 Michael B. Ranke
Michael B. Ranke- Children’s Hospital, University of Tuebingen, Tuebingen, Germany
The syndrome of impaired GH secretion (GH deficiency) in childhood and adolescence had been identified at the end of the 19th century. Its non-acquired variant (naGHD) is, at childhood onset, a rare syndrome of multiple etiologies, predominantly characterized by severe and permanent growth failure culminating in short stature. It is still difficult to diagnose GHD and, in particular, to ascertain impaired GH secretion in comparison to levels in normally-growing children. The debate on what constitutes an optimal diagnostic process continues. Treatment of the GH deficit via replacement with cadaveric pituitary human GH (pit-hGH) had first been demonstrated in 1958, and opened an era of therapeutic possibilities, albeit for a limited number of patients. In 1985, the era of recombinant hGH (r-hGH) began: unlimited supply meant that substantial long-term experience could be gained, with greater focus on efficacy, safety and costs. However, even today, the results of current treatment regimes indicate that there is still a substantial fraction of children who do not achieve adult height within the normal range. Renewed evaluation of height outcomes in childhood-onset naGHD is required for a better understanding of the underlying causes, whereby the role of various factors - diagnostics, treatment modalities, mode of treatment evaluation - during the important phases of child growth - infancy, childhood and puberty - are further explored.
Introduction
The fundamental findings relating to the chemical structure of pituitary growth hormone and its biological effects on growth and metabolism in various animals were described in the first half of the 20th century (1). The major driving forces in this field were Herbert Evans and his collaborators (2). By the beginning of the next half of the century, when the species specificity of primate GH in humans had been discovered and methods to purify GH from pituitaries of men and monkeys had been refined, the first studies to prove the efficacy of this peptide hormone were conducted. In 1958, human pituitary GH (pit-hGH) was shown to promote growth in a GH-deficient adolescent over a period of several months (3). Human and monkey pituitary GH revealed a variety of short term (days) metabolic effects in adolescents and adults with hypopituitary disorders (4). The era of pit-hGH ended in 1985, when hGH produced via recombinant technology became available. This initiated the era of virtually unlimited availability of r-hGH worldwide and the expansion of its use in adults with GHD, in children with growth disorders and for other indications.
The primary aim of this article is to review the effect of GH treatment on growth, predominantly in children and adolescents with GHD and to evaluate our current understanding of the factors affecting the magnitude of the response in the short- and long-term. Such an evaluation not only requires a review of the specific literature pertaining to treated cohorts but also necessitates a discussion – from a historical perspective – of the instruments and their suitability in establishing the diagnosis of GHD, along with the tools used to analyze the growth response during different developmental phases.
Classification of Growth Hormone Deficiency
By definition, GHD is a syndrome caused by the impaired secretion of GH. This can be the consequence of a disorder at the level of the pituitary itself and/or within the cascade of function and structures of the hypothalamus or brain which regulate its secretion. However, the wider understanding of the term GHD also includes disorders resulting from impaired action of GH at the cellular level. After recognizing that GH-dependent components of the IGF-family were involved in mediating the effects of GH, the concept was nurtured that IGF was at the center of a GH–IGF regulatory system (5). On the basis on this concept, a distinction between secondary IGF-deficiency [IGFD] (as in GHD) and primary IGFD (=non-GHD) was proposed (6, 7).
Although this was a logical approach and suited for the clinical sub-classification of the GHD syndrome, it was simplistic and did not do justice to the complexity of the IGF-system (5–14). The major peptides of the IGF system in blood - IGF-I, IGFBP-3 and ALS - are GH-dependent and their levels in blood are quantitatively related to the GH secreted (15). But their levels in blood are also dependent on many other factors, for instance, hormonal or nutritional status (16, 17). In addition, growth promotion at the cellular level of the epiphyseal growth plate requires the local presence of both IGF and GH, whose quantitative relationship with their circulating levels is not fully understood (11, 12).
From the clinical perspective, it needs to be understood that GHD is also classified according to descriptive characteristics rather than a uniform principle (7). Some examples are:
•the onset of its origin: congenital/non-acquired vs. acquired;
•the hormonal extent of a pituitary defect: isolated (GH deficiency only) vs. combined [with other pituitary hormone deficits];
•the known cause: causal [specific cause known] vs. idiopathic [cause unknown];
•the extent of GH impairment: complete vs. incomplete (partial);
•its existence over the lifespan: permanent vs. transient;
•the age of disease discovery: during infancy, childhood, adolescence, or adult life.
Prevalence – Incidence of GHD
Reports on the incidence or prevalence of GHD in children are scarce. In the pit-hGH era, when very short children (height < -3 SDS) used to be diagnosed and a GH cut-off to tests of < 5 ng/mL was applied, an incidence of 1:4000 and a prevalence of 1:5,000-30,000 were reported (18–20). During the r-hGH era, when the test cut-off was at 10 ng/ml, an incidence of 1:3,400 and a prevalence of 1:29,000 were reported (21). After r-hGH became available in 1987, a doubling of the incidence in childhood-onset GHD in Denmark was observed, which was similar to that in southern Germany (22, 23).
Diagnosing GHD in Children and Adolescents
There is no single tool to confirm GHD. Thus, the diagnosis must be established by means of a variety of symptoms, signs and test results. The interpretation of non-clinical investigations must always be in accordance with clinical findings. Quantitative results need to be based on methodologically correct procedures and must be compared with appropriate normative references. Abnormal test results should always be repeated, particularly if they do not correspond with other findings.
The diagnostic path to establishing GHD involves several steps:
•history (family, gestation and birth, individual),
•clinical investigation,
•anthropometrical (growth) evaluation,
•static biochemical tests,
•GH-related basic biochemical investigations,
•evaluation of GH secretion,
•imaging techniques,
•molecular genetics.
Commonly, the initial suspicion of GHD is proposed by a general practitioner or family physician, who observes signs of impaired growth; while the conclusive diagnosis of GHD is confirmed by paediatric endocrinologists in tertiary institutions. Therefore, in some medical environments, the criteria for referring such children from a lower level of child care to experts may differ from the criteria used by specialists to confirm GHD (24, 25).
Due to the complexity related to diagnosing and classifying GHD in childhood and adolescence, in particular in the less severe, non-acquired forms, a number of controversies had arisen which led to numerous publications by specialists, societies and expert groups over the years (26–30). In the author’s view, there are only a few aspects which are of particular significance in diagnosing and treating GHD during the main phases of growth - infancy, childhood, and puberty (which partly overlap) (31) – these will be considered in detail here.
GHD in Infancy and Very Early Childhood
While – in simplistic terms - postnatal growth during the childhood phase is apparently driven by parameters of the GH-IGF system, prenatal growth is primarily influenced by the insulin-nutrition environment. During the first months of life, GH blood levels are high, while those of IGF-I are low, presumably due to lower GH sensitivity during the growth phase of infancy, which, when it fades, is accompanied by an inverse trend: decline of GH and increase in levels of GH-dependent hormones (e.g., IGF-I, IGFBP-3) (32, 33). The dynamics of growth and the GH-IGF system during infancy and early childhood pose specific problems when diagnosing GHD during this period of life. In contrast to later childhood, the suspicion of GHD in the neonatal period is commonly neither driven by severe smallness at birth (34) nor by poor postnatal growth, but often by normo-insulinemic hypoglycaemia or/and protracted postnatal icterus (with elevated direct bilirubin), or/and underdeveloped external genitalia (phallus, clitoris, maldescensus testis). Besides hypoglycaemia, the other signs are commonly only present in the additional absence (also prenatally) of other pituitary (TSH, LH, FSH, ACTH) hormones.
Although conventional techniques to quantify GHD secretion as described below are generally not applicable during this phase of life, the diagnosis of GHD in suspected cases can be established without dynamic tests. Indications of GHD can be ascertained by means of basal IGF-I measurements and/or IGFBP-3 of < - 2 SD (sensitivity of 80%) (35) and via tests of GH levels using single serum drawn during hypoglycaemia (GH < 20 ng/mL) (36). In infants and toddlers very low normal levels of IGF-I make it difficult to distinguish normal from GHD (16). Therefore IGFBP-3 is the preferred diagnostic tool at this age. Additionally, filter paper samples used for neonatal screening also offer clues (GH < 7 ng/mL) (37), as does a series of low, randomly-measured GH levels.
Growth in infancy is very dynamic: body length at 2 years is about 40 cm greater than at birth. Height velocity (HV) decreases from about 25 cm/year during the first year to about 12 cm/year during the second year (38). About 50% of infants with congenital GHD deviate from the infancy component of growth (39) and height after one year declines below normal limits (40). However in many cases in which GHD was detected during childhood, low height velocity could have previously been observed in infancy (41, 42). On the other hand, feeding difficulties and failure to thrive may be misleading symptoms in terms of GHD. The careful evaluation of length and weight during regular post-natal care could thus lead to an increase in the fraction of children with suspected/diagnosed GHD at an early age.
Children who are diagnosed very early in life often suffer from a congenital disorder (cGHD), such as anatomical defects in the hypothalamic-pituitary region (e.g., pituitary stalk interruption syndrome [PSIS]), which can be visualized by means of neuroimaging (43) or by identifying other genetically-caused disorders (44, 45). Such cases are often associated with combined pituitary hormone deficiencies. Whether or not perinatal head trauma is a possibly relevant cause of GHD acquired at birth, as suggested in the past (46), is yet to be clarified. Differences in the characteristics of very young children with GHD as compared to those during childhood have been documented in a few series (47–49) and are listed in Table 1.
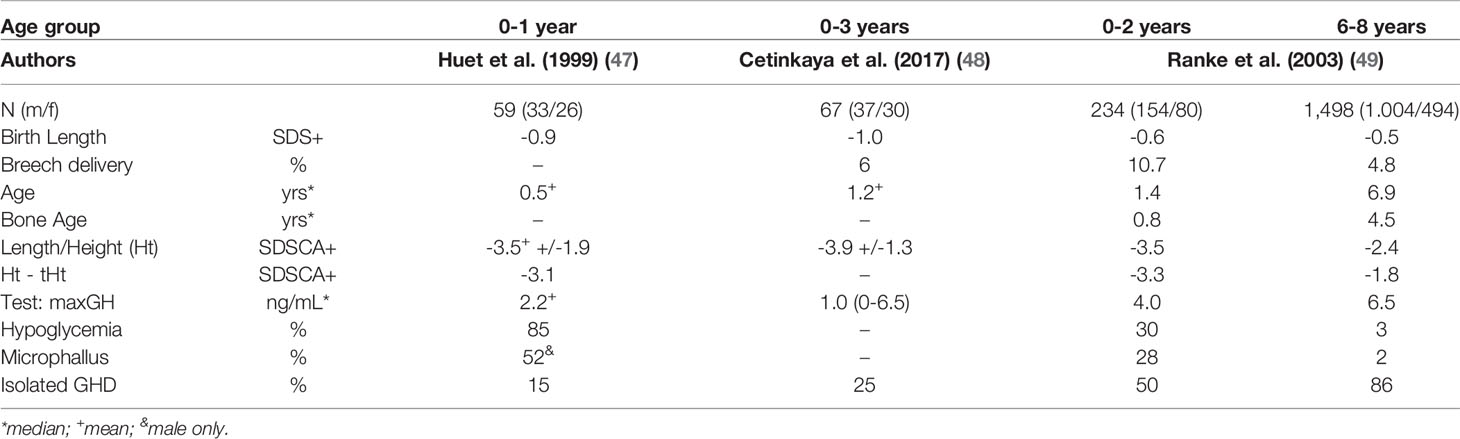
Table 1 Characteristics of very early onset of GHD compared to childhood onset.
Non-Acquired GHD During Childhood
Anthropometry
It is the observed deviation from normal growth – from about two years of age to the onset of puberty – that typically initiates exploratory steps towards diagnosing GHD. A comprehensive analysis of growth must include measurements of height, weight, head circumference, and other anthropometrical data to determine body proportions (e.g., sitting height, arm span); in addition, it is also imperative to apply methods to estimate the relative amount of fat mass (e.g., BMI, fat fold thickness, DXA). In order to visualize and/or calculate the extent of any deviation from normal values, appropriate references need to be applied. For the assessment of height, there are up-to-date and ethnically-appropriate references, which are commonly available for the corresponding population; and, in parallel, SD scores for chronological age (Ht SDSCA) should be calculated. By convention, a height measurement below –2.0 SDSCA defines short stature for a given population. In order to determine height in relationship to parental height, a familial “target height” must be calculated and transformed into an SD-score (THt SDS) based on the same references (50, 51). This information is then used to calculate the child’s height, corrected for its parental target height (cHt SDSCA = Ht SDSCA - THt SDS). A cHt below –1.3 SDSCA (equivalent to about the 10th centile), roughly denotes shortness outside the familial range. It is remarkable many recent national guidelines do not recommend cHT as a diagnostic criterium (25).
Height velocity [HV] - the change of height over time (cm/year) – expresses the dynamic growth process and is considered the “golden parameter” for any growth evaluation. However the calculation of HV necessitates taking a minimum of two height measurements in 3, 6 and 12-month intervals. The time required between two measurements, in order to obtain an accurate result, is a function of the underlying HV [the greater, the shorter] and the error of Ht measurement [the smaller, the shorter]. The HV SDSCA is calculated on the basis of appropriate numerical HV references, deriving from (difficult-to-obtain) longitudinal investigations (52). Moreover, the complex dynamics of height velocity over time, plus the common delay in developmental tempo in GHD, as evidenced by a delay in bone age [BA], makes HV - and even more so HV SDSCA - a diagnostic tool prone to error. Therefore, it is difficult to clearly distinguish between normal HV and one that is too low in children with suspected GHD. However, a HV SDSCA > -1.0 SDS (approx. 25th centile) is considered to be unlikely during childhood, in the context of non-acquired GHD (25, 26). A practical and probably more robust surrogate measure for HV is the change in height, expressed in terms of delta HtSDSCA, derived from two Ht measurements taken 6-12 months apart. A decrease in delta Ht SDS (deflection) of >0.25 SD over one year is considered to be a strong indicator of true growth disorder during childhood (53, 54). Since the diagnostic procedure for childhood non-acquired GHD often takes several months, and considering that height measurements were frequently documented in the past, it became evident that the inclusion of HV parameters strengthens the diagnostic process without unduly delaying treatment.
The appearance of a child with severe GHD can be conspicuous: there may be puppet-like features, with a relatively large neurocranium, slight truncal obesity, and small hands and feet, among other characteristics. However less attention has been given to the measurement of various relevant anthropometrical features and to compare them with the height data (in terms of SDSCA) of normal and short children (55, 56). Only few comprehensive references have documented a great variety of anthropometrical variables in children simultaneously (38, 57). Although such references may not match the population of the child in question, they need to be applied in order to ensure complex anthropometrical analyses. If different normative references for each parameter (e.g., height, weight, arm span) are used in calculating SD scores, a false picture will emerge. Investigations of body composition with the help of modern tools, such as DXA, BIA, CT and MRI, provide evidence of the negative change in the muscle to fat mass ratio that is typical for GHD children.
An x-ray of the hand and wrist is done to evaluate bone maturity [transformed into bone age (BA)]. If possible, it should be determined automatically in order to avoid a rater bias (58), but also to detect a primary bone disorder, as part of the evaluation for GHD. It is important to remember that, in GHD, a BA [yrs] > (CA – 1) [yrs] is not likely to be found in true GHD during childhood (59, 60).
Insulin-Like Growth Factors
The two most important GH-dependent static peptide hormones in blood that must be measured during the diagnostic work-up of GHD are insulin-like growth factor-1 (IGF-I) and the IGF-binding protein-3 (IGFBP-3). They are part of a complex system that regulates cellular growth (13). The immunoassay is a well-established method for measuring these peptides in body fluids (61, 62) and reference values of basal blood levels over the whole human age spectrum in both sexes have been established by means of various assays (16, 63, 64). Based on the results of IGF levels in blood, further GH testing may be required in short children in order to obtain compelling evidence for the true existence of GHD. The interpretation of IGF levels measured by means of this biochemical diagnostic process must include the results of the above-mentioned clinical and anthropometrical investigations (65).
There is a wealth of literature on the diagnostic utility of IGF-I and/or IGFBP-3 measurements in the case of childhood GHD (16, 66, 67). In most of these studies, groups of children with GHD, based on various results of diagnostic tests, were analyzed. The IGF results in groups with (often isolated idiopathic) GHD were compared with groups of children with similar clinical characteristics but who had been classified as non-GHD (e.g., idiopathic short stature [ISS] (68, 69). The criteria for the anthropometric work-up and the static biochemistry in the studies with patients during mid- to late childhood were not uniform; in addition, the modalities of GH quantification (assays, test procedures) and cut-off levels to tests (commonly between 5 and 10 ng/mL) varied between studies. Nevertheless, the overall results from studies in which a cut-off of 10 ng/mL of GH (maximum) was implemented show a rather uniform qualitative picture: For both IGF-I and IGFBP-3 (expressed as an SD score for age), a cut-off of about -2.0 SDS denoted lower sensitivity (the power to correctly confirm GHD) than specificity (the power to correctly exclude GHD) (16). Thus a normal level is likely to exclude GHD, but below normal levels do not prove GHD. When a GH test cut-off of 7-8 ng/mL was accepted as evidence of GHD in childhood, IGF-I levels of < -1.4 SDS demonstrated a sensitivity of 100% and a specificity of 33%. In the same cohort investigated, a IGFBP-3 level of < -0.2 SDS showed a sensitivity of 100% at a specificity of 14% (70). In many countries, an IGF-I level of < - 2.0 SDS is a requirement for the diagnosis of GHD during childhood (25). However, a note of caution should be given here: the reference ranges reported for children ensued from a number of different assays, which is why the derived SDSCA values of IGF-I or IGFBP-3 may differ considerably. New approaches for establishing multidimensional references may be developed in the future (17).
Defining Impaired hGH Secretion
The core issue for the diagnosis of GHD is to obtain proof of impaired GH secretion. This entails determining hGH in blood as well as exactly quantifying GH secretion in normal and short children. The possibility to measure minute quantities of hGH in blood, for clinical purposes, started with the first immunoassays in 1963; and a process of methodological refinement has followed ever since (71–73). This process has involved, among others, the development of international reference preparations - from pit-hGH [IRP 66/217; specific activity approx. 2 I.U./mg] to authentic r-hGH of the 22 kD variety [IRP 98/574, specific activity 3 I.U./mg] (74), in addition, it has advanced from the use of polyclonal antibodies to very specific monoclonal antibodies for (22 kD hGH) detection. Modern assays do not determine all GH variants, which may have biological functions different from 22 kD hGH (75).
The discovery of the pulsatility of pituitary GH secretion led to the recognition that it is not possible for single measurements to represent the overall amount of GH secreted. Consequently, the total daily amount of GH secreted began to be quantitated by means of various procedures over the whole age range (76, 77). Groups which used spontaneous GH secretion for the evaluation of the GH secretory status in children mostly took a frequent sampling approach (e.g., every 20 or 30 minutes) over 8-12 hours of sleep; and considered a maximum GH level of > 7 ng/mL and/or an integrated level of > 3 ng/mL) to be the approximate borders of normality in prepubertal children (70, 78). However this approach was not held to be feasible by most physicians involved in diagnosing GHD proper in pediatric endocrine practices (26). Nevertheless, the quantitation of spontaneous GH secretion remains a prerequisite for diagnosing one variety of GHD, namely, neurosecretory dysfunction (79).
The discovery that hypoglycemia can provoke a GH release, the magnitude of which can be taken as a surrogate for the secretion capacity (80, 81) initiated the identification of many such stimuli (36) which found their way into our clinical routine. However the mechanism of GH stimulation through such agents differs from their “stimulatory power”, due to the fact that their effects may also vary, depending on their susceptibility to metabolic and other influences (36, 82–84). In the search for a parameter that reveals normal/too low GH secretion in patients, clinicians opted for a plain and simple answer: the maximum level observed during a test. This set off the ongoing debate about “cut-off” levels, which basically depend on the GH assay and test procedure used. The low repeatability of all types of stimulation tests was acknowledged and the medical community agreed upon accepting only the maximum level of two tests in differentiating between GHD and non-GHD. In “standard” tests, a maximum level of >5- 10 ng/mL was accepted as normal in prepubertal children; on the other hand, it was recognized that test procedures involving GH-releasing hormone (GHRH) provoked a release of pituitary GH, which is about 2-4 fold higher than that seen in “classic” tests (85).
Since the amount of GH secreted spontaneously or through stimulation depends on other factors, such as age, sex, pubertal stage, body composition and nutritional stage; and also varies individually from day to day, it remains a very difficult task to establish normal references. Moreover, each child may also have an inherent set point of GH secretion for maintaining physiology. Thus, in order to define GHD in children by means of a complex diagnostic process, it is expedient to apply a cut-off range for GH levels rather than use a single cut-off.
GHD: Diagnosis at Early Pubertal Age
Anthropometry
At the time when puberty can be expected in normal children (86, 87), short children do not exhibit signs of puberty. Thus during this period, it is particularly difficult to differentiate between true GHD and idiopathic short stature (of the variety with pubertal delay) or hypogonadism (88). The diagnostic problems are mainly related to (1) establishing the onset of puberty, (2) the evaluation of growth, and (3) the issue of how to determine an impairment in GH secretion.
Tanner introduced the globally-used standards for the clinical stages of puberty (89). The onset of puberty in girls can be determined by palpating breast tissue, not by inspecting the breast, since breast tissue growth is an effect of estrogen. In boys, the onset of puberty is assumed at a mean testis volume of ≥4 ml, the volume being predominantly an indicator of an increase in the testicular seminiferous structures and not testosterone production. Testis volume is commonly estimated by comparison with an orchidometer (90). These procedures are prone to inaccuracies, which are not eliminated by applying new methods like sonography. The analysis of a pubertal growth spurt by means of mathematical algorithms (91–93) has shown that the onset (“take-off) of puberty - which is driven by hormones - is an exact indicator and may occur 6-36 months before the clinical signs mentioned above are evident.
For the diagnosis and quantification of a growth disorder, it particularly relevant to adequately compare an individual’s height with normative height references. According to historical data devised by Marshall and Tanner (86, 87), the pubertal stage B2 in girls normally occurs between about 8 und 13 years of age, whereas the pubertal stage G2 in boys normally occurs between about 10 and 14 years of age. The normal take-off of the pubertal growth spurt occurs at about 8-11 years of age in girls and 10-12 years of age in boys (38). Thus, in clinically prepubertal children, a height deviation from normal at pubertal age – expressed in terms of HtSDSCA - is falsely exaggerated, since the normal growth curve has left the childhood path and is dominated by the pubertal component of growth (93). A Belgian survey showed that 19% of 295 children diagnosed with IGHD were ≥ 11 years of age; similar results - 17% of 156 children - were found in a German study (21, 23). In these children, height should rather be compared with data based on childhood references that have been extrapolated (adjusted) into the pubertal age range (94, 95). It is not known whether bone age - instead of CA – would be suitable to correct the error of HtSDS calculations based on CA. This aspect is even more relevant in terms of height velocity, for which adjusted HV references are available (96). Height velocity shows a marked prepubertal nadir which is more pronounced the longer puberty is delayed (38, 97). This is why, in the author’s view, a low HV should be interpreted with great caution in children during the pubertal age. These anthropometrical considerations can be effective in correcting the calculated growth parameters for delayed puberty and may increase the likelihood of classifying short children correctly before biochemical testing is done. For the static GH-dependent parameters, IGF-I and IGFBP-3, which also increase during hormonal puberty take-off, similar considerations should apply; in addition, adjusted references should be published in order to avoid the falsely low calculations of SD scores for age. This may avert inappropriate treatment being given on the basis of incorrect (false positive) classification of isolated naGHD during the pubertal age.
Impaired GH Secretion and Priming
The next and even more strongly debated issue is the question of how to interpret GH test results during the pubertal age. Puberty onset varies between populations, but as discussed above, starts at the earliest at about 8 years in girls and 10 years in boys and is accompanied by marked hormonal changes (98, 99). We know today that the amount of GH secreted is augmented during puberty, as a result of estrogens secreted in both sexes (100). While there seems to be no major change in GH secretion during mid-childhood, the total amount of spontaneously secreted GH during puberty is increased (78) as are the maximal levels of GH observed in varying test procedures (36, 101, 102). Logically, this means that higher cut-off levels should mark subnormal GH secretion in pubertal (GHD) children. In contrast, a (short) child who is still prepubertal during the pubertal age may secrete GH amounts considered to be too low – but only on grounds of non-existing puberty. The same reasoning applies for the static IGF parameters that are not adapted for delayed puberty.
To avoid such misclassification, it was proposed that GH testing in these children should be conducted after exposing them to sex steroids (called “sex-steroid priming”) to briefly induce sex steroid augmented GH secretion (103). Unfortunately, this procedure, involving short exposure to estrogen (in girls) or aromatizable androgens or estrogen (in males), is not standardized. Nevertheless, it has been shown that such priming leads to enhanced maximal GH levels in tests (36, 103, 104). However the endocrine community is still divided on this issue (25, 26, 105, 106). It is likely that the wish to diagnose non-acquired GHD at pubertal age will diminish when the anthropometric and other tools mentioned above are valued for facilitating the correct interpretation of data in the context of naGHD. Some examples of characteristics of children at the timepoint of diagnosis, recorded over the past 50 years, are listed in Table 2 (107–110).
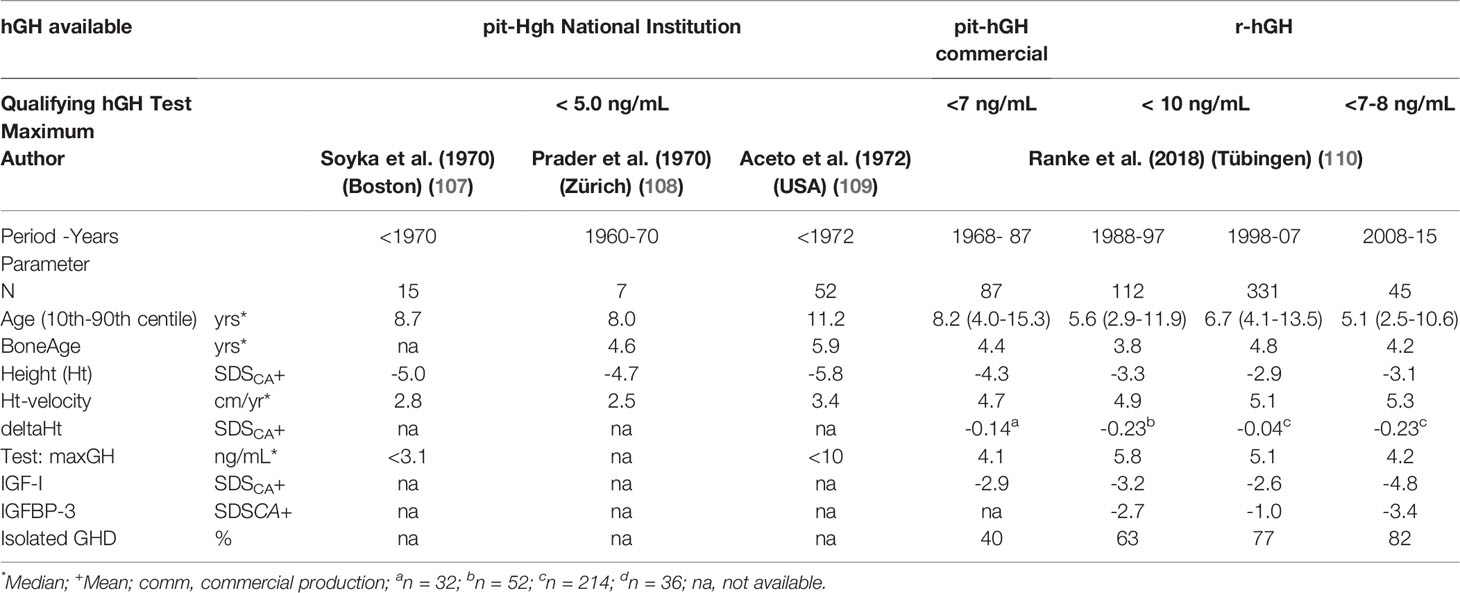
Table 2 Characteristics of children and adolescents with non-acquired GHD (idiopathic GHD [IGHD] plus congenital GHD [cGHD]) at diagnosis).
Treatment of GHD With hGH
Aims of GH Treatment
In GHD, replacement with hGH aims at the normalization of deviant aspects of growth, body composition and body function. In children and adolescents, the issue of hGH efficacy is primarily associated with growth: rapid catch-up growth, normal maintenance growth, appropriate timing and magnitude of pubertal growth, and the achievement of an adult height within the normal range. In addition, efficacy in children with GHD should also include the achievement of normal body composition and functioning, as well as the normalization of biochemical abnormalities associated with GHD during post-adolescence and throughout adult life.
Dosing and Mode of Application of hGH
The first patient to receive pit-hGH through Maurice Raben was initially given 1 mg, injected twice a week (b.i.w.) i.m.; later, the dose was raised to 3 mg, three times per week (t.i.w.). Raben administered his pit-hGH powder after reconstituting it in solvent (3). In subsequent years, pit-hGH units were devised, based on the growth response as well as the results of bio-assays using hypophysectomized female rats (111). More refined methods of purification led to a product with a potency of about 2 IU/mg (112). A dose effect in GHD – 5 IU b.i.w. vs. 10 IU b.i.w. - was observed by Preece et al. (113) Frazier described a linear-log relationship to the induced height velocity that resulted from doses ranging between at least 30 mIU/kg and 100 mIU/kg body weight t.i.w (114). The potency of recombinant hGH preparations was validated against international reference preparation with modern assays: 2.6 IU/mg for meth-r-hGH and 3.0 IU/mg authentic r-hGH. The amount of GH secreted - as evaluated by deconvolution analysis - was estimated to be about 20 µg/kg per body weight/day before puberty and about twice as high thereafter (76). The current starting doses of r-hGH, approved by authorities for prepubertal children, vary between a range of about 25-43 µg/kg body weight per day (115, 116) but may exceed this margin during puberty.
Pit-hGH was often administered using the total content of one ampule (2-4 IU), 2-3 times i.m. per week. After studies showed that the same amount could result in higher growth rates – in the long and short term - by dividing it into daily injections (117–119), daily s.c. injections became standard practice. GH doses are calculated either according to body weight (amount/kg BW) or per body surface (amount/m2 BS), with the latter precluding overdosing in obese patients. Today, exact doses can be applied easily with the help of “pens”, which may also allow monitored self-application (120). The role of long-acting GH variants for the treatment of GHD will be evaluated in the future (121).
Adherence
Adherence (compliance) is an essential prerequisite for any therapy to be effective. The risk of non-adherence in GHD is high, because GH must be injected daily (by proxy or by patients) over many years. Great differences were found – mostly in short-term growth - in studies on this subject, particularly in terms of the method of recording adherence, the characteristics of the cohorts investigated and the quantification of missed injections (122–125). Generally, the level of adherence appears to be high during the important but less dose-dependent first year of treatment (126), but it is lower thereafter, particularly in independent adolescents (127). Even one missed dose per week during the first treatment year in children results in a loss of height gain of 0.11 SD (122), a number which adds up to a substantial figure over time. Due to the great heterogeneity of causes (e.g., discrepancy to expectation, social circumstances, injection problems), strategies to prevent non-adherence must be individually adapted (121, 124, 127, 128).
Evaluation of the Growth Response and Results to GH Therapy in GHD
There have been roughly four phases of GH treatment from the time treatment with pit-hGH was first reported in 1958: (a) the experimental phase with pit-hGH (1958-approx. 1962), (b) the era of greater availability of pit-hGH (1962-1985), (c) the early era of r-hGH (1985–2000), and (d) the “consolidated” era of r-hGH (> 2000). The total growth process during GH treatment of GHD, starting with prepubertal age, can be divided into: (a) the initial phase of the first 2-3) years, which mark the phase of catch-up growth, (b) the childhood growth phase and (c) the pubertal growth phase, that ends in (d) the period in which (near) adult height is reached.
Prepubertal Growth Phase
Response Evaluation
The response to GH treatment is mostly analyzed in annual intervals and can be expressed in terms of height velocity (HV; cm/yr), change in HV in comparison to a previous period, HV SDSCA and the resulting change (delta HV SDSCA) (129) or in terms of delta HT SDSCA calculated over a certain period of time with treatment (prepubertal years, total puberty, start of GH to NAH). Pure HV (cm/yr) is a robust term and also practical as it can be visualized in a growth chart; however, it provides little exact information when measurements exceed the normal range. The expression of HV in terms of SD scores or changes over time is problematic, particularly during infancy and the pubertal age. During the catch-up phase and over longer periods of time, growth can also be described by means of mathematical algorithms (130–134).
Several cut-off levels for distinguishing between a normal and poor response during the first treatment year have been proposed: a change of ≥ 3 cm/year in HV as compared to pretreatment values (135), HV SDS ≥ mean – 1 SDS (136), HV SDS (for sex and age in normal children) ≥ + 1 SDS (52), delta Ht SDS ≥ +0.3 SDS or +0.5 SDS (137, 138). However comparisons led to inconsistent results (139).
Empirical Response Targets
Rather than using normal references for evaluating the response to GH, it was proposed that results should be compared with the response of other treated patients. Based on large numbers of treated prepubertal children, who were observed in pharmaco-epidemiological surveys (NCGS and KIGS), references for HV (cm/yr) or delta Ht SDS were published (136, 138). These “height velocity targets (HVT)” took into consideration the diagnosis, sex, and age in prepubertal children from 4-13 years of age, but examined only the mean GH dose of the cohort. Based on NCGS data (136), HV targets were devised in graphical terms for both male and female children with IGHD and OGHD (maximum GH in tests: <10 ng/mL) for the first treatment year. The mean GH dose given was 0.30 mg/kg per week. Based on KIGS data (138) references for HV and delta Ht SDS were presented as graphs as well as numerically for prepubertal children with both severe (maximum GH in tests: < 5 ng/mL) and less severe (maximum GH in tests: 5-10 ng/mL) GHD, during the 1st and 2nd treatment year. The mean GH dose given was 0.22 mg/kg per week. The HVs of the GHD cohorts in the NCGS study were very similar to the HVs of the “severe” GHD cohort in KIGS.
Growth Prediction
Another approach to evaluate the response of a treated patient is to compare the response variable (e.g., HV (cm/yr), delta Ht SDS) during a certain growth phase with the most likely expected response (and its error, at the start of each treatment phase) based on prediction algorithms derived from large cohorts. The advantage of this approach, as against using HVTs, is that validated prediction models consider a multitude of characteristics of an individual, the most important being the individual GH dose applied. The problem is to keep the error of prediction as low as possible. This error tends to rise when an increasing number of predictors that are not standardized are included. Several approaches have been used to develop prediction models (140–145). The observed and the predicted growth response can be compared and the difference can be expressed in terms of an ‘index of responsiveness’ (IOR) = [(observed response –predicted response)/error of prediction], which is a surrogate for the potential of an individual to respond (responsiveness) to GH, as compared to matched patients. An IoR below –1.0 denotes a poor response. Prediction models for various growth phases and diagnoses have been developed (145, 146) and are also available in the form of a software medical device (147). Prediction models will be developed further with the emerging field of pharmacogenomics (148, 149). Their applicability will expand with the growing importance of new electronic (self-)learning tools in medicine and in terms of optimizing cost-effective treatment.
Apart from the growth response, IGF-I targets have also been proposed as a means to guide and optimize dosing (150–153). Advocates of this approach point out that it offers a more cost-effective use of GH. Overall, the evaluation of the response to treatment, regardless of the tools used - particularly but not exclusively during the first phase of treatment - is of great importance in order to ensure an optimal outcome of growth in a treatment strategy which includes the prevention of non-adherence and an efficacious use of GH.
Pubertal Growth Phase
Clinically, pubertal growth is the phase between the first appearance of clinical pubertal markers – breast in girls, testis volume in boys – and the end of growth due to the closure of the epiphyses of the long bones (89). In practice, the near end of growth is commonly assumed if the HV is below 2 cm/year and bone age is above 14 years in girls and 16 years in boys (154). Since hormonal changes take effect before clinical markers are noticeable, the pubertal growth phase is actually longer (95, 155, 156). Pubertal growth is governed by the interaction of sex steroids (estrogens and androgens in both sexes) with the activated GH-IGF system (100, 157) and its combined effects on the skeletal growth targets (158, 159). Several specific issues exist with respect to GHD treatment during pubertal growth: the GH dosing, the timing and length of puberty (starting age vs. end of growth) and the choice of sex steroid in the case of gonadotropin deficiency.
Bourguignon (160) discovered that total pubertal growth (TPG) is inversely correlated to age at onset of puberty in normal children, but that this did not affect final adult height. This means that the partial contribution of pubertal growth to total growth is inversely correlated to the prepubertal fraction. Accordingly, in idiopathic GHD (non-acquired GHD), TPG was found to be positively correlated with HT at puberty onset and at age at the end of growth and negatively correlated with age at puberty onset and that GH dose only has a minor effect (161). Mauras showed that a doubling of the prepubertal GH dose during puberty, over four years, results in only about 4 cm of additional gain in TPG (162). Thus, the extra gain in height by means of r-hGH during puberty is much more expensive. Results of studies comparing males and females with spontaneous or induced puberty showed a smaller gain in the induced groups, since they were older at puberty onset (Table 3) (163–165). However the lower pubertal gain in females is probably the result of sub-optimal estrogen replacement, in terms of timing, dose and preparation (166). Considering the fact that TPG only accounts for about 20% of total postnatal growth, the aim should be to normalize height well before puberty onset. It is a common observation that the relative height attained in terms of SD scores for age at puberty onset can be maintained even with prepubertal GH doses. Delaying puberty onset and prolonging the whole pubertal phase - with drugs suppressing puberty, such as GnRH (167) and/or increasing GH doses at puberty onset (e.g. doubling the dose over pre-pubertal levels) - are approaches to be considered in individual cases with non-acquired GHD as a kind of “rescue attempt” to improve adult height. By doing this, however, the well-known phenomenon of the acromegaloid phenotype of puberty may also be overly augmented.
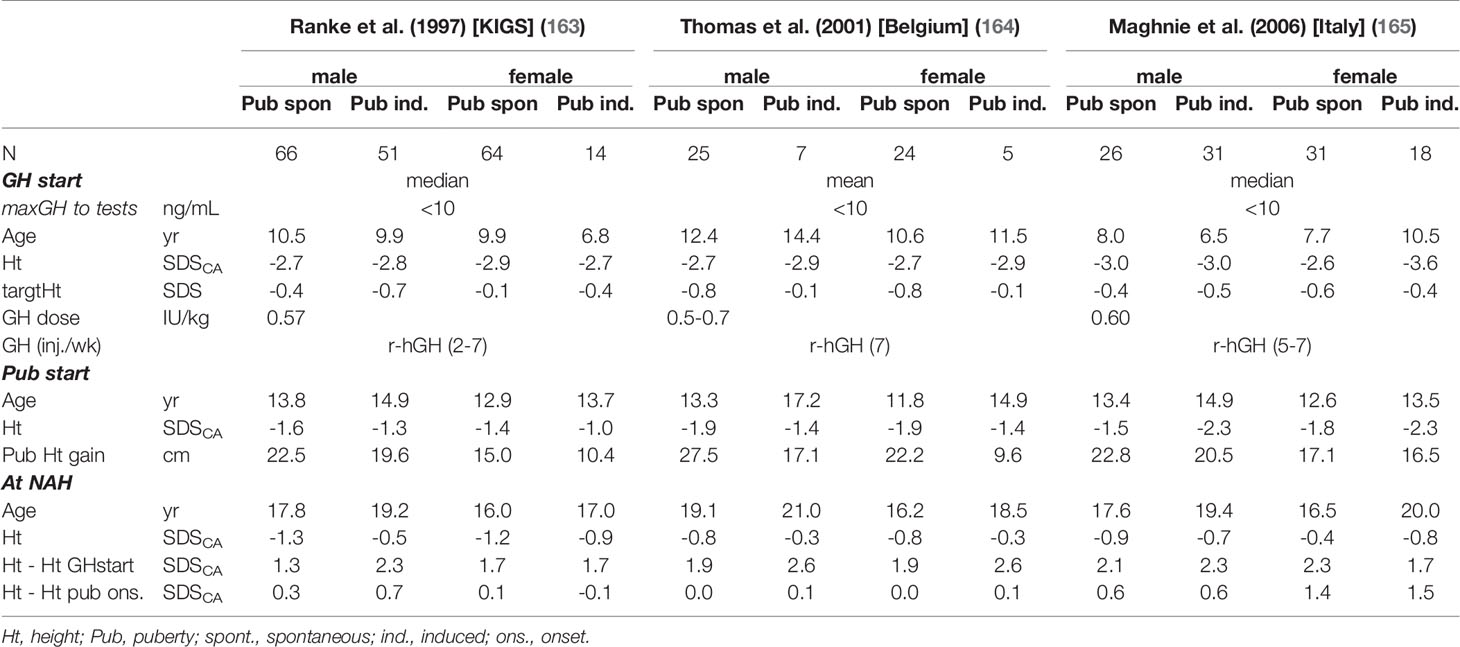
Table 3 Examples of height in children with non-acquired GHD: start GH, puberty onset (spontaneous vs. induced), near adult height (NAH).
Adult Height Reached
Several reports were published after years of treatment with pit-hGH in which the adult height outcomes achieved in non-acquired GHD (often called IGHD) were described. These results were summarized in reviews (168–170). As exemplified in Table 4, (169, 171–174, 176) these patients had been severely GH deficient (maximum in tests < 7.5 ng/mL) and were relatively old (approx. mean age: 13 yrs) at diagnosis and GH start. These characteristics were not only due to a selection bias, since the oldest patients at start are the earliest to reach their (near) end of growth. On the other hand, the patients treated during the pit-hGH era were very short (mean height at GH start < -4.0 SDS) and were given dosages of about 8–12 IU of pit-hGH from various sources, injected 2-3 times per week i.m., and the total amount of one ampule often contained 4 (2) I.U. After about > 5-6 years of treatment, an adult height of about -3.0 SDS was reached in patients with spontaneous puberty, while those with induced puberty reached a height of about -1.5 SDS. Females tended to be younger and shorter at start but reached a lower adult height.
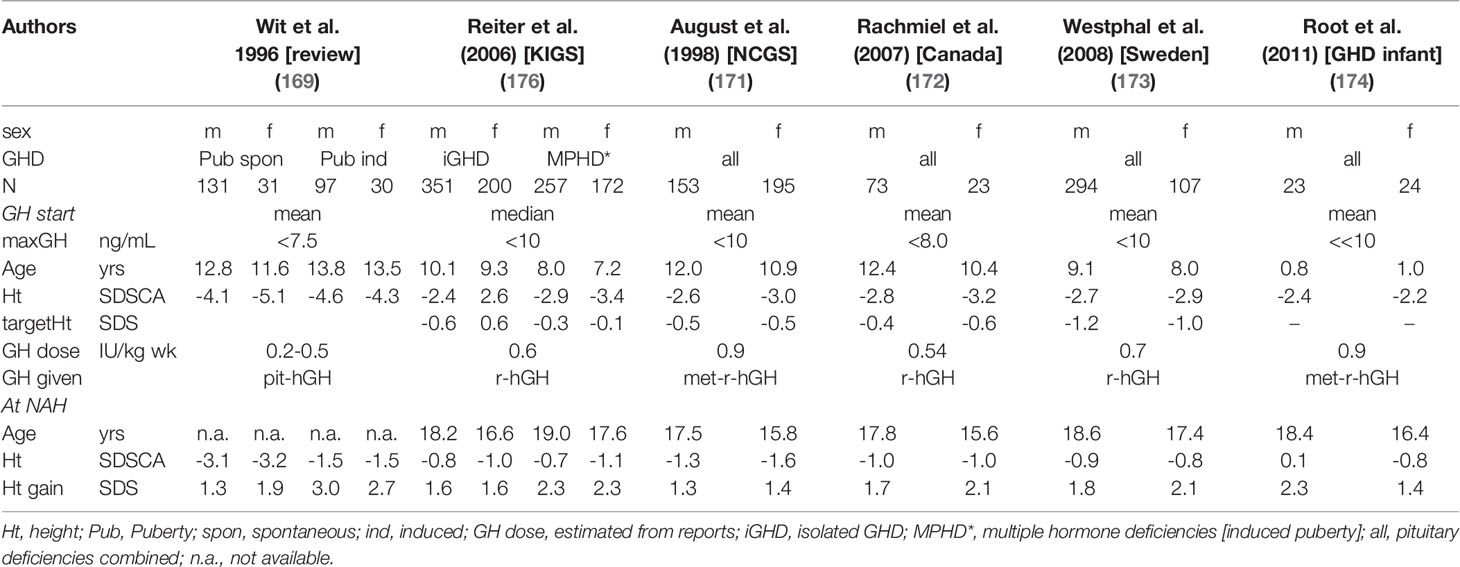
Table 4 Examples of groups of non-acquired GHD patients treated to NAH.
Patients during the early r-hGH era were reported to be less short (height about –2.9 SDS). With r-hGH doses of about 0.5 IU/mg/wk, injected in 3-7 fractions s.c. per week, they reached a height of about -1.4 SDS (164, 171, 175). As illustrated in Table 4, more recent patients who received somewhat higher doses and daily r-hGH injections reached a near adult height (NAH) mostly within the lower half of the normal range and closer to their calculated target height. However it should be remembered that in some populations there is a positive secular trend in adult height between generations in the order of about 0.4 SDS (54). Again, in such studies, females had a slightly lower height outcome. Japanese children with IGHD, who were treated with slightly lower doses compared to Europe/USA, achieved a slightly lower total height gain (176–178). Remarkably, practically all children treated as toddlers, for predominantly congenital organic GHD and MPHD, reached completely normal height (174).
Several authors have examined the factors correlating with NAH by means or regression analyses (116, 164, 165, 172, 173, 175). On the whole, the results of these studies revealed certain factors that correlated positively with NAH: height at GH start, mid-parental height, duration of treatment, GH dose, and the magnitude of the first year response to GH. On the other hand, the factors correlating negatively with NAH are: age at GH start and the severity of GHD (maxGH in tests, MPHD). In randomized studies, the long-term effect of GH dosage, in terms of NAH, was only marginally positive (179, 180). This may be due to the fact that childhood and pubertal growth are evaluated together: however, during childhood growth there is high sensitivity to GH, whereas during puberty there is low sensitivity to GH. The negative correlation of the outcome with the maximum GH level to testing may also suggest that the high GH cut-off may lead to the inclusion of non-/less severe GHD patients (e.g. ISS) who exhibit overall lower responsiveness to GH treatment. The negative effect of patients with MPHD is probably the result of an inappropriate induction and/or maintenance of puberty in children with gonadotropin deficiency. This needs further evaluation.
Safety of hGH Replacement in Children
Safety issues during GH replacement may be related to the medical substance itself, may be due to the formulation of the drug (e.g. impurities, additives for drug formulation), be the result of the genuine (normal) effects (e.g., on the growth of bones, on other tissues, or be related to its metabolic action); they may be due to inappropriate dosages or a genuine incompatibility with the patient being treated (181). During the pit-hGH era, when relatively crude GH material were applied in low doses, local effects (pain, lipoatrophy) were occasionally observed (182). Due to the transmission of prions through some pit-hGH preparations, which caused the deadly Creutzfelt-Jakob disease, this era ended (183–185).
After the approval of r-hGH preparations, the analysis, detection and prevention of adverse effects became an integral part of large surveillance studies in children (186). Detailed reviews of the safety of r-hGH in children and adolescents are available (181, 187). A rare side-effect of normal GH action on accelerated bone growth in children is the slipped capital femoral epiphysis [SCFE] (188). The normal metabolic effects of sodium and water retention may cause benign intracranial hypertension (189). The anti-insulin effect of GH may cause impaired glucose tolerance or accelerate the development of DM2 in predisposed children with GHD (190). An early report associating pit-hGD with an increased risk of colonic cancer in GHD (191) raised a critical discussion about the potential role of the GH-IGF axis in cancer pathogenesis (192). A particularly controversial multinational survey on the safety and appropriateness of GH in Europe (SAGhE), which investigated mortality in adults who had received GH treatment in childhood, however presented inconclusive results (191, 193, 194). There is strong evidence that replacement with r-hGH in children and adolescents with non-acquired GHD is safe, as they receive the usual dosage range and have a low risk of other diseases (195, 196); nevertheless, it is prudent to ensure structured long-term follow-up and monitoring of IGF parameters during GH replacement (153, 187).
Summary
For more than a century, it has been known that the growth hormone deficiency syndrome (GHD) affects the entire life span. Developments over many decades have led to the understanding of the key modalities, such as anthropometrical and biochemical methods, that facilitate the correct diagnosis of non-acquired – in particular isolated – GHD. However there are still a number of difficulties to overcome in order to arrive at the diagnosis as early and as properly as possible, particularly during the late childhood phase. The precise application of known techniques and principles in anthropometry as well as the prudent interpretation of test results is the imperative task of those entrusted with the medical care of children. During the past decades, replacement with GH has led to improvements in height gain during childhood and in final adult height. Yet a sizeable fraction of children does not achieve optimal adult height. Therefore the modalities for evaluating growth and the tools for adjusting treatment appropriately need to be further individualized and optimized, not only with regard to stature but also in terms of safety and costs. This entails combining the known principles of individual endocrine care with novel evidenced-based tools that substantiate the results of analyses before, during and after treatment.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The author would like to thank Priscilla Herrmann for her assistance in preparing this manuscript.
References
1. Ranke MB, Wit JM. Growth Hormone - Past, Present and Future. Nat Rev Endocrinol (2018) 14:285–300. doi: 10.1038/nrendo.2018.22
2. Grumbach MM. Herbert McLean Evans, Revolutionary in Modern Endocrinology: A Tale of Great Expectations. J Clin Endocrinol Metab (1982) 55:1240–7. doi: 10.1210/jcem-55-6-1240
3. Raben MS. Treatment of a Pituitary Dwarf With Human Growth Hormone. J Clin Endocrinol Metab (1958) 18:901–3. doi: 10.1210/jcem-18-8-901
4. Beck JC, Mcgarry EE, Dyrenfurth I, Venning EH. The Metabolic Effects of Human and Monkey Growth Hormone in Man. Ann Intern Med (1958) 49:1090–105. doi: 10.7326/0003-4819-49-5-1090
5. Hwa V, Oh Y, Rosenfeld RG. The Insulin-Like Growth Factor-Binding Protein (IGFBP) Superfamily. Endocr Rev (1999) 20:761–87. doi: 10.1210/edrv.20.6.0382
6. Rosenfeld RG. The IGF System: New Developments Relevant to Pediatric Practice. Endocr Dev (2005) 9:1–10. doi: 10.1159/000085716
7. Wit JM, Ranke MB, Kelnar CJH. ESPE Classification of Paediatric Endocrine Diagnoses. Horm Res (2007) 68:1–120.
8. Yakar S, Kim H, Zhao H, Toyoshima Y, Pennisi P, Gavrilova O, et al. The Growth Hormone-Insulin Like Growth Factor Axis Revisited: Lessons From IGF-1 and IGF-1 Receptor Gene Targeting. Pediatr Nephrol (2005) 20:251–4. doi: 10.1007/s00467-004-1613-y
9. Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of Insulin-Like Growth Factors in Embryonic and Postnatal Growth. Cel (1993) 75:73–82.
10. Oberbauer AM, Peng R. Growth Hormone and IGF-I Stimulate Cell Function in Distinct Zones of the Rat Epiphyseal Growth Plate. Connect Tissue Res (1995) 31:189–95. doi: 10.3109/03008209509010810
11. Isaksson OG, Jansson JO, Gause IA. Growth Hormone Stimulates Longitudinal Bone Growth Directly. Science (1982) 216:1237–39. doi: 10.1126/science.7079756
12. Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence Supporting Dual, IGF-I-Independent and IGF-I-Dependent, Roles for GH in Promoting Longitudinal Bone Growth. J Endocrinol (2004) 180:247–55. doi: 10.1677/joe.0.1800247
13. Kaplan SA, Cohen P. The Somatomedin Hypothesis 2007: 50 Years Later. J Clin Endocrinol Metab (2007) 92:4529–35. doi: 10.1210/jc.2007-0526
14. Forbes BE, Blyth AJ, Wit JM. Disorders of IGFs and IGF-1R Signaling Pathways. Mol Cell Endocrinol (2020) 518:111035. doi: 10.1016/j.mce.2020.111035
15. Blum WF, Albertsson-Wikland K, Rosberg S, Ranke MB. Serum Levels of Insulin-Like Growth Factor I (IGF-I) and IGF Binding Protein 3 Reflect Spontaneous Growth Hormone Secretion. J Clin Endocrinol Metab (1993) 76:1610–6. doi: 10.1210/jcem.76.6.7684744
16. Blum WF, Böttcher C, Wudy SA. Insulin-Like Growth Factors and Their Binding Proteins. In: Ranke MB, Mullis PE, editors. Diagnostics of Endocrine Function in Children and Adolescents. Basel: Karger (2011). p. 157–82.
17. Ranke MB. A Proposal to Develop New References for Serum IGF-I Levels in Children. J Clin Res Pediatr Endocrinol (2020) 12:140–2. doi: 10.4274/jcrpe.galenos.2020.2020.0040
18. Vimpani GV, Vimpani AF, Lidgard GP, Cameron EH, Farquhar JW. Prevalence of Severe Growth Hormone Deficiency. Br Med J (1977) 2:427–30. doi: 10.1136/bmj.2.6084.427
19. Rona RJ, Tanner JM. Aetiology of Idiopathic Growth Hormone Deficiency in England and Wales. Arch Dis Child (1977) 52:197–208. doi: 10.1136/adc.52.3.197
20. Lindsay R, Feldkamp M, Harris D, Robertson J, Rallison M. Utah Growth Study: Growth Standards and the Prevalence of Growth Hormone Deficiency. J Pediatr (1994) 125:29–35. doi: 10.1016/s0022-3476(94)70117-2
21. Thomas M, Massa G, Craen M, de Zegher F, Bourguignon JP, Heinrichs C, et al. Prevalence and Demographic Features of Childhood Growth Hormone Deficiency in Belgium During the Period 1986-2001. Eur J Endocrinol (2004) 151:67–72. doi: 10.1530/eje.0.1510067
22. Stochholm K, Gravholt CH, Laursen T, Jørgensen JO, Laurberg P, Andersen M, et al. Incidence of GH Deficiency - A Nationwide Study. Eur J Endocrinol (2006) 155:61–71. doi: 10.1530/eje.1.02191
23. Schweizer R, Blumenstock G, Mangelsdorf K, Ehehalt S, Rössner L, Dorn T, et al. Prevalence and Incidence of Endocrine Disorders in Children: Results of a Survey in Baden-Wuerttemberg and Bavaria (EndoPrIn BB) 2000-2001. Klin Padiatr (2010) 222:67–72. doi: 10.1055/s-0029-1241868
24. van Dommelen P, van Zoonen R, Vlasblom E, Wit JM, Beltman M. Expert Committee Guideline for Referring Short or Tall Children in Preventive Child Health Care. Acta Paediatr (2020) 110:1231–38. doi: 10.1111/apa.15625
25. Binder G, Reinehr T, Ibáñez L, Thiele S, Linglart A, Woelfle J, et al. GHD Diagnostics in Europe and the US: An Audit of National Guidelines and Practice. Horm Res Paediatr (2019) 92:150–6. doi: 10.1159/000503783
26. Collett-Solberg PF, Ambler G, Backeljauw PF, Bidlingmaier M, Biller BMK, Boguszewski MCS, et al. Diagnosis, Genetics, and Therapy of Short Stature in Children: A Growth Hormone Research Society International Perspective. Horm Res Paediatr (2019) 92:1–14. doi: 10.1159/000502231
27. Guidelines for the Use of Growth Hormone in Children With Short Stature. A Report by the Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. J Pediatr (1995) 127:857–67. doi: 10.1016/s0022-3476(95)70019-6
28. Growth Hormone Research Society. Consensus Guidelines for the Diagnosis and Treatment of Growth Hormone (GH) Deficiency in Childhood and Adolescence: Summary Statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab (2000) 85:3990–3. doi: 10.1210/jcem.85.11.6984
29. Wilson TA, Rose SR, Cohen P, Rogol AD, Backeljauw P, Brown R, et al. Lawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics Committee. Update of Guidelines for the Use of Growth Hormone in Children: The Lawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics Committee. J Pediatr (2003) 143:415–21. doi: 10.1067/s0022-3476(03)00246-4
30. Shalet SM, Toogood A, Rahim A, Brennan BM. The Diagnosis of Growth Hormone Deficiency in Children and Adults. Endocr Rev (1998) 19:203–23. doi: 10.1210/edrv.19.2.0329
31. Karlberg J. On the Modelling of Human Growth. Stat Med (1987) 6:185–92. doi: 10.1002/sim.4780060210
32. Leger J, Noel M, Limal JM, Czernichow P. Growth Factors and Intrauterine Growth Retardation. II. Serum Growth Hormone, Insulin-Like Growth Factor (IGF) I, and IGF-Binding Protein 3 Levels in Children With Intrauterine Growth Retardation Compared With Normal Control Subjects: Prospective Study From Birth to Two Years of Age. Study Group of IUGR. Pediatr Res (1996) 40:101–7. doi: 10.1203/00006450-199607000-00018
33. Ogilvy-Stuart AL. Growth Hormone Deficiency (GHD) From Birth to 2 Years of Age: Diagnostic Specifics of GHD During the Early Phase of Life. Horm Res (2003) 60:2–9. doi: 10.1159/000071219
34. Gluckman PD, Gunn AJ, Wray A, Cutfield WS, Chatelain PG, Guilbaud O, et al. Congenital Idiopathic Growth Hormone Deficiency Associated With Prenatal and Early Postnatal Growth Failure. J Pediatr (1992) 121:920–23. doi: 10.1016/s0022-3476(05)80342-7
35. Jensen RB, Jeppesen KA, Vielwerth S, Michaelsen KF, Main KM, Skakkebaek NE, et al. Insulin-Like Growth Factor I (IGF-I) and IGF-Binding Protein 3 as Diagnostic Markers of Growth Hormone Deficiency in Infancy. Horm Res (2005) 63:15–21. doi: 10.1159/000082456
36. Ranke MB. Growth Hormone Deficiency: Diagnostic Principles and Practice. In: Ranke MB, Mullis PE, editors. Diagnostics of Endocrine Function in Children and Adolescents. Basel: Karger (2011). p. 102–37.
37. Binder G, Weber K, Rieflin N, Steinruck L, Blumenstock G, Janzen N, et al. Diagnosis of Severe Growth Hormone Deficiency in the Newborn. Clin Endocrinol (Oxf) (2020) 93:305–11. doi: 10.1111/cen.14264
38. Prader A, Largo RH, Molinari L, Issler C. Physical Growth of Swiss Children From Birth to 20 Years of Age. First Zurich Longitudinal Study of Growth and Development. Helv Paediatr Acta Suppl (1989) 52:1–125.
39. Karlberg J, Albertsson-Wikland K. Infancy Growth Pattern Related to Growth Hormone Deficiency. Acta Paediatr Scand (1988) 77:385–91. doi: 10.1111/j.1651-2227.1988.tb10665.x
40. Wit JM, van Unen H. Growth of Infants With Neonatal Growth Hormone Deficiency. Arch Dis Child (1992) 67:920–4. doi: 10.1136/adc.67.7.920
41. Mayer M, Schmitt K, Kapelari K, Frisch H, Köstl G, Voigt M. Spontaneous Growth in Growth Hormone Deficiency From Birth Until 7 Years of Age: Development of Disease-Specific Growth Curves. Horm Res Paediatr (2010) 74:136–44. doi: 10.1159/000281020
42. Noda M, Sato N, Tanaka T. Growth Failure Starts From Early Infancy in Children With Short Stature at Age 6. Clin Pediatr Endocrinol (2015) 24:1–10. doi: 10.1297/cpe.24.1
43. Pampanini V, Pedicelli S, Gubinelli J, Scirè G, Cappa M, Boscherini B, et al. Brain Magnetic Resonance Imaging as First-Line Investigation for Growth Hormone Deficiency Diagnosis in Early Childhood. Horm Res Paediatr (2015) 84:323–30. doi: 10.1159/000439590
44. Bosch I Ara L, Katugampola H, Dattani MT. Congenital Hypopituitarism During the Neonatal Period: Epidemiology, Pathogenesis, Therapeutic Options, and Outcome. Front Pediatr (2021) 8:600962. doi: 10.3389/fped.2020.600962
45. Davis SW, Ellsworth BS, Peréz Millan MI, Gergics P, Schade V, Foyouzi N, et al. Pituitary Gland Development and Disease: From Stem Cell to Hormone Production. Curr Top Dev Biol (2013) 106:1–47. doi: 10.1016/B978-0-12-416021-7.00001-8
46. Bierich JR. Aetiology and Pathogenesis of Growth Hormone Deficiency. Baillieres Clin Endocrinol Metab (1992) 6:491–511. doi: 10.1016/s0950-351x(05)80108-9
47. Huet F, Carel JC, Nivelon JL, Chaussain JL. Long-Term Results of GH Therapy in GH-Deficient Children Treated Before 1 Year of Age. Eur J Endocrinol (1999) 140:29–34. doi: 10.1530/eje.0.1400029
48. Çetinkaya S, Poyrazoğlu Ş, Baş F, Ercan O, Yıldız M, Adal E, et al. Response to Growth Hormone Treatment in Very Young Patients With Growth Hormone Deficiencies and Mini-Puberty. J Pediatr Endocrinol Metab (2018) 31:175–84. doi: 10.1515/jpem-2017-0123
49. Ranke MB, Lindberg A. KIGS International Board. Early-Onset Idiopathic Growth Hormone Deficiency Within KIGS. Horm Res (2003) 60:18–21. doi: 10.1159/000071221
50. Tanner JM, Goldstein H, Whitehouse RH. Standards for Children's Height at Age 2 to 9 Years Allowing for Height of Parents. Arch Dis Child (1970) 45:819. doi: 10.1136/adc.45.244.819-b
51. Hermanussen M, Cole J. The Calculation of Target Height Reconsidered. Horm Res (2003) 59:180–3. doi: 10.1159/000069321
52. Tanner JM, Whitehouse RH. Clinical Longitudinal Standards for Height, Weight, Height Velocity, Weight Velocity, and Stages of Puberty. Arch Dis Child (1976) 51:170–9. doi: 10.1136/adc.51.3.170
53. Grote FK, van Dommelen P, Oostdijk W, de Muinck Keizer-Schrama SM, Verkerk PH, Wit JM, et al. Developing Evidence-Based Guidelines for Referral for Short Stature. Arch Dis Child (2008) 93:212–7. doi: 10.1136/adc.2007.120188
54. Stalman SE, Hellinga I, van Dommelen P, Hennekam RC, Saari A, Sankilampi U, et al. Application of the Dutch, Finnish and British Screening Guidelines in a Cohort of Children With Growth Failure. Horm Res Paediatr (2015) 84:376–82. doi: 10.1159/000440652
55. Zachmann M, Fernandez F, Tassinari D, Thakker R, Prader A. Anthropometric Measurements in Patients With Growth Hormone Deficiency Before Treatment With Human Growth Hormone. Eur J Pediatr (1980) 133:277–82. doi: 10.1007/BF00496089
56. Chen RS, Shiffman RN. Assessing Growth Patterns - Routine But Sometimes Overlooked. Clin Pediatr (Phila) (2000) 39:97–102. doi: 10.1177/000992280003900204
57. Gerver WJM, deBruin R. Paediatric Morphometrics .A Reference Manual. Maastricht: Universsitaire Pers Maastricht (2001).
58. Martin DD, Deusch D, Schweizer R, Binder G, Thodberg HH, Ranke MB. Clinical Application of Automated Greulich-Pyle Bone Age Determination in Children With Short Stature. Pediatr Radiol (2009) 39:598–607. doi: 10.1007/s00247-008-1114-4
59. Kemp SF, Sy JP. Analysis of Bone Age Data From National Cooperative Growth Study Substudy VII. Pediatrics (1999) 104:1031–36.
60. Darendeliler F, Ranke MB, Bakker B, Lindberg A, Cowell CT, Albertsson-Wikland K, et al. Bone Age Progression During the First Year of Growth Hormone Therapy in Pre-Pubertal Children With Idiopathic Growth Hormone Deficiency, Turner Syndrome or Idiopathic Short Stature, and in Short Children Born Small for Gestational Age: Analysis of Data From KIGS (Pfizer International Growth Database). Horm Res (2005) 63:40–7. doi: 10.1159/000082872
61. Clemmons DR. Consensus Statement on the Standardization and Evaluation of Growth Hormone and Insulin-Like Growth Factor Assays. Clin Chem (2011) 57:555–9. doi: 10.1373/clinchem.2010.150631
62. Junnila RK, Strasburger CJ, Bidlingmaier M. Pitfalls of Insulin-Like Growth Factor-I and Growth Hormone Assays. Endocrinol Metab Clin North Am (2015) 44:27–34. doi: 10.1016/j.ecl.2014.10.003
63. Juul A, Bang P, Hertel NT, Main K, Dalgaard P, Jørgensen K, et al. Serum Insulin-Like Growth Factor-I in 1030 Healthy Children, Adolescents, and Adults: Relation to Age, Sex, Stage of Puberty, Testicular Size, and Body Mass Index. J Clin Endocrinol Metab (1994) 78:744–52. doi: 10.1210/jcem.78.3.8126152
64. Juul A, Dalgaard P, Blum WF, Bang P, Hall K, Michaelsen KF, et al. Serum Levels of Insulin-Like Growth Factor (IGF)-Binding Protein-3 (IGFBP-3) in Healthy Infants, Children, and Adolescents: The Relation to IGF-I, IGF-II, IGFBP-1, IGFBP-2, Age, Sex, Body Mass Index, and Pubertal Maturation. J Clin Endocrinol Metab (1995) 80:2534–42. doi: 10.1210/jcem.80.8.7543116
65. Wit JM, Bidlingmaier M, de Bruin C, Oostdijk W. A Proposal for the Interpretation of Serum IGF-I Concentration as Part of Laboratory Screening in Children With Growth Failure. J Clin Res Pediatr Endocrinol (2020) 12:130–9. doi: 10.4274/jcrpe.galenos.2019.2019.0176
66. Shen Y, Zhang J, Zhao Y, Yan Y, Liu Y, Cai J. Diagnostic Value of Serum IGF-1 and IGFBP-3 in Growth Hormone Deficiency: A Systematic Review With Meta-Analysis. Eur J Pediatr (2015) 174:419–27. doi: 10.1007/s00431-014-2406-3
67. Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, et al. The Growth Hormone-Insulin-Like Growth Factor-I Axis in the Diagnosis and Treatment of Growth Disorders. Endocr Connect (2018) 7:R212–22. doi: 10.1530/EC-18-0099
68. Ranke MB. Towards a Consensus on the Definition of Idiopathic Short Stature. Horm Res (1996) 45:64–6. doi: 10.1159/000184851
69. Rapaport R, Wit JM, Savage MO. Growth Failure: "Idiopathic" Only After a Detailed Diagnostic Evaluation. Endocr Connect (2021) 1:EC–20-0585.R1. doi: 10.1530/EC-20-0585
70. Binder G, Huller E, Blumenstock G, Schweizer R. Auxology-Based Cut-Off Values for Biochemical Testing of GH Secretion in Childhood. Growth Horm IGF Res (2011) 21:212–8. doi: 10.1016/j.ghir.2011.05.007
71. Glick SM, Roth J, Yalow RS, Berson SA. Immunoassay of Human Growth Hormone in Plasma. Nature (1963) 199:784–7. doi: 10.1038/199784a0
72. Wu Z, Devany E, Balarini G, Junnila R, Bidlingmaier M, Strasburger CJ. Specific Monoclonal Antibodies and Ultrasensitive Immunoassays for 20K and 22K Human Growth Hormone. Growth Horm IGF Res (2010) 20:239–44. doi: 10.1016/j.ghir.2010.02.006
73. Arsene CG, Kratzsch J, Henrion A. Mass Spectrometry - an Alternative in Growth Hormone Measurement. Bioanalysis (2014) 6:2391–402. doi: 10.4155/bio.14.196
75. Baumann G, Shaw M, Amburn K, Jan T, Davila N, Mercado M, et al. Heterogeneity of Circulating Growth Hormone. Nucl Med Biol (1994) 21:369–79. doi: 10.1016/0969-8051(94)90060-4
76. Veldhuis JD, Keenan DM, Pincus SM. Motivations and Methods for Analyzing Pulsatile Hormone Secretion. Endocr Rev (2008) 29:823–64. doi: 10.1210/er.2008-0005
77. Albertsson-Wikland K, Rosberg S. Analysis of 24-Hour Growth Hormone Profiles in Children: Relation to Growth. J Clin Endocrinol Metab (1988) 67:493–500. doi: 10.1210/jcem-67-3-493
78. Albertsson-Wikland K, Rosberg S. Methods of Evaluating Spontaneous Growth Hormone Secretion. In: Ranke MB, Mullis M-E, editors. Diagnostics of Endocrine Function in Children and Adolescents. Basel: Karger (2011). p. 138–56.
79. Spiliotis BE, August GP, Hung W, Sonis W, Mendelson W, Bercu BB. Growth Hormone Neurosecretory Dysfunction. A Treatable Cause of Short Stature. JAMA (1984) 251:2223–30.
80. Roth J, Glick SM, Yalow RS, Berson S. Hypoglycemia: A Potent Stimulus to Secretion of Growth Hormone. Science (1963) 140:987–88. doi: 10.1126/science.140.3570.987
81. Rochiccioli P, Pienkowski C, Tauber MT, Uboldi F, Enjaume C. Association of Pharmacological Tests and Study of 24-Hour Growth Hormone Secretion in the Investigation of Growth Retardation in Children: Analysis of 257 Cases. Horm Res (1991) 35:70–5. doi: 10.1159/000181876
82. Rochiccioli P, Enjaume C, Tauber MT, Pienkowski C. Statistical Study of 5473 Results of Nine Pharmacological Stimulation Tests: A Proposed Weighting Index. Acta Paediatr (1993) 82:245–8. doi: 10.1111/j.1651-2227.1993.tb12652.x
83. Patel L, Skinner AM, Price DA, Clayton PE. The Influence of Body Mass Index on Growth Hormone Secretion in Normal and Short Statured Children. Growth Regul (1994) 4:29–34.
84. Casanueva FF, Villanueva L, Dieguez C, Diaz Y, Cabranes JA, Szoke B. Free Fatty Acids Block Growth Hormone (GH) Releasing Hormone-Stimulated GH Secretion in Man Directly at the Pituitary. J Clin Endocrinol Metab (1987) 65:634–42. doi: 10.1210/jcem-65-4-634
85. Ghigo E, Bellone J, Aimaretti G, Bellone S, Loche S, Cappa M, et al. Reliability of Provocative Tests to Assess Growth Hormone Secretory Status. Study in 472 Normally Growing Children. J Clin Endocrinol Metab (1996) 81:3323–7. doi: 10.1210/jcem.81.9.8784091
86. Marshall WA, Tanner JM. Variations in Pattern of Pubertal Changes in Girls. Arch Dis Child (1969) 44:291–303. doi: 10.1136/adc.44.235.291
87. Marshall WA, Tanner JM. Variations in the Pattern of Pubertal Changes in Boys. Arch Dis Child (1970) 45:13–23. doi: 10.1136/adc.45.239.13
88. Wit JM. Definition and Subcategorization of Idiopathic Short Stature: Between Consensus and Controversy. Horm Res Paediatr (2011) 3:3–6. doi: 10.1159/000330134
90. De Sanctis V, Marsella M, Soliman A, Yassin M. Macroorchidism in Childhood and Adolescence: An Update. Pediatr Endocrinol Rev (2014) 11 Suppl 2:263–73.
91. Preece MA, Baines MJ. A New Family of Mathematical Models Describing the Human Growth Curve. Ann Hum Biol (1978) 5:1–24. doi: 10.1080/03014467800002601
92. Gasser T, Muller HG, Kohler W, Prader A, Largo R, Molinari L. An Analysis of the Midgrowth and Adolescent Spurts of Height Based on Acceleration. Ann Hum Biol (1985) 12:129–48. doi: 10.1080/03014468500007631
93. Karlberg J. A Biologically-Oriented Mathematical Model (ICP) for Human Growth. Acta Paediatr Scand Suppl (1989) 350:70–94. doi: 10.1111/j.1651-2227.1989.tb11199.x
94. Lindberg A, Ranke MB. Data Analysis Within KIGS. In: Ranke MB, Price DA, Reiter EO, editors. Growth Hormone Therapy in Pediatrics - 20 Years of KIGS. Basel: Karger (2007). p. 23–8.
95. Preece MA. The Assessment of Growth. In: Ranke MB, Gunnarsson R, editors. Progress in Growth Hormone Therapy – 5 Years of KIGS. Mannheim: J&J (1994). p. 10–36. Q21.
96. Rikken B, Wit JM. Prepubertal Height Velocity References Over a Wide Age Range. Arch Dis Child (1992) 67:1277–80. doi: 10.1136/adc.67.10.1277
97. Tanner JM, Whitehouse RH, Takaishi M. Standards From Birth to Maturity for Height, Weight, Height Velocity, and Weight Velocity: British Children, 1965. I. Arch Dis Child (1966) 41:454–71. doi: 10.1136/adc.41.219.454
98. Lee PA, Migeon CJ. Puberty in Boys: Correlation of Plasma Levels of Gonadotropins (LH, FSH), Androgens (Testosterone, Androstenedione, Dehydroepiandrosterone and Its Sulfate), Estrogens (Estrone and Estradiol) and Progestins (Progesterone and 17-Hydroxyprogesterone). J Clin Endocrinol Metab (1975) 41:556–62. doi: 10.1210/jcem-41-3-556
99. Lee PA, Xenakis T, Winer J, Matsenbaugh S. Puberty in Girls: Correlation of Serum Levels of Gonadotropins, Prolactin, Androgens, Estrogens, and Progestins With Physical Changes. J Clin Endocrinol Metab (1976) 43:775–84. doi: 10.1210/jcem-43-4-775
100. Veldhuis JD, Metzger DL, Martha PM Jr, Mauras N, Kerrigan JR, Keenan B, et al. Estrogen and Testosterone, But Not a Nonaromatizable Androgen, Direct Network Integration of the Hypothalamo-Somatotrope (Growth Hormone)-Insulin-Like Growth Factor I Axis in the Human: Evidence From Pubertal Pathophysiology and Sex-Steroid Hormone Replacement. J Clin Endocrinol Metab (1997) 82:3414–20. doi: 10.1210/jcem.82.10.4317
101. Westphal O. Transitory hGH Deficiency and Its Consequences. Pediatr Adolesc Endocr (1987) 16:88–95.
102. Marin G, Domené HM, Barnes KM, Blackwell BJ, Cassorla FG, Cutler GB Jr. The Effects of Estrogen Priming and Puberty on the Growth Hormone Response to Standardized Treadmill Exercise and Arginine-Insulin in Normal Girls and Boys. J Clin Endocrinol Metab (1994) 79:537–41. doi: 10.1210/jcem.79.2.8045974
103. Illig R, Bucher H. Testosterone Priming of Growth Hormone Release. In: Laron Z, Butenandt O, editors. Evaluation of Growth Hormone Secretion Basel: Karger (1983). p. 75–85.
104. Martínez AS, Domené HM, Ropelato MG, Jasper HG, Pennisi PA, Escobar ME, et al. Estrogen Priming Effect on Growth Hormone (GH) Provocative Test: A Useful Tool for the Diagnosis of GH Deficiency. J Clin Endocrinol Metab (2000) 85:4168–72. doi: 10.1210/jcem.85.11.6928
105. Wetterau LA. The Pros and Cons of Sex Steroid Priming in Growth Hormone Stimulation Testing. Pediatr Endocrinol Metab (2012) 25:1049–55. doi: 10.1515/jpem.2011.327
106. Grimberg A, Allen DB. Growth Hormone Treatment for Growth Hormone Deficiency and Idiopathic Short Stature: New Guidelines Shaped by the Presence and Absence of Evidence. Curr Opin Pediatr (2017) 29:466–71. doi: 10.1097/MOP.0000000000000505
107. Soyka LF, Bode HH, Crawford JD, Flynn FJ Jr. Effectiveness of Long-Term Human Growth Hormone Therapy for Short Stature in Children With Growth Hormone Deficiency. J Clin Endocrinol Metab (1970) 30:1–14. doi: 10.1210/jcem-30-1-1
108. Ferrandez A, Zachmann M, Prader A, Illig R. Isolated Growth Hormone Deficiency in Prepubertal Children. Influence of Human Growth Hormone on Longitudinal Growth, Adipose Tissue, Bone Mass and Bone Maturation. Helv Paediatr Acta (1970) 25:566–76.
109. Aceto T Jr, Frasier SD, Hayles AB, Meyer-Bahlburg HF, Parker ML, Munschauer R, et al. Collaborative Study of the Effects of Human Growth Hormone in Growth Hormone Deficiency. I. First Year of Therapy. J Clin Endocrinol Metab (1972) 35:483–96. doi: 10.1210/jcem-35-4-483
110. Ranke MB, Schweizer R, Binder G. Basal Characteristics and First Year Responses to Human Growth Hormone (GH) Vary According to Diagnostic Criteria in Children With Non-Acquired GH Deficiency (naGHD): Observations From a Single Center Over a Period of Five Decades. J Pediatr Endocrinol Metab (2018) 31:1257–66. doi: 10.1515/jpem-2018-0025
111. Greenspan FS, Li CH, Simpson ME, Evans HM. Bioassay of Hypophyseal Growth Hormone: The Tibia Test. Endocrinol (1949) 45:455–63. doi: 10.1210/endo-45-5-455
112. Mills JB, Ashworth RB, Wilhelmi AE, Hartree AS. Improved Method for the Extraction and Purification of Human Growth Hormone. J Clin Endocr (1969) 29:1456–59. doi: 10.1210/jcem-29-11-1456
113. Preece MA, Tanner JM, Whitehouse RH, Cameron N. Dose Dependence of Growth Response to Human Growth Hormone in Growth Hormone Deficiency. J Clin Endocrinol Metab (1976) 42:477–83. doi: 10.1210/jcem-42-3-477
114. Frasier SD, Costin G, Lippe BM, Aceto T Jr, Bunger PF. A Dose-Response Curve for Human Growth Hormone. J Clin Endocrinol Metab (1981) 53:1213–17. doi: 10.1210/jcem-53-6-1213
115. MacGillivray MH, Blethen SL, Buchlis JG, Clopper RR, Sandberg DE, Conboy TA. Current Dosing of Growth Hormone in Children With Growth Hormone Deficiency: How Physiologic? Pediatr (1998) 102:527–30.
116. Ranke MB, Lindberg A, Mullis PE, Geffner M, Tanaka T, Cutfield W, et al. Towards Optimal Treatment With Growth Hormone in Short Children and Adolescents: Evidence and Theses. Horm Res Paediatr (2013) 79:51–67. doi: 10.1159/000347121
117. Kastrup DW, Christiansen JS, Anderson JK, Orskov H. Increased Growth Rate Following Transfer to Daily Sc. Administration From Three Weekly Im. Injection of hGH in Growth Hormone Deficient Children. Acta Endocrinol (1983) 104:148–52. doi: 10.1530/acta.0.1040148
118. Albertsson-Wikland K, Westphal O, Westgren U. Daily Subcutaneous Administration of Human Growth Hormone in Growth Hormone Deficient Children. Acta Paediatr Scand (1986) 75:89–97. doi: 10.1111/j.1651-2227.1986.tb10163.x
119. MacGillivray MH, Baptista J, Johanson A, Genentech Study Group. Outcome of a Four-Year Randomized Study of Daily Versus Three Times Weekly Somatropin Treatment in Prepubertal Naive Growth Hormone-Deficient Children. J Clin Endocrinol Metab (1996) 81:1806–9. doi: 10.1210/jcem.81.5.8626839
120. Rohrer TR, Horikawa R, Kappelgaard AM. Growth Hormone Delivery Devices: Current Features and Potential for Enhanced Treatment Adherence. Expert Opin Drug Deliv (2017) 14:1253–64. doi: 10.1080/17425247.2017.1243526
121. Yuen KCJ, Miller BS, Boguszewski CL, Hoffman AR. Usefulness and Potential Pitfalls of Long-Acting Growth Hormone Analogs. Front Endocrinol (Lausanne) (2021) 12:637209. doi: 10.3389/fendo.2021.637209
122. Hughes IP, Choong C, Rath S, Atkinson H, Cotterill A, Cutfield W, et al. Early Cessation and Non-Response Are Important and Possibly Related Problems in Growth Hormone Therapy: An OZGROW Analysis. Growth Horm IGF Res (2016) 29:63–70. doi: 10.1016/j.ghir.2016.04.006
123. Rosenfeld RG, Bakker B. Compliance and Persistence in Pediatric and Adult Patients Receiving Growth Hormone Therapy. Endocr Pract (2008) 14:143–54. doi: 10.4158/EP.14.2.143
124. Cutfield WS, Derraik JG, Gunn AJ, Reid K, Delany T, Robinson E, et al. Non-Compliance With Growth Hormone Treatment in Children Is Common and Impairs Linear Growth. PloS One (2011) 6:e16223. doi: 10.1371/journal.pone.0016223
125. Koledova E, Tornincasa V, van Dommelen P. Analysis of Real-World Data on Growth Hormone Therapy Adherence Using a Connected Injection Device. BMC Med Inform Decis Mak (2020) 20:176. doi: 10.1186/s12911-020-01183-1
126. van Dommelen P, Koledova E, Wit JM. Effect of Adherence to Growth Hormone Treatment on 0-2 Year Catch-Up Growth in Children With Growth Hormone Deficiency. PloS One (2018) 13:e0206009. doi: 10.1371/journal.pone.0206009
127. Fisher BG, Acerini CL. Understanding the Growth Hormone Therapy Adherence Paradigm: A Systematic Review. Horm Res Paediatr (2013) 9:189–96. doi: 10.1159/000350251
128. De Pedro S, Murillo M, Salinas I, Granada ML, Martinez M, Puig-Domingo M, et al. Variability in Adherence to r-hGH Treatment: Socioeconomic Causes and Effect on Children's Growth. Growth Horm IGF Res (2016) 26:32–5. doi: 10.1016/j.ghir.2015.12.002
129. Van den Broeck J, Hokken-Koelega A, Wit J. Validity of Height Velocity as a Diagnostic Criterion for Idiopathic Growth Hormone Deficiency and Turner Syndrome. Horm Res (1999) 51:68–73. doi: 10.1159/000023317
130. Boersma B, Wynne HJ, Wit JM. A Mathematical Model Describing Catch-Up Growth in Celiac Disease. Acta Paediatr (1994) 83:1097–9. doi: 10.1111/j.1651-2227.1994.tb12994.x
131. Wit JM, Boersma B. Catch-Up Growth: Definition, Mechanisms, and Models. J Pediatr Endocrinol Metab (2002) Suppl 5:1229–4. doi: 10.1159/000064771
132. Nierop AF, Niklasson A, Holmgren A, Gelander L, Rosberg S, Albertsson-Wikland K. Modelling Individual Longitudinal Human Growth From Fetal to Adult Life– QEPS I. Jour Theor Biol (2016) 406:143–65. doi: 10.1016/j.jtbi.2016.06.007
133. Holmgren A, Niklasson A, Nierop AFM, Gelander L, Aronson AS, Sjöberg A, et al. Estimating Secular Changes in Longitudinal Growth Patterns Underlying Adult Height With the QEPS Model: The Grow Up Gothenburg Cohorts. Pediatr Res (2018) 84:41–9. doi: 10.1038/s41390-018-0014-z
134. Wit JM, Sas TCJ, Ranke MB, van Dommelen P. Catch-Up Growth in Prepubertal Children Treated for Juvenile Hypothyroidism and Growth Hormone Deficiency can be Modelled With a Monomolecular Function. J Clin Res Pediatr Endocrinol (2020). doi: 10.4274/jcrpe.galenos.2020.2020.0130
135. Spagnoli A, Spadoni GL, Cianfarani S, Pasquino AM, Troiani S, Boscherini B. Prediction of the Outcome of Growth Hormone Therapy in Children With Idiopathic Short Stature. A Multivariate Discriminant Analysis. J Pediatr (1995) 126:905–9. doi: 10.1016/s0022-3476(95)70206-7
136. Bakker B, Frane J, Anhalt H, Lippe B, Rosenfeld RG. Height Velocity Targets From the National Cooperative Growth Study for First Year Growth Hormone Responses in Short Children. J Clin Endocrinol Metab (2008) 93:352–57. doi: 10.1210/jc.2007-1581
137. Cohen P, Rogol AD, Deal CL, Saenger P, Reiter EO, Ross JL, et al. Consensus Statement on the Diagnosis and Treatment of Children With Idiopathic Short Stature: A Summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab (2008) 93:4210–17. doi: 10.1210/jc.2008-0509
138. Ranke MB, Lindberg A, KIGS International Board. Observed and Predicted Growth Responses in Prepubertal Children With Growth Disorders: Guidance of Growth Hormone Treatment by Empirical Variables. J Clin Endocrinol Metab (2010) 95:1229–37. doi: 10.1210/jc.2009-1471
139. Bang P, Bjerknes R, Dahlgren J, Dunkel L, Gustafsson J, Juul A, et al. A Comparison of Different Definitions of Growth Response in Short Prepubertal Children Treated With Growth Hormone. Horm Res Paediatr (2011) 75:335–45. doi: 10.1159/000322878
140. Ranke MB, Guilbaud O. Growth Response in Prepubertal Children With Idiopathic Growth Hormone Deficiency During the First Two Years of Treatment With Human Growth Hormone. Analysis of the Kabi Pharmacia International Growth Study. Acta Paediatr Scand Suppl (1991) 379:109–15.
141. Ranke MB, Guilbaud O, Lindberg A, Cole T. Prediction of the Growth Response in Children With Various Growth Disorders Treated With Growth Hormone: Analyses of Data From the Kabi Pharmacia International Growth Study. International Board of the Kabi Pharmacia International Growth Study. Acta Paediatr Suppl (1993) 391:82–8. doi: 10.1111/j.1651-2227.1993.tb12936.x
142. Blethen SL, Compton P, Lippe BM, Rosenfeld RG, August GP, Johansson A. Factors Predicting the Response to Growth Hormone (GH) Therapy in Prepubertal Children With GH Deficiency. J Clin Endocrinol Metab (1993) 76:574–79. doi: 10.1210/jcem.76.3.8445013
143. Kriström B, Wikland KA. Growth Prediction Models, Concept and Use. Horm Res (2002) 57(Suppl 2):66–70. doi: 10.1159/000058104
144. Schönau E, Westermann F, Rauch F, Stabrey A, Wassmer G, Keller E, et al. A New and Accurate Prediction Model for Growth Response to Growth Hormone Treatment in Children With Growth Hormone Deficiency. Eur J Endocrinol (2001) 144:13–20. doi: 10.1530/eje.0.1440013
145. Ranke MB, Lindberg A. Predicting Growth in Response to Growth Hormone Treatment. Growth Horm IGF Res (2009) 19:1–11. doi: 10.1016/j.ghir.2008.08.001
146. Wit JM, Ranke MB, Albertsson-Wikland K, Carrascosa A, Rosenfeld RG, Van Buuren S, et al. Personalized Approach to Growth Hormone Treatment: Clinical Use of Growth Prediction Models. Horm Res Paediatr (2013) 79:257–70. doi: 10.1159/000351025
147. Loftus J, Lindberg A, Aydin F, Gomez R, Maghnie M, Rooman R, et al. Individualised Growth Response Optimisation (iGRO) Tool: An Accessible and Easy-to-Use Growth Prediction System to Enable Treatment Optimisation for Children Treated With Growth Hormone. J Pediatr Endocrinol Metab (2017) 30:1019–26. doi: 10.1515/jpem-2017-0120
148. Dauber A, Meng Y, Audi L, Vedantam S, Weaver B, Carrascosa A, et al. A Genome-Wide Pharmacogenetic Study of Growth Hormone Responsiveness. J Clin Endocrinol Metab (2020) 105:3203–14. doi: 10.1210/clinem/dgaa443
149. Steven A, Perchard R, Garner T, Clayton P, Murray P. Pharmacogenomics Applied to Recombinant Human Growth Hormone Responses in Children With Short Stature. Rev Endocr Metab Dis (2021). doi: 10.1007/s11154-021-09637-1
150. Cohen P, Rogol AD, Howard CP, Bright GM, Kappelgaard AM, Rosenfeld RG. Insulin Growth Factor-Based Dosing of Growth Hormone Therapy in Children: A Randomized, Controlled Study. American Norditropin Study Group. J Clin Endocrinol Metab (2007) 92:2480–6. doi: 10.1210/jc.2007-0204
151. Cohen P, Germak J, Rogol AD, Weng W, Kappelgaard AM, Rosenfeld RG. Variable Degree of Growth Hormone (GH) and Insulin-Like Growth Factor (IGF) Sensitivity in Children With Idiopathic Short Stature Compared With GH-Deficient Patients: Evidence From an IGF-Based Dosing Study of Short Children. American Norditropin Study Group. J Clin Endocrinol Metab (2010) 95:2089–98. doi: 10.1210/jc.2009-2139
152. Cohen P, Weng W, Rogol AD, Rosenfeld RG, Kappelgaard AM, Germak J. Dose-Sparing and Safety-Enhancing Effects of an IGF-I-Based Dosing Regimen in Short Children Treated With Growth Hormone in a 2-Year Randomized Controlled Trial: Therapeutic and Pharmacoeconomic Considerations. Clin Endocrinol (Oxf) (2014) 81:71–6. doi: 10.1111/cen.12408
153. Baron J. Editorial: Growth Hormone Therapy in Childhood: Titration Versus Weight-Based Dosing? J Clin Endocrinol Metab (2007) 92:2436–8. doi: 10.1210/jc.2007-0900
154. Frindik JO, Baptista J. Adult Height in Growth Hormone Deficiency: Historical Perspective and Examples From the National Cooperative Growth Study. Pediatr (1999) 104:1000–04.
155. Largo RH, Gasser T, Prader A, Stuetzle W, Huber PJ. Analysis of the Adolescent Growth Spurt Using Smoothing Spline Functions. Ann Hum Biol (1978) 5:421–34. doi: 10.1080/03014467800003071
156. Martin DD, Hauspie RC, Ranke MB. Total Pubertal Growth and Markers of Puberty Onset in Adolescents With GHD: Comparison Between Mathematical Growth Analysis and Pubertal Staging Methods. Horm Res (2005) 63:95–101. doi: 10.1159/000084156
157. Clark PA, Rogol AD. Growth Hormones and Sex Steroid Interactions at Puberty. Endocrinol Metab Clin North Am (1996) 25:665–81. doi: 10.1016/s0889-8529(05)70346-7
158. Benyi E, Sävendahl L. The Physiology of Childhood Growth: Hormonal Regulation. Horm Res Paediatr (2017) 88:6–14. doi: 10.1159/000471876
159. Karimian E, Chagin AS, Sävendahl L. Genetic Regulation of the Growth Plate. Front Endocrinol (Lausanne) (2012) 2:113. doi: 10.3389/fendo.2011.00113
160. Bourguignon JP. Growth and Timing of Puberty: Reciprocal Effects. Horm Res (1991) 36:131–5. doi: 10.1159/000182146
161. Ranke MB, Lindberg A, Martin DD, Bakker B, Wilton P, Albertsson-Wikland K, et al. The Mathematical Model for Total Pubertal Growth in Idiopathic Growth Hormone (GH) Deficiency Suggests a Moderate Role of GH Dose. J Clin Endocrinol Metab (2003) 88:4748–53. doi: 10.1210/jc.2003-030600
162. Mauras N, Attie KM, Reiter EO, Saenger P, Baptista J. High Dose Recombinant Human Growth Hormone (GH) Treatment of GH-Deficient Patients in Puberty Increases Near-Final Height: A Randomized, Multicenter Trial. Genentech Inc. Cooperative Study Group. J Clin Endocrinol Metab (2000) 85:3653–60. doi: 10.1210/jcem.85.10.6906
163. Ranke MB, Price DA, Albertsson-Wikland K, Maes M, Lindberg A. Factors Determining Pubertal Growth and Final Height in Growth Hormone Treatment of Idiopathic Growth Hormone Deficiency. Analysis of 195 Patients of the Kabi Pharmacia International Growth Study. Horm Res (1997) 48:62–71. doi: 10.1159/000185487
164. Thomas M, Massa G, Bourguignon JP, Craen M, De Schepper J, de Zegher F, et al. Final Height in Children With Idiopathic Growth Hormone Deficiency Treated With Recombinant Human Growth Hormone: The Belgian Experience. Horm Res (2001) 55:88–94. doi: 10.1159/000049976
165. Maghnie M, Ambrosini L, Cappa M, Pozzobon G, Ghizzoni L, Ubertini MG, et al. Adult Height in Patients With Permanent Growth Hormone Deficiency With and Without Multiple Pituitary Hormone Deficiencies. J Clin Endocrinol Metab (2006) 91:2900–5. doi: 10.1210/jc.2006-0050
166. Klein KO, Phillips SA. Review of Hormone Replacement Therapy in Girls and Adolescents With Hypogonadism. J Pediatr Adolesc Gynecol (2019) 32:460–8. doi: 10.1016/j.jpag.2019.04.010
167. Reiter EO. A Brief Review of the Addition of Gonadotropin-Releasing Hormone Agonists (GnRH-Ag) to Growth Hormone (GH) Treatment of Children With Idiopathic Growth Hormone Deficiency: Previously Published Studies From America. Mol Cell Endocrinol (2006) 254-255:221–5. doi: 10.1016/j.mce.2006.04.024
168. Grumbach MM, Bin-Abbas BS, Kaplan SL. The Growth Hormone Cascade: Progress and Long-Term Results of Growth Hormone Treatment in Growth Hormone Deficiency. Horm Res (1998) 49 Suppl 2:41–57. doi: 10.1159/000053087
169. Wit JM, Kamp GA, Rikken B. Spontaneous Growth and Response to Growth Hormone Treatment in Children With Growth Hormone Deficiency and Idiopathic Short Stature. Pediatr Res (1996) 39:295–302. doi: 10.1203/00006450-199602000-00018
170. Guyda HJ. Four Decades of Growth Hormone Therapy for Short Children: What Have We Achieved? J Clin Endocrinol Metab (1999) 84:4307–16. doi: 10.1210/jcem.84.12.6189
171. August GP, Julius JR, Blethen SL. Adult Height in Children With Growth Hormone Deficiency Who Are Treated With Biosynthetic Growth Hormone: The National Cooperative Growth Study Experience. Pediatr (1998) 102:512–16.
172. Rachmiel M, Rota V, Atenafu E, Daneman D, Hamilton J. Final Height in Children With Idiopathic Growth Hormone Deficiency Treated With a Fixed Dose of Recombinant Growth Hormone. Horm Res (2007) 68:236–43. doi: 10.1159/000101427
173. Westphal O, Lindberg A, Swedish KIGS National Board. Final Height in Swedish Children With Idiopathic Growth Hormone Deficiency Enrolled in KIGS Treated Optimally With Growth Hormone. Acta Paediatr (2008) 97:1698–706. doi: 10.1111/j.1651-2227.2008.01053.x
174. Root AW, Dana K, Lippe B. Treatment of Growth Hormone-Deficient Infants With Recombinant Human Growth Hormone to Near-Adult Height: Patterns of Growth. Horm Res Paediatr (2011) 75:276–83. doi: 10.1159/000322881
175. Blethen SL, Baptista J, Kuntze J, Foley T, LaFranchi S, Johanson A. Adult Height in Growth Hormone (GH)-Deficient Children Treated With Biosynthetic GH. The Genentech Growth Study Group. J Clin Endocrinol Metab (1997) 82:418–20. doi: 10.1210/jcem.82.2.3734
176. Reiter EO, Price DA, Wilton P, Albertsson-Wikland K, Ranke MB. Effect of Growth Hormone (GH) Treatment on the Near-Final Height of 1258 Patients With Idiopathic GH Deficiency: Analysis of a Large International Database. J Clin Endocrinol Metab (2006) 91:2047–54. doi: 10.1210/jc.2005-2284
177. Fujieda K, Tanaka T, Takano K, Chihara K, Seino Y, Irie M, et al. Adult Height After Growth Hormone Treatment in Japanese Children With Idiopathic Growth Hormone Deficiency: Analysis From the KIGS Japan Database. J Pediatr Endocrinol Metab (2011) 24:457–62. doi: 10.1515/jpem.2011.212
178. Tanaka T. International Comparison of Adult Height in Children With Growth Hormone Deficiency and Limitations of Growth Hormone Treatment in Japan. Pediatr Endocrinol Rev (2017) 14:216–21.
179. Sas TC, de Ridder MA, Wit JM, Rotteveel J, Oostdijk W, Reeser HM, et al. Adult Height in Children With Growth Hormone Deficiency: A Randomized, Controlled, Growth Hormone Dose-Response Trial. Horm Res Paediatr (2010) 74:172–81. doi: 10.1159/000281323
180. Coelho R, Brook CG, Preece MA, Stanhope RG, Dattani MT, Hindmarsh PC. A Randomised Study of Two Doses of Biosynthetic Human Growth Hormone on Final Height of Pubertal Children With Growth Hormone Deficiency. Horm Res (2008) 70:85–8. doi: 10.1159/000139149
181. Clayton PE, Cowell CT. Safety Issues in Children and Adolescents During Growth Hormone Therapy–a Review. Growth Horm IGF Res (2000) 10:306–17. doi: 10.1054/ghir.2000.0175
182. Underwood LE, Voina SJ, Van Wyk JJ. Restoration of Growth by Human Growth Hormone (Roos) in Hypopituitary Dwarfs Immunized by Other Human Growth Hormone Preparations: Clinical and Immunological Studies. J Clin Endocrinol Metab (1974) 38:288–97. doi: 10.1210/jcem-38-2-288
183. Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P. The Epidemiology of Creutzfeldt-Jakob Disease: Conclusion of a 15-Year Investigation in France and Review of the World Literature. Neurol (1987) 37:895–904. doi: 10.1212/wnl.37.6.895
184. Buchanan CR, Preece MA, Milner RDG. Mortality, Neoplasia and Creutzfeldt-Jakob Disease in Patients Treated With Human Pituitary Growth Hormone in the United Kingdom. Br Med J (1991) 302:824–28. doi: 10.1136/bmj.302.6780.824
185. Hintz RL. The Prismatic Case of Creutzfeldt-Jakob Disease Associated With Pituitary Growth Hormone Treatment. J Clin Endocrinol Metab (1995) 80:2298–301. doi: 10.1210/jcem.80.8.7629222
186. Aronson JK, Hauben M, Bate A. Defining 'Surveillance' in Drug Safety. Drug Saf (2012) 35:347–57. doi: 10.2165/11597590-000000000-00000
187. Miller BS, Rosenfeld RD. Monotoring r-hGH Safety: r-hGH Registries, SAGhE and Future Needs. Ped Endocrinol Rev (2018) 16:150–61. doi: 10.17458/pes.vol16.2018.mr.monitoringrhghsafety
188. Darendeliler F, Karagiannis G, Wilton P. Headache, Idiopathic Intracranial Hypertension and Slipped Capital Femoral Epiphysis During Growth Hormone Treatment: A Safety Update From the KIGS Database. Horm Res (2007) 68 Suppl 5:41–7. doi: 10.1159/000110474
189. Reeves GD. Doyle DA Growth Hormone Treatment and Pseudotumor Cerebri: Coincidence or Close Relationship? J Pediatr Endocrinol Metab (2002) 15(Suppl 2):723–30. doi: 10.1515/jpem.2002.15.s2.723
190. Cutfield WS, Wilton P, Bennmarker H, Albertsson-Wikland K, Chatelain P, Ranke MB, et al. Incidence of Diabetes Mellitus and Impaired Glucose Tolerance in Children and Adolescents Receiving Growth-Hormone Treatment. Lancet (2000) 355:610–13. doi: 10.1016/S0140-6736(99)04055-6
191. Swerdlow AJ, Cooke R, Albertsson-Wikland K, Borgström B, Butler G, Cianfarani S, et al. Description of the SAGhE Cohort: A Large European Study of Mortality and Cancer Incidence Risks After Childhood Treatment With Recombinant Growth Hormone. Horm Res Paediatr (2015) 84:172–83. doi: 10.1159/000435856
192. Cohen P, Clemmons DR, Rosenfeld RG. Does the GH-IGF Axis Play a Role in Cancer Pathogenesis? Growth Horm IGF Res (2000) 10:297–305. doi: 10.1054/ghir.2000.0171
193. Carel JC, Ecosse E, Landier F, Meguellati-Hakkas D, Kaguelidou F, Rey G, et al. Long-Term Mortality After Recombinant Growth Hormone Treatment for Isolated Growth Hormone Deficiency or Childhood Short Stature: Preliminary Report of the French SAGhE Study. J Clin Endocrinol Metab (2012) 97:416–25. doi: 10.1210/jc.2011-1995
194. Sävendahl L, Polak M, Backeljauw P, Blair JC, Miller BS, Rohrer TR, et al. Long-Term Safety of Growth Hormone Treatment in Childhood: Two Large Observational Studies NordiNet IOS and ANSWER. J Clin Endocrinol Metab (2021) 106:1728–41. doi: 10.1210/clinem/dgab080
195. Mo D, Hardin DS, Erfurth EM, Melmed S. Adult Mortality or Morbidity Is Not Increased in Childhood-Onset Growth Hormone Deficient Patients Who Received Pediatric GH Treatment: An Analysis of the Hypopituitary Control and Complications Study (HypoCCS). Pituit (2014) 17:477–85. doi: 10.1007/s11102-013-0529-6
Keywords: growth hormone deficiency (GHD), diagnosis, childhood, puberty, GH treatment, adult height
Citation: Ranke MB (2021) Short and Long-Term Effects of Growth Hormone in Children and Adolescents With GH Deficiency. Front. Endocrinol. 12:720419. doi: 10.3389/fendo.2021.720419
Received: 04 June 2021; Accepted: 19 July 2021;
Published: 01 September 2021.
Edited by:
Alan David Rogol, University of Virginia, United StatesReviewed by:
Fred Wondisford, Rutgers Robert Wood Johnson University Hospital, United StatesBenjamin Udoka Nwosu, University of Massachusetts Medical School, United States
Edward Reiter, Baystate Medical Center, United States
Copyright © 2021 Ranke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael B. Ranke, bWljaGFlbC5yYW5rZUBnbXguZGU=