 Boping Tang1†
Boping Tang1† Zhongkai Wang2†Qiuning Liu1†Huabin Zhang1†Senhao Jiang1Xinzheng Li3
Zhongkai Wang2†Qiuning Liu1†Huabin Zhang1†Senhao Jiang1Xinzheng Li3 Zhengfei Wang1Yue Sun1Zhongli Sha3Hui Jiang4,5Xugan Wu6
Zhengfei Wang1Yue Sun1Zhongli Sha3Hui Jiang4,5Xugan Wu6 Yandong Ren2Haorong Li2Fujun Xuan1Baoming Ge1Wei Jiang3Shusheng She7Hongying Sun8Qiang Qiu2Wen Wang2
Yandong Ren2Haorong Li2Fujun Xuan1Baoming Ge1Wei Jiang3Shusheng She7Hongying Sun8Qiang Qiu2Wen Wang2 Qun Wang9*
Qun Wang9* Gaofeng Qiu6*Daizhen Zhang1*
Gaofeng Qiu6*Daizhen Zhang1* Yongxin Li2*
Yongxin Li2*- 1Jiangsu Key Laboratory for Bioresources of Saline Soils, Jiangsu Provincial Key Laboratory of Coastal Wetland Bioresources and Environmental Protection, Jiangsu Synthetic Innovation Center for Coastal Bio-agriculture, Yancheng Teachers University, Yancheng, China
- 2Center for Ecological and Environmental Sciences, Northwestern Polytechnical University, Xi'an, China
- 3Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 4National Engineering Laboratory of Marine Germplasm Resources Exploration and Utilization, Zhejiang Ocean University, Zhoushan, China
- 5National Engineering Research Center for Facilitated Marine Aquaculture, Zhejiang Ocean University, Zhoushan, China
- 6Key Laboratory of Freshwater Aquatic Genetic Resources, Ministry of Agriculture and Rural Affairs, Shanghai Ocean University, Shanghai, China
- 7China-Hong Kong Ecology Consultant Company, Hong Kong, Hong Kong
- 8College of Life Sciences, Nanjing Normal University, Nanjing, China
- 9Department of Biology, School of Life Science, East China Normal University, Shanghai, China
As an important freshwater aquaculture species in China, the Chinese mitten crab (Eriocheir japonica sinensis) has high economic and nutritional value. However, limited genomic information is currently available for studying its basic development and genetic diversity. Here, we performed whole-genome sequencing on Oxford Nanopore Technologies Limited's platform using promethION. The assembled size of E. j.sinensis genome was approximately 1.27 Gb, which is close to the estimated size (1.19 Gb). Furthermore, based on assessment using Benchmarking Universal Single-Copy Orthologs (BUSCO) (Simao et al., 2015), 94.00% of the expected eukaryotic genes were completely present in the genome assembly. In addition, repetitive sequences accounted for ~61.42% of the assembled genome, and 22,619 protein-coding genes were annotated. Comparative genomics analysis demonstrated that the Chinese mitten crab diverged from Penaeus vannamei ~373.6 million years ago, with a faster evolution rate than shrimp. We anticipate that the annotated high-quality genome of E. j. sinensis will promote research on its basic development and evolution and make substantial contributions to comparative genomic analyses of crustaceans.
Introduction
The Chinese mitten crab, Eriocheir japonica sinensis (i.e., Eriocheir sinensis), which belongs to the Varunidae family of crustaceans, is a medium-sized burrowing crab containing dense patches of dark setae on its claws and preying on fish, shrimp, snails, mussels, worms, earthworms, and insects (Panning, 1939; Rudnick et al., 2005; Rybczyk et al., 2010). The mitten crab is native to rivers, estuaries, and other coastal habitats of Eastern Asia from North Korea to South China (Gilbey et al., 2008), but has also been introduced to Europe and North America, where it is considered an invasive species (Rudnick et al., 2003; Herborg et al., 2005; Gilbey et al., 2008). These crabs spend most of their life in freshwater, but migrate to shallow estuaries during the breeding season (Bentley, 2011). They mate and deposit eggs at brackish water in late summer and only once in their lifetime, laying eggs at the end of their life cycle (Panning, 1939; Tsukimura, 2008).
Chinese mitten crabs are an economically important and nutritious aquatic food in Asia (Chen and Zhang, 2007; Chen et al., 2007; Jiang et al., 2010). As such, populations have declined drastically due to overfishing, water pollution, and dam construction (Tang et al., 2000; Chang et al., 2008), although resources have been somewhat restored by artificial breeding and release (Jiang et al., 2010; Chen et al., 2015). While their diet (Jin et al., 2003), development (Ng et al., 1998), reproduction (Montú et al., 1996), and immune system (Gai et al., 2009; Zhao et al., 2009) have been studied, a high-quality mitten crab genome has yet to be reported. Importantly, with the rapid development of third-generation sequencing technology, high-quality genomes can now be obtained more easily, which will greatly promote biological research.
In this study, we examined genomic data of E. j. sinensis using newly obtained Nanopore long reads and previously made available Illumina short reads. We anticipate that the well-annotated genome and large amount of sequencing data obtained in the current study will promote research on the basic development and evolution of E. j. sinensis and also contribute to comparative genomic analysis of related species.
Materials and Methods
Sampling, Library Construction, and Sequencing
To obtain high-quality DNA for Chinese mitten crab genome sequencing using Oxford Nanopore Technologies (ONT; Oxford, UK), we sampled one individual (E. j. sinensis) from a large vertical lake in Jiangsu, China. The crab was first rinsed under flowing clean water and dissected. After removing the foreign bodies covering its surface, muscle tissue was sampled and ground into powder in liquid nitrogen. DNA was then extracted using a Qiagen Blood & Cell Culture DNA Mini Kit (Germany), with DNA quality checked by agarose gel electrophoresis. The tissue samples of hepatopancreas, muscle, and heart were used for RNA extraction using TRIzol reagent, with RNA concentration and quality assessed using a Qubit fluorometer. The extracted DNA and RNA were sequenced on the Oxford Nanopore PromethION 48 (P48) platform and Illumina NovaSeq 6000 platform, respectively. The genome short reads were downloaded from NCBI (Song et al., 2016) (SRR3081450, SRR3081453, SRR3081454, SRR3081455, and SRR3081456).
Quality Control of Sequencing Reads
Three different kinds of reads were used for this study, including Nanopore long reads (PromethION 48; Oxford Nanopore; insert size, 20 Kb), Illumina genomic short reads (HiSeq 2000; Illumina), and Illumina RNA-seq reads (NovaSeq 6000; Illumina; insert size, 250 bp). The genomic short reads (81×, produced on Illumina 2000 platform) produced in previous study were downloaded from NCBI: SRR3081450, SRR3081453, SRR3081454, SRR3081455, and SRR3081456. To ensure the reads quality, all low-quality reads, adaptor sequences, and polymerase chain reaction (PCR) duplicates which are the same reads produced by PCR were removed. For the Nanopore long reads, Guppy v2.2.2 was used as ONT basecaller and we only removed the short reads and the low-quality reads. Specifically, for short reads filtering, all the reads longer than 1 Kb were retained by an internal Perl script. For reads quality filtering, for each read longer than 1 Kb were calculated the mean quality value and only reads with mean quality ≥7 were retained using another in-house Perl script. In the RNA sequencing (RNA-seq) data were also removed the low-quality reads, adaptor sequences, and PCR duplicates produced by PCR. After filtering these data, the Illumina reads and Nanopore reads were both aligned to NR database (from NCBI) by BLAST software (blastn) (Altschul, 2012) with e value set as 10−5. Any reads that had the best blast hit to microorganism were considered as contamination and were removed.
Genome Characteristic Estimation
To estimate the genome size of the Chinese mitten crab, we implemented the count subprogram of the jellyfish software by 19-mer to generate the binary file which should be the input of the next step histogram to format the kmer histogram using all filtered Illumina short reads. Based on the histogram file, we used GenomeScope v1.0 and made the final estimation. The genome size is calculated by this formula: G = N/D, where the N is the total number of 19-mer and the D is the average depth of 19-mer.
Nanopore Long Read Correction and Genome Assembly
The error rate of the Nanopore long reads is much higher than the Illumina short reads (Ross et al., 2013). As such, all long reads underwent self-error correction before assembly using NextDenovo software (https://github.com/Nextomics/NextDenovo) with default parameters. After self-error correction, all reads were assembled into contigs using WTDBG v2.1 (Ruan and Li, 2019) with parameters -p 19 -k 1 -AS 4 -K 0.05 -s 0.5 to obtain raw genome assembly results. Then, we used Pilon (Walker et al., 2014) v1.21 with two iterations to polish the genome by filtered Illumina short reads (downloaded from NCBI). To assess the completeness of the final genome, several different strategies were used. BUSCO (Benchmarking Universal Single-Copy Orthologs) (Simao et al., 2015) was first employed using both Eukaryota and Metazoa core conserved genes as databases. To test the mapping ratio of Illumina reads, all filtered short reads were mapped to the assembled genome using BWA-MEM v0.7.12 (Li and Durbin, 2009).
Genome Annotation
The genome annotation includes the repetitive elements annotation, gene annotation, and function annotation. For repetitive elements annotation, both tandem repeats and transposable elements (TEs) were determined for the Chinese mitten crab genome. For tandem repeats, Tandem Repeat Finder v4.09 (Benson, 1999) was used for tandem repeat annotation with default parameters. For TE annotation, we searched the TEs on both DNA level and protein level. The RepeatProteinMask (RM-BLASTX) software was used to search TEs with default parameters at the protein level. On DNA level, we used both the consensus sequences produced from the RepeatModeler software and the repbase library (downloaded from RepeatMsker website: http://www.repeatmasker.org/) employed this analysis by the RepeatMasker software (Bedell et al., 2000).
Based on the repeat-masked genome of the Chinese mitten crab, we applied de novo prediction, RNA-seq-based prediction, and homology-based prediction for gene structure annotation. For de novo prediction, the assembled Chinese mitten crab genome and RNA-seq data assembled transcripts were used for Augustus v2.5.5 (Stanke and Waack, 2003) training to obtain proper gene annotation results of Chinese mitten crab. We then used Augustus v2.5.5 (Stanke and Waack, 2003) for de novo prediction of coding genes using the previous training results of Chinese mitten crab. For homology-based prediction, to obtain high-quality gene annotation results, gene set from several species that have high-quality genome or have close relationship with Chinese mitten crab were downloaded, including Bicyclus anynana (GCF_900239965.1) (Nowell et al., 2017), Bombus terrestris (GCF_000214255.1) (Sadd et al., 2015), Drosophila melanogaster (GCA_000001215.4) (Adams et al., 2000), Mus musculus (GCF_000001635.26) (Gnerre et al., 2011), Stegodyphus mimosarum (GCA_000611955.2), Penaeus vannamei (GCA_003789085.1), Mesobuthus martensii (http://lifecenter.sgst.cn/main/en/scorpion.jsp)1 (Cao et al., 2013), E. j. sinensis (GigaDB: 100186) (Song et al., 2016), and Tachypleus tridentatus (GCA_004102145.1). For each species, for each gene we chose the longest transcript and used for tblastn (Altschul, 2012) search with an e value cutoff of 1e−5. The tblastn results were used for Genewise (Birney et al., 2004) analysis to obtain the final homology-based prediction results for each species. For RNA-seq prediction, the assembled transcripts were aligned against the genome using BLAT software (Kent, 2002) (identity >90% and coverage >90%), with PASA (Haas et al., 2003) used to filter the overlaps. EvidenceModeler (Haas et al., 2008) was used to integrate all the above results.
For functional annotation, we used InterProScan v4.8 (Zdobnov and Apweiler, 2001) for InterPro and Gene Ontology (GO) analyses. In addition, the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa, 2002), UniProt/SwissProt, and UniProt/TrEMBL databases were used for BLAST (Altschul, 2012) search with an e value cutoff set as 1e−5 and other parameters set to default.
Gene Family Identification and Unique Gene Family Identification
For comparative genomics analysis, we used all the annotated genes in the Chinese mitten crab and five closely related species, including Aedes aegypti (GCF_002204515.2), D. melanogaster (GCA_000001215.4), S. mimosarum (GCA_000611955.2), P. vannamei (GCA_003789085.1), and B. terrestris (GCF_000214255.1). Genes with the longest transcripts of each species were chosen and used to run all to all BLAST (Altschul, 2012) search. Blastp were used and all the protein sequences of these genes were blasted to each other. The BLAST results were used as input for OrthoMCL (Li et al., 2003) and calculated the pairwise relationships of all genes. The reciprocal best similarity pairs (two-way BLAST best match) were considered as putative orthologous or paralogous genes, and the genes with no ortholog or paralog relationship were identified as unique genes. All the 1:1:1:1:1:1 single-copy genes identified among the six species were chosen and used for further gene family analysis. Besides, we also analyzed the unique gene family in the Chinese mitten crab genome relative to other five species for enrichment analysis. The enrichment analysis was performed using GOstat (Beissbarth and Speed, 2004). Fisher's exact test (chi-square test was used if gene numbers are large) was performed to judge whether the observed difference is significant or not. If the P value is less than 0.05, we considered that the difference is significant.
Phylogenetic Relationship and Divergence Time Estimation
To determine the phylogenetic relationship of the Chinese mitten crab and other species (A. aegypti, B. anynana, D. melanogaster, S. mimosarum, and P. vannamei), we used the previous identified single-copy genes as input and utilized RAxML software (Stamatakis, 2014) with PROTGAMMAAUTO model. S. mimosarum (spider), as the only species in Arachnida among these six species, has the furthest relationship with the five Pancrustacea species. Therefore, S. mimosarum was used as the outgroup, and the single-copy genes of these six species were used for phylogenetic analysis using default parameters. After obtaining the tree of these six species, we performed the divergence time calculation. We employed MCMCTREE analysis in the PAML package (Yang, 1997) with nucleotide substitution model set as JC69 and other parameters set as default, with fossil records of these species from the TIMETREE website (http://www.timetree.org) used for calibration. The calibration data of D. melanogaster and A. aegypti, P. vannamei and E. sinensis, P. vannamei and B. terrestris, and S. mimosarum and P. vannamei were downloaded from the TIMETREE website.
Gene Family Expansion and Contraction Analysis
We performed gene family expansion analysis of the six species using CAFE v4.0 along with the phylogenetic tree and divergence time. Considering that the expanded gene families in the Chinese mitten crab may have important functions, we conducted GO enrichment analysis subsequently. The enrichment analysis was performed using GOstat (Beissbarth and Speed, 2004) as previously described.
Relative Evolution Rate Analysis
Different environments can place different survival pressures on different species. To determine the pressures on the above species, we performed relative evolution rate analysis using LINTRE version 1 (Takezaki et al., 1995) with the tpcv model, with spider as the outgroup species. We also used the Tajima's relative rate test. The spider were also used as outgroup and MEGA8 (Kumar et al., 1994) was used for analysis using default parameters.
Results
Assembly and Characterization of E. j. sinensis Genome
To construct a high-quality genome draft of the Chinese mitten crab, we extracted DNA from the muscle tissue of the crab (Figure 1) and constructed a high-quality Nanopore library for sequencing, which yielded a total of ~53.00 Gb filtered reads (Table S1). Using the downloaded 40.50× Illumina short reads (Table S2), the genome size of this crab was estimated about 1.19 Gb (Figure S1). We performed the contamination detection step and found that 4.39% reads of Illumina short reads and 1.30% Nanopore long reads had the best alignments to the microorganisms and not to other prokaryotes genome. These reads were considered as contamination reads and were all removed. Then, we used the retained Nanopore long reads and Illumina short reads to obtain the final assembled genome with N50 3.19 Mb, which is much better than the previously published Chinese mitten crab genome (Song et al., 2016) (Table 1). To assess the completeness of the final genome, several different strategies were used. BUSCO assessment showed that 94.00% of Eukaryota and 92.90% of Metazoa core conserved genes were found in the Chinese mitten crab genome (Table 2), indicating that most genes (at least over 92% genes were assembled) were assembled. All the filtered ~18-Gb RNA-seq data (Table S3) were assembled into transcripts with N50 1.75 Kb (Table S4). The mapping ratio of Illumina reads (98.82%), Nanopore reads (94.14%), and transcripts (81.32%) to our assembled genome indicated a good quality of the assembled genome (Table 3 and Table S5). N50 of our assembled transcripts (1.76 Kb) and basic information compared to other species further indicated a good genome quality (Table S6). Besides, comparative guanine–cytosine (GC) content analysis of the genome with other species demonstrated that the GC content in the Chinese mitten crab (43%) was slightly higher than that of most related species (from 28% to 42%) (Figure 2).

Figure 1 Chinese mitten crab, Eriocheir japonica sinensis.
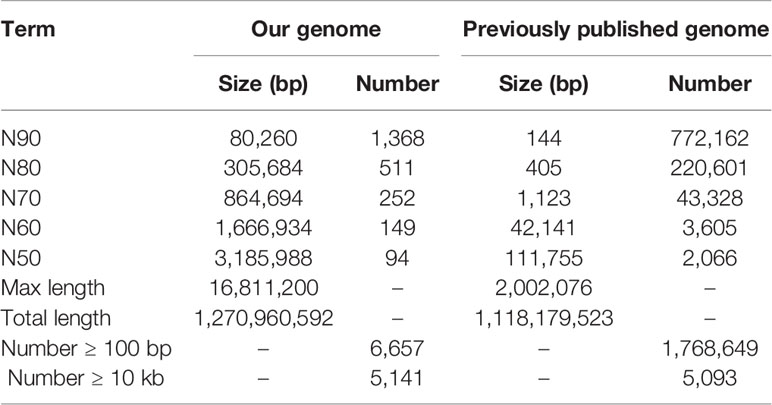
Table 1 Statistics on assembled Chinese mitten crab genome and previously published Chinese mitten crab genome.
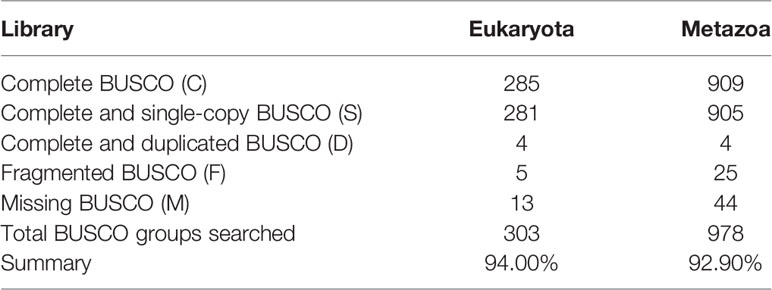
Table 2 Statistics on genome quality evaluation by BUSCO software.

Table 3 Statistics on Illumina short-read mapping ratio for assembled Chinese mitten crab genome.
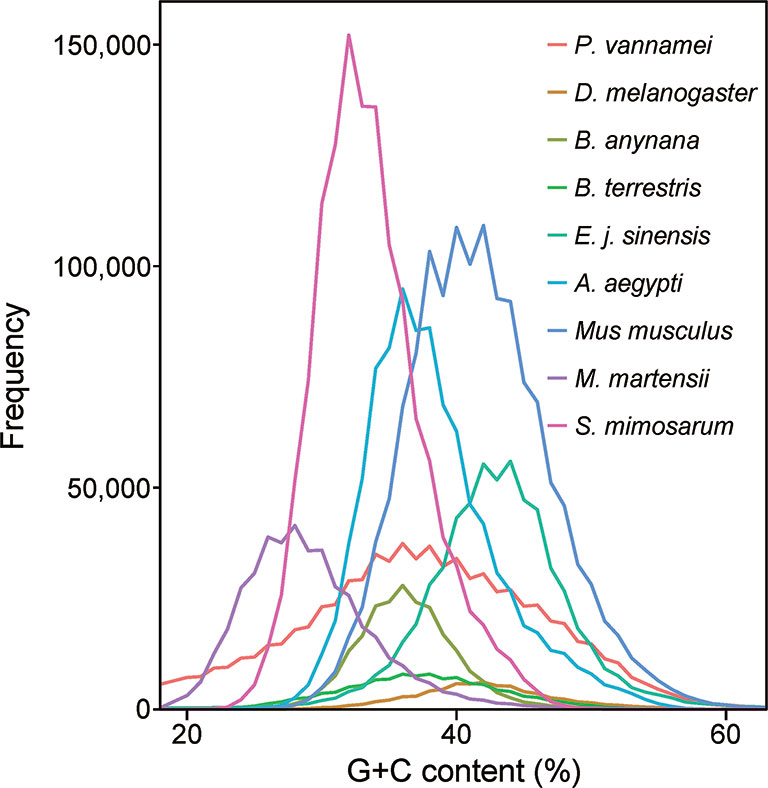
Figure 2 Guanine–cytosine (GC) content of Chinese mitten crab and other genomes. The GC content values were calculated using a sliding window method and the window size is 2 Kb. The whole-genome sequences including the coding region and non-coding region were used for analysis.
Genome Annotation
We conducted three parts of the genome annotation, including repeat annotation, gene annotation, and function annotation. In total, 780.68 Mb of repetitive sequences was identified, accounting for 61.42% of the assembled genome (Table 4). Among these repetitive sequences, 54.43% (~691.78 Mb) were TEs (Table 5). Based on the repeat-masked genome of the Chinese mitten crab, we applied gene structure annotation. And a total of 22,619 high-quality protein-coding genes were identified using the previously described annotation methods, including the de novo prediction, RNA-seq-based prediction, and homology-based prediction. The function annotation results showed that 92.43% of genes have orthology genes in the public database (Table 6). Comparison of the gene quality between the Chinese mitten crab and other published species, including A. aegypti, S. mimosarum, P. vannamei, and D. melanogaster, showed that the lengths of mRNA, coding sequences, exons, and introns were quite similar, suggesting that we acquired a reasonable gene set of the Chinese mitten crab (Figure 3 and Table S7).
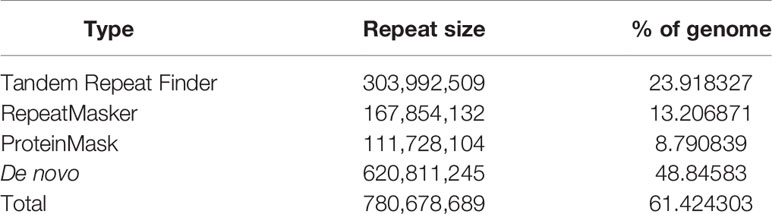
Table 4 Statistics on repetitive sequence annotation in Chinese mitten crab genome.
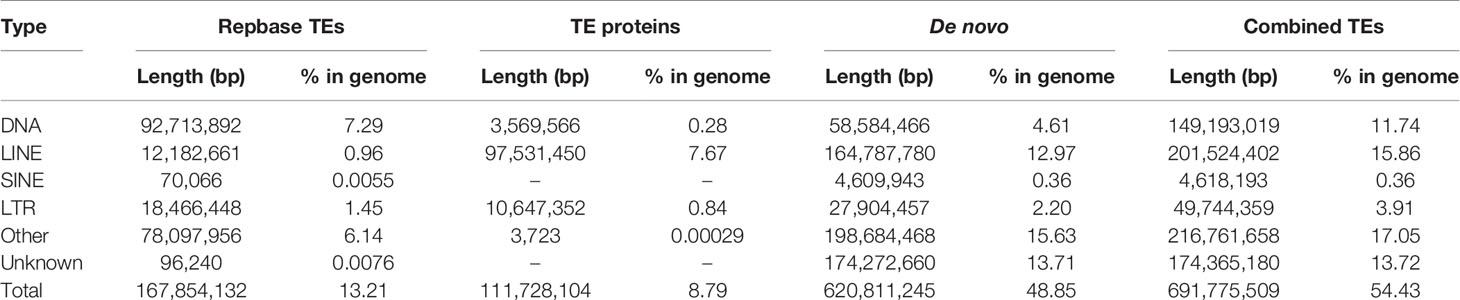
Table 5 Statistics on transposable elements (TEs) in Chinese mitten crab genome.
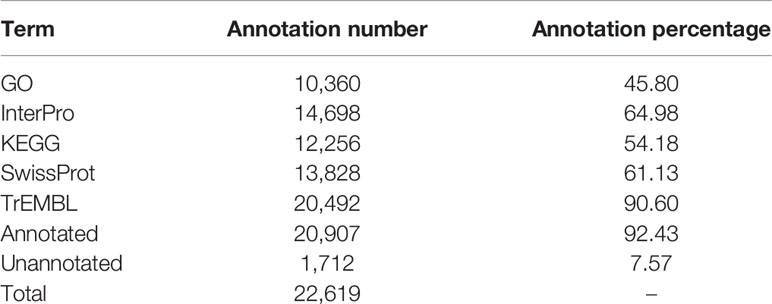
Table 6 Statistics on functional annotation of protein-coding genes in Chinese mitten crab genome.
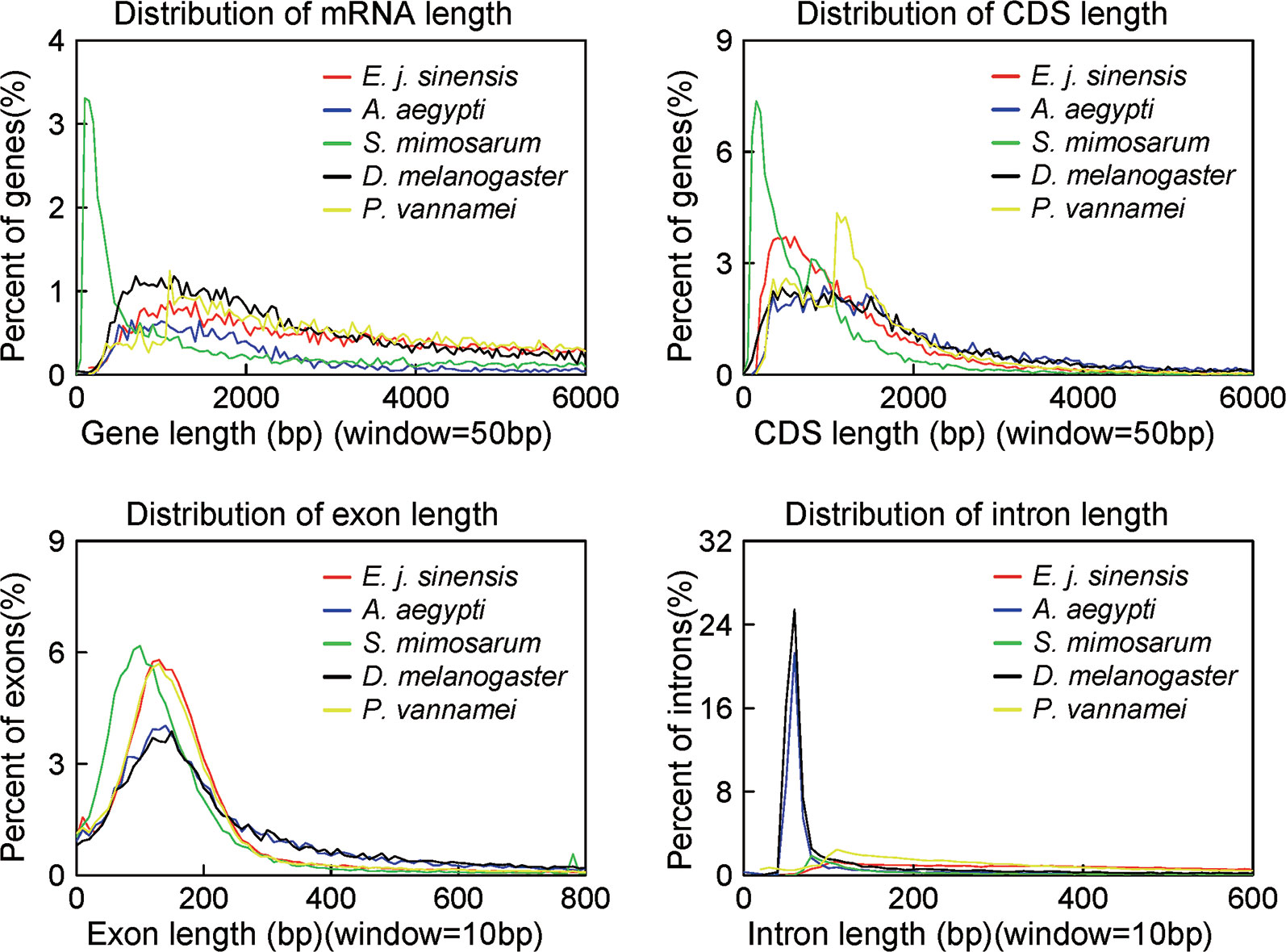
Figure 3 Quality comparison of protein-coding genes. The distribution of mRNA, CDS, exon, and intron among these species were shown. The species include Eriocheir japonica sinensis, Aedes aegypti, Stegodyphus mimosarum, Drosophila melanogaster, and Penaeus vannamei.
Orthologous Gene Identification
For comparative genomics analysis, we used the annotated genes in the Chinese mitten crab and five other species, including A. aegypti, D. melanogaster, S. mimosarum, P. vannamei, and B. terrestris. We identified 2,324 single-copy genes among the six species, which were used for further gene family analysis (Figure 4A). We analyzed the unique gene family in the Chinese mitten crab genome relative to the other five species and finally identified 726 gene families for enrichment analysis (Figure 4B). Results indicated that they were mainly related to heterocyclic compound binding (adjusted P = 9.24e−45), structural constituent of cuticle (adjusted P = 2.99e−05), signaling receptor activity (adjusted P = 0.0021) processes, detection of stimulus (adjusted P = 3.43e−2), response to external stimulus (adjusted P = 4.78e−2), and response to abiotic stimulus (adjusted P = 4.78e−2) (Table S8), suggesting the unique immunity response or ability of Chinese mitten crab.
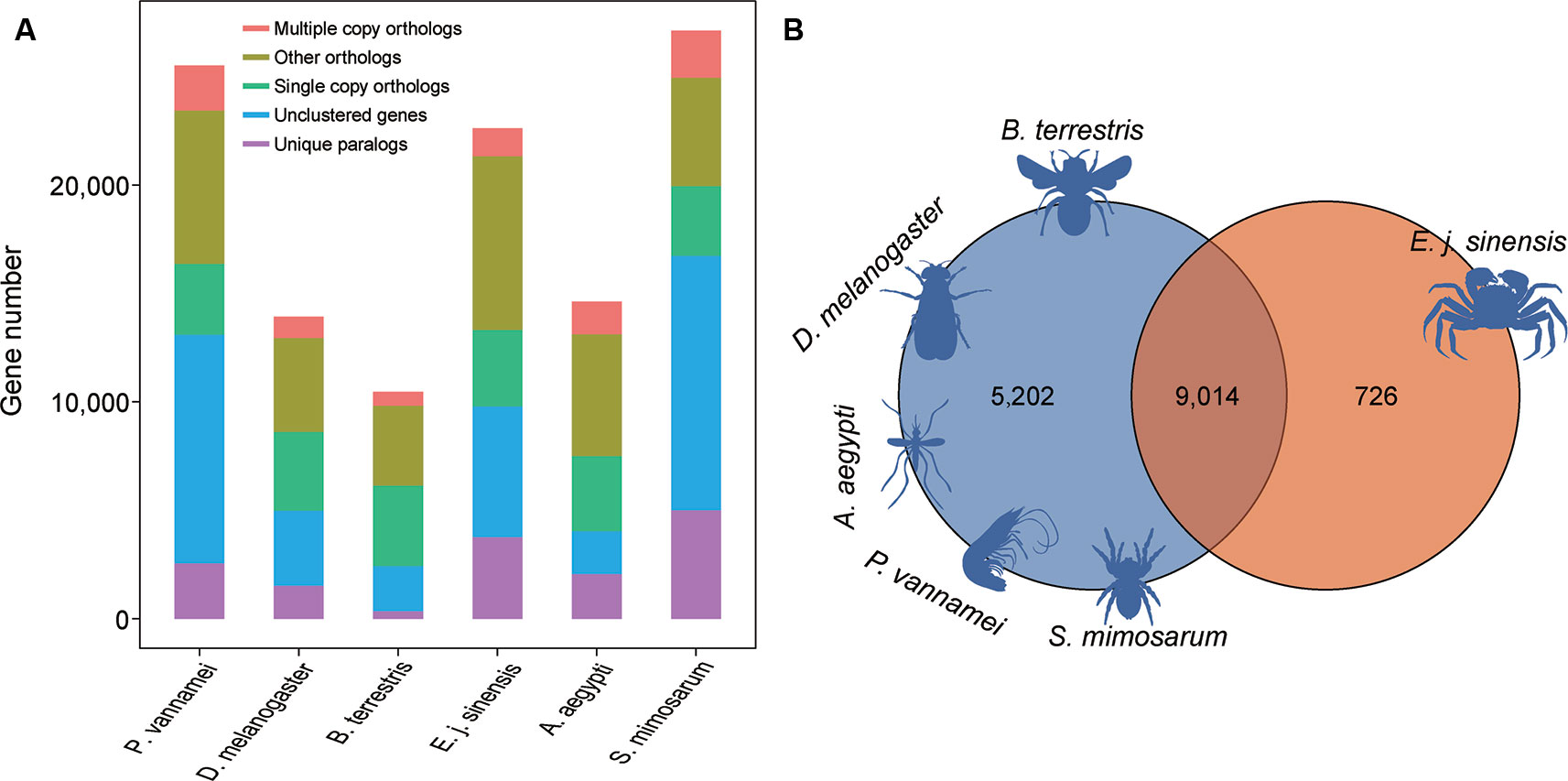
Figure 4 Gene family analysis of Chinese mitten crab. (A) Orthologous genes among species. (B) Unique and shared gene families in species.
Gene Family Expansion and Contraction Analysis
We performed gene family expansion analysis of the six species using CAFE v4.0 (De Bie et al., 2006) and found 603 expanded gene families in the Chinese mitten crab. As these gene families may have important functions in the crab, we conducted GO enrichment analysis. The results showed that the expanded genes were significantly enriched in nine GO terms (adjusted P < 0.05) (Table S9). Specifically, the expanded gene families were primarily related to structural constituent of cuticle (adjusted P = 1.11e−90), heterocyclic compound binding (adjusted P = 2.61e−30), ion binding (adjusted P = 5.39e−3), cellular metabolic process (adjusted P = 1.86e−2), and substrate-specific transporter activity (adjusted P = 3.21e−2) (Table S9). These categories are basic physiological processes, suggesting unique developmental and environmental adaptation of the Chinese mitten crab. We also checked the contracted gene families in Chinese mitten crab among these species. There are 868 contracted gene families, and the GO enrichment analysis showed that these genes are related to the macromolecular complex (adjusted P = 3.74e−24), non-membrane-bounded organelle (adjusted P = 7.62e−19), intracellular non-membrane-bounded organelle (adjusted P = 7.62e−19), intracellular organelle part (adjusted P = 2.54e−17), and organelle part (adjusted P = 2.54e−17) (Table S10).
Phylogenetic Relationship and Divergence Time Estimation
The phylogenetic relationship results showed that the Chinese mitten crab has a close relationship with P. vannamei, but a more distant relationship with B. terrestris, D. melanogaster, and A. aegypti. The five species of Pancrustacea formed two clades, i.e., Hexapoda and Crustacea (Figure 5A). The divergence time results showed that the Chinese mitten crab (Pleocyemata) and P. vannamei (Dendrobranchiata) diverged ~373.6 million years ago (Mya), and Crustacea diverged with the hexapods B. terrestris, D. melanogaster, and A. aegypti ~533.8 Mya (Figure 5A).
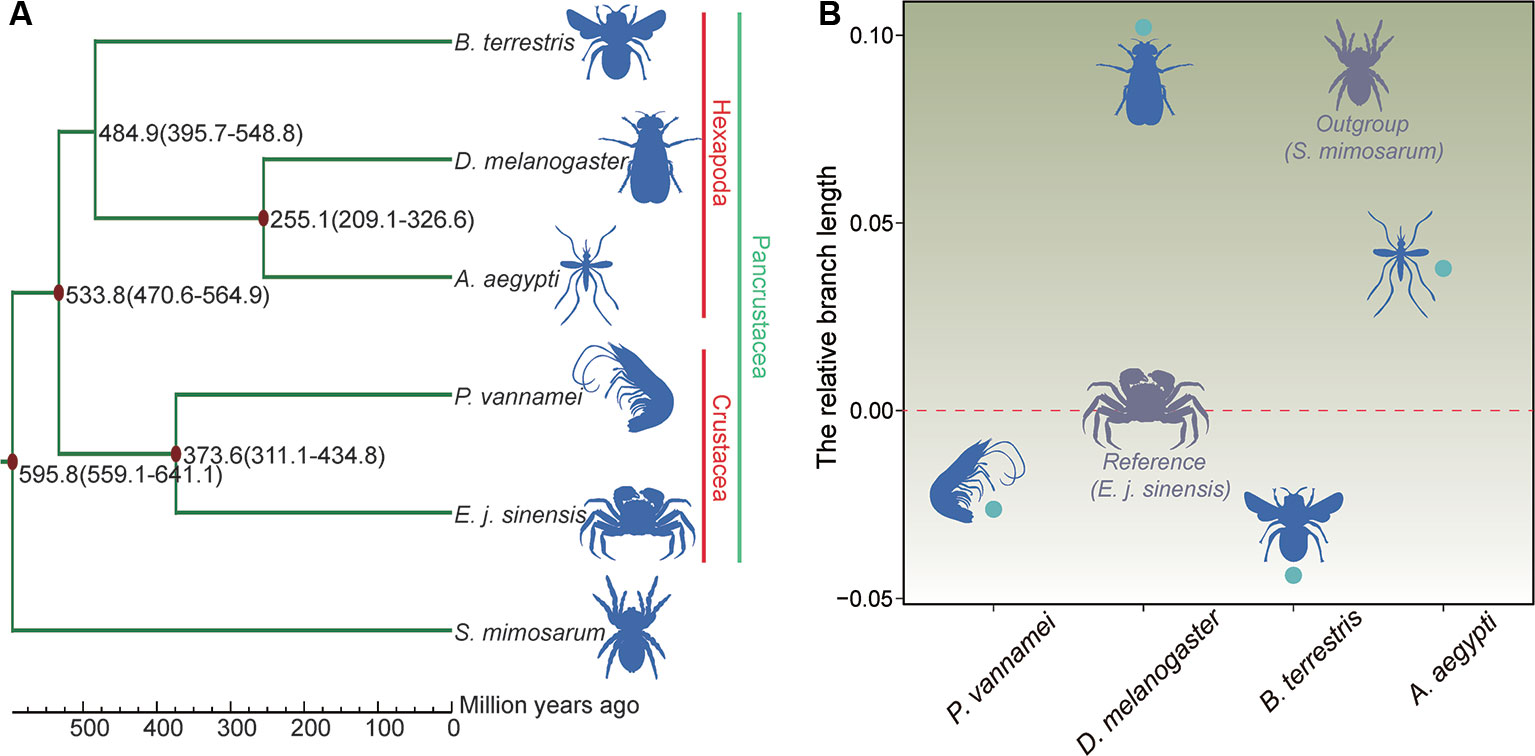
Figure 5 Phylogenetic relationships, divergence time, and evolution rate. (A) Phylogenetic relationship and divergence time of species. Red dot represents fossil record used here. (B) Relative evolution rate of species.
Relative Evolution Rate Analysis
Different environments can place different survival pressures on different species. After the relative evolution rate analysis, the results indicated that the Chinese mitten crab had a faster evolution rate than the shrimp or bumblebee, but slower rate than the other three species (Figure 5B and Table S11), indicating that the Chinese mitten crab inhabits relatively stable environment and experiences little survival pressure.
Conclusion
The size of the E. j. sinensis assembled genome was approximately 1.27 Gb, close to the estimated size (1.19 Gb), and the BUSCO results showed that 94.00% of the expected eukaryotic genes were completely present in the genome assembly. Comparative genomics analysis demonstrated that the Chinese mitten crab diverged from P. vannamei ~373.6 Mya, with a faster evolution rate than shrimp. The expanded genes of E. j. sinensis are basic physiological processes, suggesting unique developmental and environmental adaptation of the Chinese mitten crab. Compared with the previously published genome assembly of Chinese mitten crab, the assembly produced in this study has obvious improvement in genome continuity and gene completeness. We anticipate that the annotated high-quality genome of E. j. sinensis will promote research on its basic development and evolution and make substantial contributions to comparative genomic analyses of crustaceans.
Data Availability Statement
The datasets generated for this study can be found in the BioProject database under the accession number PRJNA555707.
Ethics Statement
This study was approved by the Animal Care and Use Committee, Jiangsu Provincial Key Laboratory of Coastal Wetland Bioresources and Environmental Protection, Yancheng Teachers University. The methods were carried out in accordance with approved guidelines.
Author Contributions
YL, BT, DZ, GQ, and QW conceived and supervised the project. BT, QL, DZ, HZ, SS, and HS collected the samples. ZS, YS, FX, BG, and XW designed and carried out the experiments. ZhoW, HJ, YR, ZheW, and HL performed bioinformatics analyses. YL, BT, WW, and QQ wrote the manuscript. XL, SJ, and WJ helped perform analyses with constructive discussions. All authors have read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (31672267, 31640074), the Jiangsu Agriculture Science and Technology Innovation Fund (CX(18) 3027), the Natural Science Foundation of Jiangsu Province (BK20171276, BK20160444), “Qing Lan Project” of Daizhen Zhang, China Postdoctoral Science Foundation (2018M642105) and the National Key R&D Program of China (2018YFD0900201).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://lifecenter.sgst.cn/main/en/scorpion.jsp. Last accessed June 17, 2018.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01340/full#supplementary-material
References
Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., et al. (2000). The genome sequence of Drosophila melanogaster. Science 287, 2185–2195. doi: 10.1126/science.287.5461.2185
Altschul, S. F. (2012). Basic local alignment search tool (BLAST). J. Mol. Biol. 215, 403–410. doi: 10.1006/jmbi.1990.9999
Bedell, J. A., Korf, I., Gish, W. (2000). MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16, 1040. doi: 10.1093/bioinformatics/16.11.1040
Beissbarth, T., Speed, T. P. (2004). GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 20, 1464–1465. doi: 10.1093/bioinformatics/bth088
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bentley, M. G. (2011). “The Global Spread of the Chinese Mitten Crab Eriocheir sinensis,” in In the Wrong Place – Alien Marine Crustaceans: Distribution, Biology and Impacts. Invading Nature - Springer Series in Invasion Ecology, Eds. Galil, B., Clark, P., Carlton, J. (Springer, Dordrecht), 6.
Birney, E., Clamp, M., Durbin, R. (2004). GeneWise and Genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Cao, Z., Yu, Y., Wu, Y., Hao, P., Di, Z., He, Y., et al. (2013). The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nat. Commun. 4, 2602. doi: 10.1038/ncomms3602
Chang, Y., Liang, L., Ma, H., He, J., Sun, X. (2008). Microsatellite analysis of genetic diversity and population structure of Chinese mitten crab (Eriocheir sinensis). J. Genet. Genomics 35, 171–176. doi: 10.1016/S1673-8527(08)60023-5
Chen, D. W., Zhang, M. (2007). Non-volatile taste active compounds in the meat of Chinese mitten crab (Eriocheir sinensis). Food Chem. 104, 1200–1205. doi: 10.1016/j.foodchem.2007.01.042
Chen, D. W., Zhang, M., Shrestha, S. (2007). Compositional characteristics and nutritional quality of Chinese mitten crab (Eriocheir sinensis). Food Chem. 103, 1343–1349. doi: 10.1016/j.foodchem.2006.10.047
Chen, X., Di, P., Wang, H., Li, B., Pan, Y., Yan, S., et al. (2015). Bacterial community associated with the intestinal tract of Chinese Mitten Crab (Eriocheir sinensis) farmed in Lake Tai, China. PLoS One 10, e0123990. doi: 10.1371/journal.pone.0123990
De Bie, T., Cristianini, N., Demuth, J. P., Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Gai, Y., Wang, L., Zhao, J., Qiu, L., Song, L., Li, L., et al. (2009). The construction of a cDNA library enriched for immune genes and the analysis of 7535 ESTs from Chinese mitten crab Eriocheir sinensis. Fish Shellfish Immunol. 27, 684–694. doi: 10.1016/j.fsi.2009.08.013
Gilbey, V., Attrill, M. J., Coleman, R. A. (2008). Juvenile Chinese mitten crabs (Eriocheir sinensis) in the Thames estuary: distribution, movement and possible interactions with the native crabCarcinus maenas. Biol. Invasions 10, 67–77. doi: 10.1007/s10530-007-9110-4
Gnerre, S., Maccallum, I., Przybylski, D., Ribeiro, F. J., Burton, J. N., Walker, B. J., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 108, 1513–1518. doi: 10.1073/pnas.1017351108
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. doi: 10.1186/gb-2008-9-1-r7
Herborg, L.-M., Rushton, S. P., Clare, A. S., Bentley, M. G. (2005). The invasion of the chinese mitten crab (Eriocheir sinensis) in the United Kingdom and its comparison to continental Europe. Biol. Invasions 7, 959–968. doi: 10.1007/s10530-004-2999-y
Jiang, H., Kang, L., Chew, L., Wang, Q., Liu, Q., Liu, L. (2010). Nutritional requirement of the Chinese mitten-handed Crab Eriocheir sinensis Juvenile for Arginine and Lysine. J. World Aquaculture Soc. 36, 515–520. doi: 10.1111/j.1749-7345.2005.tb00399.x
Jin, G., Xie, P., Li, Z., Jin, G., Xie, P., Li, Z. (2003). Food habits of two-year-old Chinese mitten crab (Eriocheir sinensis) stocked in Lake Bao'an, China. J. Freshwater Ecol. 18, 369–375.
Kent, W. J. (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kumar, S., Tamura, K., Nei, M. (1994). MEGA: Molecular Evolutionary Genetics Analysis software for microcomputers. Comput. Appl. Biosci 10, 189–191. doi: 10.1093/bioinformatics/10.2.189
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L. (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Montú, M., Anger, K., Bakker, C. D. (1996). Larval development of the Chinese mitten crabEriocheir sinensis H. Milne-Edwards (Decapoda: Grapsidae) reared in the laboratory. Helgoländer Meeresuntersuchungen 50, 223–252. doi: 10.11646/zootaxa.4507.1.1
Ng, N. K., Dai, A. Y., Guo, J., Ng, P. K. L. (1998). The Complete Larval Development of the Southern Chinese Mitten Crab, Eriocheir hepuensis Dai, 1991 (Decapoda, Brachyura, Grapsidae) Reared under Laboratory Conditions. Crustaceana 71, 493–517.
Nowell, R. W., Elsworth, B., Oostra, V., Zwaan, B. J., Wheat, C. W., Saastamoinen, M., et al. (2017). A high-coverage draft genome of the mycalesine butterfly Bicyclus anynana. Gigascience 6, 1–7. doi: 10.1093/gigascience/gix035
Panning, A. (1939). The Chinese mitten crab. Rep. Board Regents Smithsonian Institution 1938, 361–375.
Ross, M. G., Russ, C., Costello, M., Hollinger, A., Lennon, N. J., Hegarty, R., et al. (2013). Characterizing and measuring bias in sequence data. Genome Biol. 14, R51. doi: 10.1186/gb-2013-14-5-r51
Ruan, J., Li, H. (2019). Fast and accurate long-read assembly with wtdbg2. Nat. Methods. doi: 10.1038/s41592-019-0669-3
Rudnick, D. A., Hieb, K., Grimmer, K. F., Resh, V. H. (2003). Patterns and processes of biological invasion: the Chinese mitten crab in San Francisco Bay. Basic Appl. Ecol. 4, 249–262. doi: 10.1078/1439-1791-00152
Rudnick, D. A., Chan, V., Resh, V. H. (2005). Morphology and impacts of the burrows of the chinese mitten crab, eriocheir sinensis h. Milne Edwards (Decapoda, Grapsoidea), in South San Francisco Bay, California, U.S.A. Crustaceana 78, 787–807. doi: 10.2307/20107549
Rybczyk, A., Wawrzyniak, W., Czerniejewski, P. A. (2010). Diet of the Chinese mitten crab, Eriocheir sinensis H. Milne Edwards, 1853, and potential effects of the crab on the aquatic community in the River Odra/Oder estuary (N.-W. Poland). Crustaceana 83, 195–205. doi: 10.1163/001121609X12591347509202
Sadd, B. M., Barribeau, S. M., Bloch, G., De Graaf, D. C., Dearden, P., Elsik, C. G., et al. (2015). The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 16, 76. doi: 10.1186/s13059-015-0623-3
Simao, F. A., Waterhouse, R. M., Panagiotis, I., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Song, L., Bian, C., Luo, Y., Wang, L., You, X., Li, J., et al. (2016). Draft genome of the Chinese mitten crab, Eriocheir sinensis. GigaScience 5, 5. doi: 10.1186/s13742-016-0112-y
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, 215–225. doi: 10.1093/bioinformatics/btg1080
Takezaki, N., Rzhetsky, A., Nei, M. (1995). Phylogenetic test of the molecular clock and linearized trees. Mol. Biol. Evol. 12, 823–833. doi: 10.1093/oxfordjournals.molbev.a040259
Tang, B. P., Zhou, K. Y., Song, D. X. (2000). Biodiversity of the Mitten Crab Eriocheir. J. Hebei University. 20 (3), 304–308.
Tsukimura, B. (2008). Determination of Chinese mitten crab, Eriocheir sinensis, year-class strength through investigation of their reproductive life history. California Sea Grant Coll. Program.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963. doi: 10.1371/journal.pone.0112963
Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. In Biosci. Cabios 13, 555. doi: 10.1093/bioinformatics/13.5.555
Zdobnov, E. M., Apweiler, R. (2001). InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Keywords: Eriocheir japonica sinensis, evolution, nanopore, crab, genome assembly and annotation
Citation: Tang B, Wang Z, Liu Q, Zhang H, Jiang S, Li X, Wang Z, Sun Y, Sha Z, Jiang H, Wu X, Ren Y, Li H, Xuan F, Ge B, Jiang W, She S, Sun H, Qiu Q, Wang W, Wang Q, Qiu G, Zhang D and Li Y (2020) High-Quality Genome Assembly of Eriocheir japonica sinensis Reveals Its Unique Genome Evolution. Front. Genet. 10:1340. doi: 10.3389/fgene.2019.01340
Received: 04 September 2019; Accepted: 09 December 2019;
Published: 17 January 2020.
Edited by:
Jianxin Wang, Central South University, ChinaReviewed by:
Sergey Aganezov, Johns Hopkins University, United StatesGuilherme Corrêa De Oliveira, Vale Technological Institute (ITV), Brazil
Copyright © 2020 Tang, Wang, Liu, Zhang, Jiang, Li, Wang, Sun, Sha, Jiang, Wu, Ren, Li, Xuan, Ge, Jiang, She, Sun, Qiu, Wang, Wang, Qiu, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qun Wang, cXdhbmdAYmlvLmVjbnUuZWR1LmNu; Gaofeng Qiu, Z2ZxaXVAc2hvdS5lZHUuY24=; Daizhen Zhang, ZGFpemhlbjc5d2VueGluQDE2My5jb20=; Yongxin Li, eXhsaTI4c2NpZW5jZUBzaW5hLmNvbQ==
†These authors have contributed equally to this work.