
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 28 July 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.594285
 Meng-Che Tsai1,2Yen-Yin Chou2Chia-Yi Li1Yi-Chieh Wang1Hui-Wen Yu1Chia-Hsiang Chen1Peng-Chieh Chen1,3*
Meng-Che Tsai1,2Yen-Yin Chou2Chia-Yi Li1Yi-Chieh Wang1Hui-Wen Yu1Chia-Hsiang Chen1Peng-Chieh Chen1,3*Collagenopathy is a rare genetic condition characterized by abnormality in either collagen structure or metabolism. Variations in its clinical presentations highlight diversity in the genetic causes and potential existence of concurrent mutations. Through whole exome sequencing (WES) complemented with multiplex ligation-dependent probe amplification, we identified the genetic etiologies for six cases with osteogenesis imperfecta (OI) in COL1A1 (p.T1298N, p.Q1280Pfs∗51, and p.G557Vfs∗23) and COL1A2 (c.1-1677_133-441del) as well as three cases with spondyloepiphyseal dysplasia congenita in COL2A1 (p.G1041S, p.G654S, and p.G441A). Co-occurrence of COL1A1 and WNT1 mutations was found in a patient with a mild OI phenotype but severe osteoporosis. These findings extended the pathogenic variant spectrum of COL1A1, COL1A2, and COL2A1 for type I and type II collagenopathies. Although WES provides a fast and accurate method to identify the genetic causes in most of the patients with type I and type II collagenopathies, its limitation of detecting CNVs because of variable capturing uniformity should be kept in mind when interpreting the results. Taken together, we demonstrate that multiple genetic characterizing technologies can provide an accurate and efficient molecular diagnostic of new genetic variants in disease-causing genes that are compatible with clinical phenotypes.
Collagenopathy is a group of disorders characterized by molecular abnormality either in the structure or metabolism of collagen proteins. Its clinical findings vary widely in affected tissue types and severity according to involved collagens and mutations (Carter and Raggio, 2009). Because collagen is the most abundant structural connective tissue protein with a particularly high expression in bones and cartilages, skeletal dysplasia becomes a prominent presentation associated with defective collagen proteins (Hall, 2002). Osteogenesis imperfecta (OI) is one of the most common genetic conditions in pediatric primary collagen disease with a rare incidence estimated around 1 in 15,000–20,000 live births (Kuurila et al., 2002; Forlino and Marini, 2016). OI is an inherited connective tissue disorder due to molecular dysfunctions in type I collagen synthesis and it manifests a wide-ranging spectrum of skeletal and non-skeletal phenotypes (Forlino et al., 2011). The clinical features in skeletal dysplasia are primarily related to bone fragility and deformity, such as macrocephaly, abnormal tooth development, flat midface and triangular facies, chest wall deformities, and scoliosis or kyphosis. It also presents non-skeletal features, including blue/gray sclerae, hearing loss, decreased pulmonary function, and cardiac valvular regurgitation.
OI cases are associated with dominant, recessive or X-linked mutations in a number of genes involved in different aspects of defects in collagen synthesis, structure or processing (COL1A1, COL1A2, BMP1); or defects in post-translational modification (PLOD2, CRTAP, LEPRE1, P3H1, PPIB, TMEM38B); folding or crosslinking defects (SERPINH1, FKBP10, CREB3L1, MBTPS2); or ossification or mineralization defects (IFITM5, SERPINF1, SPARC); or osteoblast differentiation defects with collagen insufficiency (WNT1,CREB3L1, SP7, SEC24D); endoplasmic reticulum function (KDELR2, MESD); Ca2 + sensitivity (PLS3) and other mechanisms (P4HB, P4HA1, TENT5A) (Dalgleish, 1997, 1998; Morello, 2018). Obtaining genetic diagnosis with the use of conventional Sanger sequencing may not be feasible in a timely and economical manner.
Moreover, some other genetic disorders with similar skeletomuscular manifestations should be consider for differential diagnoses. For example, defective function in COL2A1, encoding type II collagen, may results in spondyloepiphyseal dysplasia congenita (SEDC) that is rare chondrodysplastic condition characterized by skeletal deformities such as short-trunk dwarfism, odontoid hypoplasia, cervical spine subluxation, scoliosis, kyphosis, lumbar lordosis, coxa vara, genu valgum, clubfoot, pes planus, and metaphyseal changes (Zhang et al., 2015). Conventional diagnostic process requires detailed clinical and radiographic findings, which is now increasingly confirmed through molecular genetic testing.
Herein, we presented a group of Taiwanese patients presenting skeletal dysplastic features that are compatible with primary collagen disease.
A total of 9 patients diagnosed as type I and type II collagenopathies were consecutively enrolled from 7 families in this study (Supplementary Table 1). The enrollment of subjects took place in a tertiary medical center where it received pediatric referrals from a catchment area of nearly 3 million residents. The entire procedure was approved by the Institutional Review Board of the Cheng Kung University Hospital. A written informed consent was obtained from the enrollees and their guardians for the publication of the identifiable data if it applicable.
The proband’s genomic DNA was extracted from peripheral blood collected in EDTA-containing tubes. The exome library was built using the Nextera Rapid Exome Capture kit (Illumina), which covered approximately 37 Mb of the exonic regions, to capture the fragmented genomic DNA. Pair-end sequencing 2 × 75 bp was performed on the Illumina NextSeq 500 sequencer. Sequencing reads were aligned to human genome reference Hg19 using BWA-mem1. Single nucleotide variants and small insertions and deletions were identified with Genome Analysis Toolkit 3.5 (GATK)2. Variants were annotated with ANNOVAR3 and novel variants were filtered against 1000 Genomes, dbSNP, and Genome Aggregation Database4. Variants were sorted according to the Combined Annotation Dependent Depletion (CADD) score (Rentzsch et al., 2019), and the functional effects of these amino acid substitutions were predicted using in silico analysis tools, such as PolyPhen 25, PROVEAN6, SIFT7, MutationTaster8, and varSEAK9 (Supplementary Table 1). Segregation analysis with Sanger sequencing on the DNA of the probands and their family members was finally used to validate the potentially pathogenic variants.
Peripheral blood mononuclear cells (PBMCs) were isolated from 5 ml of blood using Ficoll-Paque PLUS. Blood samples from the same family were collected and processed to the library construction at the same time. Total RNA was extracted from the same amount of PBMCs using RNeasy Micro Kit (QIAGEN). Sequencing libraries from mRNA was prepared with SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara Bio USA) and Nextera XT DNA Library Prep Kit (Illumina), and then sequenced with Illumina NextSeq 500 sequencer with 2 × 75 pair-end sequencing reaction with Illumina NextSeq500 system. On average, 39.46 million reads were obtained for each sample. Reads were aligned with GRCh38 using HISAT210 and the aligned reads were counted with HTSeq11 and annotation GTF file from ENSEMBL (Homo_sapiens.GRCh38.99). Genes with fewer than 10 reads in all genotypes were not included in further analyses. Differential expression of genes in probands when compared with control was analyzed with DESeq212. For gene set enrichment analysis (GSEA), normalized expression level of genes was analyzed with gene set collections from the Molecular Signatures Database v7.013.
Probands with no variants detected with WES were subjected for multiplex ligation-dependent probe amplification (MLPA) analysis (Schouten et al., 2002; van Dijk et al., 2010). SYBR Green PCR Master Mix (Thermo Fisher Scientific) and primers (Supplementary Table 2) designed to detect copy number changes of COL1A2 exon 1 and exon 4 were used to confirm the results of MLPA. Concentration of genomic DNA was measured by Qubit dsDNA BR Assay (Thermo Fisher Scientific). All reaction sets were conducted in triplicate, and a non-template control was run simultaneously. The amount of COL1A2 was normalized to the genomic GAPDH and quantified using the comparative CT method (ddCT).
The concentration of genomic DNA was quantified by Qubit dsDNA BR Assay Kit before used. Each reaction mix consisted of 2.5 μl of genomic DNA (150 ng), 0.5 μl of PfuUltra II Fusion HS DNA Polymerase (Agilent), 0.5 μl of dNTP (25 mM), 5 μl of 10X PfuUltra II reaction buffer, 1 μl of forward primer (10 μM), 1 μl of reverse primer (10 μM), and 14.5 μl nuclease- free water in a total volume of 25 μl. PCR was performed using VeritiTM Thermal Cycler (Applied Biosystems) and was run on the program 95°C for 2 m followed by 40 cycles of 20 s at 95°C, 20 s at 57°C, and 3 m at 72°C. PCR products were visualized on 1.5% agarose gel using SYBR Safe DNA gel stain (Invitrogen) and purified by QIAquick Gel Extraction Kit (QIAGEN).
Gel purified-pair 3 PCR amplicons was sequenced using the specific forward and reverse primers for break point identification (Supplementary Table 2). Sanger sequencing was performed by ABI 3730 XL DNA Analyzer (Applied Biosystems). Sanger sequencing reads were aligned to human genome reference GRCh38 to reveal the deleted region. The DNA sequence reads were performed by SnapGene Viewer.
P1 and P2 were a pair of siblings in a family, where their father and paternal aunt also presented some mild OI phenotypes, such as blue sclera, short stature, and infrequent non-fatal fractures of extremities since childhood (Figure 1A). Their father measured 153 cm in height, which corresponded to a standard deviation score (SDS) of −4.0 as compared to average male adult height in the Taiwanese population. P1 was clinically diagnosed of OI because of blue sclera and multiple fractures in her infancy. She was wheelchair-bound since early childhood despite the use of intravenous bisphosphonate therapy. Her brother, P2, was regularly followed at the pediatric endocrinology clinic for his short stature. He had infrequent bone fractures that only required external splinting. He was found to have severe osteoporosis and hence started to receive intravenous bisphosphonate therapy, shortly after he had vertebral fractures at T9-T10 level that made him unable to walk at age 12 years.
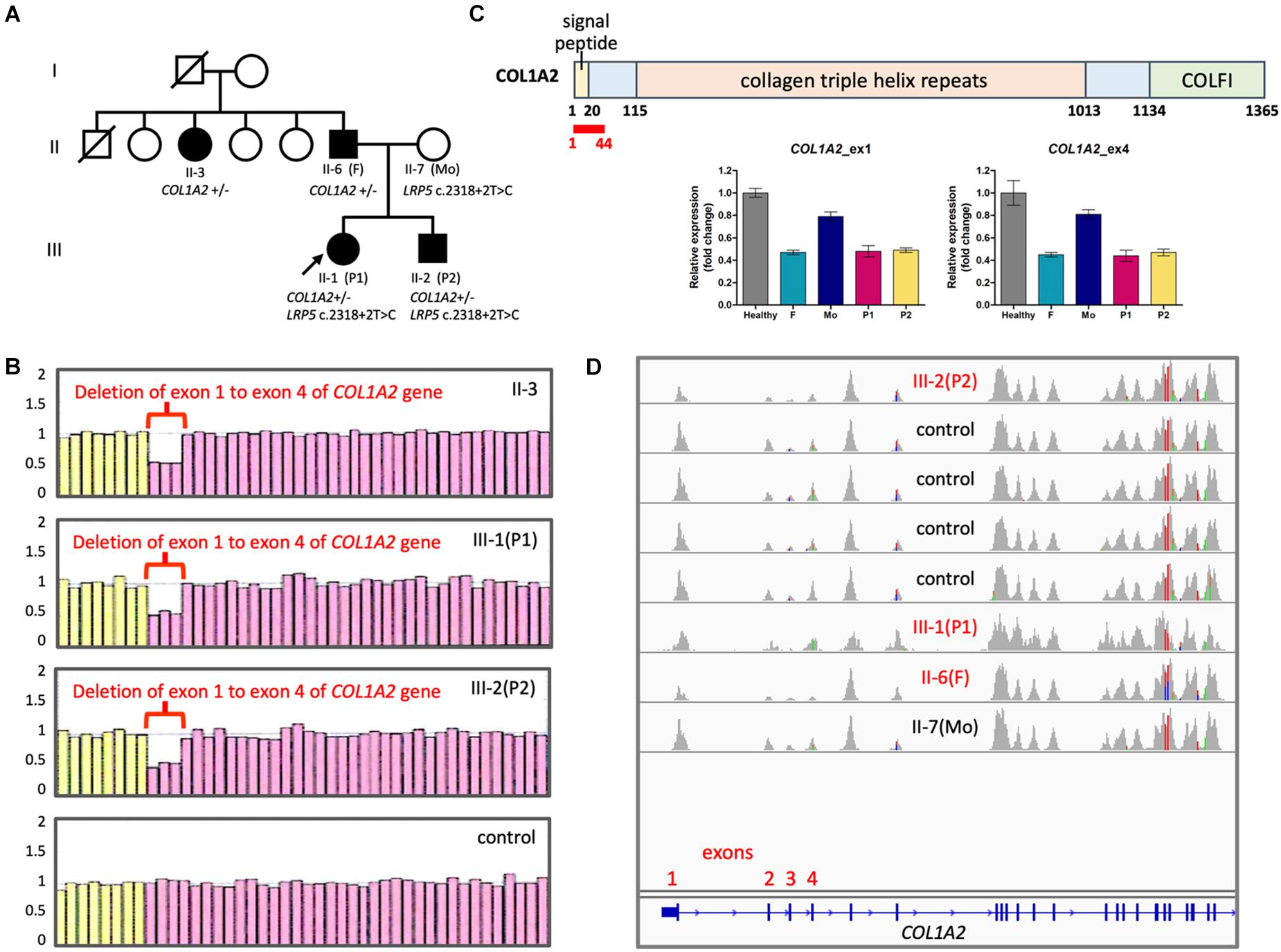
Figure 1. Copy number changes in COL1A2 in the family with OI. (A) Family pedigree. Filled symbols, affected; half filled, heterozygous carrier; unfilled, not clinically affected. (B) Results of MLPA showing fold change of probes detecting COL1A2. Pink probes are detecting COL1A2 while yellow probes are detecting control genes. (C) (Top panel) Positions of COL1A2 exon 1–4 (encodes amino acid residue 1∼44 in red) (NP_000080.2). COLFI, Fibrillar collagen C-terminal domain. (Bottom panel) Quantification of copy numbers in COL1A2 exon 1 and exon 4 by qPCR. (D) Integrative Genomics Viewer showing sashimi plots of the results of WES from OI patients and controls. Notice that read coverage of exon 2–4 of COL1A2 are lower than other exons in general.
Initially WES did not reveal pathological variants linked to OI. However, with typical phenotype of type I OI, we suspect that copy number variations (CNVs) may be the underlying cause. In order to identify potential long-range duplication or deletion in the region of COL1A1 and COL1A2, the family consented for a clinical test based on MLPA technique. The results revealed loss of heterozygosity at COL1A2 exon 1 to exon 4 that was consistently linked to the affected members within the family (Figure 1B). Quantitative PCR confirmed the copy number loss of the exons 1 and 4 (Figure 1C). Copy number change in exon 1–4 was not detected in WES because the coverage of reads was in general lower in exon 2–4 (Figure 1D).
To identify the breakpoint of the COL1A2 deletion, we designed multiple pairs of primer recognizing upstream of exon 1 and downstream of exon 4 to the assumptive breakpoints of the deleted segment. Polymerase chain reaction (PCR)-based electrophoresis revealed an aberrant product of 783 bp in the affected members (Figure 2A). Finally, Sanger sequencing revealed the dropped read coverage from exon 1 to 4, which located on chromosome 7 at genomic location 94,393,355–94,399,754 (GRCh38), in the COL1A2 gene (chromosome7: 94,394,895–94,431,227) (Figure 2B).
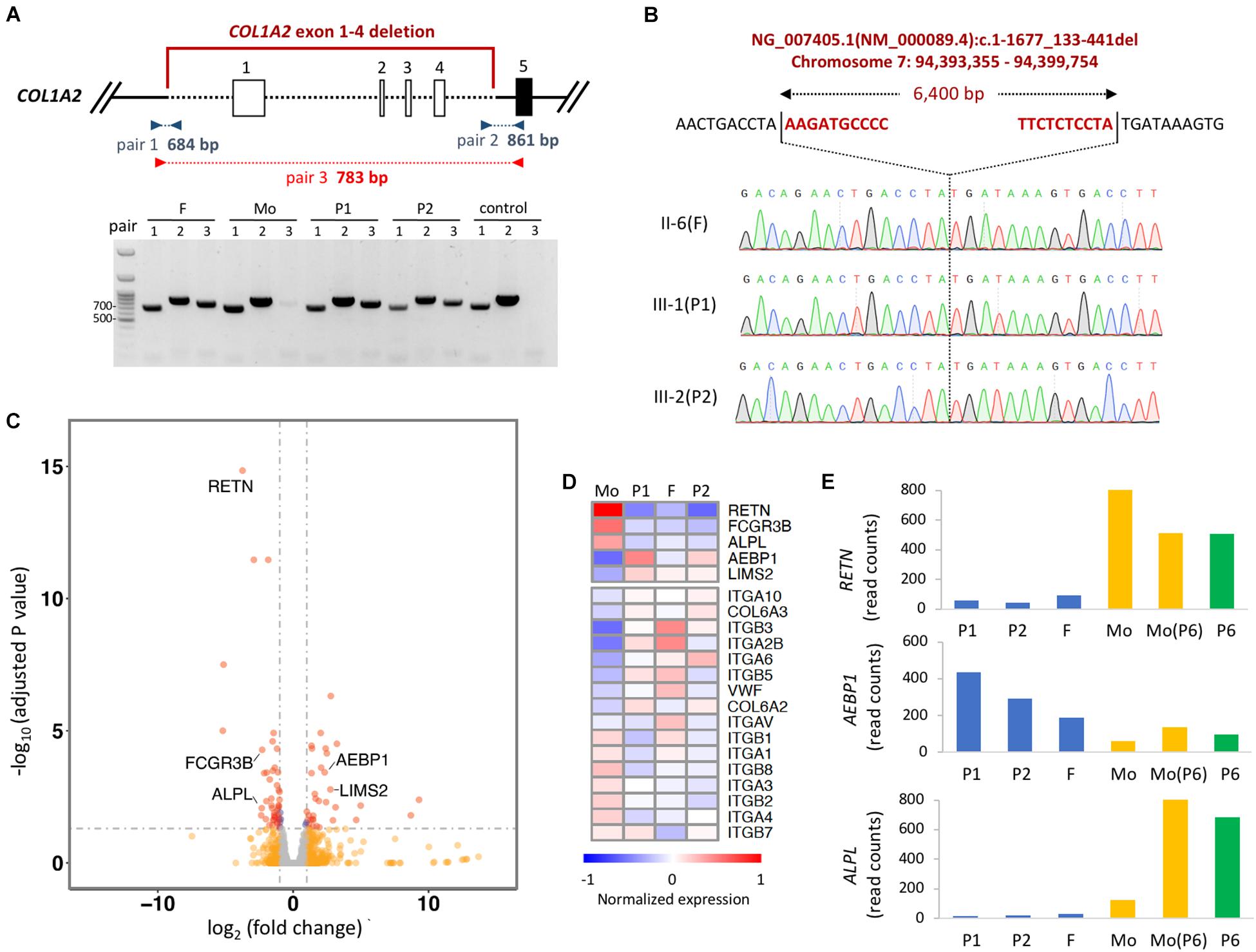
Figure 2. Detecting the breakpoint of heterozygous deletion in COL1A2. (A) Schematic representation of the deletion region of exon 1–4. Boxes represent exons, and dashed lined represent deleted regions. PCR amplicons used to validate the deletion with three set of primers are shown. Red primers only amplified the deleted region. (B) Identification of breakpoint of COL1A2 by Sanger-sequencing the product amplified with red primers shown in (A). The upper sequences show the non-deleted DNA reference sequence, while the lower sequence shows the read-out from Sanger sequencing with the breakpoint location. (C) Volcano plot showing differentially expressed gene in PBMCs when comparing samples with COL1A2 deletion (P1, P2, and father) vs. control (mother of P1 and P2). Red dots, genes with adjusted P-value < 0.05 and log2 fold change > 1 (n = 37) or < –1 (n = 48); blue dots, adjusted P-value < 0.05; orange dots: log2 fold change > 1 or < –1; gray dots, none of the above. (D) Heatmap showing DEseq2 normalized expression of genes differentially expressed when comparing P1, P2, and F vs. Mo. Mo, mother of P1 and P2; F, father of P1 and P2. (E) DEseq2 normalized read counts of genes in two families. Mo, mother of P1 and P2; F, father of P1 and P2; Mo (P6), mother of P6.
The COL1A2 deletion includes 1540 bp of the proximal promoter region, which is required for transcriptional regulation, and the transcription start site (Ramirez et al., 2006). Therefore, we would like to evaluate whether the expression level of COL1A2 in the cells of the proband’s is lower than that in cells of the family members carrying normal COL1A2. However, because patients did not consent to tissue biopsy, we chose to evaluate the gene expression in the PBMCs isolated from their blood. Given the limited amount of RNA obtained, mRNA sequencing libraries were constructed with SMART-seq protocol and sequenced to the depth of ∼40 million reads for each sample. As expected, expression of COL1A2 in PBMCs is extremely low (with reads aligned fewer than 10), making it difficult to compare the expression of COL1A2 mRNA in cells with the COL1A2 deletion to the normal cells. However, when comparing the expression profile between the family members with COL1A2 deletion (P1, P2 and father) vs. those with normal COL1A2 (mother), we identified that 5 genes out of the 27 most differentially expressed (DE) genes (adjusted p-value < 0.05 and log2 fold change > 2 or < −2) were related to bone development or remodeling (RETN, FCGR3B, ALPL, AEBP1, and LIMS2) (Figures 2C,D). The trend of these differentially expressed genes are likely to be genotype-specific because of comparable expression level detected in the mother of P1 and the mother of P6 with reference allele of COL1A2 and COL1A1, and P6 with COL1A1 and WNT1 mutation (Figure 2E). In addition, gene set enrichment analysis revealed that extracellular receptor interacting molecules, especially integrins, were also differentially expressed (Figure 2D). Taken together, these results suggest that the coding transcriptome in the circulating blood cells of patients with COL1A2 deletion shows unique gene expression profile.
We observed variability in the phenotypic expression within the P1 and P2 family. While the sibling pair manifested walking disability in their childhood, their father and aunt were less affected. Therefore, we went on to investigate rare variants associated with OI that may potentially contribute to the phenotypes of the probands. Interestingly, we identified a maternally inherited splice-site variant LRP5 c.2318 + 2T > C (Supplementary Table 3). The novel LRP5 splice-site variant in the intron 10 was predicted to cause loss of function for 5’ splice site donor and render exon skipping as well as a premature stop codon at the beginning of exon 12. Heterozygous frameshift mutation of LRP5 has been identified in patients with primary osteoporosis, suggesting haploinsufficiency of this gene (Hartikka et al., 2005). These results may help explain why the sibling pair had severe osteoporosis with other mild OI phenotypes.
Four cases (P3, P4, P5, and P6) were found to have mutations in COL1A1 (Figures 3A–D). P3 and P4 were identical twins and both had multiple perinatal fractures (Figure 3B). They also presented blue sclera and multiple fractures over their slow growth. Dual-energy x-ray absorptiometry found a decreased bone mineral density (Figure 3E). A previously known pathological mutation COL1A1 c.3893C > A (p.T1298N) was found in these twin sisters but not in their parents (Supplementary Table 1).
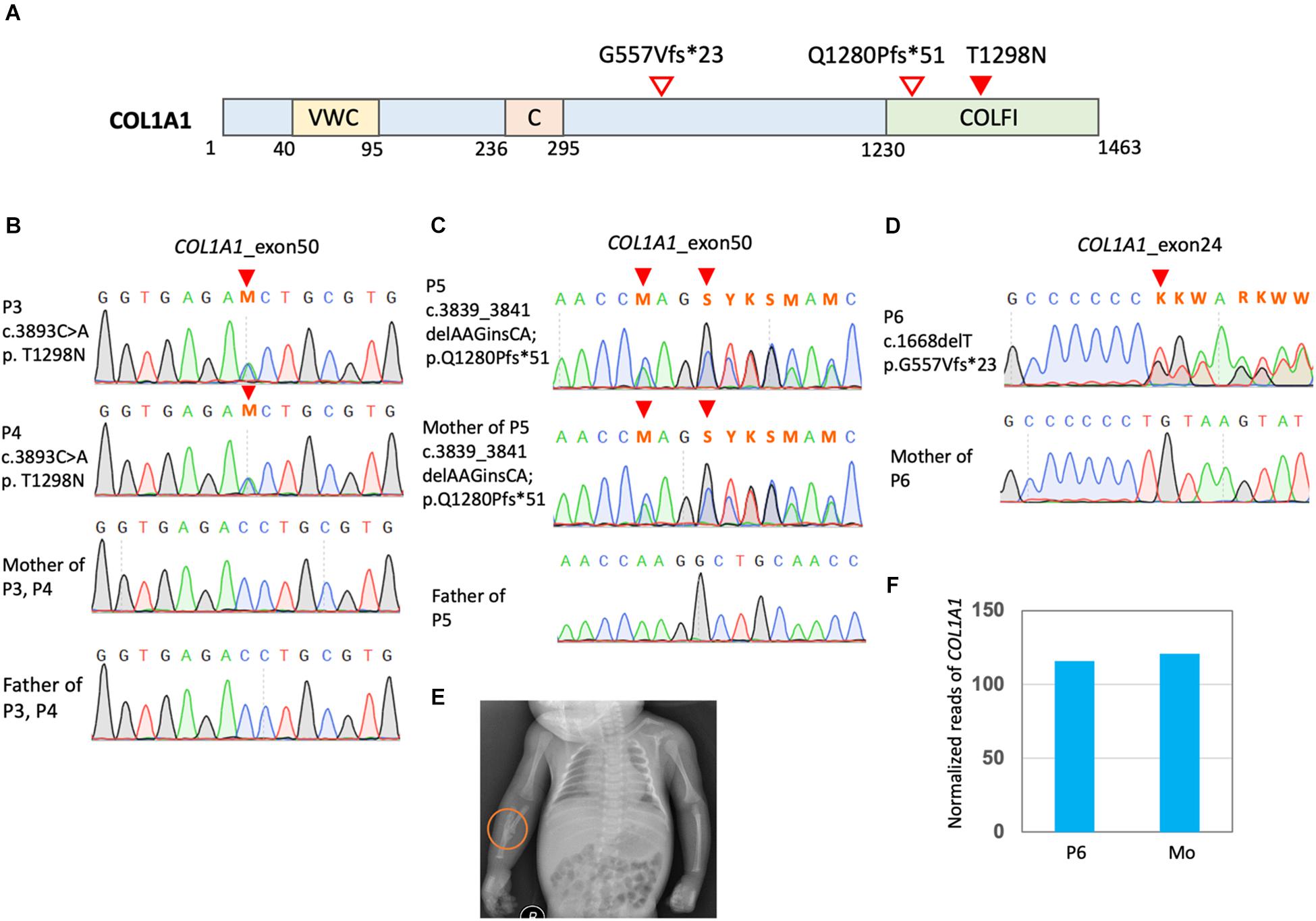
Figure 3. Mutation in COL1A1 in the OI patients. (A) Positions of COL1A1 (NP_000079.2) variants. VWC, von Willebrand factor type C domain; C, Collagen triple helix repeat (20 copies); COLFI, Fibrillar collagen C-terminal domain. (B–D) Sanger sequencing confirmed COL1A1 mutation. (E) P3 presented a greenstick fracture in the mid-shaft of the right radius and ulna. (F) Read counts of COL1A1 in RNA sequencing of PBMCs of OI patients P6 carrying COL1A1 c.1668delT. Mo, mother of P6.
P5 and her mother presented a borderline short stature with a body height at the 3rd % of the population. They also had blue sclera and several non-life-threatening fractures since birth. The mother had mild bilateral hearing impairment that did not require hearing aids. They were found to have COL1A1 c.3839_3841delAAGinsCA; p.Q1280Pfs∗51 (Figure 3C). The frameshift mutations was a novel mutation and predicted to have deleterious effects (Supplementary Table 1).
P6 was found to have COL1A1 c.1668delT (p.G557Vfs∗23) and a single allelic WNT1 c.677C > T (p.S226L) (Figure 3D and Supplementary Figures 1A,B). He presented a very short stature and a kyphotic spinal curvature. He had several non-life-threatening fractures since childhood and typically blue sclera. Dual-energy x-ray absorptiometry found severe osteoporosis (Supplementary Figure 1C). The WNT1 mutation was reported to have an allele frequency around 1.5/10,000 in the East Asian population from the gnomAD. Functionally, this mutation was predicted to be pathogenic. RNA sequencing with PBMCs detected comparable amount of COL1A1 in P6 and his mother (Figure 3F). However, no reads containing the COL1A1 c.1668delT were detected, which is evidence of nonsense mediated decay (NMD) of the mutant transcript. On the other hand, transcripts of WNT1 c.677C > T was detected with comparable amount of the reference allele (Supplementary Figure 1D).
Three cases (P7, P8, and P9) were found to have de novo heterozygous mutations, COL2A1 c.3121G > A (p.G1041S), COL2A1 c.1960G > A (p.G654S), and COL2A1 c.1322C > G (p.G441A), respectively (Supplementary Figures 2A–D). They all presented an extremely short stature with an abnormal spinal curvature, remarkably in the lumbar region. The bone X-ray found platyspondyly and epiphyseal dysplastic changes (Supplementary Figures 2E–G). Mutations c.1960G > A (p.G654S) and c.1322C > G (p.G441A) found in these patients were novel and predicted to have deleterious effects (Supplementary Table 1). COL2A1 c.3121G > A (p.G1041S) has been reported to be a pathological mutation clinically associated with spondyloepimetaphyseal dysplasia (SEMD) that was also noted in P7 (Supplementary Figure 2E; Meredith et al., 2007; Zhang et al., 2015).
In this report, we confirmed the genetic diagnosis of 9 patients with type I and type II collagenopathies. Their clinical presentations fell into two main categories: OI (6 cases) and SEDC (3 cases). The pathogenic variants included one in COL1A2, four in COL1A1, and three in COL2A1. These results extended the current knowledge of genotype-phenotype correlation in type I and type II collagenopathies.
Mutations in COL1A2 may cause symptoms ranging from severe OI to a milder Ehler-Danlos syndrome (Forlino and Marini, 2016). Less than 5% of COL1A2 mutations can cause classic OI and they usually occur in the procollagen C-propeptide, thus impairing chain association or folding, while non-sense mutations leading to decreased synthesis of normal collagen are less detrimental (Pace et al., 2008). As compared to missense or splice-site mutations, multiexon duplication or deletion in COL1A2 has less been reported in classic OI cases but it may be linked to a wide array of clinical OI phenotypes or several subtypes of Ehlers-Danlos syndrome (EDS), depending on the location of variant (Kuivaniemi et al., 1988; Byers et al., 1997; Raff et al., 2000). In our cases, MLPA analysis helped to complement WES by contributing toward the detection of copy number variations. Further using multiple PCR primers, we were able to identify the both breaking ends of deletion and qPCR analysis showed decreased levels of exons 1 and 4. Indeed, since the deletion in the proband occurred in exon 1, split reads with sequences with the breakpoint won’t be captured with exome-sequencing capture. The deletion of exon 1–4 was a novel structural mutation and presumed to have a deleterious effect on the functionality of mutant protein.
Although we could not obtain tissue samples from the probands, mRNA sequencing revealed DE genes related to bone homeostasis in individuals with COL1A2 deletion. Because of similar expression level were detected in the mother of P1 and in the family with no COL1A2 mutation, the signature of these DE genes are likely to be genotype-specific and may be the result of compensatory regulation of imbalanced level of type I α1 and α2 collagen in the cells. Further study is needed to clarify the relationship between DE genes and COL1A2. The expression level of these differentially expressed genes are likely to be regulated through trans-acting factors since we did not identify pathogenic mutations in these gene through WES. The most statistically significant DE gene, RETN, encodes for resistin, a secreted protein that regulates glucose metabolism in the cells and induces bone remodeling (Acquarone et al., 2019). In addition to circulating resistin that is produced by PBMCs, resistin is also expressed by mature osteoblasts in the bone (Acquarone et al., 2019). Moreover, resistin has been shown to stimulate osteoclast differentiation and increases osteoblast proliferation in vitro as well as to alter the composition of type I collagen α1/α2 ratio in human bone explants (Thommesen et al., 2006; Philp et al., 2017). Another DE gene, ALPL, encodes tissue non-specific alkaline phosphatase (TNAP) that is involved in mineralization of the bone (Orimo, 2010; Millan and Whyte, 2016). Loss-of-function mutations in ALPL may cause hypophosphatasia (HPP), a rare inherited skeletal dysplasia (Millan and Whyte, 2016). Interestingly, OI is one of the common differential diagnoses of HPP (Taillandier et al., 2015). Our finding of decreased ALPL expression in probands with COL1A2 deletion may be the result of aberrant type I collagen composition, and may worsen the phenotypes of OI.
Pathogenic deletion involving the first exon of COL1A2 (exon 1–18 deletion) has been reported in the database of collagen mutation in OI and EDS14. However, with the breakpoint of the deletion not determined, the expression of truncated COL1A2 protein is not clear. In our case, the deleted COL1A2 allele is likely to produce no COL1A2. While mutations in COL1A2 rendering premature stop codon usually have no phenotypes in heterozygote, modifying variant may contribute to the phenotypes of primary osteoporosis (Marini et al., 2007; Skarp et al., 2020). We also observed variable expressivity of the COL1A2 deletion within the family. A potential explanation is that the presence or the detrimental effect of the mutant protein varies in vivo within individuals or the compound variant inherited maternally aggravated the phenotypes, thus making phenotype-genotype less correlate. Functional investigation using the tissue harvested from the affected family members may be needed to verify our deduction.
Consistent with previous research, mutations in COL1A1 gene are most likely to be reported in OI patients. We identified one known missense mutation (c.3893C > A; p.T1298N), one known frameshift mutation (c.1668delT; p.G557Vfs∗23), and one novel frameshift mutation (c.3839_3841delAAGinsCA; p.Q1280Pfs∗51) in COL1A1 (Lu et al., 2014). Type I collagen is a heterotrimer, containing two α1 and one α2 chains. It is synthesized as a procollagen molecule, with N-terminal and C-terminal globular pro-peptides flanking the helical domain. Heterozygous COL1A1 mutations causing a null allele to induce haploinsufficiency and are the most common type of mutations that result in dominant OI (Ben Amor et al., 2013). The most common structural defects in type I collagen causing OI are substitutions for glycine residues in the helical domain and result in moderate to severe, and perinatally lethal OI (Marini et al., 2007; Rauch et al., 2010). In the cells, transcripts containing a premature stop codon triggers the nonsense-mediated mRNA decay (NMD) (Dyle et al., 2020). The p.Q1280Pfs∗51 leads to frameshifts and premature termination codons located > 55 bp from the start of last exon of COL1A1 that is predicted to be targeted by NMD. Hence, no truncated COL1A1 protein will be generated. As compared to missense mutations that result in expressed mutant COL1A1 proteins, frameshift mutations usually cause milder OI phenotypes due to haploinsufficiency of type 1 collagen α, which is the typical mechanism of OI type I (Wang et al., 2015).
Another co-occurrence of mutations was found in P6, who had a heterozygous COL1A1 c.1668delT (p.G557Vfs∗23) and WNT1 c.677C > T (p.S226L). This may explain that P12 had a mild OI phenotype, due to the presence of frameshift COL1A1 mutation, but severe osteoporosis. The c.1668delT (p.G557Vfs∗23) mutation also locates > 55 bp from the start of the last exon of COL1A1 and therefore is predicted to be targeted by NMD. Indeed, read with COL1A1 c.1668delT was not found in the RNA sequencing. The mutation, WNT1 c.677C > T (p.S226L), was expressed in the patient, has been recently indicated in severe recessively-inherited OI phenotypes in patients with homozygous mutations or compound heterozygous mutations (Lu et al., 2018). Modeling of the three-dimensional molecular structure of mutant proteins indicated possible changes in the binding cavity conferred by the p.S226L substitution, while functional in vitro studies also supported WNT signaling reduction in cells transfected with p.S226L variant (Lu et al., 2018). In the canonical pathway, WNT binding enables cytosolic β-catenin to escape proteasomal destruction and be translocated into the nucleus, where it activates expression of several genes implicated in bone formation (Baron and Kneissel, 2013). WNT1 is one of the key ligands to the WNT pathway in bone. Family segregation analysis found that heterozygous WNT1 mutations lead to autosomal dominant osteoporosis, while homozygous mutations, a more severe OI (Fahiminiya et al., 2013; Laine et al., 2013; Pyott et al., 2013). As reported previously, our patient also presented early onset osteoporosis and kyphotic deformity because of vertebral compression (Makitie et al., 2019). Although the extended family members were not recruited in this present study, we expected that the WNT1 mutation (c.677C > T) might exist in the local population as its compatibility with life in affected individuals. Further research may be required to understand the clinical impacts on heterozygous carriers and allele frequency in osteoporotic patients due to the founder effect.
Clinically, the presentations of the three patients found to have mutations in COL2A1 were more compatible with SEDC (p.G654S and p.G441A) and SEMD (p.G1041S) in that they were short in stature with a marked spinal kyphotic curvature. The alpha-1 chain of type II collagen encoded by COL2A1 has three domains: an N-propeptide which may be involved in the regulation of primary fibril diameters, a triple-helical domain which contains 330 Gly-X-Y repeats and is the predominant motif and a C-propeptide which is considered to be an important factor in the initiation of triple helix formation (Gelse et al., 2003). Previous literature summarized that Glycine to Serine substitution in the triple helical domain of the type 2 procollagen chain is the most common mutation in COL2A1 (Nishimura et al., 2005). In our cohort, two of the mutations were Glycine to Serine substation but none of them were located in the helical domain. As the mutant protein was predicted to have deleterious effects in protein function, we assumed that these mutations still altered the structural stability of mutant protein and thus manifested negative dominant effects in affected individuals. The recurrent mutation c.3121G > A (p.G1041S) has been reported in 4 individuals listed in the LOVD database15. Aligned with previously reported cases (Meredith et al., 2007; Zhang et al., 2015), our patient presented a marked kypholordotic curve along with platyspondyly, a mild pectus carinatum, short lone bones, with dappled metaphyses. However, there was no abnormal ocular problem in this case.
In conclusion, WES is effective to identify the genetic causes and help differentially diagnosing type I and type II collagenopathies. However, this technique is limited by its inability to detect CNVs because of insufficient uniformity of capturing. Some other genetic techniques, such as MLPA, may be required to complement the diagnostic search for genetic causes. Our case series manifested varied clinical presentations of type I and type II collagenopathies, which highlighting diversity in its genetic causes and potential existence of concurrent mutations either in the same or different genes. Knowing the genetic factors help understand the underlying mechanisms and provide further information for specific treatment and proper genetic counseling for patients and family.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Cheng Kung University Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
M-CT and Y-YC recruited the patients and collected their clinical information. C-YL, Y-CW, H-WY, and C-HC conducted molecular research. P-CC conducted bioinformatic analysis and supervised the entire research, critically reviewed and revised the manuscript. M-CT and C-HC drafted the manuscript. All authors have read and approved the final version of manuscript.
This study was supported by the research grant from National Cheng Kung University Hospital (NCKUH-10507020) to Y-YC and P-CC and Higher Education SPROUT Project to P-CC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.594285/full#supplementary-material
Supplementary Figure 1 | Co-occurring mutation in WNT1 in the OI patients. (A) Positions of WNT1 variants: TM, transmembrane region. (B) Sanger sequencing confirmed WNT1 mutation. (C) X-ray films showed marked osteoporosis and thinning of vertebral bodies. (D) IGV view of shashimi plot showing reads with WNT1 c.677C > T (p.S226L) from RNA sequencing of PBMCs of P6.
Supplementary Figure 2 | Mutation in COL2A1 in the SEDC patients. (A) Positions of COL2A1 (NP_001835.3) variants: VWC, von Willebrand factor type C domain; (C) Collagen triple helix repeat (20 copies); COLFI, Fibrillar collagen C-terminal domain. (B–D) Sanger sequencing confirmed de novo COL2A1 mutation. (E–G) X-ray films showed epiphyseal dysplasia and various degrees of flattening and notching of the vertebral bodies that aligned in kypholordotic curvatures in P7, P8, and P9.
Acquarone, E., Monacelli, F., Borghi, R., Nencioni, A., and Odetti, P. (2019). Resistin: a reappraisal. Mech. Ageing Dev. 178, 46–63. doi: 10.1016/j.mad.2019.01.004
Baron, R., and Kneissel, M. (2013). WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 19, 179–192. doi: 10.1038/nm.3074
Ben Amor, I. M., Roughley, P., Glorieux, F. H., and Rauch, F. (2013). Skeletal clinical characteristics of osteogenesis imperfecta caused by haploinsufficiency mutations in COL1A1. J. Bone Miner. Res. 28, 2001–2007. doi: 10.1002/jbmr.1942
Byers, P. H., Duvic, M., Atkinson, M., Robinow, M., Smith, L. T., Krane, S. M., et al. (1997). Ehlers-danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 72, 94–105. doi: 10.1002/(sici)1096-8628(19971003)72:1<94::aid-ajmg20>3.0.co;2-o
Carter, E. M., and Raggio, C. L. (2009). Genetic and orthopedic aspects of collagen disorders. Curr. Opin. Pediatr. 21, 46–54. doi: 10.1097/mop.0b013e32832185c5
Dalgleish, R. (1997). The human type I collagen mutation database. Nucleic Acids Res. 25, 181–187. doi: 10.1093/nar/25.1.181
Dalgleish, R. (1998). The human collagen mutation database 1998. Nucleic Acids Res. 26, 253–255. doi: 10.1093/nar/26.1.253
Dyle, M. C., Kolakada, D., Cortazar, M. A., and Jagannathan, S. (2020). How to get away with nonsense: mechanisms and consequences of escape from nonsense-mediated RNA decay. Wiley Interdiscip. Rev RNA 11:e1560.
Fahiminiya, S., Majewski, J., Mort, J., Moffatt, P., Glorieux, F. H., and Rauch, F. (2013). Mutations in WNT1 are a cause of osteogenesis imperfecta. J. Med. Genet. 50, 345–348. doi: 10.1136/jmedgenet-2013-101567
Forlino, A., Cabral, W. A., Barnes, A. M., and Marini, J. C. (2011). New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 7, 540–557.
Gelse, K., Poschl, E., and Aigner, T. (2003). Collagens–structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 55, 1531–1546. doi: 10.1016/j.addr.2003.08.002
Hall, C. M. (2002). International nosology and classification of constitutional disorders of bone (2001). Am. J. Med. Genet. 113, 65–77. doi: 10.1002/ajmg.10828
Hartikka, H., Mäkitie, O., Männikkö, M., Doria, A. S., Daneman, A., Cole, W. G., et al. (2005). Heterozygous mutations in the LDL receptor-related protein 5 (LRP5) gene are associated with primary osteoporosis in children. J. Bone Miner. Res. 20, 783–789. doi: 10.1359/jbmr.050101
Kuivaniemi, H., Sabol, C., Tromp, G., Sippola-Thiele, M., and Prockop, D. J. (1988). A 19-base pair deletion in the pro-alpha 2(I) gene of type I procollagen that causes in-frame RNA splicing from exon 10 to exon 12 in a proband with atypical osteogenesis imperfecta and in his asymptomatic mother. J. Biol. Chem. 263, 11407–11413. doi: 10.1016/s0021-9258(18)37971-7
Kuurila, K., Kaitila, I., Johansson, R., and Grénman, R. (2002). Hearing loss in finnish adults with osteogenesis imperfecta: a nationwide survey. Ann. Otol. Rhinol. Laryngol. 111, 939–946. doi: 10.1177/000348940211101014
Laine, C. M., Joeng, K. S., Campeau, P. M., Kiviranta, R., Tarkkonen, K., Grover, M., et al. (2013). WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 368, 1809–1816.
Lu, Y., Ren, X., Wang, Y., Bardai, G., Sturm, M., Dai, Y., et al. (2018). Novel WNT1 mutations in children with osteogenesis imperfecta: clinical and functional characterization. Bone 114, 144–149. doi: 10.1016/j.bone.2018.06.018
Lu, Y., Ren, X., Wang, Y., Li, T., Li, F., Wang, S., et al. (2014). Mutational and structural characteristics of four novel heterozygous C-propeptide mutations in the proalpha1(I) collagen gene in Chinese osteogenesis imperfecta patients. Clin. Endocrinol. 80, 524–531. doi: 10.1111/cen.12354
Makitie, R. E., Costantini, A., Kämpe, A., Alm, J. J., and Mäkitie, O. (2019). New insights into monogenic causes of osteoporosis. Front. Endocrinol. 10:70. doi: 10.3389/fendo.2019.00070
Marini, J. C., Forlino, A., Cabral, W. A., Barnes, A. M., San Antonio, J. D., Milgrom, S., et al. (2007). Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 28, 209–221. doi: 10.1002/humu.20429
Meredith, S. P., Richards, A. J., Bearcroft, P., Pouson, A. V., and Snead, M. P. (2007). Significant ocular findings are a feature of heritable bone dysplasias resulting from defects in type II collagen. Br. J. Ophthalmol. 91, 1148–1151. doi: 10.1136/bjo.2006.112482
Millan, J. L., and Whyte, M. P. (2016). Alkaline phosphatase and hypophosphatasia. Calcif. Tissue Int. 98, 398–416. doi: 10.1007/s00223-015-0079-1
Nishimura, G., Haga, N., Kitoh, H., Tanaka, Y., Sonoda, T., Kitamura, M., et al. (2005). The phenotypic spectrum of COL2A1 mutations. Hum. Mutat. 26, 36–43.
Orimo, H. (2010). The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nippon Med. Sch. 77, 4–12. doi: 10.1272/jnms.77.4
Pace, J. M., Wiese, M., Drenguis, A. S., Kuznetsova, N., Leikin, S., Schwarze, U., et al. (2008). Defective C-propeptides of the proalpha2(I) chain of type I procollagen impede molecular assembly and result in osteogenesis imperfecta. J. Biol. Chem. 283, 16061–16067. doi: 10.1074/jbc.m801982200
Philp, A. M., Collier, R. L., Grover, L. M., Davis, E. T., and Jones, S. W. (2017). Resistin promotes the abnormal type I collagen phenotype of subchondral bone in obese patients with end stage hip osteoarthritis. Sci. Rep. 7:4042.
Pyott, S. M., Tran, T. T., Leistritz, D. F., Pepin, M. G., Mendelsohn, N. J., Temme, R. T., et al. (2013). WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet. 92, 590–597. doi: 10.1016/j.ajhg.2013.02.009
Raff, M. L., Craigen, W. J., Smith, L. T., Keene, D. R., and Byers, P. H. (2000). Partial COL1A2 gene duplication produces features of osteogenesis imperfecta and Ehlers-Danlos syndrome type VII. Hum. Genet. 106, 19–28. doi: 10.1007/s004399900198
Ramirez, F., Tanaka, S., and Bou-Gharios, G. (2006). Transcriptional regulation of the human alpha2(I) collagen gene (COL1A2), an informative model system to study fibrotic diseases. Matrix Biol. 25, 365–372. doi: 10.1016/j.matbio.2006.05.002
Rauch, F., Lalic, L., Roughley, P., and Glorieux, F. H. (2010). Genotype-phenotype correlations in nonlethal osteogenesis imperfecta caused by mutations in the helical domain of collagen type I. Eur. J. Hum. Genet. 18, 642–647. doi: 10.1038/ejhg.2009.242
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894.
Schouten, J. P., McElgunn, C. J., Waaijer, R., Zwijnenburg, D., Diepvens, F., and Pals, G. (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 30:e57.
Skarp, S., Xia, J. H., Zhang, Q., Löija, M., Costantini, A., Ruddock, L. W., et al. (2020). Exome sequencing reveals a phenotype modifying variant in ZNF528 in primary osteoporosis with a COL1A2 deletion. J. Bone Miner. Res. 35, 2381–2392. doi: 10.1002/jbmr.4145
Taillandier, A., Domingues, C., De Cazanove, C., Porquet-Bordes, V., Monnot, S., Kiffer-Moreira, T., et al. (2015). Molecular diagnosis of hypophosphatasia and differential diagnosis by targeted next generation sequencing. Mol. Genet. Metab. 116, 215–220.
Thommesen, L., Stunes, A. K., Monjo, M., Grøsvik, K., Tamburstuen, M. V., Kjøbli, E., et al. (2006). Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J. Cell. Biochem. 99, 824–834. doi: 10.1002/jcb.20915
van Dijk, F. S., Huizer, M., Kariminejad, A., Marcelis, C. L., Plomp, A. S., Terhal, P. A., et al. (2010). Complete COL1A1 allele deletions in osteogenesis imperfecta. Genet. Med. 12, 736–741. doi: 10.1097/gim.0b013e3181f01617
Wang, X., Pei, Y., Dou, J., Lu, J., Li, J., and Lv, Z. (2015). Identification of a novel COL1A1 frameshift mutation, c.700delG, in a Chinese osteogenesis imperfecta family. Genet. Mol. Biol. 38, 1–7. doi: 10.1590/s1415-475738120130336
Keywords: osteogenesis imperfecta, spondyloepiphyseal dysplasia congenita, collagen, osteoporosis, next-generation sequencing
Citation: Tsai M-C, Chou Y-Y, Li C-Y, Wang Y-C, Yu H-W, Chen C-H and Chen P-C (2021) New Structural and Single Nucleotide Mutations in Type I and Type II Collagens in Taiwanese Children With Type I and Type II Collagenopathies. Front. Genet. 12:594285. doi: 10.3389/fgene.2021.594285
Received: 13 August 2020; Accepted: 22 June 2021;
Published: 28 July 2021.
Edited by:
Abjal Pasha Shaik, King Saud University, Saudi ArabiaReviewed by:
Yanqin Lu, Shandong First Medical University, ChinaCopyright © 2021 Tsai, Chou, Li, Wang, Yu, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng-Chieh Chen, pengchic@mail.ncku.edu.tw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.