
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Genet., 28 July 2021
Sec. Statistical Genetics and Methodology
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.699793
This article is part of the Research TopicExploring Omics Tools & Data Integration Approaches to Deepen Insights into Complex DiseasesView all 5 articles
 Lu Qi Wei1†
Lu Qi Wei1† Io Hong Cheong1†
Io Hong Cheong1† Guang Huan Yang1
Guang Huan Yang1 Xiao Guang Li1
Xiao Guang Li1 Zisis Kozlakidis2Lei Ding1Ning Ning Liu1Hui Wang1*
Zisis Kozlakidis2Lei Ding1Ning Ning Liu1Hui Wang1*Human gut microbiome research, especially gut microbiome, has been developing at a considerable pace over the last decades, driven by a rapid technological advancement. The emergence of high-throughput technologies, such as genomics, transcriptomics, and others, has afforded the generation of large volumes of data, and in relation to specific pathologies such as different cancer types. The current review identifies high-throughput technologies as they have been implemented in the study of microbiome and cancer. Four main thematic areas have emerged: the characterization of microbial diversity and composition, microbial functional analyses, biomarker prediction, and, lastly, potential therapeutic applications. The majority of studies identified focus on the microbiome diversity characterization, which is reaching technological maturity, while the remaining three thematic areas could be described as emerging.
Human microbiome research has been developing at a considerable pace over the last two decades, partly driven by technological advancement and the ability for high-throughput, culture-independent analyses, and in part because the ability to analyze and interpret the increasing quantities of data has now become possible. As in any rapidly evolving field, there can emerge differences in the definition. For the purposes of this study, which focuses on the human microbiome, especially gut microbiome, the term microbiome aligns with previously reported ones and refers to the entire habitat view, including the microorganisms, their genomes, and associated clinical metadata (Marchesi and Ravel, 2015).
The human microbiome is a dynamic collection of bacteria, viruses, and fungi. Under ideal conditions, these organisms live symbiotically with their human host in gut (Lynch and Pedersen, 2016), and individual species and/or collective bacterial functions under certain conditions may confer many benefits throughout their host's life by metabolizing dietary compounds, educating the immune system, defending against pathogens, and contributing to overall health (Kau et al., 2011; Sharon et al., 2016; Valdes et al., 2018). Therefore, it is critical to try and understand the microbiome as it impacts on a multitude of aspects, including a wide range of pathologies. Accordingly, numerous avenues of research are being pursued to understand what constitutes healthy and abnormal microbiomes (Schwartz et al., 2020), and how they relate to specific disease conditions, such as cancer.
In terms of the latter, historically the first close link between cancer research and the microbiome was achieved already a few decades ago. Specifically, Helicobacter pylori was first identified in the late 1970s by J. Robin Warren in gastric tissue samples from patients with chronic gastritis, which was an inflammatory precursor of gastric cancer (Warren and Marshall, 1983). Wotherspoon et al. found 101 (92%) H. pylori infection cases out of 110 cases of gastric mucosa-associated lymphoma using modified Giemsa or cresyl violet stain (Wotherspoon et al., 1991). Additionally, the association of H. pylori infection with the risk of gastric carcinoma was confirmed in a nested case-control study in 1991 by ELISA assay (Parsonnet et al., 1991). From the history of investigation into this relationship of H. pylori infection and chronic gastritis, leading to gastric cancer, the field moved into more extensive studies on the microbiome and its relationship with cancer.
The earliest microbial diversity detection was carried out through microscopic observation (Van Leewenhoeck, 1677) and established microbial isolation and culture technologies (Janssen et al., 2002; Kaeberlein et al., 2002). However, although pure-culture technologies were improved significantly (Browne et al., 2016), the overall knowledge and view of microbial diversity were still limited due to the natural difficulties of laboratory cultivation (Amann et al., 1995; Fredricks et al., 2005). Therefore, as an additional means to the morphological observation and selection of growth conditions, microbiologists also took advantage of the metabolic properties to distinguish different microbes (Pace, 1997). The Biolog technology was successfully developed by BIOLOG in 1989 for carrying out the biochemical reaction test of 95 unique carbon sources and was initially applied to the identification of pure microorganisms (Garland and Mills, 1991).
Beyond these early historical examples, the current era of laboratory automation has ushered-omics technologies, which are increasingly high-throughput, allowing for the detailed characterization of collected samples and specimens from patients and healthy individuals alike. Still, the efforts are mostly concentrating on the accurate characterization of the diversity of the microbiome (and its progressive changes over time in the case of sequential sampling), leading to the interpretation of these observations. While interventions have started taking place (DeFilipp et al., 2018; Smibert et al., 2019; Wing and Kremenchutzky, 2019), these are at the initial stages and not yet an established clinical practice. It is anticipated that the increased understanding in this field through high-throughput laboratory methodologies will lead to future interventions, as well as preventive actions in relation to cancer development. Previous reviews have summarized high-throughput sequencing technologies and the platforms used (Reuter et al., 2015), reviewed shotgun metagenomics process in detail (Quince et al., 2017) or its application in microbiome and several diseases (Wang and Jia, 2016), discussed the gut microbiome (virome) in health or disease situation (Carding et al., 2017), or reviewed investigations on microbiome and cancer (Contreras et al., 2016; Helmink et al., 2019). Notwithstanding the above, the current manuscript is a systematic review on the subject of the high-throughput methodologies that have been employed over the last two decades in the study of the human gut microbiome in relation to cancer. It provides a useful benchmark on current technological developments, biological interpretations, and how the latter might eventually influence clinical practice.
The systematic review followed the PRISMA guidelines (Figure 1) (Stewart et al., 2015). Two investigators (LW and GY) independently conducted literature search using as combined keywords microbiome and cancer, security on PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) and Web of Science (v. 5.35). The database search was run of all the published articles, all languages, from database inception until March 1, 2021. In both databases, the following search strategy was used: terms were searched as follows: Microbiome AND Cancer AND *omics; Microbiome AND Cancer AND high-throughput; Microbiome AND Cancer AND genomics/metabolomics. *omics was used in the search in order to identify longer forms. It is thought that these terms would be able to identify the majority of manuscripts within a narrow definition of microbiome and cancer and applied omics methodologies, though it remains likely that relevant sections might be embedded within methodology sections of particular projects and thus more challenging to identify.
Figure 1. PRISMA graph detailing the search results.
All studies reporting information on microbiome, cancer, high-throughput, and -omics were included. A total of 962 articles were identified and reviewed independently by two authors (LW and GY), and after all duplicates were removed, 673 articles were considered. After removing articles that were not in English, and those that had simply a mention of the words with no further expansion, 127 articles were considered. These articles are included in Table 1 for transparency and further reference. One hundred twenty-one articles (of the 127) devoted considerable amount of the manuscript to expand on those topics, while 6 articles had much reduced and/or incomplete analyses. Both of these latter categories were used in the current review. Any inconsistencies were resolved by consensus with a third author (ZK), while thematic groupings (Table 2) and analyses were reviewed by an additional author (IC). All outcomes were included, due to the wide range of use of the terminologies.
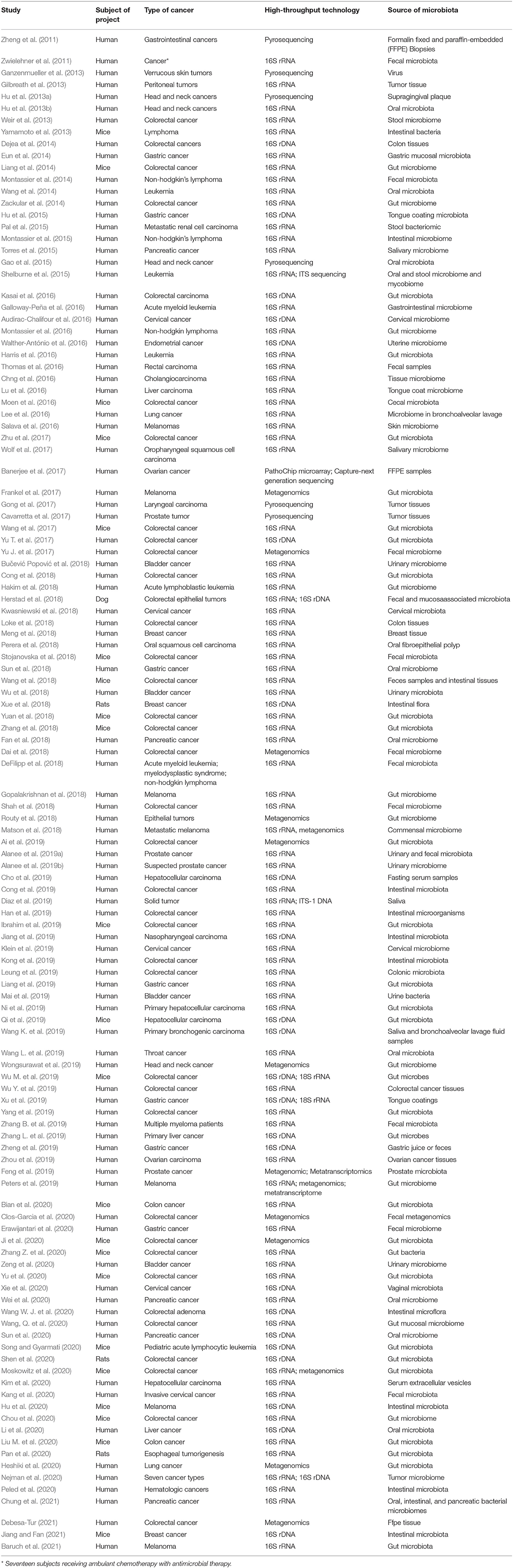
Table 1. Characteristics of included studies.

Table 2. Thematic groupings of included articles.
The manuscripts identified in this review (n = 121) followed four loosely defined thematic groups: (a) the methods used in measuring diversity (n = 121, i.e., all of the manuscripts used in this systematic review contained an element of measuring microbiome diversity), (b) the microbial functional analyses (n = 19), (c) the biomarker predictions (n = 19), and (d) microbiome in relation to cancer therapy (n = 30). They will be presented subsequently in this order, reflecting the scientific continuum, moving from the characterization and acquisition of knowledge, to the interpretation and finally toward clinical implementation. It becomes clear that both the number of technologies applied as well as the number of publications are increasing consistently, especially in the last few years, as can also be evidenced by the information on Table 2, and Table 1, with some studies deploying more than one methods in parallel. The most frequently used high-throughput technologies include in relative order of frequency amplicon sequencing, metagenomics, meta-transcriptomics, proteomics, and metabolomics. All of the above are accompanied with references, sometimes extensive, on continuously advancing bioinformatics analytical methods.
Diversity characterizing of the microbiome nowadays depends largely on cultivation-independent molecular technologies (Su et al., 2012) due to the unculturable property of the majority of microbes consisting the microbiome (Stewart, 2012; Browne et al., 2016). The sequential development of tools [including PCR-denaturing gradient gel electrophoresis (DGGE) (Scanlan and Marchesi, 2008; Zhang et al., 2010), fluorescence in situ hybridization (FISH) (Fredricks et al., 2005), quantitative dot blot hybridization, restriction fragment length polymorphism (RFLP) (Laguerre et al., 1994), terminal restriction fragment length polymorphisms (T-RFLP) (Wang et al., 2009), clone library (Bik et al., 2010; Rehman et al., 2011), and gene chip (Luo et al., 2020)] and the emergence of high-throughput sequencing technologies [16S/18S rRNA/rDNA gene sequence analysis (Fredricks et al., 2005; Scanlan and Marchesi, 2008; Rehman et al., 2011), high-throughput pyrosequencing (Rehman et al., 2011), metagenomics, meta-transcriptomics, and single-cell genomics (Lasken, 2007; Ishoey et al., 2008)] have broadened the perception of microbial diversity and evolutionary relationships of microbiota (Pace, 1997).
Initially, the sequence-based methods for analyzing microbiota relied on the first-generation sequencing technology developed by Sanger et al. (1977), which allowed culture-independent investigations (Morgan et al., 2017). However, these fingerprinting methods did not provide taxonomic information directly and were hard to detect rare or low-abundance taxa (Morgan et al., 2017). Subsequently, ribosomal RNA (rRNA) gene sequences in the conserved regions have been utilized to define and distinguish specific microbial species or populations from mixed organisms (Pace et al., 1986; Yarza et al., 2014) or to explore the bacterial diversity (Hugenholtz et al., 1998; Bik et al., 2010). Bik et al. determined the composition of oral bacterial diversity of 10 healthy individuals by constructing clone libraries from the amplified 16S rRNA gene, which was a comprehensive and high-resolution analysis of healthy human oral bacterial diversity in 2010 (Bik et al., 2010). Furthermore, combinations of two or more methods were utilized in an effort to avoid certain bias and discrepancies (Su et al., 2012), for example, deploying DGGE and ITS sequencing for analyzing the fungal diversity and richness in healthy human gut (Scanlan and Marchesi, 2008).
Responding to the high-throughput needs, the Biolog system also developed and provided phenotype microarrays specifically designed for microbiome analysis and ecological studies as a complement for traditional genomic, transcriptomic, and proteomic analyses, allowing users to conduct more targeted studies (Shea et al., 2012). PathoChip Microarray and Capture-next Generation Sequencing were also adopted to screen known pathogenic microbiomes including viruses, helminths, protozoa, fungi, and bacteria in ovarian cancer samples for investigating specific insertion sites of microbiome into the host genome, and that provided a solid association of microbiota with the ovarian cancer (Banerjee et al., 2017). Although microarray was a powerful tool to identify microbial species, only containing the known species of microbiota largely limited its application (Ehrenreich, 2006). The next-generation high-throughput sequencing avoided the system bias from the construction process of plasmid cloning library due to direct sequencing of the genome fragments (Pérez-Losada et al., 2018). The advantages of (eventual) low cost, high flux, good repeatability, and high accuracy provided a technological advantage and made it possible to profile the diversity of human gut microbiome comprehensively and to prevail in microbial ecology research (Liu Y.-X. et al., 2020).
Amplicon sequencing (Luo et al., 2020) is the most diffusely used method in microbiome analysis, as it is applicable to almost all sample types, provides vital insights into the microbial structural community, and helps to investigate the intricate and unsolved association between host and microbiome (Lynch and Pedersen, 2016). The main marker genes for amplicon sequencing include 16S rDNA for prokaryotes (Janda and Abbott, 2007) and 18S rDNA and ITS for eukaryotes (Shelburne et al., 2015; Dong et al., 2017; Diaz et al., 2019), among which 16S rDNA amplicon sequencing is currently the most commonly used method for detecting bacteria communities (Dejea et al., 2014; Hu et al., 2015; Audirac-Chalifour et al., 2016; Kasai et al., 2016; Montassier et al., 2016; Walther-António et al., 2016; Daniel et al., 2017; Herstad et al., 2018; Kwasniewski et al., 2018; Xue et al., 2018; Yuan et al., 2018; Cho et al., 2019; Leung et al., 2019; Qi et al., 2019; Zheng et al., 2019; Li et al., 2020; Sun et al., 2020; Wang W. J. et al., 2020; Zhang H. et al., 2020; Chung et al., 2021; Jiang and Fan, 2021). Some of the reasons for its wide adoptions are its ability to be used for low-biomass samples (Janda and Abbott, 2007) or for specimens contaminated with host DNA (Quince et al., 2017). Nonetheless, it does also have certain disadvantages, such as the biases and systematic errors induced during sampling, DNA extraction, library preparing, and sequencing (Hugerth and Andersson, 2017), environmental contaminations, or sample cross-talk (Edgar, 2016), potentially confusing primer sequences and limited genus-level resolution (Liu Y.-X. et al., 2020). In addition, the sensitivity to specific primers and selection of PCR cycle number may result in potential false-positive or false-negative results in downstream analysis (Liu Y.-X. et al., 2020). For analyzing the amplicon sequencing data, advanced specialized bioinformatic algorithms and pipelines were updated and adopted addressing biases, offering a better data quality, higher sensitivity, and higher specificity (Prodan et al., 2020). Collectively, operating taxonomic unit (OTU) clustering and amplicon sequence variant (ASV) analysis were two approaches for clustering and analyzing sequencing data based on either sequence identity (a threshold at 97%) or exact sequences with a statistical confidence (Zhai et al., 2020). Operating taxonomic units were normally used for evaluating the alpha-diversity of a microbial community (Hugerth and Andersson, 2017) by clustering similar sequences into a consensus sequence so as to filter and reduce noises or systematic errors in pipelines such as UPARSE (Edgar, 2010, 2013), MOTHUR (Schloss et al., 2009), or QIIME (Caporaso et al., 2010), whereas the ASVs showed great advantages when dealing with complicated samples or diminishing confounding factors that interfere with classification or analysis, especially its good performance on sensitivity and accuracy for big biomass (Caruso et al., 2019) in pipelines such as DADA2 (Callahan et al., 2016). Amplicon sequence variants have been proven to exhibit better sensitivity and specificity and distinguish microbial communities than OTUs (Callahan et al., 2016), even reaching species level or more (Callahan et al., 2017).
In recent years, metagenomics and meta-transcriptome are the two most rapidly advancing “omics” technologies (Aw and Fukuda, 2015), as they can monitor strain-level changes in microbiome and analyze potential functional activities of the gut microbiome in patients with cancer (Quince et al., 2017). For example, Yu J. et al. (2017) and Coker et al. (2019) revealed several gut species significantly associated with colorectal cancer (CRC) by metagenomics; in the oral squamous cell carcinoma, Yost et al. (2018) pointed out that Fusobacteria, Selenomonas spp., Capnocytophaga spp., and members of the genera Dialister and Johnsonella were significantly more active. Thus, high-throughput sequencing technologies have enabled the collection of comprehensive information on the gut microbiome and begun to reveal the correlation between microbiome and tumor (Zeller et al., 2014; Feng et al., 2015; Thomas et al., 2019; Yachida et al., 2019). While amplicon sequencing is a commonly used methodology for characterizing the microbiome due its lower cost, metagenomics and meta-transcriptome are more frequently applied to complex environmental samples.
Advantages and limitations of major high-throughput technologies are shown in Table 3. The integration of such multi-omic methodologies can provide further insights into cancer research (Liu Y.-X. et al., 2020). For example, Peters et al. (2019) characterized the gut microbiome for melanoma patients by 16S rRNA gene and shotgun metagenome sequencing and pointed out that the clustering of patients based on 16S microbiome composition was slightly more predictive of progression-free survival than clusters based on shotgun microbiome composition; on the other hand, species-level classification was much higher in the shotgun data, permitting researchers to identify more response-associated species than with 16S data alone.

Table 3. Advantages and limitations of major high-throughput technologies.
The application of high-throughput methodologies to the study of the human gut microbiome focuses not only on the microbiome composition but also on the functional analysis of the identified microbiome. Amplicon sequencing is a commonly used key tool for studying microbial communities as discussed above. The application of 16S rDNA (Dubin et al., 2019) or ITS rDNA using ASVs in DADA2 pipeline detected microbiome community in a high-resolution and high-accuracy way, which also helps identify the cross-kingdom dysbiosis and demonstrate the expansion and translocation pattern of pathogenic fungi during disease progression (Zhai et al., 2020). In addition, amplicon sequencing can also provide predictive functional analyses of microbial communities with quantifiable uncertainty if combined with advanced computational algorithms. PICRUSt was developed for predicting metagenomes according to amplicon sequencing data and reference genome databases (Langille et al., 2013). For example, QIIME and PICRUSt were utilized for diversity and compositional analysis and functional prediction after 16S rRNA sequencing, and it showed that proinflammatory pathways, such as lipopolysaccharide biosynthesis and peptidases, were enriched in the oral squamous cell carcinoma tissues and provided evidence for the inflammatory characteristic of bacteria related to cancer (Perera et al., 2018).
NGS-based methods provide the most common platform to explore metagenomic abundance of microbial community members at high genomic resolution (Quince et al., 2017). Specifically, shotgun metagenomics, i.e., the untargeted sequencing of all microbial genomes present in one sample, is a useful tool for quantifying microbiome and have been used to profile taxonomic composition and functional potential of microbial communities and to recover whole genome sequences (Quince et al., 2017). Databases that include combinations of manually annotated and computationally predicted proteins families, such as KEGG (Kanehisa et al., 2014; Erawijantari et al., 2020) or UniProt (UniProt Consortium., 2014), can be used for characterization of the functional potential of the microbiome. For instance, there were significantly lower catabolic pathway expression of local microbiota for responders to lung cancer therapy (Heshiki et al., 2020).
However, metagenomics still has limitations when it comes to profiling the active microbial community as measured by gene expression, a technological challenge addressed by meta-transcriptomics. Analysis of the meta-transcriptome, the mRNA of the microbiome, can reveal which organisms are active and which microbial genes are being expressed at the time of sampling under different conditions (Franzosa et al., 2014). For example, in prostate cancer, 10 Pseudomonas genes were found positively associated with eight host genes encoding small RNAs by such meta-transcriptome analysis (Feng et al., 2019). Furthermore, metagenomic functions related to progression-free survival were correlated with specific meta-transcriptomic expression patterns in melanoma patients (Peters et al., 2019). However, because of the short half-life of mRNA, such meta-transcriptome analyses represent a single time point of gene expression that may not necessarily reflect longer-term adaptations between the host and microbiota (Bikel et al., 2015). Therefore, integrating metagenomics and meta-transcriptomics enables the calculation of transcript/gene ratios, which represents an improved measure of gene transcriptional activation or repression (Bikel et al., 2015).
Additionally, different from the mRNA-based analyzing of meta-transcriptomics, meta-proteomics and metabolome are also new post-genomics high-throughput omics technologies for characterization of the whole protein component and all metabolites of microbiome at any given moment. They reveal the structural–functional diversity and dynamic changes at the protein level and metabolite level of microbes, which serves as potential biomarkers and enables an in-depth understanding of metabolic changes of microbial communities under diverse habitats (Johnson et al., 2016; Wilmanski et al., 2019; Dubey et al., 2020). The combination of metagenome and metabolome helped researchers to distinguish unique stage-specific phenotypes of the gut microbiota in CRC at the levels of species, genes, metabolic pathways, and metabolites (Yachida et al., 2019).
The number of microbes associated with the human body is estimated as at least 10 times that of human cells (Sender et al., 2016). Thus, initial investigations into microbes in cancer, such as the association between H. pylori and MALT lymphoma (Stolte, 1992), mainly focused on discovery, culture, and identification when they first emerged (Gilbert et al., 2016) using established and well-validated methodologies. Moreover, the characterization of microbiome in tumor remains challenging due to the low biomass of microbiota and methodological limitations (Nejman et al., 2020). Most microbiota was broadly considered as unculturable because of its tremendous genetic and biochemical diversity and the difficulties to mimic the natural living conditions in the laboratory (Stewart, 2012; Browne et al., 2016). High-throughput omics analyses are no longer limited to just detecting a few strains, but can detect microbiome in different microbiome niches in various cancer types (Nejman et al., 2020), thus providing a more comprehensive picture of the microbiome in relation to tumor development.
For example, such an analysis was performed in 1,526 samples from seven different types of solid tumors by applying a combination of methods including electron microscopy, H&E staining, immunohistochemistry (IHC), 16S rRNA FISH, qPCR, and culture ex vivo, coupled with high-throughput 16S rDNA sequencing (Nejman et al., 2020). This validated distinct microbial distributions in different tumor types and even across different subtypes of the same tumor type, which was also associated with bacterial prevalence and metabolic functions (Nejman et al., 2020). Accordingly, well-defined microbiome constituents can serve as a potential screen for early-stage cancer (Zackular et al., 2014) or a biomarker for prediction of cancer progression (Li et al., 2020). Re-analysis of raw 16S rRNA gene sequence data sets from nine separate studies in conjunction with a detailed meta-analysis and machine learning identified a composite microbial biomarker for diagnosing CRC consistent across studies (Shah et al., 2018). However, the low taxonomical and functional resolution of 16S rRNA sequencing limited the interpretation of the results beyond the accurate reach of species level (Shah et al., 2018). Multi-cohort metagenomic profiling studies highlighted and validated the potential of fecal metagenomic biomarkers for early non-invasive diagnosis of CRC even in different populations with distinct intestinal microbial community (Yu J. et al., 2017; Dai et al., 2018). The dynamic changes of microbial composition, gene abundance, and metabolites in gut microflora during the progression of CRC revealed by metagenomic and metabolomic analysis in a large cohort indicated microbial and metabolic shifts in the very early stages of CRC, which may contribute to a routine etiological diagnosis in the future (Yachida et al., 2019).
Besides the correlation of microbiome and cancer development, the same suite of methodologies is starting to be applied in order to characterize the therapeutic sensitivity or resistance to the treatment(s) of cancer(s). They can also contribute to discovering specific microbiota that influence the curative effects. For example, to examine the potential relationship between altered intestinal flora, CRC recurrence, and chemoresistance, investigators performed pyrophosphate sequencing and found that Fusobacterium nucleatum enriched in the CRC recurrent group promoted CRC chemoresistance via activating the cancer autophagy pathway (Yu T. et al., 2017).
16S rRNA sequencing, metagenomics, and metabolomics have been employed widely to reveal changes of intestinal or tissue microbiome in cancer patients treated with chemotherapy (Montassier et al., 2014, 2015, 2016; Wang et al., 2017; Hakim et al., 2018; Diaz et al., 2019), immune checkpoint inhibitors (Frankel et al., 2017), or surgery (Cong et al., 2018; Kong et al., 2019), and helped to predict the patient outcomes of cancer treatment. To name a few such examples, in a study based on high-depth sequencing results of 16S rRNA of fecal microbiota from children undergoing chemotherapy for newly diagnosed acute lymphoblastic leukemia, researchers linked the relative abundance of Proteobacteria before chemotherapy to the development of febrile neutropenia and found that domination of Enterococcaceae or Streptococcaceae in gut microbiome during chemotherapy predicted infection in subsequent phases of chemotherapy (Hakim et al., 2018). Moreover, immune checkpoint inhibitors targeting the programmed death 1 (PD-1) protein are important cancer therapeutics but have been reported failure for some patients probably because of dysbiosis in intestinal microbiome (Gopalakrishnan et al., 2018; Matson et al., 2018; Routy et al., 2018). In a research on patients with metastatic melanoma starting treatment with anti-PD-1 therapy, multiple high-throughput technologies, including 16S rRNA sequencing, metagenomic whole genome shotgun (WGS) sequencing, and whole exome sequencing, were utilized to reveal the association between diversity/relative abundance of Ruminococcaceae, Faecalibacterium, and Bacteroidales with the systemic and antitumor immune responses, which underlined the therapeutic potential of manipulating gut microbiome in patients with immune therapy (Gopalakrishnan et al., 2018). Additionally, a pilot study using 16S rRNA sequencing identified the changes of gut microbiota in post-surgery CRC patients and highlighted the key role of gut microbiota in the future care of surgical CRC patients (Cong et al., 2018). Shotgun metagenomic sequencing and metabolomic analysis based on capillary electrophoresis time-of-flight mass spectrometry revealed altered intestinal microbiome after gastrectomy and demonstrated its association with postoperative comorbidities (Erawijantari et al., 2020).
Application of sequencing-based high-throughput technologies enabled the scientists to observe the microbial dysbiosis at an integrated scale (Dong et al., 2018; Gopalakrishnan et al., 2018). In such a case, the microbiome and prognosis of allogeneic hematopoietic cell transplantation was investigated via 16S rRNA gene sequencing (Peled et al., 2020). The results demonstrated that higher microbial diversity during the transplantation period was associated with a reduced risk of death and increased overall survival, which can potentially be used as a biomarker to predict mortality in allogeneic hematopoietic cell transplantation patients (Peled et al., 2020). Notably, 16S rRNA gene and metagenomic sequencing of fecal samples in a phase I clinical trial suggested that performing fecal microbiota transplantation (FMT) treatment was associated with favorable changes in immune cell infiltration and gene expression profiles in the intestinal lamina propria and tumor microenvironment (Baruch et al., 2021). Overall, the integration of NGS methodologies with clinical analyses and treatment allowed one to observe the dynamic changes of gut microbiome and adjust the choice of treatment on tumor in time (Tanoue et al., 2019; Zheng et al., 2019; Erawijantari et al., 2020).
This manuscript is a systematic review of the application of high-throughput technologies to investigate both the microbiome and cancer. It is important to note that the studies identified in this review, using these high-throughput technologies, tend to focus more on characterizing the diversity of the microbiome as a whole and in cancer in particular. The large data volumes generated through -omics applications, e.g., genomics, metagenomics, and meta-transcriptomics, are frequently applied to the purposes of taxonomic composition profiling, functional annotation, and pathway enrichment analyses through computational approaches. This increasing application of omics enables also a better look into the dynamic changes and functional features of microbial communities under specific habitats, and for specific patient groups. As was evident by a number of identified publications, the latter analyses can also provide evidence for potential biomarkers or predictors for disease detection.
Lastly, a small number of publications demonstrated that avenues of applying such methodologies in the study of microbiome and cancer, in relation to therapy, have started to emerge. It is expected that the application of such high-throughput methodologies will continue, revealing the interrelationship between microbiome and cancer. The accrued understanding is anticipated to expand the potential of the microbiome as a prognostic indicator of cancer treatment, while high-throughput methodologies may also pave the way for new clinical interventions that alter composition and function of specific microbial communities in directions that might favor cancer therapeutic responsiveness.
Notwithstanding the above, the current review has some limitations. Specifically, the search included manuscripts that were identified in two online databases (PubMed and Web of Science) with parameters including year/language type/article type/keywords. This might have limited the breadth of the results. Additionally, pre-print databases, such as bioArxiv and F1000, were excluded as those manuscripts have not completed a peer-review process. In a field that is actively growing, such as the application of high-throughput technologies on microbiome and cancer, this strategy may lead to the omissions of the newest technologies currently under development. Furthermore, this review focuses on the application of these technologies without comparing potential integrating methodologies that may offer an additional layer of complexity.
The emergence of high-throughput technologies enables in-depth studies on the relationship between microbiome and cancer. The ability to profile the microbiome as a whole, as well as the complex micro-ecosystems of the microbiome, enhances the possibility to use/measure specific microbial strata as predictive markers of cancer and eventually perhaps as a guide for precise treatments. However, these high-throughput methodologies produce high volumes of data and, as such, a downstream pressure for bioinformatics component able to ingest and interpret the results. Additionally, there still exist technical detection limits, especially with processing low-biomass samples.
Having said that, the majority of identified manuscripts in this review are still focusing their efforts on characterizing the microbiome and its relationship with cancer in detail. The many mechanisms by which the microbiome has the potential to modulate cancer development provide the possibility to target the microbiome for cancer prevention strategies. Additional clinically relevant data need to be generated, before microbiota-based strategies for cancer prevention can be envisioned and integrated into routine healthcare.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
LW and GY conducted the systematic review and applied the eligibility selection criteria for the identified manuscripts. IC and ZK validated the selected manuscripts and arbitrated any queries. LW, GY, IC, and XL wrote the manuscript. NL, LD, and HW oversaw the process and provided critical input throughout. All authors were involved in the drafting of the manuscript.
This study was supported by grants from the National Key R&D Program of China (2018YFC2000700), the National Natural Science Foundation (82030099 and 81630086), Shanghai Public Health System Construction Three-Year Action Plan (GWV-10.1-XK15), the Major Science and Technology Innovation Program of Shanghai Municipal Education Commission (2019-01-07-00-01-E00059), and Innovative research team of high-level local universities in Shanghai.
Where authors are identified as personnel of the International Agency for Research on Cancer/WHO, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/WHO.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ai, D., Pan, H., Li, X., Wu, M., Xia, L. C, et al. (2019). Association network analysis identifies enzymatic components of gut microbiota that significantly differ between colorectal cancer patients and healthy controls. PeerJ 7:e7315. doi: 10.7717/peerj.7315
Alanee, S., El-Zawahry, A., Dynda, D., Dabaja, A., McVary, K., Karr, M., et al. (2019a). A prospective study to examine the association of the urinary and fecal microbiota with prostate cancer diagnosis after transrectal biopsy of the prostate using 16sRNA gene analysis. Prostate 79, 81–87. doi: 10.1002/pros.23713
Alanee, S., El-Zawahry, A., Dynda, D., McVary, K., Karr, M., and Braundmeier-Fleming, A. (2019b). Prospective examination of the changes in the urinary microbiome induced by transrectal biopsy of the prostate using 16S rRNA gene analysis. Prostate Cancer Prostatic Dis. 22, 446–452. doi: 10.1038/s41391-018-0120-3
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1128/mr.59.1.143-169.1995
Audirac-Chalifour, A., Torres-Poveda, K., Bahena-Román, M., Téllez-Sosa, J., Martínez-Barnetche, J., Cortina-Ceballos, B., et al. (2016). Cervical microbiome and cytokine profile at various stages of cervical cancer: a pilot study. PLoS ONE 11:e0153274. doi: 10.1371/journal.pone.0153274
Aw, W., and Fukuda, S. (2015). An integrated outlook on the metagenome and metabolome of intestinal diseases. Diseases 3, 341–359. doi: 10.3390/diseases3040341
Banerjee, S., Tian, T., Wei, Z., Shih, N., Feldman, M. D., Alwine, J. C., et al. (2017). The ovarian cancer oncobiome. Oncotarget 8, 36225–36245. doi: 10.18632/oncotarget.16717
Baruch, E. N., Youngster, I., Ben-Betzalel, G., Ortenberg, R., Lahat, A., Katz, L., et al. (2021). Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609. doi: 10.1126/science.abb5920
Bian, S., Wan, H., Liao, X., and Wang, W. (2020). Inhibitory effects of apigenin on tumor carcinogenesis by altering the gut microbiota. Mediators Inflamm. 2020:7141970. doi: 10.1155/2020/7141970
Bik, E. M., Long, C. D., Armitage, G. C., Loomer, P., Emerson, J., Mongodin, E. F., et al. (2010). Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J. 4, 962–974. doi: 10.1038/ismej.2010.30
Bikel, S., Valdez-Lara, A., Cornejo-Granados, F., Rico, K., Canizales-Quinteros, S., Soberón, X., et al. (2015). Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 13, 390–401. doi: 10.1016/j.csbj.2015.06.001
Browne, H. P., Forster, S. C., Anonye, B. O., Kumar, N., Neville, B. A., Stares, M. D., et al. (2016). Culturing of 'unculturable' human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546. doi: 10.1038/nature17645
Bučević Popović, V., Šitum, M., Chow, C.-E. T, Chan, L. S., Roje, B., and Terzić, J. (2018). The urinary microbiome associated with bladder cancer. Sci. Rep. 8:12157. doi: 10.1038/s41598-018-29054-w
Callahan, B. J., McMurdie, P. J., and Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643. doi: 10.1038/ismej.2017.119
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Carding, S. R., Davis, N., and Hoyles, L. (2017). Review article: the human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 46, 800–815. doi: 10.1111/apt.14280
Caruso, V., Song, X., Asquith, M., Karstens, L., and Gibbons Sean, M. (2019). Performance of microbiome sequence inference methods in environments with varying biomass. mSystems 4:e00163–18. doi: 10.1128/mSystems.00163-18
Cavarretta, I., Ferrarese, R., Cazzaniga, W., Saita, D., Lucianò, R., Ceresola, E. R., et al. (2017). The microbiome of the prostate tumor microenvironment. Eur. Urol. 72, 625–631. doi: 10.1016/j.eururo.2017.03.029
Chng, K. R., Chan, S. H., Ng, A., Li, C., Jusakul, A., Bertrand, D., et al. (2016). Tissue microbiome profiling identifies an enrichment of specific enteric bacteria in Opisthorchis viverrini associated cholangiocarcinoma. EBioMedicine 8, 195–202. doi: 10.1016/j.ebiom.2016.04.034
Cho, E. J., Leem, S., Kim, S. A., Yang, J., Lee, Y. B., Kim, S. S., et al. (2019). Circulating microbiota-based metagenomic signature for detection of hepatocellular carcinoma. Sci. Rep. 9:7536. doi: 10.1038/s41598-019-44012-w
Chou, Y. C., Ho, P. Y., Chen, W. J., Wu, S. H., and Pan, M. H. (2020). Lactobacillus fermentum V3 ameliorates colitis-associated tumorigenesis by modulating the gut microbiome. Am. J. Cancer Res. 10, 1170–1181.
Chung, M., Zhao, N., Meier, R., Koestler, D. C., Wu, G., de Castillo, E., et al. (2021). Comparisons of oral, intestinal, and pancreatic bacterial microbiomes in patients with pancreatic cancer and other gastrointestinal diseases. J. Oral Microbiol. 13:1887680. doi: 10.1080/20002297.2021.1887680
Clos-Garcia, M., Garcia, K., Alonso, C., Iruarrizaga-Lejarreta, M., D'Amato, M., Crespo, A., et al. (2020). Integrative analysis of fecal metagenomics and metabolomics in colorectal cancer. Cancers (Basel). 12:1142. doi: 10.3390/cancers12051142
Coker, O. O., Nakatsu, G., Dai, R. Z., Wu, W. K. K., Wong, S. H., Ng, S. C., et al. (2019). Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 68, 654–662. doi: 10.1136/gutjnl-2018-317178
Cong, J., Zhu, H., Liu, D., Li, T., Zhang, C., Zhu, J., et al. (2018). A pilot study: changes of gut microbiota in post-surgery colorectal cancer patients. Front. Microbiol. 9:2777. doi: 10.3389/fmicb.2018.02777
Cong, J., Zhu, J., Zhang, C., Li, T., Liu, K., Liu, D., et al. (2019). Chemotherapy alters the phylogenetic molecular ecological networks of intestinal microbial communities. Front Microbiol. 10:1008. doi: 10.3389/fmicb.2019.01008
Contreras, A. V., Cocom-Chan, B., Hernandez-Montes, G., Portillo-Bobadilla, T., and Resendis-Antonio, O. (2016). Host-microbiome interaction and cancer: potential application in precision medicine. Front. Physiol. 7:606. doi: 10.3389/fphys.2016.00606
Dai, Z., Coker, O. O., Nakatsu, G., Wu, W. K. K., Zhao, L., Chen, Z., et al. (2018). Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 6, 70–70. doi: 10.1186/s40168-018-0451-2
Daniel, S. G., Ball, C. L., Besselsen, D. G., Doetschman, T., and Hurwitz, B. L. (2017). Functional changes in the gut microbiome contribute to transforming growth factor beta-deficient colon cancer. Msystems 2:e00065–17. doi: 10.1128/mSystems.00065-17
Debesa-Tur, G., Pérez-Brocal, V., Ruiz-Ruiz, S., Castillejo, A., Latorre, A., Soto, J. L., et al. (2021). Metagenomic analysis of formalin-fixed paraffin-embedded tumor and normal mucosa reveals differences in the microbiome of colorectal cancer patients. Sci. Rep. 11:391. doi: 10.1038/s41598-020-79874-y
DeFilipp, Z., Peled, J. U., Li, S., Mahabamunuge, J., Dagher, Z., Slingerland, A. E., et al. (2018). Third-party fecal microbiota transplantation following allo-HCT reconstitutes microbiome diversity. Blood Adv. 2, 745–753. doi: 10.1182/bloodadvances.2018017731
Dejea, C. M., Wick, E. C., Hechenbleikner, E. M., White, J. R., Mark Welch, J. L., Rossetti, B. J., et al. (2014). Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 111, 18321–18326. doi: 10.1073/pnas.1406199111
Diaz, P. I., Hong, B. Y., Dupuy, A. K., Choquette, L., Thompson, A., Salner, A. L., et al. (2019). Integrated analysis of clinical and microbiome risk factors associated with the development of oral candidiasis during cancer chemotherapy. J. Fungi (Basel). 5:49. doi: 10.3390/jof5020049
Dong, M., Meng, Z., Kuerban, K., Qi, F., Liu, J., Wei, Y., et al. (2018). Diosgenin promotes antitumor immunity and PD-1 antibody efficacy against melanoma by regulating intestinal microbiota. Cell Death Dis. 9:1039. doi: 10.1038/s41419-018-1099-3
Dong, T., Feng, Q., Liu, F., Chang, L. K., Zhou, X., Han, M., et al. (2017). Alteration of stomach microbiota compositions in the progression of gastritis induces nitric oxide in gastric cell. Exp. Ther. Med. 13, 2793–2800. doi: 10.3892/etm.2017.4373
Dubey, R. K., Tripathi, V., Prabha, R., Chaurasia, R., Singh, D. P., Rao, C. S., (2020). Unravelling the Soil Microbiome: Perspectives For Environmental Sustainability. Cham: Springer International Publishing.
Dubin, K. A., Mathur, D., McKenney, P. T., Taylor, B. P., Littmann, E. R., Peled, J. U., et al. (2019). Diversification and evolution of vancomycin-resistant enterococcus faecium during intestinal domination. Infect. Immun. 87:e00102–19. doi: 10.1128/iai.00102-19
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C. (2016). UNCROSS: filtering of high-frequency cross-talk in 16S amplicon reads. bioRxiv 088666. doi: 10.1101/088666
Ehrenreich, A. (2006). DNA microarray technology for the microbiologist: an overview. Appl. Microbiol. Biotechnol. 73, 255–273. doi: 10.1007/s00253-006-0584-2
Erawijantari, P. P., Mizutani, S., Shiroma, H., Shiba, S., Nakajima, T., Sakamoto, T., et al. (2020). Influence of gastrectomy for gastric cancer treatment on faecal microbiome and metabolome profiles. Gut 69, 1404–1415. doi: 10.1136/gutjnl-2019-319188
Eun, C. S., Kim, B. K., Han, D. S., Kim, S. Y., Kim, K. M., Choi, B. Y., et al. (2014). Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter 19, 407–416. doi: 10.1111/hel.12145
Fan, X., Alekseyenko, A. V., Wu, J., Peters, B. A., Jacobs, E. J., Gapstur, S. M., et al. (2018). Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut 67, 120–127. doi: 10.1136/gutjnl-2016-312580
Feng, Q., Liang, S., Jia, H., Stadlmayr, A., Tang, L., Lan, Z., et al. (2015). Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 6:6528. doi: 10.1038/ncomms7528
Feng, Y., Ramnarine, V. R., Bell, R., Volik, S., Davicioni, E., Hayes, V. M., et al. (2019). Metagenomic and metatranscriptomic analysis of human prostate microbiota from patients with prostate cancer. BMC Genomics 20:146. doi: 10.1186/s12864-019-5457-z
Frankel, A. E., Coughlin, L. A., Kim, J., Froehlich, T. W., Xie, Y., Frenkel, E. P., et al. (2017). Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 19, 848–855. doi: 10.1016/j.neo.2017.08.004
Franzosa, E. A., Morgan, X. C., Segata, N., Waldron, L., Reyes, J., Earl, A. M., et al. (2014). Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. U.S.A. 111, E2329–E2338. doi: 10.1073/pnas.1319284111
Fredricks, D. N., Fiedler, T. L., and Marrazzo, J. M. (2005). Molecular identification of bacteria associated with bacterial vaginosis. N. Engl. J. Med. 353, 1899–1911. doi: 10.1056/NEJMoa043802
Galloway-Peña, J. R., Smith, D. P., Sahasrabhojane, P., Ajami, N. J., Wadsworth, W. D., Daver, N. G., et al. (2016). The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer 122, 2186–2196. doi: 10.1002/cncr.30039
Ganzenmueller, T., Hage, E., Yakushko, Y., Kluba, J., Woltemate, S., Schacht, V., et al. (2013). No human virus sequences detected by next-generation sequencing in benign verrucous skin tumors occurring in BRAF-inhibitor-treated patients. Exp. Dermatol. 22, 725–729. doi: 10.1111/exd.12249
Gao, L., Hu, Y., Wang, Y., Jiang, W., He, Z., Zhu, C., et al. (2015). Exploring the variation of oral microbiota in supragingival plaque during and after head-and-neck radiotherapy using pyrosequencing. Arch. Oral Biol. 60, 1222–1230. doi: 10.1016/j.archoralbio.2015.05.006
Garland, J. L., and Mills, A. L. (1991). Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl. Environ. Microbiol. 57, 2351–2359. doi: 10.1128/aem.57.8.2351-2359.1991
Gilbert, J. A., Quinn, R. A., Debelius, J., Xu, Z. Z., Morton, J., Garg, N., et al. (2016). Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 535, 94–103. doi: 10.1038/nature18850
Gilbreath, J. J., Semino-Mora, C., Friedline, C. J., Liu, H., Bodi, K. L., McAvoy, T. J., et al. (2013). A core microbiome associated with the peritoneal tumors of Pseudomyxoma peritonei. Orphanet J. Rare Dis. 8:105. doi: 10.1186/1750-1172-8-105
Gong, H., Shi, Y., Xiao, X., Wu, C., Tao, L., Hou, D., et al. (2017). Alterations of microbiota structure in the larynx relevant to laryngeal carcinoma. Sci. Rep. 7:5507. doi: 10.1038/s41598-017-05576-7
Gopalakrishnan, V., Spencer, C. N., Nezi, L., Reuben, A., Andrews, M. C., Karpinets, T. V., et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. doi: 10.1126/science.aan4236
Hakim, H., Dallas, R., Wolf, J., Tang, L., Schultz-Cherry, S., Darling, V., et al. (2018). Gut microbiome composition predicts infection risk during chemotherapy in children with acute lymphoblastic leukemia. Clin. Infect. Dis. 67, 541–548. doi: 10.1093/cid/ciy153
Han, S., Pan, Y., Yang, X., Da, M., Wei, Q., Gao, Y., et al. (2019). Intestinal microorganisms involved in colorectal cancer complicated with dyslipidosis. Cancer Biol. Ther. 20, 81–89. doi: 10.1080/15384047.2018.1507255
Harris, B., Morjaria, S. M., Littmann, E. R., Geyer, A. I., Stover, D. E., Barker, J. N., et al. (2016). Gut microbiota predict pulmonary infiltrates after allogeneic hematopoietic cell transplantation. Am. J. Respir. Crit. Care Med. 194, 450–463. doi: 10.1164/rccm.201507-1491OC
Helmink, B. A., Khan, M. A. W., Hermann, A., Gopalakrishnan, V., and Wargo, J. A. (2019). The microbiome, cancer, and cancer therapy. Nat. Med. 25, 377–388. doi: 10.1038/s41591-019-0377-7
Herstad, K. M. V., Moen, A. E. F., Gaby, J. C., Moe, L., and Skancke, E. (2018). Characterization of the fecal and mucosa-associated microbiota in dogs with colorectal epithelial tumors. PLoS ONE 13:e0198342. doi: 10.1371/journal.pone.0198342
Heshiki, Y., Vazquez-Uribe, R., Li, J., Ni, Y., Quainoo, S., Imamovic, L., et al. (2020). Predictable modulation of cancer treatment outcomes by the gut microbiota. Microbiome 8:28. doi: 10.1186/s40168-020-00811-2
Hu, H., Cui, L., Lu, J., Wei, K., Wei, J., Li, S., et al. (2020). Intestinal microbiota regulates anti-tumor effect of disulfiram combined with Cu(2+) in a mice model. Cancer Med. 9, 6791–6801. doi: 10.1002/cam4.3346
Hu, J., Han, S., Chen, Y., and Ji, Z. (2015). Variations of tongue coating microbiota in patients with gastric cancer. Biomed Res. Int. 2015:173729. doi: 10.1155/2015/173729
Hu, Y. J., Shao, Z. Y., Wang, Q., Jiang, Y. T., Ma, R., Tang, Z. S., et al. (2013a). Exploring the dynamic core microbiome of plaque microbiota during head-and-neck radiotherapy using pyrosequencing. PLoS ONE 8:e56343. doi: 10.1371/journal.pone.0056343
Hu, Y. J., Wang, Q., Jiang, Y. T., Ma, R., Xia, W. W., Tang, Z. S., et al. (2013b). Characterization of oral bacterial diversity of irradiated patients by high-throughput sequencing. Int. J. Oral Sci. 5, 21–25. doi: 10.1038/ijos.2013.15
Hugenholtz, P., Goebel, B. M., and Pace, N. R. (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180, 4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998
Hugerth, L. W., and Andersson, A. F. (2017). Analysing microbial community composition through amplicon sequencing: from sampling to hypothesis testing. Front. Microbiol. 8:1561. doi: 10.3389/fmicb.2017.01561
Ibrahim, A., Hugerth, L. W., Hases, L., Saxena, A., Seifert, M., Thomas, Q., et al. (2019). Colitis-induced colorectal cancer and intestinal epithelial estrogen receptor beta impact gut microbiota diversity. Int. J. Cancer 144, 3086–3098. doi: 10.1002/ijc.32037
Ishoey, T., Woyke, T., Stepanauskas, R., Novotny, M., and Lasken, R. S. (2008). Genomic sequencing of single microbial cells from environmental samples. Curr. Opin. Microbiol. 11, 198–204. doi: 10.1016/j.mib.2008.05.006
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Janssen, P. H., Yates, P. S., Grinton, B. E., Taylor, P. M., and Sait, M. (2002). Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl. Environ. Microbiol. 68, 2391–2396. doi: 10.1128/aem.68.5.2391-2396.2002
Ji, X., Hou, C., Gao, Y., Xue, Y., Yan, Y., and Guo, X. (2020). Metagenomic analysis of gut microbiota modulatory effects of jujube (Ziziphus jujuba Mill.) polysaccharides in a colorectal cancer mouse model. Food Funct. 11, 163–173. doi: 10.1039/C9FO02171J
Jiang, C., Wang, H., Xia, C., Dong, Q., Chen, E., Qiu, Y., et al. (2019). A randomized, double-blind, placebo-controlled trial of probiotics to reduce the severity of oral mucositis induced by chemoradiotherapy for patients with nasopharyngeal carcinoma. Cancer 125, 1081–1090. doi: 10.1002/cncr.31907
Jiang, Y., and Fan, L. (2021). The effect of Poria cocos ethanol extract on the intestinal barrier function and intestinal microbiota in mice with breast cancer. J. Ethnopharmacol. 266:113456. doi: 10.1016/j.jep.2020.113456
Johnson, C. H., Ivanisevic, J., and Siuzdak, G. (2016). Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 17, 451–459. doi: 10.1038/nrm.2016.25
Kaeberlein, T., Lewis, K., and Epstein, S. S. (2002). Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296, 1127–1129. doi: 10.1126/science.1070633
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kang, G. U., Jung, D. R., Lee, Y. H., Jeon, S. Y., Han, H. S., Chong, G. O., et al. (2020). Dynamics of fecal microbiota with and without invasive cervical cancer and its application in early diagnosis. Cancers (Basel). 12:3800. doi: 10.3390/cancers12123800
Kasai, C., Sugimoto, K., Moritani, I., Tanaka, J., Oya, Y., Inoue, H., et al. (2016). Comparison of human gut microbiota in control subjects and patients with colorectal carcinoma in adenoma: terminal restriction fragment length polymorphism and next-generation sequencing analyses. Oncol. Rep. 35, 325–333. doi: 10.3892/or.2015.4398
Kau, A. L., Ahern, P. P., Griffin, N. W., Goodman, A. L., and Gordon, J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336. doi: 10.1038/nature10213
Kim, B., Cho, E. J., Yoon, J. H., Kim, S. S., Cheong, J. Y., Cho, S. W., et al. (2020). Pathway-based integrative analysis of metabolome and microbiome data from hepatocellular carcinoma and liver cirrhosis patients. Cancers (Basel) 12:2705. doi: 10.3390/cancers12092705
Klein, C., Gonzalez, D., Samwel, K., Kahesa, C., Mwaiselage, J., Aluthge, N., et al. (2019). Relationship between the cervical microbiome, HIV status, and precancerous lesions. mBio 10:e02785–18. doi: 10.1128/mBio.02785-18
Kong, C., Gao, R., Yan, X., Huang, L., He, J., Li, H., et al. (2019). Alterations in intestinal microbiota of colorectal cancer patients receiving radical surgery combined with adjuvant CapeOx therapy. Sci. China Life Sci. 62, 1178–1193. doi: 10.1007/s11427-018-9456-x
Kwasniewski, W., Wolun-Cholewa, M., Kotarski, J., Warchol, W., Kuzma, D., Kwasniewska, A., et al. (2018). Microbiota dysbiosis is associated with HPV-induced cervical carcinogenesis. Oncol. Lett. 16, 7035–7047. doi: 10.3892/ol.2018.9509
Laguerre, G., Allard, M.-R., Revoy, F., and Amarger, N. (1994). Rapid identification of rhizobia by restriction fragment length polymorphism analysis of PCR-amplified 16S rRNA genes. Appl. Environ. Microbiol. 60, 56–63.
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lasken, R. S. (2007). Single-cell genomic sequencing using multiple displacement amplification. Curr. Opin. Microbiol. 10, 510–516. doi: 10.1016/j.mib.2007.08.005
Lee, S. H., Sung, J. Y., Yong, D., Chun, J., Kim, S. Y., Song, J. H., et al. (2016). Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 102, 89–95. doi: 10.1016/j.lungcan.2016.10.016
Leung, P. H. M., Subramanya, R., Mou, Q., Lee, K. T., Islam, F., Gopalan, V., et al. (2019). Characterization of mucosa-associated microbiota in matched cancer and non-neoplastic mucosa from patients with colorectal cancer. Front. Microbiol. 10:1317. doi: 10.3389/fmicb.2019.01317
Li, D., Xi, W., Zhang, Z., Ren, L., Deng, C., Chen, J., et al. (2020). Oral microbial community analysis of the patients in the progression of liver cancer. Microb. Pathog. 149, 104479. doi: 10.1016/j.micpath.2020.104479
Liang, W., Yang, Y., Wang, H., Wang, H., Yu, X., Lu, Y., et al. (2019). Gut microbiota shifts in patients with gastric cancer in perioperative period. Medicine (Baltimore). 98:e16626. doi: 10.1097/MD.0000000000016626
Liang, X., Li, H., Tian, G., and Li, S. (2014). Dynamic microbe and molecule networks in a mouse model of colitis-associated colorectal cancer. Sci. Rep. 4:4985. doi: 10.1038/srep04985
Liu, M., Xie, W., Wan, X., and Deng, T. (2020). Clostridium butyricum modulates gut microbiota and reduces colitis associated colon cancer in mice. Int. Immunopharmacol. 88:106862. doi: 10.1016/j.intimp.2020.106862
Liu, Y.-X., Qin, Y., Chen, T., Lu, M., Qian, X., Guo, X., et al. (2020). A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 12, 315–330. doi: 10.1007/s13238-020-00724-8
Loke, M. F., Chua, E. G., Gan, H. M., Thulasi, K., Wanyiri, J. W., Thevambiga, I., et al. (2018). Metabolomics and 16S rRNA sequencing of human colorectal cancers and adjacent mucosa. PLoS ONE 13:e0208584. doi: 10.1371/journal.pone.0208584
Lu, H., Ren, Z., Li, A., Zhang, H., Jiang, J., Xu, S., et al. (2016). Deep sequencing reveals microbiota dysbiosis of tongue coat in patients with liver carcinoma. Sci. Rep. 6:33142. doi: 10.1038/srep33142
Luo, W., Cao, Z., Qiu, J., Liu, Y., Zheng, L., and Zhang, T. (2020). Novel discoveries targeting pathogenic gut microbes and new therapies in pancreatic cancer: does pathogenic E. coli infection cause pancreatic cancer progression modulated by TUBB/Rho/ROCK signaling pathway? A bioinformatic analysis. Biomed. Res. Int. 2020:2340124. doi: 10.1155/2020/2340124
Lynch, S. V., and Pedersen, O. (2016). The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369–2379. doi: 10.1056/NEJMra1600266
Mai, G., Chen, L., Li, R., Liu, Q., Zhang, H., and Ma, Y. (2019). Common core bacterial biomarkers of bladder cancer based on multiple datasets. Biomed. Res. Int. 2019:4824909. doi: 10.1155/2019/4824909
Marchesi, J. R., and Ravel, J. (2015). The vocabulary of microbiome research: a proposal. Microbiome 3:31. doi: 10.1186/s40168-015-0094-5
Matson, V., Fessler, J., Bao, R., Chongsuwat, T., Zha, Y., Alegre, M. L., et al. (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science359, 104–108. doi: 10.1126/science.aao3290
Meng, S., Chen, B., Yang, J., Wang, J., Zhu, D., Meng, Q., et al. (2018). Study of microbiomes in aseptically collected samples of human breast tissue using needle biopsy and the potential role of in situ tissue microbiomes for promoting malignancy. Front. Oncol. 8:318. doi: 10.3389/fonc.2018.00318
Moen, B., Henjum, K., Måge, I., Knutsen, S. H., Rud, I., Hetland, R. B., et al. (2016). Effect of dietary fibers on cecal microbiota and intestinal tumorigenesis in azoxymethane treated A/J Min/+ mice. PLoS ONE 11:e0155402. doi: 10.1371/journal.pone.0155402
Montassier, E., Al-Ghalith, G. A., Ward, T., Corvec, S., Gastinne, T., Potel, G., et al. (2016). Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome Med. 8:49. doi: 10.1186/s13073-016-0301-4
Montassier, E., Batard, E., Massart, S., Gastinne, T., Carton, T., Caillon, J., et al. (2014). 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb. Ecol. 67, 690–699. doi: 10.1007/s00248-013-0355-4
Montassier, E., Gastinne, T., Vangay, P., Al-Ghalith, G. A., Bruley des Varannes, S., Massart, S., et al. (2015). Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment. Pharmacol. Ther. 42, 515–528. doi: 10.1111/apt.13302
Morgan, H. H., du Toit, M., and Setati, M. E. (2017). The grapevine and wine microbiome: insights from high-throughput amplicon sequencing. Front. Microbiol. 8:820. doi: 10.3389/fmicb.2017.00820
Moskowitz, J. E., Doran, A. G., Lei, Z., Busi, S. B., Hart, M. L., Franklin, C. L., et al. (2020). Integration of genomics, metagenomics, and metabolomics to identify interplay between susceptibility alleles and microbiota in adenoma initiation. BMC Cancer 20:600. doi: 10.1186/s12885-020-07007-9
Nejman, D., Livyatan, I., Fuks, G., Gavert, N., Zwang, Y., Geller, L. T., et al. (2020). The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980. doi: 10.1126/science.aay9189
Ni, J., Huang, R., Zhou, H., Xu, X., Li, Y., Cao, P., et al. (2019). Analysis of the relationship between the degree of dysbiosis in gut microbiota and prognosis at different stages of primary hepatocellular carcinoma. Front. Microbiol. 10:1458. doi: 10.3389/fmicb.2019.01458
Pace, N. R. (1997). A molecular view of microbial diversity and the biosphere. Science 276, 734–740. doi: 10.1126/science.276.5313.734
Pace, N. R., Stahl, D. A., Lane, D. J., and Olsen, G. J. (1986). The analysis of natural microbial populations by ribosomal RNA sequences, in Advances in Microbial Ecology. Advances in Microbial Ecology, Vol. 9, ed K. C. Marshall (Boston, MA: Springer), 1–55. doi: 10.1007/978-1-4757-0611-6_1
Pal, S. K., Li, S. M., Wu, X., Qin, H., Kortylewski, M., Hsu, J., et al. (2015). Stool bacteriomic profiling in patients with metastatic renal cell carcinoma receiving vascular endothelial growth factor-tyrosine kinase inhibitors. Clin. Cancer Res. 21, 5286–5293. doi: 10.1158/1078-0432.CCR-15-0724
Pan, F., Xu, X., Zhang, L.-L., Luo, H.-J., Chen, Y., Long, L., et al. (2020). Dietary riboflavin deficiency induces genomic instability of esophageal squamous cells that is associated with gut microbiota dysbiosis in rats. Food Funct. 11, 10070–10083. doi: 10.1039/D0FO01944E
Parsonnet, J., Friedman, G. D., Vandersteen, D. P., Chang, Y., Vogelman, J. H., Orentreich, N., et al. (1991). Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325, 1127–1131. doi: 10.1056/nejm199110173251603
Peled, J. U., Gomes, A. L. C., Devlin, S. M., Littmann, E. R., Taur, Y., Sung, A. D., et al. (2020). Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N. Engl. J. Med. 382, 822–834. doi: 10.1056/NEJMoa1900623
Perera, M., Al-Hebshi, N. N., Perera, I., Ipe, D., Ulett, G. C., Speicher, D. J., et al. (2018). Inflammatory bacteriome and oral squamous cell carcinoma. J. Dent. Res. 97, 725–732. doi: 10.1177/0022034518767118
Pérez-Losada, M., Arenas, M., and Castro-Nallar, E. (2018). Microbial sequence typing in the genomic era. Infect. Genet. Evol. 63, 346–359. doi: 10.1016/j.meegid.2017.09.022
Peters, B. A., Wilson, M., Moran, U., Pavlick, A., Izsak, A., Wechter, T., et al. (2019). Relating the gut metagenome and metatranscriptome to immunotherapy responses in melanoma patients. Genome Med. 11:61. doi: 10.1186/s13073-019-0672-4
Prodan, A., Tremaroli, V., Brolin, H., Zwinderman, A. H., Nieuwdorp, M., and Levin, E. (2020). Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 15:e0227434. doi: 10.1371/journal.pone.0227434
Qi, H., Liu, Y., Qi, X., Liang, H., Chen, H., Jiang, P., et al. (2019). Dietary recombinant phycoerythrin modulates the gut microbiota of H22 tumor-bearing mice. Mar. Drugs 17:665. doi: 10.3390/md17120665
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35, 833–844. doi: 10.1038/nbt.3935
Rehman, A., Sina, C., Gavrilova, O., Häsler, R., Ott, S., Baines, J. F., et al. (2011). Nod2 is essential for temporal development of intestinal microbial communities. Gut 60, 1354–1362. doi: 10.1136/gut.2010.216259
Reuter, J. A., Spacek, D. V., and Snyder, M. P. (2015). High-throughput sequencing technologies. Mol. Cell 58, 586–597. doi: 10.1016/j.molcel.2015.05.004
Routy, B., Le Chatelier, E., Derosa, L., Duong, C. P. M., Alou, M. T., Daillère, R., et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. doi: 10.1126/science.aan3706
Salava, A., Aho, V., Pereira, P., Koskinen, K., Paulin, L., Auvinen, P., et al. (2016). Skin microbiome in melanomas and melanocytic nevi. Eur. J. Dermatol. 26, 49–55. doi: 10.1684/ejd.2015.2696
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74, 5463–5467. doi: 10.1073/pnas.74.12.5463
Scanlan, P. D., and Marchesi, J. R. (2008). Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J. 2, 1183–1193. doi: 10.1038/ismej.2008.76
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Schwartz, D. J., Langdon, A. E., and Dantas, G. (2020). Understanding the impact of antibiotic perturbation on the human microbiome. Genome Med. 12:82. doi: 10.1186/s13073-020-00782-x
Sender, R., Fuchs, S., and Milo, R. (2016). Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 14:e1002533. doi: 10.1371/journal.pbio.1002533
Shah, M. S., DeSantis, T. Z., Weinmaier, T., McMurdie, P. J., Cope, J. L., Altrichter, A., et al. (2018). Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut 67, 882–891. doi: 10.1136/gutjnl-2016-313189
Sharon, G., Sampson, T. R., Geschwind, D. H., and Mazmanian, S. K. (2016). The central nervous system and the gut microbiome. Cell 167, 915–932. doi: 10.1016/j.cell.2016.10.027
Shea, A., Wolcott, M., Daefler, S., and Rozak, D. A. (2012). Biolog phenotype microarrays. Methods Mol. Biol. 881, 331–373. doi: 10.1007/978-1-61779-827-6_12
Shelburne, S. A., Ajami, N. J., Chibucos, M. C., Beird, H. C., Tarrand, J., Galloway-Pena, J., et al. (2015). Implementation of a pan-genomic approach to investigate holobiont-infecting microbe interaction: a case report of a leukemic patient with invasive mucormycosis. PLoS ONE 10:e0139851. doi: 10.1371/journal.pone.0139851
Shen, J., Li, P., Liu, S., Liu, Q., Li, Y., Zhang, Z., et al. (2020). The chemopreventive effects of Huangqin-tea against AOM-induced preneoplastic colonic aberrant crypt foci in rats and omics analysis. Food Funct 11, 9634–9650. doi: 10.1039/D0FO01731K
Smibert, O. C., Guo, C. W., Khoo, C., Thursky, K. A., Sandhu, S., and Slavin, M. A. (2019). Microbiome transplantation and modulation of immune related adverse events. EClinicalMedicine 8, 10–11. doi: 10.1016/j.eclinm.2019.03.003
Song, Y., and Gyarmati, P. (2020). Microbiota changes in a pediatric acute lymphocytic leukemia mouse model. Microbiologyopen 9:e982. doi: 10.1002/mbo3.982
Stewart, E. J. (2012). Growing unculturable bacteria. J. Bacteriol. 194, 4151–4160. doi: 10.1128/JB.00345-12
Stewart, L. A., Clarke, M., Rovers, M., Riley, R. D., Simmonds, M., Stewart, G., et al. (2015). Preferred reporting items for systematic review and meta-analyses of individual participant data: the PRISMA-IPD statement. JAMA 313, 1657–1665. doi: 10.1001/jama.2015.3656
Stojanovska, V., McQuade, R. M., Fraser, S., Prakash, M., Gondalia, S., Stavely, R., et al. (2018). Oxaliplatin-induced changes in microbiota, TLR4+ cells and enhanced HMGB1 expression in the murine colon. PLoS ONE. 13:e0198359. doi: 10.1371/journal.pone.0198359
Stolte, M. (1992). Helicobacter pylori gastritis and gastric MALT-lymphoma. Lancet 339, 745–746. doi: 10.1016/0140-6736(92)90645-j
Su, C., Lei, L., Duan, Y., Zhang, K. Q., and Yang, J. (2012). Culture-independent methods for studying environmental microorganisms: methods, application, and perspective. Appl. Microbiol. Biotechnol. 93, 993–1003. doi: 10.1007/s00253-011-3800-7
Sun, H., Zhao, X., Zhou, Y., Wang, J., Ma, R., Ren, X., et al. (2020). Characterization of oral microbiome and exploration of potential biomarkers in patients with pancreatic cancer. Biomed Res. Int. 2020:4712498. doi: 10.1155/2020/4712498
Sun, J. H., Li, X. L., Yin, J., Li, Y. H., Hou, B. X., and Zhang, Z. (2018). A screening method for gastric cancer by oral microbiome detection. Oncol. Rep. 39, 2217–2224. doi: 10.3892/or.2018.6286
Tanoue, T., Morita, S., Plichta, D. R., Skelly, A. N., Suda, W., Sugiura, Y., et al. (2019). A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605. doi: 10.1038/s41586-019-0878-z
Thomas, A. M., Jesus, E. C., Lopes, A., Aguiar, S. Jr., Begnami, M. D., Rocha, R. M., et al. (2016). Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling. Front. Cell Infect. Microbiol. 6:179. doi: 10.3389/fcimb.2016.00179
Thomas, A. M., Manghi, P., Asnicar, F., Pasolli, E., Armanini, F., Zolfo, M., et al. (2019). Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 667–678. doi: 10.1038/s41591-019-0405-7
Torres, P. J., Fletcher, E. M., Gibbons, S. M., Bouvet, M., Doran, K. S., and Kelley, S. T. (2015). Characterization of the salivary microbiome in patients with pancreatic cancer. PeerJ 3:e1373. doi: 10.7717/peerj.1373
UniProt Consortium. (2014). Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198. doi: 10.1093/nar/gkt1140
Valdes, A. M., Walter, J., Segal, E., and Spector, T. D. (2018). Role of the gut microbiota in nutrition and health. BMJ 361:k2179. doi: 10.1136/bmj.k2179
Van Leewenhoeck, A. (1677). Observations, communicated to the publisher by Mr. Antony van Leewenhoeck, in a dutch letter of the 9th of Octob. 1676. here English'd: concerning little animals by him observed in rain-well-sea- and snow water; as also in water wherein pepper had lain infused. Philos. Trans. 12, 821–831.
Walther-António, M. R., Chen, J., Multinu, F., Hokenstad, A., Distad, T. J., Cheek, E. H., et al. (2016). Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med. 8:122. doi: 10.1186/s13073-016-0368-y
Wang, J., and Jia, H. (2016). Metagenome-wide association studies: fine-mining the microbiome. Nat. Rev. Microbiol. 14, 508–522. doi: 10.1038/nrmicro.2016.83
Wang, H., Guan, L., Li, J., Lai, M., and Wen, X. (2018). The effects of berberine on the gut microbiota in Apc (min/+) mice fed with a high fat diet. Molecules 23:2298. doi: 10.3390/molecules23092298
Wang, K., Huang, Y., Zhang, Z., Liao, J., Ding, Y., Fang, X., et al. (2019). A preliminary study of microbiota diversity in saliva and bronchoalveolar lavage fluid from patients with primary bronchogenic carcinoma. Med. Sci. Monit. 25. 2819–2834. doi: 10.12659/MSM.915332
Wang, L., Yin, G., Guo, Y., Zhao, Y., Zhao, M., Lai, Y., et al. (2019). Variations in oral microbiota composition are associated with a risk of throat cancer. Front. Cell Infect. Microbiol. 9:205. doi: 10.3389/fcimb.2019.00205
Wang, Q., Ye, J., Fang, D., Lv, L., Wu, W., Shi, D., et al. (2020). Multi-omic profiling reveals associations between the gut mucosal microbiome, the metabolome, and host DNA methylation associated gene expression in patients with colorectal cancer. BMC Microbiol. 20(Suppl 1):83. doi: 10.1186/s12866-020-01762-2
Wang, W. J., Zhou, Y. L., He, J., Feng, Z. Q., Zhang, L., Lai, X. B., et al. (2020). Characterizing the composition of intestinal microflora by 16S rRNA gene sequencing. World J. Gastroenterol. 26, 614–626. doi: 10.3748/wjg.v26.i6.614
Wang, X., Ye, T., Chen, W. J., Lv, Y., Hao, Z., Chen, J., et al. (2017). Structural shift of gut microbiota during chemo-preventive effects of epigallocatechin gallate on colorectal carcinogenesis in mice. World J. Gastroenterol. 23, 8128–8139. doi: 10.3748/wjg.v23.i46.8128
Wang, Y., Hoenig, J. D., Malin, K. J., Qamar, S., Petrof, E. O., Sun, J., et al. (2009). 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. ISME J. 3, 944–954. doi: 10.1038/ismej.2009.37
Wang, Y., Xue, J., Zhou, X., You, M., Du, Q., Yang, X., et al. (2014). Oral microbiota distinguishes acute lymphoblastic leukemia pediatric hosts from healthy populations. PLoS ONE 9:e102116. doi: 10.1371/journal.pone.0102116
Warren, J. R., and Marshall, B. (1983). Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1, 1273–1275
Wei, A. L., Li, M., Li, G. Q., Wang, X., Hu, W. M., Li, Z. L., et al. (2020). Oral microbiome and pancreatic cancer. World J. Gastroenterol. 26, 7679–7692. doi: 10.3748/wjg.v26.i48.7679
Weir, T. L., Manter, D. K., Sheflin, A. M., Barnett, B. A., Heuberger, A. L., and Ryan, E. P. (2013). Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE. 8:e70803. doi: 10.1371/journal.pone.0070803
Wilmanski, T., Rappaport, N., Earls, J. C., Magis, A. T., Manor, O., Lovejoy, J., et al. (2019). Blood metabolome predicts gut microbiome α-diversity in humans. Nat. Biotechnol. 37, 1217–1228. doi: 10.1038/s41587-019-0233-9
Wing, A. C., and Kremenchutzky, M. (2019). Multiple sclerosis and faecal microbiome transplantation: are you going to eat that? Benef. Microbes 10, 27–32. doi: 10.3920/bm2018.0029
Wolf, A., Moissl-Eichinger, C., Perras, A., Koskinen, K., Tomazic, P. V., Thurnher, D., et al. (2017). The salivary microbiome as an indicator of carcinogenesis in patients with oropharyngeal squamous cell carcinoma: a pilot study. Sci. Rep. 7:5867. doi: 10.1038/s41598-017-06361-2
Wongsurawat, T., Nakagawa, M., Atiq, O., Coleman, H. N., Jenjaroenpun, P., Allred, J. I., et al. (2019). An assessment of Oxford Nanopore sequencing for human gut metagenome profiling: a pilot study of head and neck cancer patients. J. Microbiol. Methods 166:105739. doi: 10.1016/j.mimet.2019.105739
Wotherspoon, A. C., Ortiz-Hidalgo, C., Falzon, M. R., and Isaacson, P. G. (1991). Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 338, 1175–1176. doi: 10.1016/0140-6736(91)92035-Z
Wu, M., Li, J., An, Y., Li, P., Xiong, W., Li, J., et al. (2019). Chitooligosaccharides prevents the development of colitis-associated colorectal cancer by modulating the intestinal microbiota and mycobiota. Front. Microbiol. 10:2101. doi: 10.3389/fmicb.2019.02101
Wu, P., Zhang, G., Zhao, J., Chen, J., Chen, Y., Huang, W., et al. (2018). Profiling the urinary microbiota in male patients with bladder cancer in China. Front. Cell Infect. Microbiol. 8:167. doi: 10.3389/fcimb.2018.00167
Wu, Y., Shi, L., Li, Q., Wu, J., Peng, W., Li, H., et al. (2019). Microbiota diversity in human colorectal cancer tissues is associated with clinicopathological features. Nutr. Cancer 71, 214–222. doi: 10.1080/01635581.2019.1578394
Xie, Y., Feng, Y., Li, W., Zhan, F., Huang, G., Hu, H., et al. (2020). Revealing the disturbed vaginal micobiota caused by cervical cancer using high-throughput sequencing technology. Front. Cell Infect. Microbiol. 10:538336. doi: 10.3389/fcimb.2020.538336
Xu, J., Xiang, C., Zhang, C., Xu, B., Wu, J., Wang, R., et al. (2019). Microbial biomarkers of common tongue coatings in patients with gastric cancer. Microb. Pathog. 127, 97–105. doi: 10.1016/j.micpath.2018.11.051
Xue, M., Ji, X., Liang, H., Liu, Y., Wang, B., Sun, L., et al. (2018). The effect of fucoidan on intestinal flora and intestinal barrier function in rats with breast cancer. Food Funct. 9, 1214–1223. doi: 10.1039/c7fo01677h
Yachida, S., Mizutani, S., Shiroma, H., Shiba, S., Nakajima, T., Sakamoto, T., et al. (2019). Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976. doi: 10.1038/s41591-019-0458-7
Yamamoto, M. L., Maier, I., Dang, A. T., Berry, D., Liu, J., Ruegger, P. M., et al. (2013). Intestinal bacteria modify lymphoma incidence and latency by affecting systemic inflammatory state, oxidative stress, and leukocyte genotoxicity. Cancer Res. 73, 4222–4232. doi: 10.1158/0008-5472.CAN-13-0022
Yang, Y., Misra, B. B., Liang, L., Bi, D., Weng, W., Wu, W., et al. (2019). Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics 9, 4101–4114. doi: 10.7150/thno.35186
Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F. O., Ludwig, W., Schleifer, K. H., et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645. doi: 10.1038/nrmicro3330
Yost, S., Stashenko, P., Choi, Y., Kukuruzinska, M., Genco, C. A., Salama, A., et al. (2018). Increased virulence of the oral microbiome in oral squamous cell carcinoma revealed by metatranscriptome analyses. Int. J. Oral Sci. 10:32. doi: 10.1038/s41368-018-0037-7
Yu, J., Feng, Q., Wong, S. H., Zhang, D., Liang, Q. Y., Qin, Y., et al. (2017). Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 66, 70–78. doi: 10.1136/gutjnl-2015-309800
Yu, T., Guo, F., Yu, Y., Sun, T., Ma, D., Han, J., et al. (2017). Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170, 548.e16–563.e16. doi: 10.1016/j.cell.2017.07.008
Yu, W., Zhang, J., Chen, Z., Wang, S., Ruan, C., Zhou, W., et al. (2020). Inhibitory effect of a microecological preparation on azoxymethane/dextran sodium sulfate-induced inflammatory colorectal cancer in mice. Front. Oncol. 10:562189. doi: 10.3389/fonc.2020.562189
Yuan, L., Zhang, S., Li, H., Yang, F., Mushtaq, N., Ullah, S., et al. (2018). The influence of gut microbiota dysbiosis to the efficacy of 5-Fluorouracil treatment on colorectal cancer. Biomed. Pharmacother. 108, 184–193. doi: 10.1016/j.biopha.2018.08.165
Zackular, J. P., Rogers, M. A., Ruffin, M. T., and t. Schloss, P. D. (2014). The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. (Phila) 7, 1112–1121. doi: 10.1158/1940-6207.Capr-14-0129
Zeller, G., Tap, J., Voigt, A. Y., Sunagawa, S., Kultima, J. R., Costea, P. I., et al. (2014). Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 10:766. doi: 10.15252/msb.20145645
Zeng, J., Zhang, G., Chen, C., Li, K., Wen, Y., Zhao, J., et al. (2020). Alterations in urobiome in patients with bladder cancer and implications for clinical outcome: a single-institution study. Front. Cell Infect. Microbiol. 10:555508. doi: 10.3389/fcimb.2020.555508
Zhai, B., Ola, M., Rolling, T., Tosini, N. L., Joshowitz, S., Littmann, E. R., et al. (2020). High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat. Med. 26, 59–64. doi: 10.1038/s41591-019-0709-7
Zhang, B., Gu, J., Liu, J., Huang, B., and Li, J. (2019). Fecal microbiota taxonomic shifts in chinese multiple myeloma patients analyzed by Quantitative Polimerase Chain Reaction (QPCR) and 16S rRNA high-throughput sequencing. Med. Sci. Monit. 25, 8269–8280. doi: 10.12659/MSM.919988
Zhang, H., Lan, M., Cui, G., and Zhu, W. (2020). The influence of caerulomycin A on the intestinal microbiota in SD rats. Mar. Drugs 18:277. doi: 10.3390/md18050277
Zhang, L., Wu, Y. N., Chen, T., Ren, C. H., Li, X., and Liu, G. X. (2019). Relationship between intestinal microbial dysbiosis and primary liver cancer. Hepatobiliary Pancreat. Dis. Int. 18, 149–157. doi: 10.1016/j.hbpd.2019.01.002
Zhang, S., Song, X., Jia, J., Zhang, Z., Zhou, H., Fu, H., et al. (2018). Inhibition effect of glycyrrhiza polysaccharide (GCP) on tumor growth through regulation of the gut microbiota composition. J. Pharmacol. Sci. 137, 324–332. doi: 10.1016/j.jphs.2018.03.006
Zhang, W., Wang, H., Zhang, R., Yu, X.-Z., Qian, P.-Y., and Wong, M. H. (2010). Bacterial communities in PAH contaminated soils at an electronic-waste processing center in China. Ecotoxicology 19, 96–104. doi: 10.1007/s10646-009-0393-3
Zhang, Z., Shao, S., Zhang, Y., Jia, R., Hu, X., Liu, H., et al. (2020). Xiaoyaosan slows cancer progression and ameliorates gut dysbiosis in mice with chronic restraint stress and colorectal cancer xenografts. Biomed. Pharmacother. 132:110916. doi: 10.1016/j.biopha.2020.110916
Zheng, C., Chen, T., Wang, Y., Gao, Y., Kong, Y., Liu, Z., et al. (2019). A randomised trial of probiotics to reduce severity of physiological and microbial disorders induced by partial gastrectomy for patients with gastric cancer. J. Cancer 10, 568–576. doi: 10.7150/jca.29072
Zheng, Z., Andersson, A. F., Ye, W., Nyrén, O., Normark, S., and Engstrand, L. (2011). A method for metagenomics of Helicobacter pylori from archived formalin-fixed gastric biopsies permitting longitudinal studies of carcinogenic risk. PLoS ONE 6:e26442. doi: 10.1371/journal.pone.0026442
Zhou, B., Sun, C., Huang, J., Xia, M., Guo, E., Li, N., et al. (2019). The biodiversity composition of microbiome in ovarian carcinoma patients. Sci. Rep. 9:1691. doi: 10.1038/s41598-018-38031-2
Zhu, H., Xu, W. Y., Hu, Z., Zhang, H., Shen, Y., Lu, S., et al. (2017). RNA virus receptor Rig-I monitors gut microbiota and inhibits colitis-associated colorectal cancer. J. Exp. Clin. Cancer Res. 36:2. doi: 10.1186/s13046-016-0471-3
Keywords: microbiome, high-throughput, cancer, microbiome diversity, functional analysis, cancer therapy
Citation: Wei LQ, Cheong IH, Yang GH, Li XG, Kozlakidis Z, Ding L, Liu NN and Wang H (2021) The Application of High-Throughput Technologies for the Study of Microbiome and Cancer. Front. Genet. 12:699793. doi: 10.3389/fgene.2021.699793
Received: 24 April 2021; Accepted: 21 June 2021;
Published: 28 July 2021.
Edited by:
Gong Zhang, Jinan University, ChinaCopyright © 2021 Wei, Cheong, Yang, Li, Kozlakidis, Ding, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Wang, aHVpd2FuZ0BzaHNtdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.