 Jian Diao1†
Jian Diao1† Shuxuan Li
Shuxuan Li Ling Ma
Ling Ma Ping Zhang
Ping Zhang- 1College of Forestry, Northeast Forestry University, Harbin, China
- 2Institute of Forest Protection, Heilongjiang Academy of Forestry, Harbin, China
- 3College of Medicine, Heilongjiang University of Chinese Medicine, Harbin, China
The major facilitator superfamily (MFS) is one of the largest known membrane transporter families. MFSs are involved in many essential functions, but studies on the MFS family in poplar have not yet been reported. Here, we identified 41 MFS genes from Populus trichocarpa (PtrMFSs). We built a phylogenetic tree, which clearly divided members of PtrMFS into six groups with specific gene structures and protein motifs/domains. The promoter regions contain various cis-acting elements involved in stress and hormone responsiveness. Genes derived from segmental duplication events are unevenly distributed in 17 poplar chromosomes. Collinearity analysis showed that PtrMFS genes are conserved and homologous to corresponding genes from four other species. Transcriptome data indicated that 40 poplar MFS genes were differentially expressed when treated with Fusarium oxysporum. Co-expression networks and gene function annotations of MFS genes showed that MFS genes tightly co-regulated and closely related in function of transmembrane transport. Taken together, we systematically analyzed structure and function of genes and proteins in the PtrMFS family. Evidence indicated that poplar MFS genes play key roles in plant development and response to a biological stressor.
Introduction
The major facilitator superfamily (MFS) is one of the largest membrane transporter families currently known (Chen et al., 2019). MFS’s diverse members are found in essentially all organisms in the biosphere. According to membrane transporter database TransportDB 2.0 (http://www.membranetransport.org/transportDB2/index.html) (Elbourne et al., 2017), the Arabidopsis thaliana MFS accounts for 128 transporters. María Niño-González proposed that MFS consists of 218 members, clustered in 22 families (María et al., 2019). Setyowati T. Utamia identifified 177 putative MFS transporters and classifified them into 17 subfamilies in Penicillium marneffei (Setyowati et al., 2020). Martin Broberg identifed 232 MFS transporters in maize pathogen Cochliobolus heterostrophus (Martin et al., 2021). MFS transporters belong to a wide family and in Penicillium digitatum more than 80 MFS have been identified due to the availability of the P. digitatum genome (Marcet-Houben et al., 2012). The MFS was first characterized in 1993, when a large class of transporters with 12 transmembrane helix domains were discovered among many membrane proteins (Marger and Saier, 1993). The function of MFS is involved in many essential functions. The basic function of members of this family is to assist in transporting substances across membranes (Bagchi et al., 2020). MFS proteins can transport many small molecules, such as monosaccharides, polysaccharides, amino acids, peptides, vitamins, enzyme cofactors, drug molecules, chromophores, and nucleotide bases (Lorca et al., 2007; Chen et al., 2008; Ming et al., 2010; Saier and Paulsen, 2011). Some MFS proteins are also closely related to immunological processes, such as virus invasion and pathogenic resistance (Nicolas et al., 2005).
Based on protein functions and sequence homology, the MFS can be divided into 89 families (Saier et al., 2009). Presently, more than 10,000 membrane protein genes from different species have been defined in the MFS. Most members of the MFS are ∼400–600 amino acids in length (Chun et al., 2018). Both the N-termini and C-termini of proteins are located within cells. Secondary structures of MFS proteins mainly comprise 12 α-helix transmembrane domains, which are divided into N-terminal domains and C-terminal domains, each of which contains six α-helices (Lee, 2015). In addition to transmembrane domains, some proteins in the MFS also have intracellular domains (Lu et al., 2010), which are conserved in the superfamily and perform important functions.
MFS transporters can confer resistance to a variety of toxic compounds, including specialized metabolites, fungicidal substances, and antibiotics (Sorbo et al., 2000; Fluman et al., 2012; Zhang et al., 2020). They can be used as drug H+ antiporters in microorganisms to indirectly regulate internal pH and stress response mechanisms in fungi (Santos et al., 2014); thus they can confer a multi-drug resistance (MDR) phenotype to fungi (Omrane et al., 2015). For example, it was reported that MFS1 in Penicillium digitatum (PdMFS1) was able to confer the MDR phenotype due to efflux of fungicide (Ramon et al., 2019). Another study found that PdMFS2 and PdMFS3 could contribute to fungicidal resistance (Sorbo et al., 2000). The MFS transporter Acinetobacter baumannii Fosfomycin efflux (AbaF) actively effluxes fosfomycin, making A. baumannii resistant to this antibiotic (Atin et al., 2017). The expression level of AbaF is upregulated under fosfomycin exposure. AbaF participates in the secretion of biofilm matrix, which promotes the generation of bacterial pathogenicity and participates in expulsion of host defense molecules, leading to a significant impact on the virulence of A. baumannii (Atin et al., 2017). Most MFS transporters implicated in MDR belong to the drug:H(+) antiporter family 1 (DHA1) subfamily (Paulsen et al., 1996). DHA1-subfamily transporters of Penicillium marneffei provide resistance to various drugs, including azoles, polyene and antimalarial (Utami et al., 2020). TetA is an MFS pump which causes tetracycline resistance, and it is one of the most well-known antibiotic resistance mechanisms (Reisz et al., 2013; Grossman, 2016). In phytopathogenic fungi, MFS transporters can pump + to increase fungal invasiveness to host plants (Hayashi et al., 2002; Mathias et al., 2007; Ramin et al., 2007; Matthias et al., 2009). MDR has been found in a variety of phytopathogenic fungi, such as Zymoseptoria tritici (the pathogen of septoria leaf blotch on wheat) (Omrane et al., 2017), P. digitatum (the pathogen of green mold on citrus) (Wang et al., 2012; Wu et al., 2016; Ramon et al., 2019), and Botrytis cinerea (Matthias et al., 2009). The expression of MFS1 in Zymoseptoria tritici is related to antifungal resistance (Omrane et al., 2015; Omrane et al., 2017). The expression of MFS19 and MFS54 play an important role in the oxidative stress response, the tolerance of xenobiotics such as fungicides, and the virulence of Alternaria alternata (Chen et al., 2017; Lin et al., 2018). Overexpression of MFS1 in P. digitatum could make citrus more resistant to fungicides (Ramon et al., 2019). The MFS transporter mfsG is an important factor in determining the virulence effect of B. cinerea on Brassicaceae such as Arabidopsis thaliana (Vela-Corcía et al., 2019). MFS transporters help cells to better handle carbon sources and transport nutrients (especially sugars) to cells, which can provide advantages for the development of fungi (Ramó n-Carbonell and S á nchez-Torres, 2017). During the development of pathogens such as Colletotrichum and Botrytis, MFS transporters are responsible for the uptake of sugar in the form of glucose, mannose and fructose from environments (Monalessa et al., 2013; Vela-Corcía et al., 2019).
After host invasion, plant pathogens encounter potent plant defense compounds. MFS transporters may transport defense compounds and toxins secreted by pathogens out of host cells, thereby promoting plant resistance to pathogens. In this study, we performed systematic investigation of 41 PtrMFS genes, studying structures and functions of PtrMFS genes and proteins, respectively, as well as phylogenetic relationships, cis-acting elements, chromosomal distribution, collinearity across related species, and duplication events. Additionally, we analyzed transcriptome data to identify differentially expressed poplar MFS genes during Fusarium oxysporum infection. Finally, we performed a co-expression network analysis of poplar MFS genes, and gene set enrichment analysis revealed pathways related to a variety of biological processes. This research provides novel characterization of poplar MFS genes and establishes a theoretical basis for functional verification.
Materials and Methods
Identification of MFS Proteins in Populus trichocarpa
Genome data for P. trichocarpa were downloaded from Phytozome v12.1 (https://phytozome.jgi.doe.gov/pz/portal.html) (Goodstein et al., 2012). The typical MFS protein domains (PF07690, PF16983, PF05631, PF07672, PF05977, PF06779, PF12832, and PF13347) were obtained from Pfam (http://pfam.xfam.org/) (Finn et al., 2014). Scanning the poplar genome for potential PtrMFS proteins was conducted with HMMER3 (Jaina et al., 2013). Verification was performed using the SMART database (http://smart.embl-heidelberg.de/) (Ivica and Peer, 2018) and Pfam database to remove proteins without MFS domains.
Phylogenetic Relationship and Physicochemical Properties of MFS Proteins
MFS protein sequences of P. trichocarpa and A. thaliana were downloaded from Phytozome. We used MEGA v5.1 (Tamura et al., 2011) with the Maximum-Likelihood (ML) method to construct a phylogenetic tree of MFS proteins using the JTT (protein mutation data matrix) + G (site-specific variations in mutation rate) + F (mutation frequency data) model.
Physical and chemical properties of PtrMFS proteins were predicted with ProtParam (https://web.expasy.org/protparam/) (Gasteiger et al., 2003), including the number of amino acids, molecular weight, theoretical isoelectric point (pI), aliphatic index, grand average of hydrophilicity (GRAVY), chemical formulas, total number of atoms, and instability index.
Gene Structure and Protein Motif Analysis of PtrMFS Family
To analyze gene structures of PtrMFSs, we downloaded DNA and coding sequences of PtrMFS from Phytozome database. DNA and coding sequences for each PtrMFS gene were aligned to obtain gene structures. TBtools (Chen et al., 2018) was used to visualize gene structures of PtrMFSs, and MEME (http://meme-suite.org/) (Bailey et al., 2006) was used to identify conserved motifs in PtrMFS proteins. Annotations of identified motifs were obtained from InterProScan (https://www.ebi.ac.uk/interpro/search/sequence/) (Quevillon et al., 2005).
Secondary and Tertiary Structures Prediction of PtrMFS Proteins
Secondary structures of PtrMFS proteins were predicted by SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopma.html) (Geourjon and Delé age, 1995), and tertiary structures were constructed by SWISS-MODEL (https://swissmodel.expasy.org/) (Torsten et al., 2003).
Topological Heterogeneity Model and Subcellular Localization Prediction
Topological heterogeneity models of PtrMFS proteins were predicted with Protter (http://wlab.ethz.ch/protter/start/) (Ulrich et al., 2014). Subcellular localization was predicted using WoLF PSORT (https://wolfpsort.hgc.jp) (Paul et al., 2007).
Cis-Acting Elements Analysis
For each PtrMFS gene, sequences starting at 2,000 bp upstream of the start codon were downloaded from Phytozome. Cis-acting elements were extracted using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (Rombauts et al., 1999). TBtools was used to visualize cis-acting elements.
Chromosome Distribution and Collinearity Analysis
PtrMFS genes were mapped to the genome of P. trichocarpa, and the chromosomal distribution of PtrMFSs in poplar was visualized with TBtools. TBtools and MCScanX (Wang et al., 2012) were used to analyze tandem duplication events in the PtrMFS gene family, and Dual Synteny Plotter (Tang et al., 2008) was used to analyze segmental duplication events and collinearity between PtrMFSs and homologous gene pairs from other species (A. thaliana, Eucalyptus grandis, Oryza sativa, and Solanum lycopersicum). TBtools was used to visualize the results. The ratio of non-synonymous substitution to synonymous substitution (Ka/Ks) of duplicate gene pairs was determined with KaKs_Calculator (Zhang et al., 2006).
Sample Preparation
WT Pdpap seedlings used in this research were cultured on 0.5X Murashige and Skoog (MS) medium supplemented with 0.01 mg/ml 1-naphthaleneacetic acid (NAA). Two-month-old WT Pdpap plants at the same growth stage was selected. Fifty milliliters of F. oxysporum at a spore density of 1 × 105/ml was poured onto Pdpap roots (Kang et al., 2018). Infection times were 6, 12, 24, or 48 h. WT Pdpap treated with 50 ml ddH2O was used as control. Every treatment consisted of 3 duplicated samples. The materials involved in this research and their preparation methods have been reported in detail in our previous research (Diao et al., 2021).
Gene Expression Analysis
RNA-Seq was used to explore gene expression patterns of the MFS gene family from PdPap under F. oxysporum stress. The data were described in detail in our previous research (Diao et al., 2021). Differentially expressed genes (DEGs) were identified with DESeq2 using log2 (fold change) ≥ and adjusted p-value (padj) ≤ 0.05 as the criteria. Expression patterns of PdPapMFS genes under different treatments of F. oxysporum at 6, 12, 24, and 48 h were visualized with heatmaps. We identified upregulated and downregulated DEGs in response to F. oxysporum and displayed the data by Venn diagrams. To validate the RNA-Seq data, we further analyzed expression levels of DEGs under F. oxysporum stress by qRT-PCR. The qRT-PCR was performed on a Stratagene Mx3000P real-time PCR system (Agilent Technologies, Santa Clara, CA, United States) using the 2 × SYBR Green qPCR Master Mix kit (Bimake, Shanghai, China). Reaction systems are shown in Supplementary Table S1 and primer sequences designed by Primer5.0 (Zhai et al., 2008) are shown in Supplementary Table S2. qRT-PCR amplification conditions were as follows: initial denaturation at 94°C for 30 s; 44 cycles of 94°C for 12 s, 58°C for 30 s, and 72°C for 45 s, then 79°C for 1 s. The reaction specificity was determined by performing a melting-curve analysis from 55°C to 99°C, with fluorescence readings taken every 0.5°C for 1 s. The amplification curve was obtained after analyzing the raw data, and the cycle threshold (Ct) was set with a fluorescence threshold of 0.01 (Wang et al., 2012). Relative expression level of target genes was calculated by the 2−△△Ct method (Kenneth and Thomas, 2002). Three duplicates were set for each gene. PdpapActin and PdPapEF1-α were used as the internal control genes (Huang et al., 2008).
Gene Co-Expression Analysis and Gene Ontology Annotation
We analyzed co-expression-based gene networks using STRING (Damian et al., 2011), and visualized the results with Cytoscape (Ideker, 2011). Co-expression analysis was performed on the 41 PdPapMFS genes identified from RNA-Seq data analysis as described above. Genes were annotated with gene ontology (GO)-based functions using agriGO v2.0 (http://systemsbiology.cau.edu.cn/agriGOv2/index.php) (Du et al., 2010).
Statistical Analysis
Data were analyzed with the Statistical Software Package for Social Science (SPSS) version 17.0 (Kumar, 2014). Using Student’s t-test to compare the data, p < 0.05 was considered statistically significant (Choi et al., 2012). Significant differences (p < 0.05) are indicated by different lowercase letters in figures.
Results
Phylogenetic Relationship and Physicochemical Properties of PtrMFSs
We identified 41 MFS genes in P. trichocarpa (each named PtrMFS with a number based on position in the poplar genome; Supplementary Table S3). To determine evolutionary relationships of genes in this family, we constructed a ML phylogenetic tree (Figure 1) using protein sequences from poplar and Arabidopsis. As shown in Figure 1, we classified PtrMFSs into six groups of various sizes. Group MFS_4 is the largest with nine genes, while group MFS_5 is the smallest with five genes.

FIGURE 1. Phylogenetic analysis of MFS proteins in poplar and Arabidopsis. The dendrogram of 56 MFS proteins was performed by MEGA5 with the ML method based on JTT + G + F model. Different groups are marked with different colors.
The physicochemical properties of the identified PtrMFSs vary significantly (Supplementary Table S3). The average length of PtrMFS proteins is 498 amino acids (range = 372–698 aa) and the average molecular weight is 54.27 kDa (range = 40.20–78.23 kDa). The theoretical pI of PtrMFSs is in the range of 5.54–10.07 and the aliphatic index is in the range of 85.96–118.46. The hydrophilicity value of PtrMFSs ranges from 0.183 to 0.758. All of the proteins are predicted to be hydrophilic. PtrMFSs consist of five elements: C, H, N, O, and S. The total number of atoms in each protein range from 5,724 to 11,110. The instability indexes of PtrMFSs range from 24.80 to 52.06; an index >40 indicates an unstable protein, while <40 indicates a stable protein.
Sequence Structure Analysis of PtrMFSs
PtrMFS genes within the same groups share similar structures with respect to introns and exons (Figure 2). Four members in group MFS_2 contain 14 exons. Three members in group MFS_3 contain 2 exons and only 1 intron. Three members in group MFS_6 contain 2 exons and another three members contain 3 exons. Members with closer relationships share more similar gene structures and exon lengths.
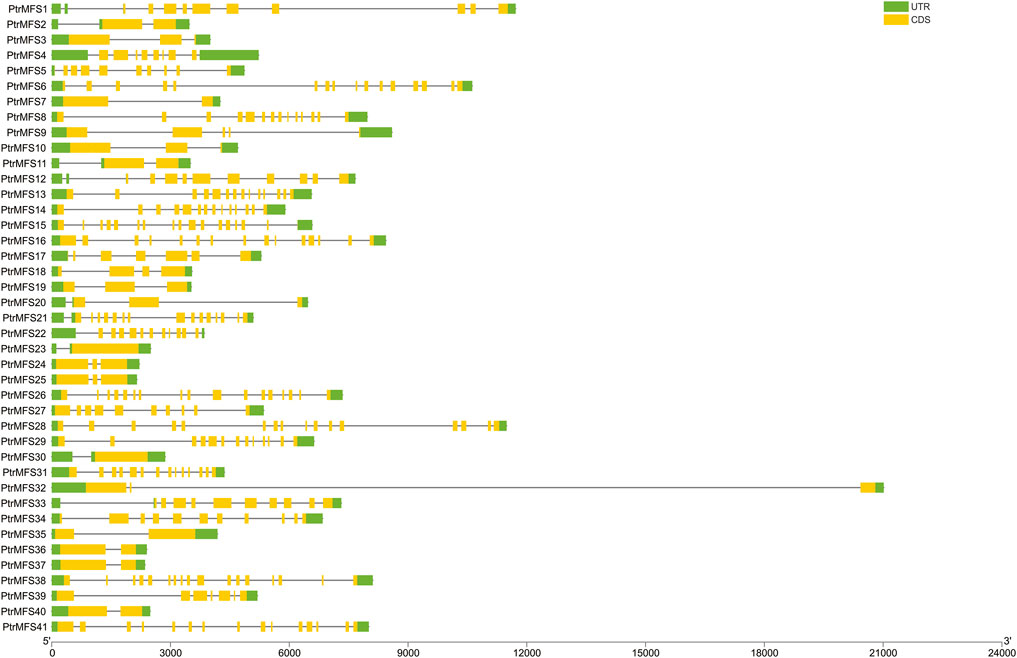
FIGURE 2. DNA structures of PtrMFS gene family in poplar. Green boxes represent untranslated regions. Yellow boxes denote coding regions. Black lines indicate introns.
PtrMFS proteins with similar evolutionary relationships have the same or similar conserved domains. We identified 20 conserved motifs using MEME, and motif annotations were predicted with Pfam and InterProScan (Supplementary Table S4). We found that motifs 2, 3, 4, 5, 6, 7, 8, 12, 13, 14, 17, and 19 were annotated as MFS transporters. Motifs 2, 3, 11, and 16 were high affinity nitrate transporter-related; motif 8 was tetracycline resistance domain. Motif 13 was annotated as protein zinc induced facilitator. Motif 15 was annotated as anion transporter and solute carrier family. Motif 1 was annotated as LytB protein and motif 20 was annotated as SPX domain. Motifs 9, 10, and 18 were unknown and could not be annotated. Results show that motifs 13 and 20 are present only in all members of group MFS_1 (Figure 3). Motif 8 is present only in all members of group MFS_2. Motif 6, 7, 12, 17, and 18 are present only in members of group MFS_3. Motif 4, 5, 14, 15, and 19 are present only in all members of group MFS_4. Motif 1, 2, 5, 7, 9, 11, and 16 only occurred in members of group MFS_6. These results demonstrate that there are many group-specific motifs, which may be correlated to specific biological functions.

FIGURE 3. Protein motifs of PtrMFS family in poplar. Different colorful boxes represent different motifs.
Secondary and Tertiary Structures of PtrMFSs
In the secondary structure of PtrMFS proteins (Supplementary Table S5), an average of 234.59 aa belonged to alpha helixes, accounting for 46.93% of total secondary structures. On average, a further 74.73 aa (15.08%) belonged to extended strands, 23.29 aa (4.71%) belonged to beta turns, and 165.37 aa (33.28%) belonged to random coils.
Tertiary structures were generated primarily using 6s4m.1.A and 6e9c.1.A as templates (Supplementary Table S6). They have functions of major facilitator superfamily domain-containing proteins. Sequence identity of the 41 PtrMFS proteins identified in this study and their corresponding templates is 20.80% (11.02–37.71%) on average.
Topological Heterogeneity Models and Subcellular Localization of PtrMFSs
Topological heterogeneity models of PtrMFSs proteins (Supplementary Table S7) showed that all members of the PtrMFS family have transmembrane helical segments. PtrMFS19, PtrMFS23, and PtrMFS26 have no N-glycosylation sites. PtrMFS8, PtrMFS14, PtrMFS18, PtrMFS19, and PtrMFS28 have signal peptides.
Subcellular localization predictions (Supplementary Table S8) showed that a majority of PtrMFSs are located at the plasma membrane. PtrMFS26 is predicted to be located at the endoplasmic reticulum, PtrMFS31 at the vacuole, and PtrMFS4, PtrMFS5, PtrMFS13, PtrMFS22, PtrMFS29, PtrMFS39, and PtrMFS41 at the chloroplast.
Cis-Acting Elements Analysis in Promoters of PtrMFS Genes
We used PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) to predict cis-acting elements within 2,000 bp upstream of PtrMFS genes (Supplementary Table S9; Figure 4). Many predicted cis-acting elements identified are involved in hormone responses, such as response to auxin, response to gibberellin, response to salicylic acid, and response to abscisic acid. Many elements are also predicted to be involved in defense and stress responses, meristem expression, root specific expression, flavonoid biosynthetic gene regulation, and wounding response.
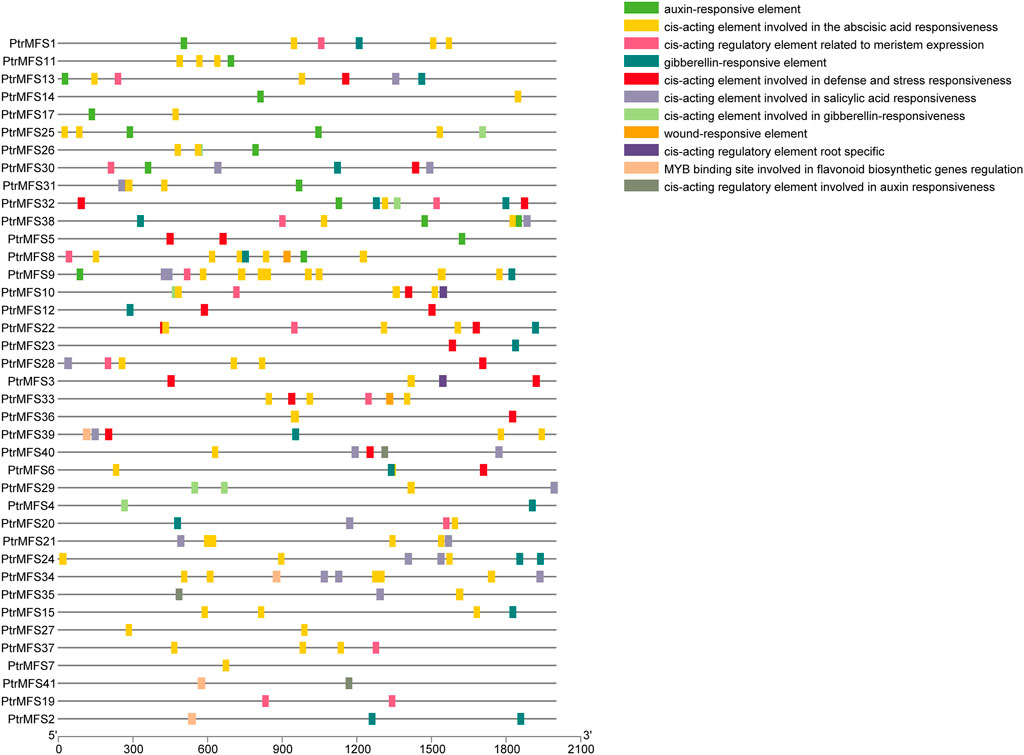
FIGURE 4. Analysis of cis-acting elements in promoters of PtrMFS genes. The patterns in different colors represent different cis-acting elements.
Chromosomal Location and Collinearity Analysis of PtrMFSs
The location of PtrMFS genes within chromosomes is variable (Figure 5); they are unevenly distributed in 16 of the 19 poplar chromosomes. The number of genes located in each chromosome is irrelevant to the size of chromosomes. Chromosome 9 contains the most PtrMFS genes, with 8. Only one PtrMFS gene each was located on chromosome 4, chromosome 5, chromosome 10, and chromosome 12, and no PtrMFS genes were found on chromosome 11, chromosome 13, chromosome 17, or chromosome 19.
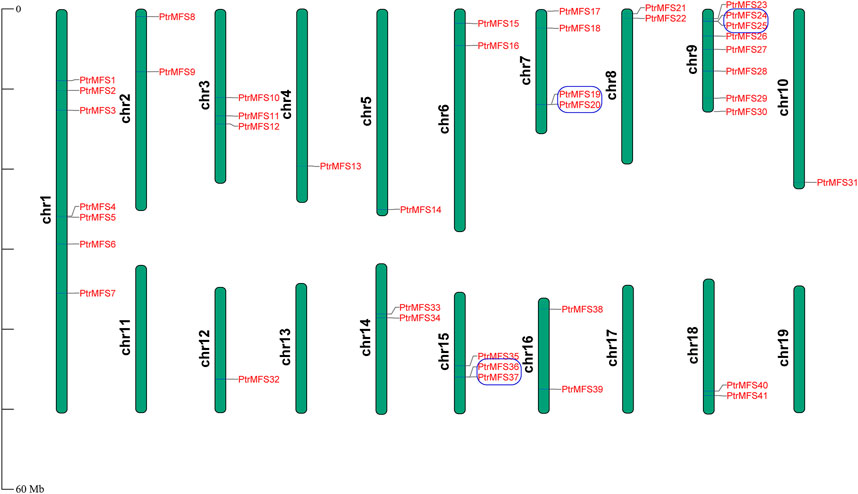
FIGURE 5. Chromosomal distribution of PtrMFS genes. Chr1–19 represent chromosome numbers 1–19. Blue boxes represent pairs of tandem repeated genes.
We analyzed within-genome duplication events of PtrMFS genes using MCScanX. PtrMFS19 and PtrMFS20 (on chromosome 7), PtrMFS24 and PtrMFS25 (on chromosome 9), PtrMFS36 and PtrMFS37 (on chromosome 15) were categorized as derived from tandem duplication events. Fourteen genes exhibit 8 pairs of segmental duplication events (Figure 6; Supplementary Table S10). They are unevenly distributed in eight of 19 chromosomes. Segmental duplication events may be the important driving force for diversity of PtrMFS genes. The ratio of Ka/Ks is an important indicator of selective pressure in evolution, with a Ka/Ks < 1 indicating negative selection. Ka/Ks values of PtrMFS duplicate genes range from 0.16 to 0.37, with an average value of 0.24, suggesting that PtrMFS genes have been subject to purifying selection during evolution.
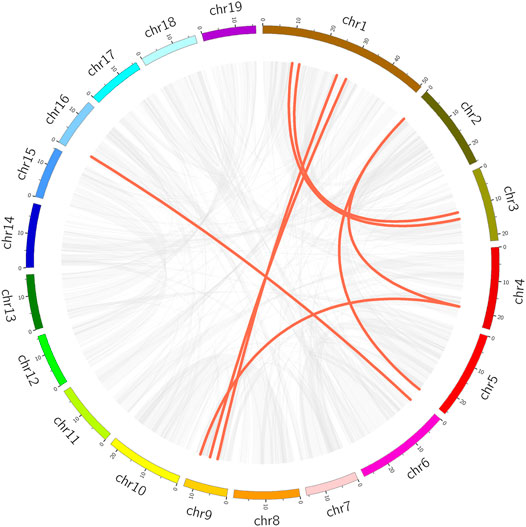
FIGURE 6. Segmental duplication events of PtrMFS genes. Chr1–19 are represented by different colorful rectangles. Red lines represent collinear pairs of PtrMFS genes. Gray lines indicate collinear pairs in all poplar genome.
To explore the DNA sequence similarity between PtrMFS genes and related genes from other representative species, we constructed collinearity relationship maps of Populus trichocarpa with three dicotyledons (E. grandis, S. lycopersicum, and A. thaliana) and one monocotyledon (O. sativa) (Figure 7). We identified 32 repetitive events in E. grandis, 26 in S. lycopersicum, 23 in A. thaliana, and seven in O. sativa (Supplementary Table S11). Collinearity blocks were mainly distributed in the first 10 chromosomes of P. trichocarpa. There was greater collinearity between PtrMFS genes and those in dicotyledons than those in the monocotyledon.
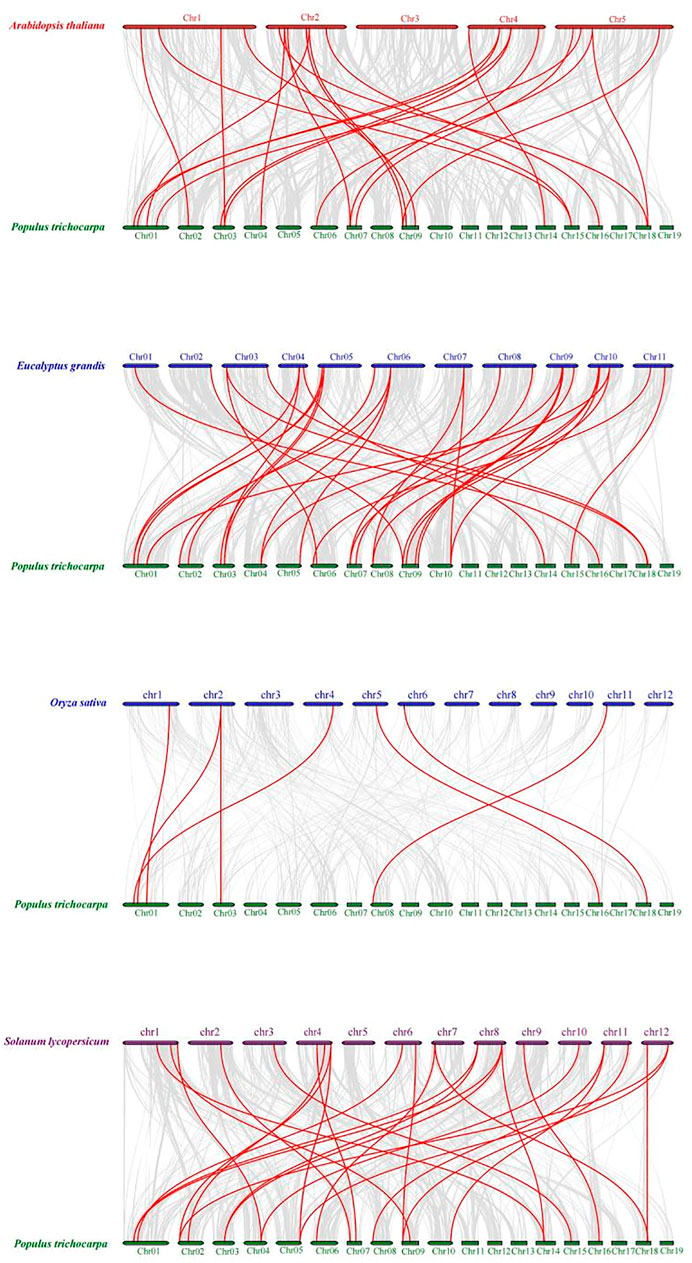
FIGURE 7. Collinearity relationship maps of PtrMFS genes in poplar to other four species. Red lines denote collinearity between PtrMFS genes and other species. Gray lines represent collinearity between poplar genome and that are orthologous to the other species.
PtrMFS Gene Expression in Response to F. oxysporum
We analyzed expression patterns of PdPapMFS genes in response to F. oxysporum with different lengths of time post-inoculation using RNA-Seq data (Supplementary Table S12). Statistical results indicated that PdPapMFSs expression are all in different fold-changes (Figure 8). The relative gene expression values of PdPapMFS are shown in Supplementary Table S13.

FIGURE 8. Heatmap of different expression levels in PdpapMFS genes under different treatments by F. oxysporum. Cluster analyses were based on log2FPKM. Red boxes represent highly expressed genes. Green boxes represent low expressed genes. The left side represents gene clusters. T1, T2, T3, and T4 indicated that PdPap were infected by F. oxysporum for 6, 12, 24, and 48 h.
In the samples collected 6 h after inoculation with F. oxysporum, we identified 40 DEGs, including 13 upregulated genes, and 27 downregulated genes. PdPapMFS38 was the most upregulated (3.14X) and PdPapMFS25 was the most downregulated (−2.07X). In the 12 h treatment group, 41 DEGs were found, including 14 upregulated genes and 27 downregulated genes. PdPapMFS38 was the most upregulated (3.22X) and PdPapMFS17 was the most downregulated (−1.43X). In the 24 h treatment group, 40 DEGs were found, including 14 upregulated genes, and 26 downregulated genes. PdPapMFS38 was the most upregulated (2.93X) and PdPapMFS17 was the most downregulated (−1.16X). In the 48 h treatment group, 40 DEGs were found, including 23 upregulated genes, and 17 downregulated genes. PdPapMFS38 was the most upregulated (2.32X) and PdPapMFS17 was the most downregulated (−1.14X). DEG expression in the four treatment groups was visualized with heatmaps (Figure 9) and Venn diagrams (Figure 10).
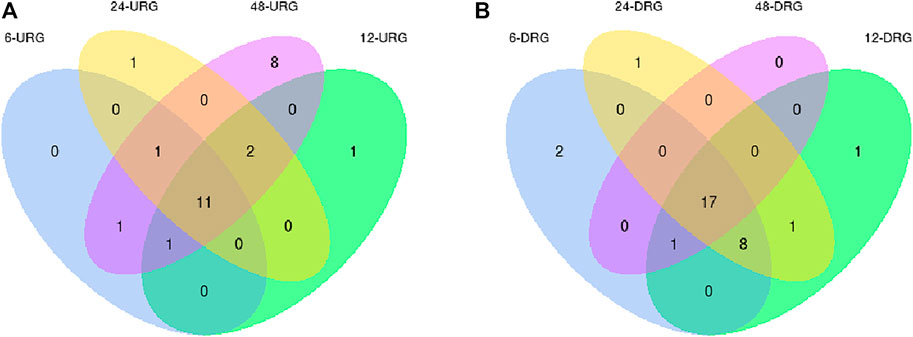
FIGURE 9. Venn diagrams of DEGs in response to F. oxysporum stress under different treatments. (A) Venn diagram of up-regulated DEGs (URGs) in response to F. oxysporum stress under different treatments. (B) Venn diagram of down-regulated DEGs (DRGs) in response to F. oxysporum stress under different treatments.
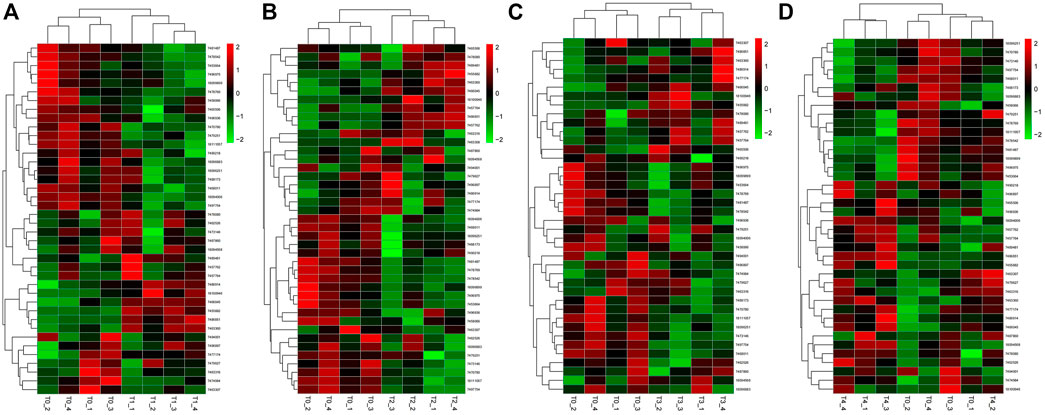
FIGURE 10. Heatmaps of DEGs in response to F. oxysporum stress under different treatments. (A) The heatmap of DEGs in response to F. oxysporum stress at 6 h. (B) The heatmap of DEGs in response to F. oxysporum stress at 12 h. (C) The heatmap of DEGs in response to F. oxysporum stress at 24 h. (D) The heatmap of DEGs in response to F. oxysporum stress at 48 h. Cluster analyses were based on log2FPKM. Red boxes represent highly expressed genes. Green boxes represent low expressed genes. The left side represents gene clusters. T1, T2, T3, and T4 indicated that PdPap were infected by F. oxysporum for 6, 12, 24, and 48 h.
Verification of PtrMFS Genes Expression by RT-qPCR
To validate the RNA-Seq data, we performed qRT-PCR to analyze expression levels of putative DEGs in response to F. oxysporum stress. Results of RNA-Seq and qRT-PCR were generally consistent, with a few exceptions such as PdPapMFS8, PdPapMFS16, PdPapMFS34, which may be caused by experimental errors of RNA-Seq or qRT-PCR (Figure 11).

FIGURE 11. DEG expression levels in Pdpap based on RNA-Seq and qRT-PCR. Expression levels in RNA-Seq was quantified by fragments per kilo-bases per million mapped reads (FPKM). T0, T1, T2, T3, and T4 indicated that PdPap were infected by F. oxysporum for 0, 6, 12, 24, and 48 h. Error bars represented standard deviation of three independent replicates. Significant differences (p < 0.05) were indicated by different lowercase letters. Gene co-expression and gene ontology analysis.
Gene Co-Expression and Gene Ontology Analysis
We constructed a co-expression network using RNA-Seq data of the 41 PtrMFS genes (Figure 12). A large proportion of the genes are shared in the networks, such as PtrMFS32 (LOC7458066), PtrMFS38 (LOC7455882), PtrMFS33 (LOC7496936), PtrMFS6 (LOC7470780), PtrMFS5 (LOC7478542), PtrMFS39 (LOC7466045), PtrMFS3 (LOC18094568), PtrMFS13 (LOC7453664), PtrMFS25 (LOC7463307). We have observed that the shared gene expression patterns in the co-expression network are not exactly the same, which indicated that MFS genes under the stress of F. oxysporum may have complex regulatory characteristics.

FIGURE 12. Co-expression-based gene networks of PtrMFS gene family. Dots represent genes. Lines indicate that they have co-expression relationships.
Using agriGO, we performed gene set enrichment analysis with PdPapMFS genes (Figure 13). Based on the annotations, these genes can participate in a variety of biological processes (Figure 14A; Supplementary Table S14), such as in process terms “transmembrane transport,” “establishment of localization,” and “cellular process.” Many PdPapMFS genes are enriched in various molecular functions (Figure 14B; Supplementary Table S14), such as in function terms “transporter activity,” and “transmembrane transporter activity.” Regarding cellular localization (Figure 14C; Supplementary Table S14), PdPapMFS genes are enriched in localization terms “integral to membrane,” “intrinsic to membrane,” and “cell part.” It is worth mentioning that, many MFS genes are enriched in “transmembrane transport,” “transport,” “transporter activity,” “transmembrane transporter activity,” “membrane,” “integral to membrane,” and “intrinsic to membrane.” That also proved our speculation that MFS genes are related to the transmembrane transport function of substances.
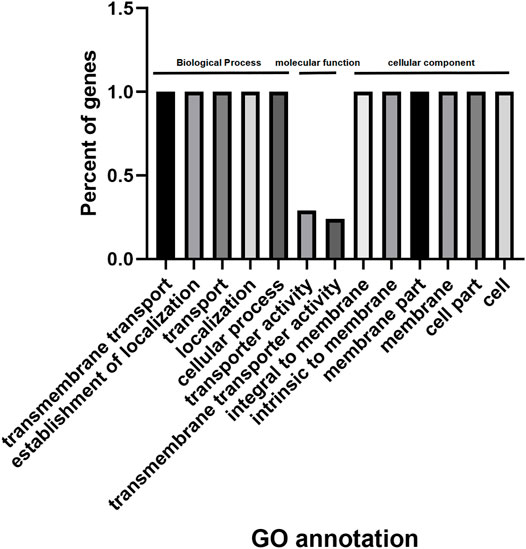
FIGURE 13. GO analysis of PdpapMFS genes.
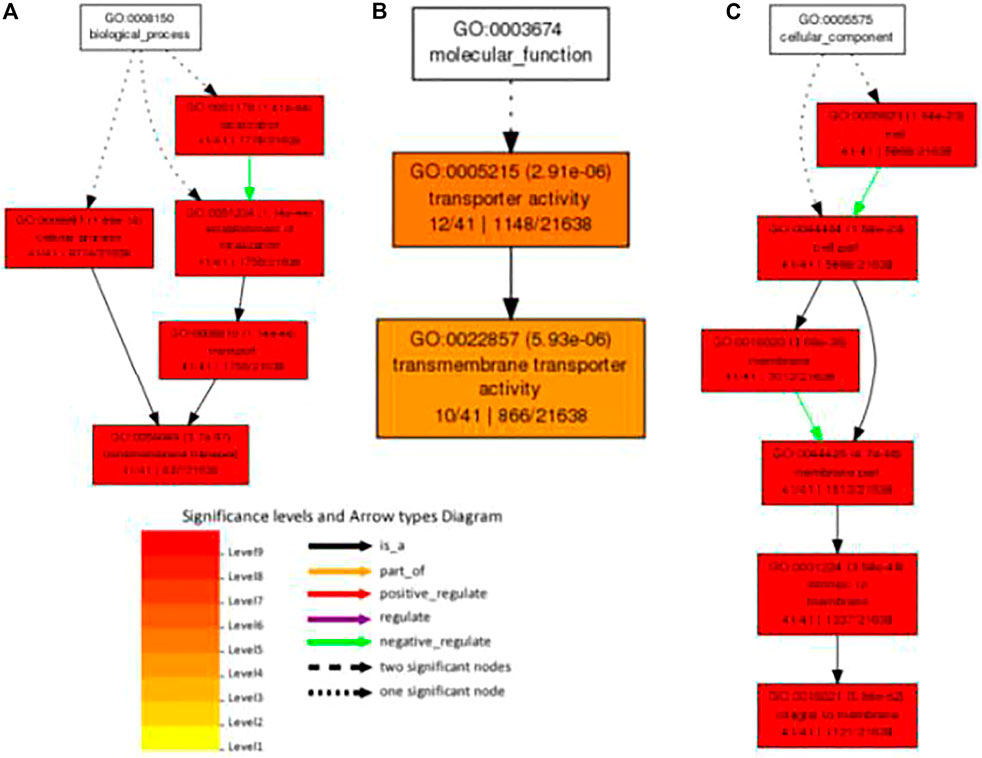
FIGURE 14. GO analysis of PdpapMFS genes in different classifications. (A) GO analysis of based on biological processes. (B) GO analysis of based on molecular functions. (C) GO analysis of based on cellular components.
Discussion
The MFS gene family exists widely in most organisms and is involved in many critical activities. Some MFS proteins play key roles in immunological processes, such as viral invasion and pathogen resistance. In this study, we identified 41 PtrMFS genes in the P. trichocarpa genome. The genes were divided into six groups based on amino acid sequence similarity. The largest group was MFS_4, which had nine genes. Analysis of PtrMFS protein domains showed that most contain the major facilitator superfamily transporter domain, while others contain a high-affinity nitrate transporter domain and a tetracycline resistance protein domain. We found that many unannotated domains exist in these proteins, which may be responsible for the diverse functions of PtrMFSs.
The number of introns varies significantly among the PtrMFS gene family. As expected, there is less variability in the structure of introns and exons in genes within the same group. Previous studies have shown that reduction in the number of introns can shorten the time from transcription to translation, thereby promoting rapid gene expression during environmental changes (Jeffares et al., 2008). We found that there are relatively few introns in groups MFS_3, MFS_5, and MFS_6. Overall, 41.46% of the PtrMFS genes contain 1–3 introns. Therefore, PtrMFS genes may be involved in rapid response to environmental changes.
PtrMFS proteins contained multiple transmembrane domains with the annotated function of material transport. PtrMFS proteins can bind with macromolecular compounds in order to transport them (Wannes and Thomas, 2014). Studies have shown that N-glycosylation sites are essential for protein folding and material transport (Fujii et al., 2006). Pathogen tolerance of poplar may be related to the presence of defensive compounds that function in transporting pathogen toxins out of host cells.
Cis-acting elements contained in promoter regions play key roles in gene regulation and expression. Our analysis of cis-acting elements helped to identify genes with specific functions, such as genes related to stress resistance and plant development. We found that elements related to hormone response and regulation of stresses are present in promoters of almost all PtrMFS genes. Thus, the results showed that PtrMFS genes may play a key role in regulating responses of poplar to multiple stressors.
Gene families can contain large subfamilies as a result of events such as segmental duplication, tandem duplication, or conversion events (Cannon et al., 2004; Kong et al., 2010). Duplication events can promote the emergence of new genes, which can contribute to increasing the diversity of gene functions, and can effectively improve the ability of plants to adapt to different environments (Flagel and Wendel, 2009). Studies have shown that poplar has undergone at least three rounds of whole genome duplications, in addition to multi-segment duplications, tandem duplications, and transposition events (Tuskan et al., 2006; Wang et al., 2019). In this study, we identified both tandem and segmental duplicates among PtrMFS genes. Results showed that segmental duplication events (8 pairs) occurred more frequently than tandem duplication events (3 pairs), suggesting a potentially important role of segmental duplication events in expansion of the PtrMFS gene family. Analysis of duplicate gene pairs found that the Ka/Ks ratios were all less than 1; therefore, we infer that PtrMFS genes have undergone a process of purification and selection during evolutionary processes.
To explore the evolutionary relationships of MFS genes among different species, we analyzed the collinearity between PtrMFS genes and counterparts from three dicotyledonous and one monocotyledonous plant. Results showed that PtrMFS genes have more collinearity with dicotyledonous plants than with monocotyledonous plants. In addition, species with relatively close evolutionary relationships have more collinear gene pairs; we found that PtrMFS genes have the most homology with genes from E. grandis and the least homology with genes from O. sativa.
Analysis of RNA-Seq data showed that poplar MFS genes were differentially expressed over time under F. oxysporum stress. There were 11 upregulated and 17 downregulated genes shared across all time points after T0. Results revealed that the plants had complex responses in the regulatory networks after different lengths of time post-inoculation. Functional annotations indicated that PtrMFS genes play an important role in regulation of material transportation. For example, AT1G08090 (homologous to PtrMFS24) encodes a main component of nitrate affinity transport system in roots. Its post-translational regulation mechanism plays a key role in the control of nitrate absorption in roots (Jacquot et al., 2019). AT1G63010 (homologous to PtrMFS12) is a tonoplast phosphate transporter, and its ectopic expression can regulate strawberry fruit ripening and quality through sucrose transport (Liu et al., 2015; Huang et al., 2019). AT3G43790 (homologous to PtrMFS26) is a functional transporter that mediates K+ and Cs+ influx when heterologously expressed in yeast (Estelle et al., 2015). AT4G00370 (homologous to PtrMFS34) encodes ascorbate transporters on chloroplast envelope membranes, which are necessary for plants to tolerate strong light (Miyaji et al., 2015). Pathogens may activate signal pathways that induce similar cellular responses.
Results of co-expression analysis is useful to find genes with similar expression patterns. These genes are tightly co-regulated and closely related in function. They can also play a role in the same signaling pathway or physiological process (Mathias et al., 2015). We constructed gene co-expression networks to explore the functional relevance of PtrMFS genes. PtrMFS genes in gene networks are cross-linked, which suggests complex regulation of PtrMFS genes in response to F. oxysporum stress. We analyzed enrichment of gene sets and found that most genes in the networks are related to transmembrane transport process and membrane part components. Evidence further indicates that poplar MFS genes play important roles in the functional regulation of transmembrane transport.
MFS genes are known to be involved in the process of material transportation and may play important roles in improving plant resistance to pathogens. In this study, we systematically analyzed properties and expression levels of poplar MFS genes. Further study should be conducted to functionally characterize these MFS genes. Additionally, a large number of genes related to pathogens resistance need to be mined to realize F. oxysporum tolerance of poplars.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
JD: Investigation, Methodology, Writing—original draft. SL: Formal analysis, Project administration, Investigation. LM: Methodology, Project administration. PZ: Validation. JB: Formal analysis. JW: Funding acquisition. XM: Funding acquisition. WM: Funding acquisition, Writing—review and editing.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities, grant number 2572019CP01; Fundamental Research Funds for the Central Universities, grant number 2572019AA05; Heilongjiang Provincial Natural Science Foundation of China, grant number C2018055.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.769888/full#supplementary-material
References
Atin, S., Rajnikant, S., Tapas, B., Timsy, B., and Ranjana, P. (2017). Fosfomycin Resistance in Acinetobacter Baumannii Is Mediated by Efflux through a Major Facilitator Superfamily (MFS) Transporter—AbaF. J. Antimicrob. Chemoth. 72, 68–74.
Bagchi, S., Perland, E., Hosseini, K., Lundgren, J., Al-Walai, N., Kheder, S., et al. (2020). Probable Role for Major Facilitator Superfamily Domain Containing 6 (MFSD6) in the Brain during Variable Energy Consumption. Int. J. Neurosci. 130, 476–489. doi:10.1080/00207454.2019.1694020
Bailey, T. L., Williams, N., Misleh, C., and Li, W. W. (2006). MEME: Discovering and Analyzing DNA and Protein Sequence Motifs. Nucleic Acids Res. 34, W369–W373. doi:10.1093/nar/gkl198
Cannon, S. B., Mitra, A., Baumgarten, A., Young, N. D., and May, G. (2004). The Roles of Segmental and Tandem Gene Duplication in the Evolution of Large Gene Families in Arabidopsis thaliana. BMC Plant Biol. 4, 10–768. doi:10.1186/1471-2229-4-10
Chen, C. J., Xia, R., Chen, H., and He, Y. H. (2018). TBtools, a Toolkit for Biologists Integrating Various HTS-Data Handling Tools with a User-Friendly Interface. Mol. Plant 3, 1–7.
Chen, D. E., Podell, S., Sauer, J.-D., Swanson, M. S., and Saier, M. H. (2008). The Phagosomal Nutrient Transporter (Pht) Family. Microbiol 154, 42–53. doi:10.1099/mic.0.2007/010611-0
Chen, L.-H., Tsai, H.-C., Yu, P.-L., and Chung, K.-R. (2017). A Major Facilitator Superfamily Transporter-Mediated Resistance to Oxidative Stress and Fungicides Requires Yap1, Skn7, and MAP Kinases in the Citrus Fungal Pathogen Alternaria alternata. Plos One 12, e0169103. doi:10.1371/journal.pone.0169103
Chen, Y., Zhou, G., and Liu, J. (2019). A Major Facilitator Superfamily Transporter inColletotrichum fructicola(CfMfs1) Is Required for Sugar Transport, Appressorial Turgor Pressure, Conidiation and Pathogenicity. For. Path. 49, e12558. doi:10.1111/efp.12558
Choi, K.-J., Kim, H.-B., and Park, S.-H. (2012). The Comparison of Postoperative Pain: Total Laparoscopic Hysterectomy versus Vaginal Hysterectomy. Korean J. Obstet. Gynecol. 55, 384–391. doi:10.5468/kjog.2012.55.6.384
Chun, L. I., Sun, C. Y., Chen, J., Lin, Y. P., Wang, Y., and Zhang, M. P. (2018). Research Advances in the Major Facilitator Superfamily. Biotechnol. Bull. 34, 43–49.
Damian, S., Andrea, F., Michael, K., Milan, S., Alexander, R., Pablo, M., et al. (2011). The STRING Database in 2011: Functional Interaction Networks of Proteins, Globally Integrated and Scored. Nucleic Acids Res. 39, 561–568. doi:10.1093/nar/gkq1331
de Ramón-Carbonell, M., López-Pérez, M., González-Candelas, L., and Sánchez-Torres, P. (2019). PdMFS1 Transporter Contributes to Penicilliun Digitatum Fungicide Resistance and Fungal Virulence during Citrus Fruit Infection. JoF 5, 100. doi:10.3390/jof5040100
Del Sorbo, G., Schoonbeek, H.-j., and De Waard, M. A. (2000). Fungal Transporters Involved in Efflux of Natural Toxic Compounds and Fungicides. Fungal Genet. Biol. 30, 1–15. doi:10.1006/fgbi.2000.1206
Diao, J., Wang, J. Q., Zhang, P., Hao, X., Wang, Y., Liang, L. W., et al. (2021). Transcriptome Analysis Reveals the Important Role of WRKY28 in Fusarium Oxysporum Resistance. Front. Plant Sci. 12, 1–17. doi:10.3389/fpls.2021.720679
dos Santos, S. C., Teixeira, M. C., Dias, P. J., and Sã¡-Correia, I. (2014). MFS Transporters Required for Multidrug/multixenobiotic (MD/MX) Resistance in the Model Yeast: Understanding Their Physiological Function through post-genomic Approaches. Front. Physiol. 5, 180. doi:10.3389/fphys.2014.00180
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and SuAgrigo, Z. (2010). agriGO: a GO Analysis Toolkit for the Agricultural Community. Nucleic Acids Res. 38, W64–W70. doi:10.1093/nar/gkq310
Elbourne, L. D. H., Tetu, S. G., Hassan, K. A., and Paulsen, I. T. (2017). TransportDB 2.0: a Database for Exploring Membrane Transporters in Sequenced Genomes from All Domains of Life. Nucleic Acids Res. 45, D320–D324. doi:10.1093/nar/gkw1068
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the Protein Families Database. Nucl. Acids Res. 42, D222–D230. doi:10.1093/nar/gkt1223
Flagel, L. E., and Wendel, J. F. (2009). Gene Duplication and Evolutionary novelty in Plants. New Phytol. 183, 557–564. doi:10.1111/j.1469-8137.2009.02923.x
Fluman, N., Ryan, C. M., Whitelegge, J. P., and Bibi, E. (2012). Dissection of Mechanistic Principles of a Secondary Multidrug eFflux Protein. Mol. Cel 47, 777–787. doi:10.1016/j.molcel.2012.06.018
Fujii, Y., Khoshnoodi, J., Takenaka, H., Hosoyamada, M., Nakajo, A., Bessho, F., et al. (2006). The Effect of Dexamethasone on Defective Nephrin Transport Caused by ER Stress: A Potential Mechanism for the Therapeutic Action of Glucocorticoids in the Acquired Glomerular Diseases. Kidney Int. 69, 1350–1359. doi:10.1038/sj.ki.5000317
Geourjon, C., and Deléage, G. (1995). SOPMA: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 11, 681–684. doi:10.1093/bioinformatics/11.6.681
Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., and Bairoch, A. (2003). ExPASy: The Proteomics Server for In-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 31, 3784–3788. doi:10.1093/nar/gkg563
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a Comparative Platform for green Plant Genomics. Nucleic Acids Res. 40, D1178–D1186. doi:10.1093/nar/gkr944
Grossman, T. H. (2016). Tetracycline Antibiotics and Resistance. Cold Spring Harb Perspect. Med. 6, a025387. doi:10.1101/cshperspect.a025387
Hayashi, K., Schoonbeek, H.-j., and De Waard, M. A. (2002). Bcmfs1 , a Novel Major Facilitator Superfamily Transporter from Botrytis Cinerea , Provides Tolerance towards the Natural Toxic Compounds Camptothecin and Cercosporin and towards Fungicides. Appl. Environ. Microbiol. 68, 4996–5004. doi:10.1128/aem.68.10.4996-5004.2002
Huang, Y.-C., Chang, Y.-L., Hsu, J.-J., and Chuang, H.-w. (2008). Transcriptome Analysis of Auxin-Regulated Genes of Arabidopsis thaliana. Gene 420, 118–124. doi:10.1016/j.gene.2008.05.017
Huang, Y., Xu, P. H., Hou, B. Z., and Shen, Y. Y. (2019). Strawberry Tonoplast Transporter, FaVPT1, Controls Phosphate Accumulation and Fruit Quality. Plant Cel Environ 42, 2715–2729. doi:10.1111/pce.13598
Ideker, T. (2011). Cytoscape 2.8: New Features for Data Integration and Network Visualization. Bioinformatics 27, 431–432.
Ivica, L., and Peer, B. (2018). 20 Years of the SMART Protein Domain Annotation Resource. Nucleic Acids Res. 46, D493–D496. doi:10.1093/nar/gkx922
Jacquot, A., Chaput, V., Mauries, A., Li, Z., and Lejay, L. (2019). NRT2.1 Phosphorylation Prevents Root High Affinity Nitrate Uptake Activity in Arabidopsis thaliana. Bioinformatics 228, 1038–432. doi:10.1101/583542
Jaina, M., Finn, R. D., Eddy, S. R., Alex, B., and Marco, P. (2013). Challenges in Homology Search: HMMER3 and Convergent Evolution of Coiled-Coil Regions. Nucleic Acids Res. 41, e121.
Jeffares, D. C., Penkett, C. J., and Bähler, J. (2008). Rapidly Regulated Genes Are Intron Poor. Trends Genet. 24, 375–378. doi:10.1016/j.tig.2008.05.006
Kang, P. Z., Bai, X. J., Zhang, L. R., Du, Y. N., and Zhang, Z. K. (2018). Biological Characteristics of Fusarium oxysporium (Schl.) f.sp.Cucumerinum Owen. Nor. Horticul. 22, 65–69.
Kenneth, J. L., and Thomas, D. S. (2002). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 25, 402–408.
Kong, H., Landherr, L. L., Frohlich, M. W., Leebens-Mack, J., Ma, H., and dePamphilis, C. W. (2010). Patterns of Gene Duplication in the Plant SKP1 Gene Family in Angiosperms: Evidence for Multiple Mechanisms of Rapid Gene Birth. Plant J. 50, 873–885. doi:10.1111/j.1365-313X.2007.03097.x
Kumar, K. (2014). SPSS for Applied Sciences: Basic Statistical Testing. J. R. Stat. Soc. A. Stat. 177, 990–990. doi:10.1111/rssa.12082_2
Lee, J. (2015). MFS Transporter Superfamily: Modelling and Dynamics. Oxford: Doctoral, University of Oxford.
Lin, H.-C., Yu, P.-L., Chen, L.-H., Tsai, H.-C., and Chung, K.-R. (2018). A Major Facilitator Superfamily Transporter Regulated by the Stress-Responsive Transcription Factor Yap1 Is Required for Resistance to Fungicides, Xenobiotics, and Oxidants and Full Virulence in Alternaria alternata. Front. Microbiol. 9, 2229. doi:10.3389/fmicb.2018.02229
Liu, J., Yang, L., Luan, M., Wang, Y., Zhang, C., Zhang, B., et al. (2015). A Vacuolar Phosphate Transporter Essential for Phosphate Homeostasis in Arabidopsis. Proc. Natl. Acad. Sci. U S A. 112, E6571–E6578. doi:10.1073/pnas.1514598112
Lorca, G. L., Barabote, R. D., Zlotopolski, V., Tran, C., Winnen, B., Hvorup, R. N., et al. (2007). Transport Capabilities of Eleven Gram-Positive Bacteria: Comparative Genomic Analyses. Biochim. Biophys. Acta (Bba) - Biomembranes 1768, 1342–1366. doi:10.1016/j.bbamem.2007.02.007
Lu, L., Giang, H., Loussaert, D. F., and Wang, H. (2010). Functional Expression of Higher Plant Nitrate Transporters in Pichia Pastoris.
Marcet-Houben, M., Ballester, A.-R., de la Fuente, B., Harries, E., Marcos, J. F., González-Candelas, L., et al. (2012). Genome Sequence of the Necrotrophic Fungus Penicillium digitatum, the Main Postharvest Pathogen of Citrus. BMC Genomics 13, 646. doi:10.1186/1471-2164-13-646
Marger, M. D., and Saier, M. H. (1993). A Major Superfamily of Transmembrane Facilitators that Catalyse Uniport, Symport and Antiport. Trends Biochem. Sci. 18, 13–20. doi:10.1016/0968-0004(93)90081-w
Maria, N. G., Esther, N. U., Dale, N. R., Pedro, M. B., and Paula, D. (2019). More Transporters, More Substrates: The Arabidopsis Major Facilitator Superfamily Revisited. Mol. Plant 12, 1182–1202.
Martin, B., Hiba, S., Michael, S., Alona, R., Magnus, K., and Benjamin, A. H. (2021). Ferulic Acid, an Abundant maize Phenolic, Regulates ABC and MFS Transporter Gene Expression in the maize Pathogen Cochliobolus Heterostrophus. J. Plant Dis. Protect. 128, 1383–1391.
Mathias, C., Miin-Huey, L., Huey-Jiunn, B., and Kuang, R. (2007). Deletion of a MFS Transporter-like Gene in Cercospora Nicotianae Reduces Cercosporin Toxin Accumulation and Fungal Virulence. Febs Lett. 581, 489–494.
Matthias, K., Michaela, L., Andreas, M., Anne-Sophie, W., and Sabine, F. (2009). Fungicide-driven Evolution and Molecular Basis of Multidrug Resistance in Field Populations of the Grey Mould Fungus Botrytis Cinerea. Plos Pathog. 5, e1000696.
Mathias, T. J., Natarajan, K., Shukla, S., Doshi, K. A., Singh, Z. N., Ambudkar, S. V., et al. (2015). The FLT3 and PDGFR Inhibitor Crenolanib is a Substrate of the Multidrug Resistance Protein ABCB1 but does not Inhibit Transport Function at Pharmacologically Relevant Concentrations. Invest. New Drug. 33, 300–309.
Miyaji, T., Kuromori, T., Takeuchi, Y., Yamaji, N., Yokosho, K., Shimazawa, A., et al. (2015). AtPHT4;4 Is a Chloroplast-Localized Ascorbate Transporter in Arabidopsis. Nat. Commun. 6, 5928. doi:10.1038/ncomms6928
Monalessa, F. P., Carolina, M. A. S., Elza, F. A., Marisa, V. Q., and Denise, M. S. B. (2013). Beginning to Understand the Role of Sugar Carriers in Colletotrichum Lindemuthianum: the Function of the Gene Mfs1. J. Microbiol. 51, 70–81.
Nicolas, M., Jean-Luc, B., Naomi, T., and Marc, S. (2005). HTLV-1 Tropism and Envelope Receptor. Oncogene 24, 6016–6025.
Omrane, S., Audéon, C., Ignace, A., Duplaix, C., Aouini, L., Kema, G., et al. (2017). Plasticity of the MFS1 Promoter Leads to Multidrug Resistance in the Wheat Pathogen Zymoseptoria Tritici. Msphere 2, 393–417. doi:10.1128/mSphere.00393-17
Omrane, S., Sghyer, H., Audéon, C., Lanen, C., Duplaix, C., Walker, A.-S., et al. (2015). Fungicide Efflux and the MgMFS1 Transporter Contribute to the Multidrug Resistance Phenotype inZymoseptoria Triticifield Isolates. Environ. Microbiol. 17, 2805–2823. doi:10.1111/1462-2920.12781
Paul, H., Keun-Joon, P., Takeshi, O., Naoya, F., Hajime, H., Adams-Collier, C. J., et al. (2007). WoLF PSORT: Protein Localization Predictor. Nucleic Acids Res. 35, 585–587. doi:10.1093/nar/gkl929
Paulsen, I. T., Brown, M. H., and Skurray, R. A. (1996). Proton-dependent Multidrug Efflux Systems. Microbiol. Rev. 60, 575–608. doi:10.1128/mr.60.4.575-608.1996
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: Protein Domains Identifier. Nucleic Acids Res. 33, W116–W120. doi:10.1093/nar/gki442
Ramin, R., Maarten, A. D. W., Gert, H. J. K., and Lute-Harm, Z. (2007). MgMfs1, a Major Facilitator Superfamily Transporter from the Fungal Wheat Pathogen Mycosphaerella Graminicola, Is a strong Protectant against Natural Toxic Compounds and Fungicides. Fungal Genet. Biol. 44, 378–388.
Ramón-Carbonell, M. d., and Sánchez-Torres, P. (2017). Involvement of Penicillium digitatum PdSUT1 in Fungicide Sensitivity and Virulence during Citrus Fruit Infection. Microbiol. Res. 203, 57–67. doi:10.1016/j.micres.2017.06.008
Reisz, J. A., Bechtold, E., King, S. B., Poole, L. B., and Furdui, C. M. (2013). Thiol-blocking Electrophiles Interfere with Labeling and Detection of Protein Sulfenic Acids. Febs J. 280, 6150–6161. doi:10.1111/febs.12535
Remy, E., Cabrito, T. R., Batista, R. A., Teixeira, M. C., Sá-Correia, I., and Duque, P. (2015). The Major Facilitator Superfamily Transporter ZIFL2 Modulates Cesium and Potassium Homeostasis in Arabidopsis. Plant Cel Physiol 56, 148–162. doi:10.1093/pcp/pcu157
Rombauts, S., Dehais, P., Van Montagu, M., and Rouze, P. (1999). PlantCARE, a Plant Cis-Acting Regulatory Element Database. Nucleic Acids Res. 27, 295–296. doi:10.1093/nar/27.1.295
Saier, M. H., and Paulsen, I. T. (2011). Phylogeny of Multidrug Transporters. Semin. Cel Dev. Biol. 12, 205–213. doi:10.1006/scdb.2000.0246
Saier, M. H., Reddy, V. S., Tamang, D. G., and Åke, V. (2009). The Transporter Classification Database: Recent Advances. Nucleic Acids Res. 44, 372–379. doi:10.1093/nar/gkn862
Setyowati, T. U., Carissa, I. I., Anom, B., Takashi, Y., Chen, X. Y., Kyoko, N., et al. (2020). Identification and Functional Characterization of Penicillium marneffei Major Facilitator Superfamily (MFS) Transporters. Biosci. Biotech. Bioch. 84, 1373–1383.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum. Mol. Biol. Evol. 28, 2731–2739. doi:10.1093/molbev/msr121
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and Collinearity in Plant Genomes. Science 320, 486–488. doi:10.1126/science.1153917
Torsten, S., Jürgen, K., Nicolas, G., and Peitsch, M. C. (2003). SWISS-MODEL: an Automated Protein Homology-Modeling Server. Nucleic Acids Res. 31, 3381–3385. doi:10.1126/science.1128691
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi:10.1126/science.1128691
Ulrich, O., Ahrens, C. H., Sebastian, M., and Bernd, W. (2014). Protter: Interactive Protein Feature Visualization and Integration with Experimental Proteomic Data. Bioinformatics 30, 884–886. doi:10.1093/bioinformatics/btt658
Utami, S. T., Indriani, C. I., Bowolaksono, A., Yaguchi, T., Chen, X., Niimi, K., et al. (2020). Identification and Functional Characterization of Penicillium marneffei Major Facilitator Superfamily (MFS) Transporters. Biosci. Biotechnol. Biochem. 84, 1373–1383. doi:10.1080/09168451.2020.1732185
Vela-Corcía, D., Aditya Srivastava, D., Dafa-Berger, A., Rotem, N., Barda, O., and Levy, M. (2019). MFS Transporter from Botrytis Cinerea Provides Tolerance to Glucosinolate-Breakdown Products and Is Required for Pathogenicity. Nat. Commun. 10, 2886. doi:10.1038/s41467-019-10860-3
Wang, D.-M., Liu, L., Fan, L., Zou, Z.-J., Zhang, L.-N., Yang, S., et al. (2012). Expression Level of DEK in Chronic Lymphocytic Leukemia Is Regulated by Fludarabine and Nutlin-3 Depending on P53 Status. Cancer Biol. Ther. 13, 1522–1528. doi:10.4161/cbt.22252
Wang, J. Y., Sun, X. P., Lin, L. Y., Zhang, T. Y., and Li, H. Y. (2012). PdMfs1, a Major Facilitator Superfamily Transporter from Penicillium digitatum, Is Partially Involved in the Imazalil-Resistance and Pathogenicity. Afr. J. Microbiol. Res. 6, 95–105. doi:10.5897/ajmr11.1045
Wang, Y., Salasini, B. C., Khan, M., Devi, B., Bush, M., Subramaniam, R., et al. (2019). Clade I TGACG-Motif Binding Basic Leucine Zipper Transcription Factors Mediate BLADE-ON-PETIOLE-dependent Regulation of Development. Plant Physiol. 180, 937–951. doi:10.1104/pp.18.00805
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 40, e49. doi:10.1093/nar/gkr1293
Wannes, D., and Thomas, V. L. (2014). The ABC Gene Family in Arthropods: Comparative Genomics and Role Ininsecticide Transport and Resistance. Insect Biochem. Mol. 45, 89–110.
Wu, Z., Wang, S., Yuan, Y., Zhang, T., Liu, J., and Liu, D. (2016). A Novel Major Facilitator Superfamily Transporter in Penicillium digitatum (PdMFS2) Is Required for Prochloraz Resistance, Conidiation and Full Virulence. Biotechnol. Lett. 38, 1349–1357. doi:10.1007/s10529-016-2113-4
Yen, M. R., Chen, J. S., Marquez, J. L., Sun, E. I., and Saier, M. H. (2010). Multidrug Resistance: Phylogenetic Characterization of Superfamilies of Secondary Carriers that Include Drug Exporters. Methods Mol. Biol. 637, 47–64. doi:10.1007/978-1-60761-700-6_3
Zhai, Z. H., Chen, X. N., and Wang, J. (2008). Primer Design with Primer Premier 5.0. Northwest. Med. Educ. 4, 695–698.
Zhang, T., Cao, Q., Li, N., Liu, D., and Yuan, Y. (2020). Transcriptome Analysis of Fungicide-Responsive Gene Expression Profiles in Two Penicillium italicum Strains with Different Response to the Sterol Demethylation Inhibitor (DMI) Fungicide Prochloraz. BMC Genomics 21, 156. doi:10.1186/s12864-020-6564-6
Keywords: Populus davidiana × P. alba var. pyramidalis Louche, Fusarium oxysporum, major facilitator superfamily, expression patterns, tissue-differential expression
Citation: Diao J, Li S, Ma L, Zhang P, Bai J, Wang J, Ma X and Ma W (2021) Genome-Wide Analysis of Major Facilitator Superfamily and Its Expression in Response of Poplar to Fusarium oxysporum. Front. Genet. 12:769888. doi: 10.3389/fgene.2021.769888
Received: 02 September 2021; Accepted: 29 September 2021;
Published: 22 October 2021.
Edited by:
Madhav P. Nepal, South Dakota State University, United StatesReviewed by:
Dawei Xue, Hangzhou Normal University, ChinaSarbottam Piya, The University of Tennessee, United States
Copyright © 2021 Diao, Li, Ma, Zhang, Bai, Wang, Ma and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Ma, bWF3ZWlAaGxqdWNtLm5ldA==
†These authors have contributed equally to this work