 Tong-Jie Sun1
Tong-Jie Sun1 Gai-Fang Dong
Gai-Fang Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 03 November 2022
Sec. Computational Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.1062576
This article is part of the Research TopicNovel Machine Learning Algorithms for Conventional Omics Data and their ApplicationView all 5 articles
Lactic acid bacteria antimicrobial peptides (LABAMPs) are a class of active polypeptide produced during the metabolic process of lactic acid bacteria, which can inhibit or kill pathogenic bacteria or spoilage bacteria in food. LABAMPs have broad application in important practical fields closely related to human beings, such as food production, efficient agricultural planting, and so on. However, screening for antimicrobial peptides by biological experiment researchers is time-consuming and laborious. Therefore, it is urgent to develop a model to predict LABAMPs. In this work, we design a graph convolutional neural network framework for identifying of LABAMPs. We build heterogeneous graph based on amino acids, tripeptide and their relationships and learn weights of a graph convolutional network (GCN). Our GCN iteratively completes the learning of embedded words and sequence weights in the graph under the supervision of inputting sequence labels. We applied 10-fold cross-validation experiment to two training datasets and acquired accuracy of 0.9163 and 0.9379 respectively. They are higher that of other machine learning and GNN algorithms. In an independent test dataset, accuracy of two datasets is 0.9130 and 0.9291, which are 1.08% and 1.57% higher than the best methods of other online webservers.
Lactic acid bacteria (LAB) is a kind of bacteria that can use fermentable carbohydrates to produce large amounts of lactic acid (Gu et al., 2022; Hu et al., 2022). Organic acids, special enzymes, lactobacilli and other substances produced by lactic acid bacteria through fermentation have special physiological functions. A large number of research data show that lactic acid bacteria can promote animal growth, regulate the normal flora of gastrointestinal tract, maintain micro ecological balance, thereby improving gastrointestinal function, improving food digestibility and biological titer, reducing serum cholesterol, controlling endotoxin, inhibiting the growth of intestinal putrefactive bacteria, and improving the immunity of the body (Teusink and Molenaar, 2017). Lactic acid bacteria have been widely used in food industry and poultry husbandry, and also have important academic value in genetic engineering (Greub et al., 2016), biochemistry (Kadomatsu, 2022), genetics (Sung Won et al., 2020) and molecular biology (Saibil, 2022).
Antimicrobial peptides of lactic acid bacteria are a kind of active peptides or proteins produced by the metabolic process of lactic acid bacteria, which can inhibit or kill pathogenic bacteria or spoilage bacteria in food. In recent years, several new methods have been developed for the screening and development of new antimicrobial peptides, including enzyme-linked immunodeficient assay (Huang X et al., 2022), biological analysis of K+ ion current (Lauger and Apell, 1988), ATP-bioluminescence method (Crouch et al., 1993; Aiken et al., 2011), Lux gene-bioluminescence method (Van Dyk et al., 1994), berberine-based fluorescence analysis method (Liu et al., 1998; Song et al., 2018) and micro-plate method (Kai et al., 2012). Although the above wet experimental methods can distinguish, they are time-consuming and expensive, so they cannot be popularized and used. To help wet lab researchers identify novel antimicrobial peptides, a variety of computational methods for antimicrobial peptide identification have been proposed. Many algorithms combine machine learning or statistical analysis techniques such as discriminant analysis (DA) (Kouw and Loog, 2021; Beck and Sharon, 2022), fuzzy K-nearest neighbors (Zhai et al., 2020), hidden Markov models (Fuentes-Beals et al., 2022), logistic regression (Fagerland and Hosmer, 2012), random forests (RF) (Ziegler and Koenig, 2014), and support vector machines (SVM) (Azar and El-Said, 2014). Although these models have made great progress in antimicrobial peptide recognition, the following challenges still exist: First, many related classification tasks based on machine learning suffer from the small number of samples. The model trained with small sample size cannot achieve robustness and is prone to the problems of over fitting and poor generalization ability. Secondly, most of the existing feature extraction technologies are aimed at specific datasets, and do not have universality.
In a word, most of the existing machine learning based classification work mainly uses the manually determined features (Jiang et al., 2021), which is highly dependent on biologists. The artificially determined features also have their shortcomings. On the one hand, the intrinsic nonlinear information of the function of some peptides cannot be obtained through this featured way; On the other hand, when the research object is changed, the adaptability of artificial features is poor. In addition, the dimension disaster caused by feature engineering brings new troubles to researchers.
In the past 10 years, deep learning has achieved extremely rapid development. In the field of text processing, achievements in the application of natural language processing to biological information prediction have been published repeatedly. In particular, graph neural network plays an excellent role in text classification (Xie et al., 2022; Zhou et al., 2022). Qu (Qu et al., 2017) proposed a method based on deep learning to identify DNA binding protein sequences. This method uses a two-stage convolutional network to detect the functional domain of protein sequences, and uses LSTM neural networks to identify context dependencies. In the independent test set, the accuracy of the model in the yeast data set is 80%; Hamid and Friedberg (Hamid and Friedberg, 2018) proposed a method used word embedding and RNN to identify bacteriocin and non bacteriocin sequences. The recall of the model in the two training data sets is 89.8% and 92.1% respectively; Veltri (Veltri et al., 2018) proposed a deep neural network model, which includes an embedded layer, a convolution layer and a recursive layer. The accuracy of the model in the independent test set is 91.01%; Zeng (Zeng et al., 2019) proposed to identify protein sequences based on the use of node2vec technology, convolution neural network and sampling technology. In this framework, node2vec technology is used to capture the semantic features and topology of each protein in the protein interaction network, and convolution layer is used to extract information from gene expression profiles. The AUC of the model in the training set is 82%; he (He et al., 2021) proposed a new Meta learning framework based on mutual information maximization. The core of the framework is ProtoNet, a classical meta learning algorithm based on metric learning, which learns the vector representation of each prototype. The accuracy of this model in the training set of antifungal peptides was 91.3%. The above five deep learning models have improved the performance of AMP prediction to a certain extent, but most of these models used convolutional neural network and LSTM neural network combination framework without significant innovation. Recently, due to the rise of graph neural networks, more and more people began to do some research on graph neural networks. Therefore, our work is based on graph convolution neural network to predict LABAMPs.
In this work, we design a graph convolution neural network framework to predict antimicrobial peptides of lactic acid bacteria. First, we construct a large heterogeneous graph based on all the samples, which contains sequences and peptides (amino acids, dipeptide, tripeptide. We can think of these peptides as words in natural language processing) as nodes. Then connect the nodes by doing that: The edge between two peptide fragments is determined by whether the two peptide fragments appear together in the fixed range (window size) of a sequence. The edge between a peptide fragment and a sequence depends on whether the peptide fragment is a substring of this sequence. Finally, the classification of nodes on the graph is realized through the calculation and transmission of information between nodes on the graph. The experimental results show that our model has great advantages over machine learning methods, deep learning models and other webservers.
We collected LABAMPs records from 25 databases (Gueguen et al., 2006; Mulvenna et al., 2006; Fjell et al., 2007; Henderson et al., 2007; Kawashima et al., 2008; Hammami et al., 2009; Hammami et al., 2010; Sundararajan et al., 2012; Gogoladze et al., 2014; Theolier et al., 2014; Pirtskhalava et al., 2021; Shi et al., 2022) according to the 30 genus classification of lactic acid bacteria in Supplementary Table S1. Finally, after removing duplicate records, 1622 LABAMPs are obtained, and their lengths are from 2 to 1619.
According to the positive raw data set obtained above, we do some processing on it: First, we remove records which contain unnatural amino acids such as B, J, O, U, X, and Z from these raw data. Second, to reduce sequence homology bias and redundancy, we used respectively the CD-HIT program (Li and Godzik, 2006) to delete peptides with 70% and 90% similarity to each other. Finally, we get 460 and 636 peptide sequences after removing redundancy, respectively.
Our negative raw datasets obtained as follows:
1 On the UniProt website (Consortium, 2021), we obtain peptide sequences between the length of 2–1619;
2 Remove sequences contain or annotated with information of antimicrobial, antibiotic, fungicide, defensin, AMP, membrane, toxic, secretory, defensive, anticancer, antiviral, antifungal, effector, and exacted;
3 Remove resulting protein sequences which include unnatural amino acids;
4 Remove peptide sequences with 70% and 90% similarity by CD-HIT program;
5 Randomly select the same number of sequences as the number of positive samples.
All positive and negative samples are shown as Table 1, with processing of 70% and 90% by CD-HIT. We called them DS-70% and DS-90% respectively. The statistics of the preprocessed datasets are summarized in Table 2. Since we classify nodes on the graph, the number of graphs is one respectively in DS-70% and DS-90%. The number of sequences is the total number of positive and negative samples of DS-70% or DS-90%. The number of words is obtained by removing stop words and the words whose frequencies are less than 5. The number of nodes is the sum of the number of sequences and the number of words. Because our work has two categories of tasks, the number of categories is two.

TABLE 1. Raw data processed through CD-HIT program.
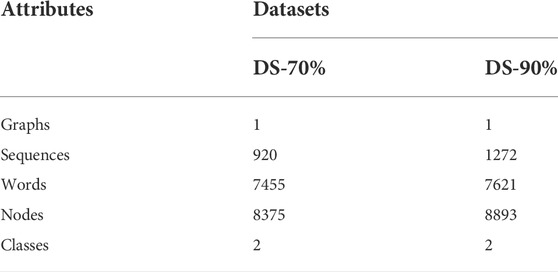
TABLE 2. Summary statistics results of datasets.
The model construction is divided into three steps: first, establish the initial graph, then conduct the convolution operation on the graph, and finally complete the node classification through the classification function.
Before the construction of initial graph, we preprocess all positive and negative samples. First, all positive and negative samples are segmented by amino acid, dipeptide or tripeptide as words. Secondly, count the words frequencies, and filter all the words whose word frequency is less than 5 times. Then, we get the required words.
Suppose the initial input graph is expressed as Graph
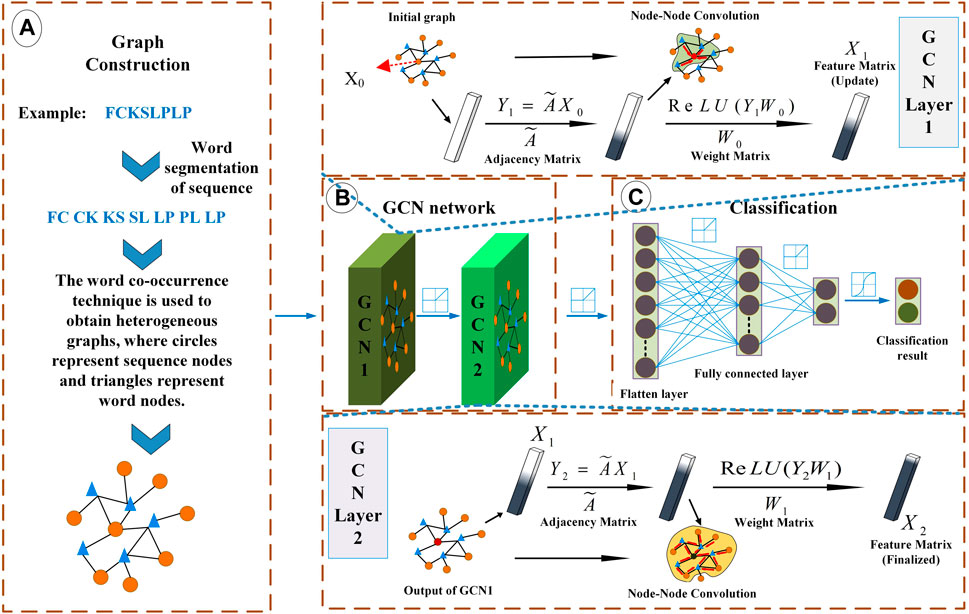
FIGURE 1. Overview of LABAMPsGCN model architecture. (A) Graph Construction. Each sequence is processed by word segmentation, and then the required graph is obtained by word co-occurrence technology. (B) Graph convolutional neural network. It mainly carries out message transmission through word-sequence relations. (C) Classification. It uses the full connection layer for classification.
In order to calculate the information on the graph through the edges, we establish the adjacency matrix A of the initial graph, that is, assign a certain weight to each edge. The calculation method is shown in Eq. 1. Where
Word embedding is a method converting a word into a vector representation. There are many methods for word embedding, such as one-hot embedding, Skip Gram model (Carrasco and Sicilia, 2018), CBOW model (Xiong et al., 2019) and GloVe word vector (Gao and Huang, 2021). In this module, we first need to determine the node features of the initial graph. We use one-hot embedding to embed each word and send it to the model together with the sequence for training. Because the initial values of node features have little influence on the graph convolution neural network, we set
Since the diagonal element of the adjacency matrix is 0, it is easy to lose the information of the nodes themselves in the calculation process, so an identity matrix is added to the adjacency matrix. In order to avoid the change of feature distribution, the adjacent matrix with an identity matrix is normalized to obtain the processed adjacent matrix
We design a graph convolution neural network framework to learn the information between nodes on the graph and transfer the related information under the supervision of labels, and finally achieve node classification. The graph convolution neural network framework of lactobacillus antibacterial peptides can be expressed as Eq. 2.
The network learning process under the supervision of sequence labels needs to calculate the loss rate, and we use the cross entropy loss function to calculate the loss (Aurelio et al., 2019). Eq. 2 is a general model of LABAMPsGCN. Figure 1B shows a two-layer LABAMPsGCN. In the following chapters, we analyze that the two-layer LABAMPsGCN has the best performance.
We use the full connection layer to integrate the feature space into the sample label space, and then use the softmax classification function to calculate the probability of nodes being classified into different categories. As is shown in Figure 1C.
To assess the performance of LABAMPsGCN, we adopt statistical metric of precision, recall, accuracy and
The parameters of a model have an important impact on the performance of the model. In our LABAMPsGCN, we set the activation function, window size, first layer convolution size, learning rate and loss rate to ReLU, 15, 200, 0.01, and 0.5 respectively. We used Adam optimizer (Shao et al., 2021) to train our model for 150 epochs.
We constructed a webserver with our prediction model embedded at the back end of website. When users submit their interested LABAMPs, the predicted percentage will be displayed based on the website prediction tool (Sim et al., 2012). Because the weight matrix of the graph convolution neural network will change with the change of the adjacency matrix and feature matrix of the input data, we embedded SVM model with accuracy lower of 3.77% than that of LABAMPsGCN.
We randomly combined the features of single peptide, dipeptide and tripeptide respectively, and obtained six feature combinations: dipeptide, dipeptide and single peptide, tripeptide, tripeptide and single peptide, tripeptide and dipeptide, tripeptide added by dipeptide and single peptide. Table 3 shows the model accuracy on the DS-70% and DS-90%.
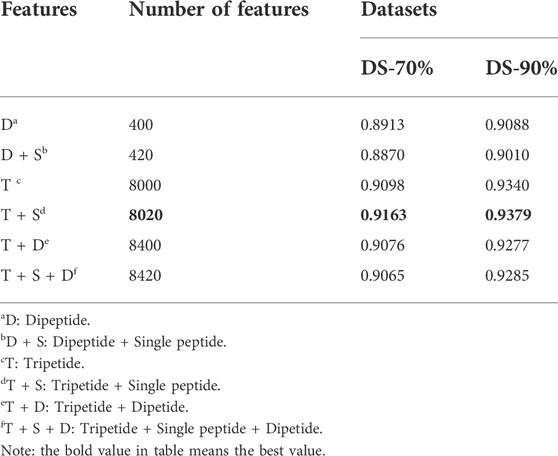
TABLE 3. The different accuracy of different features on LABAMPsGCN.
It can be seen that the features of tripeptide and single peptide are significantly better than other combinations on DS-70% and DS-90%. As the number of features continues to increase, the accuracy (ACC) of the test data is also slowly increasing, and the number of features in DS-70% and DS-90% begins to decline significantly after 8020.
Figure 2A reports the accuracy for different sliding window on DS-70% and DS-90% based on features of tripeptide and single peptides. It demonstrates that the influence of the size of the sliding window on the prediction accuracy meets the general rule—taking 15 as the dividing point, it rises first and then falls. It is further explained that small windows cannot accommodate sequence fragments that play key functions, while too large windows take some irrelevant information as key information to participate, disturbing the judgment. Therefore, in this paper, window size is set to 15.
We designed GCNs with different layers to obtain the features of LABAMPs. Figure 2B indicate the effect of the number of GCN layers on the performance of our model. In this paper, we changed the GCN layer in {1, 2, 3, 4}. It can be seen from Figure 2B that the 2-layer GCN can achieve the optimal performance. Too many GCN layers will cause the model to be over-smoothing, thus causing the learned model to collapse. Although there is no direct sequence-sequence edge connection in the graph, 2 GCNs can be connected through the middle word node, thus realizing sequence to sequence information interaction.
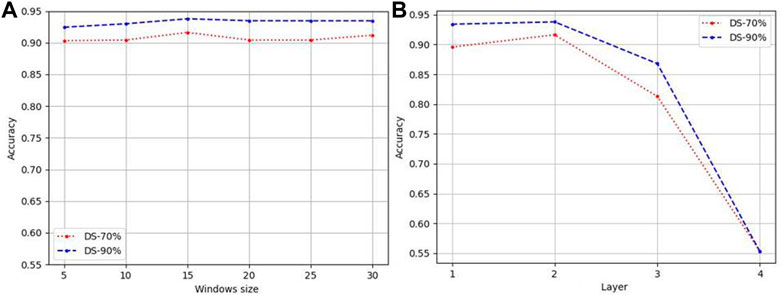
FIGURE 2. Parameter analysis of LABAMPsGCN. (A) Accuracy varied by windows size. (B) Accuracy varied by numbers of layers.
If there are too many layers, the features of a node will aggregate the features of more and more neighbors, so that these nodes become similar, which increases the similarity between classes, and the natural classification effect is poor.
In order to verify metric of LABAMPsGCN, we compare machine learning models with it on the same features. In Table 4 all results are obtained by using 10-fold cross-validation. We used Multinomial Bayesian classifier (MNB), Random forest (RF), Support vector machine (SVM), AdaBoost (Huang H et al., 2022) and XGBoost (Zhang et al., 2022). It can be seen that LABAMPsGCN show good performance no matter how features change. This is because LABAMPsGCN can obtain the information of sequence nodes through word nodes.
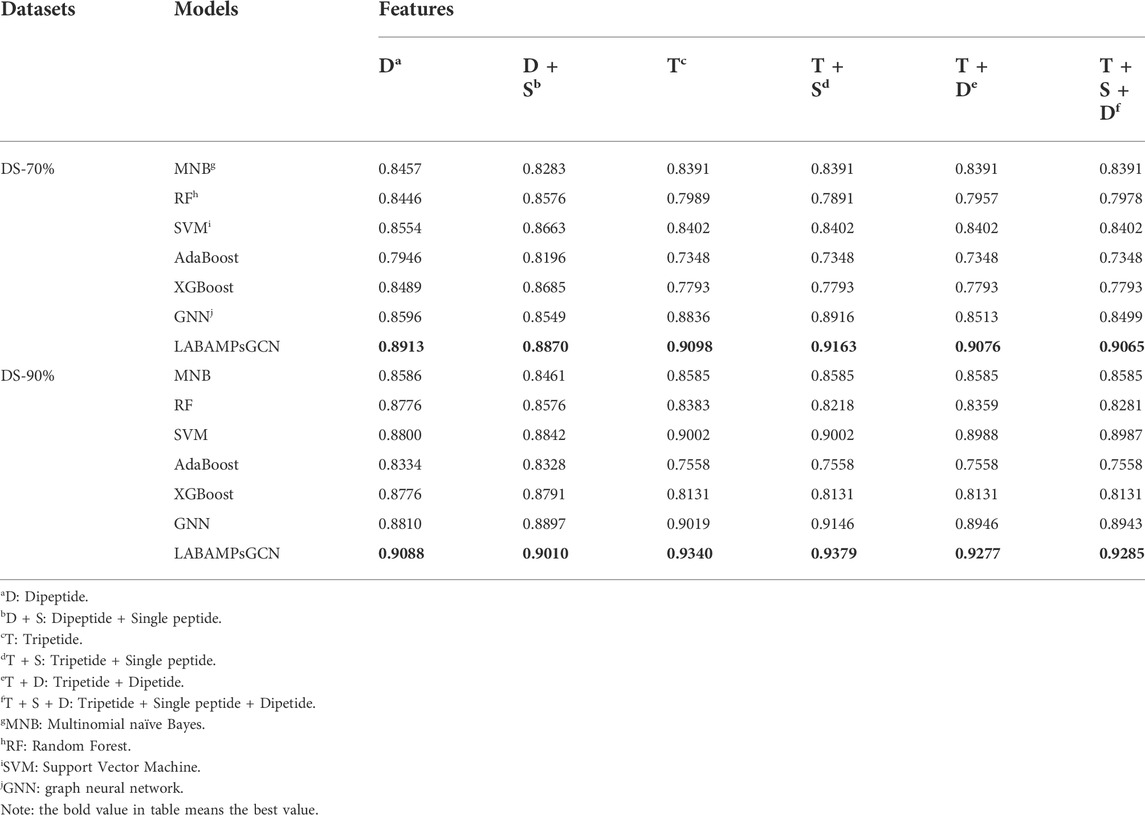
TABLE 4. Comparisons of LABAMPsGCN with machine learning and GNN models.
Table 5 compares our LABAMPsGCN model to three state-of-the-art machine learning methods which can be found publicly for AMPs recognition. Table 5 shows that our LABAMPsGCN model achieves the best values of metrics for Recall, Precision and Accuracy. In DS-70%, the Recall score of AMPfun model (Chung et al., 2020) is the highest (3.42% higher than our model). In DS-90%, the metrics of our LABAMPsGCN model are significantly better than other methods.
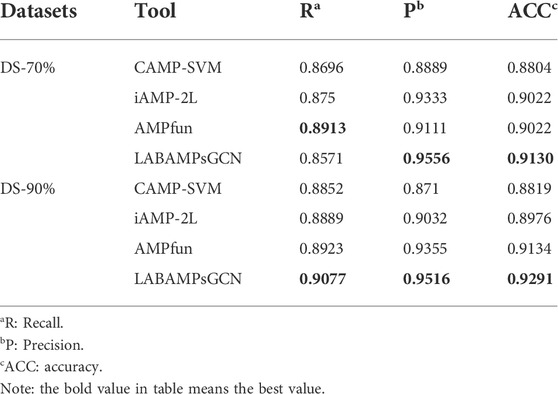
TABLE 5. Comparisons of LABAMPsGCN with three state-of-the-art webservers.
In order to judge if all the parts of our identifier are necessary, we adopt three variants of LABAMPsGCN (LABAMPsGCN-noFC, LABAMPsGCN-cheby and LABAMPsGCN-cheby-noFC) as comparison methods. Specifically, LABAMPsGCN-noFC means that we do not add a full connection layer after the GCN layer for classification, while directly use the output of the GCN layer for classifying. LABAMPsGCN-cheby adds Chebyshev polynomials (Christiansen et al., 2021), which can use polynomial expansion to approximate the convolution of graphs, that is, polynomial approximation of parameterized frequency response functions. LABAMPsGCN-cheby-noFC adds Chebyshev polynomials and there is no full connection layer after GCN layer output.
Table 6 shows the evaluation indicators obtained by training with LABAMPsGCN and its variants on DS-90%. These four groups of training were conducted on the feature of tripeptide and single peptide. For LABAMPsGCN and LABAMPsGCN-noFC, the ACC of LABAMPsGCN was significantly higher than that of LABAMPsGCN-noFC. This is because the full connection layer integrates the feature representations and maps them to the space where the sample labels are located. For LABAMPsGCN and LABAMPsGCN-cheby, the performance of LABAMPsGCN-cheby is slightly poor because the use of Chebyshev polynomials makes each sequence vertex fuse too much irrelevant information. For LABAMPsGCN and LABAMPsGCN-cheby-noFC, the performance of LABAMPsGCN with full connection layer is significantly higher than that without it.
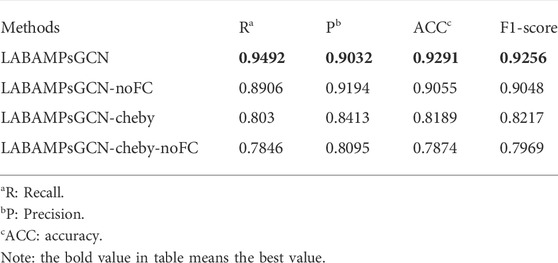
TABLE 6. Performance evaluation of LABAMPsGCN and its three variants.
LABAMPsGCN learned a lot of word features related to labels. In order to observe these words clearly, we visualized them qualitatively. Figure 3 shows the t-SNE visualization (Ruit et al., 2022) of the second layer word features learned from DS-70% and DS-90%. We set the dimension of the maximum value in the word feature vectors as the label of the word. As can be seen from Figure 3, words of the same color are clustered together, which means that a large number of words are closely related to certain specific classes. The red, green and orange in Figure 3 are used for visualization to determine whether word embedding can learn the main information of some sequences. Different colors in the figure represent different sequences. Figure 3A and Figure 3B is the results of DS-70% and DS-90%, respectively. In Table 7, we show the top representative words in each category, such as “ILE,” “TIW,” and “KLK”.
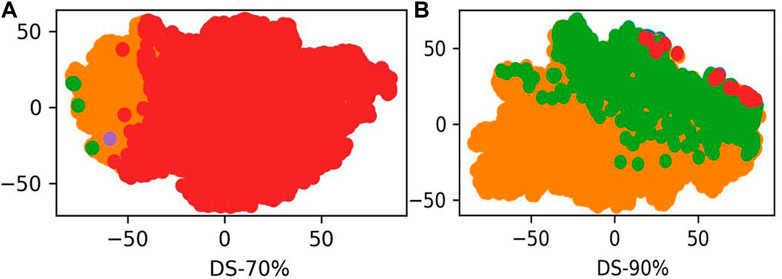
FIGURE 3. The t-SNE visualization of the second layer features learned from DS-70% and DS-90%. (A) The second word features learned from DS-70%. (B) The second word features learned from DS-90%.
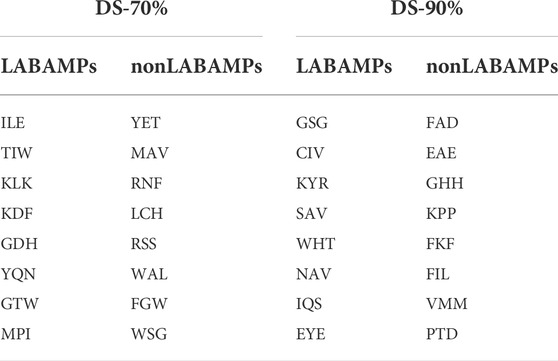
TABLE 7. Words with highest ACCs for two datasets of DS-70% and DS-90%. We used the word embedding at the last level of GCN to view the best performing words under each category.
In this study, we constructed LABAMPsGCN, a novel graph-based identifier to predict LABAMPs accurately. In this identifier, we designed a graph convolutional neural network framework to automatically learning sequence features. By retrieving and reorganizing multiple AMPs databases and Uni-Prot database, we constructed the positive and negative datasets. We organized positive and negative samples into a large heterogeneous graph, transforming the sequence classification problem into a node classification problem. Graph convolution neural network can aggregate the information of the surrounding nodes to predict the label information of the central node.
LABAMPsGCN is superior to other predictors, on the one hand, because the graph structure can effectively represent the relationship between sequences and words (when constructing a graph, an edge is established between a word and a sequence when this word belongs to this sequence), on the other hand, the label information of sequences can be transferred through the edges on the graph. Because the graph structure is a kind of many-to-many structure, the label information of sequences can be transferred in the whole graph. In this way, the words corresponding to positive and negative labels can be easily distinguished. These words may be the key features to determine whether a sequence is a LABAMP.
For users’ convenience, we have established a publicly accessible web server (http://www.dong-group.cn/database/dlabamp/Prediction/amplab/result/) that can help to predict LABAMPs metabolized from various Lactic acid bacteria. In the next step, we will discuss how to mine the key fragments with antimicrobial function from the whole genome sequence by combining information such as multiple sequence alignment and domain prediction. We believe LABAMPsGCN will be a competent tool for screening lactic acid strains with antimicrobial activities.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
T-JS conducted experiments, analyzed data and wrote the manuscript. H-LB and XY collected data and made the webserver. Z-HS guided the collection of data and the construction of the webserver. G-FD and M-SZ supervised the experiment and managed the whole project.
This work has been supported by the High level Talent Fund Project of Inner Mongolia Agricultural University, China (No. NDYBH 2017-1, NDYB 2018-9), Inner Mongolia Natural Science Foundation Project (No.2021MS06023), the National Natural Science Foundation Project (No.31901666), Major Project of Inner Mongolia Natural Science Foundation (2020ZD12), and 2022 Basic Scientific Research Business Fee Project of Universities Directly under the Inner Mongolia Autonomous Region—Interdisciplinary Research Fund of Inner Mongolia Agricultural University (BR22-14-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.1062576/full#supplementary-material
Aiken, Z. A., Wilson, M., and Pratten, J. (2011). Evaluation of ATP bioluminescence assays for potential use in a hospital setting. Infect. Control Hosp. Epidemiol. 32, 507–509. doi:10.1086/659761
Aurelio, Y. S., de Almeida, G. M., de Castro, C. L., and Braga, A. P. (2019). Learning from imbalanced data sets with weighted cross-entropy function. Neural process. Lett. 50, 1937–1949. doi:10.1007/s11063-018-09977-1
Azar, A. T., and El-Said, S. A. (2014). Performance analysis of support vector machines classifiers in breast cancer mammography recognition. Neural comput. Appl. 24, 1163–1177. doi:10.1007/s00521-012-1324-4
Beck, A., and Sharon, R. (2022). New results on multi-dimensional linear discriminant analysis. Operations Res. Lett. 50, 1–7. doi:10.1016/j.orl.2021.11.003
Carrasco, R. S. M., and Sicilia, M.-A. (2018). Unsupervised intrusion detection through skip-gram models of network behavior. Comput. Secur. 78, 187–197. doi:10.1016/j.cose.2018.07.003
Christiansen, J. S. L., Henriksen, C., Pedersen, H. L., and Petersen, C. L. (2021). Filled julia sets of Chebyshev polynomials. J. Geom. Anal. 31, 12250–12263. doi:10.1007/s12220-021-00716-y
Chung, C.-R., Kuo, T.-R., Wu, L.-C., Lee, T.-Y., and Horng, J.-T. (2020). Characterization and identification of antimicrobial peptides with different functional activities. Brief. Bioinform. 21, 1098–1114. doi:10.1093/bib/bbz043
Consortium, U. (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Crouch, S. P., Kozlowski, R., Slater, K. J., and Fletcher, J. (1993). The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 160, 81–88. doi:10.1016/0022-1759(93)90011-u
Fagerland, M. W., and Hosmer, D. W. (2012). A generalized Hosmer-Lemeshow goodness-of-fit test for multinomial logistic regression models. Stata J. 12, 447–453. doi:10.1177/1536867x1201200307
Fjell, C. D., Hancock, R. E. W., and Cherkasov, A. (2007). AMPer: A database and an automated discovery tool for antimicrobial peptides. Bioinformatics 23, 1148–1155. doi:10.1093/bioinformatics/btm068
Fuentes-Beals, C., Valds-Jimnez, A., and Riadi, G. (2022). Hidden Markov modeling with HMMTeacher. PLoS Comput. Biol. 18, e1009703–e1009709. doi:10.1371/journal.pcbi.1009703
Gao, W., and Huang, H. (2021). A gating context-Aware text classification model with BERT and graph convolutional networks. J. Intelligent Fuzzy Syst. 40, 4331–4343. doi:10.3233/jifs-201051
Garg, S. B., and Subrahmanyam, V. V. (2022). Sentiment analysis: Choosing the right word embedding for deep learning model. Lect. Notes Netw. Syst. 218, 417–428. doi:10.1007/978-981-16-2164-2_33
Gogoladze, G., Grigolava, M., Vishnepolsky, B., Chubinidze, M., Duroux, P., Lefranc, M.-P., et al. (2014). Dbaasp: Database of antimicrobial activity and structure of peptides. FEMS Microbiol. Lett. 357, 63–68. doi:10.1111/1574-6968.12489
Greub, G., Holliger, C., Sanglard, D., Schrenzel, J., Thiel, V., and Viollier, P. (2016). The Swiss society of microbiology: Small bugs, big questions and cool answers. Chimia 70, 874–877. doi:10.2533/chimia.2016.874
Gu, Y., Tian, J., Zhang, Y., Wu, J., and He, Y. (2022). Effect of Saccharomyces cerevisiae cell-free supernatant on the physiology, quorum sensing, and protein synthesis of lactic acid bacteria. LWT 165, 113732. doi:10.1016/j.lwt.2022.113732
Gueguen, Y., Garnier, J., Robert, L., Lefranc, M. P., Mougenot, I., de Lorgeril, J., et al. (2006). PenBase, the shrimp antimicrobial peptide penaeidin database: Sequence-based classification and recommended nomenclature. Dev. Comp. Immunol. 30, 283–288. doi:10.1016/j.dci.2005.04.003
Hamid, M. N., and Friedberg, I. (2018). Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 35, 2009–2016. doi:10.1093/bioinformatics/bty937
Hammami, R., Ben Hamida, J., Vergoten, G., and Fliss, I. (2009). PhytAMP: A database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 37, D963–D968. doi:10.1093/nar/gkn655
Hammami, R., Zouhir, A., Lay, C. L., Hamida, J. B., Fliss, I., and Forsberg, A. (2010). BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 10, 22. doi:10.1186/1471-2180-1-22
He, W., Jiang, Y., Jin, J., Li, Z., Zhao, J., Manavalan, B., et al. (2021). Accelerating bioactive peptide discovery via mutual information-based meta-learning. Brief. Bioinform. 23, bbab499–18. doi:10.1093/bib/bbab499
Henderson, K. A., Bialeschki, M. D., and James, P. A. (2007). Overview of camp research. Child. Adolesc. Psychiatr. Clin. N. Am. 16, 755–767. doi:10.1016/j.chc.2007.05.010
Hu, Y., Zhang, L., Wen, R., Chen, Q., and Kong, B. (2022). Role of lactic acid bacteria in flavor development in traditional Chinese fermented foods: A review. Crit. Rev. Food Sci. Nutr. 62, 2741–2755. doi:10.1080/10408398.2020.1858269
Huang, H., Liu, F., Zeng, S., Zeng, X., Qian, Z., Shi, D., et al. (2022). Preparation of trifluralin antibody and development of enzyme linked immunosorbent assay. Mod. Food Sci. Technol. 38, 345–354. doi:10.13982/j.mfst.1673-9078.2022.1.0470
Huang, X., Li, Z., Jin, Y., and Zhang, W. (2022). Fair-AdaBoost: Extending AdaBoost method to achieve fair classification. Expert Syst. Appl. 202, 117240. doi:10.1016/j.eswa.2022.117240
Jiang, J., Wang, C., Yu, M., Jia, C., Li, G., Liu, C., et al. (2021). Nfdd: A dynamic malicious document detection method without manual feature dictionary. Lect. Notes Comput. Sci. 12938, 147–159. doi:10.1007/978-3-030-86130-8_12
Kadomatsu, K., Kishida, S., and Tsubota, S. (2022). The heparin-binding growth factor midkine: The biological activities and candidate receptors. J. Biochem. 172, 511–521. doi:10.1093/jb/mvt035
Kai, J., Puntambekar, A., Santiago, N., Lee, S. H., Sehy, D. W., Moore, V., et al. (2012). A novel microfluidic microplate as the next generation assay platform for enzyme linked immunoassays (ELISA). Lab. Chip 12, 4257–4262. doi:10.1039/c2lc40585g
Kawashima, S., Pokarowski, P., Pokarowska, M., Kolinski, A., Katayama, T., and Kanehisa, M. (2008). AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 36, D202–D205. doi:10.1093/nar/gkm998
Kouw, W. M., and Loog, M. (2021). Target robust discriminant analysis. Lect. Notes Comput. Sci. 12644, 3–13. doi:10.1007/978-3-030-73973-7_1
Lauger, P., and Apell, H. J. (1988). Transient behaviour of the Na+/K+-pump: Microscopic analysis of nonstationary ion-translocation. Biochim. Biophys. Acta 944, 451–464. doi:10.1016/0005-2736(88)90516-0
Li, W., and Godzik, A. (2006). Cd-Hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi:10.1093/bioinformatics/btl158
Liu, W. H., Wang, Y., Tang, J. H., Shen, G. L., and Yu, R. Q. (1998). An optical fiber sensor for berberine based on immobilized 1, 4-bis(naphth 2, 1-d oxazole-2-yl)benzene in a new copolymer. Talanta 46, 679–688. doi:10.1016/s0039-9140(97)00330-5
Mulvenna, J. P., Wang, C., and Craik, D. J. (2006). CyBase: A database of cyclic protein sequence and structure. Nucleic Acids Res. 34, D192–D194. doi:10.1093/nar/gkj005
Pirtskhalava, M., Amstrong, A. A., Grigolava, M., Chubinidze, M., Alimbarashvili, E., Vishnepolsky, B., et al. (2021). DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 49, D288–D297. doi:10.1093/nar/gkaa991
Qu, Y. H., Yu, H., Gong, X. J., Xu, J. H., and Lee, H. S. (2017). On the prediction of DNA-binding proteins only from primary sequences: A deep learning approach. PLoS One 12, e0188129. doi:10.1371/journal.pone.0188129
Ruit, M. v. d., Billeter, M., and Eisemann, E. (2022). An efficient dual-hierarchy t-SNE minimization. IEEE Trans. Vis. Comput. Graph. 28, 614–622. doi:10.1109/tvcg.2021.3114817
Saibil, H. R. (2022). Cryo-EM in molecular and cellular biology. Mol. Cell. 82, 274–284. doi:10.1016/j.molcel.2021.12.016
Shao, Y., Dietrich, F., Nettelblad, C., and Zhang, C. (2021). Training algorithm matters for the performance of neural network potential: A case study of Adam and the kalman filter optimizers. J. Chem. Phys. 155, 204108. doi:10.1063/5.0070931
Shi, G., Kang, X., Dong, F., Liu, Y., Zhu, N., Hu, Y., et al. (2022). Dramp 3.0: An enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res. 50, D488–D496. doi:10.1093/nar/gkab651
Sim, N.-L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. doi:10.1093/nar/gks539
Song, X., Fu, B., Lan, Y., Chen, Y., Wei, Y., and Dong, C. (2018). Label-free fluorescent aptasensor berberine-based strategy for ultrasensitive detection of Hg2+ ion. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 204, 301–307. doi:10.1016/j.saa.2018.06.058
Sundararajan, V. S., Gabere, M. N., Pretorius, A., Adam, S., Christoffels, A., Lehvaeslaiho, M., et al. (2012). Dampd: A manually curated antimicrobial peptide database. Nucleic Acids Res. 40, D1108–D1112. doi:10.1093/nar/gkr1063
Sung Won, C., Jaewoo, Y., and Sang Woo, S. (2020). Engineering tools for the development of recombinant lactic acid bacteria. Biotechnol. J. 15, e1900344. doi:10.1002/biot.201900344
Teusink, B., and Molenaar, D. (2017). Systems biology of lactic acid bacteria: For food and thought. Curr. Opin. Syst. Biol. 6, 7–13. doi:10.1016/j.coisb.2017.07.005
Theolier, J., Fliss, I., Jean, J., and Hammami, R. (2014). MilkAMP: A comprehensive database of antimicrobial peptides of dairy origin. Dairy Sci. Technol. 94, 181–193. doi:10.1007/s13594-013-0153-2
Van Dyk, T. K., Majarian, W. R., Konstantinov, K. B., Young, R. M., Dhurjati, P. S., and LaRossa, R. A. (1994). Rapid and sensitive pollutant detection by induction of heat shock gene-bioluminescence gene fusions. Appl. Environ. Microbiol. 60, 1414–1420. doi:10.1128/aem.60.5.1414-1420.1994
Veltri, D., Kamath, U., and Shehu, A. (2018). Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747. doi:10.1093/bioinformatics/bty179
Xie, L., Lu, Y., Furuhata, T., Yamakawa, S., Zhang, W., Regmi, A., et al. (2022). Graph neural network-enabled manufacturing method classification from engineering drawings. Comput. Industry 142, 103697. doi:10.1016/j.compind.2022.103697
Xiong, Z., Shen, Q., Xiong, Y., Wang, Y., and Li, W. (2019). New generation model of word vector representation based on CBOW or skip-gram. Comput. Mat. Contin. 60, 259–273. doi:10.32604/cmc.2019.05155
Zeng, M., Li, M., Wu, F.-X., Li, Y., and Pan, Y. (2019). DeepEP: A deep learning framework for identifying essential proteins. BMC Bioinforma. 20, 506. doi:10.1186/s12859-019-3076-y
Zhai, J., Qi, J., and Zhang, S. (2020). An instance selection algorithm for fuzzy K-nearest neighbor. J. Intelligent Fuzzy Syst. 40, 521–533. doi:10.3233/jifs-200124
Zhang, P., Jia, Y., Shang, Y., Zhao, Z., Yang, L., Zheng, F., et al. (2022). Altered dynamic functional connectivity in rectal cancer patients with and without chemotherapy: A resting-state fMRI study. Int. J. Neurosci. 18, 1–11. doi:10.1080/00207454.2022.2130295
Zhou, Y., Zheng, H., Huang, X., Hao, S., Li, D., and Zhao, J. (2022). Graph neural networks: Taxonomy, advances, and trends. ACM Trans. Intell. Syst. Technol. 13, 1–54. doi:10.1145/3495161
Keywords: lactic acid bacteria antimicrobial peptides, word embedding, tripeptide, graph convolution neural network, deep learning
Citation: Sun T-J, Bu H-L, Yan X, Sun Z-H, Zha M-S and Dong G-F (2022) LABAMPsGCN: A framework for identifying lactic acid bacteria antimicrobial peptides based on graph convolutional neural network. Front. Genet. 13:1062576. doi: 10.3389/fgene.2022.1062576
Received: 06 October 2022; Accepted: 24 October 2022;
Published: 03 November 2022.
Edited by:
Wenzheng Bao, Xuzhou University of Technology, ChinaReviewed by:
Hao Wu, School of Software, Shandong University, ChinaCopyright © 2022 Sun, Bu, Yan, Sun, Zha and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gai-Fang Dong, ZG9uZ2dmQGltYXUuZWR1LmNu; Mu-Su Zha, bXN6aGExOTg4QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.