 Roozbeh Akbari Motlagh
Roozbeh Akbari Motlagh Jasmine Pipella1,2
Jasmine Pipella1,2 Peter J. Thompson
Peter J. Thompson- 1Department of Physiology and Pathophysiology, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
- 2Diabetes Research Envisioned and Accomplished in Manitoba (DREAM) Theme, Children’s Hospital Research Institute of Manitoba, Winnipeg, MB, Canada
Type 1 Diabetes (T1D) is a chronic metabolic disease resulting from insulin deficiency due to autoimmune loss of pancreatic β cells. In addition to β cell destruction, it is now accepted that β cell stress and dysfunction, such as senescence, plays a crucial role in the development of the disease. Accumulation of senescent β cells occurs during development of T1D in humans and contributes to the progression of T1D in the nonobese diabetic (NOD) mouse model. Senescent β cells are thought to exacerbate the inflammatory response within the islets by production and secretion of senescence-associated secretory phenotype (SASP). Extracellular vesicles (EVs) from β cells have been shown to carry protein and microRNAs (miRNAs), influencing cellular signaling and may contribute to the development of T1D but it remains to be addressed how senescence impacts β cell EV cargo. In this minireview, we discuss emerging evidence that EV cargo proteins and miRNAs associated with senescence could contribute to the development of T1D and could suggest potential biomarkers and therapeutic targets for the regulation of SASP and elimination of senescent β cells in T1D. Future investigation exploring the intricate relationship between β cell senescence, EVs and miRNAs could pave the way for the development of novel diagnostic techniques and therapeutic interventions.
1 Introduction
Type 1 diabetes (T1D) is a chronic metabolic disease resulting from severe destruction of β cells by an autoimmune T-cell-mediated response, manifested by insulin deficiency (1). T1D is a complex disease driven by genetic susceptibility, environmental factors, and epigenetic changes (2) that progresses through three stages. At the first stage, the onset of autoimmunity is accompanied by seropositivity against two or more specific proteins, such as glutamate decarboxylase 65 (GAD65), insulin, insulinoma antigen-2 (IA-2) and zinc transporter 8 (ZnT8) (3). A clinical manifestation of T1D typically occurs months or years before two or more of these autoantibodies are detected (4). Stage 2 is characterized by glucose intolerance, but the patient is otherwise asymptomatic. The third and final stage is marked by the emergence of diabetes symptoms, accompanied by a sustained decline in β cell function for several years (5). Although significant progress has been made in understanding T1D pathogenesis and the clinical implementation of the first FDA-approved therapy for use prior to symptoms onset, Teplizumab (6), a bona fide cure has remained elusive and major questions remain about how the disease develops, hindering advancement of therapies for the disease (7, 8).
A growing body of evidence has now shown that β cell dysfunction contributes to T1D. β cell dysfunction in T1D involves various stressed states, including the Unfolded Protein Response (UPR), type I interferon (IFN) response and senescence (9, 10). While investigations into the therapeutic potential of modulating β cell UPR and IFN response have progressed into phase II clinical trials with encouraging results (11–14), preclinical investigations into β cell senescence are still required to pave the way for clinical translation. The interactions between β cells and immune cells during T1D remains a puzzle (15), but exciting efforts have also suggested roles for small secreted extracellular vesicles (EVs) as important conveyors of signaling between β cells and immune cells during the pathogenesis of T1D (16). EVs are a heterogeneous group of nanoparticle-sized membrane-bound cargo carriers secreted from most cell types, containing DNA, RNA, proteins, and metabolites from their host cell. EVs consist of exosomes (30-150 nm) in addition to larger particles termed microvesicles (200-1000 nm) and apoptotic bodies (>1000 nm) (17). Remarkably, cellular senescence has been shown to dramatically increase small EV biogenesis and alter EV RNA and protein cargoes as a component of the senescence-associated secretory phenotype (SASP) (18–20). However, it remains unclear how senescence alters EV biogenesis and cargo in the β cell and what role, if any, senescent β cell EVs may play in T1D development.
In this minireview, we discuss the potential for senescent β cell EVs to contribute to the pathogenesis of T1D. We propose a novel approach to therapy, wherein the utilization of miRNA packaged into therapeutic EVs could target senescent β cells to mitigate their inflammatory effects.
2 Relationships between β cell EVs, senescence and miRNAs: implications for development of T1D and novel therapies
2.1 β cell EVs and development of T1D
Early evidence of EVs secretion by pancreatic β cells was reported by Sheng H. and colleagues in 2011, which showed EVs released from the mouse β cell line MIN6 (21). As with many other cell types, β cells produce and secrete mainly the small exosome-sized EVs rather than microvesicles (MVs) as demonstrated by culture studies using ultracentrifugation-based approaches for EV isolation (22) on rodent β cell lines or isolated islets from rodents and humans (23–26). While highly proliferative rodent β cell lines such as MIN6 and the rat β cell line INS-1, showed robust EV secretion, isolated rodent, and human primary islets, which generally do not proliferate, secreted few EVs in comparison, suggesting that EV release may be associated with β cell proliferation. Proteomic analysis of β cell EVs from mouse and human sources showed that they contain canonical exosomal markers, including endosomal sorting complex required for transport (ESCRT) machinery (Alix, Flotillin-1, Tsg101), tetraspanins (CD9, CD81, CD63), as well as housekeeping proteins (GAPDH, Actin) (Figure 1). Interestingly, primary mouse and human islet β cell EVs also contained putative autoantigens, including GAD65, proinsulin and ZnT8, which were shown to be released in a caspase-independent manner and at higher rates during proinflammatory cytokine exposure (23, 24) (Figure 1). Notably in regards to tetraspannin markers, a recent study showed that CD63 levels discriminate between two different β cell subsets in mice and humans, where CD63High β cells had higher metabolic and functional capacity compared with CD63Low (27), suggesting possible differences in EV markers or biogenesis associated with β cell function. However, further studies to test this hypothesis are clearly warranted.
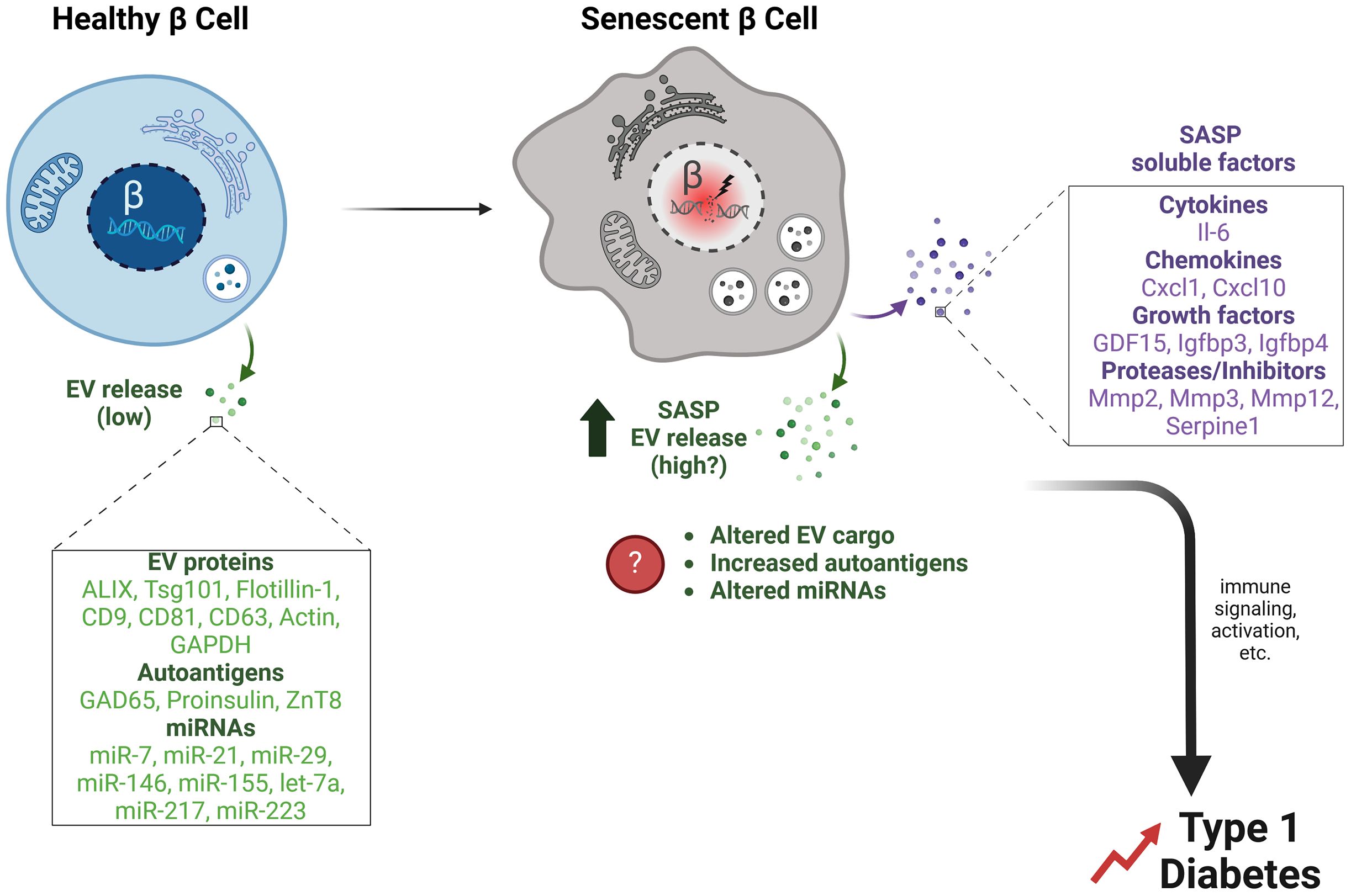
Figure 1. β cell EVs and potential effects of senescence on EV release and cargo. Healthy primary islet β cells release a low quantity of exosomes under typical culture conditions. β cell EVs contain typical EV proteins (Alix, Tsg101, Flotillin-1, CD9, CD81, CD63, Actin, GAPDH), along with autoantigens (GAD65, Proinsulin, ZnT8), and miRNAs (miR-7, miR-21, miR-29, miR-146, miR-155 let-7a, miR-217, miR-223). In contrast, senescent β cells that accumulate during T1D develop a SASP, which we propose involves not only classical SASP factors (SASP soluble factors) but also increased release of EVs (SASP EV release). Senescence may also alter β cell EV cargo, such as increasing the release of autoantigens and changing the miRNA profile. Together these changes in β cell EVs during senescence could facilitate immune signaling and activation events that accelerate the development of T1D.
The observation that β cell EVs contain autoantigens has raised the intriguing possibility that β cell EVs convey autoantigens directly to the immune system to activate and drive autoimmunity. Indeed, one study found that human islet EVs are internalized by monocytes and B cells, provoking T cell activation and memory responses in peripheral blood mononuclear cells (PBMCs) from T1D donors (28). EVs containing GAD65 triggered the production of anti-GAD antibodies from B cells (28) providing an explanation as to how β cell autoantigens are being sampled and provoke immune responses during T1D progression. In other work, it was demonstrated that the proinflammatory chemokine CXCL10 is enriched on the surface of stressed β cell EVs, promoting β cell dysfunction via paracrine effects in addition to immune cell chemotaxis (26). Inhibition of small EV release rescued defects in proinflammatory cytokine-stressed primary islet function and reduced macrophage and CD8+ T cell infiltration (26), suggesting a role for stressed β EVs in promoting islet dysfunction and immune recruitment.
Despite these advances and the insights learned from tissue culture studies, the question of how and whether β cell EV release actually contributes to autoimmunity in vivo during the development of T1D has not been addressed. In particular, it remains a major challenge in the field to specifically determine the cell type of origin for isolated EV populations from blood or even islets in culture, since EV populations are a heterogeneous mixture produced by multiple cell types and cell-type specific surface markers on EVs for β cells have not yet been identified. To tackle this challenge, recently developed conditional reporter mouse models could be used to examine the impact of senescence on β cell EVs Specifically, a tissue-specific EV reporter mouse model (e.g., lox-STOP-lox-CD9-GFP) could be employed, where the conditional activation occurs in β cells (e.g., by crossing with an Ins1-Cre driver line).These allow for tissue-specific fluorescent protein tagging of EVs, such as the Transgenic inducible GFP extracellular vesicle reporter (TIGER) mouse which uses a CD9-GFP (29), a truncated CD9-GFP EV conditional reporter that allows for affinity isolation (30), or the CD63-GFP conditional reporter mouse models (31, 32). Senescent beta cells could then be ablated by treating the animals with senolytics (compounds the trigger apoptosis preferentially in senescent cells (33), and comparing beta cell EV cargo from control and senolytic-treated animal islets. Given that β cell EVs naturally express these tetraspanin markers, these mouse models could provide key tools for addressing how β cells release EVs in vivo, elucidating the trafficking of these EVs and the impact of stress on β cell EV release during the development of T1D.
2.2 The role of senescent β cells and the potential role of senescence-derived EVs in T1D
Cellular senescence is a stress response leading to various hallmark phenotypes including an irreversible cell cycle arrest, resistance to apoptosis, metabolic adaptation and acquisition of a SASP (34). Although senescence is beneficial in certain contexts such as during embryogenesis and wound healing, when senescent cells are not efficiently removed by the immune system, their accumulation leads to chronic inflammation and tissue dysfunction (35). Senescent β cells that accumulate during T1D development in the NOD autoimmune diabetes mouse model and human donor pancreas tissue exhibit the major hallmarks of senescence in other cell types. These include a DNA damage response (DDR), apoptosis resistance, the lysosomal senescence-associated β-galactosidase (SA-βgal) and a SASP (Cha et al., 2023) although the precise triggers responsible for the induction of senescence in β cells during T1D are still unknown. Senescent β cells in NOD mice upregulate the prosurvival protein Bcl-2, rendering them sensitive to Bcl-2 inhibitor senolytics, which can selectively trigger apoptosis in this subset of β cells (36).
Senescent cells activate the release of a proinflammatory secretome known as SASP, which in β cells comprises cytokines and chemokines (e.g., IL-6, Cxcl10, Ccl2, Ccl4, Cxcl1, Cxcl8), matrix metalloproteinases and inhibitors (MMP-2, MMP-3, MMP-12, Serpine1), and growth factors (Gdf15/GDF15, Igfbp3, Igfbp4/IGFBP4) (15) (Figure 1). The various biological activities elicited by the SASP are intimately connected to the microenvironment in which senescent cells reside. In β cells, SASP can also exert detrimental paracrine effects on adjacent non-senescent healthy β cells, such as triggering senescence (36–38). Additionally, the induction of SASP in cultured human islets causes changes in glucagon secretion from α cells, implying that the SASP originating from β cells can have an impact on the normal functioning of α cells (39). Suppression of SASP from senescent β cells in NOD mice protects against diabetes (40), supporting a deleterious role for SASP in T1D development.
In addition to the secretion of soluble immunogenic proteins, recent studies have also shown that SASP involves increased secretion of EVs and altered cargo compared to the EVs released from non-senescent cells (19, 20). Notably, senescent cells are more sensitive to inhibition of EV release compared with non-senescent cells (41), and loss of EV release machinery proteins can accelerate senescence onset, leading to speculation that EV release may provide an adaptive mechanism for removal of unwanted cargo in senescent cells (42). Senescence also alters EV miRNA cargo (43, 44). However, it remains to be determined how senescence alters β cell EV release and cargo (Figure 1). Although proteomic studies have been carried out to identify β cell SASP factors, EVs were not specifically isolated in those studies (45). Similarly, while it was recently shown that miRNAs have coding motifs that direct their sorting into exosomes (46), the extent to which this mechanism operates to direct miRNA sorting in β cell EVs in the context of senescence remains an open question. We postulate that, aside from its effects through the direct release of soluble paracrine factors, β cell SASP could also cause detrimental impacts on neighboring or distant cells through increased release of EVs with modified protein and/or miRNA cargo (Figure 1). For instance, senescence may increase autoantigen cargo or lead to the export of immune-activating miRNAs. The broader scope of SASP influence is facilitated by the transmission of EVs, allowing it to potentially affect cells that are distant from islets.
2.3 β cell EV miRNAs and impact of senescence on EV miRNA
In addition to protein cargo, miRNAs are also enriched in β cell EVs. miRNAs are non-coding RNAs 21-23 nucleotides long, regulate gene expression post-transcriptionally. They inhibit mRNA translation or reduce mRNA stability, playing key roles in genetic regulation and various biological processes (47). Cataloging more than 2500 miRNAs in humans has revealed their significant role in regulating around 60% of protein-coding genes within the genome. While miRNAs are predominantly generated and function within cells, recent studies have shed light on their ability to be secreted outside the cell in micro-vesicles or exosomes. This intriguing finding has expanded our understanding of miRNAs, revealing their capacity to transmit their regulatory functions to other cell types. β cell EVs contain: miR-7, miR-21, miR-29, miR-146a, miR-223, let-7a, miR-217 and miR-155 (25, 48–52) (Figure 1). Interestingly, some of the targets for these miRNAs include genes involved in senescence/SASP, including Bcl2 CXCR2, MMP2, MMP9, Cdkn2a (encoding p16Ink4a), TP53, TNF, and IL6. For example, miR-21 increases in abundance during proinflammatory cytokine exposure in the MIN6 β cell line and primary isolated mouse and human islets (25). miR-21 increases β cell susceptibility to apoptosis by downregulating Bcl2 mRNA levels (53), and overexpression of miR-21 precursor pre-miRNA in β cells leads to defects in glucose-stimulated insulin secretion (54), suggesting a novel link between proinflammatory conditions and impairment of β cell function via miRNA expression.
Upon entering senescence, the expression patterns of miRNAs undergo notable alteration (55). This alteration in miRNA expression is also manifested in the composition of EVs (55). Alibhai et al. revealed that in aging mice, the levels of miR-146a, miR-21, miR-223, and let-7a in circulating EVs in plasma increased, indicating a correlation with modifications in immune function (56). Another study observed that EVs released by replicative senescent human umbilical vein endothelial cells, enriched with elevated levels of miR-21-5p and miR-217, were observed to reduce proliferation and promote senescence in neighboring endothelial cells (57). Salama et al. have observed that miR-29 in β cell-derived exosomes induced TNFa, and IL-6 in splenocytes of NOD mice (49). Up-regulation of miR-29 correlated with DNA damage response, increased levels of the DNA damage marker γ-H2A.X, and accumulation of SA-βGal (58). miR-29 was also upregulated in aged mice (59). Moreover, EVs containing miR-29 were considered controls for aging and the induction of an inflammatory environment in white adipose tissue (60). Davis et al. utilized a miRNA array to showcase a variation in miRNA expression within EVs obtained from the bones of aged and youthful mice. The EVs originating from the bones of older mice exhibited a distinct elevation in the levels of miR-183-5p. Moreover, the overexpression of a miR-183-5p mimic exhibited the ability to stimulate senescence and hinder the proliferation and differentiation of Bone Marrow Mesenchymal Stromal Cells (BMSCs) (61). Thus, it is apparent that in pathological situations, the isolated EVs can have cell type specific effects in the microenvironment. Although the impact of senescence has not been studied on islet EVs, a recent preprint reported that isolated EVs from human islets treated with inflammatory cytokines led to the upregulation of miR-155-5p (52). These findings were in line with the analysis of EVs plasma derived from children with recent T1D and autoantibody-positivity (52) suggesting inflammatory stress could provoke β cell miR-155 EV release into circulation.
In summary, despite the considerable number of studies conducted on senescent cell EVs and their miRNA cargo and impacts of inflammatory stress on β cell EVs, there remains a gap in understanding senescent β cells and identifying EVs produced by senescent β cells. To address this question, genetic tools to disrupt the β cell senescence program in vivo or ablate p21-expressing senescent cells, such as in the p21-Cre mouse (62) or the p21 promoter-driven suicide gene mouse model (p21-ATTAC) (63) could be utilized. These models would allow for ablation of senescent beta cells, thereby also eliminating their potentially altered EVs. In addition, now that reliable approaches for inducing senescence in mouse and human β cells in vitro have been established by us (64) and others (45), they provide suitable culture models to investigate the impact of senescence on β cell EV release and cargo. These strategies will enable the isolation of EVs from control versus senescent β cells for transcriptomic analyses to gain a deeper comprehension of how senescent β cell EV miRNAs could promote the progression of autoimmune β cell loss in the islet microenvironment.
The heterogeneity in EVs complicates the task of isolating and identifying specific EV subtypes (e.g., islets-derived EVs) amid other non-target EVs circulating in the body. Therefore, the exploration of novel biomarkers on the surface of EVs is of great interest. The application of flow cytometry has been used to address the heterogeneity of senescent cell EVs, both in vitro and in vivo. Meng et al. (65) identified the proteins present on extracellular vesicles associated with senescence (S-EVs) across various senescence models. Their findings revealed that the proteins DPP4, ANXA1, ANXA6, S10AB, AT1A1, and EPHB2 were predominantly present on the surface of EVs from senescent cells. DPP4 is distinguished by its unique capability to impede EV uptake by proliferating cells. Furthermore, it was noted that there was a marked increase in DPP4-rich EVs in the blood of women with gestational diabetes mellitus (GDM) (66). DDP4-rich EVs were also reported in the urine of patients with diabetic kidney disease (67). This raises the question of whether DPP-4-bearing EVs could be used as a unique biomarker for applying flow cytometry to identify senescent β cell-derived EVs in vivo. Future efforts in this area could use a screening approach for plasma membrane proteins that are selectively expressed on the surface of EVs from senescent β cells. By performing proteomics analysis to identify new potential markers of β cell senescence found on the surface of EVs, the effectiveness of drugs targeting senescent β cells could be more effectively evaluated.
2.4 Therapeutic EVs to improve targeting of senescent β cells in T1D
Pharmacological elimination of senescent cells predominantly hinges on two specific types of compounds known as senolytics and senomorphics (68). Senolytic compounds are designed to specifically target and eliminate senescent cells while minimizing any impact on the proliferating and healthy cells, while senomorphics act to modify senescent cell phenotypes in a beneficial manner (68, 69). While there are not yet available tools to specifically manipulate the EV release from senescent cells (while not impacting EV release generally from other cell types) both senolytics and senomorphics could mitigate senescent β cell EV release, since senolytics would eliminate senescent β cells themselves along with their EVs and senomorphics could suppress SASP which also involves altered EV release. Most efforts have focused on use of senolytic compounds which have been extensively tested both in vitro and in vivo, including inhibitors targeting the BCL-2 family such as ABT-263 and ABT-737 (68). Recently clinical trials suggest the senolytic cocktail dasatinib and quercetin (D+Q) is safe and promising for treating senescence-associated diseases in humans (70–72). While senolytics have not reached the clinic for treating T1D, preclinical studies support the positive influence of senolytic compounds ABT-199 and ABT-737 in depleting senescent β cells in NOD mice, which protected against T1D (36). However, senolytic compounds are administered systemically and can have off-target effects. Senolytics also display significant variability in efficacy across various cell lines and tissues, making it problematic to identify a single senolytic as a commonly implemented method for eliminating senescent cells in vivo (69).
Another approach to targeting senescence is using senomorphics to directly target and inhibit the SASP process. Suppression of β cell SASP using Bromodomain Extra-Terminal domain (BET) protein inhibitor iBET-762 protects against T1D in NOD mice (40). However, BET proteins are ubiquitously expressed and employed in a variety of inflammatory and housekeeping processes unrelated to SASP, making it highly problematic to adapt this senomorphic approach to T1D in humans. On the other hand, miRNAs may provide an under-appreciated mechanism to regulate and modify the SASP. Numerous studies have highlighted the role of miRNAs in the regulation of senescence signaling pathways, and specifically in controlling SASP (47, 73). For example, Qi et al. indicated that miR-204 suppresses IL-6, IL-18, and TNF-α by upregulation of Bcl-2 and SIRT1 in rats with diabetic retinopathy (74). Studies have demonstrated that miR-107 triggers apoptosis and downregulates the expression of CXCR2, MMP-2, and MMP-9, leading to a decrease in SASP secretion (75). Furthermore, miRNAs are involved in the modulation of senescence through the key cell cycle arrest regulators p16Ink4a and p53 (73). Indeed, miR-24 effectively hinders replicative senescence by repressing Cdkn2a expression through binding to the 3′UTR of its mRNA (76). EVs have been proposed as next generation treatment alternatives and designer nanoparticles to transport medications (77). For example, EV-delivered miRNAs have reached clinical trials for cancer therapy (78). Therefore, EVs may present a natural, cell/tissue-derived avenue for efficient transport of senolytic or senomorphic miRNAs to improve targeting efficiency of senescent β cells in T1D, acknowledging that other cargo of β cell EVs may play roles in T1D as well (Figure 2). EVs may have lower immunogenicity as compared with other delivery reagents, as shown in recent studies where no toxic effects were observed in mice after three weeks of treatment with EVs from the human embryonic kidney cell line HEK293T (77). EVs also may provide a suitable barrier to prevent the degradation of therapeutic miRNAs. Surface proteins on EVs may act like distinct barcodes, helping delivered EVs to reach their intended targets in vivo. In this regard, in previous studies, engineered EVs with surface localization of a β cell protein p88 fused to EV protein lactoadherin showed increased uptake to the pancreas upon intravenous administration in mice (79). In the future, it will be crucial to identify and characterize miRNAs that suppress SASP in β cells. The use of targeted EV surface proteins (such as p88 or ligands for GLP1R that is enriched on β cells) along with loading of miRNA cargo to inhibit SASP could provide precision suppression of senescent β cell SASP in vivo, while reducing the impacts of off-target effects associated with systemic senomorphic use (Figure 2).
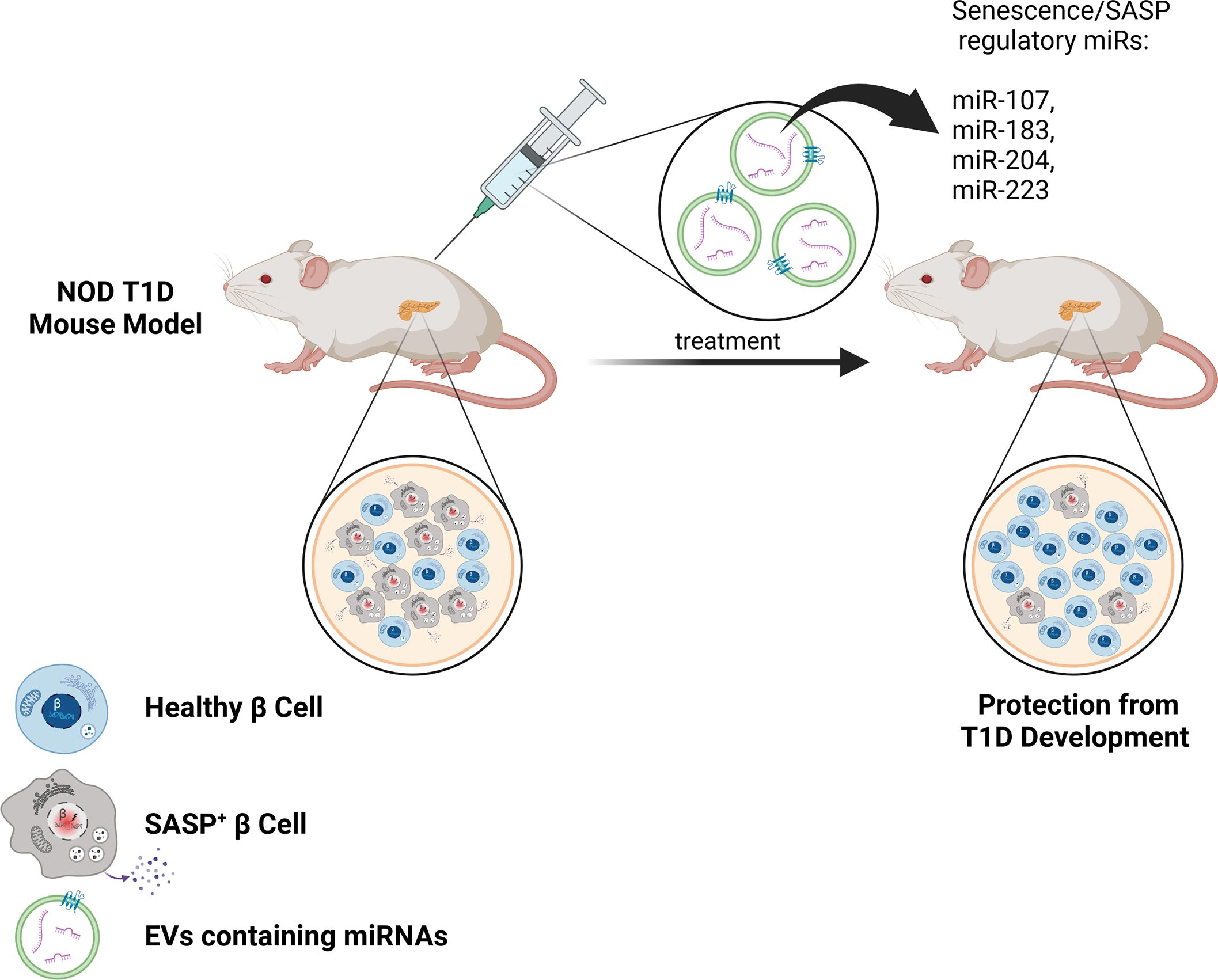
Figure 2. Therapeutic EV delivery of senotherapeutic miRNAs for improved targeting of SASP in β cells during T1D. The use of specially designed EVs with surface molecules to target β cells (such as GLP1R agonists) and harboring miRNAs to suppress SASP (such as miR-107, miR-183, miR-204, miR-223) could be a powerful approach for improved targeting of SASP+ β cells and sparing healthy β cells in vivo during the development of T1D. This approach could downregulate SASP preferentially in β cells, thereby reducing off-target effects of other senotherapeutics and protecting against T1D.
3 Conclusion
In conclusion, current evidence suggests that β cell EVs could play a critical role in T1D progression. However, the specific models have been lacking to definitively address this question in vivo. The advent of conditional EV reporter mouse models has furnished the islet biology field with tools that will undoubtedly provide insights into how β cell EVs contribute to T1D pathogenesis. We propose that β cell senescence alters EV production along with the protein and miRNA cargo in a manner that accelerates T1D (Figure 1). As methods to engineer EVs continue to improve, we also anticipate that EV technology will enable the use of surface ligands with high β cell/islet specificity and these tools could be deployed to improve upon systemically administered therapeutics for targeting senescent β cells (Figure 2). As EVs have already been used to effectively package and deliver miRNA cargo in clinical trials, we suggest that a similar approach applied to miRNAs that regulate SASP, could prove highly effective for limiting the deleterious impact of accumulated senescent β cells in diabetes.
Author contributions
RM: Writing – review & editing, Writing – original draft, Conceptualization. JP: Writing – review & editing, Visualization. PT: Writing – review & editing, Writing – original draft, Visualization, Supervision, Resources, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. JP was funded by a Canada Graduate Scholarship – Master’s Award (CGS-M). Work in this research area in the laboratory of PT is funded by the Canadian Institutes of Health Research (PJT-309479641, PJT-485915), the Juvenile Diabetes Research Foundation (4-SRA-2023-1184-S-N) and the Manitoba Medical Services Foundation Allen Rouse Basic Science Career Development Award.
Acknowledgments
Figures were created with BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
2. Zajec A, Trebusak Podkrajsek K, Tesovnik T, Sket R, Cugalj Kern B, Jenko Bizjan B, et al. Pathogenesis of type 1 diabetes: established facts and new insights. Genes (Basel). (2022) 13:706. doi: 10.3390/genes13040706
3. Kawasaki E. Anti-islet autoantibodies in type 1 diabetes. Int J Mol Sci. (2023) 24:10012. doi: 10.3390/ijms241210012
4. Jia X, Yu L. Understanding islet autoantibodies in prediction of type 1 diabetes. J Endocr Soc. (2023) 8:bvad160. doi: 10.1210/jendso/bvad160
5. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. (2015) 38:1964–74. doi: 10.2337/dc15-1419
6. Herold KC, Bundy BN, Long SA, Bluestone JA, Dimeglio LA, Dufort MJ, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. (2019) 381:603–13. doi: 10.1056/NEJMoa1902226
7. Brawerman G, Thompson PJ. Beta cell therapies for preventing type 1 diabetes: from bench to bedside. Biomolecules. (2020) 10:1681. doi: 10.3390/biom10121681
8. Dayan CM, Besser REJ, Oram RA, Hagopian W, Vatish M, Bendor-Samuel O, et al. Preventing type 1 diabetes in childhood. Science. (2021) 373:506–10. doi: 10.1126/science.abi4742
9. Mallone R, Halliez C, Rui J, Herold KC. The beta-cell in type 1 diabetes pathogenesis: A victim of circumstances or an instigator of tragic events? Diabetes. (2022) 71:1603–10. doi: 10.2337/dbi21-0036
10. Thompson PJ, Pipella J, Rutter GA, Gaisano HY, Santamaria P. Islet autoimmunity in human type 1 diabetes: initiation and progression from the perspective of the beta cell. Diabetologia. (2023) 66:1971–82. doi: 10.1007/s00125-023-05970-z
11. Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, et al. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med. (2018) 24:1108–12. doi: 10.1038/s41591-018-0089-4
12. Gitelman SE, Bundy BN, Ferrannini E, Lim N, Blanchfield JL, Dimeglio LA, et al. Imatinib therapy for patients with recent-onset type 1 diabetes: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. (2021) 9:502–14. doi: 10.1016/S2213-8587(21)00139-X
13. Forlenza GP, McVean J, Beck RW, Bauza C, Bailey R, Buckingham B, et al. Effect of verapamil on pancreatic beta cell function in newly diagnosed pediatric type 1 diabetes: A randomized clinical trial. JAMA. (2023) 329:990–9. doi: 10.1001/jama.2023.2064
14. Waibel M, Wentworth JM, So M, Couper JJ, Cameron FJ, Macisaac RJ, et al. Baricitinib and beta-cell function in patients with new-onset type 1 diabetes. N Engl J Med. (2023) 389:2140–50. doi: 10.1056/NEJMoa2306691
15. Rampazzo Morelli N, Pipella J, Thompson PJ. Establishing evidence for immune surveillance of beta-cell senescence. Trends Endocrinol Metab. (2024) 35:576–85. doi: 10.1016/j.tem.2024.01.003
16. Negi S, Rutman AK, Paraskevas S. Extracellular vesicles in type 1 diabetes: messengers and regulators. Curr Diabetes Rep. (2019) 19:69. doi: 10.1007/s11892-019-1193-7
17. Liu YJ, Wang C. A review of the regulatory mechanisms of extracellular vesicles-mediated intercellular communication. Cell Commun Signal. (2023) 21:77. doi: 10.1186/s12964-023-01103-6
18. Urbanelli L, Buratta S, Sagini K, Tancini B, Emiliani C. Extracellular vesicles as new players in cellular senescence. Int J Mol Sci. (2016) 17:1408. doi: 10.3390/ijms17091408
19. Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun. (2017) 8:15729. doi: 10.1038/ncomms15728
20. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PloS Biol. (2020) 18:e3000599. doi: 10.1371/journal.pbio.3000599
21. Sheng H, Hassanali S, Nugent C, Wen L, Hamilton-Williams E, Dias P, et al. Insulinoma-released exosomes or microparticles are immunostimulatory and can activate autoreactive T cells spontaneously developed in nonobese diabetic mice. J Immunol. (2011) 187:1591–600. doi: 10.4049/jimmunol.1100231
22. Chhoy P, Brown CW, Amante JJ, Mercurio AM. Protocol for the separation of extracellular vesicles by ultracentrifugation from in vitro cell culture models. STAR Protoc. (2021) 2:100303. doi: 10.1016/j.xpro.2021.100303
23. Cianciaruso C, Phelps EA, Pasquier M, Hamelin R, Demurtas D, Alibashe Ahmed M, et al. Primary human and rat beta-cells release the intracellular autoantigens GAD65, IA-2, and proinsulin in exosomes together with cytokine-induced enhancers of immunity. Diabetes. (2017) 66:460–73. doi: 10.2337/db16-0671
24. Hasilo CP, Negi S, Allaeys I, Cloutier N, Rutman AK, Gasparrini M, et al. Presence of diabetes autoantigens in extracellular vesicles derived from human islets. Sci Rep. (2017) 7:5000. doi: 10.1038/s41598-017-04977-y
25. Lakhter AJ, Pratt RE, Moore RE, Doucette KK, Maier BF, Dimeglio LA, et al. Beta cell extracellular vesicle miR-21-5p cargo is increased in response to inflammatory cytokines and serves as a biomarker of type 1 diabetes. Diabetologia. (2018) 61:1124–34. doi: 10.1007/s00125-018-4559-5
26. Javeed N, Her TK, Brown MR, Vanderboom P, Rakshit K, Egan AM, et al. Pro-inflammatory beta cell small extracellular vesicles induce beta cell failure through activation of the CXCL10/CXCR3 axis in diabetes. Cell Rep. (2021) 36:109613. doi: 10.1016/j.celrep.2021.109613
27. Rubio-Navarro A, Gomez-Banoy N, Stoll L, Dundar F, Mawla AM, Ma L, et al. A beta cell subset with enhanced insulin secretion and glucose metabolism is reduced in type 2 diabetes. Nat Cell Biol. (2023) 25:565–78. doi: 10.1038/s41556-023-01103-1
28. Rutman AK, Negi S, Gasparrini M, Hasilo CP, Tchervenkov J, Paraskevas S. Immune response to extracellular vesicles from human islets of langerhans in patients with type 1 diabetes. Endocrinology. (2018) 159:3834–47. doi: 10.1210/en.2018-00649
29. Neckles VN, Morton MC, Holmberg JC, Sokolov AM, Nottoli T, Liu D, et al. A transgenic inducible GFP extracellular-vesicle reporter (TIGER) mouse illuminates neonatal cortical astrocytes as a source of immunomodulatory extracellular vesicles. Sci Rep. (2019) 9:3094. doi: 10.1038/s41598-019-39679-0
30. Norgard MO, Steffensen LB, Hansen DR, Fuchtbauer EM, Engelund MB, Dimke H, et al. A new transgene mouse model using an extravesicular EGFP tag enables affinity isolation of cell-specific extracellular vesicles. Sci Rep. (2022) 12:496. doi: 10.1038/s41598-021-04512-0
31. Men Y, Yelick J, Jin S, Tian Y, Chiang MSR, Higashimori H, et al. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat Commun. (2019) 10:4136. doi: 10.1038/s41467-019-11534-w
32. McCann JV, Bischoff SR, Zhang Y, Cowley DO, Sanchez-Gonzalez V, Daaboul GD, et al. Reporter mice for isolating and auditing cell type-specific extracellular vesicles in vivo. Genesis. (2020) 58:e23369. doi: 10.1002/dvg.23369
33. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. (2022) 28:1556–68. doi: 10.1038/s41591-022-01923-y
34. Di Micco R, Krizhanovsky V, Baker D, D'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
35. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging. (2021) 1:870–9. doi: 10.1038/s43587-021-00121-8
36. Thompson PJ, Shah A, Ntranos V, van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. (2019) 29:1045–60.e10. doi: 10.1016/j.cmet.2019.01.021
37. Aguayo-Mazzucato C, Andle J, Lee TB Jr., Midha A, Talemal L, Chipashvili V, et al. Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. (2019) 30:129–42.e4. doi: 10.1016/j.cmet.2019.05.006
38. Walker EM, Cha J, Tong X, Guo M, Liu JH, Yu S, et al. Sex-biased islet beta cell dysfunction is caused by the MODY MAFA S64F variant by inducing premature aging and senescence in males. Cell Rep. (2021) 37:109813. doi: 10.1016/j.celrep.2021.109813
39. Brawerman G, Ntranos V, Thompson PJ. Alpha cell dysfunction in type 1 diabetes is independent of a senescence program. Front Endocrinol (Lausanne). (2022) 13:932516. doi: 10.3389/fendo.2022.932516
40. Thompson PJ, Shah A, Apostolopolou H, Bhushan A. BET proteins are required for transcriptional activation of the senescent islet cell secretome in type 1 diabetes. Int J Mol Sci. (2019) 20:4776. doi: 10.3390/ijms20194776
41. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. (2017) 8:15287. doi: 10.1038/ncomms15287
42. Zou W, Lai M, Jiang Y, Mao L, Zhou W, Zhang S, et al. Exosome release delays senescence by disposing of obsolete biomolecules. Adv Sci (Weinh). (2023) 10:e2204826. doi: 10.1002/advs.202204826
43. Fulzele S, Mendhe B, Khayrullin A, Johnson M, Kaiser H, Liu Y, et al. Muscle-derived miR-34a increases with age in circulating extracellular vesicles and induces senescence of bone marrow stem cells. Aging (Albany NY). (2019) 11:1791–803. doi: 10.18632/aging.v11i6
44. Yin Y, Chen H, Wang Y, Zhang L, Wang X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J Extracell Vesicles. (2021) 10:e12154. doi: 10.1002/jev2.12154
45. Midha A, Pan H, Abarca C, Andle J, Carapeto P, Bonner-Weir S, et al. Unique human and mouse beta-cell senescence-associated secretory phenotype (SASP) reveal conserved signaling pathways and heterogeneous factors. Diabetes. (2021) 70:1098–116. doi: 10.2337/db20-0553
46. Garcia-Martin R, Wang G, Brandao BB, Zanotto TM, Shah S, Kumar Patel S, et al. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature. (2022) 601:446–51. doi: 10.1038/s41586-021-04234-3
47. Wang Z, Gao J, Xu C. Tackling cellular senescence by targeting miRNAs. Biogerontology. (2022) 23:387–400. doi: 10.1007/s10522-022-09972-z
48. Figliolini F, Cantaluppi V, de Lena M, Beltramo S, Romagnoli R, Salizzoni M, et al. Isolation, characterization and potential role in beta cell-endothelium cross-talk of extracellular vesicles released from human pancreatic islets. PloS One. (2014) 9:e102521. doi: 10.1371/journal.pone.0102521
49. Salama A, Fichou N, Allard M, Dubreil L, de Beaurepaire L, Viel A, et al. MicroRNA-29b modulates innate and antigen-specific immune responses in mouse models of autoimmunity. PloS One. (2014) 9:e106153. doi: 10.1371/journal.pone.0106153
50. Guay C, Menoud V, Rome S, Regazzi R. Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun Signal. (2015) 13:17. doi: 10.1186/s12964-015-0097-7
51. Sims E, Lakhter AJ, Restrepo I, Tong X, Kono T, Anderson-Baucum E, et al. β Cell derived Mir-21 increases apoptosis via translation inhibition of the antiapoptotic protein Bcl2 and could serve as a of type 1 diabetes mellitus. J Invest Med. (2016) 64:927.2-928. doi: 10.1136/jim-2016-000120.35
52. Syed F, Krishnan P, Chang G, Langlais SR, Hati S, Yamada K, et al. beta cell microRNAs function as molecular hubs of type 1 diabetes pathogenesis and as biomarkers of diabetes risk. bioRxiv. (2023) 15:2023.06.15.545170. doi: 10.1101/2023.06.15.545170
53. Sims EK, Lakhter AJ, Anderson-Baucum E, Kono T, Tong X, Evans-Molina C. MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells. Diabetologia. (2017) 60:1057–65. doi: 10.1007/s00125-017-4237-z
54. Ibrahim S, Johnson M, Stephens CH, Xu J, Moore R, Mariani A, et al. beta-Cell pre-mir-21 induces dysfunction and loss of cellular identity by targeting transforming growth factor beta 2 (Tgfb2) and Smad family member 2 (Smad2) mRNAs. Mol Metab. (2021) 53:101289. doi: 10.1016/j.molmet.2021.101289
55. Estevez-Souto V, da Silva-Alvarez S, Collado M. The role of extracellular vesicles in cellular senescence. FEBS J. (2023) 290:1203–11. doi: 10.1111/febs.16585
56. Alibhai FJ, Lim F, Yeganeh A, Distefano PV, Binesh-Marvasti T, Belfiore A, et al. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell. (2020) 19:e13103. doi: 10.1111/acel.13103
57. Mensa E, Guescini M, Giuliani A, Bacalini MG, Ramini D, Corleone G, et al. Small extracellular vesicles deliver miR-21 and miR-217 as pro-senescence effectors to endothelial cells. J Extracell Vesicles. (2020) 9:1725285. doi: 10.1080/20013078.2020.1725285
58. Ugalde AP, Ramsay AJ, de la Rosa J, Varela I, Marino G, Cadinanos J, et al. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. (2011) 30:2219–32. doi: 10.1038/emboj.2011.124
59. Xu S, Wu W, Huang H, Huang R, Xie L, Su A, et al. The p53/miRNAs/Ccna2 pathway serves as a novel regulator of cellular senescence: Complement of the canonical p53/p21 pathway. Aging Cell. (2019) 18:e12918. doi: 10.1111/acel.12918
60. Kern F, Kuhn T, Ludwig N, Simon M, Groger L, Fabis N, et al. Ageing-associated small RNA cargo of extracellular vesicles. RNA Biol. (2023) 20:482–94. doi: 10.1080/15476286.2023.2234713
61. Davis C, Dukes A, Drewry M, Helwa I, Johnson MH, Isales CM, et al. MicroRNA-183-5p increases with age in bone-derived extracellular vesicles, suppresses bone marrow stromal (Stem) cell proliferation, and induces stem cell senescence. Tissue Eng Part A. (2017) 23:1231–40. doi: 10.1089/ten.tea.2016.0525
62. Wang B, Wang L, Gasek NS, Zhou Y, Kim T, Guo C, et al. An inducible p21-Cre mouse model to monitor and manipulate p21-highly-expressing senescent cells in vivo. Nat Aging. (2021) 1:962–73. doi: 10.1038/s43587-021-00107-6
63. Chandra A, Lagnado AB, Farr JN, Doolittle M, Tchkonia T, Kirkland JL, et al. Targeted clearance of p21- but not p16-positive senescent cells prevents radiation-induced osteoporosis and increased marrow adiposity. Aging Cell. (2022) 21:e13602. doi: 10.1111/acel.13602
64. Brawerman G, Pipella J, Thompson PJ. DNA damage to beta cells in culture recapitulates features of senescent beta cells that accumulate in type 1 diabetes. Mol Metab. (2022) 62:101524. doi: 10.1016/j.molmet.2022.101524
65. Meng Q, Chen C, Yang N, Gololobova O, Shi C, Dunn CA, et al. Surfaceome analysis of extracellular vesicles from senescent cells uncovers uptake repressor DPP4. Proc Natl Acad Sci U.S.A. (2023) 120:e2219801120. doi: 10.1073/pnas.2219801120
66. Kandzija N, Zhang W, Motta-Mejia C, Mhlomi V, McGowan-Downey J, James T, et al. Placental extracellular vesicles express active dipeptidyl peptidase IV; levels are increased in gestational diabetes mellitus. J Extracell Vesicles. (2019) 8:1617000. doi: 10.1080/20013078.2019.1617000
67. Sun AL, Deng JT, Guan GJ, Chen SH, Liu YT, Cheng J, et al. Dipeptidyl peptidase-IV is a potential molecular biomarker in diabetic kidney disease. Diabetes Vasc Dis Res. (2012) 9:301–8. doi: 10.1177/1479164111434318
68. Rossi M, Abdelmohsen K. The emergence of senescent surface biomarkers as senotherapeutic targets. Cells. (2021) 10:1740. doi: 10.3390/cells10071740
69. Robbins PD, Jurk D, Khosla S, Kirkland JL, Lebrasseur NK, Miller JD, et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol. (2021) 61:779–803. doi: 10.1146/annurev-pharmtox-050120-105018
70. Justice JN, Nambiar AM, Tchkonia T, Lebrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
71. Gonzales MM, Garbarino VR, Kautz TF, Palavicini JP, Lopez-Cruzan M, Dehkordi SK, et al. Senolytic therapy in mild Alzheimer's disease: a phase 1 feasibility trial. Nat Med. (2023) 29:2481–8. doi: 10.1038/s41591-023-02543-w
72. Nambiar A, Kellogg D 3rd, Justice J, Goros M, Gelfond J, Pascual R, et al. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine. (2023) 90:104481. doi: 10.1016/j.ebiom.2023.104481
73. Ma X, Zheng Q, Zhao G, Yuan W, Liu W. Regulation of cellular senescence by microRNAs. Mech Ageing Dev. (2020) 189:111264. doi: 10.1016/j.mad.2020.111264
74. Qi F, Jiang X, Tong T, Chang H, Li RX. MiR-204 inhibits inflammation and cell apoptosis in retinopathy rats with diabetic retinopathy by regulating Bcl-2 and SIRT1 expressions. Eur Rev Med Pharmacol Sci. (2020) 24:6486–93. doi: 10.26355/eurrev_202006_21631
75. Xiao D, Gao HX. Mechanism of miR-107-targeting of regulator of G-protein signaling 4 in hepatocellular carcinoma. Oncol Lett. (2019) 18:5145–54. doi: 10.3892/ol
76. Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann R Jr., Srikantan S, et al. p16(INK4a) translation suppressed by miR-24. PloS One. (2008) 3:e1864. doi: 10.1371/journal.pone.0001864
77. Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol. (2021) 16:748–59. doi: 10.1038/s41565-021-00931-2
78. Martellucci S, Orefice NS, Angelucci A, Luce A, Caraglia M, Zappavigna S. Extracellular vesicles: new endogenous shuttles for miRNAs in cancer diagnosis and therapy? Int J Mol Sci. (2020) 21:6486. doi: 10.3390/ijms21186486
Keywords: type 1 diabetes, cellular senescence, extracellular vesicles, microRNAs, SASP (senescence-associated secretory phenotype)
Citation: Motlagh RA, Pipella J and Thompson PJ (2024) Exploring senescence as a modifier of β cell extracellular vesicles in type 1 diabetes. Front. Endocrinol. 15:1422279. doi: 10.3389/fendo.2024.1422279
Received: 23 April 2024; Accepted: 05 August 2024;
Published: 22 August 2024.
Edited by:
Alok Raghav, Gachon University, Republic of KoreaReviewed by:
Ping Guo, Steadman Philippon Research Institute, United StatesAngelica Giuliani, Marche Polytechnic University, Italy
Copyright © 2024 Motlagh, Pipella and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter J. Thompson, cGV0ZXIudGhvbXBzb25AdW1hbml0b2JhLmNh