 Benzhi Dong1
Benzhi Dong1 Bo Gao
Bo Gao Tianjiao Zhang
Tianjiao Zhang- 1College of Information and Computer Engineering, Northeast Forestry University, Harbin, China
- 2Tianjin Second People’s Hospital, Tianjin Institute of Hepatology, Tianjin, China
- 3Department of Radiology, The Second Affiliated Hospital of Harbin Medical University, Harbin, China
Antimicrobial peptides (AMPs) are alkaline substances with efficient bactericidal activity produced in living organisms. As the best substitute for antibiotics, they have been paid more and more attention in scientific research and clinical application. AMPs can be produced from almost all organisms and are capable of killing a wide variety of pathogenic microorganisms. In addition to being antibacterial, natural AMPs have many other therapeutically important activities, such as wound healing, antioxidant and immunomodulatory effects. To discover new AMPs, the use of wet experimental methods is expensive and difficult, and bioinformatics technology can effectively solve this problem. Recently, some deep learning methods have been applied to the prediction of AMPs and achieved good results. To further improve the prediction accuracy of AMPs, this paper designs a new deep learning method based on sequence multidimensional representation. By encoding and embedding sequence features, and then inputting the model to identify AMPs, high-precision classification of AMPs and Non-AMPs with lengths of 10–200 is achieved. The results show that our method improved accuracy by 1.05% compared to the most advanced model in independent data validation without decreasing other indicators.
1 Introduction
Antimicrobial peptides (AMPs) are host defense molecules produced by the innate immune system in a variety of organisms and have many advantages, such as rapid killing, low toxicity, and broad activity (Fjell et al., 2009), and their drug resistance is relatively low. About 50% of the amino acids in AMP are hydrophobic, and they can adopt an amphiphilic structure, which enables them to interact with and penetrate cell membranes, which then lead to disruption of membrane potential, changes in membrane permeability, and permeation of metabolites leakage, eventually leading to bacterial cell death (Kumar et al., 2018). AMPs not only exhibit synergy with antibiotics, but may also synergize with the immune system (Pasupuleti et al., 2012). At present, there are corresponding drug-resistant pathogenic strains of conventional antibiotics, and the drug-resistant problem of pathogenic bacteria has increasingly threatened people’s health. Finding new antibiotics is an effective way to solve the drug-resistant problem. The characteristics of high antibacterial activity, broad antibacterial spectrum, and wide selection range are considered to be an effective way to solve the problem of drug resistance (Hancock and Sahl, 2006). Given the multiple advantages of AMPs, there is an urgent need to identify new AMPs.
In recent years, the rapid development of bioinformatics has provided a rational design method for the acquisition of AMPs. We can predict AMPs based on their sequence information. At present, the research on sequence classification algorithms mainly focuses on the combination of classification algorithms and biological sequence features. Various applied machine learning models have also been applied in AMPs prediction, for example, support vector machines (SVM) (Lata et al., 2010; Meher et al., 2017; Agrawal et al., 2018; Gong et al., 2021; Zou et al., 2021; Zhang Q. et al., 2022), random forest (RF) (Bhadra et al., 2018; Veltri, 2015; Nakayama et al., 2021; Yang et al., 2021; Ao et al., 2022; Lv et al., 2022a), discriminant analysis (DA) (Thomas et al., 2010; Waghu et al., 2016), Hidden Markov (Fjell et al., 2009), k-nearest neighbors (Xiao et al., 2013), etc. The core problem of such methods is how to perform feature extraction on protein sequences, which is greatly affected by the feature extraction method, which limits the maximum performance of the model. In addition, artificial feature engineering is often required when machine learning builds a classification model. In this process, important information is likely to be lost. Deep learning methods that have developed rapidly in recent years can effectively solve this problem.
Deep learning methods can automatically learn features from the raw data through convolution operations, avoiding the loss of data features. Various deep learning methods have been applied in protein sequence classification, such as bidirectional long short-term memory network (Bi-LSTM) (Tng et al., 2021; Zhang Y. et al., 2022; Zhang et al., 2022c; Li et al., 2022; Qiao et al., 2022; Wang et al., 2022), two-dimensional convolutional neural network (2D CNN) (Le et al., 2021), deep residual network (ResNet) (Xu et al., 2021), graph convolutional network (GCN) (Chen et al., 2021), deep neural network (DNN) (Gao et al., 2019; Han et al., 2019; Le et al., 2019; Hathaway et al., 2021), and Recurrent Neural Network (RNN) (Zheng et al., 2020; Yun et al., 2021). These research methods have generally achieved good classification results and have attracted increasing attention. In the prediction of AMPs, deep learning methods have also received attention, such as deep neural network (DNN) (Veltri et al., 2018; Su et al., 2019; Fu et al., 2020; Yan et al., 2020), bidirectional long short-term memory network (Bi-LSTM) (Sharma et al., 2021a; Xiao et al., 2021; Sharma et al., 2022), and Transformer (Zhang et al., 2021). These models all demonstrate the superiority of deep learning in AMPs prediction.
Whether it is a machine learning method or a deep learning method, the first step of these methods is to represent protein sequences as machine-readable and to encode biological sequences with features, that is, to map biological sequences to digital sequences using digital signal processing methods. It is widely used in biological sequence classification. As an important biological sequence analysis method, biological sequence encoding has been studied by many scholars, for example, the interaction of protein sequences (Moretta et al., 2020; Khabbaz et al., 2021; Wani et al., 2021; Söylemez et al., 2022), sparse coding (binary coding) (Spänig and Heider, 2019; Akbar et al., 2021; Jain et al., 2021; Ren et al., 2022). In addition, pre-trained models in natural language processing (NLP) have been used in protein sequence analysis, for example, the word2vec method (Zhang et al., 2019; Dao et al., 2021) and the N-gram method (Li et al., 2018; Wu and Yu, 2021) showed excellent performance in prediction.
The AMPs classification methods are usually based on machine learning or deep learning consider the interaction between protein sequences and represents the sequences as a matrix, ignoring the upstream and downstream information of the sequences, and the prediction accuracy will be reduced during the classification process. In this paper, deep learning-based feature combinations of N-gram encoding, K-space amino acid pair composition (CKSAAP), position-weighted amino acid composition (PWAA), and raw sequence number encoding were selected to predict AMPs. The CKSAAP encoding effectively describes the short-range interactions between amino acids, the PWAA encoding determines the positional information of amino acids in the protein sequence, and considers the upstream and downstream information of the protein sequence, and the N-gram encoding enhances the expression of the protein sequence and reduces the training process. Information is lost. It not only considers the interaction and positional weight of amino acids in the protein sequence but also combines the upstream and downstream information in the sequence and enhances the expression of the AMPs sequence, avoiding the above problems and improving the prediction performance. To evaluate the model, we use a 10-fold cross-validation method. Figure 1 shows our workflow.
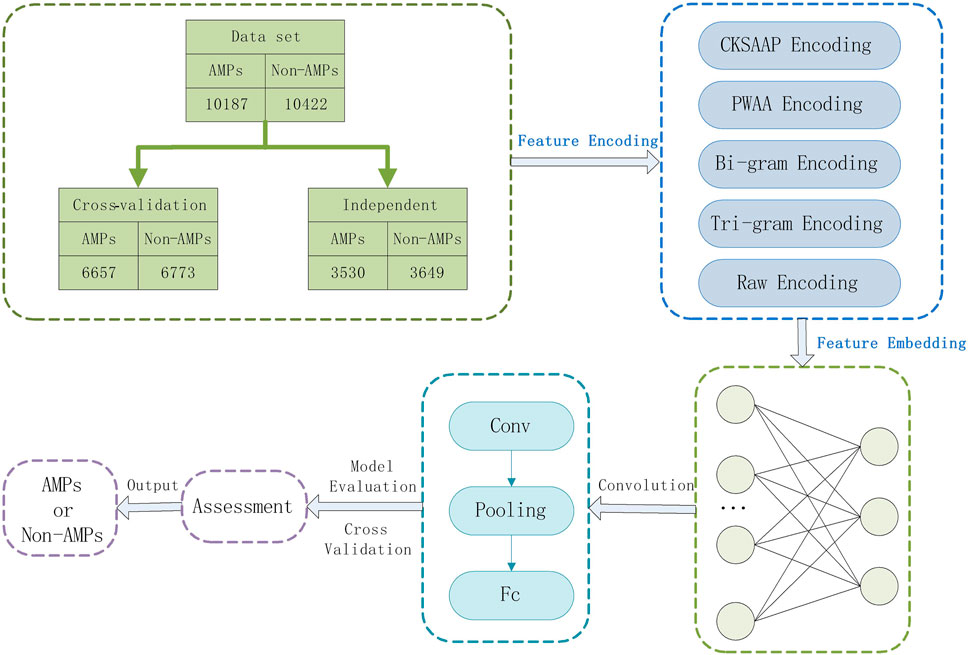
FIGURE 1. The workflow.
2 Materials and methods
2.1 Baseline datasets
In this study, we used the dataset of (Sharma et al., 2021b), which collected AMPs data belonging to 13 phyla and 41 kingdoms (animal kingdom) categories from NCBI and StarPepDB databases and obtained Non-AMPs data from the UniProt database. This dataset considers all AMPs of suitable length in the animal kingdom to train the model. After the data is de-redundant, the dataset finally consists of 10,187 AMPs and 10,422 Non-AMPs, shown in supplementary materialthe, which contains about 65% of AMPs and non-AMPs. AMPs were used as the cross-validation dataset to train our model, and the rest contained about 35% of AMPs and non-AMPs as independent datasets for evaluating model performance, whose composition is shown in Table 1.
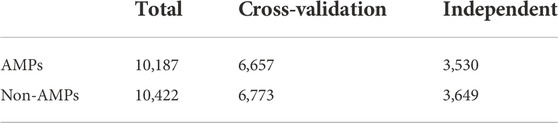
TABLE 1. Statistics for datasets.
2.2 Encoding method of sequence
2.2.1 Raw sequence encoding
Protein is composed of 20 kinds of amino acids, each amino acid is represented by a character, and the sequences represented by these 20 kinds of characters contain important biological genetic information. The raw sequence encoding, that is, mapping the sequence to a set of numbers, reflects the selection bias of the AMPs sequence at each amino acid position. If given a protein sequence of length n,
2.2.2 Composition of k-space amino acid pairs (CKSAAP) encoding
CKSAAP is a coding scheme based on the interaction between amino acid pairs, which has been widely used in protein prediction (Yuan et al., 2022). CKSAAP can represent amino acids as a combination of multiple amino acid pairs with spacing K (Chen et al., 2011), reflecting the short-range interaction between amino acid pairs. If K = 0, there are 400 residue pairs with spacing 0 (AA, AC, AD, AE, …, YY). The eigenvector can be calculated by Eq. 1:
Where, NTotal = L-K-1, NTotal represents the total number of residue pairs in the protein sequence, L represents the sequence length, and K represents the amino acid spacing. For example, when the sequence length is 200 and K = 0, 1, 2, 3, the values of NTotal are 199, 198, 197, and 196. In this paper, we take K as 0, 1, 2, 3, 4, and 5, so the total dimension of this feature is 2,400.
2.2.3 Position weighted amino acid composition (PWAA) encoding
To determine the position information of amino acids in the protein sequences, we used the PWAA method for encoding. Given amino acid residue ai (i = 1, 2, 3,..., 20), we can calculate the positional information of ai in a protein sequence by Eq. 2:
Where L represents the data of upstream residue or downstream residue at the central site of the protein sequence fragment, if ai is the residue at the jth position of the protein sequence fragment, then x (i, j) = 1, otherwise x (i, j) = 0. Generally, the closer ai is to the center position (position 0), The smaller the absolute value of Ci. The PWAA encoding involves 20 kinds of amino acid residues, so this method encodes a dimension of 20.
2.2.4 N-gram encoding
N-gram is a statistical language model, which can be applied to protein sequence analysis to enhance the expression of protein sequences (Sharma and Srivastava, 2021). We treat each amino acid residue of a protein sequence of length L-N+1 as a word and each sequence as a sentence. In this study, our data length is short, and the Bi-gram (binary model) and tri-gram (ternary model) we used are enough to enhance the expression of AMPs sequences. For an raw sequence of length n S= (s1, s2, … sn), Bi-gram can be expressed as S2=(s1s2,s2s3, … ,s(n-1)sn), whose length is n-1, and the coding process is shown in Figure 2. Similarly, Tri-gram can be expressed as S3=(s1s2s3,s2s3s4, … ,s(n-2)s(n-1)sn), whose length is n-2. To align the encoding length of the N-gram, we set the encoding length of the N-gram to 200, and the encodings shorter than 200 are padded with 0 at the end, so the dimensions of the Bi-gram and Tri-gram are 200 respectively.
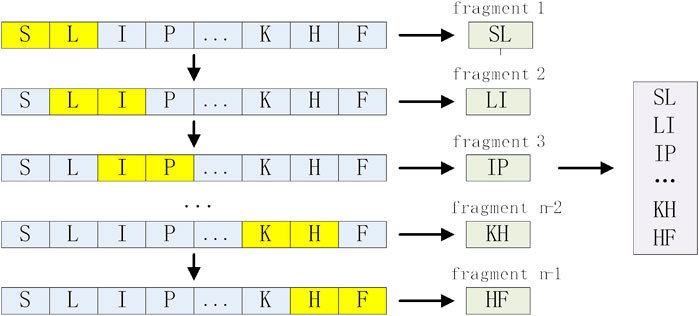
FIGURE 2. Bi-gram encoding process.
2.3 Deep learning model
Our deep learning model consists of three parts: encoding layer, embedding layer, and convolutional layer. The model architecture is shown in Figure 3.
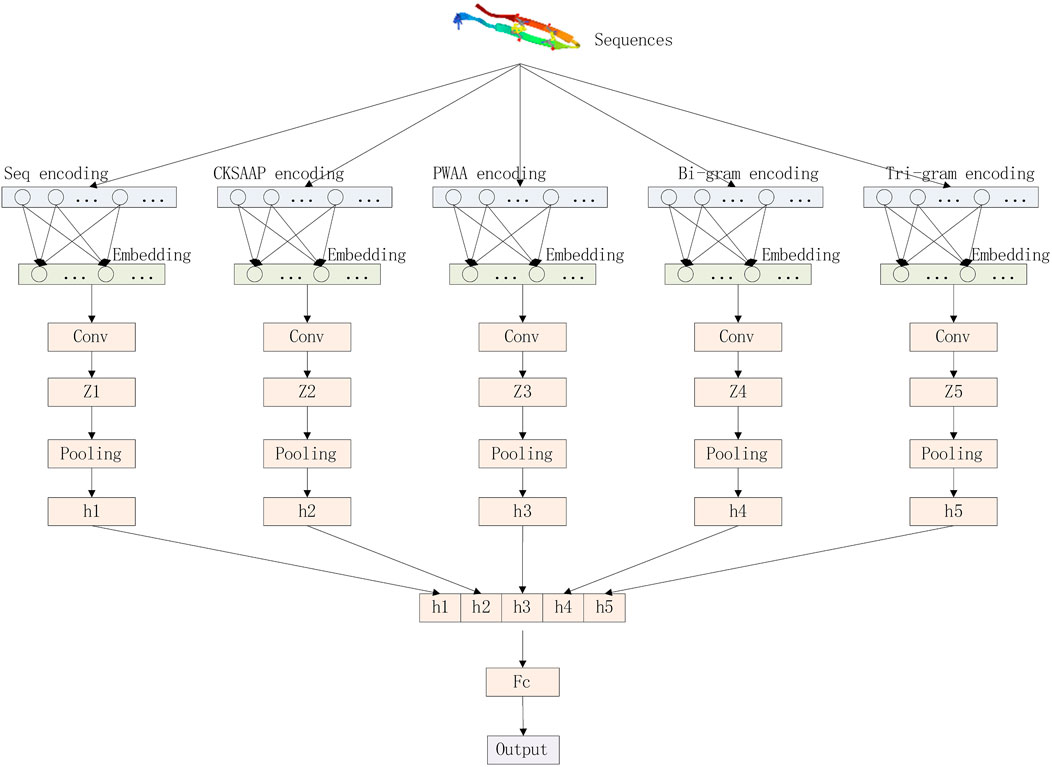
FIGURE 3. Model architecture diagram.
We convert protein sequences into numerical vectors using CKSAAP, PWAA, N-grams, and the numerical encoding of the raw sequence and then pass these vectors into the embedding layer. The embedding layer converts the sparse vector into a dense vector and reduces the dimension of the vector to facilitate the processing of the upper neural network. The processing process of the embedding layer can be represented by the following matrix operations. The first matrix represents the input feature matrix, the middle matrix represents the weight of this layer, and the multiplied result matrix represents the dimension-reduced feature matrix.
The convolution layer convolutes the embedded matrix E with N parallel convolution blocks, which can be composed of a set of triples {(sk, qk, rk)}(k=1,..., N), where sK represents the size of the convolution filter, qk represents the number of convolution filters in the convolution block, and rk represents the activation function corresponding to the convolution block. The convolution direction is one-dimensional convolution along the direction of the sequence, and the convolution block will output a set of feature maps
Where, q = 1, … , qk, C∈
Global average pooling integrates global spatial information, while CKSAAP and PWAA codes encode protein sequences as sparse matrices (with many 0s). Choosing global average pooling may reduce the accuracy of prediction, while global pooling can preserve more Boundary information. Therefore, after obtaining each feature map, we perform a global maximum pooling operation to reduce the number of features in the training phase to prevent overfitting. The vector hk can be calculated by Eq. 4:
Finally, the vector h = [h1; h2; …; hN] is obtained by fully connecting all hk, and the prediction results are output.
Because the learning rate is greatly affected by the output error, the cross-entropy loss function has a larger parameter adjustment range in the early stage of model training, which can make the model training converge faster. To improve the classification efficiency, we use the binary cross-entropy function as our loss function, which can be expressed by Eq. 5:
Where, y represents the binary label 0 or 1, and p(y) represents the probability that the output belongs to the y label. If the predicted value p(y) approaches 1, then the value of the loss function should approach 0. Conversely, if the predicted value p(y) approaches 0 at this point, the value of the loss function should be very large.
2.4 Model evaluation
To objectively evaluate the performance of this method, we train the model using a 10-fold cross-validation method, which randomly divides the negative and positive samples into k (k = 10) equal-sized subsamples. Among the k subsamples, one sub-sample is reserved as validation data for testing the model, and the remaining k-1 subsamples are used as training data (Lv et al., 2022b; Zhang et al., 2022d). Then repeat the cross-validation process for K (k = 10) times (folds), and each sub-sample is used only once as validation data.
To evaluate the precision of the results, we use 7 metrics of accuracy (Acc), sensitivity (Sn), precision (Pr), specificity (Sp), F1 score (Fs), balance accuracy (Ba), and area under the curve (AUROC) on independent datasets, as shown in Formulas 6 to 12.
Where, TP is the true positive, FP is the false positive, TN is the true negative, FN is the false negative, TPR is the true positive and FPR is the false positive.
3 Results and discussion
3.1 Sequence composition analysis based on benchmark datasets
All proteins are made up of 20 amino acid residues, but the frequency of amino acid residues in each protein varies and the lengths of the amino acid sequences that make up the protein vary. During model training, the composition of peptides in the benchmark dataset is very important to analyze the properties of antimicrobial peptides. By counting the centralized peptide lengths of the AMPs and Non-AMPs data, the peptide lengths of our AMPs and Non-AMPs data sets are between 10 and 200, and most of the peptides are below 100 in length, as shown in Figure 4.
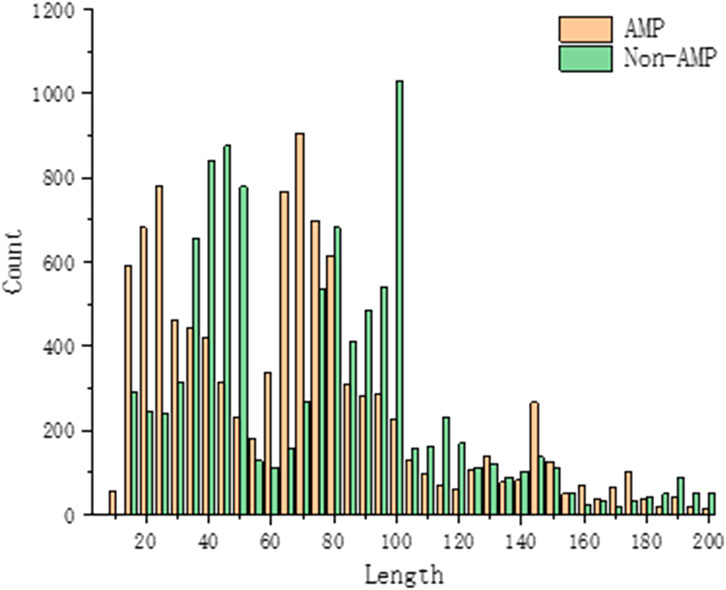
FIGURE 4. Benchmark dataset protein sequence length statistics.
To analyze the sequence consisting of the benchmark dataset, we counted the occurrence frequency of different amino acids at each sequence position. Since the length of AMPs sequences is mainly concentrated in 10–100, we only draw the sequence logo diagram of the first 100 positions, as shown in Figure 5. It can be seen from the figure that specific amino acids belonging to AMPs and Non-AMPs have different positional preferences. In the AMPs sequence, the positions 22–42 are often occupied by glutamic acid (E), and in the Non-AMPs sequence, the positions 22–42 are often occupied by glutamic acid (E). The positions 4–33 are often occupied by leucine (L), and this difference may be due to their belonging to different protein families.
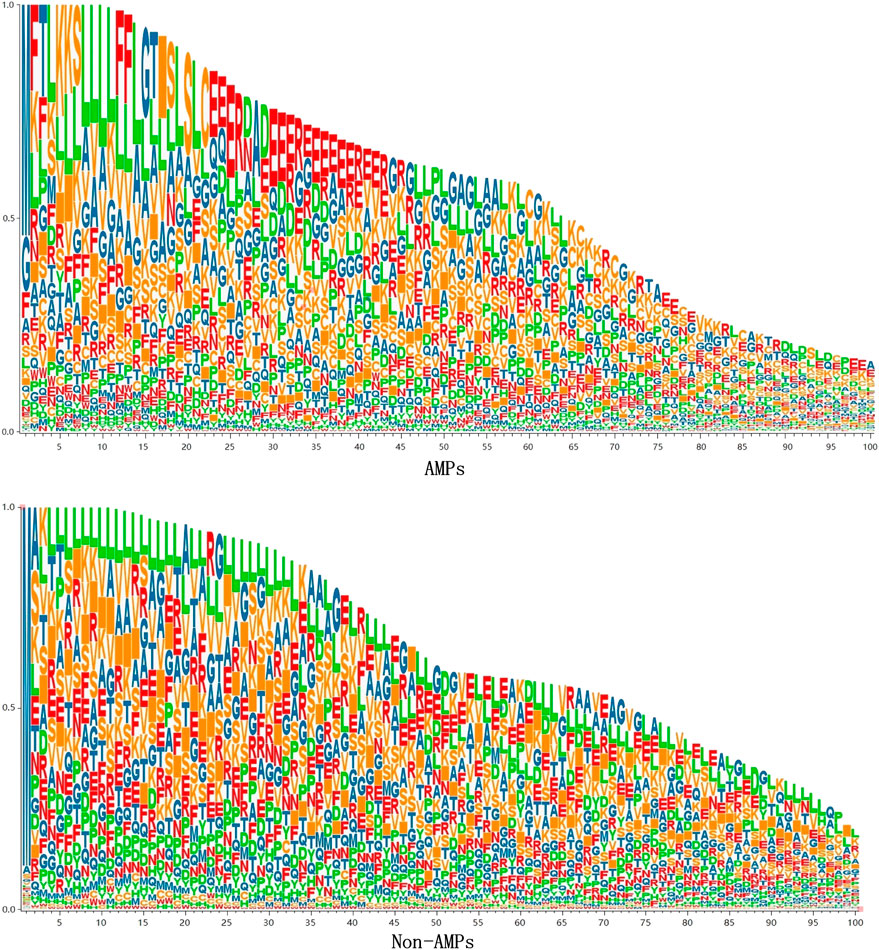
FIGURE 5. sequence logo diagram.
3.2 Comparison of feature coding methods for different combinations
To study the prediction effect of different feature encodings, we conducted experiments on the combination of these three feature encodings with the original sequences based on the verification set. We treat the Bi-gram and Tri-gram encodings as independent feature encoding methods, and finally, combine all the features for experiments, so we did five sets of comparative experiments. CKSAAP encoding and PWAA encoding only extract amino acid combination and position information. The feature encoding is a sparse matrix with many 0 elements. When it is used alone, the prediction accuracy is relatively low, so the original sequence encoding is added to the experiment to make up. The experimental results are shown in Table 2.
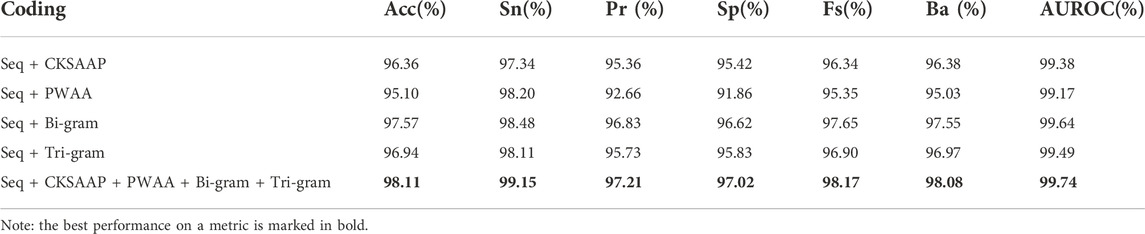
TABLE 2. Comparison of different combination feature coding methods.
It can be found by observation that in the combination with the original sequence, Bi-gram encoding has the best prediction effect, and the sizes of various indicators after combination are most similar to Bi-gram encoding. Bi-gram encoding combines two adjacent amino acids to enhance sequence expression. Compared with Tri-gram encoding, Bi-gram encoding has stronger local association expression. PWAA encoding has the worst prediction effect and the various indicators are not as balanced as the other three encoding methods. This encoding method considers the upstream and downstream information of the sequence and does not consider the interaction between amino acids. It has only 20 dimensions and is a sparse matrix, which contains data Relatively few, even if there is a supplementary prediction effect encoded by the raw sequence, the effect is not good enough. CKSAAP encoding describes short-range interactions between amino acids. Although its form is also a sparse encoding, it has higher dimensions and more information, so the prediction effect is better than PWAA encoding. The prediction results of this study are most affected by Bi-gram encoding and less affected by PWAA encoding. After we combine these kinds of codes, the prediction effect is improved. As can be seen from Figure 6, this feature combination combines the advantages of these kinds of feature codes and considers the interaction of amino acids in protein sequences, position weights, and upstream and downstream information. And it is not affected by the imbalance of PWAA encoding indicators.
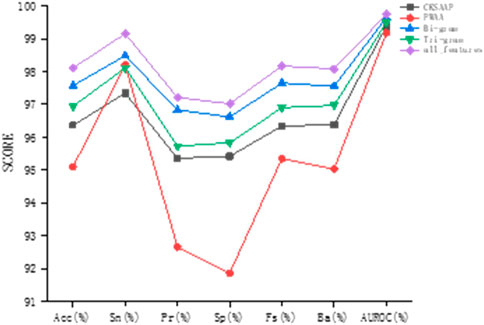
FIGURE 6. Comparison of different combination feature coding methods.
To judge the recognition ability of various encoding combinations for AMPs, we plotted the ROC curves of various combinations, as shown in Figure 7.
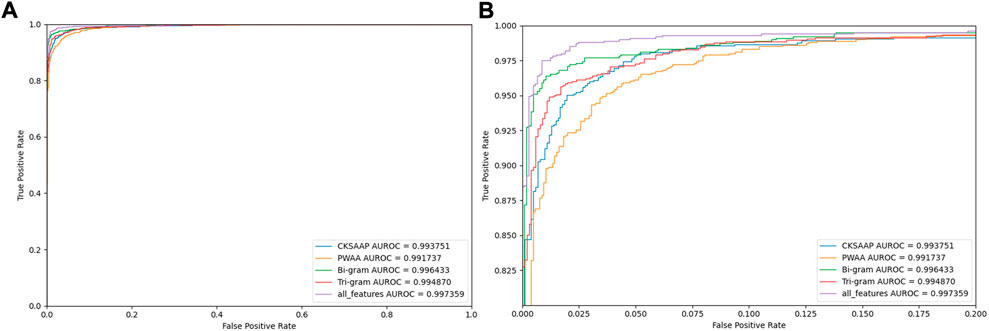
FIGURE 7. Comparing ROC curves with different feature codes. Note: (B) is a partially enlarged view of (A).
3.3 Comparison with other methods
To prove the effectiveness of our method, we compared the prediction results of the method proposed in this paper with other most advanced models (AMPFUN (Chung et al., 2020), AMP Scanner vr.2 (Veltri et al., 2018), CAMPR3 (Waghu et al., 2016), ADAM (Lee et al., 2015), ANIAMPpred (Sharma et al., 2021b)) based on independent test sets. The results are shown in Table 3 and Figure 8. It can be seen from the figure that the performance of ANIAMPpred and the method proposed in this paper is far superior to other models. In terms of PR and SP indicators, ANIAMPpred is slightly higher than our method, but we are the highest in other indicators. The accuracy of our model is 1.05% higher than that of the most advanced method.
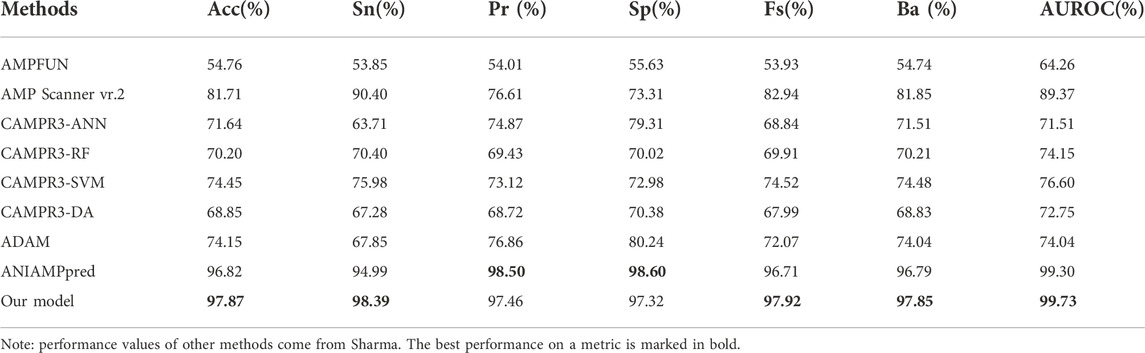
TABLE 3. Performance comparison of different models.
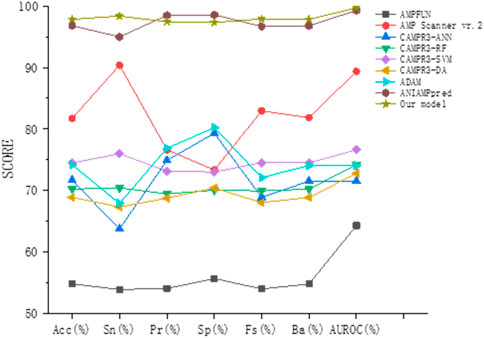
FIGURE 8. Performance comparison of different models.
4 Discussion
In this paper, we combine CKSAAP, PWAA, N-gram, and raw sequence encoding and apply a deep learning approach to predict AMPs. First, we analyzed the benchmark dataset and compared the differences. Then, we separately evaluated and analyzed the prediction effects of CKSAAP, PWAA, N-gram encoding, and raw sequence encoding combination. Finally, we compare state-of-the-art methods, and the results show that this method has the best performance. We combined CKSAAP, PWAA, N-gram encoding, and original sequence encoding, which not only considered the interaction between amino acids commonly used by other methods, but also considered the upstream and downstream information ignored by other methods, and enhanced the AMPs sequence. Therefore, this method has better performance.
Our method achieves high-precision classification of AMPs based on protein sequence information and yields good performance. But AMPs may have undesirable properties as a drug, including instability and toxicity. In studies of synthesizing and modifying AMPs, even small changes can alter the function of AMPs. This method can only identify AMPs and does not consider the functional characteristics of AMPs. Further research can be carried out according to the functions of AMPs, which will help to better understand the mode of action of AMPs and predict their activities.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
Writing—Original Draft, BD; Writing—Review ML, BJ; Writing—Editing, BG, DL,TZ; Funding Acquisition, TZ.
Funding
This work was supported by National Natural Science Foundation of China (62272095, 62172087, 62172129); the Fundamental Research Funds for the Central Universities (2572021BH01);
Acknowledgments
We would like to thank the reviewers for valuable suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1069558/full#supplementary-material
References
Agrawal, P., Bhalla, S., Chaudhary, K., Kumar, R., Sharma, M., and Raghava, G. P. (2018). In silico approach for prediction of antifungal peptides. Front. Microbiol. 9, 323. doi:10.3389/fmicb.2018.00323
Akbar, S., Ahmad, A., Hayat, M., Rehman, A. U., Khan, S., and Ali, F. (2021). iAtbP-Hyb-EnC: Prediction of antitubercular peptides via heterogeneous feature representation and genetic algorithm-based ensemble learning model. Comput. Biol. Med. 137, 104778. doi:10.1016/j.compbiomed.2021.104778
Ao, C., Zou, Q., and Yu, L. (2022). NmRF: Identification of multispecies RNA 2’-O-methylation modification sites from RNA sequences. Brief. Bioinform. 23 (1), bbab480. doi:10.1093/bib/bbab480
Bhadra, P., Yan, J., Li, J., Fong, S., and Siu, S. W. (2018). AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 8 (1), 1697–1710. doi:10.1038/s41598-018-19752-w
Chen, J., Zheng, S., Zhao, H., and Yang, Y. (2021). Structure-aware protein solubility prediction from sequence through graph convolutional network and predicted contact map. J. Cheminform. 13 (1), 7–10. doi:10.1186/s13321-021-00488-1
Chen, Z., Chen, Y.-Z., Wang, X.-F., Wang, C., Yan, R.-X., and Zhang, Z. (2011). Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PloS one 6 (7), e22930. doi:10.1371/journal.pone.0022930
Chung, C.-R., Kuo, T.-R., Wu, L.-C., Lee, T.-Y., and Horng, J.-T. (2020). Characterization and identification of antimicrobial peptides with different functional activities. Briefings Bioinforma. 21 (3), 1098–1114. doi:10.1093/bib/bbz043
Dao, F.-Y., Lv, H., Zhang, D., Zhang, Z.-M., Liu, L., and Lin, H. (2021). DeepYY1: A deep learning approach to identify YY1-mediated chromatin loops. Brief. Bioinform. 22 (4), bbaa356. doi:10.1093/bib/bbaa356
Fjell, C. D., Jenssen, H., Hilpert, K., Cheung, W. A., Panté, N., Hancock, R. E., et al. (2009). Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 52 (7), 2006–2015. doi:10.1021/jm8015365
Fu, H., Cao, Z., Li, M., and Wang, S. (2020). Acep: Improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC genomics 21 (1), 597–614. doi:10.1186/s12864-020-06978-0
Gao, R., Wang, M., Zhou, J., Fu, Y., Liang, M., Guo, D., et al. (2019). Prediction of enzyme function based on three parallel deep CNN and amino acid mutation. Int. J. Mol. Sci. 20 (11), 2845. doi:10.3390/ijms20112845
Gong, Y., Liao, B., Wang, P., and Zou, Q. (2021). DrugHybrid_BS: Using hybrid feature combined with bagging-SVM to predict potentially druggable proteins. Front. Pharmacol. 12, 771808. doi:10.3389/fphar.2021.771808
Han, X., Zhang, L., Zhou, K., and Wang, X. (2019). ProGAN: Protein solubility generative adversarial nets for data augmentation in DNN framework. Comput. Chem. Eng. 131, 106533. doi:10.1016/j.compchemeng.2019.106533
Hancock, R. E., and Sahl, H.-G. (2006). Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557. doi:10.1038/nbt1267
Hathaway, Q. A., Yanamala, N., Budoff, M. J., Sengupta, P. P., and Zeb, I. (2021). Deep neural survival networks for cardiovascular risk prediction: The Multi-Ethnic Study of Atherosclerosis (MESA). Comput. Biol. Med. 139, 104983. doi:10.1016/j.compbiomed.2021.104983
Jain, P., Tiwari, A. K., and Som, T. (2021). Enhanced prediction of anti-tubercular peptides from sequence information using divergence measure-based intuitionistic fuzzy-rough feature selection. Soft Comput. 25 (4), 3065–3086. doi:10.1007/s00500-020-05363-z
Khabbaz, H., Karimi-Jafari, M. H., Saboury, A. A., and BabaAli, B. (2021). Prediction of antimicrobial peptides toxicity based on their physico-chemical properties using machine learning techniques. BMC Bioinforma. 22 (1), 549–611. doi:10.1186/s12859-021-04468-y
Kumar, P., Kizhakkedathu, J. N., and Straus, S. K. (2018). Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 8, 4. doi:10.3390/biom8010004
Lata, S., Mishra, N. K., and Raghava, G. P. (2010). AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinforma. 11 (1), 199–S27. doi:10.1186/1471-2105-11-S1-S19
Le, N. Q. K., Ho, Q.-T., Nguyen, T.-T.-D., and Ou, Y.-Y. (2021). A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information. Brief. Bioinform. 22 (5), bbab005. doi:10.1093/bib/bbab005
Le, N. Q. K., Yapp, E. K. Y., Nagasundaram, N., and Yeh, H.-Y. (2019). Classifying promoters by interpreting the hidden information of DNA sequences via deep learning and combination of continuous fasttext N-grams. Front. Bioeng. Biotechnol. 7, 305. doi:10.3389/fbioe.2019.00305
Lee, H.-T., Lee, C.-C., Yang, J.-R., Lai, J. Z., and Chang, K. Y. (2015). A large-scale structural classification of antimicrobial peptides. Biomed. Res. Int. 2015, 475062. doi:10.1155/2015/475062
Li, M., Ling, C., Xu, Q., and Gao, J. (2018). Classification of G-protein coupled receptors based on a rich generation of convolutional neural network, N-gram transformation and multiple sequence alignments. Amino acids 50 (2), 255–266. doi:10.1007/s00726-017-2512-4
Li, Z., Fang, J., Wang, S., Zhang, L., Chen, Y., and Pian, C. (2022). Adapt-kcr: A novel deep learning framework for accurate prediction of lysine crotonylation sites based on learning embedding features and attention architecture. Brief. Bioinform. 23 (2), bbac037. doi:10.1093/bib/bbac037
Lv, H., Dao, F. Y., and Lin, H. (2022a). DeepKla: An attention mechanism-based deep neural network for protein lysine lactylation site prediction. iMeta 1 (1), e11. doi:10.1002/imt2.11
Lv, H., Zhang, Y., Wang, J.-S., Yuan, S.-S., Sun, Z.-J., Dao, F.-Y., et al. (2022b). iRice-MS: an integrated XGBoost model for detecting multitype post-translational modification sites in rice. Brief. Bioinform. 23 (1), bbab486. doi:10.1093/bib/bbab486
Meher, P. K., Sahu, T. K., Saini, V., and Rao, A. R. (2017). Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 7 (1), 42362–42412. doi:10.1038/srep42362
Moretta, A., Salvia, R., Scieuzo, C., Di Somma, A., Vogel, H., Pucci, P., et al. (2020). A bioinformatic study of antimicrobial peptides identified in the Black Soldier Fly (BSF) Hermetia illucens (Diptera: Stratiomyidae). Sci. Rep. 10 (1), 16875–16914. doi:10.1038/s41598-020-74017-9
Nakayama, J. Y., Ho, J., Cartwright, E., Simpson, R., and Hertzberg, V. S. (2021). Predictors of progression through the cascade of care to a cure for hepatitis C patients using decision trees and random forests. Comput. Biol. Med. 134, 104461. doi:10.1016/j.compbiomed.2021.104461
Pasupuleti, M., Schmidtchen, A., and Malmsten, M. (2012). Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 32 (2), 143–171. doi:10.3109/07388551.2011.594423
Qiao, Y., Zhu, X., and Gong, H. (2022). BERT-kcr: Prediction of lysine crotonylation sites by a transfer learning method with pre-trained BERT models. Bioinformatics 38 (3), 648–654. doi:10.1093/bioinformatics/btab712
Ren, Y., Chakraborty, T., Doijad, S., Falgenhauer, L., Falgenhauer, J., Goesmann, A., et al. (2022). Prediction of antimicrobial resistance based on whole-genome sequencing and machine learning. Bioinformatics 38 (2), 325–334. doi:10.1093/bioinformatics/btab681
Sharma, A. K., and Srivastava, R. (2021). Protein secondary structure prediction using character bi-gram embedding and bi-LSTM. Curr. Bioinform. 16 (2), 333–338. doi:10.2174/15748936mta3imdeu1
Sharma, R., Shrivastava, S., Kumar Singh, S., Kumar, A., Saxena, S., and Kumar Singh, R. (2021b). AniAMPpred: Artificial intelligence guided discovery of novel antimicrobial peptides in animal kingdom. Brief. Bioinform. 22 (6), bbab242. doi:10.1093/bib/bbab242
Sharma, R., Shrivastava, S., Kumar Singh, S., Kumar, A., Saxena, S., and Kumar Singh, R. (2021a). Deep-ABPpred: Identifying antibacterial peptides in protein sequences using bidirectional LSTM with word2vec. Brief. Bioinform. 22 (5), bbab065. doi:10.1093/bib/bbab065
Sharma, R., Shrivastava, S., Kumar Singh, S., Kumar, A., Saxena, S., and Kumar Singh, R. (2022). Deep-AFPpred: Identifying novel antifungal peptides using pretrained embeddings from seq2vec with 1DCNN-BiLSTM. Brief. Bioinform. 23 (1), bbab422. doi:10.1093/bib/bbab422
Söylemez, Ü. G., Yousef, M., Kesmen, Z., Büyükkiraz, M. E., and Bakir-Gungor, B. (2022). Prediction of linear cationic antimicrobial peptides active against gram-negative and gram-positive bacteria based on machine learning models. Appl. Sci. 12 (7), 3631. doi:10.3390/app12073631
Spänig, S., and Heider, D. (2019). Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Min. 12 (1), 7–29. doi:10.1186/s13040-019-0196-x
Su, X., Xu, J., Yin, Y., Quan, X., and Zhang, H. (2019). Antimicrobial peptide identification using multi-scale convolutional network. BMC Bioinforma. 20 (1), 730–810. doi:10.1186/s12859-019-3327-y
Thomas, S., Karnik, S., Barai, R. S., Jayaraman, V. K., and Idicula-Thomas, S. (2010). Camp: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 38 (1), D774–D780. doi:10.1093/nar/gkp1021
Tng, S. S., Le, N. Q. K., Yeh, H.-Y., and Chua, M. C. H. (2021). Improved prediction model of protein lysine Crotonylation sites using bidirectional recurrent neural networks. J. Proteome Res. 21 (1), 265–273. doi:10.1021/acs.jproteome.1c00848
Veltri, D., Kamath, U., and Shehu, A. (2018). Deep learning improves antimicrobial peptide recognition. Bioinformatics 34 (16), 2740–2747. doi:10.1093/bioinformatics/bty179
Veltri, D. P. (2015). A computational and statistical framework for screening novel antimicrobial peptides. George Mason University.
Waghu, F. H., Barai, R. S., Gurung, P., and Idicula-Thomas, S. (2016). CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 44 (D1), D1094–D1097. doi:10.1093/nar/gkv1051
Wang, Y., Xu, L., Zou, Q., and Lin, C. (2022). prPred-DRLF: Plant R protein predictor using deep representation learning features. Proteomics 22 (1-2), 2100161. doi:10.1002/pmic.202100161
Wani, M. A., Garg, P., and Roy, K. K. (2021). Machine learning-enabled predictive modeling to precisely identify the antimicrobial peptides. Med. Biol. Eng. Comput. 59 (11), 2397–2408. doi:10.1007/s11517-021-02443-6
Wu, X., and Yu, L. (2021). Epsol: Sequence-based protein solubility prediction using multidimensional embedding. Bioinformatics 37 (23), 4314–4320. doi:10.1093/bioinformatics/btab463
Xiao, X., Shao, Y.-T., Cheng, X., and Stamatovic, B. (2021). iAMP-CA2L: a new CNN-BiLSTM-SVM classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Brief. Bioinform. 22 (6), bbab209. doi:10.1093/bib/bbab209
Xiao, X., Wang, P., Lin, W.-Z., Jia, J.-H., and Chou, K.-C. (2013). iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 436 (2), 168–177. doi:10.1016/j.ab.2013.01.019
Xu, Z., Luo, M., Lin, W., Xue, G., Wang, P., Jin, X., et al. (2021). DLpTCR: An ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief. Bioinform. 22 (6), bbab335. doi:10.1093/bib/bbab335
Yan, J., Bhadra, P., Li, A., Sethiya, P., Qin, L., Tai, H. K., et al. (2020). Deep-AmPEP30: Improve short antimicrobial peptides prediction with deep learning. Mol. Ther. Nucleic Acids 20, 882–894. doi:10.1016/j.omtn.2020.05.006
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk prediction of diabetes: Big data mining with fusion of multifarious physical examination indicators. Inf. Fusion 75, 140–149. doi:10.1016/j.inffus.2021.02.015
Yuan, S.-S., Gao, D., Xie, X.-Q., Ma, C.-Y., Su, W., Zhang, Z.-Y., et al. (2022). IBPred: A sequence-based predictor for identifying ion binding protein in phage. Comput. Struct. Biotechnol. J. 20, 4942–4951. doi:10.1016/j.csbj.2022.08.053
Yun, H.-R., Lee, G., Jeon, M. J., Kim, H. W., Joo, Y. S., Kim, H., et al. (2021). Erythropoiesis stimulating agent recommendation model using recurrent neural networks for patient with kidney failure with replacement therapy. Comput. Biol. Med. 137, 104718. doi:10.1016/j.compbiomed.2021.104718
Zhang, H., Liao, L., Cai, Y., Hu, Y., and Wang, H. (2019). IVS2vec: A tool of inverse virtual screening based on word2vec and deep learning techniques. Methods 166, 57–65. doi:10.1016/j.ymeth.2019.03.012
Zhang, Q., Li, H., Liu, Y., Li, J., Wu, C., and Tang, H. (2022a). Exosomal non-coding RNAs: New insights into the biology of hepatocellular carcinoma. Curr. Oncol. 29 (8), 5383–5406. doi:10.3390/curroncol29080427
Zhang, Y., Lin, J., Zhao, L., Zeng, X., and Liu, X. (2021). A novel antibacterial peptide recognition algorithm based on BERT. Brief. Bioinform. 22 (6), bbab200. doi:10.1093/bib/bbab200
Zhang, Y., Zhu, G., Li, K., Li, F., Huang, L., Duan, M., et al. (2022b). Hlab: Learning the BiLSTM features from the ProtBert-encoded proteins for the class I HLA-peptide binding prediction. Briefings Bioinforma. 23. doi:10.1093/bib/bbac173
Zhang, Z.-Y., Ning, L., Ye, X., Yang, Y.-H., Futamura, Y., Sakurai, T., et al. (2022c). iLoc-miRNA: extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief. Bioinform. 23 (5), bbac395. doi:10.1093/bib/bbac395
Zhang, Z.-Y., Sun, Z.-J., Yang, Y.-H., and Lin, H. (2022d). Towards a better prediction of subcellular location of long non-coding RNA. Front. Comput. Sci. 16 (5), 165903–165907. doi:10.1007/s11704-021-1015-3
Zheng, X., Fu, X., Wang, K., and Wang, M. (2020). Deep neural networks for human microRNA precursor detection. BMC Bioinforma. 21 (1), 17–7. doi:10.1186/s12859-020-3339-7
Keywords: deep learning, feature encoding, feature embedding, N-gram encoding, antimicrobial peptides
Citation: Dong B, Li M, Jiang B, Gao B, Li D and Zhang T (2022) Antimicrobial Peptides Prediction method based on sequence multidimensional feature embedding. Front. Genet. 13:1069558. doi: 10.3389/fgene.2022.1069558
Received: 14 October 2022; Accepted: 02 November 2022;
Published: 17 November 2022.
Edited by:
Quan Zou, University of Electronic Science and Technology of China, ChinaCopyright © 2022 Dong, Li, Jiang, Gao, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Li, bGQ3MjU3MjVAMTI2LmNvbQ==; Tianjiao Zhang, dGlhbmppYW96aGFuZ0BuZWZ1LmVkdS5jbg==