 Meghan Blair Turner
Meghan Blair Turner Carolina Dalmasso
Carolina Dalmasso Analia S. Loria
Analia S. Loria- Department of Pharmacology and Nutritional Sciences, University of Kentucky, Lexington, KY, United States
Adverse Childhood Experiences (ACEs) refer to early life stress events, including abuse, neglect, and other psychosocial childhood traumas that can have long-lasting effects on a wide range of physiological functions. ACEs provoke sex-specific effects, whereas women have been shown to display a strong positive correlation with obesity and cardiometabolic disease. Notably, rodent models of chronic behavioral stress during postnatal life recapitulate several effects of ACEs in a sex-specific fashion. In this review, we will discuss the potential mechanisms uncovered by models of early life stress that may explain the greater susceptibility of females to obesity and metabolic risk compared with their male counterparts. We highlight the early life stress-induced neuroendocrine shaping of the adrenal-adipose tissue axis as a primary event conferring sex-dependent heightened sensitivity to obesity.
1 Introduction
Adverse childhood experiences (ACEs) are traumatic events that happen before the age of 17, such as abuse, neglect, parental separation, and household instability. Cumulative number of ACEs have been associated with an increased risk for premature death (1, 2). ACEs are highly prevalent worldwide, as six out of 10 adults report experiencing at least one adverse event in childhood (3). Based on 2011–2020 Behavioral Risk Factor Surveillance System (BRFSS) data, the CDC estimates that the prevalence of ACEs among U.S. adults is similar. Overall, 63.9% of U.S. adults reported at least one ACE and 17.3% reported exposure to four or more ACEs (4). In 1998, a large health maintenance organization clinic in San Diego published a seminal study on ACEs. This report revealed that increased ACE exposures corresponded to increased rates of severe obesity, ischemic heart disease, and liver disease (1). Specifically, participants who experienced more than three ACEs had a 1.5 to 1.9-fold risk for severe obesity (Body Mass Index, BMI, ≥ 35 kg/m2) in adulthood (1, 5).
Since that publication, numerous epidemiological and population-based studies have replicated the link between ACE exposures and increased BMI, obesogenic behaviors, and cardiometabolic risk factors in adulthood. A recent systematic review found that four or more ACE exposures results in increased risk for obesity, diabetes, heart disease, and risk-taking behaviors (6). Moreover, a study of Finnish adolescents found that multiple ACE exposures confer a 46% increased likelihood of adult obesity (7), while The Growing Up in Ireland cohort found that ACE exposure before age nine was associated with early adolescent obesity (8). Also, ACE exposure was found to be associated with type 2 diabetes, whereas neglect has the strongest influence increasing the risk for metabolic disease (9). The seminal ACE study was based on a retrospective survey comprised of 10 binary questions regarding childhood physical and emotional abuse, however, validated variations are widely used in clinical and epidemiological settings (1, 10). Notably, a cumulative effect of ACE exposure was demonstrated, as each positive response on the ACE questionnaire corresponded to a one-point increase in BMI (10).
Other factors such as sex, race, and type of ACE exposure also contribute to the stratification of the effects of ACEs on BMI and metabolic dysregulation. The Trondelag Health Study in Norway found that parental divorce and negative childhood memories were associated with higher women’s pre-pregnancy BMI (11). In a longitudinal U.S. cohort of women, two or more ACE exposures corresponded to increased adolescent and adult BMI, where sexual and physical abuse specifically affected Black women (12). The UK-based Millennium cohort found that BMI in females was increased by a single ACE exposure, and was most sensitive to parental separation and physical punishment, while in males, elevated BMI emerged after the occurrence of three or more ACEs (13). The number of ACEs needed to increase BMI in both Black and white women was reduced compared with white men (14). In contrast, Black men showed a negative association between ACEs and BMI; however, increased waist circumference suggested greater central adiposity (14).
Animal models have recapitulated the sex-specific effects of early life stress on metabolic function and body composition, providing a translational tool to identify therapeutic targets and effective approaches to treat disease in this vulnerable population. Modeling ACEs in rodents is possible based on the premise that the mammalian maternal bond is crucial for modulating the stress response in early life (15). Critical windows of stress susceptibility have been observed across mammalian species, from rodents to non-human primates, as well as in human infants, which are protected by maternal sensory input (16). In rodents, a stress hypo-responsive period (SHRP) that occurs during the first two postnatal weeks is characterized by lower corticosterone levels and corticosterone unresponsiveness (16, 17). This SHRP is critical for the optimal development and maturation of the brain and other physiological systems. Thus, high levels of stress hormones during this critical period result in the long-term dysregulation of the stress response (18–21).
Investigations of the effects of early life stress have found significant impairment of the adrenal-adipose tissue axis function in women and pre-clinical models using female rodents. Notably, aldosterone has emerged as an adrenal hormone associated with a sex-specific cardiometabolic risk, where females with early life stress experience increased susceptibility (22–24). In women, plasma aldosterone correlates positively with visceral adipose tissue and negatively with insulin sensitivity, and these associations are independent of plasma renin activity (25, 26). It is known that aldosterone synthesis is stimulated by adrenocorticotrophic hormone (ACTH), arginine-vasopressin (AVP), angiotensin II (Ang II), leptin, and potassium (27). Subsequently, high circulating aldosterone binds to mineralocorticoid receptors expressed in adipocytes, immune cells, endothelial cells, and brain areas controlling the metabolic function. Along these lines, female mice exposed to early life stress have shown increased plasma and adrenal-derived aldosterone, as well as a mineralocorticoid-dependent expansion of adipose tissue and metabolic dysregulation (24, 28, 29). Early life stress exposure does not increase basal adrenal glucocorticoids in adult women or rodents (24, 30); however, corticosterone response to stress is exacerbated (31). Adrenal androgens are not well-studied in this context due to limited translational potential, as rodents do not display a defined period of adrenarche and secrete adrenal androgens in minuscule quantities (32). Additionally, adrenal androgens are difficult to study in humans, as gonadal androgens are systemically prominent (32).
In his New York Times best-selling book, The Body Keeps the Score, Dr. Bessel van der Kolk discusses how traumatic experiences commonly induce neurobiological, immune, and cardiovascular changes in the body. However, we put forth that in women, the adipose tissue is truly the scorecard marked upon by early life stress, as the dysregulated hypothalamic-pituitary-adrenal (HPA) axis predisposes to obesity and metabolic syndromes.
2 Early life stress models
Animal models that recapitulate the long-term effects of early life stress aim to disrupt or limit access to maternal care during early postnatal life. In rodents, this is commonly achieved by separating the mother from her litter of pups during the SHRP, which is known as maternal separation (MS) (33, 34). A more aggressive model of neglect in early life is to combine MS and early weaning (MSEW) (28, 35). This model is widely used in mice because murine offspring exhibit greater stress resilience than rats. Maternal care disruptions induce a stress response during a time when the HPA axis is not fully developed, which programs the sensitivity threshold for subsequent stressors. As such, maladapted HPA axis sensitivity is a hallmark of early life stress models that leads to neuroendocrine, behavioral, and cardiometabolic outcomes that mimic those observed in children and adults exposed to ACEs (36).
Different MS paradigms exist that involve removing the dam from the whole litter in the home cage, for durations ranging from 10 minutes to 3- 8 hours daily (34). Across MS models, separations are typically performed daily for 3-14 days, either consecutively or intermittently. Litters from an undisturbed cage often serve as a control group, while in other studies, half of the litter identified by tail snip or ink marks is subjected to separations and the other half remains with the dam, serving as controls. MS is typically sufficient to induce long-term HPA disruptions in rats; however, MSEW is a more effective approach to induce long-lasting neuroendocrine dysfunctions in mice (35). In MSEW paradigms, pups are not only separated from their mothers, typically for longer periods but are also weaned prematurely at postnatal day (PND)17 compared to controls weaned at PND21 (28, 35). Another paradigm of early life stress involves maternal deprivation (MD), a form of MS where offspring are separated individually for up to 24 hours, which induces higher levels of stress on the pups (20, 37). When pups are subjected to MD for shorter periods, the protocol can also be referred to as early handling (15 minutes) or early deprivation (four hours) (38).
Maternal care disruption induced by MS or MSEW promotes fragmented nursing, anxiety, depression, aggression, and changing licking/grooming frequency that have been shown to sensitize the offspring’s response to subsequent stressors in the short and long term (39–42). Another model of early life stress uses this approach by providing limited nesting and bedding (LNB) materials for the dam. The LBN model leads to disorganized maternal care in addition to poor nest construction which disrupts thermoregulation, an environmental stressor affecting the dam and pups (41, 43).
3 Early life stress and the HPA axis
Activation of the HPA axis is triggered by stressful events that induce the release of corticotropin-releasing hormone (CRH) and AVP from magnocellular neurons in the paraventricular nucleus of the hypothalamus (PVN) to the pituitary gland (Figure 1). CRH stimulates ACTH synthesis and release into the bloodstream in the anterior pituitary gland, while AVP is released directly into the portal veins of the posterior pituitary (Figure 1). Upon stimulation, both hormones are released into the systemic circulation and bind to specific receptors in the adrenal glands to promote rapid corticosteroid synthesis (27). ACTH binding to melanocortin type 2 receptors (Mc2R) results in calcium-mediated increase in transcription and activity of rate-limiting steroidogenic acute regulatory protein (StAR) (44, 45). AVP binds to AVP receptor type 1 (V1R), inducing a similar intracellular signaling pathway (46, 47). StAR is responsible for supplying the steroidogenic substrate, cholesterol, to the mitochondrial membrane for downstream conversion to steroid hormones based on cell-specific enzymes (44). In cells of the zonas fasciculata and glomerulosa of the adrenal gland, enzymes, including CYP11B1, are present for the synthesis of corticosteroids, corticosterone, and cortisol, specifically in humans (48). However, activation of CYP11B2 transcription in the zona glomerulosa, further converts corticosterone into the primary mineralocorticoid, aldosterone (49). This pattern of enzyme expression defines the zone-specificity of glucocorticoid and mineralocorticoid production (50).
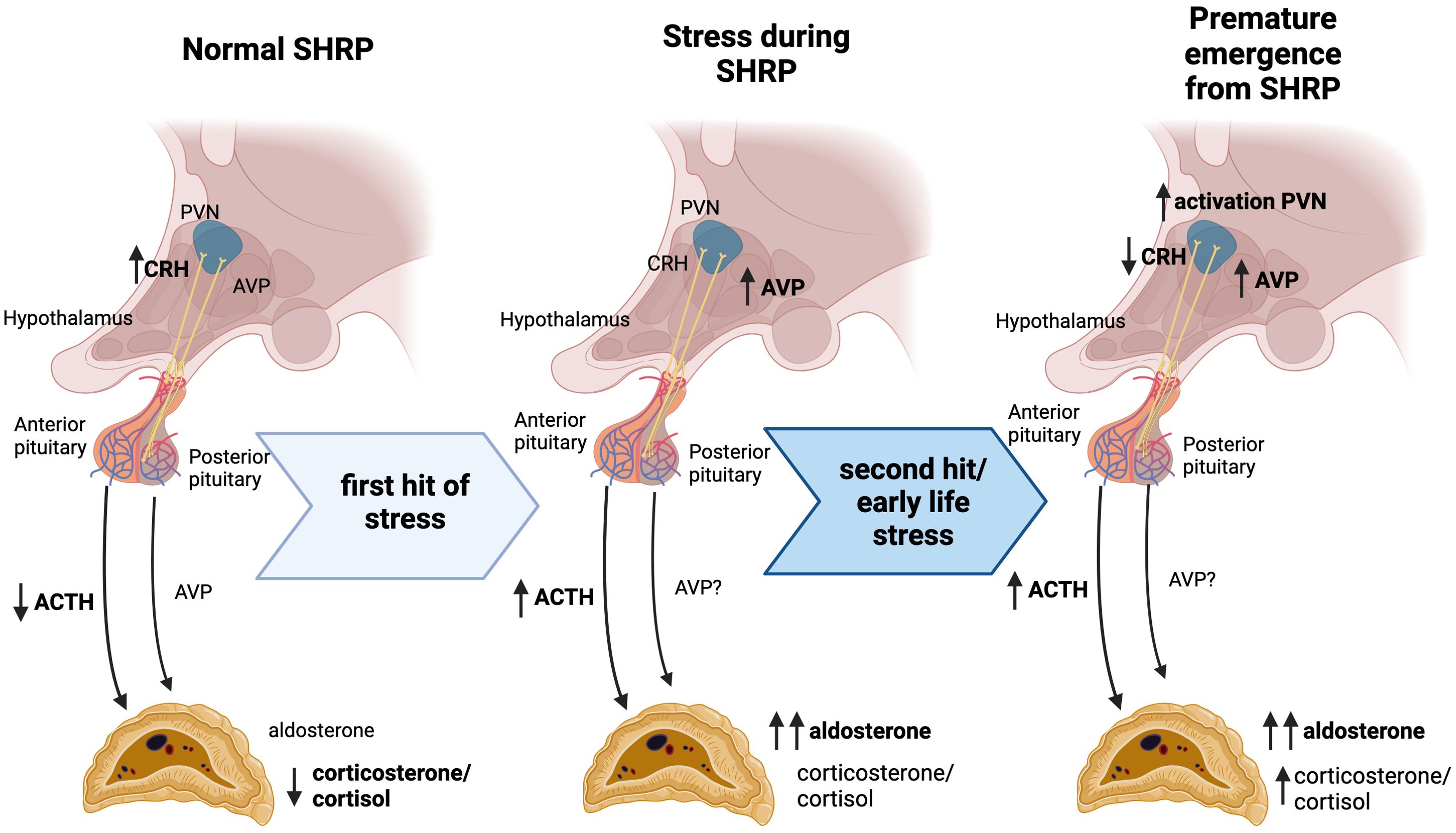
Figure 1. Progressive stress exposure during the stress hyporesponsive period alters HPA function. In rodent models, SHRP is characterized by decreased ACTH and corticosterone production despite increased CRH expression in the hypothalamus. A first hit of stress during this period elicits a modest elevation in AVP and ACTH, coupled with a significant increase in aldosterone. After early life stress exposure, a subsequent hit of stress leads to increased activation in the PVN accompanied by increased AVP expression and decreased CRH expression. This “second hit” elicits an increase in ACTH, followed by elevated corticosterone and significant increases in aldosterone. Priming of the HPA axis during this period of altered stress response mechanisms may underlie the predisposition for a hyper-aldosteronogeneic response to stressors such as high fat diet in adulthood.
The SHRP is marked by a relatively quiescent stress response compared to immediately after birth or following the SHRP (16, 17), and is well documented to occur between PND4-14 in rats and PND1-12 in mice (16, 21, 51, 52). Expression of CRH in the PVN is exceptionally high during this time, only downstream ACTH and corticosterone release in response to mild stress are dampened (20, 21) (Figure 1). Sex differences in HPA function are in part due to differences in the circulating gonadal steroid hormone. While testosterone seems to inhibit HPA function, estrogens show stimulatory effects. One mechanism by which androgens and estrogens modulate stress responses is through the binding to their cognate receptors in the central nervous system showing sex and site-specific distribution. In the case of androgens, data suggest that the control of the hypothalamic paraventricular nucleus is mediated trans-synaptically. For estrogen, modulation of the HPA axis may be due to changes in glucocorticoid receptor-mediated negative feedback mechanisms (53). Glucocorticoid-receptor-mediated negative feedback suppresses ACTH response to mild stressors. However, if the stressor is sufficient to reach a threshold the neonatal pituitary is able to producing a robust ACTH response (16).
Exposure to a more severe stressors during early postnatal life can cause premature emergence from the SHRP (Figure 1). For instance, although early life stress induces an attenuated CRH expression in the PVN, plasma corticosterone remains elevated (19, 20, 54). Artificially-reared PND12 rat pups remained hypo-responsive as long as rat milk substitute was supplemented via gastrostomy (55). Dietary glucose supplementation or blocking ghrelin signaling were also able to attenuate MS effects (56). Interestingly, early life stress did not alter body weight or growth at weaning (24, 28, 55). Taken together, this data indicates that hunger-induced stress could play a critical role in the metabolic programing of the obesogenic response regardless significant effects in growth and weight gain.
Most studies of early life stress focus on corticosterone as the major output of the HPA-mediated stress-response; however, pituitary hormones are also potent stimulators of aldosterone. Rat pups subjected to lipopolysaccharide and hypoglycemic challenges during the SHRP show increased circulating aldosterone compared to non-challenged pups or adults. This increase in aldosterone levels cannot be attributed to elevations in ACTH alone (57). Additionally, primary adrenal cultures from MD rat pups produce more aldosterone than cultures from non-separated pups; however, corticosterone production is not different between groups (58). Early life stress has been shown to methylate the AVP enhancer, leading to increased expression in adult rodents (59, 60), and AVP is clinically elevated in adults with early life stress exposures (61). Taken together, these data suggest that AVP may play a role in mediating increased aldosterone production following early life stress exposure.
Activation of the PVN is similar between maternally deprived and non-deprived pups after a “second hit” of restraint stress; however, maternally deprived pups show reduced CRH and increased AVP expression in this brain area (20). This suggests that deprivation shifts the hypothalamic stress-response from primarily CRH-mediated to AVP-mediated. Neither AVP nor CRH, however, entirely account for steroidogenic stimulation in early postnatal rats (62, 63). Taken together, these studies suggest that while the anterior pituitary and zona fasciculata stress responses are dampened during the SHRP, the posterior pituitary and zona glomerulosa are preferentially activated during this period. These insights open up the discussion concerning whether the early neonatal period may not be hypo-responsive at all, rather the stress response has been historically measured in the context of a mature neuroendocrine system. Further, priming of these systems in early life leads to long-term differential activation that can be pathogenic during adult life.
4 Sex differences in the HPA axis
Sexually dimorphic responses are documented in a wide variety of stressors. Female rodents generally display increased baseline HPA axis activity, and exaggerated HPA responses to stress (64). In a resting state, females have increased CRH mRNA expression in the PVN (65, 66) and higher circulating corticosterone compared to male counterparts (66–68) (Figure 2). Although stress increases the CRH expression in the PVN in both males and females, the magnitude and duration of the response are greater in females (69, 70). Female mice are also more sensitized to CRH-induced ACTH production (71, 72), and show elevated AVP expression in the stress-responsive medial parvocellular dorsal division of the PVN (66, 70) (Figure 2). Despite that females display increases in CRH and AVP synthesis in response to stressful stimuli (73), differences in neuronal activation are rarely reported (66, 74, 75).
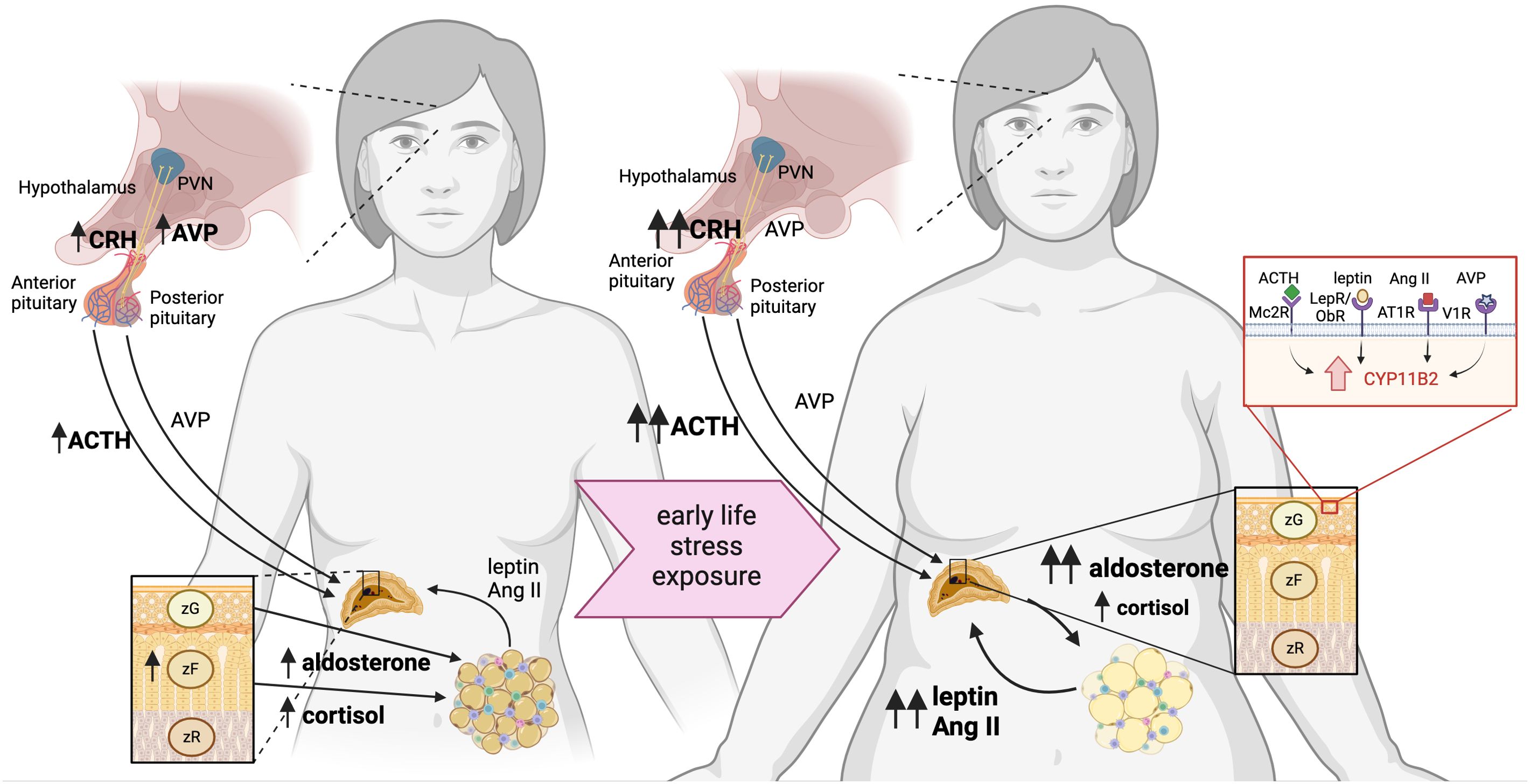
Figure 2. Neuroendocrine mechanism linking early life stress with increased susceptibility of women to obesity. Women have heightened HPA axis activation and responsiveness, which is exacerbated by early life stress, predisposing to obesity. Females have increased activation of the hypothalamus, delineated by increased CRH and AVP expression, compared to males. Both women and female rodents have elevated ACTH in plasma, as well as aldosterone and cortisol/corticosterone. Increased cortisol/corticosterone is attributed to an expanded zona fasciculata in the adrenal glands, which are larger in females than males. Exposure to early life stress leads to sensitization of the HPA axis, which increases CRH in the hypothalamus of rodent models and increased plasma ACTH and AVP in women and female rodents. These secretagogues elicit an elevated basal corticosterone/cortisol, but moreso, elevated aldosterone from the adrenal glands via binding to MC2R and V1R. Adrenal aldosterone acts on the mineralocorticoid receptor in adipose tissue to exacerbate production of adipokines leptin and AngII production, which stimulate further aldosterone production via LepR and AT1R, respectively.
Both the total size and size relative to body mass of the adrenal gland are increased in females, which is attributed to hyperplasia of the corticosterone-producing zona fasciculata (76, 77). Furthermore, the female adrenal also produces more corticosterone in response to in vivo ACTH stimulation compared to adrenal-derived production in males (67, 78). Both central and peripheral mechanisms have been shown to sensitize the stress response in female rodents and humans. Additionally, female rodents have an increased capacity to synthesize adrenal steroid hormones peripherally, even at resting conditions.
Gonadectomy studies have elucidated striking and consistent effects of sex hormones on the developmental organization and function of the HPA axis. Testosterone is overall protective against HPA axis activation, whereas orchidectomized rodents display increased basal CRH expression in the PVN (79–81) and plasma corticosterone (70, 82, 83) compared to intact males. Stress-induced CRH expression (70) and plasma ACTH and corticosterone levels (83–87) are also elevated after testis removal. Conversely, testosterone administration reverses the effects of gonadectomy, dampening HPA activation (83, 85–87). Interestingly, orchidectomy does not appear to alter the sensitivity of the pituitary to CRH or the adrenal to ACTH (85, 87). On the other hand, female sex hormones, primarily estrogen and progesterone, tend to exacerbate the activation of the HPA axis. Ovariectomy leads to decreased adrenal weight (88, 89) and reduced HPA output (71, 87, 90, 91) while estrogen or estrogen combined with progesterone increases ACTH-stimulated corticosterone responses in male and ovariectomized female rodents (92–94). Similarly, estradiol treatment in men leads to increased ACTH and cortisol in response to psychological stress (95). Gonadal hormones have broad and well-studied effects on all aspects of the HPA axis, which extend beyond the scope of this review but are extensively covered elsewhere (64).
5 Early life stress mediates sex-specific cardiometabolic outcomes in adulthood
The average American diet is high in both calories and fats, which is associated with the ever-rising obesity epidemic (96). In rodents and humans exposed to early life stress, hypercaloric diet acts as a secondary metabolic stressor. Dalmasso et al. discovered that male rodents exposed to MSEW and fed a HFD, display fat-derived sympathoextitatory contributing to the exacerbated neurogenic hypertension, despite showing similar levels of adiposity compared to male controls (97, 98). The mechanism underlying the increases in blood pressure in this model involves the activation of the PVN in response to the stimulation of gonadal fat-derived afferent signals, a mechanism referred as adipose-afferent reflex. Unlike males, female MSEW mice fed a HFD develop an exacerbated cardiometabolic phenotype that is independent of the overactivation of the sympathetic system (24). Female MSEW mice weaned to a HFD display increased fat mass in adulthood compared to normally reared and diet-matched controls (29). Further, this sex-specific increases in adiposity were due to greater adipocyte size and triglyceride deposition compared to controls (24). Obese MSEW female mice also showed reduced glucose tolerance, and increased expression of adipogenic genes in pre-adipocytes including the glucocorticoid receptor (Nr3c1), peroxisome proliferator-activated receptor alpha (Pparα), and fatty acid binding protein 3 (Fabp3), and dramatic downregulation of aquaporin 3 (Aqp3), a glycerol efflux channel, and increased hepatic lipogenic gene expression (24, 28, 29, 99).
As reported in female mice, female rats subjected to MS showed increased adiposity in response to a chronic HFD (99). This phenotype was reverted by metyrapone administration prior to the daily MS exposure, inhibiting both corticosterone and aldosterone synthase enzymes. This data supports the notion that blunting adrenal corticosteroid production in response to early life stress prevents the long-lasting effects on the metabolic adaptations to HFD. Additional studies are needed to elucidate the role of the adrenal-adipose tissue axis as a mechanism by which more severe cardiometabolic derangements are observed in female rodents exposed to early life stress.
6 Early life stress primes the adrenal-adipose tissue axis in female rodents
Adipose tissue, composed of adipocytes, preadipocytes, and various resident immune cell populations, has come to light in recent years as a dynamic endocrine organ (100). Adrenal hormones, particularly aldosterone and corticosterone, have direct effects on adipose tissue homeostasis via the mineralocorticoid and glucocorticoid receptors, respectively. In turn, adipose tissue-secreted adipokines such as leptin and angiotensin II stimulate adrenal steroidogenesis (101–105) (Figure 2). Exacerbated adipose tissue expansion in obese MSEW female mice is associated with increased visceral adipose tissue-derived adipokines, showing that the adrenal-adipose tissue axis is sensitized in response to early life stress.
6.1 Aldosterone
Plasma aldosterone concentration and its robust association with obesity and metabolic syndromes has been well-reviewed elsewhere (106). This is particularly true in women, as visceral fat mass positively correlates with aldosterone and negatively correlates with insulin sensitivity in women, but not in men (107). More recently, a Chinese cohort demonstrated that increased neck circumference, an indicator of visceral body fat, is associated with elevated aldosterone in women (26). Another study showed that adults who reported childhood trauma had increased aldosterone, while trauma experienced for the first time in adulthood did not elicit a significant response (23). Childhood trauma exposure was associated with increased plasma aldosterone, particularly in individuals who reported physical or emotional maltreatment (22). While additional clinical and population-based studies are needed to strengthen the relationship between aldosterone and ACEs, findings from human studies correlate with rodent models of early life stress, highlighting their value in elucidating potentially translational therapeutics.
Female MSEW mice fed a HFD show exacerbated obesity and aldosterone production compared with diet-matched control mice, while chronic treatment in adulthood with spironolactone, a mineralocorticoid receptor antagonist, resulted in loss of fat mass coupled with increased glycerol efflux and decreased adipocyte size in MSEW females, but not in HFD controls (24). Tissue explants showed that in obese MSEW female mice, the source of increased aldosterone is predominantly the adrenal gland, although adipose tissue has a local renin-angiotensin-aldosterone system (RAAS) capable of synthesizing the hormone (24). Thus, adrenal-derived aldosterone may be mediating obesogenic effects through two candidate targets within adipose tissue, as illustrated in Figure 2. First, the MR expressed in adipocytes, the principal cell type of adipose tissue, could be driving transcriptional alterations in the cell leading to hypertrophy as a consequence of triglyceride accumulation. Second, the MR expressed in resident adipose-tissue dendritic cells can alter the pro-inflammatory milieu of the tissue promoting a similar phenotype.
6.1.1 Adipocyte MR
Over-expression and over-activation of the adipocyte MR is directly related to obesity and metabolic syndrome-like phenotype in pre-clinical models. In an adipocyte-specific inducible MR overexpression model, Urbanet and colleagues found that adipocyte-MR upregulation correlated with progressive increases in body weight, visceral adipose tissue mass, and adipocyte size over 12 weeks without differences in food or calorie intake (108). Additionally, these mice showed markers of metabolic dysregulation, such as insulin resistance, hypertriglyceridemia, and hypercholesterolemia. Moreover, the metabolic dysregulation was further exacerbated by HFD (108). In primary mouse adipocyte cultures, MR stimulation with aldosterone resulted in an increased pro-inflammatory and obesogenic adipokine profile including elevated leptin, IL-6, PAI-1, and chemerin (109). Notably, MR-knockout adipocytes completely failed to accumulate lipids and had a 90% reduction in fatty acid binding protein aP2 (FABP4) expression, a carrier protein for fatty acids that promotes lipid storage. Similarly, Urbanet et al. found that aldosterone acts through lipocalin-like prostaglandin D2 synthase (PTGDS) downstream of the adipocyte MR to increase leptin, FABP4, and PPARy expression. While later studies showed that knocking out MR in adipocytes did not significantly change the metabolic phenotype of diet-induced obese male mice, further studies are needed to elucidate its role in the MSEW cardiometabolic phenotype, particularly in females. Notably, in female mice, adipocyte-derived leptin is a positive regulator of aldosterone synthesis in the zona glomerulosa of the adrenal glands (104) independent of obesity.
6.1.2 Dendritic cell MR
Dendritic cells are a population of antigen-presenting immune cells that influence the phenotypic differentiation of T-cells (110). Aldosterone activates MR in dendritic cells and induces secretion of IL-6, and TGF-b, which promotes the polarization of naive T-cells to T-helper 17 (Th17) cells (111). Dendritic cells which are resident in adipose tissue are directly correlated to increased Th17 responses, obesity, and insulin resistance in both mice and clinical populations (112–114). In obesity, dendritic cells are abundant in visceral adipose tissue, and are particularly dense in fat-associated lymphoid clusters of murine gonadal adipose tissue (115). Blockade of dendritic cell migration into visceral adipose tissue prevents diet-induced weight gain and metabolic disturbances (115). Mice with loss of function specifically in fat-resident dendritic cells were similarly protected (116). In human adipose tissue, CD1c, a dendritic cell marker, has been positively associated with insulin resistance (113), and murine CD11c+CD64- dendritic cells are specifically cited as independent contributors to obesity and insulin resistance in diet-induced obesity models (112).
6.2 Leptin production during obesity
Leptin is an anorexigenic peptide hormone primarily produced by the adipose tissue (117) and is elevated in obesity (118). Leptin dampens ACTH-mediated steroidogenesis in the adrenal cortex (58), but is also an independent stimulator of aldosterone (104). Leptin deficient mice (ob/ob) have disrupted hypothalamic networks between the arcuate nucleus and the PVN that are only restored by leptin administration during the SHRP (119). MD in rat pups at PND10 abolishes the plasma leptin surge, while corticosterone, aldosterone, and ACTH are increased (58). Three- or twelve-hour MD in PND8 mouse pups was not sufficient to induce changes in leptin or corticosterone levels compared to non-handled mice (120, 121). Interestingly, leptin stimulation in primary adrenal cultures from PND10 MD rat pups produced more aldosterone than non-separated cultures (58).
Female rat offspring exposed to both western diet and LBN showed higher increases in plasma leptin than males after weaning (121). Female rats exposed to MS and high fat, high sucrose diet had increased leptin mRNA in periovarian adipose tissue, in addition to increased PPARy mRNA, serum insulin, and HOMA index (122). Exogenous leptin administration to adult MS rats does not suppress food intake or weight gain as it does in non-handled rats (123). Further, leptin was elevated plasma of young adults who reported either childhood maltreatment or parental loss compared to those who did not report early life stress exposure (124). Mechanistically, it has been shown that obese MSEW female mice showed increased gonadal white adipose tissue-derived leptin that was not observed in males or dietary controls. This increased leptin production was associated with Hypomethylated CpG sites in the leptin promoter region (29).
6.3 Angiotensin II
Angiotensin II (AngII) is a known for its actions as a vasoactive peptide regulating vascular tone and water and electrolyte homeostasis, as well as for being a pathogenic mediator of obesity-related comorbidities. Angiotensinogen is the sole precursor for AngII, abundantly produced by the liver, serving as substrate for renin to produce angiotensin I. Cleavage of Angiotensin I by angiotensin-converting enzyme 1 (ACE1). AngII is a potent stimulator of aldosterone from the adrenal glands (105), which mediates the sodium and water reabsorption in the distal bneprhon and renal collecting duct. As such, the expression of all the renin-angiotensin-aldosterone system (RAAS) componetns have been shown in several tissues, including brain, heart, vasculature and kidney. Yet, AngII can be generated in the adipose tissue by both local ACE1 cleavage, and through non-canonical generation by cathepsin D and cathepsin G (125, 126). AngII acts locally on adipocytes via the AT1 and AT2 receptors, directly regulating adipose tissue homeostasis (101). Anti-adipogenic effects have been observed, but also lipogenic effects including triglyceride accumulation and activation of fatty acid synthase and glycerol-3-phosphate dehydrogenase (127).
Female mice fed a HFD develop exacerbated adiposity that is associated with increased circulating AngII, most likely due to increased fat-derived AngII production in visceral adipose tissue (98). However, the pressor response to acute doses of AngII was similar between control and MSEW obese female mice before and after treatment with the ACE1 inhibitor enalapril, indicating that the sensitivity to this peptide is similar between MSEW and control mice. Conversely, male MSEW mice fed a HFD display similar adiposity and circulating AngII levels when compared to obese controls (98). Furthermore, blocking AngII production with enalapril lowered blood pressure in obese MSEW male mice without decreasing the sympathetic activation (98). Taken together, this data suggests that, in the settings of obesity, MSEW activates the RAAS in a sex-specific manner. Thus, AngII could play an important role in obesity expansion experienced by female mice exposed to early life stress.
7 Discussion
Epidemiological studies on ACE exposures that are not stratified by sex still typically find an association with BMI. In women, this association is often found to show a “dose-dependent” effect, where each additional reported ACE exposure contributes to further increases in BMI (13, 14). Male mice exposed to early life stress do not show exacerbated weight-gain as their female counterparts, likely because current behavioral paradigms do not reach a persistently high enough level of stress to recapitulate the cumulative effect of ACEs needed to promote increases in adiposity. Additionally, male offspring may require exposure to different types of stress during the SHRP to elicit a physiological response that mimics what is observed in humans.
The balance between adrenal steroid production and adipokine production is likely a key mediator of obesity in women who have experienced ACEs. Aldosterone from the adrenal gland drives the expression of leptin and other key adipogenic mediators in the adipose tissue, as well as promoting a broad pro-inflammatory milieu. Similarly, leptin is an important neuroendocrine mediator that feeds back on the hypothalamus, as well as the adrenal gland. Leptin plays an inhibitory role on the PVN of the hypothalamus, suppressing HPA axis activation (128). Studies have found that leptin increases aldosterone production from the adrenal gland (104), but also that it dampens the effects of ACTH on production of aldosterone and corticosterone (58, 129). It is likely that both increased leptin and increased aldosterone perpetuate one another in conditions of metabolic stress.
There are emerging areas of study that may shed light in the mechanisms underlying sex-specific shaping of the HPA sensitivity in response to early life stress. Steenblock et al. have characterized a distinct population of stress-responsive adrenocortical precursor cells marked by positivity for nestin (Nes+) (130). This population resides in quiescence under the adrenal capsule, and migrates centripetally in response to stress, differentiating into steroidogenic cells in their fated zones (130). Isolated in vitro cultures of these cells show a propensity to become aldosterone-producing when stimulated. Though sex differences in Nes+ cell activation have not been explored, aldosterone-producing cells may become enriched long-term in the adrenal glands of female rodents exposed to early life stress due to their innate stress susceptibility.
GLP-1 is a gut-derived peptide that activates the HPA axis, whereas infusion of GLP-1 in healthy adults reduced aldosterone in a small clinical cohort (131). GLP-1 receptor agonists such as Ozempic (semaglutide) and Trulicity (dulaglutide) have become massively popular drugs for the management of weight, diabetes, and cardiovascular health. To the authors’ best knowledge, there have been no trials evaluating the differential effects of GLP-1 receptor agonists in women exposed to ACEs. A small clinical study concluded that dulaglutide did not activate the HPA axis, however measurements were limited to cortisol measurements after dexamethasone suppression and ACTH stimulation. It is possible that an already-approved GLP-1 receptor agonist could target mechanisms linked to cardiometabolic dysregulation in women exposed to ACEs, opening new venus of precision medicine that may potentially aid this vulnerable population.
While various paradigms of early life stress in rodents and other animal models have been extensively studied, few research groups have emphasized obesity and metabolic disruptions as outcomes in any of these models. Considering the high prevalence of ACEs in the population and the robustness of these associations, further investigation of mechanistic approaches to cardiometabolic therapeutics could prove highly valuable to public health. This review highlights and discusses how the adipose tissue keeps the score of early life stress in the female body. Because prevention is often not a feasible strategy for mitigating the risks of early life stress exposures, continued animal research offers hope for targeted therapeutics that could help to even that score.
Author contributions
MT: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. CD: Validation, Visualization, Writing – review & editing. AL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Institutes of Health grant R01 HL111354 to ASL.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Felitti VJ, Anda RF, Nordenberg D, Williamson DF, Spitz AM, Edwards V, et al. Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. Am J Prev Med. (1998) 14:245–58. doi: 10.1016/S0749-3797(98)00017-8
2. Yu J, Patel RA, Haynie DL, Vidal-Ribas P, Govender T, Sundaram R, et al. Adverse childhood experiences and premature mortality through mid-adulthood: A five-decade prospective study. Lancet Regional Health - Americas. (2022) 15:100349. doi: 10.1016/j.lana.2022.100349
3. Madigan S, Deneault AA, Racine N, Park J, Thiemann R, Zhu J, et al. Adverse childhood experiences: a meta-analysis of prevalence and moderators among half a million adults in 206 studies. World Psychiatry. (2023) 22:463–71. doi: 10.1002/wps.21122
4. Swedo EA, Aslam MV, Dahlberg LL, Niolon PH, Guinn AS, Simon TR, et al. Prevalence of adverse childhood experiences among U.S. Adults — Behavioral risk factor surveillance system, 2011–2020. MMWR Morbidity Mortality Weekly Rep (MMWR). (2023) 72:707–15. doi: 10.15585/mmwr.mm7226a2
5. Anda RF, Felitti VJ, Bremner JD, Walker JD, Whitfield C, Perry BD, et al. The enduring effects of abuse and related adverse experiences in childhood. A convergence of evidence from neurobiology and epidemiology. Eur Arch Psychiatry Clin Neurosci. (2006) 256:174–86. doi: 10.1007/s00406-005-0624-4
6. Hughes K, Bellis MA, Hardcastle KA, Sethi D, Butchart A, Mikton C, et al. The effect of multiple adverse childhood experiences on health: a systematic review and meta-analysis. Lancet Public Health. (2017) 2:e356–66. doi: 10.1016/S2468-2667(17)30118-4
7. Isohookana R, Marttunen M, Hakko H, Riipinen P, Riala K. The impact of adverse childhood experiences on obesity and unhealthy weight control behaviors among adolescents. Compr Psychiatry. (2016) 71:17–24. doi: 10.1016/j.comppsych.2016.08.002
8. Gardner R, Feely A, Layte R, Williams J, McGavock J. Adverse childhood experiences are associated with an increased risk of obesity in early adolescence: a population-based prospective cohort study. Pediatr Res. (2019) 86:522–8. doi: 10.1038/s41390-019-0414-8
9. Huang H, Yan P, Shan Z, Chen S, Li M, Luo C, et al. Adverse childhood experiences and risk of type 2 diabetes: A systematic review and meta-analysis. Metabolism. (2015) 64:1408–18. doi: 10.1016/j.metabol.2015.08.019
10. Mahmood S, Li Y, Hynes M. Adverse childhood experiences and obesity: A one-to-one correlation? Clin Child Psychol Psychiatry. (2023) 28:785–94. doi: 10.1177/13591045221119001
11. Sandsæter HL, Eik-Nes TT, Getz LO, Magnussen EB, Bjerkeset O, Rich-Edwards JW, et al. Adverse childhood experiences and pre-pregnancy body mass index in the HUNT study: A population-based cohort study. PloS One. (2023) 18:e0285160. doi: 10.1371/journal.pone.0285160
12. Chiu DT, Brown EM, Tomiyama AJ, Brownell KE, Abrams B, Mujahid MS, et al. Adverse childhood experiences and BMI: lifecourse associations in a black-white U.S. Women cohort. Am J Prev Med. (2024) 66:73–82. doi: 10.1016/j.amepre.2023.09.004
13. Deng K, Lacey RE. Adverse childhood experiences, child poverty, and adiposity trajectories from childhood to adolescence: evidence from the Millennium Cohort Study. Int J Obes (Lond). (2022) 46:1792–800. doi: 10.1038/s41366-022-01185-1
14. Leachman JR, Heier K, Lei F, Ahmed N, Dalmasso C, Duncan MS, et al. Sex and race define the effects of adverse childhood experiences on self-reported BMI and metabolic health biomarkers. Biol Sex Dif. (2022) 13(1). doi: 10.1186/s13293-022-00439-x
15. Driscoll CA, Barr CS. Studying longitudinal trajectories in animal models of psychiatric illness and their translation to the human condition. Neurosci Res. (2016) 102:67–77. doi: 10.1016/j.neures.2015.08.001
16. Schmidt MV. Chapter 4 - Stress-Hyporesponsive Period. In: Fink G, editor. Stress: Physiology, Biochemistry, and Pathology. San Diego, California: Academic Press (2019). p. 49–56.
17. Schapiro S, Geller E, Eiduson S. Neonatal adrenal cortical response to stress and vasopressin. Proc Soc Exp Biol Med. (1962) 109:937–41. doi: 10.3181/00379727-109-27384
18. Russell AL, Tasker JG, Lucion AB, Fiedler J, Munhoz CD, Wu TYJ, et al. Factors promoting vulnerability to dysregulated stress reactivity and stress-related disease. J Neuroendocrinol. (2018) 30:e12641. doi: 10.1111/jne.2018.30.issue-10
19. Biagini G, Pich EM, Carani C, Marrama P, Agnati LF. Postnatal maternal separation during the stress hyporesponsive period enhances the adrenocortical response to novelty in adult rats by affecting feedback regulation in the CA1 hippocampal field. Int J Dev Neurosci. (1998) 16:187–97. doi: 10.1016/S0736-5748(98)00019-7
20. Dent GW, Okimoto DK, Smith MA, Levine S. Stress-induced alterations in corticotropin-releasing hormone and vasopressin gene expression in the paraventricular nucleus during ontogeny. Neuroendocrinology. (2000) 71:333–42. doi: 10.1159/000054554
21. Schmidt MV, Enthoven L, van der Mark M, Levine S, de Kloet ER, Oitzl MS. The postnatal development of the hypothalamic-pituitary-adrenal axis in the mouse. Int J Dev Neurosci. (2003) 21:125–32. doi: 10.1016/S0736-5748(03)00030-3
22. Terock J, Hannemann A, Klinger-König J, Janowitz D, Grabe HJ, Murck H. The neurobiology of childhood trauma-aldosterone and blood pressure changes in a community sample. World J Biol Psychiatry. (2022) 23:622–30. doi: 10.1080/15622975.2021.2018724
23. Terock J, Hannemann A, Janowitz D, Van der Auwera S, Bahls M, Völzke H, et al. Differential activation of the renin-angiotensin-aldosterone-system in response to childhood and adulthood trauma. Psychoneuroendocrinology. (2019) 107:232–40. doi: 10.1016/j.psyneuen.2019.05.026
24. Leachman JR, Cincinelli C, Ahmed N, Dalmasso C, Xu M, Gatineau E, et al. Early life stress exacerbates obesity in adult female mice via mineralocorticoid receptor-dependent increases in adipocyte triglyceride and glycerol content. Life Sci. (2022) 304:120718. doi: 10.1016/j.lfs.2022.120718
25. Lee G, Kluwe B, Zhao S, Kline D, Nedungadi D, Brock GN, et al. Adiposity, aldosterone and plasma renin activity among African Americans: The Jackson Heart Study. Endocr Metab Sci. (2023) 11. doi: 10.1016/j.endmts.2023.100126
26. Maitituersun A, Heizhati M, Li N, Gan L, Li M, Yao L, et al. Associated lifestyle factors of elevated plasma aldosterone concentration in community population, gender-stratified analysis of a cross-sectional survey. BMC Public Health. (2024) 24:1370. doi: 10.1186/s12889-024-18796-0
28. Murphy MO, Herald JB, Leachman J, Villasante Tezanos A, Cohn DM, Loria AS. A model of neglect during postnatal life heightens obesity-induced hypertension and is linked to a greater metabolic compromise in female mice. Int J Obes. (2018) 42:1354–65. doi: 10.1038/s41366-018-0035-z
29. Leachman JR, Rea MD, Cohn DM, Xu X, Fondufe-Mittendorf YN, Loria AS. Exacerbated obesogenic response in female mice exposed to early life stress is linked to fat depot-specific upregulation of leptin protein expression. Am J Physiol Endocrinol Metab. (2020) 319:E852–62. doi: 10.1152/ajpendo.00243.2020
30. Wong KE, Wade TJ, Moore J, Marcellus A, Molnar DS, O'Leary DD, et al. Examining the relationships between adverse childhood experiences (ACEs), cortisol, and inflammation among young adults. Brain Behav Immun Health. (2022) 25:100516. doi: 10.1016/j.bbih.2022.100516
31. Counts CJ, Ginty AT, Larsen JM, Kampf TD, John-Henderson NA. Childhood trauma and cortisol reactivity: an investigation of the role of task appraisals. Front Psychol. (2022) 13. doi: 10.3389/fpsyg.2022.803339
32. Cumberland AL, Hirst JJ, Badoer E, Wudy SA, Greaves RF, Zacharin M, et al. The enigma of the adrenarche: identifying the early life mechanisms and possible role in postnatal brain development. Int J Mol Sci. (2021) 22(9):4296. doi: 10.3390/ijms22094296
33. Vetulani J. Early maternal separation: a rodent model of depression and a prevailing human condition. Pharmacol Rep. (2013) 65:1451–61. doi: 10.1016/S1734-1140(13)71505-6
34. Nishi M. Effects of early-life stress on the brain and behaviors: implications of early maternal separation in rodents. Int J Mol Sci. (2020) 21:7212. doi: 10.3390/ijms21197212
35. Carlyle BC, Duque A, Kitchen RR, Bordner KA, Coman D, Doolittle E, et al. Maternal separation with early weaning: a rodent model providing novel insights into neglect associated developmental deficits. Dev Psychopathol. (2012) 24:1401–16. doi: 10.1017/S095457941200079X
36. Babicola L, Ventura R, D'Addario SL, Ielpo D, Andolina D, Di Segni M. Long term effects of early life stress on HPA circuit in rodent models. Mol Cell Endocrinol. (2021) 521:111125. doi: 10.1016/j.mce.2020.111125
37. van Oers HJJ, de Kloet ER, Levine S. Early vs. late maternal deprivation differentially alters the endocrine and hypothalamic responses to stress. Dev Brain Res. (1998) 111:245–52. doi: 10.1016/S0165-3806(98)00143-6
38. Pryce CR, Bettschen D, Nanz-Bahr NI, Feldon J. Comparison of the effects of early handling and early deprivation on conditioned stimulus, context, and spatial learning and memory in adult rats. Behav Neurosci. (2003) 117:883–93. doi: 10.1037/0735-7044.117.5.883
39. Pryce CR, Bettschen D, Feldon J. Comparison of the effects of early handling and early deprivation on maternal care in the rat. Dev Psychobiol. (2001) 38:239–51. doi: 10.1002/dev.v38:4
40. Alves RL, Portugal CC, Summavielle T, Barbosa F, Magalhães A. Maternal separation effects on mother rodents’ behaviour: A systematic review. Neurosci Biobehav Rev. (2020) 117:98–109. doi: 10.1016/j.neubiorev.2019.09.008
41. Gallo M, Shleifer DG, Godoy LD, Ofray D, Olaniyan A, Campbell T, et al. Limited bedding and nesting induces maternal behavior resembling both hypervigilance and abuse. Front Behav Neurosci. (2019) 13:167. doi: 10.3389/fnbeh.2019.00167
42. Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ. Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology. (2006) 147:2909–15. doi: 10.1210/en.2005-1119
43. Walker CD, Bath KG, Joels M, Korosi A, Larauche M, Lucassen PJ, et al. Chronic early life stress induced by limited bedding and nesting (LBN) material in rodents: critical considerations of methodology, outcomes and translational potential. Stress. (2017) 20:421–48. doi: 10.1080/10253890.2017.1343296
44. Gallo-Payet N, Martinez A, Lacroix A. Editorial: ACTH action in the adrenal cortex: from molecular biology to pathophysiology. Front Endocrinol (Lausanne). (2017) 8:101. doi: 10.3389/fendo.2017.00101
45. Novoselova TV, King PJ, Guasti L, Metherell LA, Clark AJL, Chan LF. ACTH signalling and adrenal development: lessons from mouse models. Endocrine Connections. (2019) 8:R122–30. doi: 10.1530/EC-19-0190
46. Guillon G, Trueba M, Joubert D, Grazzini E, Chouinard L, Côté M, et al. Vasopressin stimulates steroid secretion in human adrenal glands: comparison with angiotensin-II effect. Endocrinology. (1995) 136:1285–95. doi: 10.1210/endo.136.3.7867583
47. Perraudin V, Delarue C, Lefebvre H, Contesse V, Kuhn JM, Vaudry H. Vasopressin stimulates cortisol secretion from human adrenocortical tissue through activation of V1 receptors. J Clin Endocrinol Metab. (1993) 76:1522–8. doi: 10.1210/jcem.76.6.7684742
48. Thau L, Gandhi J, Sharma S. Physiology, Cortisol. Treasure Island (FL): StatPearls Publishing. (2023).
49. Dutt M, Wehrle CJ, Jialal I. Physiology, Adrenal Gland. Treasure Island (FL): StatPearls Publishing. (2023).
50. Werdermann M, Berger I, Scriba LD, Santambrogio A, Schlinkert P, Brendel H, et al. Insulin and obesity transform hypothalamic-pituitary-adrenal axis stemness and function in a hyperactive state. Mol Metab. (2021) 43:101112. doi: 10.1016/j.molmet.2020.101112
51. Nagasawa M, Shibata Y, Yonezawa A, Takahashi T, Kanai M, Ohtsuka H, et al. Basal cortisol concentrations related to maternal behavior during puppy development predict post-growth resilience in dogs. Horm Behav. (2021) 136:105055. doi: 10.1016/j.yhbeh.2021.105055
52. Gunnar MR, Donzella B. Social regulation of the cortisol levels in early human development. Psychoneuroendocrinology. (2002) 27:199–220. doi: 10.1016/S0306-4530(01)00045-2
53. Handa RJ, Burgess LH, Kerr JE, O'Keefe JA. Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Hormones Behav. (1994) 28:464–76. doi: 10.1006/hbeh.1994.1044
54. Schmidt MV, Levine S, Oitzl MS, van der Mark M, Müller MB, Holsboer F, et al. Glucocorticoid receptor blockade disinhibits pituitary-adrenal activity during the stress hyporesponsive period of the mouse. Endocrinology. (2005) 146:1458–64. doi: 10.1210/en.2004-1042
55. Ward G, Xing HC, Carnide N, Slivchak J, Wainwright P. Adrenocortical response to stress in fasted and unfasted artificially reared 12-day-old rat pups. Dev Psychobiol. (2004) 45:245–50. doi: 10.1002/dev.20029
56. Schmidt MV, Levine S, Alam S, Harbich D, Sterlemann V, Ganea K, et al. Metabolic signals modulate hypothalamic-pituitary-adrenal axis activation during maternal separation of the neonatal mouse. J Neuroendocrinol. (2006) 18:865–74. doi: 10.1111/j.1365-2826.2006.01482.x
57. Varga J, Ferenczi S, Kovacs KJ, Garafova A, Jezova D, Zelena D. Comparison of stress-induced changes in adults and pups: is aldosterone the main adrenocortical stress hormone during the perinatal period in rats? PloS One. (2013) 8:e72313. doi: 10.1371/journal.pone.0072313
58. Salzmann C, Otis M, Long H, Roberge C, Gallo-Payet N, Walker CD. Inhibition of steroidogenic response to adrenocorticotropin by leptin: implications for the adrenal response to maternal separation in neonatal rats. Endocrinology. (2004) 145:1810–22. doi: 10.1210/en.2003-1514
59. Murgatroyd C, Patchev A, Fischer D, Almeida O, Spengler D. Epigenetic programming of sustained AVP expression by early-life stress in mice. Pharmacopsychiatry. (2007) 40:A044. doi: 10.1055/s-2007-991719
60. Murgatroyd C, Spengler D. Polycomb binding precedes early-life stress responsive DNA methylation at the Avp enhancer. PloS One. (2014) 9:e90277. doi: 10.1371/journal.pone.0090277
61. Brydges NM, Hall J, Best C, Rule L, Watkin H, Drake AJ, et al. Childhood stress impairs social function through AVP-dependent mechanisms. Transl Psychiatry. (2019) 9:330. doi: 10.1038/s41398-019-0678-0
62. Grino M, Oliver C. Ontogeny of insulin-induced hypoglycemia stimulation of adrenocorticotropin secretion in the rat: role of catecholamines. Endocrinology. (1992) 131:2763–8. doi: 10.1210/endo.131.6.1359963
63. Varga J, Ferenczi S, Kovács KJ, Csáno Á, Prokopova B, Jezova D, et al. Dissociation of adrenocorticotropin and corticosterone as well as aldosterone secretion during stress of hypoglycemia in vasopressin-deficient rats. Life Sci. (2016) 166:66–74. doi: 10.1016/j.lfs.2016.10.011
64. Goel N, Workman JL, Lee TT, Innala L, Viau V. Sex differences in the HPA axis. Compr Physiol. (2014) 4(3):1121–55. doi: 10.1002/cphy.c130054
65. Duncko R, Kiss A, Skultétyová I, Rusnák M, Jezová D. Corticotropin-releasing hormone mRNA levels in response to chronic mild stress rise in male but not in female rats while tyrosine hydroxylase mRNA levels decrease in both sexes. Psychoneuroendocrinology. (2001) 26:77–89. doi: 10.1016/S0306-4530(00)00040-8
66. Viau V, Bingham B, Davis J, Lee P, Wong M. Gender and puberty interact on the stress-induced activation of parvocellular neurosecretory neurons and corticotropin-releasing hormone messenger ribonucleic acid expression in the rat. Endocrinology. (2005) 146:137–46. doi: 10.1210/en.2004-0846
67. Kitay JI. Sex differemces in adrenal cortical secretion in the rat. Endocrinology. (1961) 68:818–24. doi: 10.1210/endo-68-5-818
68. Smith KB, Murack M, Ismail N. The sex-dependent and enduring impact of pubertal stress on health and disease. Brain Res Bull. (2023) 200:110701. doi: 10.1016/j.brainresbull.2023.110701
69. Iwasaki-Sekino A, Mano-Otagiri A, Ohata H, Yamauchi N, Shibasaki T. Gender differences in corticotropin and corticosterone secretion and corticotropin-releasing factor mRNA expression in the paraventricular nucleus of the hypothalamus and the central nucleus of the amygdala in response to footshock stress or psychological stress in rats. Psychoneuroendocrinology. (2009) 34:226–37. doi: 10.1016/j.psyneuen.2008.09.003
70. Seale JV, Wood SA, Atkinson HC, Bate E, Lightman SL, Ingram CD, et al. Gonadectomy reverses the sexually diergic patterns of circadian and stress-induced hypothalamic-pituitary-adrenal axis activity in male and female rats. J Neuroendocrinol. (2004) 16:516–24. doi: 10.1111/j.1365-2826.2004.01195.x
71. Rivier C. Gender, sex steroids, corticotropin-releasing factor, nitric oxide, and the HPA response to stress. Pharmacol Biochem Behav. (1999) 64:739–51. doi: 10.1016/S0091-3057(99)00148-3
72. Spinedi E, Salas M, Chisari A, Perone M, Carino M, Gaillard RC. Sex differences in the hypothalamo-pituitary-adrenal axis response to inflammatory and neuroendocrine stressors. Evidence for a pituitary defect in the autoimmune disease-susceptible female Lewis rat. Neuroendocrinology. (1994) 60:609–17. doi: 10.1159/000126804
73. Demaestri C, Pisciotta M, Altunkeser N, Berry G, Hyland H, Breton J, et al. Central amygdala CRF+ neurons promote heightened threat reactivity following early life adversity in mice. Nat Commun. (2024) 15:5522. doi: 10.1038/s41467-024-49828-3
74. Desbonnet L, Garrett L, Daly E, McDermott KW, Dinan TG. Sexually dimorphic effects of maternal separation stress on corticotrophin-releasing factor and vasopressin systems in the adult rat brain. Int J Dev Neurosci. (2008) 26:259–68. doi: 10.1016/j.ijdevneu.2008.02.004
75. Goel N, Plyler KS, Daniels D, Bale TL. Androgenic influence on serotonergic activation of the HPA stress axis. Endocrinology. (2011) 152:2001–10. doi: 10.1210/en.2010-0964
76. Bielohuby M, Herbach N, Wanke R, Maser-Gluth C, Beuschlein F, Wolf E, et al. Growth analysis of the mouse adrenal gland from weaning to adulthood: time- and gender-dependent alterations of cell size and number in the cortical compartment. Am J Physiology-Endocrinology Metab. (2007) 293:E139–46. doi: 10.1152/ajpendo.00705.2006
77. Majchrzak M, Malendowicz LK. Sex differences in adrenocortical structure and function. Cell Tissue Res. (1983) 232:457–69. doi: 10.1007/BF00213800
78. Osborn JA, Yu C, Stelzl GE, Weinberg J. Effects of fetal ethanol exposure on pituitary-adrenal sensitivity to secretagogues. Alcoholism: Clin Exp Res. (2000) 24(7):1110–9. doi: 10.1111/j.1530-0277.2000.tb04657.x
79. Almeida OFX, Hassan AHS, Harbuz MS, Linton EA, Lightman SL. Hypothalamic corticotropin-releasing hormone and opioid peptide neurons: functional changes after adrenalectomy and/or castration. Brain Res. (1992) 571(2):189–98. doi: 10.1016/0006-8993(92)90654-R
80. Patchev VK, Almeida OFX. Gonadal steroids exert facilitating and “Buffering” Effects on glucocorticoid-mediated transcriptional regulation of corticotropin-releasing hormone and corticosteroid receptor genes in rat brain. J Neurosci. (1996) 16:7077. doi: 10.1523/JNEUROSCI.16-21-07077.1996
81. Viau V, Soriano L, Dallman MF. Androgens alter corticotropin releasing hormone and arginine vasopressin mRNA within forebrain sites known to regulate activity in the hypothalamic-pituitary-adrenal axis. J Neuroendocrinol. (2001) 13:442–52. doi: 10.1046/j.1365-2826.2001.00653.x
82. Critchlow V, Liebelt RA, Bar-Sela M, Mountcastle W, Lipscomb HS. Sex difference in resting pituitary-adrenal function in the rat. Am J Physiology-Legacy Content. (1963) 205(5):807–15. doi: 10.1152/ajplegacy.1963.205.5.807
83. Seale JV, Wood SA, Atkinson HC, Harbuz MS, Lightman SL. Gonadal steroid replacement reverses gonadectomy-induced changes in the corticosterone pulse profile and stress-induced hypothalamic-pituitary-adrenal axis activity of male and female rats. J Neuroendocrinol. (2004) 16:989–98. doi: 10.1111/j.1365-2826.2004.01258.x
84. Viau V, Lee P, Sampson J, Wu J. A testicular influence on restraint-induced activation of medial parvocellular neurons in the paraventricular nucleus in the male rat. Endocrinology. (2003) 144:3067–75. doi: 10.1210/en.2003-0064
85. Handa RJ, Nunley KM, Lorens SA, Louie JP, McGivern RF, Bollnow MR. Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol Behav. (1994) 55:117–24. doi: 10.1016/0031-9384(94)90018-3
86. McCormick CM, Furey BF, Child M, Sawyer MJ, Donohue SM. Neonatal sex hormones have `organizational’ effects on the hypothalamic-pituitary-adrenal axis of male rats. Dev Brain Res. (1998) 105:295–307. doi: 10.1016/S0165-3806(97)00155-7
87. Spinedi E, Suescun MO, Hadid R, Daneva T, Gaillard RC. Effects of gonadectomy and sex hormone therapy on the endotoxin-stimulated hypothalamo-pituitary-adrenal axis: evidence for a neuroendocrine-immunological sexual dimorphism. Endocrinology. (1992) 131:2430–6. doi: 10.1210/endo.131.5.1330501
88. Kitay JI. Effects of estradiol on pituitary-adrenal function in male and female rats. Endocrinology. (1963) 72:947–54. doi: 10.1210/endo-72-6-947
89. Leśniewska B, Nowak M, Malendowicz L. Sex differences in adrenocortical structure and function. Hormone Metab Res. (1990) 22:378–81. doi: 10.1055/s-2007-1004926
90. Ogilvie KM, Rivier C. Gender difference in hypothalamic–pituitary–adrenal axis response to alcohol in the rat: activational role of gonadal steroids. Brain Res. (1997) 766:19–28. doi: 10.1016/S0006-8993(97)00525-8
91. Guo AL, Petraglia F, Criscuolo M, Ficarra G, Nappi RE, Palumbo M, et al. Acute stress- or lipopolysaccharide-induced corticosterone secretion in female rats is independent of the oestrous cycle. Eur J Endocrinol. (1994) 131:535–9. doi: 10.1530/eje.0.1310535
92. Figueiredo HF, Ulrich-Lai YM, Choi DC, Herman JP. Estrogen potentiates adrenocortical responses to stress in female rats. Am J Physiology-Endocrinology Metab. (2007) 292:E1173–82. doi: 10.1152/ajpendo.00102.2006
93. Lund TD, Munson DJ, Haldy ME, Handa RJ. Dihydrotestosterone may inhibit hypothalamo–pituitary–adrenal activity by acting through estrogen receptor in the male mouse. Neurosci Lett. (2004) 365:43–7. doi: 10.1016/j.neulet.2004.04.035
94. Nowak KW, Neri G, Nussdorfer GG, Malendowicz LK. Effects of sex hormones on the steroidogenic activity of dispersed adrenocortical cells of the rat adrenal cortex. Life Sci. (1995) 57:833–7. doi: 10.1016/0024-3205(95)02015-B
95. Kirschbaum C, Schommer N, Federenko I, Gaab J, Neumann O, Oellers M, et al. Short-term estradiol treatment enhances pituitary-adrenal axis and sympathetic responses to psychosocial stress in healthy young men. J Clin Endocrinol Metab. (1996) 81:3639–43. doi: 10.1210/jcem.81.10.8855815
96. Statistics N.C.f.H. Mean macronutrient intake among adults aged 20 and over, by sex and age: United States, selected years 1988-1994 through 2015-2018, a. Mean macronutrient intake among adults, b.s. over, and s.y.t. age: United States. Hyattsville, MD: CDC (2021).
97. Cao W, Shi M, Wu L, Li J, Yang Z, Liu Y, et al. Adipocytes initiate an adipose-cerebral-peripheral sympathetic reflex to induce insulin resistance during high-fat feeding. Clin Sci. (2019) 133:1883–99. doi: 10.1042/CS20190412
98. Dalmasso C, Leachman JR, Ensor CM, Yiannikouris FB, Giani JF, Cassis LA, et al. Female mice exposed to postnatal neglect display angiotensin II–dependent obesity-induced hypertension. J Am Heart Assoc. (2019) 8(23):F611–21. doi: 10.1161/JAHA.119.012309
99. Murphy MO, Herald JB, Wills CT, Unfried SG, Cohn DM, Loria AS. Postnatal treatment with metyrapone attenuates the effects of diet-induced obesity in female rats exposed to early-life stress. Am J Physiol Endocrinol Metab. (2017) 312:E98–E108. doi: 10.1152/ajpendo.00308.2016
100. Yang ZH, Chen FZ, Zhang YX, Ou MY, Tan PC, Xu XW, et al. Therapeutic targeting of white adipose tissue metabolic dysfunction in obesity: mechanisms and opportunities. MedComm (2020). (2024) 5:e560. doi: 10.1002/mco2.v5.6
101. Mosquera-Sulbarán J, Ryder E, Pedreáñez A, Vargas R. Angiotensin II and human obesity. A narrative review of the pathogenesis. Investigación Clínica. (2022) 63:435–53. doi: 10.54817/ic.v63n4a09
102. Faulkner JL, Bruder-Nascimento T, Belin de Chantemèle EJ. The regulation of aldosterone secretion by leptin. Curr Opin Nephrol Hypertension. (2018) 27:63–9. doi: 10.1097/MNH.0000000000000384
103. Glasow A, Bornstein SR. Leptin and the adrenal gland. Eur J Clin Invest. (2000) 30:39–45. doi: 10.1046/j.1365-2362.2000.0300s3039.x
104. Huby A-C, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. (2015) 132:2134–45. doi: 10.1161/CIRCULATIONAHA.115.018226
105. Mulrow PJ. Angiotensin II and aldosterone regulation. Regul Pept. (1999) 80:27–32. doi: 10.1016/S0167-0115(99)00004-X
106. Kawarazaki W, Fujita T. The role of aldosterone in obesity-related hypertension. Am J Hypertens. (2016) 29:415–23. doi: 10.1093/ajh/hpw003
107. Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. (1999) 7:355–62. doi: 10.1002/j.1550-8528.1999.tb00418.x
108. Urbanet R, Nguyen Dinh Cat A, Feraco A, Venteclef N, El Mogrhabi S, Sierra-Ramos C, et al. Adipocyte mineralocorticoid receptor activation leads to metabolic syndrome and induction of prostaglandin D2 synthase. Hypertension. (2015) 66:149–57. doi: 10.1161/HYPERTENSIONAHA.114.04981
109. Hoppmann J, Perwitz N, Meier B, Fasshauer M, Hadaschik D, Lehnert H, et al. The balance between gluco- and mineralo-corticoid action critically determines inflammatory adipocyte responses. J Endocrinol. (2010) 204:153–64. doi: 10.1677/JOE-09-0292
110. Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. (2001) 106:255–8. doi: 10.1016/S0092-8674(01)00449-4
111. Herrada AA, Contreras FJ, Marini NP, Amador CA, Gonzalez PA, Cortes CM, et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol. (2010) 184:191–202. doi: 10.4049/jimmunol.0802886
112. Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, et al. Adipose tissue dendritic cells are independent contributors to obesity-induced inflammation and insulin resistance. J Immunol. (2016) 197:3650–61. doi: 10.4049/jimmunol.1600820
113. Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. (2012) 61:2238–47. doi: 10.2337/db11-1274
114. Ferreira NS, Tostes RC, Paradis P, Schiffrin EL. Aldosterone, inflammation, immune system, and hypertension. Am J Hypertens. (2021) 34:15–27. doi: 10.1093/ajh/hpaa137
115. Stutte S, Ishikawa-Ankerhold H, Lynch L, Eickhoff S, Nasiscionyte S, Guo C, et al. High-fat diet rapidly modifies trafficking, phenotype, and function of plasmacytoid dendritic cells in adipose tissue. J Immunol. (2022) 208:1445–55. doi: 10.4049/jimmunol.2100022
116. Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. (2012) 61:2330–9. doi: 10.2337/db11-1523
117. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. (1994) 372(6505):425–32. doi: 10.1038/372425a0
118. Obradovic M, Sudar-Milovanovic E, Soskic S, Essack M, Arya S, Stewart AJ, et al. Leptin and obesity: role and clinical implication. Front Endocrinol (Lausanne). (2021) 12:585887. doi: 10.3389/fendo.2021.585887
119. Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. (2004) 304:108–10. doi: 10.1126/science.1095004
120. Ahima RS, Prabakaran D, Flier JS. Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest. (1998) 101:1020–7. doi: 10.1172/JCI1176
121. Strzelewicz AR, Vecchiarelli HA, Rondón-Ortiz AN, Raneri A, Hill MN, Kentner AC. Interactive effects of compounding multidimensional stressors on maternal and male and female rat offspring outcomes. Horm Behav. (2021) 134:105013. doi: 10.1016/j.yhbeh.2021.105013
122. Paternain L, Martisova E, Milagro FI, Ramírez MJ, Martínez JA, Campión J, et al. Postnatal maternal separation modifies the response to an obesogenic diet in adulthood in rats. Dis Models Mech. (2012) 5:691–7. doi: 10.1242/dmm.009043
123. Lee JH, Yoo SB, Kim JY, Lee JY, Kim BT, Park K, et al. Early life stress experience may blunt hypothalamic leptin signalling. J Biosci. (2017) 42:131–8. doi: 10.1007/s12038-016-9656-3
124. Daniels TE, Mathis KJ, Gobin AP, Lewis-de Los Angeles WW, Smith EM, Chanthrakumar P, et al. Associations of early life stress with leptin and ghrelin in healthy young adults. Psychoneuroendocrinology. (2023) 149:106007. doi: 10.1016/j.psyneuen.2022.106007
125. Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension. (2004) 43:1126–32. doi: 10.1161/01.HYP.0000122875.91100.28
126. Saint-Marc P, Kozak LP, Ailhaud G, Darimont C, Negrel R. Angiotensin II as a trophic factor of white adipose tissue: stimulation of adipose cell formation. Endocrinology. (2001) 142:487–92. doi: 10.1210/endo.142.1.7883
127. Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology. (1997) 138:1512–9. doi: 10.1210/endo.138.4.5038
128. Shin AC, MohanKumar SMJ, Balasubramanian P, Sirivelu MP, Linning K, Woolcock A, et al. Responsiveness of hypothalamo-pituitary-adrenal axis to leptin is impaired in diet-induced obese rats. Nutr Diabetes. (2019) 9:10. doi: 10.1038/s41387-019-0076-y
129. Keirns BH, Keirns NG, Tsotsoros CE, Layman HM, Stout ME, Medlin AR, et al. Adverse childhood experiences and obesity linked to indicators of gut permeability and inflammation in adult women. Physiol Behav. (2023) 271:114319. doi: 10.1016/j.physbeh.2023.114319
130. Steenblock C, Rubin de Celis MF, Delgadillo Silva LF, Pawolski V, Brennand A, Werdermann M, et al. Isolation and characterization of adrenocortical progenitors involved in the adaptation to stress. Proc Natl Acad Sci USA. (2018) 115:12997–3002. doi: 10.1073/pnas.1814072115
Keywords: adverse childhood experience, sex differences, obesity, aldosterone, HPA axis
Citation: Turner MB, Dalmasso C and Loria AS (2024) The adipose tissue keeps the score: priming of the adrenal-adipose tissue axis by early life stress predisposes women to obesity and cardiometabolic risk. Front. Endocrinol. 15:1481923. doi: 10.3389/fendo.2024.1481923
Received: 16 August 2024; Accepted: 26 September 2024;
Published: 18 October 2024.
Edited by:
Rodrigo O. Maranon, CCT CONICET Tucuman, ArgentinaReviewed by:
Stella Honoré, CONICET Higher Institute of Biological Research (INSIBIO), ArgentinaAnalia Tomat, University of Buenos Aires, Argentina
Copyright © 2024 Turner, Dalmasso and Loria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Analia S. Loria, YW5hbGlhLmxvcmlhQHVreS5lZHU=