 Jonathan de Fallois1
Jonathan de Fallois1 Ria Schönauer
Ria Schönauer Bernt Popp
Bernt Popp Jan Halbritter
Jan Halbritter
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 25 June 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.682565
This article is part of the Research Topic Next Generation Sequencing (NGS) for Rare Diseases Diagnosis View all 11 articles
Background: Autosomal polycystic kidney disease is distinguished into dominant (ADPKD) and recessive (ARPKD) inheritance usually caused by either monoallelic (PKD1/PKD2) or biallelic (PKHD1) germline variation. Clinical presentations are genotype-dependent ranging from fetal demise to mild chronic kidney disease (CKD) in adults. Additionally, exemptions from dominant and recessive inheritance have been reported in both disorders resulting in respective phenocopies. Here, we comparatively report three young adults with microcystic-hyperechogenic kidney morphology based on unexpected genetic alterations beyond typical inheritance.
Methods: Next-generation sequencing (NGS)-based gene panel analysis and multiplex ligation-dependent probe amplification (MLPA) of PKD-associated genes, familial segregation analysis, and reverse phenotyping.
Results: Three unrelated individuals presented in late adolescence for differential diagnosis of incidental microcystic-hyperechogenic kidneys with preserved kidney and liver function. Upon genetic analysis, we identified a homozygous hypomorphic PKHD1 missense variant causing pseudodominant inheritance in a family, a large monoallelic PKDH1-deletion with atypical transmission, and biallelic PKD1 missense hypomorphs with recessive inheritance.
Conclusion: By this report, we illustrate clinical presentations associated with atypical PKD-gene alterations beyond traditional modes of inheritance. Large monoallelic PKHD1-alterations as well as biallelic hypomorphs of both PKD1 and PKHD1 may lead to mild CKD in the absence of prominent macrocyst formation and functional liver impairment. The long-term renal prognosis throughout life, however, remains undetermined. Increased detection of atypical inheritance challenges our current thinking of disease ontology not only in PKD but also in Mendelian disorders in general.
Polycystic kidney disease (PKD) is a genetically and clinically heterogeneous condition. In recent years, an increasing number of genetic alterations were associated with PKD (McConnachie et al., 2020). Apart from ongoing detection of new disease-causing alleles in known PKD genes, novel disease genes, such as GANAB (Porath et al., 2016), ALG9 (Besse et al., 2019), ALG8 (Besse et al., 2017), DNAJB11 (Cornec-Le Gall et al., 2018a), and DZIP1L (Lu et al., 2017), have been identified. By mode of inheritance, PKD is categorized into autosomal dominant (ADPKD, MIM#173900/MIM#613095) and autosomal recessive (ARPKD, MIM#263200). While ADPKD is a quite frequent adult-onset condition due to monoallelic PKD1 or PKD2 variation, ARPKD is a rare childhood-onset disorder.
With an estimated prevalence of about 1:1,000, ADPKD is indeed the leading genetic cause of end-stage kidney disease (ESKD) among adults (Cornec-Le Gall et al., 2018b; Lanktree et al., 2018). Patients with protein truncating PKD1 variants are at higher risk of developing ESKD early in life than patients with non-truncating PKD1 variants, and PKD2 carriers express an even milder form of ADPKD often without requirement of renal replacement therapies throughout life (Hwang et al., 2016; Bergmann et al., 2018). However, even in patients with truncating PKD1 variants, exceptionally mild renal phenotypes can be found similar to patients with PKD2 disease (Lanktree et al., 2021).
On the contrary, ARPKD affects about 1:20,000 live births and results from biallelic variants in PKHD1, encoding the ciliary protein fibrocystin. Although perinatal manifestation is most common, genetic studies in adults demonstrated that ARPKD is also diagnosed later in life with an almost ADPKD-like phenotype (Cornec-Le Gall et al., 2018b; Schönauer et al., 2020). Similar to ADPKD, genotype–phenotype correlations have been established in ARPKD. While biallelic truncating variants are mostly associated with fetal demise (Bergmann et al., 2005; Erger et al., 2017), combinations of truncating and missense variants can be found in children surviving the first year of life, and biallelic missense variants are commonly associated with milder forms of disease compatible with ESKD in adulthood (Bergmann et al., 2018; Schönauer et al., 2020). Nevertheless, exceptional cases with biallelic truncation surviving the perinatal period (Ebner et al., 2017; Burgmaier et al., 2018) and biallelic missense variants linked to severe courses (Furu et al., 2003; Bergmann et al., 2005) were reported. Other studies suggested an impact of the type of affected PKHD1-protein domain on the clinical phenotype (Furu et al., 2003; Bergmann et al., 2005).
Clinical presentations of ADPKD and ARPKD are hence genotype-dependent ranging from fetal demise to mild chronic kidney disease (CKD) in adults (Bergmann et al., 2018). In addition to CKD, hepatic involvement presents differently in ADPKD and ARPKD. While in ADPKD, cystic liver enlargement overlapping with autosomal dominant polycystic liver disease (ADPLD) is a characteristic hallmark, ARPKD often leads to rather microcystic liver fibrosis and more often results in consecutive liver transplantation (Cornec-Le Gall et al., 2018b).
Besides classic ADPKD or ARPKD, other rare cystic kidney disorders include nephronophthisis-related ciliopathies, based on biallelic variants in multiple genes encoding ciliary and centrosomal proteins (Bergmann et al., 2018; Adamiok-Ostrowska and Piekiełko-Witkowska, 2020).
Before the advent of next-generation sequencing (NGS) techniques, establishing the correct diagnosis was mainly based on clinical presentation and kidney imaging. NGS has since increased diagnostic accuracy, also in PKD, and led to an improved understanding of the underlying disease mechanisms (Obeidova et al., 2020).
While exemptions from dominant and recessive inheritance in ADPKD and ARPKD have been reported before (Mantovani et al., 2020; Obeidova et al., 2020), we aim at illustrating the complexity of PKD genetics through peculiar genetic alterations of both PKD1 and PKHD1 in three young adult females and one male infant. The presented examples demonstrate unusual modes of inheritance and exceptional clinical presentations, challenging the given disease ontology in PKD.
Index patients and their families were recruited from the outpatient clinic for Hereditary Kidney Disorders at the University of Leipzig Medical Center. All families were included upon written informed consent approved by the local Institutional Review Board (IRB) at the University of Leipzig, Germany (IRB00001750; #402/16-ek).
All study participants underwent genetic analysis conducted from blood-derived DNA samples by NGS-based gene-panel analysis and multiplex ligation-dependent probe amplification (MLPA). Gene panels included all known PKD-associated genes displayed in Supplementary Table 1 (PKD1, PKD2, and PKHD1, among others). Further segregation analysis was done by Sanger-sequencing of respective gene variants in available family members. MLPA was used to detect copy number variations (CNVs) and to validate the NGS data set in complex gene regions, ensuring that no structural alteration was overlooked. In silico prediction with combined annotation-dependent depletion score (CADD-PHRED v1.6: 23.80) (Rentzsch et al., 2019) was used to categorize newly detected variants in terms of pathogenicity. Variant analysis and interpretation was carried out in accordance with the criteria established by the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015).
Reverse phenotyping was done in all family members available. Laboratory tests (e.g., serum-creatinine, urea, urine albumin/creatinine ratio) and kidney imaging [renal ultrasound, magnetic resonance imaging (MRI)] were ascertained to derive genotype–phenotype correlations.
The asymptomatic index patient (ID 1, female, 18 years) of Family 1 was from non-consanguineous Romany origin without any former medical conditions. She was seeking medical advice during her first pregnancy at 17 years of age. Pregnancy had been complicated by oligohydramnios and fetal hyperechogenic kidneys upon prenatal ultrasound. After birth, the newborn (ID 1.1) demonstrated small cystic alterations of both kidneys in accordance with infantile ARPKD. Extrarenally, bilateral pneumothoraces but no hepatic involvement were diagnosed in the newborn. Upon closer examination, kidney ultrasound in the mother (ID 1) revealed hyperechogenic parenchyma and microcystic alterations at the age of 18 years (Figure 1). Laboratory analysis showed normal serum creatinine with an increased estimated glomerular filtration rate (eGFR CKD-EPI: 122 ml/min/1.73 m2), mild proteinuria (albumin/creatinine ratio: 174 mg/g creatinine), and normal liver function (Table 1).
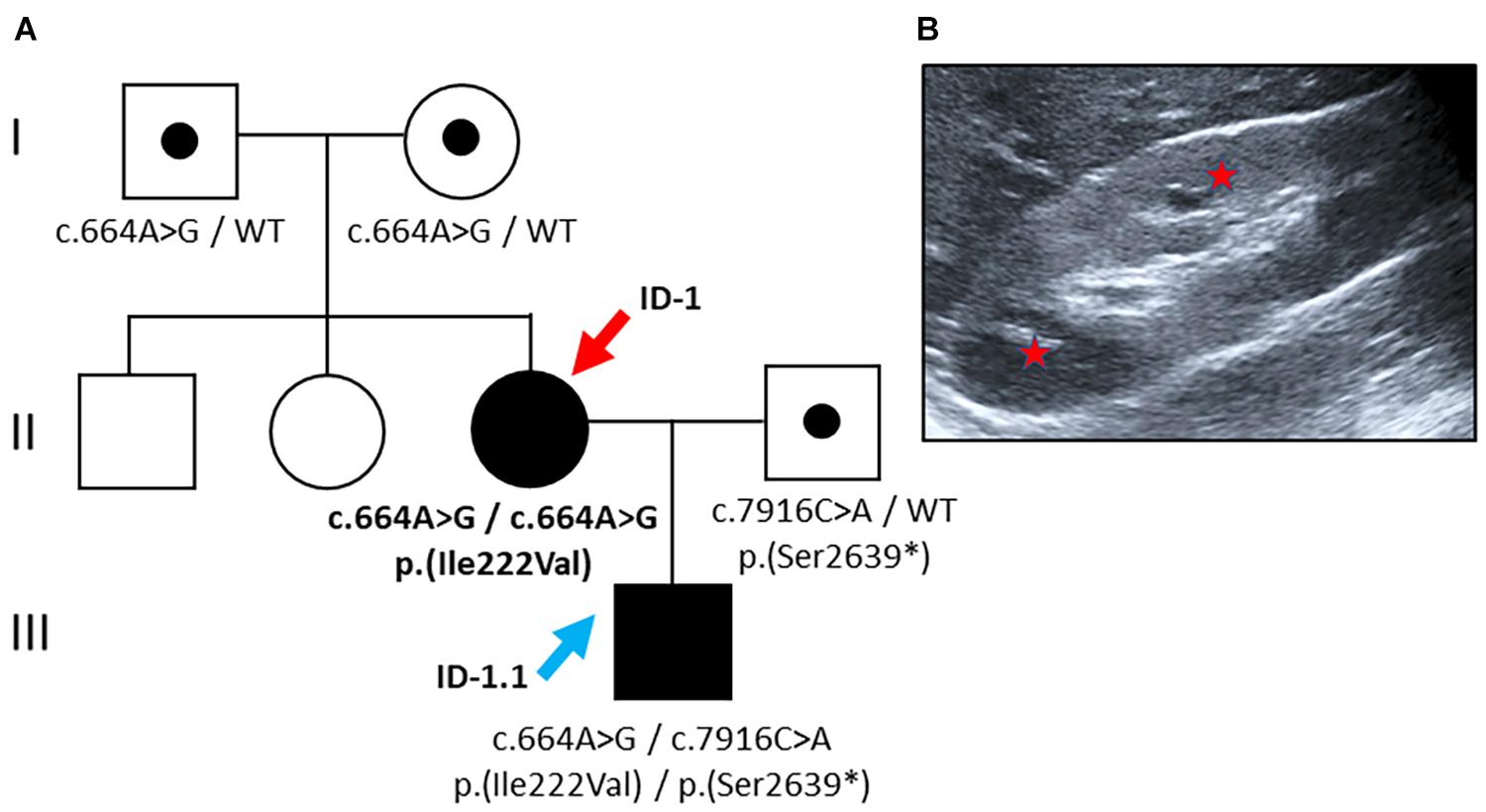
Figure 1. Pedigree and renal imaging (Family 1). (A) Family pedigree: Individuals suffering from PKD are indicated as black symbols, and variant carriers are denoted with a black dot. The female index patient (red arrow) carries a homozygous missense variant [NM_138694.3: c.664A > G, p.(Ile222Val)]. Her son (blue arrow) carries compound heterozygous PKDH1-variants: maternal missense variant plus a putative paternally transmitted pathogenic nonsense variant [NM_138694.3: c.7916C > A, p.(Ser2639∗)]. WT: wild-type. (B) Kidney ultrasound of ID-1 at the age of 18 years displaying mild renal phenotype with hyperechogenic parenchyma and microcystic alterations (red stars highlight small cysts).
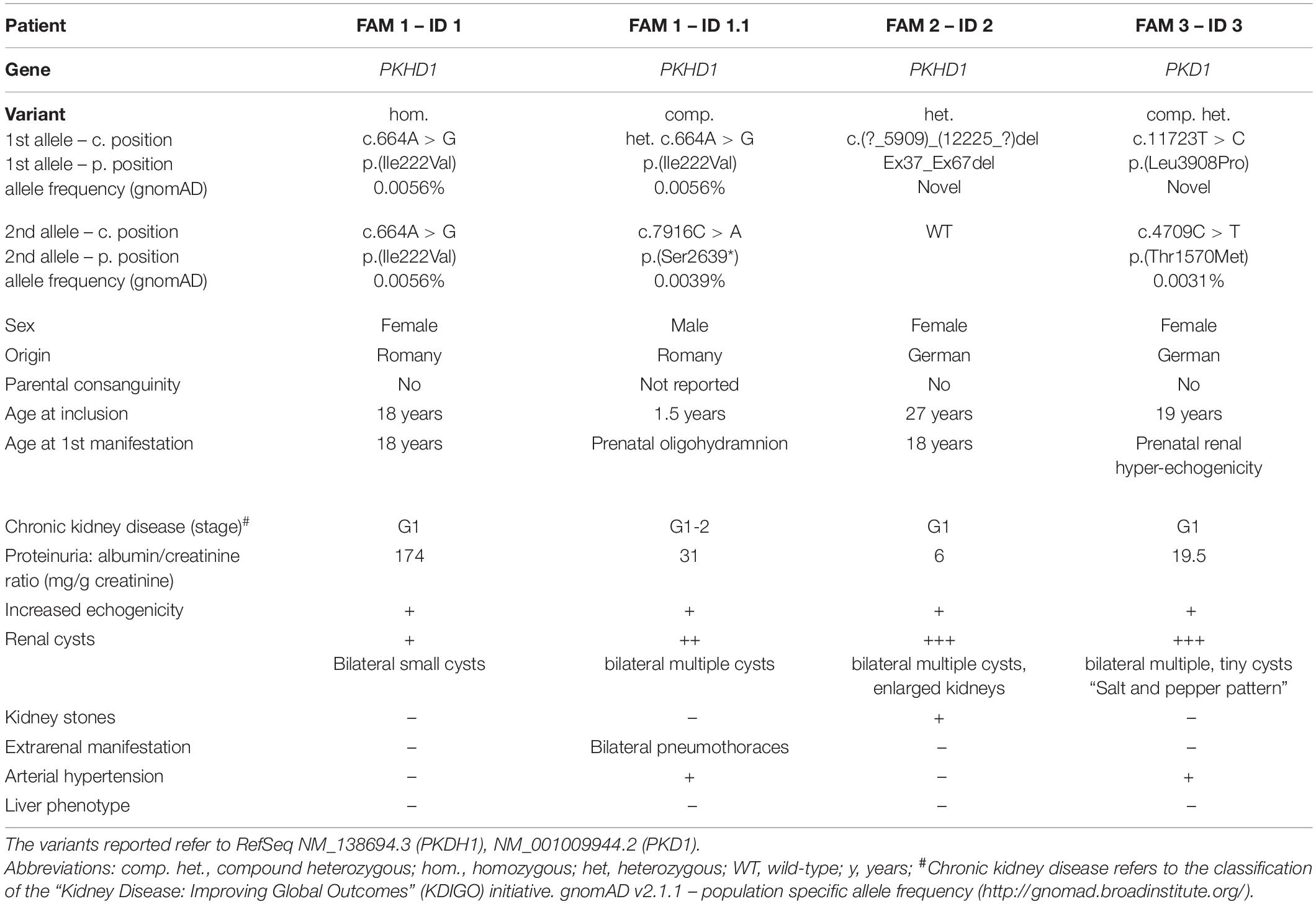
Table 1. Phenotypic and genotypic characteristics of patients included in this case series.
With affected individuals in two subsequent generations, the pedigree suggested dominant inheritance (Figure 1). Genetic analyses of the index patient (ID 1), however, revealed a known pathogenic PKHD1 missense variant [NM_138694.3: c.664A > G, p.(Ile222Val)] in the homozygous state. This variant affects an evolutionary conserved amino acid residue located near the N-terminal extracellular tail of fibrocystin within the second IPT/TIG (Ig-like, plexins, and transcription factors) domain1 (Gunay-Aygun et al., 2010). Allele frequency in Europeans (non-Finish) amounts to 0.0056% with no homozygous cases reported in population databases (gnomAD). In silico prediction using CADD yielded a score of 16.14 (CADD-PHRED v1.6) (Rentzsch et al., 2019), compatible with pathogenicity in a recessive disease model.
Surprisingly, the newborn, who was diagnosed with congenital kidney disease in utero, was found to carry compound heterozygous PKDH1 variants: the aforementioned maternal missense variant [c.664A > G, p.(Ile222Val)] in exon 9 plus another pathogenic PKHD1 nonsense variant in exon 50 out of 67 [NM_138694.3: c.7916C > A, p.(Ser2639∗)] on the paternal allele (CADD-PHRED v1.6: 40.00) (Denamur et al., 2010; Rentzsch et al., 2019). For the latter variant, population allele frequency is estimated at 0.0039% (gnomAD). Its deduced impact on the protein level leads to the introduction of a stop codon after the sixth PbH1 (parallel beta-helix) repeat resulting in the loss of the critical second G8 domain containing eight conserved glycine residues likely important for protein stability (see text foot note 1) (He et al., 2006).
In family 2 from central Europe, the index patient (ID 2; female, 27 years) came to obtain medical advice after previous genetic testing. At the age of 18 years, ID 2 was hospitalized due to repeated urinary tract infections and conspicuous renal morphology with increased parenchymal echogenicity. Abdominal MRI showed symmetric enlargement of both kidneys [diameter (length × width): right: 130 × 60 mm; left: 146 × 72 mm] with small cystic alterations but regular hepatic morphology (Figure 2). At the time of presentation in our clinic, the kidney function was preserved (eGFR CKD-EPI: 130 ml/min/1.73 m2), and no proteinuria was detected (albumin/creatinine ratio: 6 mg/g) (Table 1). The siblings and parents of the patient showed no renal or liver phenotype.
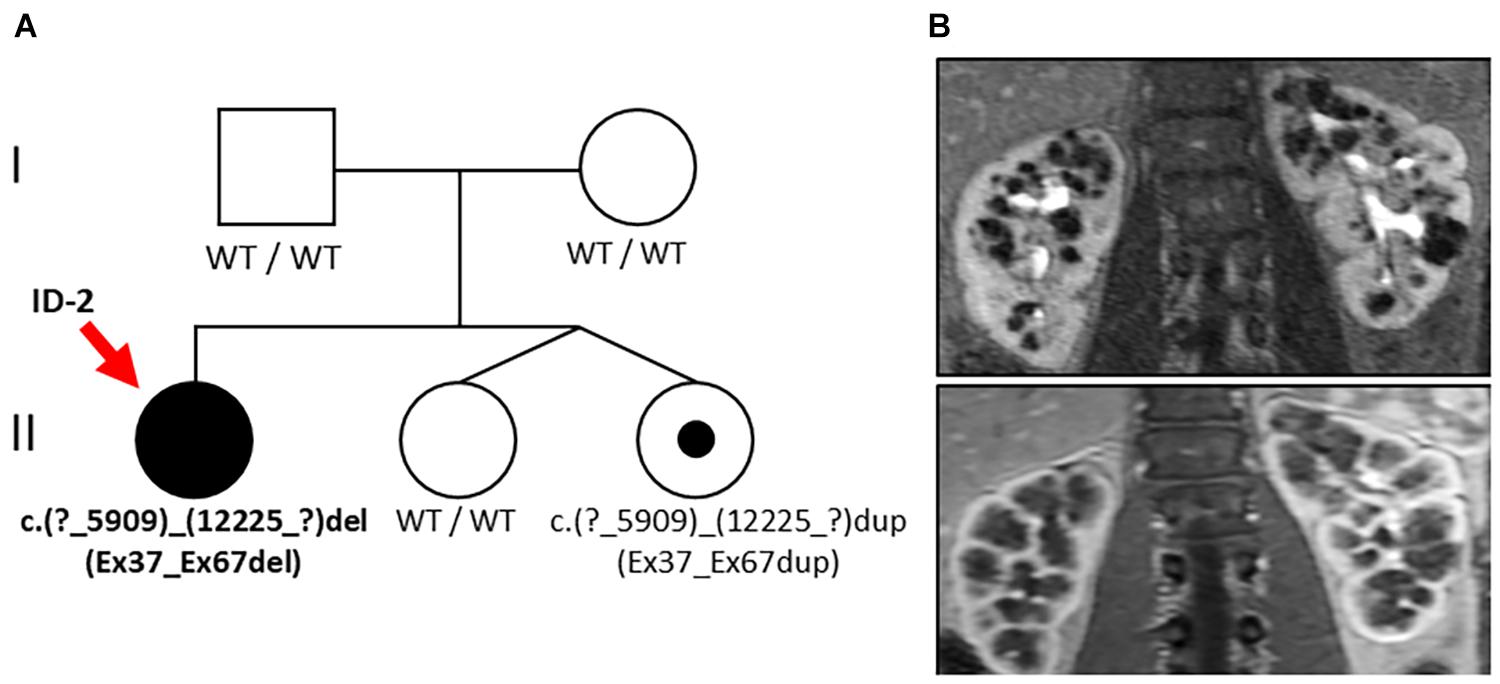
Figure 2. Pedigree and renal imaging (Family 2). (A) Family pedigree: Individuals suffering from PKD are indicated as black symbols and variant carriers with a black dot. Genetic analysis in the index patient (ID-2, red arrow) revealed the large heterozygous deletion NM_138694.3: c.(?_5909)_(12225_?)del of PKHD1 (Ex37_Ex67del) and familial segregation analysis in one younger sibling yielded duplication of the same alteration c.(?_5909)_(12225_?)dup (Ex37_Ex67dup). (B) Abdominal MRI of ID 2 at the age of 18 years showing bilateral cystic kidney enlargement.
Genetic analysis revealed a large heterozygous deletion of PKHD1 c.(?_5909)_(12225_?)del (designated as Ex37_Ex67del) encompassing the extracellular domains of PbH1-9 and one of the critical G8 repeats of fibrocystin close to the cell membrane (see text foot note 1). Familial segregation analysis of both parents showed no genetic alteration of PKHD1, suggesting the de novo nature of this finding. However, further segregation analysis including the younger siblings of ID 2 who were non-identical twins yielded duplication of the same region c.(?_5909)_(12225_?)dup (designated as Ex37_Ex67dup) in one of the twin sisters without clinical manifestation.
This index patient (ID 3, female, 19 years) of central European descent had been congenitally diagnosed with infantile PKD and suspected ARPKD because of early-onset cystic kidneys. The kidney function, however, was completely preserved (eGFR CKD-EPI 133 ml/min/1.73 m2) with merely mild proteinuria (albumin/creatinine ratio: 29 mg/g creatinine) (Table 1). While liver imaging presented normal, kidney ultrasound showed bilaterally enlarged kidneys [diameter (length × width): right: 117 × 52 mm; left: 120 × 57 mm] with increased parenchymal echogenicity and abnormal corticomedullary differentiation (the so-called “salt and pepper” pattern) (Iorio et al., 2020) (Figure 3).
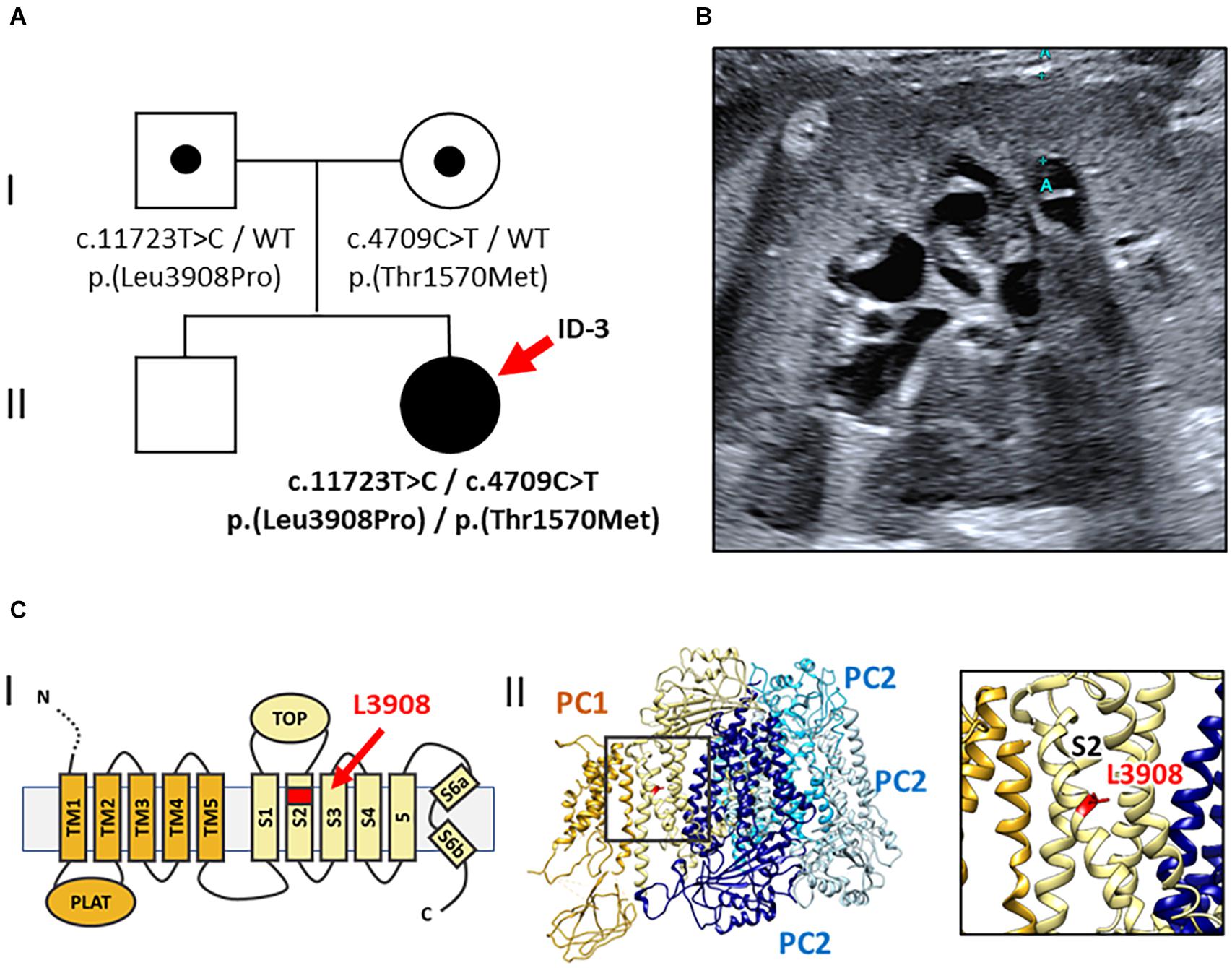
Figure 3. Pedigree, renal imaging, and 3D-PC1-protein structure (Family 3). (A) Family pedigree: ID-3 (red arrow) with compound heterozygous rare PKD1 missense variants both of uncertain significance [NM_001009944.2: c.11723T > C, p.(Leu3908Pro) and c.4709C > T, p.(Thr1570Met)]. WT: wild-type. (B) Kidney ultrasound of ID-3 at the age of 19 years shows enlarged, microcystic-hyperechogenic kidneys. (C) Localization of the novel variant p.(Leu3908Pro) in the PC1 protein structure. (I) Schematic illustration of the PC1 transmembrane region; L3908 is located in transmembrane helix S2 (red arrow). (II) Cryo-EM structure of human PC1/PC2 complex (pdb 6A70) (left) and zoom-in view (right) showing the position of L3908 in helix S2 (red) (Su et al., 2018).
Gene-panel analysis of PKD-associated genes in the index patient yielded two rare heterozygous PKD1 missense variants. Diagnostic variants in PKHD1 or other known kidney cyst genes were not detected. Both PKD1 variants were of uncertain significance by ACMG classification [NM_001009944.2: c.11723T > C, p.(Leu3908Pro) and NM_001009944.2: c.4709C > T, p.(Thr1570Met)] (Kane et al., 2006; Richards et al., 2015). Parental segregation analysis confirmed compound heterozygosity in the index patient with c.11723T > C [p.(Leu3908Pro)] being paternally inherited and c.4709C > T [p.(Thr1570Met)] being maternally transmitted. Of note, clinical examination of both parents was unremarkable, including absence of kidney and liver cysts at the age of 32 years. The first PKD1 variant is novel, neither reported in population databases (gnomAD) nor disease databases [HGMD Professional Version 2020.4/Mayo PKD-database2 (Stenson et al., 2020)]. The respective amino acid residue Leu3908 is evolutionary conserved in vertebrates belonging to the transmembrane helix S2 domain of polycystin 1 (Figure 3)(Su et al., 2018). Introduction of a proline residue into the alpha-helix is likely deleterious, leading to conformational impairments. Hence, in silico prediction using CADD also suggested pathogenicity (CADD-PHRED v1.6: 23.80)(Rentzsch et al., 2019).
The second PKD1 variant [c.4709C > T, p.(Thr1570Met)] was previously reported in the literature (Vujic et al., 2010), where it was found to be associated with in utero onset disease in the homozygous state but incomplete penetrance in heterozygous carriers. Thr1570 constitutes a highly conserved residue in vertebrates and localizes to the PKD11 domain (CADD-PHRED v1.6: 26.30)(Rentzsch et al., 2019). Its allele frequency in Europeans is 0.0031% without homozygous findings in population databases (gnomAD).
Genetic variant interpretation and its clinical correlation has become one of the major challenges of our times. Clinical geneticists and treating physicians are increasingly confronted with new rare variants that require meaningful translation into clinical practice. Furthermore, genetic results often question our traditional categories and force us to take a new perspective on disease classification and terminology. This dilemma is exemplified by the presented cases of three young females with mild cystic kidney alterations illustrating the difficulties and chances of adequate genetic counseling and disease terminology in the era of broadly accessible genetic testing. All index patients presented clinically asymptomatic with preserved kidney function as young adults.
The common denominator is hampered disease prediction through atypical and novel genetic findings with little clinical experience derivable from previously published cases. When it comes to disease ontology, neither single heterozygous PKHD1 germline variants nor biallelic PKD1 hypomorphs do match the given terminology of autosomal-recessive and autosomal-dominant PKD but mimic each other’s renal manifestation. Therefore, common risk stratification tools like the Mayo imaging classification (Irazabal et al., 2015) or the PROPKD-score (Cornec-Le Gall et al., 2016) are not applicable, complicating genetic counseling and decision-making in respective families.
The pedigree of family 1 revealed affected family members in two subsequent generations suggesting dominant inheritance. Genetic testing, however, showed PKHD1 variants defining ARPKD and thereby led to correction of previous assumptions. While pseudodominant inheritance was reported in other Mendelian kidney diseases, e.g., Gitelman’s disease (de La Faille et al., 2011), this is the first report of pseudodominant inheritance in ARPKD. The two affected family members, the index patient (Fam 1 – ID 1) and her newborn son (ID 1.1), impressively illustrate the broad clinical spectrum of ARPKD ranging from severe congenital manifestation to mild CKD in adults depending on the nature and combination of respective PKHD1 alleles (Burgmaier et al., 2019). The homozygous pathogenic PKHD1 missense variant [p.(Ile222Val)] in the index patient leads to a mild course of disease potentially even compatible with normal life expectancy. The very missense variant has been reported frequently in compound heterozygous state in patients with ARPKD before (Bergmann et al., 2005; Denamur et al., 2010; Gunay-Aygun et al., 2010; Brinkert et al., 2013; Obeidova et al., 2020), but has not yet been reported homozygously to the best of our knowledge. Therefore, long-term prognoses remain speculative. Asymptomatic presentation with normal renal and liver function at age 18 suggests that biallelic occurrence of the Ile222Val missense variant is linked to a mild renal phenotype and that only additional second/third hits result in more severe courses of ARPKD. This theory is supported by the clinical presentation of her newborn carrying an additional known PKHD1 null allele (p.Ser2639∗), which is obviously associated with aggravated disease severity (Denamur et al., 2010; Melchionda et al., 2016; Szabó et al., 2018).
The second family represents another example of mild ARPKD in adults. This time, the index patient (Fam 2 – ID 2) was found to carry a large heterozygous deletion in PKHD1 (Ex37_Ex67del). As clinically healthy parents showed PKHD1 wild-type sequences, de novo nature of the deletion was assumed until further segregation analysis in the sister yielded duplication of the very same region without clinical manifestation. This unexpected finding leads to verification of our initial assumption and established yet another mechanism of inheritance in terms of recurrent unequal meiotic crossovers or an unbalanced cryptic chromosomal translocation. This case demonstrates sporadic manifestation of ARPKD based on single heterozygous alterations of PKHD1, an observation that was increasingly reported recently (Besse et al., 2017). Additionally, data in mice suggest that heterozygous PKHD1 carriers may develop ARPKD-associated liver cysts only later in life after the occurrence of a second hit affecting the PKHD1 wild-type allele in somatic tissues (Shan et al., 2019; Besse et al., 2020). However, given the large size and complex exon structure of PKHD1, we cannot exclude a deep intronic splice variant in trans with the herein described deletion as an alternative explanation (Michel-Calemard et al., 2009; Chen et al., 2019). PKHD1 is not sufficiently expressed in blood to be easily accessible to RNA sequencing approaches, but future analyses from cultured urinary renal epithelial cells might enable screening for these rare variants in atypical cases with single PKHD1 variants.
Lastly, the third case is an example for biallelic hypomorphic PKD1 variants mimicking ARPKD. First, the index patient presented in childhood with enlarged kidneys and morphological signs of ARPKD upon kidney ultrasound (Iorio et al., 2020); second, the pedigree of the family indicated recessive inheritance. Surprisingly, genetic analysis in the index patient yielded no diagnostic findings in PKHD1, but compound heterozygosity for two rare PKD1 missense variants, both of uncertain significance. One of them [c.4709C > T, p.(Thr1570Met)], however, was previously associated with early-onset ADPKD in the homozygous state (Vujic et al., 2010). In this case, two PKD1 variants in trans manifested as phenocopy of ARPKD. One of these hypomorphic alleles alone likely results in a normal phenotype, but two alleles in trans cause early-onset cystic kidney disease. As cyst initiation is assumed to depend on the dosage of functional Polycystin 1 (Rossetti et al., 2009), the combination of two hypomorphic variants could additively reduce the PKD1 function below a critical threshold to result in clinically apparent disease. Coincidence of biallelic hypomorphic PKD1 variants represents an example for PKD without an apparent family history (Iliuta et al., 2017).
Given these examples, we suggest revisiting and adapting the classification of genetic PKD to enable the description of the apparent continuum of AR/ADPKD genotypes and phenotypes. In a subset of cases, clinical presentation and associated genetic findings do not fit into the current ontology. As a first step, a more flexible disease terminology including the respective gene name was suggested in 2018 without being widely adopted to date (Cornec-Le Gall et al., 2018b). In other renal conditions, however, such as autosomal-dominant tubulointerstitial kidney disease (ADTKD), a new disease classification was successfully established, combining the mode of inheritance, major clinical manifestation, and underlying genetic defect in one term (ADTKD-UMOD; ADTKD-REN, ADTKD-MUC1, and ADTKD-HNF1B) (Bleyer et al., 2017).
In summary, this report demonstrates the genetic complexity of PKD. Attributing the correct diagnosis and providing adequate genetic counseling to affected patients and their families is not self-evident. Atypical presentations should prompt immediate genetic testing to increase diagnostic accuracy by verification or correction of clinically assumed diagnoses.
The data analyzed in this study is subject to the following licenses/restrictions: requests to access these datasets should be directed to JH, amFuLmhhbGJyaXR0ZXJAbWVkaXppbi51bmktbGVpcHppZy5kZQ==.
The studies involving human participants were reviewed and approved by local Institutional Review Board (IRB) at the Universities of Leipzig, Germany (IRB00001750; #402/16-ek). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
JF and JH contributed to the conception and design of the study, and helped in investigation. JF wrote the first draft of the manuscript. RS, JM, and BP wrote the sections of the manuscript. JF, RS, MN, BP, and JH contributed to the analysis. JF and RS helped in visualization. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.682565/full#supplementary-material
Adamiok-Ostrowska, A., and Piekiełko-Witkowska, A. (2020). Ciliary genes in renal cystic diseases. Cells 9:907. doi: 10.3390/cells9040907
Bergmann, C., Guay-Woodford, L. M., Harris, P. C., Horie, S., Peters, D. J. M., and Torres, V. E. (2018). Polycystic kidney disease. Nat. Rev. Dis. Primers 4:50. doi: 10.1038/s41572-018-0047-y
Bergmann, C., Senderek, J., Windelen, E., Küpper, F., Middeldorf, I., Schneider, F., et al. (2005). Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 67, 829–848. doi: 10.1111/j.1523-1755.2005.00148.x
Besse, W., Chang, A. R., Luo, J. Z., Triffo, W. J., Moore, B. S., Gulati, A., et al. (2019). ALG9 mutation carriers develop kidney and liver cysts. J. Am. Soc. Nephrol. 30, 2091–2102. doi: 10.1681/ASN.2019030298
Besse, W., Dong, K., Choi, J., Punia, S., Fedeles, S. V., Choi, M., et al. (2017). Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Invest. 127, 1772–1785. doi: 10.1172/JCI90129
Besse, W., Roosendaal, C., Tuccillo, L., Roy, S. G., Gallagher, A.-R., and Somlo, S. (2020). Adult inactivation of the recessive polycystic kidney disease gene causes polycystic liver disease. Kidney360 1, 1068–1076. doi: 10.34067/kid.0002522020
Bleyer, A. J., Kidd, K., Živná, M., and Kmoch, S. (2017). Autosomal dominant tubulointerstitial kidney disease. Adv. Chronic Kidney Dis. 24, 86–93. doi: 10.1053/j.ackd.2016.11.012
Brinkert, F., Lehnhardt, A., Montoya, C., Helmke, K., Schaefer, H., Fischer, L., et al. (2013). Combined liver-kidney transplantation for children with autosomal recessive polycystic kidney disease (ARPKD): indication and outcome. Transpl. Int. 26, 640–650. doi: 10.1111/tri.12098
Burgmaier, K., Kilian, S., Bammens, B., Benzing, T., Billing, H., Büscher, A., et al. (2019). Clinical courses and complications of young adults with autosomal recessive polycystic kidney disease (ARPKD). Sci. Rep. 9:7919. doi: 10.1038/s41598-019-43488-w
Burgmaier, K., Kunzmann, K., Ariceta, G., Bergmann, C., Buescher, A. K., Burgmaier, M., et al. (2018). Risk factors for early dialysis dependency in autosomal recessive polycystic kidney disease. J. Pediatr. 199, 22–28.e6. doi: 10.1016/j.jpeds.2018.03.052
Chen, J., Ma, N., Zhao, X., Li, W., Zhang, Q., Yuan, S., et al. (2019). A rare deep intronic mutation of PKHD1 gene, c.8798-459 C > A, causes autosomal recessive polycystic kidney disease by pseudoexon activation. J. Hum. Genet. 64, 207–214. doi: 10.1038/s10038-018-0550-8
Cornec-Le Gall, E., Audrézet, M.-P., Rousseau, A., Hourmant, M., Renaudineau, E., Charasse, C., et al. (2016). The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 942–951. doi: 10.1681/ASN.2015010016
Cornec-Le Gall, E., Olson, R. J., Besse, W., Heyer, C. M., Gainullin, V. G., Smith, J. M., et al. (2018a). Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Genet. 102, 832–844. doi: 10.1016/j.ajhg.2018.03.013
Cornec-Le Gall, E., Torres, V. E., and Harris, P. C. (2018b). Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J. Am. Soc. Nephrol. 29, 13–23. doi: 10.1681/ASN.2017050483
de La Faille, R., Vallet, M., Venisse, A., Nau, V., Collet-Gaudillat, C., Houillier, P., et al. (2011). A pseudo-dominant form of Gitelman’s syndrome. Clin. Kidney J. 4, 386–389. doi: 10.1093/ndtplus/sfr094
Denamur, E., Delezoide, A. L., Alberti, C., Bourillon, A., Gubler, M. C., Bouvier, R., et al. (2010). Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int. 77, 350–358. doi: 10.1038/ki.2009.440
Ebner, K., Dafinger, C., Ortiz-Bruechle, N., Koerber, F., Schermer, B., Benzing, T., et al. (2017). Challenges in establishing genotype–phenotype correlations in ARPKD: case report on a toddler with two severe PKHD1 mutations. Pediatr. Nephrol. 32, 1269–1273. doi: 10.1007/s00467-017-3648-x
Erger, F., Brüchle, N. O., Gembruch, U., and Zerres, K. (2017). Prenatal ultrasound, genotype, and outcome in a large cohort of prenatally affected patients with autosomal-recessive polycystic kidney disease and other hereditary cystic kidney diseases. Arch. Gynecol. Obstet. 295, 897–906. doi: 10.1007/s00404-017-4336-6
Furu, L., Onuchic, L. F., Gharavi, A., Hou, X., Esquivel, E. L., Nagasawa, Y., et al. (2003). Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J. Am. Soc. Nephrol. 14, 2004–2014. doi: 10.1097/01.asn.0000078805.87038.05
Gunay-Aygun, M., Tuchman, M., Font-Montgomery, E., Lukose, L., Edwards, H., Garcia, A., et al. (2010). PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol. Genet. Metab. 99, 160–173. doi: 10.1016/j.ymgme.2009.10.010
He, Q., Liu, X., Li, Q., Studholme, D. J., Li, X., and Liang, S. (2006). G8: a novel domain associated with polycystic kidney disease and non-syndromic hearing loss. Bioinformatics 22, 2189–2191. doi: 10.1093/bioinformatics/btl123
Hwang, Y. H., Conklin, J., Chan, W., Roslin, N. M., Liu, J., He, N., et al. (2016). Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 1861–1868. doi: 10.1681/ASN.2015060648
Iliuta, I. A., Kalatharan, V., Wang, K., Cornec-Le Gall, E., Conklin, J., Pourafkari, M., et al. (2017). Polycystic kidney disease without an apparent family history. J. Am. Soc. Nephrol. 28, 2768–2776. doi: 10.1681/ASN.2016090938
Iorio, P., Heidet, L., Rutten, C., Garcelon, N., Audrézet, M.-P., Morinière, V., et al. (2020). The “salt and pepper” pattern on renal ultrasound in a group of children with molecular-proven diagnosis of ciliopathy-related renal diseases. Pediatr. Nephrol. 35, 1033–1040. doi: 10.1007/s00467-020-04480-z
Irazabal, M. V., Rangel, L. J., Bergstralh, E. J., Osborn, S. L., Harmon, A. J., Sundsbak, J. L., et al. (2015). Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J. Am. Soc. Nephrol. 26, 160–172. doi: 10.1681/ASN.2013101138
Kane, I., Lindley, R., Lewis, S., and Sandercock, P. (2006). Impact of stroke syndrome and stroke severity on the process of consent in the third international stroke trial. Cerebrovasc. Dis. 21, 348–352. doi: 10.1159/000091541
Lanktree, M. B., Guiard, E., Akbari, P., Pourafkari, M., Iliuta, I.-A., Ahmed, S., et al. (2021). Patients with protein-truncating PKD1 mutations and mild ADPKD. Clin. J. Am. Soc. Nephrol. 16, 374–383. doi: 10.2215/CJN.11100720
Lanktree, M. B., Haghighi, A., Guiard, E., Iliuta, I. A., Song, X., Harris, P. C., et al. (2018). Prevalence estimates of polycystic kidney and liver disease by population sequencing. J. Am. Soc. Nephrol. 29, 2593–2600. doi: 10.1681/ASN.2018050493
Lu, H., Galeano, M. C. R., Ott, E., Kaeslin, G., Kausalya, P. J., Kramer, C., et al. (2017). Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 49, 1025–1034. doi: 10.1038/ng.3871
Mantovani, V., Bin, S., Graziano, C., Capelli, I., Minardi, R., Aiello, V., et al. (2020). Gene panel analysis in a large cohort of patients with autosomal dominant polycystic kidney disease allows the identification of 80 potentially causative novel variants and the characterization of a complex genetic architecture in a subset of families. Front. Genet. 11:464. doi: 10.3389/fgene.2020.00464
McConnachie, D. J., Stow, J. L., and Mallett, A. J. (2020). Ciliopathies and the kidney: a review. Am. J. Kidney Dis. 66, 50–62. doi: 10.1053/j.ajkd.2020.08.012
Melchionda, S., Palladino, T., Castellana, S., Giordano, M., Benetti, E., De Bonis, P., et al. (2016). Expanding the mutation spectrum in 130 probands with ARPKD: identification of 62 novel PKHD1 mutations by sanger sequencing and MLPA analysis. J. Hum. Genet. 61, 811–821. doi: 10.1038/jhg.2016.58
Michel-Calemard, L., Dijoud, F., Till, M., Lambert, J. C., Vercherat, M., Tardy, V., et al. (2009). Pseudoexon activation in the PKHD1 gene: a French founder intronic mutation IVS46+653A>G causing severe autosomal recessive polycystic kidney disease. Clin. Genet. 75, 203–206. doi: 10.1111/j.1399-0004.2008.01106.x
Obeidova, L., Seeman, T., Fencl, F., Blahova, K., Hojny, J., Elisakova, V., et al. (2020). Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS One 15:e0235071. doi: 10.1371/journal.pone.0235071
Porath, B., Gainullin, V. G., Cornec-Le Gall, E., Dillinger, E. K., Heyer, C. M., Hopp, K., et al. (2016). Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am. J. Hum. Genet. 98, 1193–1207. doi: 10.1016/j.ajhg.2016.05.004
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi: 10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rossetti, S., Kubly, V. J., Consugar, M. B., Hopp, K., Roy, S., Horsley, S. W., et al. (2009). Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 75, 848–855. doi: 10.1038/ki.2008.686
Schönauer, R., Baatz, S., Nemitz-Kliemchen, M., Frank, V., Petzold, F., Sewerin, S., et al. (2020). Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet. Med. 22, 1374–1383. doi: 10.1038/s41436-020-0816-3
Shan, D., Rezonzew, G., Mullen, S., Roye, R., Zhou, J., Chumley, P., et al. (2019). Heterozygous Pkhd1C642∗ mice develop cystic liver disease and proximal tubule ectasia that mimics radiographic signs of medullary sponge kidney. Am. J. Physiol. Renal Physiol. 316, F463–F472. doi: 10.1152/ajprenal.00181.2018
Stenson, P. D., Mort, M., Ball, E. V., Chapman, M., Evans, K., Azevedo, L., et al. (2020). The human gene mutation database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139, 1197–1207. doi: 10.1007/s00439-020-02199-3
Su, Q., Hu, F., Ge, X., Lei, J., Yu, S., Wang, T., et al. (2018). Structure of the human PKD1-PKD2 complex. Science 361:eaat9819. doi: 10.1126/science.aat9819
Szabó, T., Orosz, P., Balogh, E., Jávorszky, E., Máttyus, I., Bereczki, C., et al. (2018). Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr. Nephrol. 33, 1713–1721. doi: 10.1007/s00467-018-3992-5
Keywords: PKD1, PKHD1, ADPKD, ARPKD, cystic kidneys, chronic kidney disease
Citation: de Fallois J, Schönauer R, Münch J, Nagel M, Popp B and Halbritter J (2021) Challenging Disease Ontology by Instances of Atypical PKHD1 and PKD1 Genetics. Front. Genet. 12:682565. doi: 10.3389/fgene.2021.682565
Received: 18 March 2021; Accepted: 05 May 2021;
Published: 25 June 2021.
Edited by:
Xiu-An Yang, Chengde Medical College, ChinaReviewed by:
Gloria Pérez-Rubio, Instituto Nacional de Enfermedades Respiratorias, MexicoCopyright © 2021 de Fallois, Schönauer, Münch, Nagel, Popp and Halbritter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Halbritter, amFuLmhhbGJyaXR0ZXJAbWVkaXppbi51bmktbGVpcHppZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.