-10-102
Olbrich, H., Fliegauf, M., Hoefele, J., Kispert, A., Otto, E., Volz, A., et al. (2003). Mutations in a Novel Gene, NPHP3, Cause Adolescent Nephronophthisis, Tapeto-Retinal Degeneration and Hepatic Fibrosis. Nat. Genet. 34, 455–459. doi:10.1038/ng1216
Onoufriadis, A., Paff, T., Antony, D., Shoemark, A., Micha, D., Kuyt, B., et al. (2013). Splice-Site Mutations in the Axonemal Outer Dynein Arm Docking Complex Gene CCDC114 Cause Primary Ciliary Dyskinesia. Am. J. Hum. Genet. 92, 88–98. doi:10.1016/j.ajhg.2012.11.002
Paff, T., Kooi, I. E., Moutaouakil, Y., Riesebos, E., Sistermans, E. A., Daniels, H. J. M. A., et al. (2018). Diagnostic Yield of a Targeted Gene Panel in Primary Ciliary Dyskinesia Patients. Hum. Mutat. 39, 653–665. doi:10.1002/humu.23403
Paulussen, A. D. C., Steyls, A., Vanoevelen, J., Van Tienen, F. H., Krapels, I. P. C., Claes, G. R., et al. (2016). Rare Novel Variants in the ZIC3 Gene Cause X-Linked Heterotaxy. Eur. J. Hum. Genet. 24, 1783–1791. doi:10.1038/ejhg.2016.91
Pennekamp, P., Karcher, C., Fischer, A., Schweickert, A., Skryabin, B., Horst, J., et al. (2002). The Ion Channel Polycystin-2 Is Required for Left-Right axis Determination in Mice. Curr. Biol. 12, 938–943. doi:10.1016/s0960-9822(02)00869-2
Pennekamp, P., Menchen, T., Dworniczak, B., and Hamada, H. (2015). Situs Inversus and Ciliary Abnormalities: 20 Years Later, what Is the Connection? Cilia 4, 1. doi:10.1186/s13630-014-0010-9
Plagnol, V., Curtis, J., Epstein, M., Mok, K. Y., Stebbings, E., Grigoriadou, S., et al. (2012). A Robust Model for Read Count Data in Exome Sequencing Experiments and Implications for Copy Number Variant Calling. Bioinformatics 28, 2747–2754. doi:10.1093/bioinformatics/bts526
Postema, M. C., Carrion-Castillo, A., Fisher, S. E., Vingerhoets, G., and Francks, C. (2020). The Genetics of Situs Inversus without Primary Ciliary Dyskinesia. Sci. Rep. 10, 3677. doi:10.1038/s41598-020-60589-z
Rodriguez, S., Chaturvedi, R., Blanchette, V., Dell, S., Axford, M., Cada, M., et al. (2019). PKD1L1‐Related Situs Inversus Associated with Sideroblastic Anemia. Clin. Genet. 95, 629–630. doi:10.1111/cge.13512
Saund, R. S., Kanai-Azuma, M., Kanai, Y., Kim, I., Lucero, M. T., and Saijoh, Y. (2012). Gut Endoderm Is Involved in the Transfer of Left-Right Asymmetry from the Node to the Lateral Plate Mesoderm in the Mouse Embryo. Development 139, 2426–2435. doi:10.1242/dev.079921
Sempou, E., and Khokha, M. K. (2019). Genes and Mechanisms of Heterotaxy: Patients Drive the Search. Curr. Opin. Genet. Develop. 56, 34–40. doi:10.1016/j.gde.2019.05.003
Shamseldin, H. E., Al Mogarri, I., Alqwaiee, M. M., Alharbi, A. S., Baqais, K., Alsaadi, M., et al. (2020). An Exome-First Approach to Aid in the Diagnosis of Primary Ciliary Dyskinesia. Hum. Genet. 139, 1273–1283. doi:10.1007/s00439-020-02170-2
Shiratori, H., and Hamada, H. (2006). The Left-Right Axis in the Mouse: from Origin to Morphology. Development 133, 2095–2104. doi:10.1242/dev.02384
Simons, M., Gloy, J., Ganner, A., Bullerkotte, A., Bashkurov, M., Krönig, C., et al. (2005). Inversin, the Gene Product Mutated in Nephronophthisis Type II, Functions as a Molecular Switch between Wnt Signaling Pathways. Nat. Genet. 37, 537–543. doi:10.1038/ng1552
Splitt, M. P., Burn, J., and Goodship, J. (1995). Connexin43 Mutations in Sporadic and Familial Defects of Laterality. N. Engl. J. Med. 333, 941–942. doi:10.1056/NEJM199510053331415
Storm Van's Gravesande, K., and Omran, H. (2005). Primary Ciliary Dyskinesia: Clinical Presentation, Diagnosis and Genetics. Ann. Med. 37, 439–449. doi:10.1080/07853890510011985
Su, V., Nakagawa, R., Koval, M., and Lau, A. F. (2010). Ubiquitin-Independent Proteasomal Degradation of Endoplasmic Reticulum-Localized Connexin43 Mediated by CIP75. J. Biol. Chem. 285, 40979–40990. doi:10.1074/jbc.m110.170753
Ta-Shma, A., Hjeij, R., Perles, Z., Dougherty, G. W., Abu Zahira, I., Letteboer, S. J. F., et al. (2018). Homozygous Loss-Of-Function Mutations in MNS1 Cause Laterality Defects and Likely Male Infertility. Plos Genet. 14, e1007602. doi:10.1371/journal.pgen.1007602
Takeuchi, K., Kitano, M., Kiyotoshi, H., Ikegami, K., Ogawa, S., Ikejiri, M., et al. (2018). A Targeted Next-Generation Sequencing Panel Reveals Novel Mutations in Japanese Patients with Primary Ciliary Dyskinesia. Auris Nasus Larynx 45, 585–591. doi:10.1016/j.anl.2017.09.007
Tamplin, O. J., Kinzel, D., Cox, B. J., Bell, C. E., Rossant, J., and Lickert, H. (2008). Microarray Analysis of Foxa2 Mutant Mouse Embryos Reveals Novel Gene Expression and Inductive Roles for the Gastrula Organizer and its Derivatives. BMC Genomics 9, 511. doi:10.1186/1471-2164-9-511
Tan, S. Y., Rosenthal, J., Zhao, X. Q., Francis, R. J., Chatterjee, B., Sabol, S. L., et al. (2007). Heterotaxy and Complex Structural Heart Defects in a Mutant Mouse Model of Primary Ciliary Dyskinesia. J. Clin. Invest. 117, 3742–3752. doi:10.1172/JCI33284
Tanaka, Y., Okada, Y., and Hirokawa, N. (2005). FGF-Induced Vesicular Release of Sonic Hedgehog and Retinoic Acid in Leftward Nodal Flow Is Critical for Left-Right Determination. Nature 435, 172–177. doi:10.1038/nature03494
Tu, C., Nie, H., Meng, L., Yuan, S., He, W., Luo, A., et al. (2019). Identification of DNAH6 Mutations in Infertile Men with Multiple Morphological Abnormalities of the Sperm Flagella. Sci. Rep. 9, 15864. doi:10.1038/s41598-019-52436-7
Van Norstrand, D. W., Asimaki, A., Rubinos, C., Dolmatova, E., Srinivas, M., Tester, D. J., et al. (2012). Connexin43 Mutation Causes Heterogeneous Gap Junction Loss and Sudden Infant Death. Circulation 125, 474–481. doi:10.1161/circulationaha.111.057224
Vetrini, F., D’Alessandro, L. C. A., Akdemir, Z. C., Braxton, A., Azamian, M. S., Eldomery, M. K., et al. (2016). Bi-Allelic Mutations in PKD1L1 Are Associated with Laterality Defects in Humans. Am. J. Hum. Genet. 99, 886–893. doi:10.1016/j.ajhg.2016.07.011
Keywords: laterality defect, situs inversus, exome, cilium, primary ciliary dyskinesia, dynein, PIFO, PKD1L1
Citation: Antony D, Gulec Yilmaz E, Gezdirici A, Slagter L, Bakey Z, Bornaun H, Tanidir IC, Van Dinh T, Brunner HG, Walentek P, Arnold SJ, Backofen R and Schmidts M (2022) Spectrum of Genetic Variants in a Cohort of 37 Laterality Defect Cases. Front. Genet. 13:861236. doi: 10.3389/fgene.2022.861236
Received: 24 January 2022; Accepted: 03 March 2022;
Published: 13 April 2022.
Edited by:
Steven LC Pei, Yale University, United StatesReviewed by:
Naoto Keicho, Japan Anti-tuberculosis Association, JapanRichard Francis, James Cook University, Australia
Copyright © 2022 Antony, Gulec Yilmaz, Gezdirici, Slagter, Bakey, Bornaun, Tanidir, Van Dinh, Brunner, Walentek, Arnold, Backofen and Schmidts. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam Schmidts, bWlyaWFtLnNjaG1pZHRzQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==
†Present Address: Elif Yilmaz Gulec, Department of Medical Genetics, Istanbul Medeniyet University, School of Medicine, Istanbul Goztepe Prof Dr Suleyman Yalcin City Hospital, Istanbul, TurkeyAlper Gezdirici, Department of Medical Genetics, Istanbul Cam and Sakura City Hospital, Istanbul, Turkey