 Yanhua Li
Yanhua Li Yaheng Luo1
Yaheng Luo1- 1Department of Endocrinology and Metabolism, The Third Hospital of Changsha, Changsha, Hunan, China
- 2Department of Endocrinology and Metabolism, Want Want Hospital, Changsha, Hunan, China
Sclerostin, a protein synthesized by bone cells, is a product of the SOST gene. Sclerostin is a potent soluble inhibitor of the WNT signaling pathway, and is known to inhibit bone formation by inhibiting osteocyte differentiation and function. Currently, sclerostin has been the subject of numerous animal experiments and clinical investigations. By conducting a literature review, we have gained insights into the most recent advancements in research. Patients with both type 1 diabetes and type 2 diabetes have high levels of serum sclerostin. Patients with type 1 diabetes and type 2 diabetes are both more likely to suffer from osteoporosis, and serum sclerostin levels are elevated in osteoporosis. Many studies have confirmed that sclerostin has been implicated in the pathogenesis of osteoporosis, so we speculate that sclerostin plays an important role in osteoporosis through the glucose metabolism pathway, which may promote the osteoporosis of morbidity in type 1 diabetes and type 2 diabetes. Based on this, we propose whether serum sclerostin can predict type 1 diabetes and type 2 diabetes-induced osteoporosis, and whether it can be a new target for the prevention and treatment of type 1 diabetes and type 2 diabetes-induced osteoporosis, providing new ideas for clinicians and researchers.
1 Introduction
The prevalence of diabetes is escalating, making it a significant chronic epidemic. The International Diabetes Federation reports that approximately 537 million individuals worldwide are currently afflicted with diabetes, constituting 10.5% of the global population (1). The prevalence of diabetes is projected to increase to 783 million individuals by 2045 (1). Another metabolic disease with increasing prevalence is osteoporosis. Osteoporosis, marked by reduced bone density, alterations in bone microstructure, and subsequent fractures, leads to substantial disability and mortality, and has now become a global health concern (2, 3). According to the International Osteoporosis Foundation, with the global population aging, it is projected that over 200 million individuals currently endure the condition of osteoporosis, and one in three women and one in five men over the age of 50 will experience an osteoporotic fracture (4).
Through literature review and previous research, we have learned that sclerostin plays an important role in the pathogenesis of osteoporosis, especially when accompanied by abnormal glucose metabolism. Based on this, this article reviews the clinical evidence regarding serum sclerostin in diabetes and osteoporosis, and delve into the underlying mechanisms involved. This will provide a basis for sclerostin as a new biomarker for diabetes-induced osteoporosis, as well as for potential therapeutic targets. It also provides reference for further clinical research and scientific basis for new drug development.
2 Structure, expression, functions, and signaling pathway related to sclerostin
Sclerostin is a secreted glycoprotein composed of 213 amino acid residues, originally derived from high bone mass disorders sclerosis and van Buchem’s disease (5, 6). Sclerostin is a 22-kDa protein characterized by a core disulfide-bonded structure composed of three distinct domains: ring 1, ring 2, and ring 3 (7); the protein’s side chain features a highly flexible N-terminal domain (amino acids 1-55) and a C-terminal domain (amino acids 145-189); it contains four disulfide bonds formed by four p-cysteine residues (8). Unlike the majority of proteins containing cystine junction motifs, sclerostin exists as a monomer (9). Sclerostin is synthesized through the expression of the SOST gene on human chromosome 17q12-q21 (10). The SOST gene contains two distinct transcription sites. The first transcription site is evolutionary conserve region 5, and monocyte enhancer factor 2 can promote the expression of sclerostin by binding to evolutionary conserve region 5 (11, 12). However, histone deacetylases 4 and 5 are capable of inhibiting the transcription of the SOST gene by binding to monocyte enhancer factor 2 (13). The second transcription site is the upstream promoter region, where runt-related transcription factor 2 binds and represses sclerostin expression (14, 15). Histone deacetylases 3 inhibits the transcription of the SOST gene by targeting the promoter region (16, 17). Sclerostin is mainly secreted by osteocytes and acts in a paracrine manner (18). It is detectable in plasma and expressed in tissues such as bone, cartilage, kidney, liver, pancreas, heart and blood vessels (19). Sclerostin expression is markedly reduced in newly embedded osteocytes and undetectable in mature osteoblasts and bone lining cells (20). Sclerostin, an inhibitor of the WNT signaling pathway, antagonizes bone formation by binding to low-density lipoprotein receptor-related protein (LRP) 5/6, functioning as an antianabolic agent (21, 22). It is also implicated in skeletal muscle regeneration, insulin resistance, and glucose metabolism (23, 24). Recent investigations have demonstrated that sclerostin potentiates the inhibitory effect on bone formation by mediating binding to LRP6 through its interaction with LRP4 (25).
3 Expression of sclerostin in diabetes
In recent years, the incidence of type 1 diabetes (T1D) has increased rapidly at a rate of 3%-5% per year worldwide. Wedrychowicz, et al. (26) demonstrated that serum sclerostin levels are significantly elevated in patients with T1D and exhibit an inverse correlation with glycosylated hemoglobin (HbA1c). Rubin et al. (27) conducted a cross-sectional study, which found that sclerostin was significantly increased in T1D patients; However, sclerostin is not associated with HbA1c. This may be because the participants are older, and the level of sclerostin increases with age, with higher values masking the relationship with HbA1c. Neumann et al. (28) conducted a study on T1D and healthy individuals, found that the level of sclerostin in T1D was significantly higher than that in the control group, and was not associated with bone metabolism markers. Therefore, we speculate that sclerostin may be involved in osteoporosis independently of bone metabolism markers. Kurban et al. (29) conducted a cross-sectional study comparing the levels of sclerostin between 40 T1D and 40 healthy controls, and found that sclerostin was elevated in T1D, but the difference was not statistically significant, possibly due to the small sample size. Employing recent research, it has been demonstrated that the serum level of sclerostin is markedly elevated in patients with T1D in comparison to healthy controls (30). Faienza’s research also confirmed the same results (31).
A lot of studies have also been conducted on the expression of sclerostin in type 2 diabetes (T2D). Sclerostin levels were reported to be higher in patients with T2D in an age-matched randomized controlled study (32). Garcia-Martin et al. (33) conducted a cross-sectional study, revealing that sclerostin levels are elevated in patients with T2D and demonstrating a correlation between sclerostin levels and the duration of T2D, HbA1c. The findings of a clinical investigation conducted by Singh et al. (34), involving a cohort of 171 individuals divided into three categories—healthy individuals, individuals with pre-T2D, and patients with T2D—demonstrated a gradual increase in sclerostin levels and sclerostin mRNA expression from healthy to pre-T2D to T2D. In addition, elevated circulating sclerostin levels were positively correlated with insulin resistance and fat mass (35). Frysz et al. (36) conducted a meta-analysis, revealing a significant association between elevated levels of sclerostin and an increased risk of diabetes. A cross-sectional analysis of femoral head bone tissue from postmenopausal women with T2D revealed a significant increase in the expression of SOST, when compared to healthy women (37) (Table 1).
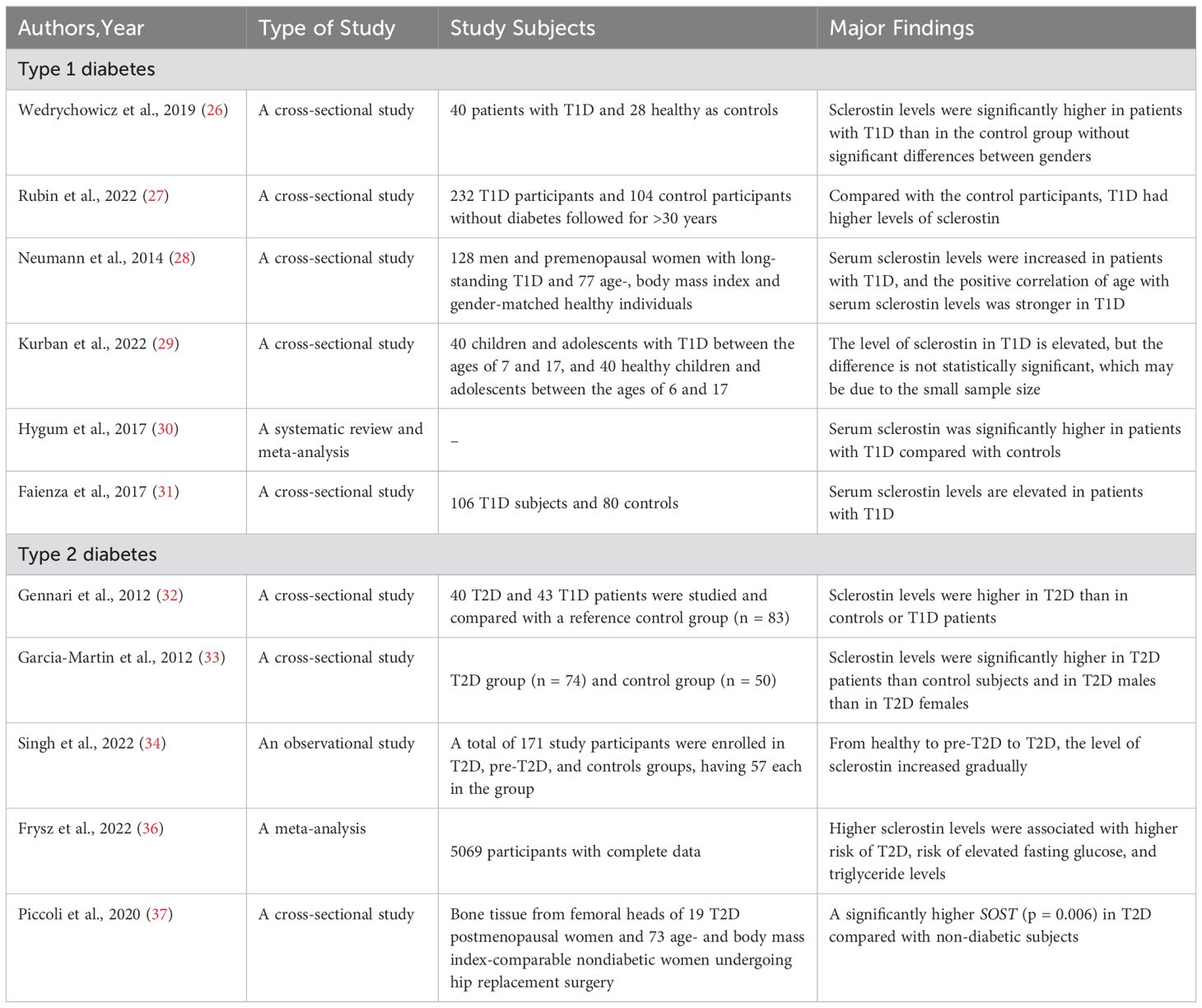
Table 1. Changes of sclerostin in patients with T1D and T2D.
Therefore, we speculate that sclerostin is not only involved in the pathogenesis of diabetes, but also plays an important role in osteoporosis through glucose metabolism.
4 Association of sclerostin with osteoporosis in diabetes
4.1 Association of diabetes with osteoporosis
Diabetes is considered a significant risk factor for osteoporosis, as an increasing body of evidence supports its association with an elevated risk of osteoporotic fractures (38–41). Glycolysis can promote osteoblast differentiation, may alter the levels of important intermediate metabolites that regulate gene expression (42). In their review, Vadivalagan et al. identified aerobic glycolysis as an effective way to accelerate the treatment of osteoporosis (43). Poor blood sugar control is an important risk factor for diabetic osteoporosis fracture (44). Nirwan and Vohora (45) performed experiments in C57BL/6 mice, which were fed a high-fat diet for 22 weeks to induce diabetic osteoporosis. Subsequently, linagliptin combined with metformin was used for intervention, and the results suggested that diabetic osteoporosis could be treated by increasing the level of bone morphogenetic protein-2 and reducing the level of sclerostin.
Previous studies have confirmed that T1D is closely related to decreased bone mineral density (BMD) and bone quality (46, 47). Studies have reported that children and adolescents with T1D compared with healthy controls, the BMD value is low (48), lead to adult peak bone mass was lower than those of healthy people, thus prone to osteopenia and osteoporosis (49). The longer the duration of T1D and the worse the blood sugar control, the higher the risk of fractures in patients (46, 47, 50). Two previous cross-sectional studies have demonstrated that poorer glycemic control was associated with lower BMD scores in patients with T1D (51). This is due to reduced bone production and formation of mineralized matrix in T1D (52, 53). It is known that T1D has a negative effect on osteoblast differentiation and function and a positive effect on osteoclast differentiation and function, thereby reducing bone formation and increasing bone resorption (54). The reason may be that hyperglycemia inhibits osteogenic differentiation of mesenchymal stem cells (55), and inhibits the ability of osteoblasts to resist mechanical load (56, 57). It has been established that the bone phenotype of patients with T1D is characterized by the following four characteristics: reduced BMD (58), disrupted bone microstructure (59), increased risk of fractures (60), reduced the conversion rate of bone (61). And risk of fracture in patients with T1D is six times that of the healthy adults (62). A meta-analysis of 46 studies with 2617 and 3851 controls showed that children with T1D had significantly lower BMD measured by dual-energy X-ray absorptiometry (whole body, lumbar spine, femur), peripheral bone quantitative CT scanning (radius and tibia), and quantitative ultrasound of calcaneus and phalanges compared with controls (63). The findings from the study conducted by Weber et al. (64) demonstrated a significant reduction in bone mass gain among individuals diagnosed with T1D one year after diagnosis. Kalaitzoglou et al. (65) used streptozotocin to induce T1D in mice. The results suggest that chronic hyperglycemia and pro-inflammatory bone microenvironment in T1D mice enhance osteoclast activity, which leads to enhanced bone resorption and decreased bone mass.
A state of low bone turnover has been demonstrated in T2D (66–68). Hygum et al. (30) conducted a meta-analysis and found that levels of bone formation markers (N-terminal propeptide of type 1 procollagen (P1NP) and osteocalcin) and bone resorption markers (C-terminal cross-linked telopeptide of type I collagen (CTX) and tartrate-resistant acid phosphatase 5b isoform) were reduced in T2D. The study by Napoli et al. (69) showed that P1NP was reduced by about 13% and CTX was reduced by about 43% in patients with T2D compared with non-diabetic subjects. It has also been confirmed that the risk of fragility fractures is increased in patients with T2D (33, 70). Strotmeyer et al. (71) conducted a cohort study, results show that the adult T2D patients than about 64% higher risk of fracture in patients without T2D.
Both types of diabetes are susceptible to osteoporosis, suggesting that the two forms of diabetes induce the development of osteoporosis through different mechanisms (72). Insufficient osteoblast differentiation is considered to be an important cause of osteoporosis in T1D patients (73); The inhibition of bone remodeling is considered a significant contributing factor of osteoporosis in T2D patients (74). However, the specific pathogenesis of osteoporosis in diabetes is not clear.
4.2 Expression of sclerostin in diabetes-induced osteoporosis
Recent studies have shown that sclerostin is involved in bone metabolism in T1D (31). Clinical data from Wędrychowicz et al. (26) could point to increased sclerostin levels as a potential cause of reduced bone formation in T1D. Yee et al. (75) conducted an animal study to establish a streptozotocin-induced fracture model of T1D mice. The researchers injected sclerostin antibody into the mouse model, and on days 21 and 42, a large number of early osteoblasts were seen, and bone quality was significantly improved.
The same changes were also observed in T2D. Yamamoto’s study not only confirmed the elevation of sclerostin in T2D, but also revealed that sclerostin increases vertebral fractures, which may be due to sclerostin mediated deterioration of bone quality (76). Ardawi et al. (77) conducted a cross-sectional study on 482 T2D patients and 482 healthy individuals, and the results showed that sclerostin was elevated in T2D, and sclerostin was associated with increased bone fragility. Wang et al. (78) included 95 T2D patients and divided them into normal bone group, osteopenia group and osteoporosis group according to bone mineral density, and found that the osteoporosis group had the highest level of sclerostin. The researchers propose that sclerostin mediates osteoporosis in T2D by inhibiting WNT signaling. In a cross-sectional study conducted by Ahmad (79), we learned that sclerostin was significantly higher in T2D compared to healthy people; Moreover, the incidence of osteopenia and osteoporosis in T2D is higher than that in the healthy people. Animal study by Hamann et al. (80) concluded that sclerostin antibody therapy reversed the adverse effects of T2D on bone mass and strength in rats and improved bone defect regeneration, suggesting that sclerostin could be used as a biomarker for early detection of osteoporosis in diabetes patients.
From the above studies, we learned that sclerostin may be involved in the pathogenesis of osteoporosis through glucose metabolism.
5 Relationship between sclerostin and clinical outcome in osteoporosis
Owing to its recognized role as a negative regulator of the WNT signaling pathway, sclerostin binds to LRP5/6 coreceptors, thereby inhibiting bone formation and promoting bone resorption (81). It has also been shown that sclerostin stimulates bone resorption through receptor activator of nuclear factor kappa-B ligand-dependent pathway, thereby promoting BMD reduction (82). Sclerostin has been demonstrated to suppress the proliferation of osteoblasts while simultaneously promoting their apoptosis (83). Cosman et al. (84) performed a randomized, controlled clinical trial on postmenopausal women with osteoporosis and demonstrated that treatment with anti-sclerostin antibodies enhances bone formation and increases BMD, thereby mitigating the risk of fracture. A 12-month phase IIb trial, involving postmenopausal women randomized to receive either teriparatide (20ug/day) or anti-sclerostin antibody (210mg/month), demonstrated a significant increase in lumbar BMD with the administration of anti-sclerostin antibody therapy (85). In a meta-analysis published in 2022 (86), Poutoglidou et al. observed that both a 6-month and a 12-month treatment course with anti-sclerostin antibodies were capable of enhancing BMD at the lumbar spine, total hip, and femoral neck, and lowering the incidence of fractures. Concurrently, anti-sclerostin antibody therapy also demonstrated a reduction in CTX levels and an elevation in P1NP levels. Therefore, the usage of anti-sclerostin monoclonal antibodies has progressively become a significant component in the management of osteoporosis (87). In a phase III clinical trial, the subcutaneous administration of antibodies to sclerostin (such as romosozumab, a monoclonal antibody that binds sclerostin) demonstrated efficacy in enhancing BMD compared to the placebo in postmenopausal women with osteoporosis and men with osteoporosis (84, 88). The findings of Recker et al. (89), Cosman et al. (84) and Kaveh et al. (90) corroborated this observation. A case report suggests that the nonunion of humeral shaft fractures in males and postmenopausal females might be addressed through the administration of anti-sclerostin medications (91) (Table 2).
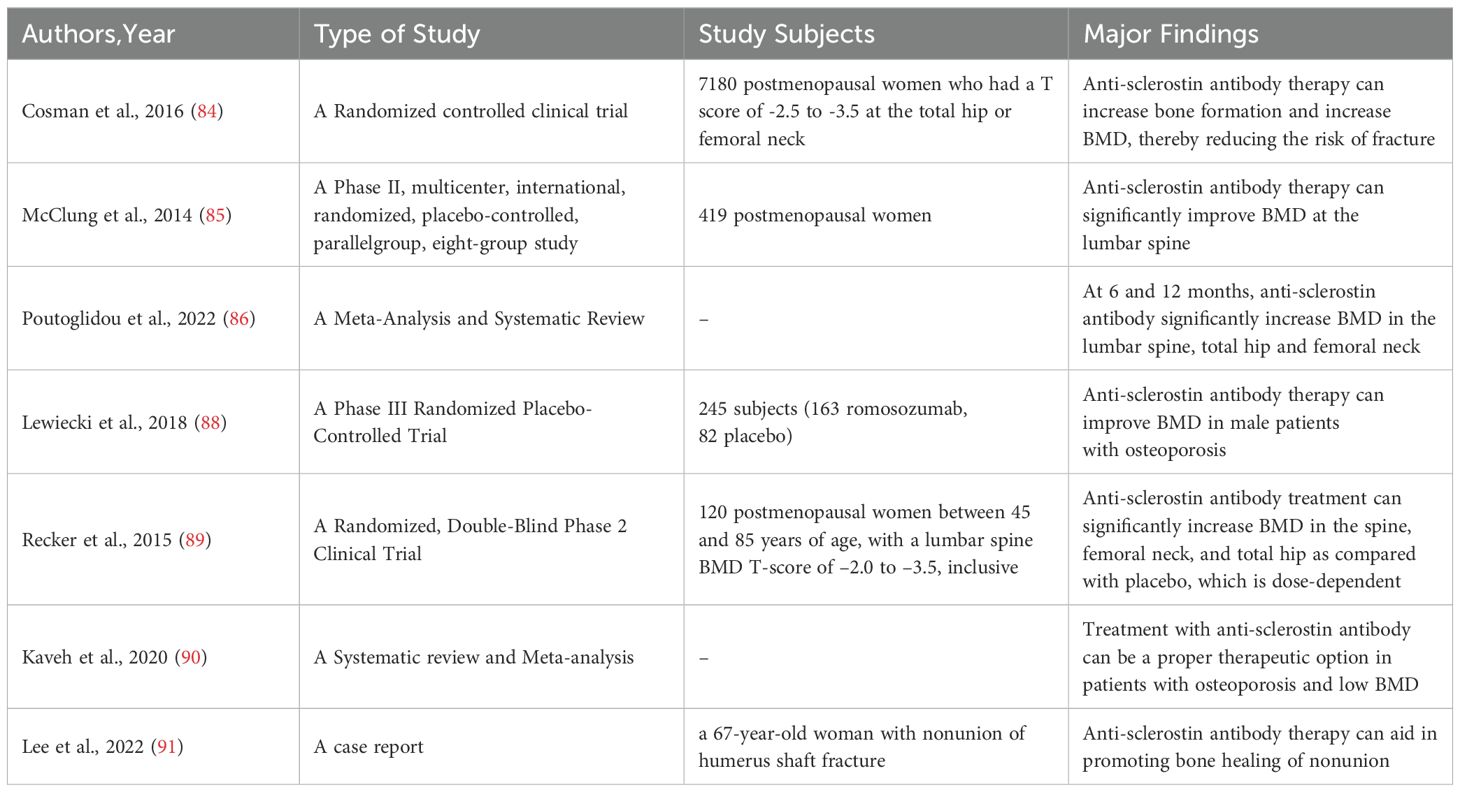
Table 2. Changes in the serum sclerostin levels in osteoporosis.
Enhanced bone formation has been observed in patients or mice lacking sclerostin, leading to bone sclerosis, as demonstrated by the results of several studies (10, 92, 93); In contrast, mice with an excessive expression of SOST exhibited reduced bone mass (94). In vitro experiments by Wang et al. (95) revealed that overexpression of SOST significantly inhibited WNT signaling and messenger ribonucleic acid levels of osteogenic markers in Col1a2+/G610C mouse osteoblasts. The domain in which sclerostin plays a major role in this process is loop 3. The elimination of SOST led to an increase in bone mass and strength (96, 97). In the mouse studies conducted by Oh et al. (98), schnurri-3 was observed to suppress SOST expression in osteoblasts, while the absence of SOST had no impact on schnurri-3. Specifically, targeted inhibition of both schnurri-3 and SOST effectively mitigated bone loss and stimulated bone formation in mice. Li et al. (81) developed a male rat model of osteoporosis and demonstrated that anti-sclerostin antibodies could significantly enhance bone mass and preserve bone quality in rats, primarily by stimulating bone formation and inhibiting bone resorption. The study conducted by Boyce et al. (99) demonstrated that administration of anti-sclerostin antibodies enhanced BMD and elevated serum concentrations of bone formation markers, but failed to affect levels of bone resorption markers. Brent et al. (100) employed a rat model, utilizing either anti-sclerostin antibody, abaloparatide, or a combination of both, and demonstrated that anti-sclerostin antibody enhanced bone strength in the mid-diaphysis, neck, and metaphysis of long bones. In contrast, the combination of anti-sclerostin antibody and abaloparatide led to a significant increase in bone strength at all sites, augmented markers of bone remodeling, and reduced trabecular bone spacing. This suggests that the combined treatment is significantly more effective than either agent alone. In a mouse model of streptozotocin-induced T1D, osteoblastic defects and reduced levels of osteocalcin and alkaline phosphatase were found to be associated with increased expression of WNT signaling inhibitors Dickkopf-1 and SOST (101). Interestingly, treatment with anti-sclerostin antibodies accelerates fracture healing by facilitating osteoblast differentiation and enhancing callus mineralization, thereby ameliorating bone microstructure (75). Maillard et al. (102) artificially induced skull defects in mice, and subsequently employed mesenchymal stem cells to counteract sclerostin. Eight weeks post-intervention, it was discovered that this approach not only increased bone formation and promoted bone repair, but also demonstrated comparable efficacy to SOST knockout mice. Hamann et al. (80) and Kruck et al. (103), respectively, demonstrated in animal models that the administration of anti-sclerostin antibodies accelerates bone formation. We are informed that anti-sclerostin antibodies act as an stimulator of bone formation in the short term and as an inhibitor of bone resorption in the long term, jointly leading to an increase in bone mass by both means (104). In a study employing SOST knockout mice, we observed that glucocorticoid administration led to a reduction in osteoprotegerin levels and an increase in the receptor activator of nuclear factor kappa-B ligand/osteoprotegerin ratio; Conversely, administration of an anti-sclerostin antibody inhibited bone resorption by augmenting osteoprotegerin levels (105). The research conducted by Lin et al. (106) demonstrated that mice lacking sclerostin exhibited resistance to bone mass decline. Carro Vazquez at el (107). demonstrated that treatment with anti-sclerostin antibodies could enhance bone quality and facilitate bone healing in rats, using the Zucker Diabetic Fatty rat model. The mechanism involves directly influencing bone by down-regulating miR-145-5p/p transcription in bone tissue, up-regulating osteoprotegerin target expression, resulting in decreased osteoclast production (108), and up-regulating Sp7 (109) and signal transduction 3A target levels (110), leading to enhanced osteogenic differentiation. The expression of the SOST gene and protein was suppressed by mechanical loading, leading to an enhancement in bone formation (106, 111, 112). The mechanism may be that mechanical stimulation activates connexin 43 hemichannells to release prostaglandin E2 from osteocytes, thereby inhibiting the expression of SOST in osteocytes and enhancing the activity of osteoblasts and bone formation (113). The research findings by Kim et al. (114) demonstrate that SOST-/- mice exhibit enhanced bone formation and reduced visceral and subcutaneous fat deposition, primarily attributed to the absence of sclerostin protein which inhibits the differentiation of progenitor cells into mature adipocytes. Thus inhibit sclerostin can also help the treatment of obesity. In Zhou et al.’s study (115), 23-month-old male rats were randomly divided into a orchiectomy group and a sham operation group. Eight weeks after surgery, the results showed that the serum sclerostin level in the orchiectomy group was significantly higher than that in the sham operation group, and was negatively correlated with trabecular BMD. This also suggests that sclerostin may be a potential therapeutic target for male osteoporosis. In both animal models and clinical studies, sclerostin antibody-induced bone formation was reactivated upon exposure to physical stimuli (84, 116, 117). Many non clinical pharmacology research results show that the sclerostin antibody can inhibit sclerostin to form, which can promote the fracture healing and callus formation (118). The mechanism of action is that sclerostin antibody inhibits the binding of sclerostin to LRP5/6, thereby weakening the antagonistic activity of sclerostin against WNT-induced responses (119).
Taking the above findings together, sclerostin is emerging as a potent inhibitor of bone formation by reducing osteoblast differentiation and activity. As a result, we speculate that sclerostin can be used as a biomarker for osteoporosis.
6 Conclusion and prospects
Sclerostin is a powerful protein molecules involved in bone metabolism and skeletal muscle regeneration, mainly related to osteoporosis (Figure 1). A large number of studies have revealed that sclerostin is elevated in T1D and T2D, and diabetic patients are more susceptible to osteoporosis. Numerous clinical studies have also demonstrated that patients with osteoporosis exhibit elevated levels of serum sclerostin. Existing evidence suggests that sclerostin antibodies such as romosozumab reduce sclerostin expression, leading to improvement in osteoporosis. Therefore, early exploration of sclerostin targets in diabetes patients plays a vital role in the prevention and treatment of diabetes-induced osteoporosis. However, sclerostin can be used in clinical medicine is still to be solved.
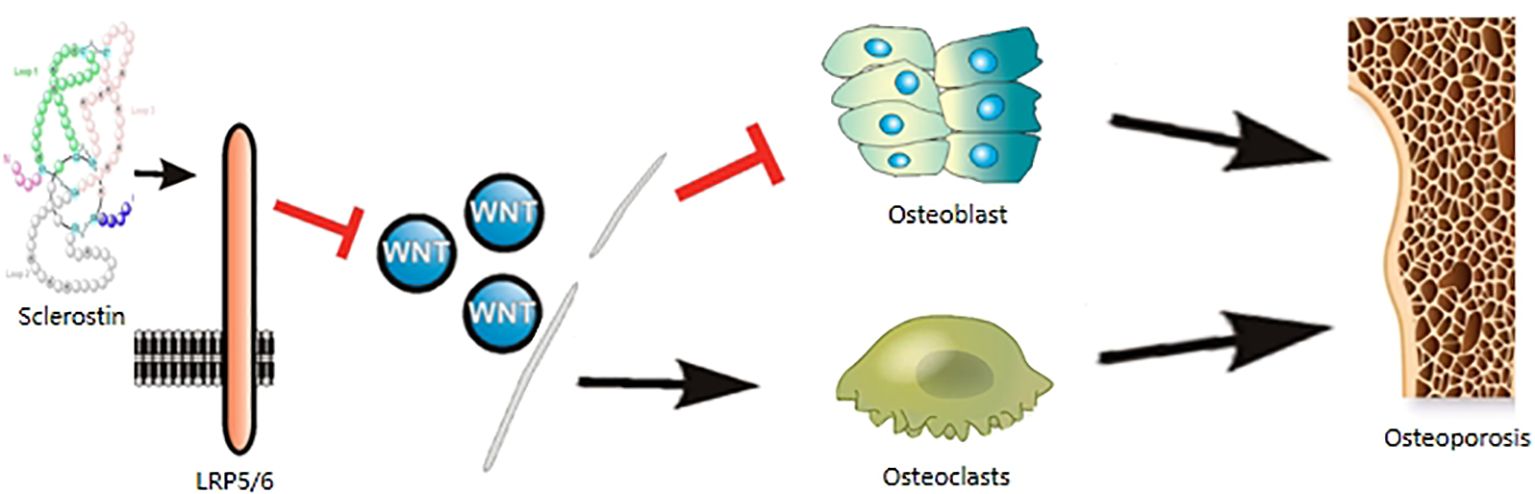
Figure 1. Sclerostin are involved in the mechanism of osteoporosis. Sclerostin binds to LRP5/6 coreceptors, acts on the WNT signaling pathway, inhibits osteoblasts and promotes osteoclastogenesis, leading to osteoporosis.
To fill the current gap, the following research is needed: To further clarify the specific mechanism of sclerostin in diabetes-induced osteoporosis, remove obstacles for clinical study; To develop safe and effective sclerostin inhibitors, prevent and treat osteoporosis induced by diabetes is warranted.
Author contributions
YHL: Writing – original draft, Writing – review & editing. YL: Writing – review & editing. DH: Writing – review & editing. LP: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank all authors for their contributions to the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BMD, bone mineral density; CTX, C-terminal cross-linked telopeptide of type I collagen; HbA1c, glycosylated hemoglobin; LRP, low-density lipoprotein receptor-related protein; P1NP, N-terminal propeptide of type 1 procollagen; T1D, type 1 diabetes; T2D, type 2 diabetes.
References
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Janner M, Saner C. Impact of type 1 diabetes mellitus on bone health in children. Horm Res Paediatr. (2022) 95:205–14. doi: 10.1159/000521627
3. Zhang N, Zhang ZK, Yu Y, Zhuo Z, Zhang G, Zhang BT. Pros and cons of denosumab treatment for osteoporosis and implication for RANKL aptamer therapy. Front Cell Dev Biol. (2020) 8:325. doi: 10.3389/fcell.2020.00325
4. Sozen T, Ozisik L, Basaran NC. An overview and management of osteoporosis. Eur J Rheumatol. (2017) 4:46–56. doi: 10.5152/eurjrheum.2016.048
5. Kim J, Han W, Park T, Kim EJ, Bang I, Lee HS, et al. Sclerostin inhibits Wnt signaling through tandem interaction with two LRP6 ectodomains. Nat Commun. (2020) 11:5357. doi: 10.1038/s41467-020-19155-4
6. Vasiliadis ES, Evangelopoulos DS, Kaspiris A, Benetos IS, Vlachos C, Pneumaticos SG. The role of sclerostin in bone diseases. J Clin Med. (2022) 11:806. doi: 10.3390/jcm11030806
7. De Mare A, Opdebeeck B, Neven E, D'Haese PC, Verhulst A. Sclerostin protects against vascular calcification development in mice. J Bone Miner Res. (2022) 37:687–99. doi: 10.1002/jbmr.4503
8. Veverka V, Henry AJ, Slocombe PM, Ventom A, Mulloy B, Muskett FW, et al. Characterization of the structural features and interactions of sclerostin: molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. (2009) 284:10890–900. doi: 10.1074/jbc.M807994200
9. Bourhis E, Wang W, Tam C, Hwang J, Zhang Y, Spittler D, et al. Wnt antagonists bind through a short peptide to the first beta-propeller domain of LRP5/6. Structure. (2011) 19:1433–42. doi: 10.1016/j.str.2011.07.005
10. Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. (2001) 10:537–43. doi: 10.1093/hmg/10.5.537
11. Van Buchem FS, Hadders HN, Hansen JF, Woldring MG. Hyperostosis corticalis generalisata. Report of seven cases. Am J Med. (1962) 33:387–97. doi: 10.1016/0002-9343(62)90235-8
12. Koide M, Kobayashi Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J Bone Miner Metab. (2019) 37:9–17. doi: 10.1007/s00774-018-0971-7
13. Sun N, Uda Y, Azab E, Kochen A, Santos R, Shi C, et al. Effects of histone deacetylase inhibitor Scriptaid and parathyroid hormone on osteocyte functions and metabolism. J Biol Chem. (2019) 294:9722–33. doi: 10.1074/jbc.RA118.007312
14. Komori T. Regulation of proliferation, differentiation and functions of osteoblasts by runx2. Int J Mol Sci. (2019) 20:1694. doi: 10.3390/ijms20071694
15. Delgado-Calle J, Sanudo C, Bolado A, Fernandez AF, Arozamena J, Pascual-Carra MA, et al. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J Bone Miner Res. (2012) 27:926–37. doi: 10.1002/jbmr.1491
16. Cohen-Kfir E, Artsi H, Levin A, Abramowitz E, Bajayo A, Gurt I, et al. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology. (2011) 152:4514–24. doi: 10.1210/en.2011-1128
17. Genetos DC, Toupadakis CA, Raheja LF, Wong A, Papanicolaou SE, Fyhrie DP, et al. Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J Cell Biochem. (2010) 110:457–67. doi: 10.1002/jcb.22559
18. Yavropoulou MP, Xygonakis C, Lolou M, Karadimou F, Yovos JG. The sclerostin story: from human genetics to the development of novel anabolic treatment for osteoporosis. Hormones (Athens). (2014) 13:323–37. doi: 10.14310/horm.2002.1552
19. Pietrzyk B, Smertka M, Chudek J. Sclerostin: Intracellular mechanisms of action and its role in the pathogenesis of skeletal and vascular disorders. Adv Clin Exp Med. (2017) 26:1283–91. doi: 10.17219/acem/68739
20. Delgado-Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone. (2017) 96:29–37. doi: 10.1016/j.bone.2016.10.007
21. Galea GL, Lanyon LE, Price JS. Sclerostin's role in bone's adaptive response to mechanical loading. Bone. (2017) 96:38–44. doi: 10.1016/j.bone.2016.10.008
22. Wang JS, Mazur CM, Wein MN. Sclerostin and osteocalcin: candidate bone-produced hormones. Front Endocrinol (Lausanne). (2021) 12:584147. doi: 10.3389/fendo.2021.584147
23. Urano T, Shiraki M, Ouchi Y, Inoue S. Association of circulating sclerostin levels with fat mass and metabolic disease–related markers in Japanese postmenopausal women. J Clin Endocrinol Metab. (2012) 97:E1473–7. doi: 10.1210/jc.2012-1218
24. Huang J, Romero-Suarez S, Lara N, Mo C, Kaja S, Brotto L, et al. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/beta-catenin pathway. JBMR Plus. (2017) 1:86–100. doi: 10.1002/jbm4.10015
25. Katchkovsky S, Chatterjee B, Abramovitch-Dahan CV, Papo N, Levaot N. Competitive blocking of LRP4-sclerostin binding interface strongly promotes bone anabolic functions. Cell Mol Life Sci. (2022) 79:113. doi: 10.1007/s00018-022-04127-2
26. Wedrychowicz A, Sztefko K, Starzyk JB. Sclerostin and its significance for children and adolescents with type 1 diabetes mellitus (T1D). Bone. (2019) 120:387–92. doi: 10.1016/j.bone.2018.08.007
27. Rubin MR, de Boer IH, Backlund JC, Arends V, Gubitosi-Klug R, Wallia A, et al. Biochemical markers of bone turnover in older adults with type 1 diabetes. J Clin Endocrinol Metab. (2022) 107:e2405–e16. doi: 10.1210/clinem/dgac099
28. Neumann T, Hofbauer LC, Rauner M, Lodes S, Kastner B, Franke S, et al. Clinical and endocrine correlates of circulating sclerostin levels in patients with type 1 diabetes mellitus. Clin Endocrinol (Oxf). (2014) 80:649–55. doi: 10.1111/cen.12364
29. Kurban S, Selver Eklioglu B, Selver MB. Investigation of the relationship between serum sclerostin and dickkopf-1 protein levels with bone turnover in children and adolescents with type-1 diabetes mellitus. J Pediatr Endocrinol Metab. (2022) 35:673–9. doi: 10.1515/jpem-2022-0001
30. Hygum K, Starup-Linde J, Harslof T, Vestergaard P, Langdahl BL. MECHANISMS IN ENDOCRINOLOGY: Diabetes mellitus, a state of low bone turnover - a systematic review and meta-analysis. Eur J Endocrinol. (2017) 176:R137–R57. doi: 10.1530/EJE-16-0652
31. Faienza MF, Ventura A, Delvecchio M, Fusillo A, Piacente L, Aceto G, et al. High sclerostin and dickkopf-1 (DKK-1) serum levels in children and adolescents with type 1 diabetes mellitus. J Clin Endocrinol Metab. (2017) 102:1174–81. doi: 10.1210/jc.2016-2371
32. Gennari L, Merlotti D, Valenti R, Ceccarelli E, Ruvio M, Pietrini MG, et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrinol Metab. (2012) 97:1737–44. doi: 10.1210/jc.2011-2958
33. Garcia-Martin A, Rozas-Moreno P, Reyes-Garcia R, Morales-Santana S, Garcia-Fontana B, Garcia-Salcedo JA, et al. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2012) 97:234–41. doi: 10.1210/jc.2011-2186
34. Singh PK, Naithani M, Pathania M, Mirza AA, Saha S. An insight into the association of sclerostin with insulin sensitivity and glycemic parameters in male Indian prediabetic and diabetic population. Cureus. (2022) 14:e27123. doi: 10.7759/cureus.27123
35. Ukita M, Yamaguchi T, Ohata N, Tamura M. Sclerostin enhances adipocyte differentiation in 3T3-L1 cells. J Cell Biochem. (2016) 117:1419–28. doi: 10.1002/jcb.25432
36. Frysz M, Gergei I, Scharnagl H, Smith GD, Zheng J, Lawlor DA, et al. Circulating sclerostin levels are positively related to coronary artery disease severity and related risk factors. J Bone Miner Res. (2022) 37:273–84. doi: 10.1002/jbmr.4467
37. Piccoli A, Cannata F, Strollo R, Pedone C, Leanza G, Russo F, et al. Sclerostin regulation, microarchitecture, and advanced glycation end-products in the bone of elderly women with type 2 diabetes. J Bone Miner Res. (2020) 35:2415–22. doi: 10.1002/jbmr.4153
38. Jia P, Bao L, Chen H, Yuan J, Liu W, Feng F, et al. Risk of low-energy fracture in type 2 diabetes patients: a meta-analysis of observational studies. Osteoporos Int. (2017) 28:3113–21. doi: 10.1007/s00198-017-4183-0
39. Koromani F, Oei L, Shevroja E, Trajanoska K, Schoufour J, Muka T, et al. Vertebral fractures in individuals with type 2 diabetes: more than skeletal complications alone. Diabetes Care. (2020) 43:137–44. doi: 10.2337/dc19-0925
40. Vilaca T, Schini M, Harnan S, Sutton A, Poku E, Allen IE, et al. The risk of hip and non-vertebral fractures in type 1 and type 2 diabetes: A systematic review and meta-analysis update. Bone. (2020) 137:115457. doi: 10.1016/j.bone.2020.115457
41. Hofbauer LC, Busse B, Eastell R, Ferrari S, Frost M, Muller R, et al. Bone fragility in diabetes: novel concepts and clinical implications. Lancet Diabetes Endocrinol. (2022) 10:207–20. doi: 10.1016/S2213-8587(21)00347-8
42. Esen E, Long F. Aerobic glycolysis in osteoblasts. Curr Osteoporos Rep. (2014) 12:433–8. doi: 10.1007/s11914-014-0235-y
43. Vadivalagan C, Krishnan A, Chen SJ, Hseu YC, Muthu S, Dhar R, et al. The Warburg effect in osteoporosis: Cellular signaling and epigenetic regulation of energy metabolic events to targeting the osteocalcin for phenotypic alteration. Cell Signal. (2022) 100:110488. doi: 10.1016/j.cellsig.2022.110488
44. Puar TH, Khoo JJ, Cho LW, Xu Y, Chen YT, Chuo AM, et al. Association between glycemic control and hip fracture. J Am Geriatr Soc. (2012) 60:1493–7. doi: 10.1111/j.1532-5415.2012.04052.x
45. Nirwan N, Vohora D. Linagliptin in combination with metformin ameliorates diabetic osteoporosis through modulating BMP-2 and sclerostin in the high-fat diet fed C57BL/6 mice. Front Endocrinol (Lausanne). (2022) 13:944323. doi: 10.3389/fendo.2022.944323
46. Shah VN, Carpenter RD, Ferguson VL, Schwartz AV. Bone health in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. (2018) 25:231–6. doi: 10.1097/MED.0000000000000421
47. Weber DR. Bone accrual in children and adolescents with type 1 diabetes: current knowledge and future directions. Curr Opin Endocrinol Diabetes Obes. (2021) 28:340–7. doi: 10.1097/MED.0000000000000638
48. Heap J, Murray MA, Miller SC, Jalili T, Moyer-Mileur LJ. Alterations in bone characteristics associated with glycemic control in adolescents with type 1 diabetes mellitus. J Pediatr. (2004) 144:56–62. doi: 10.1016/j.jpeds.2003.10.066
49. Weber DR, Haynes K, Leonard MB, Willi SM, Denburg MR. Type 1 diabetes is associated with an increased risk of fracture across the life span: a population-based cohort study using The Health Improvement Network (THIN). Diabetes Care. (2015) 38:1913–20. doi: 10.2337/dc15-0783
50. Leanza G, Maddaloni E, Pitocco D, Conte C, Palermo A, Maurizi AR, et al. Risk factors for fragility fractures in type 1 diabetes. Bone. (2019) 125:194–9. doi: 10.1016/j.bone.2019.04.017
51. Dongare-Bhor S, Lohiya N, Maheshwari A, Ekbote V, Chiplonkar S, Padidela R, et al. Muscle and bone parameters in underprivileged Indian children and adolescents with T1DM. Bone. (2020) 130:115074. doi: 10.1016/j.bone.2019.115074
52. Hu P, McKenzie JA, Buettmann EG, Migotsky N, Gardner MJ, Silva MJ. Type 1 diabetic Akita mice have low bone mass and impaired fracture healing. Bone. (2021) 147:115906. doi: 10.1016/j.bone.2021.115906
53. Jiao H, Xiao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep. (2015) 13:327–35. doi: 10.1007/s11914-015-0286-8
54. Roy B. Biomolecular basis of the role of diabetes mellitus in osteoporosis and bone fractures. World J Diabetes. (2013) 4:101–13. doi: 10.4239/wjd.v4.i4.101
55. Gopalakrishnan V, Vignesh RC, Arunakaran J, Aruldhas MM, Srinivasan N. Effects of glucose and its modulation by insulin and estradiol on BMSC differentiation into osteoblastic lineages. Biochem Cell Biol. (2006) 84:93–101. doi: 10.1139/o05-163
56. Parajuli A, Liu C, Li W, Gu X, Lai X, Pei S, et al. Bone's responses to mechanical loading are impaired in type 1 diabetes. Bone. (2015) 81:152–60. doi: 10.1016/j.bone.2015.07.012
57. Seref-Ferlengez Z, Maung S, Schaffler MB, Spray DC, Suadicani SO, Thi MM. P2X7R-panx1 complex impairs bone mechanosignaling under high glucose levels associated with type-1 diabetes. PLoS One. (2016) 11:e0155107. doi: 10.1371/journal.pone.0155107
58. Pan H, Wu N, Yang T, He W. Association between bone mineral density and type 1 diabetes mellitus: a meta-analysis of cross-sectional studies. Diabetes Metab Res Rev. (2014) 30:531–42. doi: 10.1002/dmrr.2508
59. Shanbhogue VV, Hansen S, Frost M, Jorgensen NR, Hermann AP, Henriksen JE, et al. Bone geometry, volumetric density, microarchitecture, and estimated bone strength assessed by HR-pQCT in adult patients with type 1 diabetes mellitus. J Bone Miner Res. (2015) 30:2188–99. doi: 10.1002/jbmr.2573
60. Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL, et al. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol. (2017) 13:208–19. doi: 10.1038/nrendo.2016.153
61. Murray CE, Coleman CM. Impact of diabetes mellitus on bone health. Int J Mol Sci. (2019) 20:4873. doi: 10.3390/ijms20194873
62. Janghorbani M, Feskanich D, Willett WC, Hu F. Prospective study of diabetes and risk of hip fracture: the Nurses' Health Study. Diabetes Care. (2006) 29:1573–8. doi: 10.2337/dc06-0440
63. Loxton P, Narayan K, Munns CF, Craig ME. Bone mineral density and type 1 diabetes in children and adolescents: A meta-analysis. Diabetes Care. (2021) 44:1898–905. doi: 10.2337/dc20-3128
64. Weber DR, Gordon RJ, Kelley JC, Leonard MB, Willi SM, Hatch-Stein J, et al. Poor glycemic control is associated with impaired bone accrual in the year following a diagnosis of type 1 diabetes. J Clin Endocrinol Metab. (2019) 104:4511–20. doi: 10.1210/jc.2019-00035
65. Kalaitzoglou E, Popescu I, Bunn RC, Fowlkes JL, Thrailkill KM. Effects of type 1 diabetes on osteoblasts, osteocytes, and osteoclasts. Curr Osteoporos Rep. (2016) 14:310–9. doi: 10.1007/s11914-016-0329-9
66. Shu A, Yin MT, Stein E, Cremers S, Dworakowski E, Ives R, et al. Bone structure and turnover in type 2 diabetes mellitus. Osteoporos Int. (2012) 23:635–41. doi: 10.1007/s00198-011-1595-0
67. Manavalan JS, Cremers S, Dempster DW, Zhou H, Dworakowski E, Kode A, et al. Circulating osteogenic precursor cells in type 2 diabetes mellitus. J Clin Endocrinol Metab. (2012) 97:3240–50. doi: 10.1210/jc.2012-1546
68. Farr JN, Drake MT, Amin S, Melton LJ 3rd, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J Bone Miner Res. (2014) 29:787–95. doi: 10.1002/jbmr.2106
69. Napoli N, Strollo R, Defeudis G, Leto G, Moretti C, Zampetti S, et al. Serum sclerostin and bone turnover in latent autoimmune diabetes in adults. J Clin Endocrinol Metab. (2018) 103:1921–8. doi: 10.1210/jc.2017-02274
70. Grontved A, Rimm EB, Willett WC, Andersen LB, Hu FB. A prospective study of weight training and risk of type 2 diabetes mellitus in men. Arch Intern Med. (2012) 172:1306–12. doi: 10.1001/archinternmed.2012.3138
71. Strotmeyer ES, Cauley JA, Schwartz AV, Nevitt MC, Resnick HE, Bauer DC, et al. Nontraumatic fracture risk with diabetes mellitus and impaired fasting glucose in older white and black adults: the health, aging, and body composition study. Arch Intern Med. (2005) 165:1612–7. doi: 10.1001/archinte.165.14.1612
72. Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am J Epidemiol. (2007) 166:495–505. doi: 10.1093/aje/kwm106
73. Motyl KJ, McCauley LK, McCabe LR. Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. J Cell Physiol. (2012) 227:1326–34. doi: 10.1002/jcp.22844
74. Starup-Linde J, Vestergaard P. Management of endocrine disease: Diabetes and osteoporosis: cause for concern? Eur J Endocrinol. (2015) 173:R93–9. doi: 10.1530/EJE-15-0155
75. Yee CS, Xie L, Hatsell S, Hum N, Murugesh D, Economides AN, et al. Sclerostin antibody treatment improves fracture outcomes in a Type I diabetic mouse model. Bone. (2016) 82:122–34. doi: 10.1016/j.bone.2015.04.048
76. Yamamoto M, Yamauchi M, Sugimoto T. Elevated sclerostin levels are associated with vertebral fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. (2013) 98:4030–7. doi: 10.1210/jc.2013-2143
77. Ardawi MS, Akhbar DH, Alshaikh A, Ahmed MM, Qari MH, Rouzi AA, et al. Increased serum sclerostin and decreased serum IGF-1 are associated with vertebral fractures among postmenopausal women with type-2 diabetes. Bone. (2013) 56:355–62. doi: 10.1016/j.bone.2013.06.029
78. Wang N, Xue P, Wu X, Ma J, Wang Y, Li Y. Role of sclerostin and dkk1 in bone remodeling in type 2 diabetic patients. Endocr Res. (2018) 43:29–38. doi: 10.1080/07435800.2017.1373662
79. Ahmad IH, Elhamed Gbr SSA, Ali El Naggar BMM, Abdelwahab MK, El-Saghier EOA, Mohammed DS, et al. Relation between serum sclerostin and CTRP3 levels and bone mineral density in diabetic postmenopausal women. BMC Womens Health. (2024) 24:490. doi: 10.1186/s12905-024-03311-9
80. Hamann C, Rauner M, Hohna Y, Bernhardt R, Mettelsiefen J, Goettsch C, et al. Sclerostin antibody treatment improves bone mass, bone strength, and bone defect regeneration in rats with type 2 diabetes mellitus. J Bone Miner Res. (2013) 28:627–38. doi: 10.1002/jbmr.1803
81. Li X, Ominsky MS, Villasenor KS, Niu QT, Asuncion FJ, Xia X, et al. Sclerostin antibody reverses bone loss by increasing bone formation and decreasing bone resorption in a rat model of male osteoporosis. Endocrinology. (2018) 159:260–71. doi: 10.1210/en.2017-00794
82. Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. (2011) 6:e25900. doi: 10.1371/journal.pone.0025900
83. Sutherland MK, Geoghegan JC, Yu C, Turcott E, Skonier JE, Winkler DG, et al. Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone. (2004) 35:828–35. doi: 10.1016/j.bone.2004.05.023
84. Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. (2016) 375:1532–43. doi: 10.1056/NEJMoa1607948
85. McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. (2014) 370:412–20. doi: 10.1056/NEJMoa1305224
86. Poutoglidou F, Samoladas E, Raikos N, Kouvelas D. Efficacy and safety of anti-sclerostin antibodies in the treatment of osteoporosis: A meta-analysis and systematic review. J Clin Densitom. (2022) 25:401–15. doi: 10.1016/j.jocd.2021.11.005
87. Funck-Brentano T, Cohen-Solal M. Anti-sclerostin antibodies in osteoporosis and other bone diseases. J Clin Med. (2020) 9:3439. doi: 10.3390/jcm9113439
88. Lewiecki EM, Blicharski T, Goemaere S, Lippuner K, Meisner PD, Miller PD, et al. A phase III randomized placebo-controlled trial to evaluate efficacy and safety of romosozumab in men with osteoporosis. J Clin Endocrinol Metab. (2018) 103:3183–93. doi: 10.1210/jc.2017-02163
89. Recker RR, Benson CT, Matsumoto T, Bolognese MA, Robins DA, Alam J, et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. (2015) 30:216–24. doi: 10.1002/jbmr.2351
90. Kaveh S, Hosseinifard H, Ghadimi N, Vojdanian M, Aryankhesal A. Efficacy and safety of Romosozumab in treatment for low bone mineral density: a systematic review and meta-analysis. Clin Rheumatol. (2020) 39:3261–76. doi: 10.1007/s10067-020-04948-1
91. Lee SY, Kawasaki K, Inagaki K. Successful treatment of humeral shaft nonunion with romosozumab: A case report. Trauma Case Rep. (2022) 37:100595. doi: 10.1016/j.tcr.2021.100595
92. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. (2001) 68:577–89. doi: 10.1086/318811
93. Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. (2008) 23:860–9. doi: 10.1359/jbmr.080216
94. Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. (2010) 25:178–89. doi: 10.1359/jbmr.090730
95. Wang L, Yu Y, Ni S, Li D, Liu J, Xie D, et al. Therapeutic aptamer targeting sclerostin loop3 for promoting bone formation without increasing cardiovascular risk in osteogenesis imperfecta mice. Theranostics. (2022) 12:5645–74. doi: 10.7150/thno.63177
96. van Lierop AH, Hamdy NA, Hamersma H, van Bezooijen RL, Power J, Loveridge N, et al. Patients with sclerosteosis and disease carriers: human models of the effect of sclerostin on bone turnover. J Bone Miner Res. (2011) 26:2804–11. doi: 10.1002/jbmr.474
97. Piters E, Culha C, Moester M, Van Bezooijen R, Adriaensen D, Mueller T, et al. First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. Hum Mutat. (2010) 31:E1526–43. doi: 10.1002/humu.21274
98. Oh WT, Yang YS, Xie J, Ma H, Kim JM, Park KH, et al. WNT-modulating gene silencers as a gene therapy for osteoporosis, bone fracture, and critical-sized bone defects. Mol Ther. (2023) 31:435–53. doi: 10.1016/j.ymthe.2022.09.018
99. Boyce RW, Niu QT, Ominsky MS. Kinetic reconstruction reveals time-dependent effects of romosozumab on bone formation and osteoblast function in vertebral cancellous and cortical bone in cynomolgus monkeys. Bone. (2017) 101:77–87. doi: 10.1016/j.bone.2017.04.005
100. Brent MB, Bruel A, Thomsen JS. Anti-sclerostin antibodies and abaloparatide have additive effects when used as a countermeasure against disuse osteopenia in female rats. Bone. (2022) 160:116417. doi: 10.1016/j.bone.2022.116417
101. Hie M, Iitsuka N, Otsuka T, Tsukamoto I. Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int J Mol Med. (2011) 28:455–62. doi: 10.3892/ijmm.2011.697
102. Maillard S, Sicard L, Andrique C, Torrens C, Lesieur J, Baroukh B, et al. Combining sclerostin neutralization with tissue engineering: An improved strategy for craniofacial bone repair. Acta Biomater. (2022) 140:178–89. doi: 10.1016/j.actbio.2021.11.046
103. Kruck B, Zimmermann EA, Damerow S, Figge C, Julien C, Wulsten D, et al. Sclerostin neutralizing antibody treatment enhances bone formation but does not rescue mechanically induced delayed healing. J Bone Miner Res. (2018) 33:1686–97. doi: 10.1002/jbmr.3454
104. Appelman-Dijkstra NM, Oei H, Vlug AG, Winter EM. The effect of osteoporosis treatment on bone mass. Best Pract Res Clin Endocrinol Metab. (2022) 36:101623. doi: 10.1016/j.beem.2022.101623
105. Sato AY, Cregor M, Delgado-Calle J, Condon KW, Allen MR, Peacock M, et al. Protection from glucocorticoid-induced osteoporosis by anti-catabolic signaling in the absence of sost/sclerostin. J Bone Miner Res. (2016) 31:1791–802. doi: 10.1002/jbmr.2869
106. Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. (2009) 24:1651–61. doi: 10.1359/jbmr.090411
107. Carro Vazquez D, Emini L, Rauner M, Hofbauer C, Grillari J, Diendorfer AB, et al. Effect of anti-osteoporotic treatments on circulating and bone microRNA patterns in osteopenic ZDF rats. Int J Mol Sci. (2022) 23:6534. doi: 10.3390/ijms23126534
108. Chen Y, Wang X, Yang M, Ruan W, Wei W, Gu D, et al. miR-145-5p increases osteoclast numbers in vitro and aggravates bone erosion in collagen-induced arthritis by targeting osteoprotegerin. Med Sci Monit. (2018) 24:5292–300. doi: 10.12659/MSM.908219
109. Jia J, Tian Q, Ling S, Liu Y, Yang S, Shao Z. miR-145 suppresses osteogenic differentiation by targeting Sp7. FEBS Lett. (2013) 587:3027–31. doi: 10.1016/j.febslet.2013.07.030
110. Jin Y, Hong F, Bao Q, Xu Q, Duan R, Zhu Z, et al. MicroRNA-145 suppresses osteogenic differentiation of human jaw bone marrow mesenchymal stem cells partially via targeting semaphorin 3A. Connect Tissue Res. (2020) 61:577–85. doi: 10.1080/03008207.2019.1643334
111. Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. (2008) 283:5866–75. doi: 10.1074/jbc.M705092200
112. Spatz JM, Wein MN, Gooi JH, Qu Y, Garr JL, Liu S, et al. The wnt inhibitor sclerostin is up-regulated by mechanical unloading in osteocytes in vitro. J Biol Chem. (2015) 290:16744–58. doi: 10.1074/jbc.M114.628313
113. Zhao D, Riquelme MA, Guda T, Tu C, Xu H, Gu S, et al. Connexin hemichannels with prostaglandin release in anabolic function of bone to mechanical loading. Elife. (2022) 11:e74365 doi: 10.7554/eLife.74365
114. Kim SP, Da H, Wang L, Taketo MM, Wan M, Riddle RC. Bone-derived sclerostin and Wnt/beta-catenin signaling regulate PDGFRalpha(+) adipoprogenitor cell differentiation. FASEB J. (2021) 35:e21957. doi: 10.1096/fj.202100691R
115. Zhou BN, Zhang Q, Lin XY, Hu J, Zhao DC, Jiang Y, et al. The roles of sclerostin and irisin on bone and muscle of orchiectomized rats. BMC Musculoskelet Disord. (2022) 23:1049. doi: 10.1186/s12891-022-05982-7
116. Ma YL, Hamang M, Lucchesi J, Bivi N, Zeng Q, Adrian MD, et al. Time course of disassociation of bone formation signals with bone mass and bone strength in sclerostin antibody treated ovariectomized rats. Bone. (2017) 97:20–8. doi: 10.1016/j.bone.2016.12.003
117. Gerbaix M, Ammann P, Ferrari S. Mechanically driven counter-regulation of cortical bone formation in response to sclerostin-neutralizing antibodies. J Bone Miner Res. (2021) 36:385–99. doi: 10.1002/jbmr.4193
118. Yu S, Li D, Zhang N, Ni S, Sun M, Wang L, et al. Drug discovery of sclerostin inhibitors. Acta Pharm Sin B. (2022) 12:2150–70. doi: 10.1016/j.apsb.2022.01.012
Keywords: sclerostin, type 1 diabetes, type 2 diabetes, osteoporosis, pathogenesis, target
Citation: Li Y, Luo Y, Huang D and Peng L (2024) Sclerostin as a new target of diabetes-induced osteoporosis. Front. Endocrinol. 15:1491066. doi: 10.3389/fendo.2024.1491066
Received: 04 September 2024; Accepted: 25 November 2024;
Published: 10 December 2024.
Edited by:
Antonino Catalano, University of Messina, ItalyReviewed by:
Ram Naresh Yadav, Henry Ford Hospital, United StatesCopyright © 2024 Li, Luo, Huang and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanhua Li, bGl5YW5odWE5NDEwMDNAMTYzLmNvbQ==