 Yuan Xiang
Yuan Xiang Shi-Qiang Fang
Shi-Qiang Fang Hui Wang
Hui Wang Zhong-Xin Lu
Zhong-Xin Lu- Department of Medical Laboratory, Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
Waldenström Macroglobulinemia (WM) is a rare chronic lymphoproliferative disease, accounting for less than 2% of hematological malignancies. It is characterized by plasma cytoid lymphocyte infiltration in bone marrow and abnormal increase of monoclonal IgM in peripheral blood. Only 5%–10% of cases of WM secrete monoclonal IgG and IgA components or do not secrete monoclonal long immunoglobulin. This case is the first to report of serum protein recombination from lgM and Igkappa band mutation to abnormal lgG and Igkappa band after 6 years of treatment in a male patient with WM.
Introduction
WM is a rare indolent mature B-cell lymphoma with an annual incidence of five in one million (Kaiser et al., 2021). Human immunoglobulin is synthesized and secreted by plasma cells. It is a group of globulin with antibody activity. It mainly exists in the serum and body fluid of the body, and its content reflects the humoral immune function of the body. Immunoglobulin is a tetrapeptide chain structure consisting of two identical light chains and two identical heavy chains connected by interchain disulfide bonds. There are five categories of immunoglobulins, namely IgG, IgA, IgM, IgD and IgE. The peptide chain with small molecular weight in immunoglobulin in human body is called light chain, which is divided into two types, Igkappa(κ) and Iglambda(γ), according to the difference of its structure and antigenicity in the constant region.
The peripheral blood of patients with WM contains a large amount of immunoglobulin IgM, which is called macroglobulinemia, because IgM is a pentameric immunoglobulin with the largest molecular weight. At present, the clinical tendency is to classify lymphoplasmacytoid lymphoma and WM as the same disease. WM is more common in elderly men, with an onset age of 63–73 years (Gertz, 2021). The clinical manifestations of WM are atypical and easy to be misdiagnosed. The clinical features include anemia, fever, weight loss, night sweating, etc (Dimopoulos and Anagnostopoulos, 2005; Wang and Lin, 2020). The main laboratory indicators of WM were: infiltration of small B lymphocytes and plasmacytoid lymphocytes in bone marrow, liver and splenic lymph nodes; Peripheral blood cytopenia; MYD88L265P gene mutation; Serum monoclonal IgM>10 g/L (Dimopoulos and Kastritis, 2019). According to the existing literature, 90%–95% of patients with WM have high IgM immunoglobulin, and only less than 5% of patients have high IgG and IgA. Such patients are often not accompanied by hyperviscoemia or autoimmune neuropathy (Voisin et al., 2011). This is a case report of WM with low IgM and hypermucemia. By sorting out the course of the patient’s disease, statistical examination results and medication status, it can provide a reference for the diagnosis and treatment of WM in the future.
Case presentation
The patient was a 68-year-old man with fever for 10 days, jaundice for 7 days, a history of bronchitis and gastric ulcer, BP138/71 mmHg, severe yellow staining of skin and sclera, palpable enlargement of the left cervical lymph nodes, coarse breath sounds in both lungs, no obvious rals, HR57 bpm, rhythmic, no murmurs, soft abdomen, no tenderness, liver, spleen, and subrib. Murphy’s sign was negative, percussion pain in the renal area was negative, and there was no edema in the lower extremities. Immunofixation electrophoresis (IFE) refers to the separation of various protein components in blood serum, Serum IFE can detect IgG, IgM, IgA, κ, γ, etc. IFE is often used in the clinical diagnosis of multiple immune system diseases such as monoclonal immunoglobulin proliferation disease, Benzhou protein, free light chain disease, heavy chain disease, and polyclonal immunoglobulin. This patient’s serum IFE results revealed an abnormal monoclonal band in the lgG and κ lanes of immunofixation electrophoresis (serum) (Figure 1A). The immunohistochemical results of the patient in 2016 were: CD20 (+), PAX5 (+), CD5 (+), CD10 (-), CD23 (+), KI-67 (+10%), CyclinH (-)CD3 (+), bcl-6 (-), bcl-2 (+), SOX11 (-), Kappa (-), Lambda (-), EBER(-), the proportion of tumor cells expressing CD3 and CD5 positive is small. Left neck lymph node non-Hodgkin’s lymphoma (B-small cell). The patient was diagnosed as WM according to the clinical manifestations and laboratory results. WM is a plasma cell malignant hyperplasia disease, mainly manifested by abnormal elevation of human immunoglobulin lgM (Tomowiak et al., 2019).
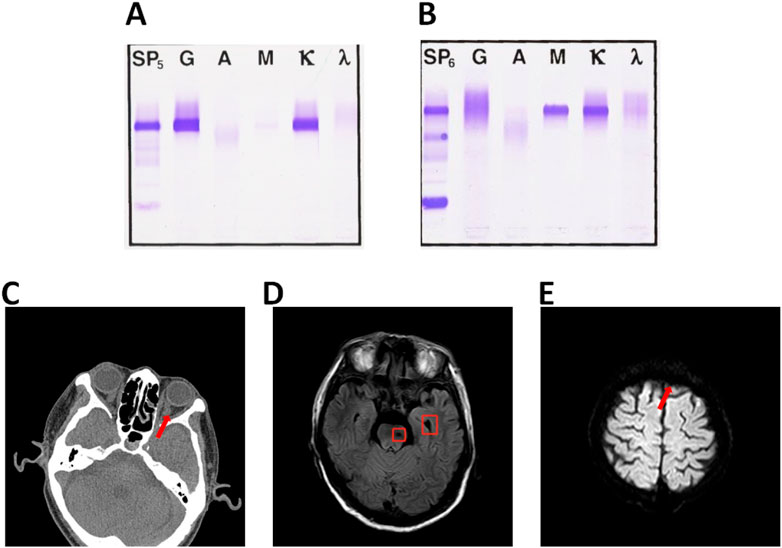
FIGURE 1. (A) Immunofixation electrophoresis (serum) abnormal monoclonal bands found in lgG and κ lane. (B) Immunofixation electrophoresis (serum) lgM and κ lane. (C) Abnormal signal foci in left pontine and left cerebellar Angle. (D) Left frontal hemorrhage foci. (E)Left frontal infarction foci of pontine.
The monoclonal immunoglobulin type of this patient was IgM- κ, 6 years ago. (Figure 1B) The common symptoms of WM are bleeding tendency, fatigue, anemia, visual impairment, emaciation, thrombocytopenia, increased blood viscosity, low fibrinogen, enlarged lymph nodes, liver and kidney function damage and proteinuria (Vijay and Gertz, 2007). The results of routine bone marrow examination showed that 20.9% of the patients had lymphocytic system and 7.0% had lymphoid plasma cells. MYD88 L256P gene mutation was positive. The patient did not have CXCR4 gene mutation. Blood tumor immunoassay report sheet: lymphocytes accounted for about 30% of nuclear cells, and B lymphocytes accounted for about 44% of lymphocytes, expressing HLA-DR, CD19, CD20, CD22, CD79b, CD200, sKappa, Bcl-2 and cKappa. Bone marrow histopathological diagnosis report: After the treatment of WM, the hematopoietic tissue hyperplasia was obviously active, and the three lines of granulosa, red and giant hyperplasia were active, and the lymphocytes were scattered or focal hyperplasia. The patient’s fibrinogen level was 0.83, which was the critical value. The abdominal CT (Computed Tomography) results of the patient showed that the infection of the right lung (middle lobe) was more progressive, and Klebsiella pneumoniae infection was considered. The common symptoms of WM combined with hyperviscosity were visual impairment, suspicious light perception in the left eye, smaller left eyelid fissure than the right eye, lateral deviation of the left eye, poor coordination of eye movement examination, and aggravated numbness in the left face. Ocular CT examination showed that the medial orbital segment of bilateral optic nerve was slightly tortuous and bilateral symmetry. Brain MR (Magnetic Resonance) examination was performed, and the results showed abnormal signal foci in the left pontine and left pontine cerebellar Angle, and acute cerebral infarction in the left frontal lobe. (Figures 1C–E).
Partial blood routine results of the patient in the past 6 years are shown in Table 1, which showed that the blood cells of the patient’s three lines were lower than normal. The white blood cell count results were: neutrophil-lobulated nuclei 85.00% (high), monocytes 5.00%, lymphocytes 10.00% (low). Peripheral blood cell morphology part of the rouleau red blood cells were rouleau shaped arrangement, other significant abnormalities were not seen. Partial serum biochemical examination results of the patients in the past 6 years are shown in Table 2. The patient’s liver function was abnormal. Color Doppler ultrasound showed diffuse non-specific damage to the liver parenchyma, and slightly hyperechoic nodules in the liver. Combined with the clinical history, the possibility of liver abscess was not excluded. The pathogenesis of WM may be related to hepatitis B. Pre-transfusion examination of patients: Hepatitis B virus surface antigen (chemiluminescence method) > 250.00 IU/mLPos, hepatitis B virus E antigen (chemiluminescence method) 21.94 S/COPos, anti-hepatitis B virus core antibody (chemiluminescence method) 4.47 S/COPos, Anti-hepatitis B virus E antibody (chemiluminescence method) 0.38 S/COPos, other results were negative. The results of hepatitis B virus deoxyribonucleic acid determined by PCR fluorescent probe are as follows: 1.63 × 10^4 IU/ml.
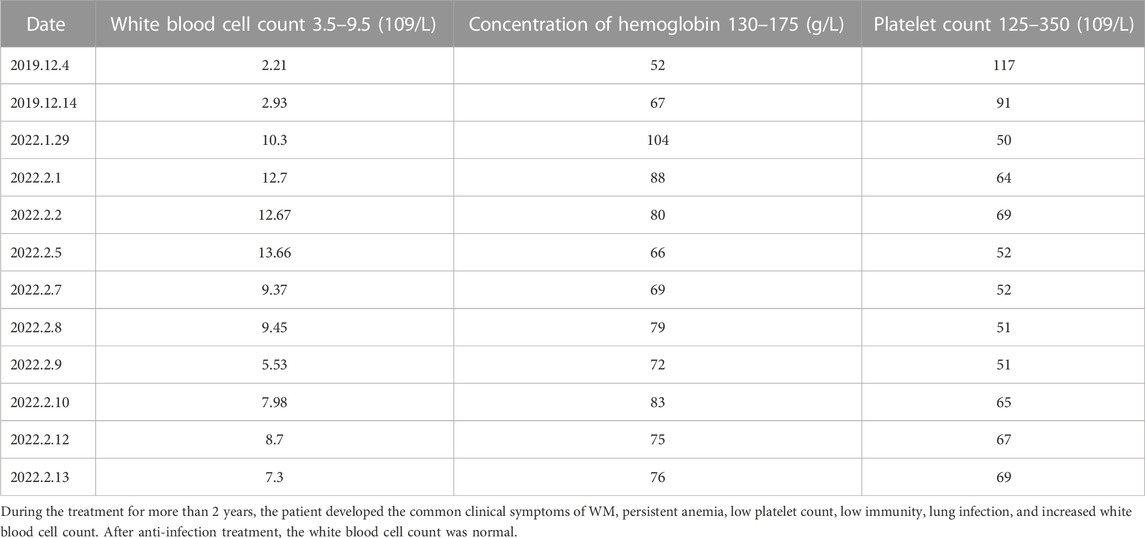
TABLE 1. Results of some blood routine examinations of patients.
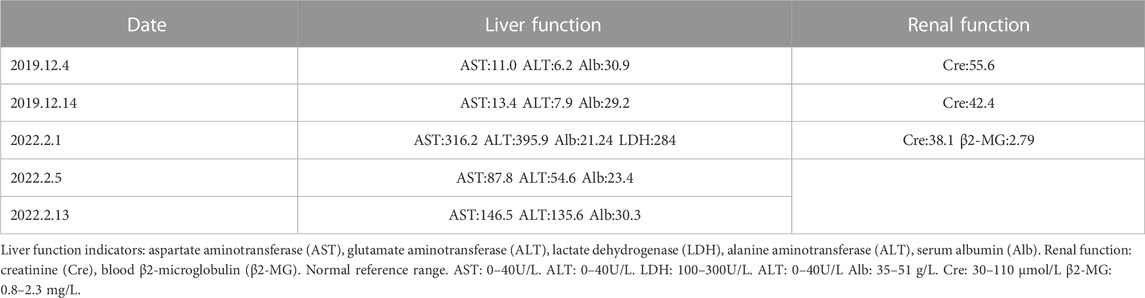
TABLE 2. Results of partial liver and renal function tests.
The results of lactate dehydrogenase, serum albumin and serum β2-microglobulin were all abnormal. These results indicate that the patient’s liver and renal function were impaired in 2022 and the patient’s prognosis was poor.
The patient was treated with VAD regimen for four courses of chemotherapy after the diagnosis of WM in 2016. From 31 August 2016 to 31 December 2016, the patient received RCOP regimen for 6 times. Rituximab 0.6 single agent consolidation chemotherapy was given from 22 February 2017 to 3 June 2017. Chemotherapy with RCD regimen was given from 4 June 2018. On 23 August 2018, the patient began to take ivitinib maleate for treatment, but the drug was stopped due to thrombocytopenia and multiple oral ulcers. In January 2019, the patient began to take ibrutinib capsules orally (3 tablets/day), and the platelet decline was obvious. The medication of ibrutinib was adjusted according to the blood test results. Blood routine examination in April 2019: white blood cell 3.2 × 10^9/L, hemoglobin 102 g/L, platelet 43 × 10^9/L, ibrutinib dose was gradually reduced to one tablet/day. The patient began long-term oral ibrutinib (2 tablets/day) targeted therapy to date. Entecavir anti-hepatitis B virus therapy and half-volume plasma exchange therapy were given until the patient was discharged from hospital in February 2022.
Literature review and discussion
WM was first reported in 1944 by a Swedish physician named Jan G. Waldenstrom, who reported six cases of bleeding from the mouth and nose, anemia, decreased levels of fibrinogen in the blood, and enlarged lymph nodes. On closer examination, Waldenstrom found that their bone marrow had proliferating plasma cells and that their blood was very sticky because of the increased amount of macroglobulin (Richter and Ambrosius, 1981). According to the WHO classification criteria, the patient had an increase in serum monoclonal immunoglobulin IgM, hyperviscosity syndrome, cytopenia, and plasmacytoid lymphocyte infiltration in the bone marrow, which was diagnosed as WM. The incidence of WM is low, and the pathogenesis is not clear, which may be related to genetic susceptibility and viral infection of hepatitis B and C. Genetic mutations may be one of the causes of WM. Researchers found that 90% of patients with WM had mutations in the MYD88 L256P gene, followed by 27% with mutations in CXCR4 (Yu et al., 2018; Ullah, 2019).
WM accounts for 1%–2% of hematological tumors. The incidence of this disease is relatively high in Caucasians, and it tends to occur in the elderly, mostly in males over 60 years old (Kastritis et al., 2015a; Teras et al., 2016; Gertz, 2021). The course of the disease is slow. About 19%–28% of the patients have no symptoms and are easy to be misdiagnosed at an early stage (Garcia-Sanz et al., 2001; Pophali et al., 2019). The survival time of the disease is only five to 7 years (Labreuche et al., 2022). For early asymptomatic patients, it can be based on the percentage of plasma cell like lymphocyte infiltration in bone marrow, the level of serum monoclonal immunoglobulin IgM, β2-microglobulin and albumin were scored, and clinical follow-up was conducted every 3 months to understand the development speed of the disease (Dimopoulos and Kastritis, 2019).
The main clinical features of WM are hypermucemia, hepatosplenomegaly, lymphadenopathy, anemia, susceptibility to infection, Raynaud’s phenomenon, and so on. In addition to the detection of bone marrow cells by bone marrow aspiration and the detection of immunoglobulins in serum by immunofixation electrophoresis, the diagnosis of WM can also be made by immunohistochemistry or flow cytometry (Wang and Lin, 2020). Patients with WM may present with B cell populations: CD20+,sIgM+,CD22+ (weak),CD79+,CD25+,CD27+,FMC7+,BCL-2+,CD52+,CD5+/−,CD10+/−,CD23+/−,CD103−; Plasma cell population: CD138+CD38+,CD19+,CD45+,CD56−.At the same time, WM can also be diagnosed by genetic methods. About 30%–50% of patients with WM have chromosome six deletion (Garcia-Sanz et al., 2021).
Plasma separation is the current treatment for the treatment of WM with hyperviscosity. Rituximab combined with oral or intravenous cyclophosphamide and dexamethasone is a common protocol for this disease (Kastritis et al., 2015b). The combination of bendamustine and rituximab has also been shown to be effective in the long term, but attention should be paid to the dose of bendamustine (Rummel et al., 2013; Laribi et al., 2019; Gertz, 2021). Bortezomib alone or in combination with rituximab has also been shown to be effective, but has some toxic side effects (Treon et al., 2007; Ghobrial et al., 2010; Dimopoulos et al., 2013; Gavriatopoulou et al., 2017; An et al., 2021). Chemotherapy drugs such as cyclophosphamide and doxorubicin can be considered when the transformation of diffuse large B-cell lymphoma occurs (Leblond et al., 2013). Ibrutinib has been shown to be effective in patients refractory to rituximab. If organ enlargement and lymph node enlargement occur, ibrutinib/rituximab may be considered (Dimopoulos et al., 2017; Treon et al., 2021). CAR-T cell immunotherapy targeting CD19+and CD20+may have a good therapeutic effect.
The transformation of WM into other blood diseases is rare. There are only a few reports that WM can transform into diffuse large B-cell lymphoma or mantle cell lymphoma. The bone marrow of diffuse large B-cell lymphoma is a low-grade lymphoma, and its immunophenotype usually expresses CD45 and whole B-cell antigen (Ollila and Olszewski, 2018). The bone marrow of mantle cell lymphoma showed monomorphic, small and medium-sized lymphocytes with abnormal nuclei (Vose, 2017). Immunohistochemical results showed CD5+, CD10−, CD23−, CD25−, CD45+, CD103−, and CD138−. The clinical manifestations, myelogram and immunotyping of this case are consistent with the diagnosis of WM.
This case is a rare case of WM, which is recombined from the mutation of lgM and κbands into an abnormal double clone band of lgG and κ. It is worth noting that, as far as we know, this is the first time that this mutation is reported in high lgM WM. This mutation may be related to physiological reasons, or it may be the mutation of immunoglobulin gene in lymphocytes and plasma cells caused by long-term administration of ibutinib. As such patients are rare, and the test samples 6 years ago have been destroyed, we do not have enough samples for DNA sequencing to further study the specific situation of gene mutation, but the report of this case can provide reference for the diagnosis and treatment of such patients in the future.
Conclusion
This is a rare case of WM with abnormal clonal bands of lgG and κ. The patient was diagnosed with WM for more than 6 years. After a variety of treatment schemes, he took ibutinib for a long time. After 6 years of treatment, he developed hyperviscosity, visual impairment, community acquired pneumonia, hypofibrinogenemia, etc. The case will provides valuable reference information for clinical diagnosis, disease transformation and treatment of WM.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Designed and conceived the experiments: Z-XL, HW, Y-WL, and YX. Analyzed the data: YX and S-QF. Wrote and reviewed the manuscript: YX, HW, and Z-XL.
Funding
This work was supported by Wuhan Municipal Health Commission Project (Project no. WX21Q49), Hubei Natural Science Foundation (2021CFB230), and by one local grant from The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology to YX.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
WM, Waldenström Macroglobulinemia; MR, Magnetic Resonance; CT, Computed Tomography; IFE, Immunofixation electrophoresis; κ, Igkappa; γ, Iglambda; AST, Aspartate aminotransferase; ALT, Glutamate aminotransferase; LDH, Lactate dehydrogenase; ALT, Alanine aminotransferase; Alb, Serum albumin; Cre, Renal function: creatinine; β2-MG, Blood β2-microglobulin.
References
An, G., Zhou, D., Cheng, S., Zhou, K., Li, J., Zhou, J., et al. (2021). A phase II trial of the bruton tyrosine-kinase inhibitor zanubrutinib (BGB-3111) in patients with relapsed/refractory Waldenstrom macroglobulinemia. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 27, 5492–5501. doi:10.1158/1078-0432.CCR-21-0539
Dimopoulos, M. A., and Anagnostopoulos, A. (2005). Waldenström's macroglobulinemia. Clin. Haematol. 18, 747–765. doi:10.1016/j.beha.2005.01.028
Dimopoulos, M. A., Garcia-Sanz, R., Gavriatopoulou, M., Morel, P., Kyrtsonis, M. C., Michalis, E., et al. (2013). Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): Long-term results of a phase 2 study of the European myeloma network (EMN). Blood 122, 3276–3282. doi:10.1182/blood-2013-05-503862
Dimopoulos, M. A., and Kastritis, E. (2019). How I treat Waldenstrom macroglobulinemia. Blood 134, 2022–2035. doi:10.1182/blood.2019000725
Dimopoulos, M. A., Trotman, J., Tedeschi, A., Matous, J. V., Macdonald, D., Tam, C., et al. (2017). Ibrutinib for patients with rituximab-refractory waldenstrom's macroglobulinaemia (iNNOVATE): An open-label substudy of an international, multicentre, phase 3 trial. Lancet. Oncol. 18, 241–250. doi:10.1016/S1470-2045(16)30632-5
Garcia-Sanz, R., Dogliotti, I., Zaccaria, G. M., Ocio, E. M., Rubio, A., Murillo, I., et al. (2021). 6q deletion in Waldenstrom macroglobulinaemia negatively affects time to transformation and survival. Br. J. Haematol. 192, 843–852. doi:10.1111/bjh.17028
Garcia-Sanz, R., Montoto, S., Torrequebrada, A., de Coca, A. G., Petit, J., Sureda, A., et al. (2001). Waldenström macroglobulinaemia: Presenting features and outcome in a series with 217 cases. Br. J. Haematol. 115, 575–582. doi:10.1046/j.1365-2141.2001.03144.x
Gavriatopoulou, M., Garcia-Sanz, R., Kastritis, E., Morel, P., Kyrtsonis, M. C., Michalis, E., et al. (2017). BDR in newly diagnosed patients with WM: Final analysis of a phase 2 study after a minimum follow-up of 6 years. Blood 129, 456–459. doi:10.1182/blood-2016-09-742411
Gertz, M. A. (2021). Waldenstrom macroglobulinemia: 2021 update on diagnosis, risk stratification, and management. Am. J. Hematol. 96, 258–269. doi:10.1002/ajh.26082
Ghobrial, I. M., Xie, W., Padmanabhan, S., Badros, A., Rourke, M., Leduc, R., et al. (2010). Phase II trial of weekly bortezomib in combination with rituximab in untreated patients with Waldenstrom Macroglobulinemia. Am. J. Hematol. 85, 670–674. doi:10.1002/ajh.21788
Kaiser, L. M., Hunter, Z. R., Treon, S. P., and Buske, C. (2021). CXCR4 in waldenstrom's macroglobulinema: Chances and challenges. Leukemia 35, 333–345. doi:10.1038/s41375-020-01102-3
Kastritis, E., Gavriatopoulou, M., Kyrtsonis, M. C., Roussou, M., Hadjiharissi, E., Symeonidis, A., et al. (2015). Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenstrom macroglobulinemia: Final analysis of a phase 2 study. Blood 126, 1392–1394. doi:10.1182/blood-2015-05-647420
Kastritis, E., Kyrtsonis, M. C., Morel, P., Gavriatopoulou, M., Hatjiharissi, E., Symeonidis, A. S., et al. (2015). Competing risk survival analysis in patients with symptomatic Waldenstrom macroglobulinemia: The impact of disease unrelated mortality and of rituximab-based primary therapy. Haematologica 100, e446–e449. doi:10.3324/haematol.2015.124149
Labreuche, J., Assouan, D., Durot, E., Tomowiak, C., Roos-Weil, D., Toussaint, E., et al. (2022). Innovative Leukaemia Organisation Chronic Lymphocytic Leukaemia, Does early disease progression predict survival after first line-treatment of Waldenstrom macroglobulinemia? Hematol. Oncol. 40, 400–408. doi:10.1002/hon.2996
Laribi, K., Poulain, S., Willems, L., Merabet, F., Le Calloch, R., Eveillard, J. R., et al. (2019). Bendamustine plus rituximab in newly-diagnosed Waldenstrom macroglobulinaemia patients. A study on behalf of the French Innovative Leukaemia Organization (FILO). Br. J. Haematol. 186, 146–149. doi:10.1111/bjh.15718
Leblond, V., Johnson, S., Chevret, S., Copplestone, A., Rule, S., Tournilhac, O., et al. (2013). Results of a randomized trial of chlorambucil versus fludarabine for patients with untreated Waldenstrom macroglobulinemia, marginal zone lymphoma, or lymphoplasmacytic lymphoma. J. Clin. Oncol. official J. Am. Soc. Clin. Oncol. 31, 301–307. doi:10.1200/JCO.2012.44.7920
Ollila, T. A., and Olszewski, A. J. (2018). Extranodal diffuse large B cell lymphoma: Molecular features, prognosis, and risk of central nervous system recurrence. Curr. Treat. options Oncol. 19, 38. doi:10.1007/s11864-018-0555-8
Pophali, P. A., Bartley, A., Kapoor, P., Gonsalves, W. I., Ashrani, A. A., Marshall, A. L., et al. (2019). Prevalence and survival of smouldering Waldenstrom macroglobulinaemia in the United States. Br. J. Haematol. 184, 1014–1017. doi:10.1111/bjh.15201
Richter, R. F., and Ambrosius, H. (1981). Detection of a monoclonal human myeloma protein of IgM and IgG class by carp anti-idiotypic sera. Acta Biol. medica Ger. 40, 861–865.
Rummel, M. J., Niederle, N., Maschmeyer, G., Banat, G. A., von Grunhagen, U., Losem, C., et al. (2013). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381, 1203–1210. doi:10.1016/S0140-6736(12)61763-2
Teras, L. R., DeSantis, C. E., Cerhan, J. R., Morton, L. M., Jemal, A., and Flowers, C. R. (2016). 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA a cancer J. Clin. 66, 443–459. doi:10.3322/caac.21357
Tomowiak, C., Poulain, S., Debiais, C., Guidez, S., and Leleu, X. (2019). Waldenstrom macroglobulinaemia. Presse medicale. 48, 832–841. doi:10.1016/j.lpm.2019.07.020
Treon, S. P., Hunter, Z. R., Matous, J., Joyce, R. M., Mannion, B., Advani, R., et al. (2007). Multicenter clinical trial of bortezomib in relapsed/refractory waldenstrom's macroglobulinemia: Results of WMCTG trial 03-248. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 13, 3320–3325. doi:10.1158/1078-0432.CCR-06-2511
Treon, S. P., Meid, K., Gustine, J., Yang, G., Xu, L., Liu, X., et al. (2021). Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenstrom macroglobulinemia. J. Clin. Oncol. official J. Am. Soc. Clin. Oncol. 39, 565–575. doi:10.1200/JCO.20.00555
Ullah, T. R. (2019). The role of CXCR4 in multiple myeloma: Cells' journey from bone marrow to beyond. J. bone Oncol. 17, 100253. doi:10.1016/j.jbo.2019.100253
Vijay, A., and Gertz, M. A. (2007). Waldenstrom macroglobulinemia. Blood 109, 5096–5103. doi:10.1182/blood-2006-11-055012
Voisin, S., Hamidou, M., Lefrancois, A., Sigaud, M., Mahe, B., and Trossaert, M. (2011). Acquired von Willebrand syndrome associated with monoclonal gammopathy: A single-center study of 36 patients. Medicine 90, 404–411. doi:10.1097/MD.0b013e3182397166
Vose, J. M. (2017). Mantle cell lymphoma: 2017 update on diagnosis, risk-stratification, and clinical management. Am. J. Hematol. 92, 806–813. doi:10.1002/ajh.24797
Wang, W., and Lin, P. (2020). Lymphoplasmacytic lymphoma and Waldenstrom macroglobulinaemia: Clinicopathological features and differential diagnosis. Pathology 52, 6–14. doi:10.1016/j.pathol.2019.09.009
Keywords: waldenström macroglobulinemia, abnormal increase of IgM, mutation, waldenström macroglobulinemia/lymphoplasmacytoid lymphoma, rare chronic lymphoproliferative
Citation: Xiang Y, Fang S-Q, Liu Y-W, Wang H and Lu Z-X (2023) A rare case report of waldenström macroglobulinemia converted to serum low IgM. Front. Genet. 13:1051917. doi: 10.3389/fgene.2022.1051917
Received: 23 September 2022; Accepted: 27 December 2022;
Published: 19 January 2023.
Edited by:
Zelin Yang, Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, ChinaReviewed by:
Joaquim Carreras, Tokai University, JapanZhang Shuyao, Guangzhou Red Cross Hospital, China
Nan Wang, Tianjin University of Science and Technology, China
Copyright © 2023 Xiang, Fang, Liu, Wang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Wen Liu, NzY3MDQxNTNAcXEuY29t; Hui Wang, MDI3d2h3aEBzaW5hLmNvbQ==; Zhong-Xin Lu, THV6aG9uZ3hpbkB6eGhvc3BpdGFsLmNvbQ==