
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 04 January 2023
Sec. Epigenomics and Epigenetics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1085391
This article is part of the Research Topic Metabolic Dysregulations and Epigenetics/Epigenomics in Cancer View all 6 articles
 Liwen Ren1,2
Liwen Ren1,2 Yihui Yang1,2
Yihui Yang1,2 Wan Li1,2Hong Yang1,2Yizhi Zhang1,2Binbin Ge1,2Sen Zhang1,2
Wan Li1,2Hong Yang1,2Yizhi Zhang1,2Binbin Ge1,2Sen Zhang1,2 Guanhua Du1,2
Guanhua Du1,2 Jinhua Wang1,2*
Jinhua Wang1,2*Tumor development is frequently accompanied by abnormal expression of multiple genomic genes, which can be broadly viewed as decreased expression of tumor suppressor genes and upregulated expression of oncogenes. In this process, epigenetic regulation plays an essential role in the regulation of gene expression without alteration of DNA or RNA sequence, including DNA methylation, RNA methylation, histone modifications and non-coding RNAs. Therefore, drugs developed for the above epigenetic modulation have entered clinical use or preclinical and clinical research stages, contributing to the development of antitumor drugs greatly. Despite the efficacy of epigenetic drugs in hematologic caners, their therapeutic effects in solid tumors have been less favorable. A growing body of research suggests that epigenetic drugs can be applied in combination with other therapies to increase efficacy and overcome tumor resistance. In this review, the progress of epigenetics in tumor progression and oncology drug development is systematically summarized, as well as its synergy with other oncology therapies. The future directions of epigenetic drug development are described in detail.
Cancers are the second leading cause of human death, second only to cardiovascular disease (Siegel et al., 2021). The origination and development of cancer is usually a synergistic effect of epigenetic alterations, genetic mutations, accompanied with environmental factors. Epigenetic regulation is distinguished from genetic mutation and refers to a form of regulation that can regulate gene expression without alteration of DNA sequence (Bird, 2007). Epitranscriptomics has emerged as another level of epigenetic regulation similar to DNA and histone modifications. The epitranscriptomic regulation refers to the relevant functional changes of the transcriptome without any alteration of the RNA sequence (Meyer et al., 2012). Recent studies have found that epigenetic regulation and relevant therapeutics play an irreplaceable role in the mechanism research of cancer occurrence and development and in the process of cancer treatment.
Due to the heterogeneity of tumor cells, tumor recurrence and drug resistance frequently occur, which are the main reasons for the high mortality of cancer. In the early stage of tumor development, numbers of epigenetic changes occur in tumor cells (Ka-Yue Chow et al., 2022; Zandieh et al., 2022). Therefore, it is essential to find drugs that can regulate the abnormal epigenetic regulation of tumor cells. Epigenetic regulation of genes includes DNA methylation, RNA methylation, histone modifications and non-coding RNAs. At present, great progress has been made in the development of antitumor drugs targeting various epigenetic regulation, and multiple drugs have entered the clinical use or clinical research stage.
Although epigenetic drugs have made great progress in the treatment of hematological tumors, they are less effective in solid tumors. With the proposal of drug combination regimens, the combination of epigenetic drugs and other therapies has achieved good efficacy in several solid tumors, such as radiation therapy, chemotherapy, hormone therapy, targeted therapy and immunotherapy. Numerous completed and ongoing clinical trials have been conducted to evaluate the plausibility of combination schemes integrating epigenetic drugs.
It is of great significance to analyze the mechanism of gene irregulation in cancer cells and to identify agents that could modify the abnormal expression of genes. In this review, we reviewed the recent progress of epigenetics in tumor progression and anticancer therapeutics development. In addition, the combination of epigenetic drugs and other oncology therapies are specially reviewed.
DNA methylation is the firstly recognized epigenetic alterations and it is closely connected with the development of cancer. When the promoter region of genes was methylated, the accessibility to regulatory regions in the DNA was blocked and the transcription factors or other transcriptional regulators can't bind with the promoter of genes, which lead to the repression of gene transcription (Jurmeister et al., 2022). Specifically, various tumor suppressor genes (TSGs) were identified to be hypermethylated thus facilitating the development of cancer via TSGs silencing, such as BRCA1 (Das et al., 2022) and CDKN2A (Maeda et al., 2003). On the contrary, hypomethylation of the DNA will lead to the overexpression of genes. It will turn on the expression of oncogenes which contributes to the tumorigenesis (Beetch et al., 2021). Furthermore, the abnormal DNA methylation, such as site-specific hypermethylation and genome-wide hypomethylation, are frequently recognized in the CpG islands of the gene regulatory region of tumor cells (Saghafinia et al., 2018).
The process of DNA methylation is modulated by the DNA methyltransferases (DNMTs) family, which contains DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L (Tajima et al., 2016). DNMT1 is the most plentiful enzyme in the DNMTs family which accounts for modulating the methylation of newly synthesized DNA (Lee et al., 2001), while the DNMT3 enzymes primarily participates in de novo methylation (Chedin, 2011). Abnormal alterations of 5-methylcytosine (5 mC) could indorse unrestrained cell propagation thus promoting tumor progression. The ten-eleven translocation (TET) family of DNA hydroxylases could catalyze 5 mC to various oxidative mediates, such as 5-formylcytosine (5 fC), 5-hydroxymethylcytosine (5hmC) and 5-carboxylcytosine (5caC) and unmethylated cytosine (Strzyz, 2022). The related enzymes of methylation are recognized as potential targets of cancer.
The methylation in the N6-position of adenosine on eucaryotic mRNA (N6-methyladenosine, m6A) could regulate the metabolism of RNA, such as splice, transport, degradation, translation and miRNA modulation (Wang et al., 2020a). Recent studies suggested that m6A could modulate the proliferation, apoptosis and metastasis of cancer cell, through regulating the cancer-associated genes (He et al., 2019). There are three main types of regulators responsible for m6A regulation, including writers, readers and erasers. The methyltransferase complex (MTC) is the writer to catalyze the methylation of mRNA, whereas the demethylase erases the m6A. The MTC takes charge of the catalysis of m6A, which include METTL3 and other assistant units (Jansens et al., 2022). And the RNA reader protein identified the m6A to exert relevant effects (Zhou et al., 2020). The eraser is demethylase which eliminates m6A with α-ketoglutarate as co-substrate and ferrous iron as cofactor. FTO and ALKBH5 are the identified m6A erasers so far. FTO could regulate the splicing of mRNA via blocking the binding of SRSF2 at RNA splice sites (Zhao et al., 2014). Many FTO inhibitors have been found to have antitumor effects and are currently in preclinical studies.
Multiple evidence suggested that the m6A modification has dual role in cancer. The m6A modification promote tumor progression via upregulating the expression of oncogenes or inhibiting the expression of tumor suppressor genes. On the contrary, the m6A modification could also inhibit the expression of oncogenes and elevate the expression of tumor suppressor genes (He et al., 2019).
Histone Modifications could regulate the accessibility and conformation of chromatin thereby modulating gene expression (Morgan and Shilatifard, 2020). The positively charged histone proteins offer competent integration with DNA of negative charge. The N-terminal of histone proteins are abundant in arginine and lysine residues that could be frequently modified (Zhou et al., 2019). The related histone-modifying enzymes modify the relevant residues of the tails of histone via methylation, acetylation, phosphorylation. Besides, histone modifications are being discovered gradually, such as the ubiquitination, citrullination, ADP-ribosylation, formylation, deamination, propionylation, O-GlcNAcylation, butyrylation, proline isomerization, crotonylation and lactylation. There are three types of proteins interacted with the histone, (I) the readers which identify the modifications of histone, (II) the writers which regulate the modifications of histone, (III) the erasers which remove the modifications of histone (Millan-Zambrano et al., 2022).
Histone acetylation is modulated via histone deacetylases (HDACs) and histone acetyltransferases (HATs) in a reversible and dynamic way (Icardi et al., 2012). The primary function of HATs is adding the acetyl group (-CH3CO) to lysine residues which are related to the activation of gene transcription. On the contrary, the HDACs are erasers which are responsible for removal of the acetyl groups (Shvedunova and Akhtar, 2022).
The charge neutralization model was applied for the explanation of the mechanism of histone acetylation. Histones tightly bind with negatively charged DNA through the lysine residues with positive charge on H3/H4. When histones are acetylated, chromatin configuration will no longer be tight and transformed to euchromatin with loose state. Therefore, the transcriptional factors (TFs) will be recruited for activation of gene transcription (Nicolas et al., 2018). In contrast, HDACs could remove the acetylation of lysine residues and the configuration of chromatin converts to condensed heterochromatin. The acetylation of H4 at the lysine-16 (H4K16) is essential for the chromatin folding and the transition of euchromatin to heterochromatin (Wang et al., 2020b). In addition, the acetylation of histone could provide the binding site for the proteins which participate in the activation of genes, such as the proteins of the bromodomain-containing family (Qin et al., 2019).
Bromodomain and extraterminal domain (BET) proteins are readers of the acetylated proteins, which contains a couple of tandem bromodomains, a C-terminal domain and an extra-terminal domain. The BRD family includes BRD2, BRD3, BRD4 and BRDT. The first three are commonly distributed in tissues, and BRDT is only expressed in the testis (Boyson et al., 2021). The BET families are principally responsible for the recognition of the acetylation of histone H4, but also recognize the acetylation of non-histone proteins, like transcription factors. For example, BRD4 could bind with the TWIST which is an essential transcription factor in the metastasis of cancer (Shi et al., 2014). It also plays an important part in the regulation of oncogene MYC (Devaiah et al., 2020). The inhibitors of BET (BETi) are recognized as an important item for the research and development of antitumor drugs.
Histones can be methylated at the arginine or lysine residues which are mediated via the histone methyl transferases (HMTs), whereas the histone demethylases (HDMs) regulate the elimination of methylation. The consequence of histone methylation can be repression or activation of transcription, depending on the methylated residues (Black et al., 2012). In general, trimethylation of lysine 4 on H3 (H3K4me3) (Hughes et al., 2020) signifies activation of gene transcription, whereas the trimethylation of lysine 9 (H3K9me3) (Feng et al., 2020) and 27 (H3K27me3) (Raas et al., 2022) on H3 represents inhibition of gene transcription. EZH2 belongs to the polycomb repressive complex 2 (PRC2), which is responsible for the catalysis of methylation of lysine 27 of histone H3 (Pan et al., 2016; Jiang et al., 2021). EZH2 is an essential therapeutic target of various cancers, and multiple inhibitors of EZH2 have entered clinical or preclinical studies. Furthermore, the levels of lysine methylation are also related to the transcription repression or activation, which could be identified via diverse methyl-lysine-binding domains. Tumor cells are usually found to possess abnormal histone modifications at single gene or global nuclei levels (Cornett et al., 2019).
The sequencing of the entire human genome has shown that only ∼2% of the genome is translated. The non-coding RNAs (ncRNAs) could be generally characterized to small and large ncRNAs (lncRNA, more than 200 nucleotides) (Anastasiadou et al., 2018). These ncRNAs were identified as an essential regulator in the development of various disease including cancer (Esteller, 2011). The small ncRNAs comprise small interfering RNAs (siRNAs), PIWI interacting RNAs (piRNAs), microRNAs (miRNAs) and small nucleolar RNAs (snoRNAs). The small ncRNAs are participated in the silencing of targeted gene with high level of sequence conservation among different species (Matsui and Corey, 2017). On the contrary, the lncRNAs possess low level of sequence conservation across species and the mechanisms in the transcription regulation are more complicated (Zhu et al., 2013). Particularly, the lncRNAs is identified as molecular scaffolds for the multiple regulators of chromatin (Rinn, 2014), whereas the function is disrupted in the various cancers. The lncRNA HOTAIR was found to be upregulated in multiple cancers (Qu et al., 2019) and act as a molecular scaffold for the PRC2 complex to target the chromatin (Tsai et al., 2010). Silencing of HOTAIR could inhibit the metastasis of colorectal cancer and breast cancer via regulating PRC2 occupancy (Kogo et al., 2011).
The DNMT inhibitors are classified to two types generally: nucleoside analogues and non-nucleoside analogues. The nucleoside analogues are modified molecule of cytidine which could covalently interact with the catalytic positions of DNMTs in an irreversible way (Yu et al., 2019). Two DNA methyltransferase inhibitors (DNMTi), 5-azacitidine (Vidaza) and its deoxyanalogue decitabine (Dacogen), have been approved for clinical use, which increases survival time and ameliorates life quality of patients. Azacitidine (Cogle et al., 2015) and decitabine (Dhillon, 2020) are usually used for the treatment of myelodysplastic syndrome (MDS), acute myeloid leukemia (AML) or chronic myelomonocytic leukemia (CMML). The derivate of decitabine, SGI-110, is a novel hypomethylating compound for the treatment of AML and MDS that has undergone phase II clinical trial (Garcia-Manero et al., 2019a). CP-4200 was designed as a pro-drug of azacytidine. It was an elaidic acid ester for azacytidine, which exerted better therapeutic effect than azacytidine (Brueckner et al., 2010). Besides, RX-3117 was also a nucleoside analogue which could suppress DNMT1 and could inhibit the proliferation of cancer in vivo (Balboni et al., 2019). Unfortunately, overall hypomethylation of genome could happen due to the non-specificity of nucleoside analogues (Flausino et al., 2021). Therefore, some non-nucleoside inhibitors of DNMTs are exploited. The non-nucleoside inhibitors can bind the catalytic site of DNMTs without binding the DNA directly. Hydralazine which is indicated for the management of hypertension has been studied for its potential as a DNMT inhibitor. It was demonstrated that in prostate cancer cells hydralazine treatment lowered the production of DNMT1, DNMT3a and DNMT3b mRNA suggesting its potential in reducing the malignant growth through epigenetic alteration (Graca et al., 2014). An antisense oligonucleotide designed to bind with the 3′ untranslated region of DNMT1 mRNA and hindering with its transcription is MG98. It is a second generation DNMT inhibitor specifically inhibiting DNMT1 without altering DNMT3 expression. Clinical study has been carried out with MG98 in combination with interferon for the treatment of metastatic renal cell carcinoma and was proven to be safe at a particular dosage (Amato et al., 2012). SGI-1027 is a derivative of quinoline which could suppress DNMT1, DNMT3A and DNMT3B without binding with DNA. SGI-1027 could upregulate the TSGs of which the transcription is blocked in tumor cells (Sun et al., 2018) (Table 1).

TABLE 1. Epigenetic anticancer therapeutics.
Studies suggested that inhibition of m6A was able to facilitate development of various cancers. So far, the first METTL3 inhibitor, STC-15, has entered phase I clinical trials for the treatment of advanced malignancies (Holz, 2022). STM2457, which is also an inhibitor of METTL3, leads to reduced AML growth, and an increase in differentiation and apoptosis of AML cells in vitro. Furthermore, STM2457 could also contribute to impaired engraftment and prolonged survival in various AML mouse models (Yankova et al., 2021). Meclofenamic acid (MA) is a selective inhibitor of FTO via preempting binding sites of FTO (Huang et al., 2015). MA2 is an ethyl ester derivative of MA and it could inhibit the proliferation of glioblastoma stem-like cell both in vitro and in vivo (Xiao et al., 2020). FB23-2 was also identified as an inhibitor of FTO. It could promote the differentiation and inhibit the proliferation of AML cells (Huang et al., 2019). R-2- hydroxyglutarate (R-2HG) is a metabolite of mutant IDH1/2 enzymes, which increased the m6A level and accelerated the degradation of oncogenes (Dang et al., 2009). The research of m6A is an emerging field. Currently, the research and development of m6A inhibitors are in the pre-clinical stage. It is believed that many m6A inhibitors will enter the clinical trials or even market stage in the future (Table 1).
HMTs are identified to be highly expressed in a variety of cancers, indicating HMTs to become latent therapeutic target for cancers (Liu and Wang, 2016). The inhibitor of lysine methyltransferase DOT1L (Vatapalli et al., 2020), EPZ004777, was designed basing on the S-adenosyl methionine binding domain. It could suppress the activity of DOT1L enzyme, thus downregulating the methylation level at H3K79 (Gao and Ge, 2018). Besides, EPZ-5676 was also a DOT1L inhibitor which could significantly inhibit the progression of leukemia via reducing the methylation of H3K27 (Waters et al., 2015). EZH2 is the main element of PRC2 which is related to the H3K27 methylation, contributing to the inhibition of TSGs. EZH2 was found to upregulate in various cancers, such as breast cancer and prostate cancer (Duan et al., 2020). EZH2 inhibitor tazemetostat have been proven effective in patients with relapsed or refractory, BAP1-inactivated malignant pleural mesothelioma in a multicentre, open-label, phase 2 study (Zauderer et al., 2022). The S-adenosyl-L-homocysteine hydrolase inhibitor DZNep could degrade the expression of EZH2 and inhibit the proliferation and metastasis of chondrosarcoma (Girard et al., 2014). EPZ005687 and EPZ-6438 (Knutson et al., 2014) are selective inhibitors of EZH2 which possess excellent inhibitory activity against lymphoma. SMYD2 is another lysine methyltransferase which mainly modulate the methylation of H2B, H3 and H4. Several inhibitors of SMYD2, like LLY-507 (Kojima et al., 2020), AZ505 (Pan et al., 2022) and A-893 (Sweis et al., 2015) could significantly suppress the proliferation of various cancer cells. The methyltransferase G9a is responsible for the methylation of H3K9 (Padeken et al., 2022). It is overexpressed in various cancers and its inhibitors, BIX-01294 (Chae et al., 2019) and UNC0638 (Li et al., 2021) are able to inhibit the activity of G9a selectively with anti-tumor effects. Studies have found that Set 7/9 could both regulate the methylation of H3K4 and estrogen receptor (ER). Cyproheptadine was recognized as a Set 7/9 inhibitor which could inhibit the proliferation of breast cancer cells by modulating the expression of ER (Takemoto et al., 2016) (Table 1).
There are two main categories of the inhibitors of HDMs. One type is the Lysine-specific demethylases LSD1/2 with the amine oxidases properties, belonging to the HDM1 subgroup. The remaining HDM2-8 subgroups contains jumonji C domain which is α-ketoglutarate and iron dependent (Nowak et al., 2016).
A variety of LSD1 inhibitors are currently in clinical or preclinical studies (Fang et al., 2019). LSD1 inhibitor pargyline was reported to suppress the growth and epithelial-to - mesenchymal transformation (EMT) of prostate carcinoma cells (Ojha et al., 2021). The antidepressant drug tranylcypromine was also identified as a LSD1 inhibitor with antineoplastic activity (Wass et al., 2021). Besides, there are several LSD1 inhibitors derivated on tranylcypromine structure undergoing clinical research for the treatment of Leukemia (Dai et al., 2020). The derivatives of polyamine could upregulate the methylation of H3K4 in triple negative breast cancer cells via inhibiting LSD1 (Zhu et al., 2012). Similarly, derivatives of biguanides or guanidines could also suppress the activity of LSD1, thus inhibiting the proliferation of lung cancer cells through upregulating H3K4 methylation (Sharma et al., 2010). In addition, LSD1 inhibitors Namolineand HCI-2509 can inhibit the proliferation of prostate cancer in like manner (Willmann et al., 2012).
On the other hand, the derivative of hydroxamic acid SAHA (vorinostat) was proved as an effective inhibitor of KDM4E and its derivative IOX1 was also demonstrated to inhibit various types of HDMs (Siegel et al., 2009). In particular, various flavonoid compounds, such as caffeic acid and myricetin, have presented inhibitory activity on numerous jumonji C HDMs (Li et al., 2022) (Table 1).
The HATs play an essential role in the modulation of transcription and are promising therapeutic target of cancer. The HATs inhibitor C646, which could competitively inhibit the activity of p300, could significantly block cell cycle and induce cell apoptosis of acute myeloid leukemia (AML) cells (Gao et al., 2013). The isothiazolone is both the inhibitor of p300 and PCAF and was demonstrated effectively in inhibiting colorectal cancer (Ghizzoni et al., 2009). The natural product anacardic acid was demonstrated as the inhibitor of MYST family (Wu et al., 2009) and its analogs 6-alkylsalicylate was identified as the inhibitor of Tip60 (Ghizzoni et al., 2012). They have been found to inhibit the growth of pancreatic cancer, breast cancer and prostate cancer. Especially, PU139 and PU141, which are derivatives of pyridoisothiazolone, could suppress the activity of p300, CBP, Gcn5 and PCAF both in vitro and in vivo (Ramakrishnan et al., 2022). The antineoplastic activity of the above compounds was also proved both in neuroblastoma cells and xenografts models in mice (Table 1).
The application of inhibitors of HDAC (HDACis) was successfully proved in the treatment of cancer in clinical practice. The HDACis are able to inhibit the proliferation of cancer cells through inducing cell apoptosis and suppressing the process of EMT by inhibiting the expression related to the cell migration and angiogenesis (Ho et al., 2020).
The derivative of hydroxamic acid Vorinostat was the first HDACi authorized by the Food and Drug Administration (FDA) for the therapeutic of cutaneous T cell lymphoma (CTCL) (Siegel et al., 2009). Since then, numerous derivatives of hydroxamic acid have been developed for preclinical or clinical studies, such as Pracinostat, Abexinostat, Givinosta, Resminostat and Panobinostat (Bird et al., 2020). Pracinostat has been approved for the treatment of AML as a breakthrough therapy, combined with azacytidine (Garcia-Manero et al., 2019b). Besides, Abexinostat has also been approved for the treatment of follicular lymphoma after achieving favorable treatment results in clinical trials (Ribrag et al., 2017). Givinostat has been undergoing phase II clinical trial for the treatment of multiple myeloma (Galli et al., 2010). Similarly, Resminostat has been evaluated for the treatment of relapsed Hodgkin lymphoma in phase II clinical study now (Walewski et al., 2019). In addition, 4SC-202 was a novel HDACi and undergoing in the phase I clinical trial for the treatment of advanced hematological cancers (von Tresckow et al., 2019). Tasquinimod, which is an anti-angiogenic compound for the therapeutic of castration resistant prostate cancer, is identified as an allosteric regulator of HDAC4 (Isaacs et al., 2013). AR-42 is a pan-HDAC inhibitor which is effectively demonstrated in phase I research for the treatment of B-, T-cell lymphomas and multiple myeloma (Sborov et al., 2017).
Another major class of HDACi is the derivatives of benzamide, such as entinostat, mocetinostat and tacedinaline (CI-994). Entinostat was effectively examined without severe toxic effects in breast cancer (Connolly et al., 2021), melanoma (Ny et al., 2021) and metastatic non-small cell lung cancer (NSCLC) (Witta et al., 2012) in phase II/III clinical trials, either alone or in combination with other drugs. Mocetinostat is also in the phase II clinical study which is applied for the metastatic leiomyosarcoma (Choy et al., 2018) and relapsed classical Hodgkin’s lymphoma (Younes et al., 2011) with promising activity with manageable toxicity as single agent. Tacedinaline could inhibit the proliferation of cell lines of NSCLC in vitro and tacedinaline will exert better effect when it with combined with other anticancer agents, like docetaxel and gemcitabine (Loprevite et al., 2005). However, results of a phase II multicenter study suggested that gemcitabine combined with tacedinaline presented no benefit than gemcitabine alone in advanced pancreatic cancer patients with advanced pancreatic cancer (Richards et al., 2006).
The valproic acid (VPA) and phenylbutyrate, which belongs to the short chain fatty acid type, are also found to inhibit the activity of HDAC with anti-cancer activity. VPA was effectively demonstrated in the neuroendocrine tumors (Arvidsson et al., 2016). In vitro studies have suggested that phenylbutyrate is able to inhibit the proliferation of glioblastomas cells (Ye et al., 2019). Various natural products were found to exert HDAC suppressing activity, such as cyclopeptide, amamistatin and chlamydocin (Byun et al., 2019) (Table 1).
There are numerous BETis undergoing the clinical or preclinical studies currently. JQ1 is the first designed BETi which could bind with the bromodomains or the acetyl-lysine competitively (Filippakopoulos et al., 2010). JQ1 could arrest cell cycle and induce cell senescence in multiple myeloma through inhibiting the expression of c-Myc (Delmore et al., 2011). Thienotriazolodiazepine OTX015 is the first BETi which enters clinical trials. OTX015 could arrest cell cycle, induce apoptosis of cell and inhibit the growth of acute leukemia cell lines by downregulating BRD2, BRD4 and MYC expression (Coude et al., 2015). In the clinical trials, OTX015 presented favorable therapeutic effects within the tolerable dose in the treatment of AML (Berthon et al., 2016). CPI-0610 is also a BETi with benzoisoxazoloazepine structure which is undergoing phase I clinical trial for the treatment of refractory or relapsed lymphomas (Albrecht et al., 2016) (Table 1).
The combinations of radiotherapy and inhibitors of DNMT, HDAC, BET and EZH2 have been demonstrated to increase the sensitivity of radiotherapy to patients in preclinical research through arresting cell cycle, upregulating oxidative stress and preventing DNA-damage repair. The above studies suggest the great potential of epigenetic drugs in combination with radiotherapy. In a phase I trials, the combination of vorinostat and radiotherapy with capecitabin significantly increased the overall survival of patients of pancreatic ductal adenocarcinoma (Tinari et al., 2012). In addition, the combination of vorinostat and radiotherapy could improve the objective response rate of refractory neuroblastoma (Mueller et al., 2011) and gastrointestinal carcinoma (Ree et al., 2010), compared with using the radiotherapy alone. Unfortunately, not all the epigenetic drugs in combination with radiotherapy will exert the above effect, whereas leading to severe toxic effect sometimes.
In preclinical research, the combinations of chemotherapy with the DNMT (Gravina et al., 2010) and HDAC inhibitors (Arrighetti et al., 2015) significantly strengthen the killing effect of chemotherapeutics on tumor cells by promoting DNA damage and inhibiting the repair of DNA damage. Besides, the drug resistance of chemotherapeutics could be overcome when in combinations of DNMT or HDAC inhibitors (Wang et al., 2020c). Unfortunately, although the preclinical experiments suggested that the combination of chemotherapy and epigenetic drugs could improve the efficacy of chemotherapy, the clinical trials frequently presented unfavourable results due to no significant improvement in efficacy accompanied by serious adverse effects (Choy et al., 2015).
In the preclinical studies, the HDACi could exaggerate the therapeutic effect and overcome drug resistance of the hormone therapy in breast cancer animal models (Bijian et al., 2018). Besides, the inhibitors of BET could be used in combination with fulvestrant to inhibit the proliferation of tamoxifen-resistant breast cancer cells both in vitro and in vivo (Li et al., 2020). CPI-1 is a specific inhibitor of CBP and p300 which could combine with the anti-oestrogen therapies for the treatment of breast cancer by inhibiting the ERα pathway (Waddell et al., 2021).
Not only that, the combination of epigenetic drugs and hormone therapy is also proven safely and effectively in the clinical trials. A phase II trial suggested that the patients with endocrine-resistant metastatic breast cancer that treated tamoxifen combined with vorinostat had high therapeutic responses with favorable tolerability (Peterson et al., 2021). Similarly, exemestane combined with entinostat (HDAC inhibitor) significantly improved the progression free survival (PFS) of hormone receptor-positive, advanced-stage, endocrine -resistant breast cancer in postmenopausal women, which was approved by the FDA as a breakthrough therapy (Connolly et al., 2021).
The combination of BET inhibitor JQ1 and anti-androgen enzalutamide could significantly inhibit the proliferation of prostate cancer xenografts which is enzalutamide-resistant (Asangani et al., 2014). Similar results were obtained for the combination of BET inhibitor OTX-015 and AR-agonist ARN-509 (Asangani et al., 2016). At present, these drug combinations are in clinical trial studies. In addition, the addition of HDAC inhibitor (Panobinostat) could overcome the resistance of the second-line anti-androgen therapy of prostate cancer, which remarkably improve the PFS of patients (Ferrari et al., 2019).
Preclinical studies suggested that the application of epigenetic drugs could overcome the drug resistance to the HER family receptor tyrosine kinases (RTKs). Using BETs remarkably upregulated the sensitivity of head and neck squamous cell cancer (HNSCC) to anti-EGFR antibody (Leonard et al., 2018) and the sensitivity of HER2-positive breast carcinoma to lapatinib (Stuhlmiller et al., 2015). Unfortunately, the combination of RTK inhibitors and epigenetic drugs usually exhibited greater toxicity in the clinical experiments, which made it difficult to achieve the desired efficacy (Pili et al., 2017).
In addition, numerous clinical trials have demonstrated that the combination of anti-angiogenic therapeutics and HDAC inhibitors could remarkably improve the efficacy of the treatment for various cancers with favorable safety profile. The drug combinations have achieved good efficacy and safety, such as sorafenib and resminostat (HDACi) in treatment of hepatocellular cancer (Bitzer et al., 2016), bevacizumab and vorinostat (HDACi) in clear cell renal cell cancer (RCC) (Pili et al., 2017), bevacizumab and panobinostat (HDACi) in high-grade glioma (Lee et al., 2015). Besides, the epigenetic drugs could also be combined with the MEK/BRAF inhibitors and PARP inhibitors (Thy et al., 2021). However, the toxicity and tolerability of these drug combinations is the biggest question in clinical trials.
The epigenetic regulation was found to overcome drug resistance of the immune-checkpoint blockade (ICB). The combinations of epigenetic drugs and immune-checkpoint inhibitors were demonstrated effective for the treatment of cancers which were refractory or resistant to ICB, both in preclinical and clinical studies. The combinations of HDACi and ICB have received favorable clinical effect in the clinical trials, such as vorinostat and pembrolizumab (anti-PD-1 antibody) for the treatment of ICB-resistant metastatic NSCLC (Rodriguez et al., 2020), entinostat and pembrolizumab for the treatment of microsatellite-stable CRC (Medina Lopez et al., 2022). It is worth mentioning that the combinations of epigenetic drugs and immune-checkpoint inhibitors are usually well tolerated without severe toxic effects, which are superior to the combinations with targeted therapy.
Nonetheless, extended application of epigenetic drugs could induce harmful influence in the antitumor immunity. For instance, the BETi could cause severe depletion of T cells in the tumor environment (Wu et al., 2021). Therefore, the sequential or intermittent dosage regimen was adopted to induce the initiation of the epigenetic regulation and create an anti-cancer microenvironment during the treatment.
Epigenetic biomarkers are able to provide relevant information for diagnosis, prognosis and therapy optimization in routine clinical treatment and drug discovery. Epigenetic biomarkers may provide a rationale for patient stratification and precision medicine, thus maximizing the chances of treatment success while minimizing unwanted effects. Epigenetic biomarkers can also provide extra advantages, including low patient invasiveness. For example, variations in DNA methylation can be detected in body fluids and liquid biopsies (Liu et al., 2018). The development of accurate measurements of epigenetic alterations of specific targets in patients will greatly guide the clinical application of epigenetic drugs. The DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) methylation status is the first discovered biomarker in neuro-oncology. The promoter methylation of MGMT in glioblastomas could predict the therapeutic effect of temozolomide (Hegi et al., 2005). It has been shown in vitro that azacytidine and decitabine use different human nucleoside transporters (hNTs), and that cytotoxicity is dependent on hNT presence. These observations suggest that hNTs may be useful biomarkers for the efficacy of DNMTis, but clinical data are still not available (Damaraju et al., 2012). Unfortunately, the most extensively studied biomarker for HDACi activity is acetylation levels of the target proteins before and after treatment in peripheral blood or tumor tissue, but no correlation to clinical response has been found. Indeed, hyperacetylation was generally observed in all patients irrespective of response to HDACi (Ellis et al., 2008; Haigentz et al., 2012). The application of patient-stratified epigenetic biomarker, along with predictive models, will take our understanding and use of cancer epigenetics to a new level in the diagnosis, prognosis and treatment of cancer patients.
Although epigenetic drugs have made great progress in cancer drug development, the problems that arise are not to be underestimated. The first and most serious problem is that the low selectivity of epigenetic drugs leads to serious adverse reactions, such as the HDACi. Therefore, the search for epigenetic drugs with better selectivity that can target more elaborate isoform of epigenetic target may be one of the significant development directions in the future. In addition, monotherapy of epigenetic drugs presented favorable efficacy in hematologic cancer rather than in solid tumor. Therefore, the combination of epigenetic drugs and other antitumor therapies in the treatment of insensitive solid tumors and drug-resistant recurrent tumors is in active development. Unfortunately, the occurrence of serious toxic effects is still the main reason that disturbs the application of combined therapy. Hence, the exploration of optimizing the combination regimen and reducing the administered dosage may be promising directions for the extensive application of epigenetic drugs in the future. In summary, this review systematically concluded the recent progress of the epigenetic therapeutics in the treatment of cancers (Table 1) and the combination strategy with other therapies (Figure 1). Epigenetic drugs still have a broad prospect in the treatment of cancers. Optimizing the combination administration regimen to reduce toxic side effects and developing new epigenetic drugs with less toxicity may be two significant directions in the future.
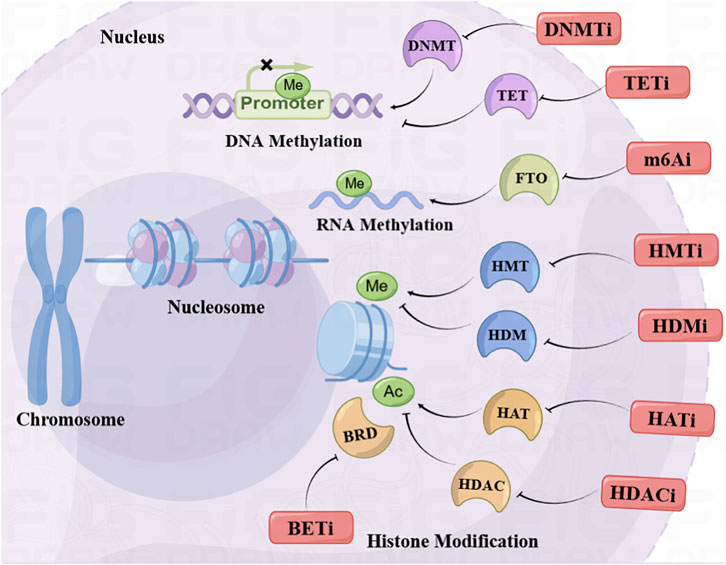
FIGURE 1. Recent advances in epigenetic anticancer therapeutics.
JW conceived the idea for the manuscript. LR drafted the manuscript, and YY, WL, HY, YZ, BG, SZ and GD revised it. All authors agreed to the final submitted version of the manuscript.
This research was funded by Beijing Natural Science Foundation (7212157), CAMS Initiative for Innovative Medicine (2021-1-I2M-029, 2022-I2M-JB-011), National Natural Science Foundation of China (81803584, 81703536), Technology Major Projects for “Major New Drugs Innovation and Development” (2018ZX09711001-005-025, 2018ZX09711001-012).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Albrecht, B. K., Gehling, V. S., Hewitt, M. C., Vaswani, R. G., Cote, A., Leblanc, Y., et al. (2016). Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J. Med. Chem. 59, 1330–1339. doi:10.1021/acs.jmedchem.5b01882
Amato, R. J., Stephenson, J., Hotte, S., Nemunaitis, J., Belanger, K., Reid, G., et al. (2012). MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Invest. 30, 415–421. doi:10.3109/07357907.2012.675381
Anastasiadou, E., Jacob, L. S., and Slack, F. J. (2018). Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18. doi:10.1038/nrc.2017.99
Arrighetti, N., Corno, C., and Gatti, L. (2015). Drug combinations with HDAC inhibitors in antitumor therapy. Crit. Rev. Oncog. 20, 83–117. doi:10.1615/critrevoncog.2014012378
Arvidsson, Y., Johanson, V., Pfragner, R., Wangberg, B., and Nilsson, O. (2016). Cytotoxic effects of valproic acid on neuroendocrine tumour cells. Neuroendocrinology 103, 578–591. doi:10.1159/000441849
Asangani, I. A., Dommeti, V. L., Wang, X., Malik, R., Cieslik, M., Yang, R., et al. (2014). Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510, 278–282. doi:10.1038/nature13229
Asangani, I. A., Wilder-Romans, K., Dommeti, V. L., Krishnamurthy, P. M., Apel, I. J., Escara-Wilke, J., et al. (2016). BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol. Cancer Res. 14, 324–331. doi:10.1158/1541-7786.MCR-15-0472
Balboni, B., El Hassouni, B., Honeywell, R. J., Sarkisjan, D., Giovannetti, E., Poore, J., et al. (2019). RX-3117 (fluorocyclopentenyl cytosine): A novel specific antimetabolite for selective cancer treatment. Expert Opin. Investig. Drugs 28, 311–322. doi:10.1080/13543784.2019.1583742
Beetch, M., Boycott, C., Harandi-Zadeh, S., Yang, T., Martin, B. J. E., Dixon-McDougall, T., et al. (2021). Pterostilbene leads to DNMT3B-mediated DNA methylation and silencing of OCT1-targeted oncogenes in breast cancer cells. J. Nutr. Biochem. 98, 108815. doi:10.1016/j.jnutbio.2021.108815
Berthon, C., Raffoux, E., Thomas, X., Vey, N., Gomez-Roca, C., Yee, K., et al. (2016). Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 3, e186–e195. doi:10.1016/S2352-3026(15)00247-1
Bijian, K., Kaldre, D., Wang, T. T., Su, J., Bouttier, M., Boucher, A., et al. (2018). Efficacy of hybrid vitamin D receptor agonist/histone deacetylase inhibitors in vitamin D-resistant triple-negative 4T1 breast cancer. J. Steroid Biochem. Mol. Biol. 177, 135–139. doi:10.1016/j.jsbmb.2017.08.010
Bird, S., Pawlyn, C., Nallamilli, S., Sriskandarajah, P., Kaiser, M., Yong, K., et al. (2020). A real-world study of panobinostat, weekly bortezomib and dexamethasone in a very heavily pretreated population of multiple-myeloma patients. Br. J. Haematol. 191, 927–930. doi:10.1111/bjh.17076
Bitzer, M., Horger, M., Giannini, E. G., Ganten, T. M., Worns, M. A., Siveke, J. T., et al. (2016). Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma - the SHELTER study. J. Hepatol. 65, 280–288. doi:10.1016/j.jhep.2016.02.043
Black, J. C., Van Rechem, C., and Whetstine, J. R. (2012). Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 48, 491–507. doi:10.1016/j.molcel.2012.11.006
Boyson, S. P., Gao, C., Quinn, K., Boyd, J., Paculova, H., Frietze, S., et al. (2021). Functional roles of bromodomain proteins in cancer. Cancers (Basel) 13, 3606. doi:10.3390/cancers13143606
Brueckner, B., Rius, M., Markelova, M. R., Fichtner, I., Hals, P. A., Sandvold, M. L., et al. (2010). Delivery of 5-azacytidine to human cancer cells by elaidic acid esterification increases therapeutic drug efficacy. Mol. Cancer Ther. 9, 1256–1264. doi:10.1158/1535-7163.MCT-09-1202
Byun, M. R., Lee, D. H., Jang, Y. P., Lee, H. S., Choi, J. W., and Lee, S. K. (2019). Repurposing natural products as novel HDAC inhibitors by comparative analysis of gene expression profiles. Phytomedicine 59, 152900. doi:10.1016/j.phymed.2019.152900
Chae, Y. C., Kim, J. Y., Park, J. W., Kim, K. B., Oh, H., Lee, K. H., et al. (2019). FOXO1 degradation via G9a-mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res. 47, 1692–1705. doi:10.1093/nar/gky1230
Chedin, F. (2011). The DNMT3 family of mammalian de novo DNA methyltransferases. Prog. Mol. Biol. Transl. Sci. 101, 255–285. doi:10.1016/B978-0-12-387685-0.00007-X
Choy, E., Ballman, K., Chen, J., Dickson, M. A., Chugh, R., George, S., et al. (2018). SARC018_SPORE02: Phase II study of mocetinostat administered with gemcitabine for patients with metastatic leiomyosarcoma with progression or relapse following prior treatment with gemcitabine-containing therapy. Sarcoma 2018, 2068517. doi:10.1155/2018/2068517
Choy, E., Flamand, Y., Balasubramanian, S., Butrynski, J. E., Harmon, D. C., George, S., et al. (2015). Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer 121, 1223–1230. doi:10.1002/cncr.29175
Cogle, C. R., Scott, B. L., Boyd, T., and Garcia-Manero, G. (2015). Oral azacitidine (CC-486) for the treatment of myelodysplastic syndromes and acute myeloid leukemia. Oncologist 20, 1404–1412. doi:10.1634/theoncologist.2015-0165
Connolly, R. M., Zhao, F., Miller, K. D., Lee, M. J., Piekarz, R. L., Smith, K. L., et al. (2021). E2112: Randomized phase III trial of endocrine therapy plus entinostat or placebo in hormone receptor-positive advanced breast cancer. A trial of the ECOG-ACRIN cancer research group. J. Clin. Oncol. 39, 3171–3181. doi:10.1200/JCO.21.00944
Cornett, E. M., Ferry, L., Defossez, P. A., and Rothbart, S. B. (2019). Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell 75, 1092–1101. doi:10.1016/j.molcel.2019.08.026
Coude, M. M., Braun, T., Berrou, J., Dupont, M., Bertrand, S., Masse, A., et al. (2015). BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 6, 17698–17712. doi:10.18632/oncotarget.4131
Dai, X. J., Liu, Y., Xiong, X. P., Xue, L. P., Zheng, Y. C., and Liu, H. M. (2020). Tranylcypromine based lysine-specific demethylase 1 inhibitor: Summary and perspective. J. Med. Chem. 63, 14197–14215. doi:10.1021/acs.jmedchem.0c00919
Damaraju, V. L., Mowles, D., Yao, S., Ng, A., Young, J. D., Cass, C. E., et al. (2012). Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids 31, 236–255. doi:10.1080/15257770.2011.652330
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. doi:10.1038/nature08617
Das, J., Chandra, L., Gandhi, G., Amle, D. B., Patnayak, R. L., Khurana, N., et al. (2022). Evaluation of promoter hypermethylation of tumor suppressor gene BRCA1 in epithelial ovarian cancer. J. Cancer Res. Ther. 18, 1578–1582. doi:10.4103/jcrt.JCRT_390_20
Delmore, J. E., Issa, G. C., Lemieux, M. E., Rahl, P. B., Shi, J., Jacobs, H. M., et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. doi:10.1016/j.cell.2011.08.017
Devaiah, B. N., Mu, J., Akman, B., Uppal, S., Weissman, J. D., Cheng, D., et al. (2020). MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. U. S. A. 117, 13457–13467. doi:10.1073/pnas.1919507117
Dhillon, S. (2020). Decitabine/Cedazuridine: First approval. Drugs 80, 1373–1378. doi:10.1007/s40265-020-01389-7
Duan, R., Du, W., and Guo, W. (2020). EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 13, 104. doi:10.1186/s13045-020-00937-8
Ellis, L., Pan, Y., Smyth, G. K., George, D. J., McCormack, C., Williams-Truax, R., et al. (2008). Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin. Cancer Res. 14, 4500–4510. doi:10.1158/1078-0432.CCR-07-4262
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874. doi:10.1038/nrg3074
Fang, Y., Liao, G., and Yu, B. (2019). LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 12, 129. doi:10.1186/s13045-019-0811-9
Feng, Y., Wang, Y., Wang, X., He, X., Yang, C., Naseri, A., et al. (2020). Simultaneous epigenetic perturbation and genome imaging reveal distinct roles of H3K9me3 in chromatin architecture and transcription. Genome Biol. 21, 296. doi:10.1186/s13059-020-02201-1
Ferrari, A. C., Alumkal, J. J., Stein, M. N., Taplin, M. E., Babb, J., Barnett, E. S., et al. (2019). Epigenetic therapy with panobinostat combined with bicalutamide rechallenge in castration-resistant prostate cancer. Clin. Cancer Res. 25, 52–63. doi:10.1158/1078-0432.CCR-18-1589
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. doi:10.1038/nature09504
Flausino, C. S., Daniel, F. I., and Modolo, F. (2021). DNA methylation in oral squamous cell carcinoma: From its role in carcinogenesis to potential inhibitor drugs. Crit. Rev. Oncol. Hematol. 164, 103399. doi:10.1016/j.critrevonc.2021.103399
Galli, M., Salmoiraghi, S., Golay, J., Gozzini, A., Crippa, C., Pescosta, N., et al. (2010). A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann. Hematol. 89, 185–190. doi:10.1007/s00277-009-0793-8
Gao, X. N., Lin, J., Ning, Q. Y., Gao, L., Yao, Y. S., Zhou, J. H., et al. (2013). A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS One 8, e55481. doi:10.1371/journal.pone.0055481
Gao, Y., and Ge, W. (2018). The histone methyltransferase DOT1L inhibits osteoclastogenesis and protects against osteoporosis. Cell Death Dis. 9, 33. doi:10.1038/s41419-017-0040-5
Garcia-Manero, G., Abaza, Y., Takahashi, K., Medeiros, B. C., Arellano, M., Khaled, S. K., et al. (2019). Pracinostat plus azacitidine in older patients with newly diagnosed acute myeloid leukemia: Results of a phase 2 study. Blood Adv. 3, 508–518. doi:10.1182/bloodadvances.2018027409
Garcia-Manero, G., Roboz, G., Walsh, K., Kantarjian, H., Ritchie, E., Kropf, P., et al. (2019). Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: Phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol. 6, e317–e327. doi:10.1016/S2352-3026(19)30029-8
Ghizzoni, M., Haisma, H. J., and Dekker, F. J. (2009). Reactivity of isothiazolones and isothiazolone-1-oxides in the inhibition of the PCAF histone acetyltransferase. Eur. J. Med. Chem. 44, 4855–4861. doi:10.1016/j.ejmech.2009.07.025
Ghizzoni, M., Wu, J., Gao, T., Haisma, H. J., Dekker, F. J., and George Zheng, Y. (2012). 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 47, 337–344. doi:10.1016/j.ejmech.2011.11.001
Girard, N., Bazille, C., Lhuissier, E., Benateau, H., Llombart-Bosch, A., Boumediene, K., et al. (2014). 3-Deazaneplanocin A (DZNep), an inhibitor of the histone methyltransferase EZH2, induces apoptosis and reduces cell migration in chondrosarcoma cells. PLoS One 9, e98176. doi:10.1371/journal.pone.0098176
Graca, I., Sousa, E. J., Costa-Pinheiro, P., Vieira, F. Q., Torres-Ferreira, J., Martins, M. G., et al. (2014). Anti-neoplastic properties of hydralazine in prostate cancer. Oncotarget 5, 5950–5964. doi:10.18632/oncotarget.1909
Gravina, G. L., Festuccia, C., Marampon, F., Popov, V. M., Pestell, R. G., Zani, B. M., et al. (2010). Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol. Cancer 9, 305. doi:10.1186/1476-4598-9-305
Haigentz, M., Kim, M., Sarta, C., Lin, J., Keresztes, R. S., Culliney, B., et al. (2012). Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 48, 1281–1288. doi:10.1016/j.oraloncology.2012.05.024
He, L., Li, H., Wu, A., Peng, Y., Shu, G., and Yin, G. (2019). Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18, 176. doi:10.1186/s12943-019-1109-9
Hegi, M. E., Diserens, A. C., Gorlia, T., Hamou, M. F., de Tribolet, N., Weller, M., et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003. doi:10.1056/NEJMoa043331
Ho, T. C. S., Chan, A. H. Y., and Ganesan, A. (2020). Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 63, 12460–12484. doi:10.1021/acs.jmedchem.0c00830
Holz, J. (2022). ClinicalTrials.gov. NCT05584111. Available at: https://clinicaltrials.gov/ct2/show/record/NCT05584111. [Acessed at October 18, 2022].
Huang, Y., Su, R., Sheng, Y., Dong, L., Dong, Z., Xu, H., et al. (2019). Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35, 677–691. doi:10.1016/j.ccell.2019.03.006
Huang, Y., Yan, J., Li, Q., Li, J., Gong, S., Zhou, H., et al. (2015). Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 43, 373–384. doi:10.1093/nar/gku1276
Hughes, A. L., Kelley, J. R., and Klose, R. J. (2020). Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Biochim. Biophys. Acta Gene Regul. Mech. 1863, 194567. doi:10.1016/j.bbagrm.2020.194567
Icardi, L., De Bosscher, K., and Tavernier, J. (2012). The HAT/HDAC interplay: Multilevel control of STAT signaling. Cytokine Growth Factor Rev. 23, 283–291. doi:10.1016/j.cytogfr.2012.08.002
Isaacs, J. T., Antony, L., Dalrymple, S. L., Brennen, W. N., Gerber, S., Hammers, H., et al. (2013). Tasquinimod Is an Allosteric Modulator of HDAC4 survival signaling within the compromised cancer microenvironment. Cancer Res. 73, 1386–1399. doi:10.1158/0008-5472.CAN-12-2730
Jansens, R. J. J., Verhamme, R., Mirza, A. H., Olarerin-George, A., Van Waesberghe, C., Jaffrey, S. R., et al. (2022). Alphaherpesvirus US3 protein-mediated inhibition of the m6A mRNA methyltransferase complex. Cell Rep. 40, 111107. doi:10.1016/j.celrep.2022.111107
Jiang, Y., Xiang, C., Zhong, F., Zhang, Y., Wang, L., Zhao, Y., et al. (2021). Histone H3K27 methyltransferase EZH2 and demethylase JMJD3 regulate hepatic stellate cells activation and liver fibrosis. Theranostics 11, 361–378. doi:10.7150/thno.46360
Jurmeister, P., Gloss, S., Roller, R., Leitheiser, M., Schmid, S., Mochmann, L. H., et al. (2022). DNA methylation-based classification of sinonasal tumors. Nat. Commun. 13, 7148. doi:10.1038/s41467-022-34815-3
Ka-Yue Chow, L., Lai-Shun Chung, D., Tao, L., Chan, K. F., Tung, S. Y., Cheong Ngan, R. K., et al. (2022). Epigenomic landscape study reveals molecular subtypes and EBV-associated regulatory epigenome reprogramming in nasopharyngeal carcinoma. EBioMedicine 86, 104357. doi:10.1016/j.ebiom.2022.104357
Knutson, S. K., Kawano, S., Minoshima, Y., Warholic, N. M., Huang, K. C., Xiao, Y., et al. (2014). Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854. doi:10.1158/1535-7163.MCT-13-0773
Kogo, R., Shimamura, T., Mimori, K., Kawahara, K., Imoto, S., Sudo, T., et al. (2011). Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326. doi:10.1158/0008-5472.CAN-11-1021
Kojima, M., Sone, K., Oda, K., Hamamoto, R., Kaneko, S., Oki, S., et al. (2020). The histone methyltransferase SMYD2 is a novel therapeutic target for the induction of apoptosis in ovarian clear cell carcinoma cells. Oncol. Lett. 20, 153. doi:10.3892/ol.2020.12014
Lee, E. Q., Reardon, D. A., Schiff, D., Drappatz, J., Muzikansky, A., Grimm, S. A., et al. (2015). Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 17, 862–867. doi:10.1093/neuonc/nou350
Lee, P. P., Fitzpatrick, D. R., Beard, C., Jessup, H. K., Lehar, S., Makar, K. W., et al. (2001). A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774. doi:10.1016/s1074-7613(01)00227-8
Leonard, B., Brand, T. M., O'Keefe, R. A., Lee, E. D., Zeng, Y., Kemmer, J. D., et al. (2018). BET inhibition overcomes receptor tyrosine kinase-mediated cetuximab resistance in HNSCC. Cancer Res. 78, 4331–4343. doi:10.1158/0008-5472.CAN-18-0459
Li, Q., Qu, B., Shen, H., Deng, H., and Sun, L. (2022). Histone demethylase GASC1 inhibitor targeted GASC1 gene to inhibit the malignant transformation of esophageal cancer through the NOTCH-MAPK signaling pathway. Ann. Clin. Lab. Sci. 52, 240–248.
Li, R. G., Deng, H., Liu, X. H., Chen, Z. Y., Wan, S. S., and Wang, L. (2021). Histone methyltransferase G9a promotes the development of renal cancer through epigenetic silencing of tumor suppressor gene SPINK5. Oxid. Med. Cell Longev. 2021, 6650781. doi:10.1155/2021/6650781
Li, Y., Zhao, J., Gutgesell, L. M., Shen, Z., Ratia, K., Dye, K., et al. (2020). Novel pyrrolopyridone bromodomain and extra-terminal motif (BET) inhibitors effective in endocrine-resistant ER+ breast cancer with acquired resistance to fulvestrant and palbociclib. J. Med. Chem. 63, 7186–7210. doi:10.1021/acs.jmedchem.0c00456
Liu, L., Toung, J. M., Jassowicz, A. F., Vijayaraghavan, R., Kang, H., Zhang, R., et al. (2018). Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann. Oncol. 29, 1445–1453. doi:10.1093/annonc/mdy119
Liu, Q., and Wang, M. W. (2016). Histone lysine methyltransferases as anti-cancer targets for drug discovery. Acta Pharmacol. Sin. 37, 1273–1280. doi:10.1038/aps.2016.64
Loprevite, M., Tiseo, M., Grossi, F., Scolaro, T., Semino, C., Pandolfi, A., et al. (2005). In vitro study of CI-994, a histone deacetylase inhibitor, in non-small cell lung cancer cell lines. Oncol. Res. 15, 39–48. doi:10.3727/096504005775082066
Maeda, K., Kawakami, K., Ishida, Y., Ishiguro, K., Omura, K., and Watanabe, G. (2003). Hypermethylation of the CDKN2A gene in colorectal cancer is associated with shorter survival. Oncol. Rep. 10, 935–938. doi:10.3892/or.10.4.935
Matsui, M., and Corey, D. R. (2017). Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 16, 167–179. doi:10.1038/nrd.2016.117
Medina Lopez, R. A., Rivero Belenchon, I., Mazuecos-Quiros, J., Congregado-Ruiz, C. B., and Counago, F. (2022). Update on the treatment of metastatic renal cell carcinoma. World J. Clin. Oncol. 13, 1–8. doi:10.5306/wjco.v13.i1.1
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149, 1635–1646. doi:10.1016/j.cell.2012.05.003
Millan-Zambrano, G., Burton, A., Bannister, A. J., and Schneider, R. (2022). Histone post-translational modifications - cause and consequence of genome function. Nat. Rev. Genet. 23, 563–580. doi:10.1038/s41576-022-00468-7
Morgan, M. A. J., and Shilatifard, A. (2020). Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 52, 1271–1281. doi:10.1038/s41588-020-00736-4
Mueller, S., Yang, X., Sottero, T. L., Gragg, A., Prasad, G., Polley, M. Y., et al. (2011). Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: Efficacy and underlying mechanisms. Cancer Lett. 306, 223–229. doi:10.1016/j.canlet.2011.03.010
Nicolas, D., Zoller, B., Suter, D. M., and Naef, F. (2018). Modulation of transcriptional burst frequency by histone acetylation. Proc. Natl. Acad. Sci. U. S. A. 115, 7153–7158. doi:10.1073/pnas.1722330115
Nowak, R. P., Tumber, A., Johansson, C., Che, K. H., Brennan, P., Owen, D., et al. (2016). Advances and challenges in understanding histone demethylase biology. Curr. Opin. Chem. Biol. 33, 151–159. doi:10.1016/j.cbpa.2016.06.021
Ny, L., Jespersen, H., Karlsson, J., Alsen, S., Filges, S., All-Eriksson, C., et al. (2021). The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 12, 5155. doi:10.1038/s41467-021-25332-w
Ojha, R., Chen, I. C., Hsieh, C. M., Nepali, K., Lai, R. W., Hsu, K. C., et al. (2021). Installation of pargyline, a LSD1 inhibitor, in the HDAC inhibitory template culminated in the identification of a tractable antiprostate cancer agent. J. Med. Chem. 64, 17824–17845. doi:10.1021/acs.jmedchem.1c00966
Padeken, J., Methot, S. P., and Gasser, S. M. (2022). Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 23, 623–640. doi:10.1038/s41580-022-00483-w
Pan, K., Hu, B., Wang, L., Yuan, J., and Xu, W. (2022). STUB1-SMYD2 Axis regulates drug resistance in glioma cells. J. Mol. Neurosci. 72, 2030–2044. doi:10.1007/s12031-022-02051-5
Pan, Y. M., Wang, C. G., Zhu, M., Xing, R., Cui, J. T., Li, W. M., et al. (2016). STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol. Cancer 15, 79. doi:10.1186/s12943-016-0561-z
Peterson, L. M., Kurland, B. F., Yan, F., Jiresova, A. N., Gadi, V. K., Specht, J. M., et al. (2021). 18F-Fluoroestradiol PET imaging in a phase II trial of vorinostat to restore endocrine sensitivity in ER+/HER2− metastatic breast cancer. J. Nucl. Med. 62, 184–190. doi:10.2967/jnumed.120.244459
Pili, R., Liu, G., Chintala, S., Verheul, H., Rehman, S., Attwood, K., et al. (2017). Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br. J. Cancer 116, 874–883. doi:10.1038/bjc.2017.33
Qin, Z. Y., Wang, T., Su, S., Shen, L. T., Zhu, G. X., Liu, Q., et al. (2019). BRD4 promotes gastric cancer progression and metastasis through acetylation-dependent stabilization of snail. Cancer Res. 79, 4869–4881. doi:10.1158/0008-5472.CAN-19-0442
Qu, X., Alsager, S., Zhuo, Y., and Shan, B. (2019). HOX transcript antisense RNA (HOTAIR) in cancer. Cancer Lett. 454, 90–97. doi:10.1016/j.canlet.2019.04.016
Raas, M. W. D., Zijlmans, D. W., Vermeulen, M., and Marks, H. (2022). There is another: H3K27me3-mediated genomic imprinting. Trends Genet. 38, 82–96. doi:10.1016/j.tig.2021.06.017
Ramakrishnan, J., Magudeeswaran, S., Suresh, S., and Poomani, K. (2022). Investigation of intermolecular interactions and binding mechanism of PU139 and PU141 molecules with p300 HAT enzyme via molecular docking, molecular dynamics simulations and binding free energy analysis. J. Biomol. Struct. Dyn. 2022, 1–15. doi:10.1080/07391102.2021.2020164
Ree, A. H., Dueland, S., Folkvord, S., Hole, K. H., Seierstad, T., Johansen, M., et al. (2010). Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: The pelvic radiation and vorinostat (PRAVO) phase 1 study. Lancet Oncol. 11, 459–464. doi:10.1016/S1470-2045(10)70058-9
Ribrag, V., Kim, W. S., Bouabdallah, R., Lim, S. T., Coiffier, B., Illes, A., et al. (2017). Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-hodgkin lymphoma and chronic lymphocytic leukemia: Results of a phase II study. Haematologica 102, 903–909. doi:10.3324/haematol.2016.154377
Richards, D. A., Boehm, K. A., Waterhouse, D. M., Wagener, D. J., Krishnamurthi, S. S., Rosemurgy, A., et al. (2006). Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: Results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann. Oncol. 17, 1096–1102. doi:10.1093/annonc/mdl081
Rinn, J. L. (2014). lncRNAs: linking RNA to chromatin. Cold Spring Harb. Perspect. Biol. 6, a018614. doi:10.1101/cshperspect.a018614
Rodriguez, C. P., Wu, Q. V., Voutsinas, J., Fromm, J. R., Jiang, X., Pillarisetty, V. G., et al. (2020). A phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin. Cancer Res. 26, 837–845. doi:10.1158/1078-0432.CCR-19-2214
Saghafinia, S., Mina, M., Riggi, N., Hanahan, D., and Ciriello, G. (2018). Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep. 25, 1066–1080. doi:10.1016/j.celrep.2018.09.082
Sborov, D. W., Canella, A., Hade, E. M., Mo, X., Khountham, S., Wang, J., et al. (2017). A phase 1 trial of the HDAC inhibitor AR-42 in patients with multiple myeloma and T- and B-cell lymphomas. Leuk. Lymphoma 58, 2310–2318. doi:10.1080/10428194.2017.1298751
Sharma, S. K., Wu, Y., Steinbergs, N., Crowley, M. L., Hanson, A. S., Casero, R. A., et al. (2010). (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J. Med. Chem. 53, 5197–5212. doi:10.1021/jm100217a
Shi, J., Wang, Y., Zeng, L., Wu, Y., Deng, J., Zhang, Q., et al. (2014). Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25, 210–225. doi:10.1016/j.ccr.2014.01.028
Shvedunova, M., and Akhtar, A. (2022). Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 23, 329–349. doi:10.1038/s41580-021-00441-y
Siegel, D., Hussein, M., Belani, C., Robert, F., Galanis, E., Richon, V. M., et al. (2009). Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2, 31. doi:10.1186/1756-8722-2-31
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2021). Cancer statistics, 2017. CA Cancer J. Clin. 71, 7–30. doi:10.3322/caac.21387
Strzyz, P. (2022). Cellular role of TETs in early embryos. Nat. Rev. Mol. Cell Biol. 23, 581. doi:10.1038/s41580-022-00526-2
Stuhlmiller, T. J., Miller, S. M., Zawistowski, J. S., Nakamura, K., Beltran, A. S., Duncan, J. S., et al. (2015). Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404. doi:10.1016/j.celrep.2015.03.037
Sun, N., Zhang, J., Zhang, C., Zhao, B., and Jiao, A. (2018). DNMTs inhibitor SGI-1027 induces apoptosis in Huh7 human hepatocellular carcinoma cells. Oncol. Lett. 16, 5799–5806. doi:10.3892/ol.2018.9390
Sweis, R. F., Wang, Z., Algire, M., Arrowsmith, C. H., Brown, P. J., Chiang, G. G., et al. (2015). Discovery of A-893, A new cell-active benzoxazinone inhibitor of lysine methyltransferase SMYD2. ACS Med. Chem. Lett. 6, 695–700. doi:10.1021/acsmedchemlett.5b00124
Tajima, S., Suetake, I., Takeshita, K., Nakagawa, A., and Kimura, H. (2016). Domain structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA methyltransferases. Adv. Exp. Med. Biol. 945, 63–86. doi:10.1007/978-3-319-43624-1_4
Takemoto, Y., Ito, A., Niwa, H., Okamura, M., Fujiwara, T., Hirano, T., et al. (2016). Identification of cyproheptadine as an inhibitor of SET domain containing lysine methyltransferase 7/9 (Set7/9) that regulates estrogen-dependent transcription. J. Med. Chem. 59, 3650–3660. doi:10.1021/acs.jmedchem.5b01732
Thy, S., Hommel, A., Meneceur, S., Bartkowiak, A. L., Schulz, W. A., Niegisch, G., et al. (2021). Epigenetic treatment of urothelial carcinoma cells sensitizes to cisplatin chemotherapy and PARP inhibitor treatment. Cancers (Basel) 13, 1376. doi:10.3390/cancers13061376
Tinari, N., De Tursi, M., Grassadonia, A., Zilli, M., Stuppia, L., Iacobelli, S., et al. (2012). An epigenetic approach to pancreatic cancer treatment: The prospective role of histone deacetylase inhibitors. Curr. Cancer Drug Targets 12, 439–452. doi:10.2174/156800912800190884
Tsai, M. C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., et al. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. doi:10.1126/science.1192002
Vatapalli, R., Sagar, V., Rodriguez, Y., Zhao, J. C., Unno, K., Pamarthy, S., et al. (2020). Histone methyltransferase DOT1L coordinates AR and MYC stability in prostate cancer. Nat. Commun. 11, 4153. doi:10.1038/s41467-020-18013-7
von Tresckow, B., Sayehli, C., Aulitzky, W. E., Goebeler, M. E., Schwab, M., Braz, E., et al. (2019). Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur. J. Haematol. 102, 163–173. doi:10.1111/ejh.13188
Waddell, A., Mahmud, I., Ding, H., Huo, Z., and Liao, D. (2021). Pharmacological inhibition of CBP/p300 blocks estrogen receptor alpha (ERα) function through suppressing enhancer H3K27 acetylation in luminal breast cancer. Cancers (Basel) 13, 2799. doi:10.3390/cancers13112799
Walewski, J., Paszkiewicz-Kozik, E., Borsaru, G., Hellmann, A., Janikova, A., Warszewska, A., et al. (2019). Resminostat in patients with relapsed or refractory Hodgkin lymphoma: Results of the phase II SAPHIRE study. Leuk. Lymphoma 60, 675–684. doi:10.1080/10428194.2018.1492122
Wang, P., Wang, Z., and Liu, J. (2020). Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 19, 5. doi:10.1186/s12943-019-1127-7
Wang, T., Kong, S., Tao, M., and Ju, S. (2020). The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 19, 88. doi:10.1186/s12943-020-01204-7
Wang, W., Zhao, M., Cui, L., Ren, Y., Zhang, J., Chen, J., et al. (2020). Characterization of a novel HDAC/RXR/HtrA1 signaling axis as a novel target to overcome cisplatin resistance in human non-small cell lung cancer. Mol. Cancer 19, 134. doi:10.1186/s12943-020-01256-9
Wass, M., Gollner, S., Besenbeck, B., Schlenk, R. F., Mundmann, P., Gothert, J. R., et al. (2021). A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia 35, 701–711. doi:10.1038/s41375-020-0892-z
Waters, N. J., Daigle, S. R., Rehlaender, B. N., Basavapathruni, A., Campbell, C. T., Jensen, T. B., et al. (2015). Exploring drug delivery for the DOT1L inhibitor pinometostat (EPZ-5676): Subcutaneous administration as an alternative to continuous IV infusion, in the pursuit of an epigenetic target. J. Control Release 220, 758–765. doi:10.1016/j.jconrel.2015.09.023
Willmann, D., Lim, S., Wetzel, S., Metzger, E., Jandausch, A., Wilk, W., et al. (2012). Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int. J. Cancer 131, 2704–2709. doi:10.1002/ijc.27555
Witta, S. E., Jotte, R. M., Konduri, K., Neubauer, M. A., Spira, A. I., Ruxer, R. L., et al. (2012). Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 30, 2248–2255. doi:10.1200/JCO.2011.38.9411
Wu, J., Xie, N., Wu, Z., Zhang, Y., and Zheng, Y. G. (2009). Bisubstrate inhibitors of the MYST HATs Esa1 and Tip60. Bioorg Med. Chem. 17, 1381–1386. doi:10.1016/j.bmc.2008.12.014
Wu, S., Jiang, Y., Hong, Y., Chu, X., Zhang, Z., Tao, Y., et al. (2021). BRD4 PROTAC degrader ARV-825 inhibits T-cell acute lymphoblastic leukemia by targeting 'Undruggable' Myc-pathway genes. Cancer Cell Int. 21, 230. doi:10.1186/s12935-021-01908-w
Xiao, L., Li, X., Mu, Z., Zhou, J., Zhou, P., Xie, C., et al. (2020). FTO inhibition enhances the antitumor effect of temozolomide by targeting MYC-miR-155/23a cluster-MXI1 feedback circuit in glioma. Cancer Res. 80, 3945–3958. doi:10.1158/0008-5472.CAN-20-0132
Yankova, E., Blackaby, W., Albertella, M., Rak, J., De Braekeleer, E., Tsagkogeorga, G., et al. (2021). Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601. doi:10.1038/s41586-021-03536-w
Ye, T., Wei, L., Shi, J., Jiang, K., Xu, H., Hu, L., et al. (2019). Sirtuin1 activator SRT2183 suppresses glioma cell growth involving activation of endoplasmic reticulum stress pathway. BMC Cancer 19, 706. doi:10.1186/s12885-019-5852-5
Younes, A., Oki, Y., Bociek, R. G., Kuruvilla, J., Fanale, M., Neelapu, S., et al. (2011). Mocetinostat for relapsed classical Hodgkin's lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 12, 1222–1228. doi:10.1016/S1470-2045(11)70265-0
Yu, J., Xie, T., Wang, Z., Wang, X., Zeng, S., Kang, Y., et al. (2019). DNA methyltransferases: Emerging targets for the discovery of inhibitors as potent anticancer drugs. Drug Discov. Today 24, 2323–2331. doi:10.1016/j.drudis.2019.08.006
Zandieh, M. A., Farahani, M. H., Rajabi, R., Avval, S. T., Karimi, K., Rahmanian, P., et al. (2022). Epigenetic regulation of autophagy by non-coding RNAs in gastrointestinal tumors: Biological functions and therapeutic perspectives. Pharmacol. Res. 187, 106582. doi:10.1016/j.phrs.2022.106582
Zauderer, M. G., Szlosarek, P. W., Le Moulec, S., Popat, S., Taylor, P., Planchard, D., et al. (2022). EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1-inactivated malignant pleural mesothelioma: A multicentre, open-label, phase 2 study. Lancet Oncol. 23, 758–767. doi:10.1016/S1470-2045(22)00277-7
Zhao, X., Yang, Y., Sun, B. F., Shi, Y., Yang, X., Xiao, W., et al. (2014). FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419. doi:10.1038/cr.2014.151
Zhou, K., Gaullier, G., and Luger, K. (2019). Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 26, 3–13. doi:10.1038/s41594-018-0166-x
Zhou, Z., Lv, J., Yu, H., Han, J., Yang, X., Feng, D., et al. (2020). Mechanism of RNA modification N6-methyladenosine in human cancer. Mol. Cancer 19, 104. doi:10.1186/s12943-020-01216-3
Zhu, J., Fu, H., Wu, Y., and Zheng, X. (2013). Function of lncRNAs and approaches to lncRNA-protein interactions. Sci. China Life Sci. 56, 876–885. doi:10.1007/s11427-013-4553-6
Zhu, Q., Huang, Y., Marton, L. J., Woster, P. M., Davidson, N. E., and Casero, R. A. (2012). Polyamine analogs modulate gene expression by inhibiting lysine-specific demethylase 1 (LSD1) and altering chromatin structure in human breast cancer cells. Amino Acids 42, 887–898. doi:10.1007/s00726-011-1004-1
Keywords: epigenetics, cancer, histone, epigenetic drug, combined pharmacotherapy
Citation: Ren L, Yang Y, Li W, Yang H, Zhang Y, Ge B, Zhang S, Du G and Wang J (2023) Recent advances in epigenetic anticancer therapeutics and future perspectives. Front. Genet. 13:1085391. doi: 10.3389/fgene.2022.1085391
Received: 31 October 2022; Accepted: 12 December 2022;
Published: 04 January 2023.
Edited by:
Gaël Roué, Vall d'Hebron Institute of Oncology (VHIO), SpainReviewed by:
Miguel F. Segura, Vall d'Hebron Research Institute (VHIR), SpainCopyright © 2023 Ren, Yang, Li, Yang, Zhang, Ge, Zhang, Du and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhua Wang, d2poQGltbS5hYy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.