 Manoocher Soleimani
Manoocher Soleimani- 1Department of Medicine, University of New Mexico, Albuquerque, NM, United States
- 2Research Services, New Mexico Veteran's Healthcare System, Albuquerque, NM, United States
Cystic fibrosis (CF) is the most common life-threatening genetic disease in the United States and among people of European descent. Despite the widespread distribution of the cystic fibrosis transmembrane conductance regulator (CFTR) along kidney tubules, specific renal phenotypes attributable to CF have not been well documented. Recent studies have demonstrated the downregulation of the apical Cl-/HCO3- exchanger pendrin (Slc26a4) in kidney B-intercalated cells of CF mouse models. These studies have shown that kidneys of both mice and humans with CF have an impaired ability to excrete excess HCO3-, thus developing metabolic alkalosis when subjected to excess HCO3- intake. The purpose of this minireview is to discuss the latest advances on the role of pendrin as a molecule with dual critical roles in acid base regulation and systemic vascular volume homeostasis, specifically in CF. Given the immense prevalence of vascular volume depletion, which is primarily precipitated via enhanced chloride loss through perspiration, we suggest that the dominant presentation of metabolic alkalosis in CF is due to the impaired function of pendrin, which plays a critical role in systemic vascular volume and acid base homeostasis.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP/PKA-activated and ATP-regulated apical chloride channel that is widely expressed in epithelial tissues (1–3). Patients with CF are very prone to developing lung injury/infection, pancreatitis, and intestinal obstruction (1–3). While CFTR expression is detected in multiple nephron segments ranging from the proximal tubule to the thick ascending limb of Henle to the collecting duct, there is no discernible kidney phenotype attributed to CF. An original study in 2018 demonstrated that the HCO3- secreting exchanger, pendrin, is downregulated in B-intercalated cells in CF mouse kidneys, making the animals prone to develop metabolic alkalosis in response to oral HCO3- loading (4). More recent studies have confirmed and expanded on those findings by demonstrating the dysregulation of the kidney secretin receptor as a link between the oral bicarbonate load and the impaired renal HCO3- excretion, thus promoting the development of metabolic alkalosis (5, 6). The lack of HCO3- response to secretin (5, 6) is in complete agreement with the original studies in children with cystic fibrosis (7).
This mini review explores the pathogenesis of metabolic alkalosis in CF, specifically during vascular volume depletion, a phenomenon that is observed frequently in patients with CF. This can occur independent of HCO3- loading and is primarily dependent on the severe loss of chloride ions through perspiration.
Case presentation
A 24-year-old woman with CF was experiencing nausea, weakness, and general malaise, along with a decreased appetite for one week. She was admitted to the hospital following an abnormal lab test at an outpatient facility.
The patient had a BP of 100/60; PR of 82/min; BUN of 22 and serum creatinine of 1.2 mg/dl. The blood chemistry panel showed the following: Na+138 (reference 135-144), K+ 3.4 (reference 3.5 to 5.4), HCO3- 33 (reference 22-29), and Cl- 93 (reference 96-106) mEq/l.
A venous blood gas analysis showed: pH of 7.45 (reference for venous pH 7.32-7.42) and pCO2 of 45 mm Hg. The urine electrolytes were: Na+ 44, Cl- 36, and K+ 35 meq/l. The above presentation is consistent with metabolic alkalosis (elevated serum HCO3- concentration and blood pH values) along with respiratory compensation.
Discussion
Acid base abnormalities in CF
Although the primary acid base disorder in patients with CF is caused by impaired ventilatory gas exchanges leading to CO2 retention and chronic respiratory acidosis, the presence of metabolic alkalosis has been well documented in patients with CF for the last 5 decades (8–10). The preponderance of data has pointed to vascular volume depletion as the driving force in the generation of metabolic alkalosis in CF (8–10).
Recent studies have examined the pathogenesis of metabolic alkalosis in animal models and individuals with CF. These studies were mainly focused on acid base homeostasis in response to HCO3- loading. The first study in 2018 demonstrated that kidneys of CF mice had an impaired ability to excrete excess HCO3- during bicarbonate loading (4). In addition, there was a reduced expression of the HCO3- secreting transporter, pendrin (Slc26a4), in the kidneys of CF mice (4). Separate experiments in CF mice showed impaired HCO3- secretion in microperfused kidney collecting duct tubules (5). Follow up studies revealed that the kidney secretin receptor (Scrt) in the collecting duct is dysregulated in CF mice (5, 6), thus impairing the signal between the secretin secretion from the small intestine to a change in function of the kidney collecting ducts; and therefore, hampering the ability of the kidney to eliminate excess HCO3- (5–7). Whether the expression levels of secretin receptor are decreased in kidneys of either CF mice or patients with CF remain unknown.
There are no studies that have comprehensively looked at those CF individuals who have not been subjected to an oral HCO3- load, and yet present with metabolic alkalosis with increased frequency.
Published studies indicate that the two most common acid base disorders in cystic fibrosis are:
1. Chronic respiratory acidosis. This is presumed to be the most frequent acid base disorder owing to the impaired gas exchange at the alveolar level due to the damage caused by CF (8).
2. Metabolic alkalosis. Metabolic alkalosis is a frequent occurrence in adults with CF (8–13). Indeed, metabolic alkalosis could be the first manifestation of CF in toddlers and adolescents (9, 13).
The authors of the above study (8) concluded that metabolic alkalosis contributes to acute hypercapnic respiratory failure in adult CF individuals.
In a study examining acid base disorders in patients with chronic pulmonary dysfunction, the authors investigated acid base homeostasis in individuals with CF vs. chronic obstructive pulmonary disease (COPD) (12). The authors reported that in patients with comparable forced expiratory volume in 1 second (FEV1), majority of patients with CF, but not those with COPD, had metabolic alkalosis (12).
The analysis of acid base parameters in individuals with CF (12) revealed the presence of a primary metabolic alkalosis in 57% (8/14) of subjects, and a mixed metabolic alkalosis along with respiratory acidosis in 28% (4/14) of participants. The authors concluded that stable patients with CF lung are more prone to display metabolic alkalosis than in COPD patients (12).
In an original study, children in the Tucson area that had been diagnosed as having CF before the age of 12 months were examined to ascertain the prevalence of metabolic alkalosis as a major presenting manifestation of the disease (13). Five of eleven infants (46%) in whom CF had been diagnosed between 1 and 12 months of age were initially seen with metabolic alkalosis, hypokalemia, and hypochloremia, unassociated with major pulmonary and/or gastrointestinal symptoms. Two infants had repeated episodes of metabolic alkalosis; for one of these infants, both episodes of metabolic alkalosis occurred before the diagnosis of CF (13). Ground breaking studies had convincingly demonstrated that individuals with cystic fibrosis could lose vast amounts of salt (Cl- and Na+) in sweat, specifically in hot weather (14–16), which would precipitate vascular volume depletion. The enhanced loss of salt through sweat in individuals with CF was shown to be primarily due to a defective route of transcellular Cl- uptake in sweat duct epithelium (17). It was postulated that chronic loss of sweat electrolytes together with mild gastrointestinal or respiratory illness may predispose young individuals with CF to develop a severe electrolyte and acid-base disturbance (13). Taken together the above studies indicate that metabolic alkalosis is a frequent occurrence in cystic fibrosis and may be independent of the presence of lung damage or infection.
Cystic fibrosis and Pseudo Barter (Bartter-Like) syndrome
The excess loss of chloride (salt) through sweat (CF), gastric content (vomiting), or kidney (in individuals with Bartter Syndrome or treated with loop diuretics) is a well-known factor that results in vascular volume depletion and precipitates metabolic alkalosis (18–20). Unlike patients who suffer from gastric acid loss (vomiting) and present with volume depletion and a very low urine chloride, patients with Bartter Syndrome present with volume depletion, metabolic alkalosis, and increased urine chloride (18, 21). Patients with CF can exhibit volume depletion and increased (but not decreased) urine chloride, a phenomenon which has been referred to as Pseudo Bartter Syndrome (22). It should be noted that in severe instances of volume depletion, patients with CF could present with a low urine chloride consistent with enhanced absorption of chloride in more proximal nephron segments (11).
CFTR and kidney collecting duct
The expression of CFTR in collecting duct cells varies according to cell type. It is most abundantly expressed in HCO3- secreting B-intercalated cells, followed by principal cells, and with the lowest levels found in acid-secreting A-intercalated cells (23). In an original study in 2018, the expression of the apical Cl-/HCO3- exchanger, pendrin, was found to be significantly reduced, which resulted in impaired HCO3- excretion in response to an oral HCO3- load (4). These studies were confirmed by other investigators (5, 6). It was concluded that patients with CF are prone to the development of metabolic alkalosis when subjected to oral HCO3- loading secondary to the inactivation of the bicarbonate secreting transporter, pendrin in their kidneys (4–6). Additional investigations have shed more light on the link between oral HCO3- loading and the impaired ability of the CF kidney to eliminate excess HCO3- (5, 6). These experiments conducted in CF mice demonstrated dysregulation of Scrt (secretin receptor) in kidney B-intercalated cells (6). Taken together, these studies indicated that SCTR plays an important role in linking the circulating secretin with pendrin activation in kidney B-intercalated cells (5, 6).
Cystic fibrosis, vascular volume depletion, and pendrin
Pendrin plays a dual role in acid-base and vascular volume regulation by secreting HCO3- into the lumen of the collecting duct in exchange for absorbing chloride (24–32). As such, pendrin is critical to both vascular volume regulation and acid base homeostasis (28–32). Increased circulating aldosterone, either as a primary event or consequent to vascular volume depletion, increases pendrin function in part through its translocation to the apical membrane from the subapical region (28, 30–32). In pendrin-deficient mice, imposing vascular volume depletion was associated with the inability to absorb chloride and secret HCO3-, thus resulting in the generation of metabolic alkalosis (28, 30–33). In a remarkably similar manner, CF mice developed a significantly higher serum HCO3- concentration vs. wildtype (WT) animals when placed on a salt deficient diet (with the serum HCO3- concentration increasing to 29.2 mEq/L in CF mice vs. 26.7 in WT; p<0.03) further supporting the critical role that pendrin plays in both volume regulation and acid base balance (4).
Figure 1, left panel, is a schematic diagram depicting acid (H+) and base (HCO3-) secretion in A and B intercalated cells, respectievely, and electrolyte and water transport in principal cells in kidney collecting duct cells. As indicated in Figure 1, right panel, pendrin mediates chloride absorption and HCO3- secretion in B intercalated cells and shows overactivation in volume depleted state. As such, the impaired ability of pendrin to absorb luminal chloride and secret HCO3- results in an excessive loss of chloride into urine in the setting of vascular volume depletion, thus worsening the magnitude of alkalosis (Pseudo Bartter Syndrome).
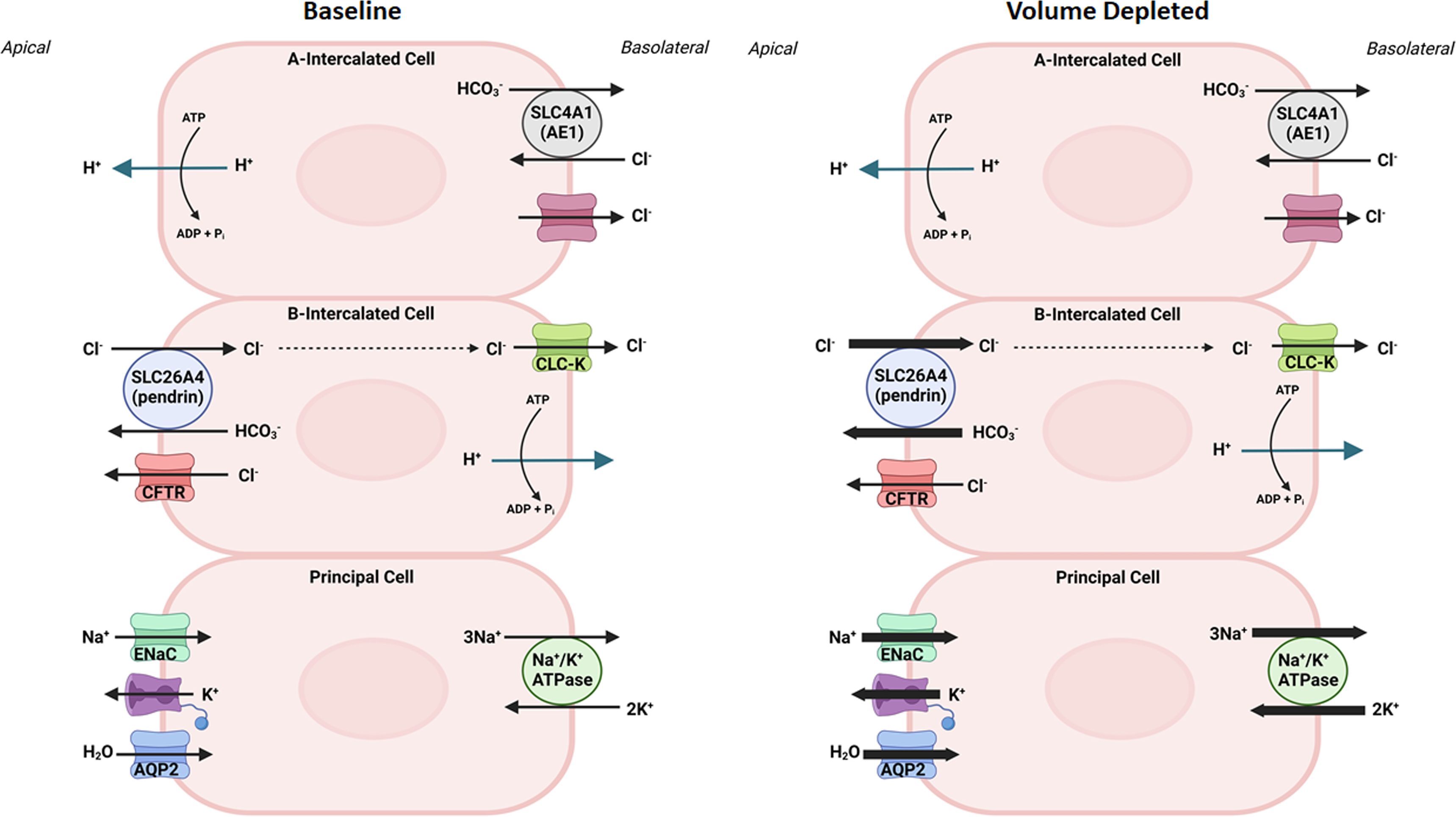
Figure 1 The role of collecting duct cells in acid base and vascular volume homeostasis. Left panel, is a schematic diagram indicating acid (H+) and base (HCO3-) secretion in A- and B- intercalated cells, and electrolyte and water transport in principal cells in kidney collecting duct cells at baseline state. Right panel is a schematic diagram depicting the activation of pendrin in chloride absorption and HCO3- secretion in the kidney B-intercalated cells, and ENaC in sodium absorption in principal cells in volume depleted states.
The role of CFTR in epithelial physiology
Cystic fibrosis affects the integrity and functional properties of epithelial cells of several organs, including the respiratory tract, exocrine pancreas, intestine, vas deferens, hepatobiliary system, and exocrine sweat glands.
Published studies indicate that:
1. CFTR plays a critical role in regulating luminal pH and maintaining epithelial surface hydration (34, 35).
2. The activation of CFTR enhances HCO3- secretion across the luminal membrane in airway epithelia, pancreatic ducts, intestines, and several other epithelial tissues (1–3, 34–41). It was suggested that bicarbonate transport mediated via an anion exchange mechanism may be defective in tissues from individuals with cystic fibrosis (42). A critical discovery revealed that in addition to a defective Cl- transport, CFTR mutations disrupt the transport of HCO3- in various tissues in cystic fibrosis (43), highlighting the pathophysiological role of impaired HCO3- secretion in cystic fibrosis (44).
3. Luminal pH plays an important role in epithelial barrier function and innate defense, particularly in the airways and GI tract (34, 35, 40, 41, 45).
The role of SLC26 family of Cl-/HCO3- exchangers in CFTR-activated HCO3- secretion in epithelial tissues
The activation of CFTR in pancreatic duct cells, airway epithelia, and the intestine enhances HCO3- secretion. Intense research on the role of CFTR activation in HCO3- secretion has identified several apical Cl-/HCO3- exchangers from the anion transporting SLC26 family that physically interact and function in tandem with CFTR, thus contributing to enhanced HCO3- transport into the lumen of epithelial cells (46, 47). These apical Cl-/HCO3- exchangers include SLC26A3 (DRA), SLC26A4 (pendrin) and SLC26A6 (PAT1) (48–50) and interact with the CFTR molecule through their STAS domain (46–49). The schematic diagram in Figure 2 depicts a remarkable similarity between the molecular machineries that facilitate HCO3- and Cl- secretion in response to CFTR activation in airway epithelia (A), pancreatic ducts (B), and enterocytes of small intestine (C). It is plausible that the kidney B-intercalated cells may exhibit a similar activating interaction between CFTR and the Cl-/HCO3- exchanger, pendrin (Figure 2D). As noted, CFTR mutations result in a widespread HCO3- secretion defect in the epithelial cells of the airways, pancreatic ducts, intestines, and the kidney collecting duct (4–6, 34–45). In addition to interacting with and stimulating the SLC26 Cl-/HCO3- exchangers, a large number of studies support the conclusion that CFTR, when activated, becomes permeant to both Cl- and HCO3- (reviewed in 51). Recent publications indicate that permeability of CFTR to various anions can be modulated by the WNK-SPAK pathway (51). The schematic diagrams in Figures 2A–C depicts the dual functional roles of CFTR in transporting Cl- and HCO3- ions in an activated state in the pancreatic duct, enterocytes, and alveolar cells.
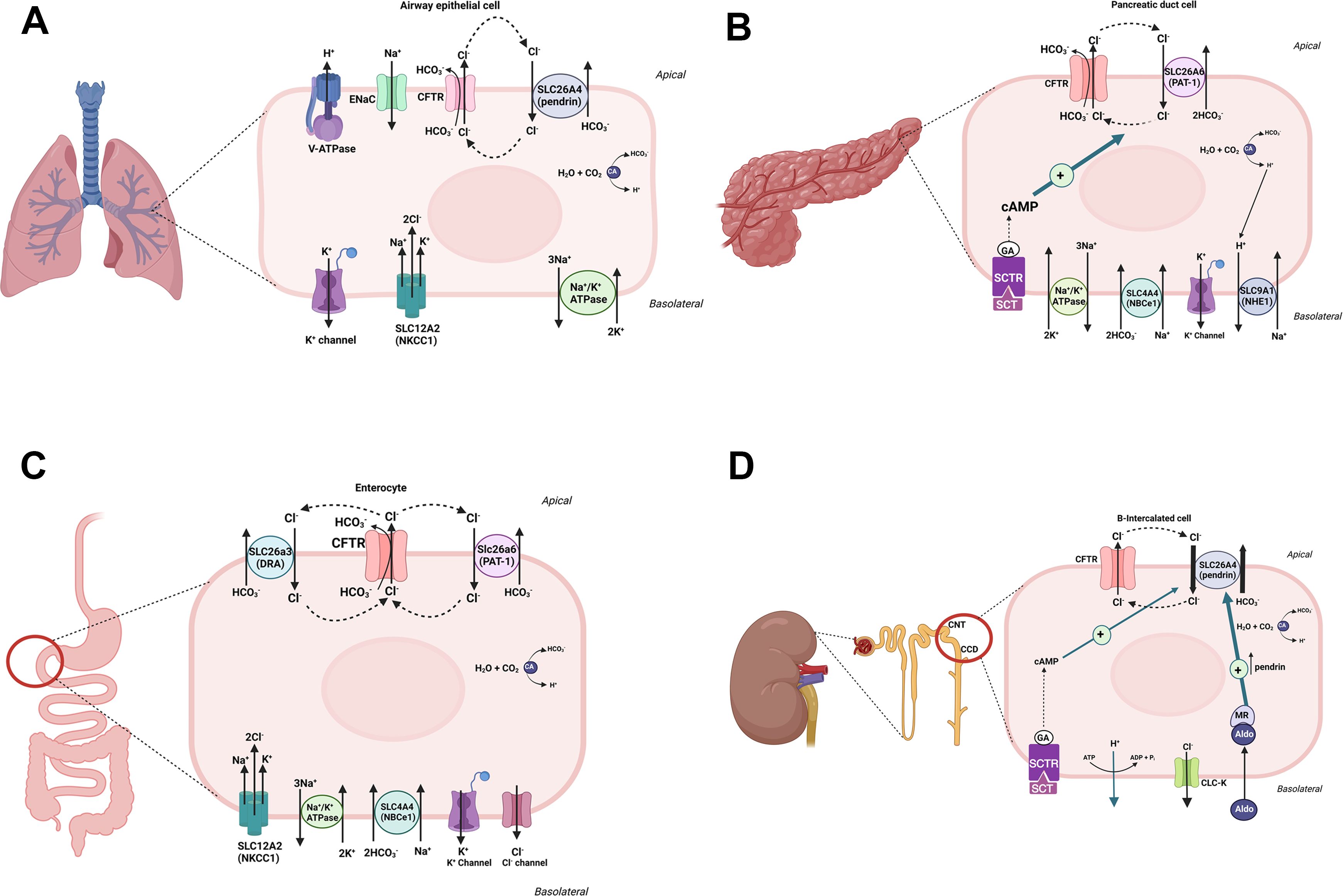
Figure 2 A schematic diagram depicting CFTR-dependent HCO3- secretion in alveolar cells (A), pancreatic duct (B), small intestine (C) and kidney B-intercalated cells (D). As noted, CFTR, when activated, may become permeant to both Cl- and HCO3- ions in alveolar, pancreatic duct and enterocytes. In kidney B intercalated cells, the role of pendrin in absorbing chloride and secreting HCO3- is paramount. The roles of aldosterone, which is critical in pendrin translocation and activation in volume depleted states, and the SCTR, which links secretin to pendrin-mediated HCO3- secretion are highlighted in kidney B-intercalated cells (D).
Cystic fibrosis, pendrin and oral HCO3- load
Excess HCO3- intake in WT mice results in an enhanced expression of pendrin along with increased HCO3- excretion (26, 27). A similar maneuver in CF mice, which display significant downregulation of pendrin, resulted in impaired HCO3- excretion along with the development of metabolic alkalosis (4–6, 36). These results indicate that a functional pendrin plays a sizeable role in preventing the generation of metabolic alkalosis in response to oral HCO3- loading. Based on the critical role of SCTR in activating pendrin-mediated HCO3- secretion in kidney B-intercalated (B-IC) cells in response to oral base (e.g., HCO3-) loading, recent studies suggest that loss of SCTR in CF impairs the appropriate increase of renal base excretion during acute base loading and that SCTR is necessary for rapid correction of metabolic alkalosis (5, 36). Extrapolated from these studies (5–7), it was proposed that the prevalent metabolic alkalosis in patients with CF could be explained by the absence of secretin-induced urinary HCO3- excretion (36). It was further suggested that patients with CF could be given a urine pH test after oral HCO3- loading to validate the efficacy of CFTR modulators in treating cystic fibrosis. Recent studies point to the impaired ability of CF patients to increase their urine HCO3- excretion during the presence of metabolic alkalosis (52).
It should be noted that while CF mice do not recapitulate several phenotypic presentations of individuals with CF (such as airway defect or male infertility), the GI and kidney manifestations in CF mice mirror those defects in human CF. In a search for non-primate models of CF, studies show that the porcine CF model exhibits a strong phenotypic resemblance to humans with CF in that the porcine model shows bacterial colonization, as well as airway anomalies at birth and the impaired ability of CF piglets to clear bacteria (53, 54). It would be enlightening to examine the role of pendrin and HCO3- secretion pathways in kidneys of porcine CF models. This issue becomes critical because it can shed light on whether lung infection/injury plays any role in the generation of metabolic alkalosis in cystic fibrosis.
As discussed in this minireview, the major determinant of metabolic alkalosis generation in CF is the development of vascular volume depletion, a process that is unencumbered by and independent of oral HCO3- load (8–13). It should further be noted that due to multiple medical ailments, patients with CF may exhibit a false positive response (impaired excretion of HCO3-) to the oral base loading test due to CF-independent tubular abnormalities. Amongst these medical maladies are:
1. Frequent lung infections that require multiple and aggressive antimicrobial therapy, which can impact kidney tubular function and the glomerular filtration rate (GFR).
2. Development of pancreatic insufficiency and diabetes mellitus, which can lead to malabsorption in patients with CF.
3. Moreover, patients with CF are at an increased risk for acute kidney injury due to the use of nephrotoxic agents, development of vascular volume depletion, or both, which can impair the physiological functions of kidney tubules and produce false positive (impaired HCO3- excretion) results when testing for HCO3- excretion.
4. Severe vascular volume depletion and the resulting decreased kidney perfusion can interefer with excess HCO3- excretion in individuals subjected to enhanced base load, whether or not they have CF.
5. Furthermore, known electrolyte abnormalities such as hypokalemia consequent to vascular volume depletion or due to the development of hypomagnesemia –either secondary to malabsorption or aminoglycoside-induced pseudo–Bartter Syndrome (14, 55–57)- can decrease pendrin expression and impair HCO3- excretion following base loading.
6. Lastly, pharmaceutical agents such as carbonic anhydrase inhibitors can downregulate kidney pendrin in a CF-independent manner (55).
Taken together, there are multiple medical maladies, as listed above, that are independent of CF that can interfere with enhanced excretion of oral HCO3- load.
Conclusion
In the kidney collecting duct, the Cl-/HCO3- exchanger, pendrin (SLC26A4), significantly contributes to the maintenance of systemic vascular volume and blood pH during vascular volume depletion by absorbing luminal chloride in exchange for HCO3- secretion. This important defensive mechanism is hampered in individuals with CF, thus making them prone to the development of metabolic alkalosis and worsening of vascular volume depletion by excessive loss of salt in their kidneys (Pseudo Bartter Syndrome). In addition, kidneys of patients with CF may be ill-equipped to unload excess blood HCO3- concentration via pendrin due to the impaired cross talk between the circulating secretin and kidney B-intercalated cells. Please see the schematic diagram in kidney B-intercalated cells (Figure 2D). While the optimal treatment for the correction of metabolic alkalosis would be via the rectification of the underlying defect in the CFTR, the treatment of systemic volume depletion will go a long way to ameliorate existing metabolic alkalosis in patients with CF.
Author contributions
MS: Conceptualization, Formal analysis, Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. These studies were supported by a Merit Review Award 5I01BX001000-11.
Acknowledgments
Figures were created by Sharon Barone on Biorender. The author acknowledges the critical review of this manuscript by Sharon Barone and Kamyar Zahedi. Manoocher Soleimani is a Senior Clinician Scientist Investigator with the Veterans Health Administration Department.
Conflict of interest
The authors declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Castellani C, Assael BM. Cystic fibrosis: a clinical view. Cell Mol Life Sci. (2017) 74:129–40. doi: 10.1007/s00018-016-2393-9
2. Angyal D, Bijvelds MJC, Bruno MJ, Peppelenbosch MP, de Jonge HR. Bicarbonate transport in cystic fibrosis and pancreatitis. Cells. (2023) 11:54. doi: 10.3390/cells11010054
3. Kelly T, Buxbaum J. Gastrointestinal manifestations of cystic fibrosis. Dig Dis Sci. (2015) 60:1903–13. doi: 10.1007/s10620-015-3546-7
4. Varasteh Kia M, Barone S, McDonough AA, Zahedi K, Xu J, Soleimani M. Downregulation of the cl-/HCO3-exchanger pendrin in kidneys of mice with cystic fibrosis: role in the pathogenesis of metabolic alkalosis. Cell Physiol Biochem. (2018) 45:1551–65. doi: 10.1159/000487691
5. Berg P, Svendsen SL, Sorensen MV, Larsen CK, Andersen JF, Jensen-Fangel S, et al. Impaired renal HCO(3)(-) excretion in cystic fibrosis. J Am Soc Nephrol. (2020) 31:1711–27. doi: 10.1681/ASN.2020010053
6. Berg P, Andersen JF, Sørensen MV, Wang T, Malte H, Leipziger J. Alkalosis-induced hypoventilation in cystic fibrosis: The importance of efficient renal adaptation. Proc Natl Acad Sci U.S.A. (2022) 119:e2116836119. doi: 10.1073/pnas.2116836119
7. Bretscher D, Schneider A, Hagmann R, Hadorn B, Howald B, Luthi C, et al. Response of renal handling of sodium (Na) and bicarbonate (HCO3-) to secretin (S) in normals and patients with cystic fibrosis (CF). Pediatr Res. (1974) 8:899. doi: 10.1203/00006450-197411000-00031
8. Holland AE, Wilson JW, Kotsimbos TC, Naughton MT. Metabolic Alkalosis contributes to acute hypercapnic respiratory failure in adult cystic fibrosis. Chest Aug;. (2003) 124:490–3. doi: 10.1378/chest.124.2.490
9. Bates CM, Baum M, Quigley R. Cystic fibrosis presenting with hypokalemia and metabolic alkalosis in a previously healthy adolescent. J Am Soc Nephrol. (1997) 8:352–5. doi: 10.1681/ASN.V82352
10. Scurati-Manzoni E, Fossali EF, Agostoni C, Riva E, Simonetti GD, Zanolari-Calderari M, et al. Electrolyte abnormalities in cystic fibrosis: systematic review of the literature. Pediatr Nephrol. (2014) 29:1015–23. doi: 10.1007/s00467-013-2712-4
11. Ghimire S, Yerneni H, Oyadomari TA, Sedlacek M. Metabolic alkalosis and cystic fibrosis: A case report. Ann Intern Med. (2020) 173:315. doi: 10.7326/L20-0038
12. Al-Ghimlas F, Faughnan ME, Tullis E. Metabolic alkalosis in adults with stable cystic fibrosis. Open Respir Med J. (2012) 6:59–62. doi: 10.2174/1874306401206010059
13. Beckerman RC, Taussig LM. Hypoelectrolytemia and metabolic alkalosis in infants with cystic fibrosis. Pediatr Apr;. (1979) 63:580–3. doi: 10.1542/peds.63.4.580
14. Kessler WR, Andersen DH. Heat prostration in fibrocystic disease of the pancreas and other conditions. Pediatrics. (1951) 8:648–56. doi: 10.1542/peds.8.5.648
15. Di Sant’Agnese PS, Darling RC, Perera GA, Shea E. Sweat and electrolyte disrutbances associated with childhood pancreatic disease. Am J Med. (1953) 15:777–84. doi: 10.1016/0002-9343(53)90169-7
16. Webb BW, Flute PT, Smith MJ. The electrolyte content of the sweat in fibrocystic disease of the pancreas. Arch Dis Child.(1957) 32(162):82–4. doi: 10.1136/adc.32.162.82
17. Bijman J, Quinton PM. Influence of abnormal Cl- impermeability on sweating in cystic fibrosis. Am J Physiol. (1984) 247:C3–9. doi: 10.1152/ajpcell.1984.247.1.C3
18. Do C, Vasquez PC, Soleimani M. Metabolic alkalosis pathogenesis, diagnosis, and treatment: core curriculum 2022. Am J Kidney Dis. (2022) 80:536–51. doi: 10.1053/j.ajkd.2021.12.016
19. Nagami GT, Kraut JA. The role of the endocrine system in the regulation of acid-base balance by the kidney and the progression of chronic kidney disease. Int J Mol Sci. (2024) 25:2420. doi: 10.3390/ijms25042420
20. Kurtz I, Kraut J, Ornekian V, Nguyen MK. Acid-base analysis: a critique of the Stewart and bicarbonate-centered approaches. Am J Physiol Renal Physiol. (2008) 294:F1009–31. doi: 10.1152/ajprenal.00475.2007
21. Florea L, Caba L, Gorduza EV. Genetic heterogeneity in bartter syndrome: clinical and practical importance. Front Pediatr Jun 3;. (2022) 10:908655. doi: 10.3389/fped.2022.908655
22. Lopes CF, Almeida V, Gomes S, Cruz C. Pseudo-Bartter syndrome as the initial presentation of cystic fibrosis in children: an important diagnosis not to be missed. BMJ Case Rep. (2024) 17:e257348. doi: 10.1136/bcr-2023-257348
23. Todd-Turla KM, Rusvai E, Náray-Fejes-Tóth A, Fejes-Tóth G. CFTR expression in cortical collecting duct cells. J Physiol. (1996) 270:F237–44. doi: 10.1152/ajprenal.1996.270.1.F237
24. Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, et al. Pendrin: an apical Cl-/OH-/HCO3– exchanger in the kidney cortex. Am J Physiol Renal Physiol. (2001) 280:F356–64. doi: 10.1152/ajprenal.2001.280.2.F356
25. Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, et al. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA. (2001) 98:4221–6. doi: 10.1073/pnas.071516798
26. Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, et al. Regulation of the expression of the Cl-/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. (2002) 62:2109–17. doi: 10.1046/j.1523-1755.2002.00671.x
27. Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschênes G, et al. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol. (2006) 17:2153–63. doi: 10.1681/ASN.2005101054
28. Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, et al. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension. (2004) 44:982–7. doi: 10.1161/01.HYP.0000145863.96091.89
29. Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, et al. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(-)/HCO3(-) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol. (2010) 299:C33–41. doi: 10.1152/ajpcell.00033.2010
30. Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, et al. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoidinduced hypertension. Hypertension. (2003) 42:356–62. doi: 10.1161/01.HYP.0000088321.67254
31. Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, et al. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA. (2012) 109:13368–73. doi: 10.1073/pnas.1202671109
32. Wall SM. Regulation of blood pressure and salt balance by pendrin-positive intercalated cells: donald seldin lecture 2020. Hypertension. (2022) 79:706–16. doi: 10.1161/HYPERTENSIONAHA.121.16492
33. Alshahrani S, Soleimani M. Ablation of the cl-/HCO3- exchanger pendrin enhances hydrochlorothiazide-induced diuresis. Kidney Blood Press Res. (2017) 42:444–55. doi: 10.1159/000479296
34. Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cell Mol Life Sci. (2017) 74:93–115. doi: 10.1007/s00018-016-2391-y
35. Ahn W, Kim KH, Lee JA, Kim JY, Choi JY, Moe OW, et al. Regulatory interaction between the cystic fibrosis transmembrane conductance regulator and HCO3- salvage mechanisms in model systems and the mouse pancreatic duct. J Biol Chem. (2001) 276:17236–43. doi: 10.1074/jbc.M011763200
36. Berg P, Svendsen SL, Ayasse N, Sorensen MV, Leipziger J. Secretin: a hormone for HCO3- homeostasis. J.Pflugers Arch. (2024) 476(4):545–54. doi: 10.1007/s00424-024-02906-3
37. Shumaker H, Amlal H, Frizzell R, Ulrich CD II, Soleimani M. CFTR drives Na+-nHCO-3 cotransport in pancreatic duct cells: a basis for defective HCO-3 secretion in CF. Am J Physiol. (1996) 276:C16–25. doi: 10.1152/ajpcell.1999.276.1.C16
38. Greeley T, Shumaker H, Wang Z, Schweinfest CW, Soleimani M. Downregulated in adenoma and putative anion transporter are regulated by CFTR in cultured pancreatic duct cells. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G1301–8. doi: 10.1152/ajpgi.2001.281.5.G1301
39. Song Y, Yamamoto A, Steward MC, Ko SB, Stewart AK, Soleimani M, et al. Deletion of Slc26a6 alters the stoichiometry of apical Cl-/HCO3- exchange in mouse pancreatic duct. Am J Physiol Cell Physiol. (2012) 303:C815–24. doi: 10.1152/ajpcell.00151.2012
40. Tse CM, Yin J, Singh V, Sarker R, Lin R, Verkman AS, et al. cAMP stimulates SLC26A3 activity in human colon by a CFTR-dependent mechanism that does not require CFTR activity. Cell Mol Gastroenterol Hepatol. (2019) 7:641–53. doi: 10.1016/j.jcmgh.2019.01.002
41. Seidler U, Nikolovska K. Slc26 family of anion transporters in the gastrointestinal tract: expression, function, regulation, and role in disease. Compr Physiol. (2019) 9:839–72. doi: 10.1002/cphy.c180027
42. Quinton PM. Suggestion of an abnormal anion exchange mechanism in sweat glands of cystic fibrosis patients. Pediatr Res Jul;. (1982) 16:533–7. doi: 10.1203/00006450-198207000-00008
43. Choi JY, Muallem D, Kiselyov K, Lee MG, Thomas PJ, Muallem S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature. (2001) 410:94–7. doi: 10.1038/35065099
45. Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. (2008) 372:415–7. doi: 10.1016/S0140-6736(08)61162-9
46. Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. (2004) 6:343–50. doi: 10.1038/ncb1115
47. Gray MA. Bicarbonate secretion: it takes two to tango. Nat Cell Biol. (2004) 6:292–4. doi: 10.1038/ncb0404-292
48. Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. (2013) 34:494–515. doi: 10.1016/j.mam.2012.07.009
49. Soleimani M. SLC26 Cl-/HCO3- exchangers in the kidney: roles in health and disease. Kidney Int. (2013) 84:657–66. doi: 10.1038/ki.2013.138
50. Aronson PS. Ion exchangers mediating Na+, HCO3 - and Cl- transport in the renal proximal tubule. J Nephrol. (2006) 19:S3–S10.
51. Angyal D, Bijvelds MJC, Bruno MJ, Peppelenbosch MP, de Jonge HR. Bicarbonate transport in cystic fibrosis and pancreatitis. Cells. (2021) 11:54. doi: 10.3390/cells11010054
52. Spoletini G, Fitch G, Gillgrass L, Etherington C, Clifton I, Peckham DG. Urinary bicarbonate and metabolic alkalosis during exacerbations in cystic fibrosis. ERJ Open Res. (2022) 8(2):00669-2021. doi: 10.1183/23120541.00669-2021
53. Stolz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. (2010) 2:29ra31. doi: 10.1126/scitranslmed.3000928
54. Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, et al. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med. (2010) 182:1251–61. doi: 10.1164/rccm.201004-0643OC
55. Soleimani M. The multiple roles of pendrin in the kidney. Nephrol Dial Transplant. (2015) 30:1257–66. doi: 10.1093/ndt/gfu307
56. Rao R, Bhalla V, Pastor-Soler NM. Intercalated cells of the kidney collecting duct in kidney physiology. Semin Nephrol. (2019) 39:353–67. doi: 10.1016/j.semnephrol.2019.04.005
Keywords: hypokalemia, Barter-like syndrome, diabetes mellitus, nephrotoxicity, acute kidney injury, kidney tubules
Citation: Soleimani M (2024) Metabolic alkalosis in cystic fibrosis: from vascular volume depletion to impaired bicarbonate excretion. Front. Endocrinol. 15:1411317. doi: 10.3389/fendo.2024.1411317
Received: 02 April 2024; Accepted: 23 July 2024;
Published: 07 August 2024.
Edited by:
Patryk Lipiński, Maria Sklodowska-Curie Medical Academy, PolandReviewed by:
Bruce D. Schultz, Kansas State University, United StatesJens Leipziger, Aarhus University, Denmark
Copyright © 2024 Soleimani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoocher Soleimani, TVNvbGVpbWFuaUBzYWx1ZC51bm0uZWR1; TWFub29jaGVyLlNvbGVpbWFuaUBWYS5Hb3Y=