 Kevin C. J. Yuen1*†
Kevin C. J. Yuen1*† Rikke Hjortebjerg2,3,4
Rikke Hjortebjerg2,3,4 Ashok Ainkaran Ganeshalingam2,3,4
Ashok Ainkaran Ganeshalingam2,3,4 David R. Clemmons5
David R. Clemmons5 Jan Frystyk2,3
Jan Frystyk2,3- 1Department of Neuroendocrinology and Neurosurgery, Barrow Neurological Institute, University of Arizona College of Medicine and Creighton School of Medicine, Phoenix, AZ, United States
- 2Department of Endocrinology, Odense University Hospital, Odense, Denmark
- 3Department of Clinical Medicine, Faculty of Health Sciences, University of Southern Denmark, Odense, Denmark
- 4Steno Diabetes Center Odense, Odense University Hospital, Odense, Denmark
- 5Department of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC, United States
Growth hormone (GH) is the key regulator of insulin-like growth factor I (IGF-I) generation in healthy states. However, portal insulin delivery is also an essential co-player in the regulation of the GH/IGF-I axis by affecting and regulating hepatic GH receptor synthesis, and subsequently altering hepatic GH sensitivity and IGF-I generation. Disease states of GH excess (e.g., acromegaly) and GH deficiency (e.g., congenital isolated GH deficiency) are characterized by increased and decreased GH, IGF-I and insulin levels, respectively, where the GH/IGF-I relationship is reflected by a “primary association”. When intra-portal insulin levels are increased (e.g., obesity, Cushing’s syndrome, or due to treatment with glucocorticoids and glucagon-like peptide 1 receptor agonists) or decreased (e.g., malnutrition, anorexia nervosa and type 1 diabetes mellitus), these changes secondarily alter hepatic GH sensitivity resulting in a “secondary association” with discordant GH and IGF-I levels (e.g., high GH/low IGF-I levels or low GH/high IGF-I levels, respectively). Additionally, intra-portal insulin regulates hepatic secretion of IGFBP-1, an inhibitor of IGF-I action. Through its effects on IGFBP-1 and subsequently free IGF-I, intra-portal insulin exerts its effects to influence endogenous GH secretion via the negative feedback loop. Therefore, it is important to understand the effects of changes in intra-portal insulin when interpreting the GH/IGF-I axis in disease states. This review summarizes our current understanding of how changes in intra-portal insulin delivery to the liver in health, disease states and drug therapy use and misuse that leads to alterations in GH/IGF-I secretion that may dictate management decisions in afflicted patients.
Introduction
The growth hormone (GH)-insulin-like growth factor I (IGF-I) axis plays a critical role in promoting linear growth in children (1), whereas in adults, its role is primarily of metabolic relevance (2). Growth hormone is secreted from the pituitary gland and stimulates IGF-I synthesis and secretion, which in turn inhibits GH secretion via the negative feedback loop (3). The liver is the major contributor to the circulation pool of IGF-I (4), the six IGF-binding proteins (IGFBP-1 to -6) (5) and acid labile subunit (ALS) (6). In the circulation, approximately 99% of the IGF-I pool is bound with high affinity to the IGFBPs that circulate in molar excess of IGF-I, thus explaining that less than 1% circulates as free, unbound IGF-I (7). By serving as carriers of circulating IGF-I, the IGFBPs and ALS prolong IGF-I half-life and modulate tissue access, thereby controlling IGF-I action (5, 8, 9).
Insulin, when secreted from pancreatic β-cells, is transported by the portal vein directly to the liver resulting in high hepatocyte exposure. Notably, the liver is a major target organ for the metabolic effects of insulin and exerts a first-pass extraction of up to 85% of insulin delivered by the portal vein (10). Additionally, because of its ability to regulate the hepatic sensitivity to GH, intra-portal insulin delivery has turned out to be an essential co-player for effective GH-induced hepatic IGF-I synthesis (11–15) (Figure 1A). Indeed, one may consider the ability of insulin to control the hepatic GH sensitivity to be a normal physiological response to changes in nutrition. During fasting with decreased intra-portal insulin levels, hepatic GH sensitivity decreases leading to decreased serum IGF-I levels despite a compensatory increase in GH secretion. Conversely, overfeeding increases intra-portal insulin levels that enhances hepatic GH sensitivity to a degree where relatively low GH secretion is sufficient to maintain ambient hepatic IGF-I synthesis and secretion, and normal circulating IGF-I levels (7). This GH response to nutritional changes is physiologically appropriate as it leads to increased GH-mediated insulin resistance that prevents hypoglycemia during fasting (16) and decreased GH-mediated insulin resistance during periods of overfeeding (17). However, disease states (e.g., obesity, Cushing’s syndrome, type 1 diabetes [T1D] and anorexia nervosa) and some commonly used drugs [e.g., glucocorticoids and glucagon-like peptide 1 receptor agonists (GLP-1RAs)] that impact pancreatic β-cell insulin secretion can also alter the GH/IGF-I axis. As GH, IGF-I, and insulin continuously modulate each other’s secretion and actions during health and disease, a good understanding of the mechanisms behind these interactions is of clinical importance.
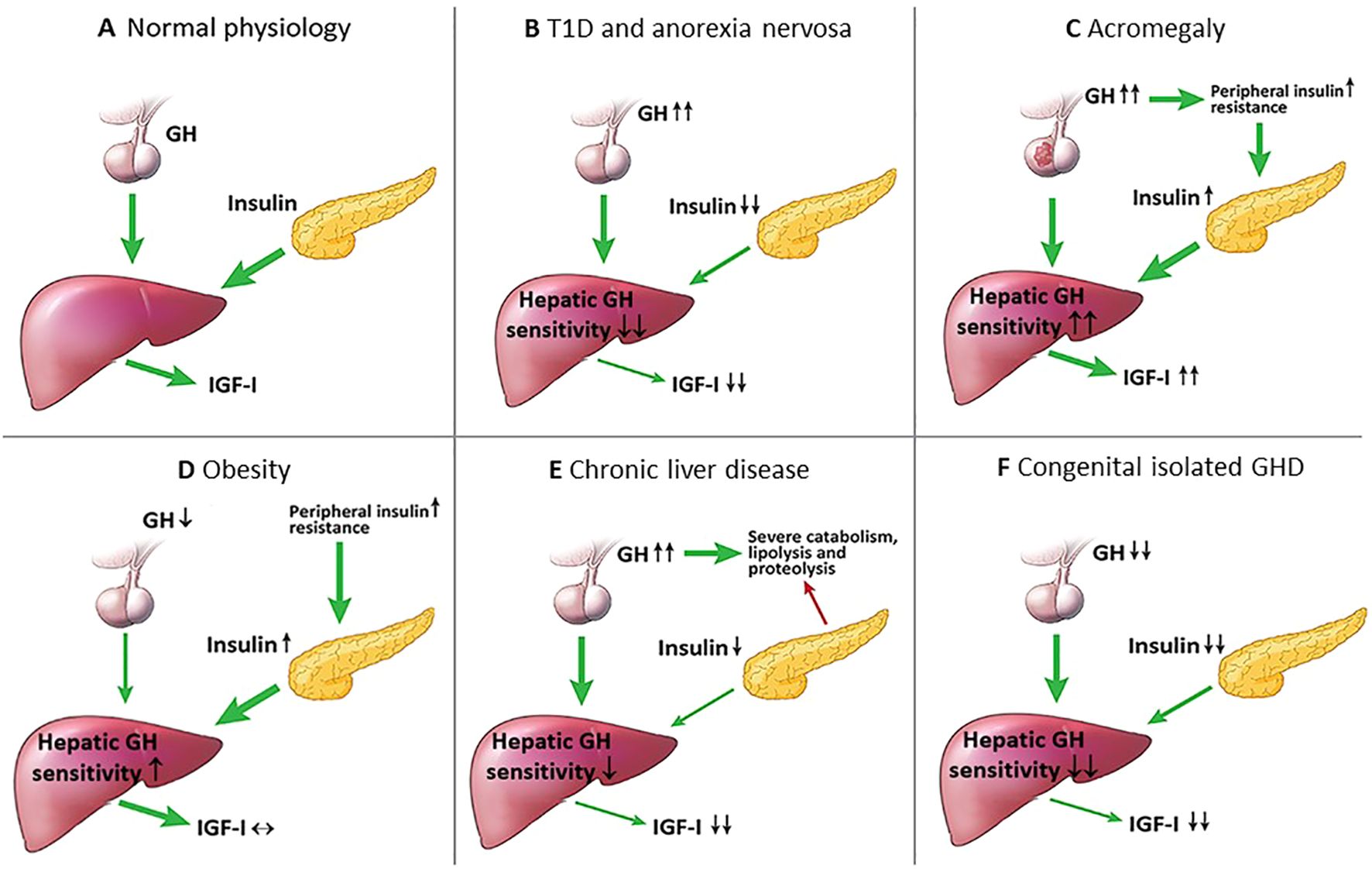
Figure 1. Intra-portal insulin changes in health and several disease states. (A) Normal physiological state: intra-portal insulin regulates hepatic GH sensitivity in the generation of IGF-I; (B) T1D and anorexia nervosa: intra-portal hypoinsulinemia decreases hepatic GH sensitivity resulting in low IGF-I levels, and due to the lack of negative feedback by IGF-I on the hypothalamus, GH levels increase; (C) Acromegaly: GH excess increases insulin resistance causing intra-portal hyperinsulinemia that leads to increased hepatic GH sensitivity, and the combination of increased GH and hepatic GH sensitivity leads to further increase in IGF-I levels; (D) Obesity: peripheral insulin resistance causes compensatory intra-portal hyperinsulinemia that increases hepatic GH sensitivity resulting in high normal IGF-I levels and low GH levels; (E) Chronic liver disease: the liver fails to produce sufficient IGF-I resulting in high GH levels due to the lack of negative feedback by IGF-I on the hypothalamus that causes a state of catabolism, lipolysis and proteolysis, lipolysis and decreased β-cell insulin secretion; (F) Congenital isolated GHD: severe lifelong GHD results in decreased β-cell mass and insulin secretion, and the combination of decreased GH and intra-portal hypoinsulinemia leads to markedly low IGF-I levels.
This review summarizes how changes in hepatic exposure to intra-portal insulin in health and disease states modulates the GH/IGF-I axis. We also discuss the relevance of kidney function, another confounder that may alter the GH/IGF-I axis in a manner that can replicate findings seen in acromegaly. Finally, we briefly describe how the GH/IGF-I axis is impacted by liver disease, weight loss, GH misuse for recreational and athletic performance-enhancing, GH-releasing hormone (GHRH) analog use in HIV lipodystrophy, and the alterations of the GH/IGF-I axis in subjects with congenital isolated GH and IGF-I deficiencies.
Physiology of the GH/IGF-I axis
Physiology
Pituitary secretion of GH is under the control of GHRH stimulation and somatostatin inhibition, both secreted from the hypothalamus (3). Growth hormone secretion is episodic and pulsatile, with levels varying between peaks and troughs, and very low levels between pulses (18). Growth hormone stimulates IGF-I synthesis and secretion, which feeds back on the pituitary to inhibit GH secretion (19, 20). Insulin-like growth factor-I affects the regulation of GH secretion at the hypothalamus by inhibiting the GHRH gene expression (19) and stimulating somatostatin secretion (20), whereas at the pituitary, IGF-I inhibits spontaneous and GHRH-stimulated GH secretion (3). Unlike the episodic and pulsatile nature of GH secretion, IGF-I is secreted continuously, has a longer half-life and exhibits more stable concentrations in the blood (21). Hence, IGF-I is utilized as a biomarker of GH secretory status, as its levels reflect the 24-hour integrated GH secretion (22–24).
Apart from insulin, there are other hormones that can influence pituitary GH secretion, such as ghrelin, estrogens and androgens. Ghrelin, a gastric peptide with potent GH secretagogue properties, amplifies hypothalamic GHRH secretion and synergizes its pituitary GH-stimulation effects (25). Estrogens stimulate pituitary GH secretion, but inhibit GH action on the liver by suppressing GHR signalling (26). Additionally, estrogens can potentiate ghrelin action (27), while androgens enhance peripheral actions of GH (28). Finally, pituitary GH secretion inversely correlates with intra-abdominal visceral adiposity via mechanisms that depend on free fatty acid (FFA) fluxes (3).
Insulin
Insulin suppresses GH secretion, but the underlying mechanisms are not well-understood, and are postulated to occur at several different levels. At the level of the pituitary, in vitro studies of pituitary cells and in vivo findings in mice harvesting a somatotroph-specific knockout of the insulin receptor have demonstrated that insulin directly suppressed pituitary GH secretion independent of the IGF-I receptor (IGF-IR) (29, 30). At the level of the liver, in vitro studies using the human hepatoma cell line (HuH7) as a model have shown that insulin stimulates GHR protein synthesis and GHR binding of GH in a dose-dependent manner (31). Similarly, in vivo studies based on rodents have shown reduced hepatic GHR expression (32) and GH binding (32, 33) following induction of diabetes, and that insulin treatment facilitated GH binding (33). These findings highlight the key role insulin plays in modulating hepatic responsiveness to GH and support the notion that insulin plays an important part in regulating hepatic GH sensitivity via its ability to directly influence GHR expression and GH binding. However, to the best of our knowledge, this notion is yet to be demonstrated in humans.
Furthermore, besides regulating circulating IGF-I levels via its impact on the hepatic GH sensitivity, insulin also inhibits hepatic synthesis and secretion of IGF-binding protein-1 (IGFBP-1); an inhibitor of IGF-I action (7). Thus, the relationship between insulin and IGFBP-I is inverse (34), where IGFBP-1 is low during states of hyperinsulinemia but elevated during states of hypoinsulinemia (35). Because IGFBP-1 levels fluctuate throughout the day (36), this has led to the hypothesis that IGFBP-1 links IGF-I action to nutritional intake. However, present data indicate that IGFBP-1 serves more as a “brake” on IGF-I action by inhibiting anabolic signaling during nutritional depletion than an “accelerator” of IGF-I action during overfeeding. The rationale is that under normal physiological circumstances, the decline in IGFBP-1 from its maximally stimulated overnight fasting level (34) to the level seen after food intake on a molar basis is relatively modest, and only leads to minor increases in serum free IGF-I (7, 37, 38). Another explanation is that the molar concentrations of the other five IGFBPs far exceeds the concentration of IGFBP-1 (39), and as they are not fully saturated, the other five IGFBPs are fully capable of sequestering the increase in free IGF-I secondary to the reductions in circulating IGFBP-1. However, when IGFBP-1 levels are chronically lowered during prolonged intra-portal hyperinsulinemia (e.g., obesity, Cushing’s syndrome and glucocorticoid therapy), the “accelerator” effect of IGFBP-1 is at play to increase serum free IGF-I (35, 40). Conversely, during prolonged intra-portal hypoinsulinemia (e.g., malnutrition, anorexia nervosa, and T1D), IGFBP-1 levels increase several-fold (7), leading to the increased complex formation between IGF-I and IGFBP-1 and clear reductions in free IGF-I (“brake” effect) (Figure 2).
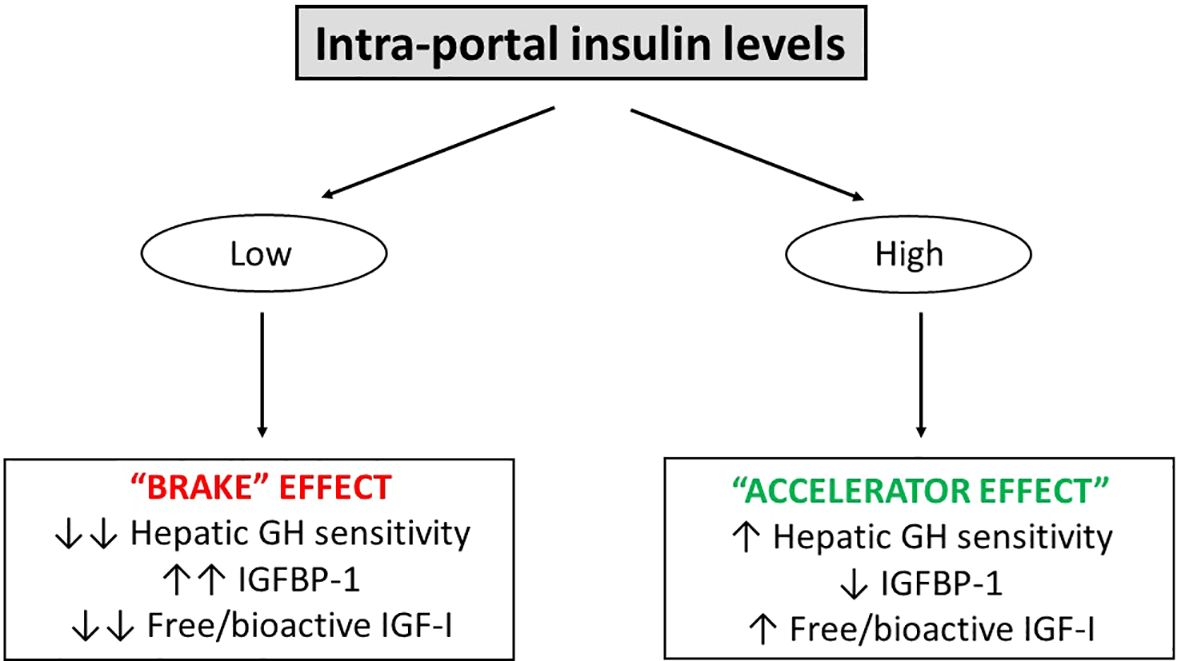
Figure 2. The ability of intra-portal insulin to serve as an “accelerator” and “brake” on hepatic GH sensitivity and free/bioactive IGF-I. In the setting of low intra-portal insulin levels (e.g., overnight fasting and T1D), there is a “brake” effect leading to reduced hepatic GH sensitivity, increased IGFBP-1 and reduced free/bioactive IGF-I. In the setting of high intra-portal insulin levels (e.g., feeding and obesity), an “accelerator” effect takes place leading to increased hepatic GH sensitivity. However, with regards to the “accelerator” and “brake” effects of insulin on serum free/bioactive IGF-I, it appears that the ability of high intra-portal insulin levels to increase (“accelerate”) serum free/bioactive IGF-I activity is less pronounced compared to the ability of low intra-portal insulin levels to decrease (“brake”) serum free/bioactive IGF-I activity. Two arrows indicate a marked effect, one arrow indicates a milder effect.
There is also now ample evidence demonstrating that insulin reaching the liver via the portal system plays an important role in making the liver more GH sensitive to IGF-I generation (23, 41, 42). Disease states of GH excess (e.g., acromegaly) and GH deficiency (GHD) (e.g., congenital isolated GHD) are characterized by increased and decreased GH and IGF-I levels, where the GH/IGF-I relationship is reflected by a “primary association” (high GH and high IGF-I for acromegaly and low GH and low IGF-I for GHD). However, certain disease states and drug therapies can influence intra-portal insulin levels and invariably alter the relationship between GH and IGF-I. For instance, when intra-portal insulin levels are increased [e.g., obesity, Cushing’s syndrome or due to treatment with glucocorticoids and glucagon-like peptide-1 receptor agonists (GLP-1RA)] or decreased (e.g., anorexia nervosa and T1D) in disease states, these secondary changes (“secondary association”) alters hepatic GH sensitivity resulting in discordant GH and IGF-I levels (high GH/low IGF-I levels and low GH/high IGF-I levels, respectively).
Effects of disease states on intra-portal insulin levels and changes in the GH/IGF-I axis
High GH and low IGF-I
Type 1 diabetes is an autoimmune disease characterized by pancreatic β-cell destruction leading to intra-portal hypoinsulinemia. This disease state provides an ideal model to illustrate the pivotal role of intra-portal insulin as the key regulator of the GH/IGF-I axis. Patients with uncontrolled T1D have high GH, low total and free IGF-I levels (Table 1; Figures 1B, 3), and elevated IGFBP-1 levels (2, 43, 44). Following subcutaneous insulin treatment, IGFBP-1 decreases while serum total and free IGF-I increases (43, 44). Notably, changes in IGFBP-1 and free IGF-I are inversely correlated (44), thereby emphasizing the “brake” effect of IGFBP-1 on IGF-I action. The “brake” effect of IGFBP-1 is further substantiated by Attia et al. (45), who paused continuous subcutaneous insulin infusion (CSII) for 8 hours in patients with T1D, and found that IGFBP- 1 levels increased by 6-fold, whereas total and free IGF-I levels decreased. Importantly, intra-portal hypoinsulinization in T1D and its dysregulatory effect on GH/IGF-I axis (Table 1; Figures 1B, 3) is not fully ameliorated by conventional subcutaneous insulin administration, most likely because subcutaneous administration of insulin cannot fully compensate for the normal portal delivery of insulin to the liver. This notion is supported by several clinical studies of patients with T1D. Shishko et al. (13) reported that intra-portal insulin infusion (IPII) was superior to CSII with regards to suppression of IGFBP-1, and normalization of IGF-I and 24-hour GH profiles. These findings were corroborated by Hannaire-Broutin et al. (11), who switched patients from CSII to intra-peritoneal insulin infusion and reported improvements in serum levels of total IGF-I and GH-binding protein (GHBP); the latter serving as a proxy of hepatic GHR density. These findings indicate that intra-peritoneal insulin delivery stimulates hepatic GH sensitivity and improves hepatic IGF-I generation. Hedman et al. (12) demonstrated that intra-peritoneal insulin increased total and bioactive IGF-I, and decreased IGFBP-1 levels. In a study by Frystyk et al. (46) comparing T1D patients, who received combined kidney and pancreas transplantation, one group received a pancreatic graft allowing portal delivery of insulin, while the other, a graft with systemic pancreas drainage. Despite obtaining similar hemoglobin A1c levels, serum total and free IGF-I were elevated and IGFBP-1 were suppressed in patients receiving a pancreas with portal drainage. In another study, when comparing T1D C-peptide positive pre-pubertal children and adults vs their C-peptide negative counterparts, patients with a residual β-cell function had higher IGF-I levels despite similar glycemic control (47, 48). In alignment with these findings, the existence of residual pancreatic β-cell function is of fundamental metabolic importance in T1D. Collectively, these findings indicate that glycemic control does not solely influence the GH/IGF-I axis in T1D and that small changes in hepatic insulinization can exert more meaningful systemic effects (49). Abnormalities in the GH/IGF-I axis have also been observed in patients with type 2 diabetes (T2D) (50). Studies have shown that exogenous administration of recombinant human IGF-I (51, 52) and IGF-I/IGFBP-3 complex (53–55) improved insulin sensitivity and decreased insulin requirements in both T1D and T2D patients. Nevertheless, the clinical implications of high GH and low IGF-I on long-term metabolic and vascular outcomes in patients with T2D remain unclear.
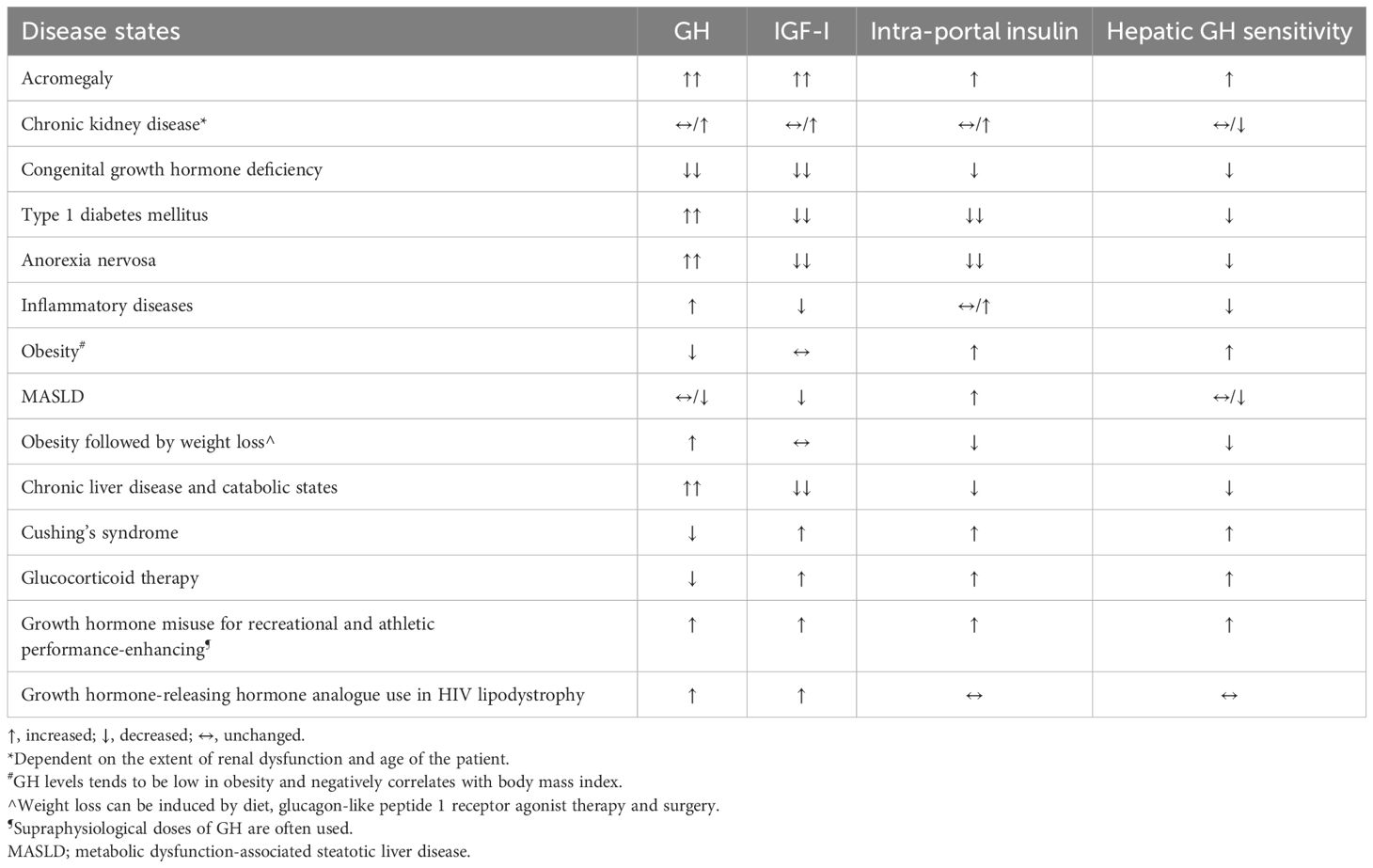
Table 1. Summary of the changes in intra-portal insulin levels in different disease states, after weight loss and after drug therapy on the GH/IGF-I axis.
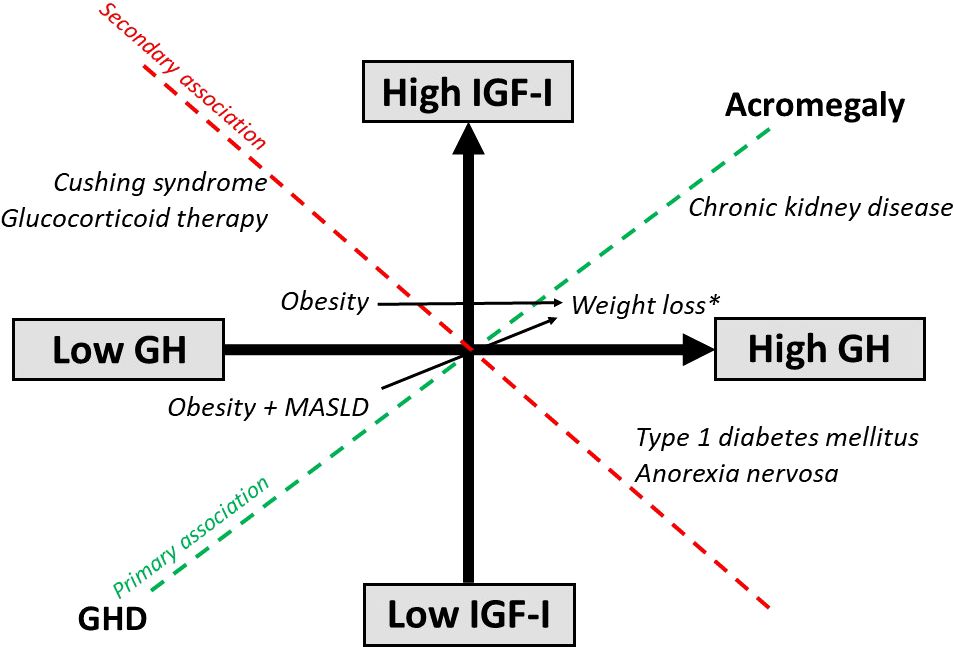
Figure 3. Effects of disease states and weight loss on the GH/IGF-I axis. *Weight loss induced by diet, glucagon-like peptide 1 receptor agonist therapy and surgery.
Anorexia nervosa is a condition of severe undernutrition characterized by GH resistance secondary to chronic nutritional deprivation with resultant high GH and low serum levels of free, bioactive and total IGF-I (56). Patients with the lowest BMI and fat mass tended to have the highest serum GH levels (57, 58). The low IGF-I levels is caused by nutritionally acquired hepatic GH resistance from intra-portal hypoinsulinemia (Table 1; Figures 1B, 3), resulting in down-regulation of the hepatic GHR expression, as inferred from reductions in circulating levels of GHBP (59), and decreased GH binding (60). The state of hepatic GH resistance in anorexia nervosa is further corroborated by the lack of increase in IGF-I following administration of supraphysiological doses of exogenous GH in women with anorexia nervosa (61), and only refeeding appears to able to restore IGF-I levels (56, 59, 62). Therefore, in states of chronic under-nutrition, hepatic GH resistance is an adaptive response, as high GH levels are necessary to maintain euglycemia, while low IGF-I levels conserves energy during periods of nutritional deprivation. Other non-insulin mechanisms of hepatic GH resistance in anorexia nervosa have also been described, including fibroblast growth factor-21, sirtuin 1, triiodothyronine, leptin and testosterone (63).
In the setting of chronic inflammatory disease states (e.g., Crohn’s disease or juvenile chronic arthritis), these diseases can also disturb the physiological synergy of the GH/IGF-I axis. The presence of insulin resistance and elevated pro-inflammatory cytokines (e.g., tumor necrosis factor-α) induce hepatic GH resistance (64), causing increases in GH and decreases in IGF-I levels (Table 1). However, the GH/IGF-I axis in these disease states generally resets itself following the resolution of the inflammatory trigger (65).
High GH and high IGF-I
One of the most well-described primary GH disorders of high GH and high IGF-I is acromegaly (66). In active disease, cellular responses elicited by high GH levels can overwhelm the intracellular mechanisms thus attenuating GH signaling (67). Additionally, the GH stimulatory effect on the liver to increase IGF-I secretion is accentuated by the direct GH effects in antagonizing insulin action and suppressing the anti-lipolytic actions of insulin (68). This leads to peripheral and hepatic insulin resistance and compensatory intra-portal hyperinsulinemia, which results in increased hepatic GH sensitivity that aggravates the condition further (Table 1; Figures 1C, 3). Supportive of this notion, a proof-of-concept study performed by Coopmans et al. (69) demonstrated that 14-day exposure to a eucaloric very-low-carbohydrate ketogenic diet as adjuvant treatment to first-generation somatostatin receptor ligands decreased IGF-I without increasing GH levels in acromegaly patients. Although measures to study insulin secretion were not performed, this study suggested that even in the context of uncontrolled acromegaly and insulin resistance, insulin still plays a key role in the regulation of circulating IGF-I levels. The role of insulin in controlling hepatic IGF-I was further illustrated in a study of moderately obese subjects undergoing caloric restriction and receiving GH. In this study, two groups of subjects received a severely energy-restricted diet. In one group, most of the calories were supplied as carbohydrate and in the other, as lipid. The subjects receiving most of their energy as fat had a lesser IGF-I response to GH, whereas subjects receiving the same caloric intake as carbohydrate demonstrated greater increases in IGF-I to GH stimulation (70, 71). When serum insulin or 24-hour urinary C-peptide were measured, those receiving carbohydrate had higher serum insulin levels and 24-hour urinary C-peptide. It is also possible that in active acromegaly, chronic exposure to elevated IGF-I levels resets IGF-IR responsiveness via the induction of IGF-IR resistance (70), as it has been demonstrated that prolonged stimulation with IGF-I induces functional IGF-IR resistance in vitro through the negative feedback loop (72). Thus, in the setting of IGF-IR resistance in the pituitary and hypothalamus of acromegaly patients, the negative feedback loop by IGF-I may be defective resulting in further IGF-I elevations.
Chronic kidney disease (CKD) is a condition that mimics some components of the biochemical picture of acromegaly with high serum GH and total IGF-I levels. The kidneys contribute to GH degradation and as a result, GH half-life is increased in patients with CKD due to decreased renal clearance (73) (Figure 4). Some investigators have suggested that the uremia of CKD is associated with decreased GH signalling contributing to the state of hepatic GH resistance (74) (Figure 4). Hence, depending on the extent of renal dysfunction manifested by the degree of uremia and age of the patient (75, 76), random fasting GH levels can either be increased or normal. Impaired kidney function is also associated with increased IGFBP levels (e.g., IGFBP-1, -2, -3, -4, and -6) (75, 77) and IGFBP-3 protease activity, which subsequently increases low-molecular-weight IGFBP-3 fragments (78), most likely due to a compromised renal IGFBP-clearance (Figure 4). The increase in IGFBP-1 and -2 leads to secondary increases in circulating IGF-I binding capacity that results in high total IGF-I, but decreased IGF-I bioactivity (77, 78) (Figure 4). It is also noteworthy that due to the excess circulating IGFBPs in end-stage renal failure patients on hemodialysis, GH is only approximately 50% efficient in increasing bioactive IGF-I compared with healthy subjects (79). Furthermore, peripheral insulin resistance triggered by uremia, metabolic acidosis and low-grade inflammation is common in CKD (80), leading to intra-portal hyperinsulinemia, but does not necessarily increase hepatic sensitivity to GH because of the reduced hepatic expression of IGF-I and GHR mRNA (81). Thus, high total IGF-I levels in CKD is inadequate in suppressing the increased GH levels because of the lower IGF-I bioactivity resulting in the lack of IGF-I feedback on pituitary GH secretion (75, 77) (Table 1; Figure 3).
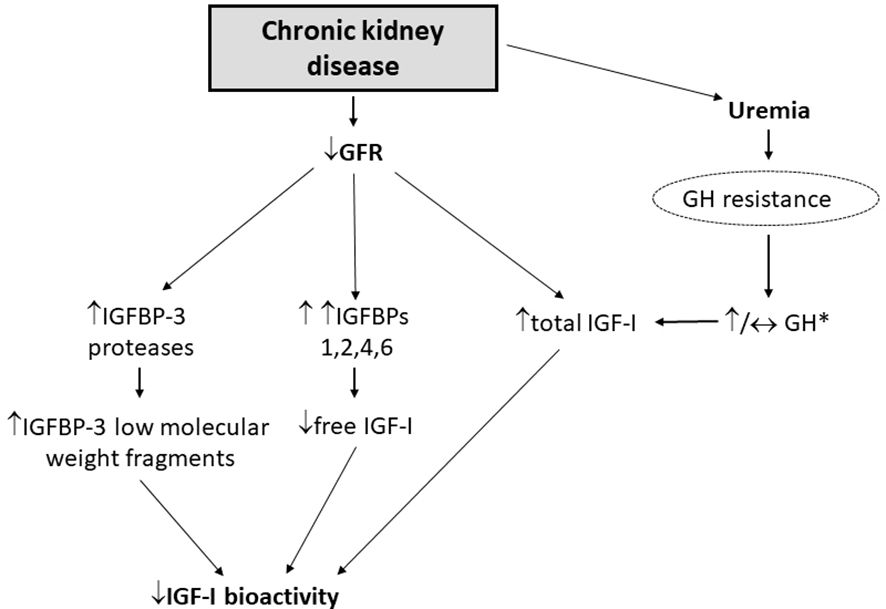
Figure 4. Deranged GH/IGF-I axis in patients with CKD [adapted from Kamenicky et al. (77)]. *Random fasting GH levels can either be increased or normal, depending on the extent of renal dysfunction manifested by the degree of uremia and age of the patient.
Low GH and low IGF-I
Like acromegaly, GHD can be considered as a primary GH disorder, where changes on the GH/IGF-I axis are reflected by a primary association with low GH and low IGF-I (Table 1; Figure 3). Patients with GHD can present as either childhood-onset (CO-GHD) or adult-onset (AO-GHD). Furthermore, GHD may occur as isolated GHD or with multiple pituitary hormone deficiencies. The most common cause of CO-GHD is idiopathic and can be isolated or associated with other pituitary hormone deficiencies, while AO-GHD is frequently acquired from hypothalamic-pituitary tumors and/or secondary to their treatment (82). Because IGF-I exerts anti-inflammatory actions and is important for peripheral glucose uptake, metabolic disturbances in these patients can, in part, be explained by the low IGF-I levels (83). Furthermore, due to deficient GH secretion, the ability of GH to antagonize the anti-lipolytic effect of insulin is reduced, leading to accumulation of visceral adipose tissue (VAT). These effects promote insulin resistance and intra-portal hyperinsulinemia that increases hepatic GH sensitivity to maintain some degree of IGF-I secretion. Conversely, CO-GHD patients tend to be less insulin-resistant than AO-GHD patients (84), and their IGF-I levels are generally lower than their AO-GHD counterparts due to relatively lower intra-portal insulin levels. Thus, insulin resistance leading to elevated portal insulin levels may explain, at least partially, the overlap in individual IGF-I levels between older GH-deficient subjects and GH-sufficient subjects (85, 86). This is clinically relevant, as IGF-I is generally less reliable of a biomarker for screening adults above 60 years with GHD and is a better screening and diagnostic biomarker in GHD in subjects with disease onset in childhood or early adulthood (86).
Low GH and normal/high IGF-I
Obesity is a common yet serious condition associated with blunted spontaneous and stimulated GH secretion (87, 88), i.e., a state of relative GHD. However, serum total and bioactive IGF-I are relatively preserved despite reductions in GH levels (89) (Table 1; Figures 1D, 3). Previous studies have found that GH levels in patients with obesity was decreased nearing the lowest levels of normal range in men and women (90), with levels in those with morbid obesity comparable to adults with GHD (91). As weight loss restores GH levels (92–94), this indicates that the blunted GH secretion is an effect rather than a cause of obesity. This paradox may be explained by the increased insulin resistance and the compensatory intra-portal hyperinsulinemia causing increased hepatic GH sensitivity that increases serum IGF-I and blunts pituitary GH secretion via the negative feedback loop. The increased hepatic exposure to insulin may also explain why obese subjects following exogenous GH administration respond better with higher increases of IGF-I than lean subjects; the main concept behind the IGF-I generation test (95). Additionally, elevations in FFAs in obesity contributes to GH suppression, as experimental lowering of FFA levels reverses obesity-associated impairment of GH secretion (96). Collectively, it may be hypothesized that obesity induces feedback inhibition of pituitary GH secretion by increasing non-fasting serum FFA levels and the associated insulin resistance that causes compensatory intra-portal hyperinsulinemia and IGFBP-1 suppression, with the net result being low GH and normal IGF-I levels. Whether this unique pattern worsens the obese state or constitutes a favorable adaptation remains unclear, and the physiological and pathophysiological implications of the altered GH/IGF-I axis in obesity remain uncertain with GH neither being the cause nor the solution to reverse obesity. Notably, in mice studies, GH secretagogue receptor suppression in adipose tissues protects against obesity and insulin resistance (97), whereas IGF-I increases skeletal muscle insulin sensitivity (98). Thus, it is possible that the combination of low GH and normal IGF-I serves as a physiological mechanism aimed at preserving the GH/IGF-I anabolic effects and minimizing the insulin-antagonistic effects of GH without compromising the positive IGF-I metabolic effects.
The hypothalamic-pituitary-adrenal and the GH/IGF-I axis are also closely associated, as cortisol is required for physiological GH secretion. In states of hypercortisolemia (e.g., Cushing’s syndrome), decreased GH secretion with normal to elevated serum IGF-I levels have been reported (99, 100) (Table 1; Figure 3). English et al. (101) assessed serum IGF-I levels in patients with Cushing’s disease and any changes following post-operative remission compared to matched controls being investigated for suspected pituitary dysfunction (e.g., nonfunctioning pituitary adenomas). These investigators found that untreated Cushing’s disease patients had low GH and high IGF-I levels without clinical features of acromegaly, and after surgery with disease remission, serum IGF-I levels normalized. Magiakou et al. (100) demonstrated decreased 24-hour GH secretion despite the mean IGF-I SDS being +1.0 in patients with Cushing’s disease, and Bang et al. (102) reported elevated IGF-I levels in 40% of patients with pituitary and adrenal Cushing’s syndrome. Similar to obese patients, patients with Cushing’s syndrome also responded with supranormal IGF-I levels following administration of exogenous GH (95, 103). An interesting observation that deserves further exploration is the inherent ability of intra-portal insulin to regulate hepatic GH sensitivity that remains preserved despite prevailing changes in peripheral insulin resistance. This observation has been reported in earlier studies in obese and hyperinsulinemic subjects, and in patients with Cushing’s syndrome, in whom hepatic sensitivity to GH is increased when compared to lean subjects (95, 103). However, the exact molecular mechanisms responsible for this preserved effect remains unclear.
Further evidence of hypercortisolemia decreasing GH and increasing IGF-I comes from data of exogenous glucocorticoid therapy in humans. Excess glucocorticoid exposure suppresses GH secretion mainly through the increase in hypothalamic somatostatin tone, whereas low cortisol levels causes relative GHD that is reversible by glucocorticoid therapy (“Giustina’s effect”) (Figure 5) (104). Other mechanisms include increased insulin resistance induced by high-dose glucocorticoids that increases intra-portal insulin, decreases IGFBP-1 that increases free IGF-I, and direct glucocorticoid effects in increasing hepatic IGF-I synthesis and decreasing hepatic insulin clearance (105). Prummel et al. (106) demonstrated that prednisone therapy rapidly increased serum IGF-I levels; however, these levels promptly returned to baseline after prednisone discontinuation followed by suppressed GH levels. Ramshanker et al. (107) demonstrated that even though prednisolone increased serum total and bioactive IGF-I, post-IGF-IR signaling was inhibited indicating that prednisolone-induced increases in serum IGF-I levels is not accompanied by increased IGF-I action. This observation aligns with the findings by Unterman et al. (108), who utilized a porcine cartilage bioassay and demonstrated that glucocorticoid treatment in children decreased IGF-I activity. Finally, there are also data showing that even though serum IGF-I is acutely increased following prednisolone, this is not the case in lymph-like tissue fluid (107), suggesting that glucocorticoids exert distinct, compartment-specific effects on IGF-I action. Collectively, these findings raise the possibility that the mechanism of glucocorticoids increasing serum IGF-I levels is by directly inducing post-IGF-IR signaling resistance.
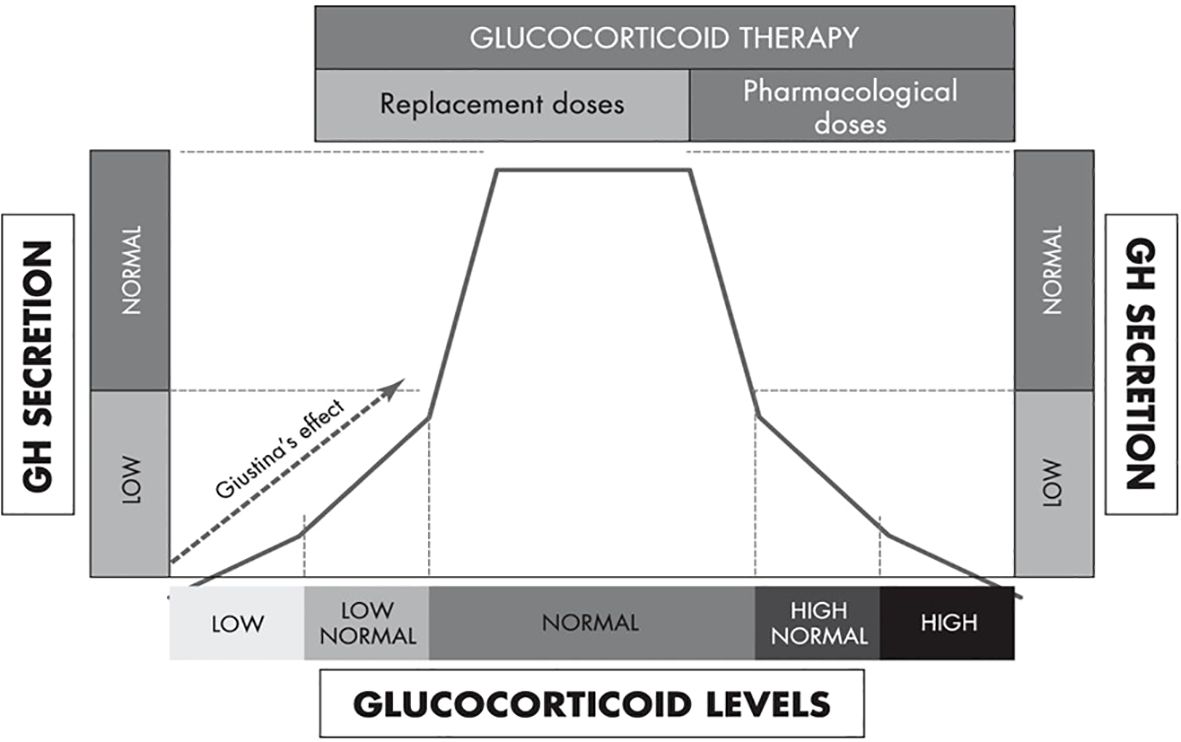
Figure 5. An integrated model of the biphasic, dose-dependent effect of glucocorticoids on GH secretion [adapted from Mazziotti et al. (104)].
Effects of weight loss
Short-term fasting is an important stimulator of GH secretion (109, 110) (Table 1; Figure 3). The increase in GH secretion precedes the reduction in IGF-I levels that is only evident by the third day of fasting, thereby ruling out the increase in GH during fasting is related to reductions in total IGF-I. However, when measuring free and bioactive IGF-I, it appears that serum levels decreased concomitantly with increases in IGFBP-1 and GH. Thus, during short-term fasting, changes in free and bioactive IGF-I rather than total IGF-I appear to be at least in part responsible for the increased GH secretion (35). Conversely, despite weight loss and concomitant improvements in insulin sensitivity, neither low caloric diets (111, 112), intermittent fasting (113), nor long-term calorie restriction (114) exerts any consistent inhibitory impact on serum IGF-I levels. One explanation for this may be that as long as the intake of protein is sufficient, serum IGF-I levels will remain relatively stable (115), i.e., the intake of calories per se does not alter serum IGF-I levels. In the last two decades, bariatric surgery has been highly successful in treating obesity (116), and changes in the GH/IGF-I axis has been extensively studied in this context. Changes in GH appear to be consistent, with several studies reporting increased GH levels following surgery (92–94), whereas the response of total IGF-I to bariatric surgery is more variable (117). We have reported that serum total IGF-I decreased 1 week and 3 months after bariatric surgery, whereas serum bioactive IGF-I remained unchanged during that period, and after 12 months, both variables returned to pre-surgery levels (117). The variability in total IGF-I may be due, in part, to major changes in selective IGFBPs during the post-bariatric surgery period, i.e. the proteolysis of IGFBP-3 (118). Another possible explanation may be the presence or absence of metabolic dysfunction-associated steatotic liver disease (MASLD), which can directly influence hepatic IGF-I generation.
Effects of MASLD, chronic liver disease and GLP-1RA therapy
Metabolic dysfunction-associated steatotic liver disease (MASLD) is highly prevalent in obese individuals with insulin resistance (119, 120). However, in contrast to simple obesity, where GH is low and IGF-I within normal range (7, 89), subjects with MASLD demonstrate variable changes in the GH/IGF-I axis depending on disease severity (121–123) (Table 1). In simple steatosis and early metabolic dysfunction-associated steatohepatitis (MASH), many of the body composition and metabolic features predisposing to MASLD/MASH contribute to decreased GH and IGF-I levels. With increasing hepatic dysfunction, the GH signaling cascade and hepatocyte IGF-I synthesis becomes impaired (124). When cirrhosis develops, a state of hepatic GH resistance ensues resulting in high GH and low IGF-I levels (125) (Figure 1E). Cianfarani et al. (126) and Dichtel et al. (127) have shown that circulating IGF-I levels are related to the histological stages of MASLD. In a recent study aimed at investigating IGF-IR and GHR physiology in patients with MASLD, Osganian et al. (128) demonstrated that GHR and IGF-IR expression hepatocytes were comparable across MASLD severity. However, the IGF-I gene expression decreased with increasing severity of MASLD, suggesting that reductions of hepatic IGF-I production in patients with MASLD may be related to the severity of the damaged hepatocytes. It is also possible that insulin resistance is the link between low GH levels and MASLD, as insulin directly suppresses pituitary GH secretion (29, 30). Furthermore, inflammation is a characteristic feature of MASLD (129). Thus, MASLD patients contrast to patients that have progressed to liver cirrhosis, as these patients are more susceptible to developing sarcopenia, a chronic catabolic state with increased energy expenditure, decreased appetite, low protein synthesis, and the development of ascites or portosystemic shunts that contribute to muscle atrophy (130). In these patients, when total hepatocyte mass decreases, IGF-I synthesis and glycogen stores decrease (131) resulting in low glucose and intra-portal insulin levels, and a state of hepatic GH resistance ensues. This inevitably results in low IGF-I and high GH that may stray into the acromegaly range (132, 133) and this catabolic state is comparable to prolonged fasting and malnutrition states (Figure 1E). Therefore, IGF-I deprivation contributes to the progressive malnutrition in chronic liver disease patients, increasing the vulnerability of the liver to an inflammatory and oxidative microenvironment that increases the risk of cirrhosis development.
Obesity is currently a growing public health concern (134) and as weight loss improves many obesity-related complications (135), there is a great deal of interest in developing weight-loss drugs. One of the most commonly studied weight-loss drugs, GLP-1RAs, were initially developed to treat T2D by reducing postprandial glucose excursions, augmenting glucose-dependent insulin release, inhibiting glucagon secretion and delaying gastric emptying (136, 137). However, GLP-1RAs are also effective in promoting weight loss by decreasing appetite and hunger, and increasing satiety (91). Heinla et al. (138) demonstrated that administration of a single dose short-acting GLP-1RA exenatide increased GH levels, whereas administration of long-acting GLP-1RA liraglutide daily for 21 days elicited increased GH levels with no changes in IGF-I levels in healthy subjects. Acute GLP-1RA infusion has been shown to decrease IGFBP-3 levels in humans, and this effect may explain the observed decrease in total IGF-1 levels (139). Alternatively, changes in GH and IGF-I levels may secondarily reflect the decrease in body weight following liraglutide treatment. Taken together these results provide further support that GLP-1RAs can alter the functioning of the GH/IGF-I axis.
Effects of GH misuse for recreational and athletic performance and GHRH analogue use in HIV lipodystrophy
The notion that GH enhances physical function due to its anabolic and lipolytic properties, albeit unproven, and difficulty of detection is what drives its misuse for recreational and athletic performance enhancement to confer a competitive advantage. Athletic performance depends on muscle strength and the energy required to increase muscle function. In recreational athletes, supraphysiological GH doses are often used either alone or in combination with other doping substances (140), and studies have shown that GH improves anaerobic sprint capacity, but not strength and endurance (141), and increases GH, IGF-I and fasting insulin levels (142, 143) mimicking those seen in acromegaly patients (Table 1).
Given the evidence that a pulsatile pattern of GH secretion is important for many of its physiological actions, a strategy to augment physiological pulsatile GH secretion without inducing the side-effects of supraphysiological GH doses has been studied in HIV lipodystrophy with excess VAT and decreased endogenous GH secretion (144, 145). Tesamorelin, a synthetic GHRH analog, that stimulates the synthesis and release of endogenous GH, is the first and, so far, only treatment indicated for the reduction of excess abdominal fat in HIV lipodystrophy (144). In a systematic review of 10 clinical trials in patients with HIV lipodystrophy, tesamorelin was effective in reducing VAT and increasing lean body mass and did not alter subcutaneous adipose tissue (146), and increased GH and IGF-I while insulin and glucose levels are unchanged indicating preservation of insulin sensitivity (147–150) (Table 1). However, this effect is transient and reversed upon discontinuation of therapy, and the long-term risk-benefit analysis of its administration is still unclear.
Learning lessons from the Ecuadorian Laron and Brazilian Itabaianinha cohorts
The state of severe GH excess and GHD exists on a spectrum from complete to partial. In states of mild GH excess and mild GHD with varying intra-portal insulin levels, the effects of insulin on the GH/IGF-I axis may not be too apparent. However, conditions caused by genetic mutations that affect growth are associated with a wide range of phenotypic abnormalities that may affect glucose handling, primarily due to diminished pancreatic β-cell mass and subsequent intra-portal hypoinsulinemia. The Ecuadorian Laron and Brazilian Itabaianinha cohorts with congenital mutations in the GH and GHRH receptors, respectively, both provide a unique and arguably the purest examples to evaluate the effects of chronic severe IGF-I and both severe GH and IGF-I deficiency, respectively, with severe hypoinsulinemia being the common denominator in both cohorts (151, 152) (Table 1; Figures 1F, 3). However, despite low intra-portal insulin levels, low IGF-I and increased fat mass and decreased lean body mass, their glucose tolerance and insulin sensitivity remains unaffected and in fact, is comparable to normal controls (151, 152). These cohorts serve as compelling examples of the lack of direct GH counter-regulatory effects on pancreatic β-cell insulin secretion, thus implying that circulating insulin exerts its glucose-lowering effects more efficiently in these subjects compared with healthy controls.
Conclusions
Changes in intra-portal insulin levels play an intricate yet permissive role in the regulation of the GH/IGF-I axis by either stimulating or suppressing hepatic GH sensitivity and hepatic IGF-I generation, probably via an insulin-mediated regulation of the hepatic GHR expression. Disease states that affect intra-portal insulin levels by increasing insulin resistance causing compensatory intra-portal hyperinsulinemia or by decreasing pancreatic β-cell insulin secretion causing intra-portal hypoinsulinemia results in changes to hepatic IGF-I synthesis and secretion, and consequently changes in GH levels via the negative feedback loop. Hence, changes in intra-portal insulin levels need to be taken into consideration to correctly interpret the alterations in the GH/IGF-I axis in healthy and disease states as this helps the clinician in formulating the decision-making process of when and how to perform appropriate diagnostic work-up and whether to offer treatment to improve any accompanying metabolic consequences.
For example, if a patient presents with elevated GH and IGF-I, it is important to rule out underlying impairment in renal function as elevated serum GH and IGF-I levels may also be present in patients with CKD, and not diagnose the patient as having acromegaly. For a patient with untreated Cushing’s disease or on high-dose glucocorticoid therapy, an elevated serum IGF-I may be observed, but levels decrease following remission or cessation of glucocorticoid therapy, respectively; hence mildly elevated IGF-I levels in these patients does not imply pathological GH excess. As for patients with GHD and concurrent diseases with intra-portal hypoinsulinemia (e.g., T1D and inflammatory diseases), higher GH doses are required to normalize IGF-I levels, which has cost implications. Conversely, in patients with anorexia nervosa, even supra-physiologic GH doses (mean maximum daily dose: 1.4 mg/day) may not be sufficient to normalize IGF-I levels (61) indicating the important role of hepatic GH sensitivity in successful IGF-I generation. In patients with obesity, there is no evidence for metabolic benefits of GH therapy in the absence of true GHD (153); instead, insulin resistance may worsen. Growth hormone secretion tends to return to normal after weight loss indicating that low GH is a consequence, not a cause, of central obesity (94), and therefore GH therapy should not be used to promote weight loss. As for individuals misusing GH for recreational and athletic performance-enhancing purposes, GH, IGF-I and insulin levels are increased suggesting that insulin sensitivity is worsened and “iatrogenic acromegaly” is induced, especially if used long-term. Conversely, for individuals with HIV lipodystrophy treated with tesamorelin, GH and IGF-I levels are increased but insulin sensitivity remains preserved.
While IGF-I remains the biomarker that best reflects endogenous GH secretion, when there is a discordancy with GH and IGF-I levels, it is important for the clinician to be mindful of the associated changes in intra-portal insulin delivery to the liver when interpreting these results in the context of health and when deciding treatment regimens in disease states. Finally, some commonly used drugs (e.g., glucocorticoids and GLP-1RAs) can also affect intra-portal insulin levels that modulate the GH/IGF-I axis either by increasing or decreasing hepatic GH sensitivity, as reflected by the discordant GH and IGF-I levels.
Author contributions
KY: Conceptualization, Writing – original draft, Writing – review & editing. RH: Writing – review & editing. AG: Writing – review & editing. DC: Supervision, Writing – review & editing. JF: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The authors declare financial support was received for the publication of this article, which was covered by a grant from the Jeppe Juhl and wife Ovita Juhl’s Memorial Trust (to JF). This research received no external funding.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Blum WF, Alherbish A, Alsagheir A, El Awwa A, Kaplan W, Koledova E, et al. The growth hormone-insulin-like growth factor-I axis in the diagnosis and treatment of growth disorders. Endocr Connect. (2018) 7:R212–22. doi: 10.1530/EC-18-0099
2. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. (2009) 30:152–77. doi: 10.1210/er.2008-0027
3. Olarescu NC, Gunawardane K, Hansen TK, Moller N, Jorgensen JOL. Normal physiology of growth hormone in adults. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc. (2000).
4. Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci U.S.A. (1999) 96:7088–92. doi: 10.1073/pnas.96.12.7088
5. Allard JB, Duan C. IGF-binding proteins: why do they exist and why are there so many? Front Endocrinol (Lausanne). (2018) 9:117. doi: 10.3389/fendo.2018.00117
6. Scott CD, Baxter RC. Synthesis of the acid-labile subunit of the growth-hormone-dependent insulin-like-growth-factor-binding protein complex by rat hepatocytes in culture. Biochem J. (1991) 275:441–6. doi: 10.1042/bj2750441
7. Frystyk J. Free insulin-like growth factors – measurements and relationships to growth hormone secretion and glucose homeostasis. Growth Horm IGF Res. (2004) 14:337–75. doi: 10.1016/j.ghir.2004.06.001
8. Guler HP, Zapf J, Schmid C, Froesch ER. Insulin-like growth factors I and II in healthy man. Estimations of half-lives and production rates. Acta Endocrinol (Copenh). (1989) 121:753–8. doi: 10.1530/acta.0.1210753
9. Ketha H, Singh RJ. Clinical assays for quantitation of insulin-like-growth-factor-1 (IGF1). Methods. (2015) 81:93–8. doi: 10.1016/j.ymeth.2015.04.029
10. Piccinini F, Polidori DC, Gower BA, Bergman RN. Hepatic but not extrahepatic insulin clearance is lower in African American than in European American women. Diabetes. (2017) 66:2564–70. doi: 10.2337/db17-0413
11. Hanaire-Broutin H, Sallerin-Caute B, Poncet MF, Tauber M, Bastide R, Chale JJ, et al. Effect of intraperitoneal insulin delivery on growth hormone binding protein, insulin-like growth factor (IGF)-I, and IGF-binding protein-3 in IDDM. Diabetologia. (1996) 39:1498–504. doi: 10.1007/s001250050604
12. Hedman CA, Frystyk J, Lindstrom T, Oskarsson P, Arnqvist HJ. Intraperitoneal insulin delivery to patients with type 1 diabetes results in higher serum IGF-I bioactivity than continuous subcutaneous insulin infusion. Clin Endocrinol (Oxf). (2014) 81:58–62. doi: 10.1111/cen.2014.81.issue-1
13. Shishko PI, Dreval AV, Abugova IA, Zajarny IU, Goncharov VC. Insulin-like growth factors and binding proteins in patients with recent-onset type 1 (insulin-dependent) diabetes mellitus: influence of diabetes control and intraportal insulin infusion. Diabetes Res Clin Pract. (1994) 25:1–12. doi: 10.1016/0168-8227(94)90155-4
14. van Dijk PR, Logtenberg SJ, Chisalita SI, Hedman CA, Groenier KH, Gans RO, et al. After 6 years of intraperitoneal insulin administration IGF-I concentrations in T1DM patients are at low-normal level. Growth Horm IGF Res. (2015) 25:316–9. doi: 10.1016/j.ghir.2015.08.007
15. van Dijk PR, Logtenberg SJ, Chisalita SI, Hedman CA, Groenier KH, Gans RO, et al. Different effects of intraperitoneal and subcutaneous insulin administration on the GH-IGF-1 axis in type 1 diabetes. J Clin Endocrinol Metab. (2016) 101:2493–501. doi: 10.1210/jc.2016-1473
16. Moller L, Dalman L, Norrelund H, Billestrup N, Frystyk J, Moller N, et al. Impact of fasting on growth hormone signaling and action in muscle and fat. J Clin Endocrinol Metab. (2009) 94:965–72. doi: 10.1210/jc.2008-1385
17. Caputo M, Pigni S, Agosti E, Daffara T, Ferrero A, Filigheddu N, et al. Regulation of GH and GH signaling by nutrients. Cells. (2021) 10. doi: 10.3390/cells10061376
18. Hartman ML, Faria AC, Vance ML, Johnson ML, Thorner MO, Veldhuis JD. Temporal structure of in vivo growth hormone secretory events in humans. Am J Physiol. (1991) 260:E101–110. doi: 10.1152/ajpendo.1991.260.1.E101
19. Ghigo MC, Torsello A, Grilli R, Luoni M, Guidi M, Cella SG, et al. Effects of GH and IGF-I administration on GHRH and somatostatin mRNA levels: I. A study on ad libitum fed and starved adult male rats. J Endocrinol Invest. (1997) 20:144–50. doi: 10.1007/BF03346893
20. Bertherat J, Bluet-Pajot MT, Epelbaum J. Neuroendocrine regulation of growth hormone. Eur J Endocrinol. (1995) 132:12–24. doi: 10.1530/eje.0.1320012
21. Bonert V, Carmichael J, Wu Z, Mirocha J, Perez DA, Clarke NJ, et al. Discordance between mass spectrometry and immunometric IGF-1 assay in pituitary disease: a prospective study. Pituitary. (2018) 21:65–75. doi: 10.1007/s11102-017-0849-z
22. Blum WF, Albertsson-Wikland K, Rosberg S, Ranke MB. Serum levels of insulin-like growth factor I (IGF-I) and IGF binding protein 3 reflect spontaneous growth hormone secretion. J Clin Endocrinol Metab. (1993) 76:1610–6. doi: 10.1210/jcem.76.6.7684744
23. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. (1995) 16:3–34. doi: 10.1210/edrv-16-1-3
24. Marzullo P, Di Somma C, Pratt KL, Khosravi J, Diamandis A, Lombardi G, et al. Usefulness of different biochemical markers of the insulin-like growth factor (IGF) family in diagnosing growth hormone excess and deficiency in adults. J Clin Endocrinol Metab. (2001) 86:3001–8. doi: 10.1210/jcem.86.7.7628
25. Cunha SR, Mayo KE. Ghrelin and growth hormone (GH) secretagogues potentiate GH-releasing hormone (GHRH)-induced cyclic adenosine 3’,5’-monophosphate production in cells expressing transfected GHRH and GH secretagogue receptors. Endocrinology. (2002) 143:4570–82. doi: 10.1210/en.2002-220670
26. Fernandez-Perez L, Guerra B, Diaz-Chico JC, Flores-Morales A. Estrogens regulate the hepatic effects of growth hormone, a hormonal interplay with multiple fates. Front Endocrinol (Lausanne). (2013) 4:66. doi: 10.3389/fendo.2013.00066
27. Anderson SM, Shah N, Evans WS, Patrie JT, Bowers CY, Veldhuis JD. Short-term estradiol supplementation augments growth hormone (GH) secretory responsiveness to dose-varying GH-releasing peptide infusions in healthy postmenopausal women. J Clin Endocrinol Metab. (2001) 86:551–60. doi: 10.1210/jcem.86.2.7240
28. Birzniece V, Ho KKY. Sex steroids and the GH axis: Implications for the management of hypopituitarism. Best Pract Res Clin Endocrinol Metab. (2017) 31:59–69. doi: 10.1016/j.beem.2017.03.003
29. Gahete MD, Cordoba-Chacon J, Lin Q, Bruning JC, Kahn CR, Castano JP, et al. Insulin and IGF-I inhibit GH synthesis and release in vitro and in vivo by separate mechanisms. Endocrinology. (2013) 154:2410–20. doi: 10.1210/en.2013-1261
30. Melmed S. Insulin suppresses growth hormone secretion by rat pituitary cells. J Clin Invest. (1984) 73:1425–33. doi: 10.1172/JCI111347
31. Leung KC, Doyle N, Ballesteros M, Waters MJ, Ho KK. Insulin regulation of human hepatic growth hormone receptors: divergent effects on biosynthesis and surface translocation. J Clin Endocrinol Metab. (2000) 85:4712–20. doi: 10.1210/jc.85.12.4712
32. Chen NY, Chen WY, Kopchick JJ. Liver and kidney growth hormone (GH) receptors are regulated differently in diabetic GH and GH antagonist transgenic mice. Endocrinology. (1997) 138:1988–94. doi: 10.1210/endo.138.5.5123
33. Baxter RC, Turtle JR. Regulation of hepatic growth hormone receptors by insulin. Biochem Biophys Res Commun. (1978) 84:350–7. doi: 10.1016/0006-291X(78)90177-8
34. Conover CA, Lee PD, Kanaley JA, Clarkson JT, Jensen MD. Insulin regulation of insulin-like growth factor binding protein-1 in obese and nonobese humans. J Clin Endocrinol Metab. (1992) 74:1355–60. doi: 10.1210/jcem.74.6.1375600
35. Chen JW, Hojlund K, Beck-Nielsen H, Sandahl Christiansen J, Orskov H, Frystyk J. Free rather than total circulating insulin-like growth factor-I determines the feedback on growth hormone release in normal subjects. J Clin Endocrinol Metab. (2005) 90:366–71. doi: 10.1210/jc.2004-0039
36. Sakai K, D’Ercole AJ, Murphy LJ, Clemmons DR. Physiological differences in insulin-like growth factor binding protein-1 (IGFBP-1) phosphorylation in IGFBP-1 transgenic mice. Diabetes. (2001) 50:32–8. doi: 10.2337/diabetes.50.1.32
37. Frystyk J, Grofte T, Skjaerbaek C, Orskov H. The effect of oral glucose on serum free insulin-like growth factor-I and -II in health adults. J Clin Endocrinol Metab. (1997) 82:3124–7. doi: 10.1210/jcem.82.9.4259
38. Frystyk J, Nyholm B, Skjaerbaek C, Baxter RC, Schmitz O, Orskov H. The circulating IGF system and its relationship with 24-h glucose regulation and insulin sensitivity in healthy subjects. Clin Endocrinol (Oxf). (2003) 58:777–84. doi: 10.1046/j.1365-2265.2003.01791.x
39. Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. (1997) 18:801–31. doi: 10.1210/edrv.18.6.0321
40. Ivarsen P, Chen JW, Tietze I, Christiansen JS, Flyvbjerg A, Frystyk J. Marked reductions in bioactive insulin-like growth factor I (IGF-I) during hemodialysis. Growth Horm IGF Res. (2010) 20:156–61. doi: 10.1016/j.ghir.2009.12.001
41. Nijenhuis-Noort EC, Berk KA, Neggers S, Lely AJV. The fascinating interplay between growth hormone, insulin-like growth factor-1, and insulin. Endocrinol Metab (Seoul). (2024) 39:83–9. doi: 10.3803/EnM.2024.101
42. Smith WJ, Underwood LE, Clemmons DR. Effects of caloric or protein restriction on insulin-like growth factor-I (IGF-I) and IGF-binding proteins in children and adults. J Clin Endocrinol Metab. (1995) 80:443–9. doi: 10.1210/jcem.80.2.7531712
43. Bereket A, Lang CH, Blethen SL, Gelato MC, Fan J, Frost RA, et al. Effect of insulin on the insulin-like growth factor system in children with new-onset insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. (1995) 80:1312–7. doi: 10.1210/jcem.80.4.7536205
44. Bereket A, Wilson TA, Blethen SL, Sakurai Y, Herndon DN, Wolfe RR, et al. Regulation of the acid-labile subunit of the insulin-like growth factor ternary complex in patients with insulin-dependent diabetes mellitus and severe burns. Clin Endocrinol (Oxf). (1996) 44:525–32. doi: 10.1046/j.1365-2265.1996.726547.x
45. Attia N, Caprio S, Jones TW, Heptulla R, Holcombe J, Silver D, et al. Changes in free insulin-like growth factor-1 and leptin concentrations during acute metabolic decompensation in insulin withdrawn patients with type 1 diabetes. J Clin Endocrinol Metab. (1999) 84:2324–8. doi: 10.1210/jcem.84.7.5861
46. Frystyk J, Ritzel RA, Maubach J, Busing M, Luck R, Klempnauer J, et al. Comparison of pancreas-transplanted type 1 diabetic patients with portal-venous versus systemic-venous graft drainage: impact on glucose regulatory hormones and the growth hormone/insulin-like growth factor-I axis. J Clin Endocrinol Metab. (2008) 93:1758–66. doi: 10.1210/jc.2007-2350
47. Hedman CA, Frystyk J, Lindstrom T, Chen JW, Flyvbjerg A, Orskov H, et al. Residual beta-cell function more than glycemic control determines abnormalities of the insulin-like growth factor system in type 1 diabetes. J Clin Endocrinol Metab. (2004) 89:6305–9. doi: 10.1210/jc.2004-0572
48. Sorensen JS, Birkebaek NH, Bjerre M, Pociot F, Kristensen K, Hoejberg AS, et al. Residual beta-cell function and the insulin-like growth factor system in Danish children and adolescents with type 1 diabetes. J Clin Endocrinol Metab. (2015) 100:1053–61. doi: 10.1210/jc.2014-3521
49. Ma Z, Christiansen JS, Laursen T, Lauritzen T, Frystyk J. Short-term effects of NPH insulin, insulin detemir, and insulin glargine on the GH-IGF1-IGFBP axis in patients with type 1 diabetes. Eur J Endocrinol. (2014) 171:471–9. doi: 10.1530/EJE-14-0258
50. Drogan D, Schulze MB, Boeing H, Pischon T. Insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in relation to the risk of type 2 diabetes mellitus: results from the EPIC-potsdam study. Am J Epidemiol. (2016) 183:553–60. doi: 10.1093/aje/kwv188
51. Acerini CL, Harris DA, Matyka KA, Watts AP, Umpleby AM, Russell-Jones DL, et al. Effects of low-dose recombinant human insulin-like growth factor-I on insulin sensitivity, growth hormone and glucagon levels in young adults with insulin-dependent diabetes mellitus. Metabolism. (1998) 47:1481–9. doi: 10.1016/S0026-0495(98)90074-9
52. Acerini CL, Patton CM, Savage MO, Kernell A, Westphal O, Dunger DB. Randomised placebo-controlled trial of human recombinant insulin-like growth factor I plus intensive insulin therapy in adolescents with insulin-dependent diabetes mellitus. Lancet. (1997) 350:1199–204. doi: 10.1016/S0140-6736(97)06467-2
53. Clemmons DR, Moses AC, Sommer A, Jacobson W, Rogol AD, Sleevi MR, et al. Rh/IGF-I/rhIGFBP-3 administration to patients with type 2 diabetes mellitus reduces insulin requirements while also lowering fasting glucose. Growth Horm IGF Res. (2005) 15:265–74. doi: 10.1016/j.ghir.2005.05.002
54. Saukkonen T, Amin R, Williams RM, Fox C, Yuen KC, White MA, et al. Dose-dependent effects of recombinant human insulin-like growth factor (IGF)-I/IGF binding protein-3 complex on overnight growth hormone secretion and insulin sensitivity in type 1 diabetes. J Clin Endocrinol Metab. (2004) 89:4634–41. doi: 10.1210/jc.2004-0243
55. Saukkonen T, Shojaee-Moradie F, Williams RM, Amin R, Yuen KC, Watts A, et al. Effects of recombinant human IGF-I/IGF-binding protein-3 complex on glucose and glycerol metabolism in type 1 diabetes. Diabetes. (2006) 55:2365–70. doi: 10.2337/db05-1646
56. Stoving RK, Chen JW, Glintborg D, Brixen K, Flyvbjerg A, Horder K, et al. Bioactive insulin-like growth factor (IGF) I and IGF-binding protein-1 in anorexia nervosa. J Clin Endocrinol Metab. (2007) 92:2323–9. doi: 10.1210/jc.2006-1926
57. Misra M, Miller KK, Bjornson J, Hackman A, Aggarwal A, Chung J, et al. Alterations in growth hormone secretory dynamics in adolescent girls with anorexia nervosa and effects on bone metabolism. J Clin Endocrinol Metab. (2003) 88:5615–23. doi: 10.1210/jc.2003-030532
58. Stoving RK, Veldhuis JD, Flyvbjerg A, Vinten J, Hangaard J, Koldkjaer OG, et al. Jointly amplified basal and pulsatile growth hormone (GH) secretion and increased process irregularity in women with anorexia nervosa: indirect evidence for disruption of feedback regulation within the GH-insulin-like growth factor I axis. J Clin Endocrinol Metab. (1999) 84:2056–63. doi: 10.1210/jc.2006-1926
59. Counts DR, Gwirtsman H, Carlsson LM, Lesem M, Cutler GB Jr. The effect of anorexia nervosa and refeeding on growth hormone-binding protein, the insulin-like growth factors (IGFs), and the IGF-binding proteins. J Clin Endocrinol Metab. (1992) 75:762–7. doi: 10.1210/jcem.75.3.1381372
60. Baxter RC, Bryson JM, Turtle JR. The effect of fasting on liver receptors for prolactin and growth hormone. Metabolism. (1981) 30:1086–90. doi: 10.1016/0026-0495(81)90052-4
61. Fazeli PK, Lawson EA, Prabhakaran R, Miller KK, Donoho DA, Clemmons DR, et al. Effects of recombinant human growth hormone in anorexia nervosa: a randomized, placebo-controlled study. J Clin Endocrinol Metab. (2010) 95:4889–97. doi: 10.1210/jc.2010-0493
62. Hotta M, Fukuda I, Sato K, Hizuka N, Shibasaki T, Takano K. The relationship between bone turnover and body weight, serum insulin-like growth factor (IGF) I, and serum IGF-binding protein levels in patients with anorexia nervosa. J Clin Endocrinol Metab. (2000) 85:200–6. doi: 10.1210/jcem.85.1.6321
63. Fazeli PK, Klibanski A. Determinants of GH resistance in malnutrition. J Endocrinol. (2014) 220:R57–65. doi: 10.1530/JOE-13-0477
64. Zhao Y, Xiao X, Frank SJ, Lin HY, Xia Y. Distinct mechanisms of induction of hepatic growth hormone resistance by endogenous IL-6, TNF-alpha, and IL-1beta. Am J Physiol Endocrinol Metab. (2014) 307:E186–198. doi: 10.1152/ajpendo.00652.2013
65. Hjortebjerg R, Thomsen KL, Agnholt J, Frystyk J. The IGF system in patients with inflammatory bowel disease treated with prednisolone or infliximab: potential role of the stanniocalcin-2/PAPP-A/IGFBP-4 axis. BMC Gastroenterol. (2019) 19:83. doi: 10.1186/s12876-019-1000-6
66. Fleseriu M, Langlois F, Lim DST, Varlamov EV, Melmed S. Acromegaly: pathogenesis, diagnosis, and management. Lancet Diabetes Endocrinol. (2022) 10:804–26. doi: 10.1016/S2213-8587(22)00244-3
67. Lanning NJ, Carter-Su C. Recent advances in growth hormone signaling. Rev Endocr Metab Disord. (2006) 7:225–35. doi: 10.1007/s11154-007-9025-5
68. Sharma R, Kopchick JJ, Puri V, Sharma VM. Effect of growth hormone on insulin signaling. Mol Cell Endocrinol. (2020) 518:111038. doi: 10.1016/j.mce.2020.111038
69. Coopmans EC, Berk KAC, El-Sayed N, Neggers S, van der Lely AJ. Eucaloric very-low-carbohydrate ketogenic diet in acromegaly treatment. N Engl J Med. (2020) 382:2161–2. doi: 10.1056/NEJMc1915808
70. Janssen J. Mechanisms of putative IGF-I receptor resistance in active acromegaly. Growth Horm IGF Res. (2020) 52:101319. doi: 10.1016/j.ghir.2020.101319
71. Snyder DK, Clemmons DR, Underwood LE. Dietary carbohydrate content determines responsiveness to growth hormone in energy-restricted humans. J Clin Endocrinol Metab. (1989) 69:745–52. doi: 10.1210/jcem-69-4-745
72. Hakuno F, Takahashi SI. IGF1 receptor signaling pathways. J Mol Endocrinol. (2018) 61:T69–86. doi: 10.1530/JME-17-0311
73. Haffner D, Schaefer F, Girard J, Ritz E, Mehls O. Metabolic clearance of recombinant human growth hormone in health and chronic renal failure. J Clin Invest. (1994) 93:1163–71. doi: 10.1172/JCI117069
74. Rabkin R, Sun DF, Chen Y, Tan J, Schaefer F. Growth hormone resistance in uremia, a role for impaired JAK/STAT signaling. Pediatr Nephrol. (2005) 20:313–8. doi: 10.1007/s00467-004-1713-8
75. Oh Y. The insulin-like growth factor system in chronic kidney disease: Pathophysiology and therapeutic opportunities. Kidney Res Clin Pract. (2012) 31:26–37. doi: 10.1016/j.krcp.2011.12.005
76. Tonshoff B, Veldhuis JD, Heinrich U, Mehls O. Deconvolution analysis of spontaneous nocturnal growth hormone secretion in prepubertal children with preterminal chronic renal failure and with end-stage renal disease. Pediatr Res. (1995) 37:86–93. doi: 10.1203/00006450-199501000-00017
77. Kamenicky P, Mazziotti G, Lombes M, Giustina A, Chanson P. Growth hormone, insulin-like growth factor-1, and the kidney: pathophysiological and clinical implications. Endocr Rev. (2014) 35:234–81. doi: 10.1210/er.2013-1071
78. Frystyk J, Ivarsen P, Skjaerbaek C, Flyvbjerg A, Pedersen EB, Orskov H. Serum-free insulin-like growth factor I correlates with clearance in patients with chronic renal failure. Kidney Int. (1999) 56:2076–84. doi: 10.1046/j.1523-1755.1999.00798.x
79. Frystyk J, Djurhuus CB, Johansen T, Lange M, Smidt K, Christiansen JS. Measurement of free GH and bioactive IGF-I in non-diabetic haemodialysis patients treated with GH for 7 days. Nephrol Dial Transplant. (2012) 27:4211–8. doi: 10.1093/ndt/gfs364
80. Rahhal MN, Gharaibeh NE, Rahimi L, Ismail-Beigi F. Disturbances in insulin-glucose metabolism in patients with advanced renal disease with and without diabetes. J Clin Endocrinol Metab. (2019) 104:4949–66. doi: 10.1210/jc.2019-00286
81. Challa A, Chan W, Krieg RJ Jr., Thabet MA, Liu F, Hintz RL, et al. Effect of metabolic acidosis on the expression of insulin-like growth factor and growth hormone receptor. Kidney Int. (1993) 44:1224–7. doi: 10.1038/ki.1993.372
82. Yuen KCJ, Biller BMK, Radovick S, Carmichael JD, Jasim S, Pantalone KM, et al. American association of clinical endocrinologists and American college of endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract. (2019) 25:1191–232. doi: 10.4158/GL-2019-0405
83. Vijayakumar A, Yakar S, Leroith D. The intricate role of growth hormone in metabolism. Front Endocrinol (Lausanne). (2011) 2:32. doi: 10.3389/fendo.2011.00032
84. Garmes HM, Castillo AR, Monte Alegre S, de Souza AL, Atala YB, Zantut-Wittmann DE. Childhood-onset GH deficiency versus adult-onset GH deficiency: relevant differences regarding insulin sensitivity. Metabolites. (2022) 12. doi: 10.3390/metabo12121251
85. Aimaretti G, Corneli G, Baldelli R, Di Somma C, Gasco V, Durante C, et al. Diagnostic reliability of a single IGF-I measurement in 237 adults with total anterior hypopituitarism and severe GH deficiency. Clin Endocrinol (Oxf). (2003) 59:56–61. doi: 10.1046/j.1365-2265.2003.01794.x
86. Hilding A, Hall K, Wivall-Helleryd IL, Saaf M, Melin AL, Thoren M. Serum levels of insulin-like growth factor I in 152 patients with growth hormone deficiency, aged 19-82 years, in relation to those in healthy subjects. J Clin Endocrinol Metab. (1999) 84:2013–9. doi: 10.1210/jcem.84.6.5793
87. Iranmanesh A, Lizarralde G, Veldhuis JD. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab. (1991) 73:1081–8. doi: 10.1210/jcem-73-5-1081
88. Veldhuis JD, Iranmanesh A, Ho KK, Waters MJ, Johnson ML, Lizarralde G. Dual defects in pulsatile growth hormone secretion and clearance subserve the hyposomatotropism of obesity in man. J Clin Endocrinol Metab. (1991) 72:51–9. doi: 10.1210/jcem-72-1-51
89. Frystyk J, Brick DJ, Gerweck AV, Utz AL, Miller KK. Bioactive insulin-like growth factor-I in obesity. J Clin Endocrinol Metab. (2009) 94:3093–7. doi: 10.1210/jc.2009-0614
90. Mittempergher F, Pata G, Crea N, Di Betta E, Vilardi A, Chiesa D, et al. Preoperative prediction of growth hormone (GH)/insulin-like growth factor-1 (IGF-1) axis modification and postoperative changes in candidates for bariatric surgery. Obes Surg. (2013) 23:594–601. doi: 10.1007/s11695-012-0820-z
91. Aldawsari M, Almadani FA, Almuhammadi N, Algabsani S, Alamro Y, Aldhwayan M. The efficacy of GLP-1 analogues on appetite parameters, gastric emptying, food preference and taste among adults with obesity: systematic review of randomized controlled trials. Diabetes Metab Syndr Obes. (2023) 16:575–95. doi: 10.2147/DMSO.S387116
92. Juiz-Valina P, Pena-Bello L, Cordido M, Outeirino-Blanco E, Pertega S, Varela-Rodriguez B, et al. Altered GH-IGF-1 axis in severe obese subjects is reversed after bariatric surgery-induced weight loss and related with low-grade chronic inflammation. J Clin Med. (2020) 9. doi: 10.3390/jcm9082614
93. Li X, Zhou B, Wu Z, Li Y, Meng H. Role of growth hormone in adipose tissue insulin resistance amelioration after bariatric surgery in adults with obesity. Metab Syndr Relat Disord. (2023) 21:345–52. doi: 10.1089/met.2023.0034
94. Rasmussen MH, Hvidberg A, Juul A, Main KM, Gotfredsen A, Skakkebaek NE, et al. Massive weight loss restores 24-hour growth hormone release profiles and serum insulin-like growth factor-I levels in obese subjects. J Clin Endocrinol Metab. (1995) 80:1407–15. doi: 10.1210/jcem.80.4.7536210
95. Gleeson HK, Lissett CA, Shalet SM. Insulin-like growth factor-I response to a single bolus of growth hormone is increased in obesity. J Clin Endocrinol Metab. (2005) 90:1061–7. doi: 10.1210/jc.2004-0501
96. Cordido F, Peino R, Penalva A, Alvarez CV, Casanueva FF, Dieguez C. Impaired growth hormone secretion in obese subjects is partially reversed by acipimox-mediated plasma free fatty acid depression. J Clin Endocrinol Metab. (1996) 81:914–8. doi: 10.1210/jcem.81.3.8772550
97. Lee JH, Fang C, Li X, Wu CS, Noh JY, Ye X, et al. GHS-R suppression in adipose tissues protects against obesity and insulin resistance by regulating adipose angiogenesis and fibrosis. Int J Obes (Lond). (2021) 45:1565–75. doi: 10.1038/s41366-021-00820-7
98. Clemmons DR. Metabolic actions of insulin-like growth factor-I in normal physiology and diabetes. Endocrinol Metab Clin North Am. (2012) 41:425–443, vii-viii. doi: 10.1016/j.ecl.2012.04.017
99. Hotta M, Shibasaki T, Masuda A, Imaki T, Sugino N, Demura H, et al. Effect of human growth hormone-releasing hormone on GH secretion in Cushing’s syndrome and non-endocrine disease patients treated with glucocorticoids. Life Sci. (1988) 42:979–84. doi: 10.1016/0024-3205(88)90427-4
100. Magiakou MA, Mastorakos G, Gomez MT, Rose SR, Chrousos GP. Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing’s disease before and after surgical cure. J Clin Endocrinol Metab. (1994) 78:131–7. doi: 10.1210/jcem.78.1.7507118
101. English K, Chikani V, Dimeski G, Inder WJ. Elevated insulin-like growth factor-1 in Cushing’s disease. Clin Endocrinol (Oxf). (2019) 91:141–7. doi: 10.1111/cen.2019.91.issue-1
102. Bang P, Degerblad M, Thoren M, Schwander J, Blum W, Hall K. Insulin-like growth factor (IGF) I and II and IGF binding protein (IGFBP) 1, 2 and 3 in serum from patients with Cushing’s syndrome. Acta Endocrinol (Copenh). (1993) 128:397–404.
103. Maccario M, Tassone F, Gauna C, Oleandri SE, Aimaretti G, Procopio M, et al. Effects of short-term administration of low-dose rhGH on IGF-I levels in obesity and Cushing’s syndrome: indirect evaluation of sensitivity to GH. Eur J Endocrinol. (2001) 144:251–6. doi: 10.1530/eje.0.1440251
104. Mazziotti G, Giustina A. Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol. (2013) 9:265–76. doi: 10.1038/nrendo.2013.5
105. Asakawa K, Takano K, Kogawa M, Hasumi Y, Shizume K. Effects of glucocorticoid on body growth and serum levels of somatomedin A in the rat. Acta Endocrinol (Copenh). (1982) 100:206–10. doi: 10.1530/acta.0.1000206
106. Prummel MF, Wiersinga WM, Oosting H, Endert E. The effect of long-term prednisone treatment on growth hormone and insulin-like growth factor-1. J Endocrinol Invest. (1996) 19:620–3. doi: 10.1007/BF03349028
107. Ramshanker N, Aagaard M, Hjortebjerg R, Voss TS, Moller N, Jorgensen JOL, et al. Effects of prednisolone on serum and tissue fluid IGF-I receptor activation and post-receptor signaling in humans. J Clin Endocrinol Metab. (2017) 102:4031–40. doi: 10.1210/jc.2017-00696
108. Unterman TG, Phillips LS. Glucocorticoid effects on somatomedins and somatomedin inhibitors. J Clin Endocrinol Metab. (1985) 61:618–26. doi: 10.1210/jcem-61-4-618
109. Ho KY, Veldhuis JD, Johnson ML, Furlanetto R, Evans WS, Alberti KG, et al. Fasting enhances growth hormone secretion and amplifies the complex rhythms of growth hormone secretion in man. J Clin Invest. (1988) 81:968–75. doi: 10.1172/JCI113450
110. Norrelund H, Frystyk J, Jorgensen JO, Moller N, Christiansen JS, Orskov H, et al. The effect of growth hormone on the insulin-like growth factor system during fasting. J Clin Endocrinol Metab. (2003) 88:3292–8. doi: 10.1210/jc.2002-021983
111. Belobrajdic DP, Frystyk J, Jeyaratnaganthan N, Espelund U, Flyvbjerg A, Clifton PM, et al. Moderate energy restriction-induced weight loss affects circulating IGF levels independent of dietary composition. Eur J Endocrinol. (2010) 162:1075–82. doi: 10.1530/EJE-10-0062
112. Espelund U, Bruun JM, Richelsen B, Flyvbjerg A, Frystyk J. Pro- and mature IGF-II during diet-induced weight loss in obese subjects. Eur J Endocrinol. (2005) 153:861–9. doi: 10.1530/eje.1.02028
113. Mattson MP, Longo VD, Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev. (2017) 39:46–58. doi: 10.1016/j.arr.2016.10.005
114. Fontana L, Villareal DT, Das SK, Smith SR, Meydani SN, Pittas AG, et al. Effects of 2-year calorie restriction on circulating levels of IGF-1, IGF-binding proteins and cortisol in nonobese men and women: a randomized clinical trial. Aging Cell. (2016) 15:22–7. doi: 10.1111/acel.2016.15.issue-1
115. Fontana L, Weiss EP, Villareal DT, Klein S, Holloszy JO. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell. (2008) 7:681–7. doi: 10.1111/j.1474-9726.2008.00417.x
116. Wolfe BM, Kvach E, Eckel RH. Treatment of obesity: weight loss and bariatric surgery. Circ Res. (2016) 118:1844–55. doi: 10.1161/CIRCRESAHA.116.307591
117. Hjortebjerg R, Bojsen-Moller KN, Soeby M, Oxvig C, Madsbad S, Frystyk J. Metabolic improvement after gastric bypass correlates with changes in IGF-regulatory proteins stanniocalcin-2 and IGFBP-4. Metabolism. (2021) 124:154886. doi: 10.1016/j.metabol.2021.154886
118. Skjaerbaek C, Frystyk J, Orskov H, Kissmeyer-Nielsen P, Jensen MB, Laurberg S, et al. Differential changes in free and total insulin-like growth factor I after major, elective abdominal surgery: the possible role of insulin-like growth factor-binding protein-3 proteolysis. J Clin Endocrinol Metab. (1998) 83:2445–9. doi: 10.1210/jcem.83.7.4966
119. Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. (2010) 28:155–61. doi: 10.1159/000282080
120. Rhee EJ, Lee WY, Cho YK, Kim BI, Sung KC. Hyperinsulinemia and the development of nonalcoholic Fatty liver disease in nondiabetic adults. Am J Med. (2011) 124:69–76. doi: 10.1016/j.amjmed.2010.08.012
121. Ichikawa T, Nakao K, Hamasaki K, Furukawa R, Tsuruta S, Ueda Y, et al. Role of growth hormone, insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 in development of non-alcoholic fatty liver disease. Hepatol Int. (2007) 1:287–94. doi: 10.1007/s12072-007-9007-4
122. Xu L, Xu C, Yu C, Miao M, Zhang X, Zhu Z, et al. Association between serum growth hormone levels and nonalcoholic fatty liver disease: a cross-sectional study. PloS One. (2012) 7:e44136. doi: 10.1371/journal.pone.0044136
123. Yao Y, Miao X, Zhu D, Li D, Zhang Y, Song C, et al. Insulin-like growth factor-1 and non-alcoholic fatty liver disease: a systemic review and meta-analysis. Endocrine. (2019) 65:227–37. doi: 10.1007/s12020-019-01982-1
124. Carotti S, Guarino MPL, Valentini F, Porzio S, Vespasiani-Gentilucci U, Perrone G, et al. Impairment of GH/IGF-1 axis in the liver of patients with HCV-related chronic hepatitis. Horm Metab Res. (2018) 50:145–51. doi: 10.1055/s-0043-118911
125. Ma IL, Stanley TL. Growth hormone and nonalcoholic fatty liver disease. Immunometabolism (Cobham). (2023) 5:e00030. doi: 10.1097/IN9.0000000000000030
126. Cianfarani S, Inzaghi E, Alisi A, Germani D, Puglianiello A, Nobili V. Insulin-like growth factor-I and -II levels are associated with the progression of nonalcoholic fatty liver disease in obese children. J Pediatr. (2014) 165:92–8. doi: 10.1016/j.jpeds.2014.01.052
127. Dichtel LE, Corey KE, Misdraji J, Bredella MA, Schorr M, Osganian SA, et al. The association between IGF-1 levels and the histologic severity of nonalcoholic fatty liver disease. Clin Transl Gastroenterol. (2017) 8:e217. doi: 10.1038/ctg.2016.72
128. Osganian SA, Subudhi S, Masia R, Drescher HK, Bartsch LM, Chicote ML, et al. Expression of IGF-1 receptor and GH receptor in hepatic tissue of patients with nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Growth Horm IGF Res. (2022) 65:101482. doi: 10.1016/j.ghir.2022.101482
129. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci. (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
130. Marasco G, Dajti E, Ravaioli F, Brocchi S, Rossini B, Alemanni LV, et al. Clinical impact of sarcopenia assessment in patients with liver cirrhosis. Expert Rev Gastroenterol Hepatol. (2021) 15:377–88. doi: 10.1080/17474124.2021.1848542
131. Yen FS, Hou MC, Liu JS, Hsu CC, Hwu CM. Severe hypoglycemia in patients with liver cirrhosis and type 2 diabetes. Front Med (Lausanne). (2022) 9:962337. doi: 10.3389/fmed.2022.962337
132. Moller S, Becker U. Insulin-like growth factor 1 and growth hormone in chronic liver disease. Dig Dis. (1992) 10:239–48. doi: 10.1159/000171362
133. Muggeo M, Tiengo A, Fedele D, Crepaldi G. Altered control of growth hormone secretion in patients with cirrhosis of the liver. Arch Intern Med. (1979) 139:1157–60. doi: 10.1001/archinte.1979.03630470065020
134. Dai H, Alsalhe TA, Chalghaf N, Ricco M, Bragazzi NL, Wu J. The global burden of disease attributable to high body mass index in 195 countries and territories, 1990-2017: An analysis of the Global Burden of Disease Study. PloS Med. (2020) 17:e1003198. doi: 10.1371/journal.pmed.1003198
135. Ryan DH, Yockey SR. Weight loss and improvement in comorbidity: differences at 5%, 10%, 15%, and over. Curr Obes Rep. (2017) 6:187–94. doi: 10.1007/s13679-017-0262-y
136. Marathe CS, Pham H, Wu T, Trahair LG, Rigda RS, Buttfield MDM, et al. Acute administration of the GLP-1 receptor agonist lixisenatide diminishes postprandial insulin secretion in healthy subjects but not in type 2 diabetes, associated with slowing of gastric emptying. Diabetes Ther. (2022) 13:1245–9. doi: 10.1007/s13300-022-01258-4
137. Muller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
138. Heinla K, Vasar E, Reppo I, Sedman T, Volke V. GLP-1 receptor agonists induce growth hormone secretion in healthy volunteers. Diabetes Ther. (2023) 14:777–86. doi: 10.1007/s13300-023-01381-w
139. Skov J, Frystyk J, Christiansen JS. GLP-1 infusion reduces IGFBP-1 serum level in humans. Growth Horm IGF Res. (2014) 24:67–70. doi: 10.1016/j.ghir.2014.01.003
140. Liu H, Bravata DM, Olkin I, Friedlander A, Liu V, Roberts B, et al. Systematic review: the effects of growth hormone on athletic performance. Ann Intern Med. (2008) 148:747–58. doi: 10.7326/0003-4819-148-10-200805200-00215
141. Holt RIG, Ho KKY. The use and abuse of growth hormone in sports. Endocr Rev. (2019) 40:1163–85. doi: 10.1210/er.2018-00265
142. Deyssig R, Frisch H, Blum WF, Waldhor T. Effect of growth hormone treatment on hormonal parameters, body composition and strength in athletes. Acta Endocrinol (Copenh). (1993) 128:313–8. doi: 10.1530/acta.0.1280313
143. Meinhardt U, Nelson AE, Hansen JL, Birzniece V, Clifford D, Leung KC, et al. The effects of growth hormone on body composition and physical performance in recreational athletes: a randomized trial. Ann Intern Med. (2010) 152:568–77. doi: 10.7326/0003-4819-152-9-201005040-00007
144. Bedimo R. Growth hormone and tesamorelin in the management of HIV-associated lipodystrophy. HIV AIDS (Auckl). (2011) 3:69–79. doi: 10.2147/HIV.S14561
145. Spooner LM, Olin JL. Tesamorelin: a growth hormone-releasing factor analogue for HIV-associated lipodystrophy. Ann Pharmacother. (2012) 46:240–7. doi: 10.1345/aph.1Q629
146. Sivakumar T, Mechanic O, Fehmie DA, Paul B. Growth hormone axis treatments for HIV-associated lipodystrophy: a systematic review of placebo-controlled trials. HIV Med. (2011) 12:453–62. doi: 10.1111/j.1468-1293.2010.00906.x
147. Falutz J, Allas S, Blot K, Potvin D, Kotler D, Somero M, et al. Metabolic effects of a growth hormone-releasing factor in patients with HIV. N Engl J Med. (2007) 357:2359–70. doi: 10.1056/NEJMoa072375
148. Falutz J, Allas S, Kotler D, Thompson M, Koutkia P, Albu J, et al. A placebo-controlled, dose-ranging study of a growth hormone releasing factor in HIV-infected patients with abdominal fat accumulation. AIDS. (2005) 19:1279–87. doi: 10.1097/01.aids.0000180099.35146.30
149. Falutz J, Mamputu JC, Potvin D, Moyle G, Soulban G, Loughrey H, et al. Effects of tesamorelin (TH9507), a growth hormone-releasing factor analog, in human immunodeficiency virus-infected patients with excess abdominal fat: a pooled analysis of two multicenter, double-blind placebo-controlled phase 3 trials with safety extension data. J Clin Endocrinol Metab. (2010) 95:4291–304. doi: 10.1210/jc.2010-0490
150. Falutz J, Potvin D, Mamputu JC, Assaad H, Zoltowska M, Michaud SE, et al. Effects of tesamorelin, a growth hormone-releasing factor, in HIV-infected patients with abdominal fat accumulation: a randomized placebo-controlled trial with a safety extension. J Acquir Immune Defic Syndr. (2010) 53:311–22. doi: 10.1097/QAI.0b013e3181cbdaff
151. Guevara-Aguirre J, Procel P, Guevara C, Guevara-Aguirre M, Rosado V, Teran E. Despite higher body fat content, Ecuadorian subjects with Laron syndrome have less insulin resistance and lower incidence of diabetes than their relatives. Growth Horm IGF Res. (2016) 28:76–8. doi: 10.1016/j.ghir.2015.08.002
152. Oliveira CR, Salvatori R, Barreto-Filho JA, Rocha IE, Mari A, Pereira RM, et al. Insulin sensitivity and beta-cell function in adults with lifetime, untreated isolated growth hormone deficiency. J Clin Endocrinol Metab. (2012) 97:1013–9. doi: 10.1210/jc.2011-2590
Keywords: growth hormone, insulin-like growth factor-I, intra-portal insulin, acromegaly, adult growth hormone deficiency, hepatic growth hormone sensitivity
Citation: Yuen KCJ, Hjortebjerg R, Ganeshalingam AA, Clemmons DR and Frystyk J (2024) Growth hormone/insulin-like growth factor I axis in health and disease states: an update on the role of intra-portal insulin. Front. Endocrinol. 15:1456195. doi: 10.3389/fendo.2024.1456195
Received: 28 June 2024; Accepted: 22 October 2024;
Published: 21 November 2024.
Edited by:
Pouneh K. Fazeli, University of Pittsburgh, United StatesReviewed by:
Max Petersen, Washington University in St. Louis, United StatesArtak Labadzhyan, Cedars Sinai Medical Center, United States
Copyright © 2024 Yuen, Hjortebjerg, Ganeshalingam, Clemmons and Frystyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin C. J. Yuen, a2V2aW4ueXVlbkBkaWduaXR5aGVhbHRoLm9yZw==
†ORCID: Kevin C. J. Yuen, orcid.org/0000-0002-8169-2728