
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 24 April 2024
Sec. Pediatric Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1385901
This article is part of the Research Topic46,XX Differences of Sex Development (DSD) outside Congenital Adrenal Hyperplasia (CAH)View all 7 articles
 Maria Tereza Martins Ferrari1
Maria Tereza Martins Ferrari1 Elinaelma Suelane do Nascimento Silva1Mirian Yumie Nishi1
Elinaelma Suelane do Nascimento Silva1Mirian Yumie Nishi1 Rafael Loch Batista1Berenice Bilharinho Mendonca1,2†
Rafael Loch Batista1Berenice Bilharinho Mendonca1,2† Sorahia Domenice1*†
Sorahia Domenice1*†In mammals, the development of male or female gonads from fetal bipotential gonads depends on intricate genetic networks. Changes in dosage or temporal expression of sex-determining genes can lead to differences of gonadal development. Two rare conditions are associated with disruptions in ovarian determination, including 46,XX testicular differences in sex development (DSD), in which the 46,XX gonads differentiate into testes, and 46,XX ovotesticular DSD, characterized by the coexistence of ovarian and testicular tissue in the same individual. Several mechanisms have been identified that may contribute to the development of testicular tissue in XX gonads. This includes translocation of SRY to the X chromosome or an autosome. In the absence of SRY, other genes associated with testis development may be overexpressed or there may be a reduction in the activity of pro-ovarian/antitesticular factors. However, it is important to note that a significant number of patients with these DSD conditions have not yet recognized a genetic diagnosis. This finding suggests that there are additional genetic pathways or epigenetic mechanisms that have yet to be identified. The text will provide an overview of the current understanding of the genetic factors contributing to 46,XX DSD, specifically focusing on testicular and ovotesticular DSD conditions. It will summarize the existing knowledge regarding the genetic causes of these differences. Furthermore, it will explore the potential involvement of other factors, such as epigenetic mechanisms, in developing these conditions.
Gonadal development is a fundamental step in forming the reproductive system, and several diseases are associated with atypical gonadal development. The determination and differentiation of gonads from the bipotential gonadal primordium can be triggered by a combination of genetic and environmental factors, making it a species-specific process among vertebrates (1).
In mammals, sex is determined by genetic heritage during fertilization. The differentiation of fetal bipotential gonads into testes or ovaries occurs through the action of specific genetic networks. These developmental pathways are typically distinct, mutually exclusive, and driven by a complex interchange of antagonistic genes (2). Changes in the dosage and/or spatiotemporal expression of sex-determining genes can lead to disruptions in the typical development of male or female gonads, causing differences in sex development (DSD). In rare conditions, testicular tissue can develop into an XX gonad, resulting in the condition called 46,XX ovotesticular or testicular DSD.
Testicular DSD (T DSD) has an estimated frequency of 1:20,000 to 1:25,000 newborn boys. These conditions account for about 2% of cases of male infertility. In about 80% of affected individuals, the genital male phenotype appears typical at birth, but diagnosis usually occurs during or after puberty due to symptoms such as gynecomastia, hypogonadism, and infertility (3). However, in some cases, individuals may present with atypical external genitalia, which enables for earlier investigation and evaluation. The severity of the condition depends on the extent of testicular tissue development.
Ovotesticular DSD (OT DSD) is a rare form of DSD, with an estimated incidence of 1:100,000 births (4). This condition is characterized by the presence of both male gonadal tissues, with well-developed seminiferous tubules, and female gonadal tissue, with primordial follicles, within the same individual. In some patients, both types of gonadal tissues may be present in the same gonad, which is referred to as an ovotestis (5). The 46,XX karyotype is the most commonly identified chromosomal pattern in OT DSD, accounting for 65 to 90% of patients (6–8).
Most of the affected individuals present with atypical genitalia at birth. Individuals assigned as males at birth might experience breast development and/or cyclic hematuria. Similarly, individuals assigned as females may exhibit breast development and menstrual irregularities and/or signs of masculinization (9–11).
Although most cases of TDSD and OTDSD are sporadic, there are reports in the literature of individuals with both conditions occurring in the same family. This suggests that a common genetic origin may contribute to the development of these conditions (12–15).
Gonad development initially follows a similar trajectory in both XX and XY fetuses, with a bipotential gonad being formed from the urogenital crest. After the formation of the bipotential gonad, the processes involved in sex determination guide the development of sex-specific gonadal structures. In male development, there is an interaction between a network of pro-testis genes that promote the differentiation of the bipotential XY gonads into testes. Conversely, in female development, a network of pro-ovarian genes interacts to differentiate the XX bipotential gonads into ovaries (16).
In humans, the genital ridge first emerges between the fourth and fifth weeks of pregnancy. During this period, coelomic epithelial cells undergo proliferation on the ventromedial surface of the mesonephros. This proliferation process is tightly regulated by numerous genes and involves coordinated activity, which leads to the formation of bipotential gonads (17–19) (Figure 1).
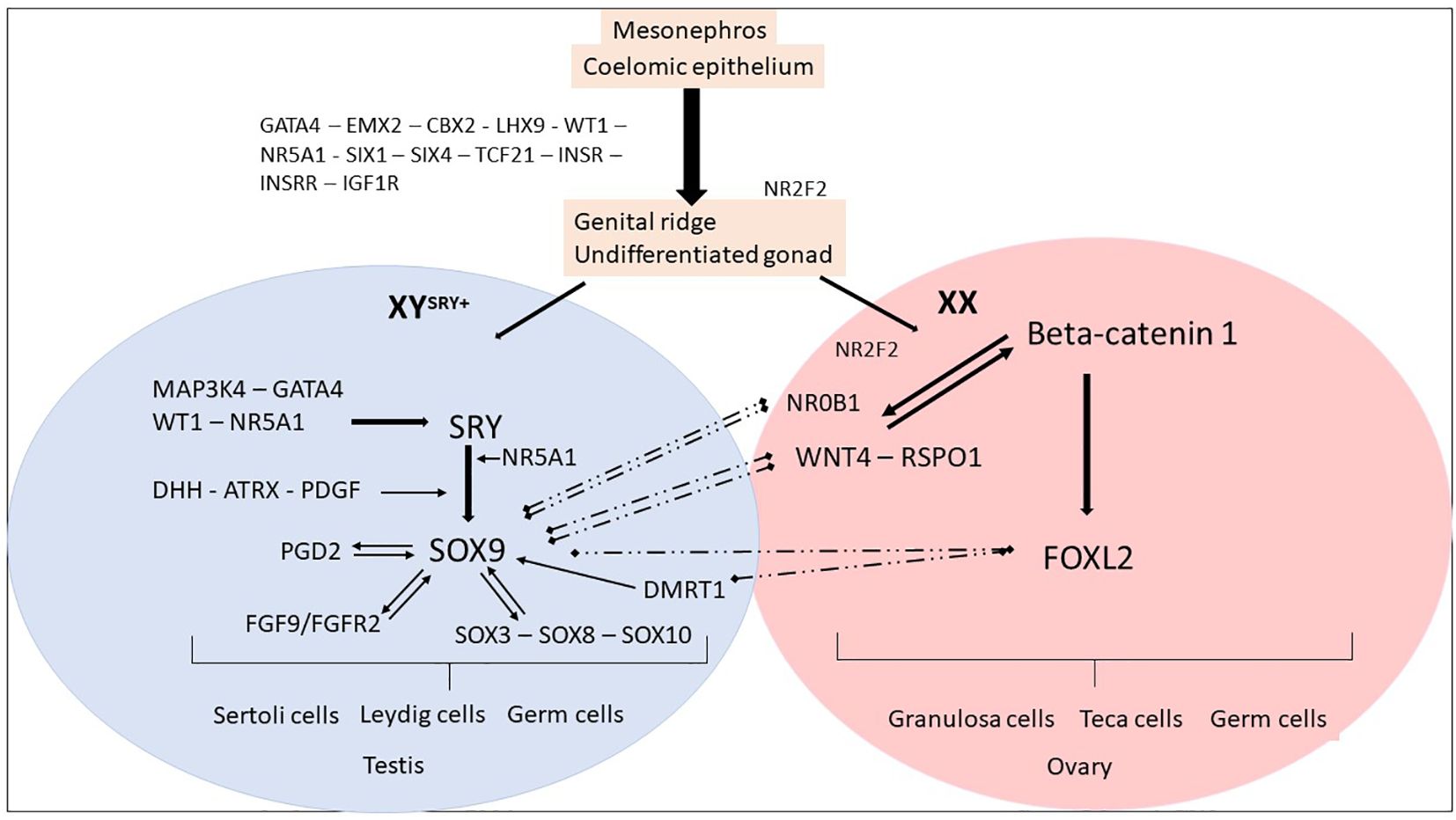
Figure 1 Genes and mechanisms of sex determination. The proliferation of cells from the mesonephros and coelomic epithelium leads to the formation of an undifferentiated and bipotential gonad. This process is regulated by several factors, including GATA4, EMX2, CBX2, LHX9, and WT1. These factors, in turn, regulate NR5A1, SIX1, SIX4, TCF21, and members of the IGF family, leading to the formation of an undifferentiated gonad. The process continues until the fate of the gonad is established, resulting in the formation of either an ovary or a testis. Ovarian differentiation. In the XX fetuses (absence of SRY), the expression of SOX9 remains low and other factors such as NR0B1, FOXL2, WNT4, and RSPO1 become dominant. The upregulation of WNT4 and RSPO1 leads to the activation of the canonical WNT signaling pathway, which in turn upregulates and stabilizes β-catenin. The activation of the WNT/β-catenin pathway plays a crucial role in the differentiation of the female gonad. NR2F2 has a role in maintaining a multipotent state in early supporting gonadal cells, which seems to be necessary for commitment to ovarian development. After birth, FOXL2 continues to suppress male-specific factors, including SOX9 and DMRT1. Testicular differentiation. In XY fetuses, the expression of SRY is triggered by MAP3K4, GATA4, WT1, and NR5A1. The presence of SRY and NR5A1 initiates the expression of SOX9, which leads to the differentiation of pre-Sertoli cells and subsequent Sertoli cells. Other members of the SOX family are also upregulated. SOX9 expression is maintained through positive feedback loops involving FGF9 and PGD2, as well as the regulation from WT1 and NR5A1. The increased expression of SOX9 prevails over NR0B1, FOXL2, WNT4, and RSPO1, promoting testicular differentiation. After birth, DMRT1 suppresses the female-specific factor FOXL2. These interactions between the male and female pathways remain essential throughout adulthood to maintain the gonadal identity.
In mice, null mutations in genes such as Emx2 (Empty Spiracles Homeobox 2), Cbx2 (Chromobox protein homolog 2), Gata4 (GATA Binding Protein 4), Lhx9 (LIM homeobox 9), Wt1 (Wilms tumor 1), and Nr5a1 (Nuclear Receptor Subfamily 5 Group A Member 1) result in regression and changes in the development of the gonadal ridge. Coelomic epithelial cells differentiate into two distinct somatic precursor lineages: supportive cell precursors and steroidogenic cell precursors (20, 21).
Concurrently, primordial germ cells migrate from the yolk sac along the hindgut and dorsal mesentery to colonize the gonad (22). The interaction between somatic and germ cells and signaling from somatic cells is essential for the proliferation and differentiation of primordial germ cells. Furthermore, the female germ cells play a role in maintaining the ovary (23). Subsequently, the bipotential gonad differentiates into testis and ovary, respectively, through a sex-related genes antagonistic network.
In bipotential gonadal tissue of XX individuals, the process of ovarian determination is initiated by a cooperative network of pro-ovarian genes, which includes WNT4 (Wingless Type MMTV integration site family, member 4), RSPO1 (R-Spondin1), and FOXL2 (Forkhead box L2) (24–26) (Table 1). These factors not only activate genes required for ovarian development but also repress pro-testis gene expression (27). In XX individuals, WNT4 and RSPO1 initially direct ovarian determination by upregulating and stabilizing the beta-catenin signaling pathway. CTNNB1 (Catenin Beta 1) essentially promotes germ cell proliferation and granulosa cell differentiation (25, 28). RSPO1, through CTNNB1, prevents WNT4 degradation to maintain ovarian fate (25). FOXL2 expression is initiated in the supporting somatic cells of bipotential gonads, in conjunction with WNT4 and RSPO1 (Figure 1).

Table 1 Genes associated with testicular development in 46,XX DSD patients.
FOXL2 is required throughout ovarian development and into adulthood to maintain granulosa cell differentiation and support folliculogenesis (2, 29). Foxl2 performs these functions through several mechanisms, such as interacting with ovarian pathway genes, Fst (Follistatin) and Cyp19a1 (cytochrome P450 family 19 subfamily A member 1) (30) and binding to a Sox9 enhancer to reduce Sox9 expression (31). CTNNB1 also promotes the repression of SOX9 expression. The genes involved in ovarian determination tend to show their expression a little later in the process of bipotential gonadal differentiation than the genes of the testicular pathway (32).
In individuals with XY chromosomes, the SRY gene triggers the cascade of testicular differentiation (33), regulated by WT1 (34), NR5A1 (35), CBX (36), GATA4 (37) and its co-factor ZFPM2 (Zinc Finger Protein, FOG Family Member 2) (38), inducing the expression of the SOX9 gene (39) (Figure 1). SOX9 expression is upregulated immediately after SRY expression in the supporting cells of the developing testis and marks their differentiation into Sertoli cells (40). Subsequently, SOX9 plays a central role in regulating the expression of various genes involved in male sexual differentiation, such as FGF9/FGF2R (Fibroblast Growth Factor/Fibroblast Growth Factor Receptor 2) (41), PTGDS (Prostaglandin D2 Synthase) (42), and AMH (Anti-Mullerian Hormone). Like SRY, the activity of SOX9 is both necessary and sufficient to induce testis development in the genital ridges (43, 44) (Figure 1). Indeed, SOX9 prevents the expression of genes inducing the ovarian differentiation, such as RSPO1 and FOXL2 (45, 46). Other genes, including MAP3K1 (Mitogen-Activated Protein Kinase Kinase Kinase 1) (47, 48), WWOX (WW Domain Containing Oxidoreductase) (49), DMRT1 (Doublesex and Mab-3 Related Transcription factor 1) (50) and DHX37 (DEAH-Box Helicase 37) (51), have been added as participants in the testicular determination pathway after the identification of deleterious point mutations or copy number alterations associated with the phenotype of differences of testicular differentiation in humans and mice (45).
WNT4 (1p36.12) encodes a glycoprotein that plays multiple roles in ovarian differentiation and Müllerian duct formation (52) (Table 1). It is modulated by RSPO1 and acts by decreasing the phosphorylation and degradation of β-catenin. Increased levels of β-catenin antagonize SOX9, leading to upregulation of DAX1, which in turn antagonizes SF1 (53) (Figure 2). In mice with Wnt4 knockout, XX individuals exhibit virilization with the presence of Leydig-like cells in their gonad. While Wolffian ducts develop typically, Müllerian ducts are absent (54). In humans, heterozygous loss-of-function pathogenic variants in WNT4 have been found in virilized 46,XX women, who presented excess ovarian androgens and atypical Mullerian duct development (55–58). Additionally, a homozygous WNT4 pathogenic variant has been reported in a consanguineous family with a rare embryonic lethal syndrome known as SERKAL (SEx Reversion, Kidneys, Adrenal and Lung dysgenesis) syndrome (Table 2). This syndrome is characterized by SRY-negative 46,XX testicular or ovotesticular DSD, as well as adrenal hypoplasia, renal agenesis, and severe defects in the lungs and cardiovascular structures (68).
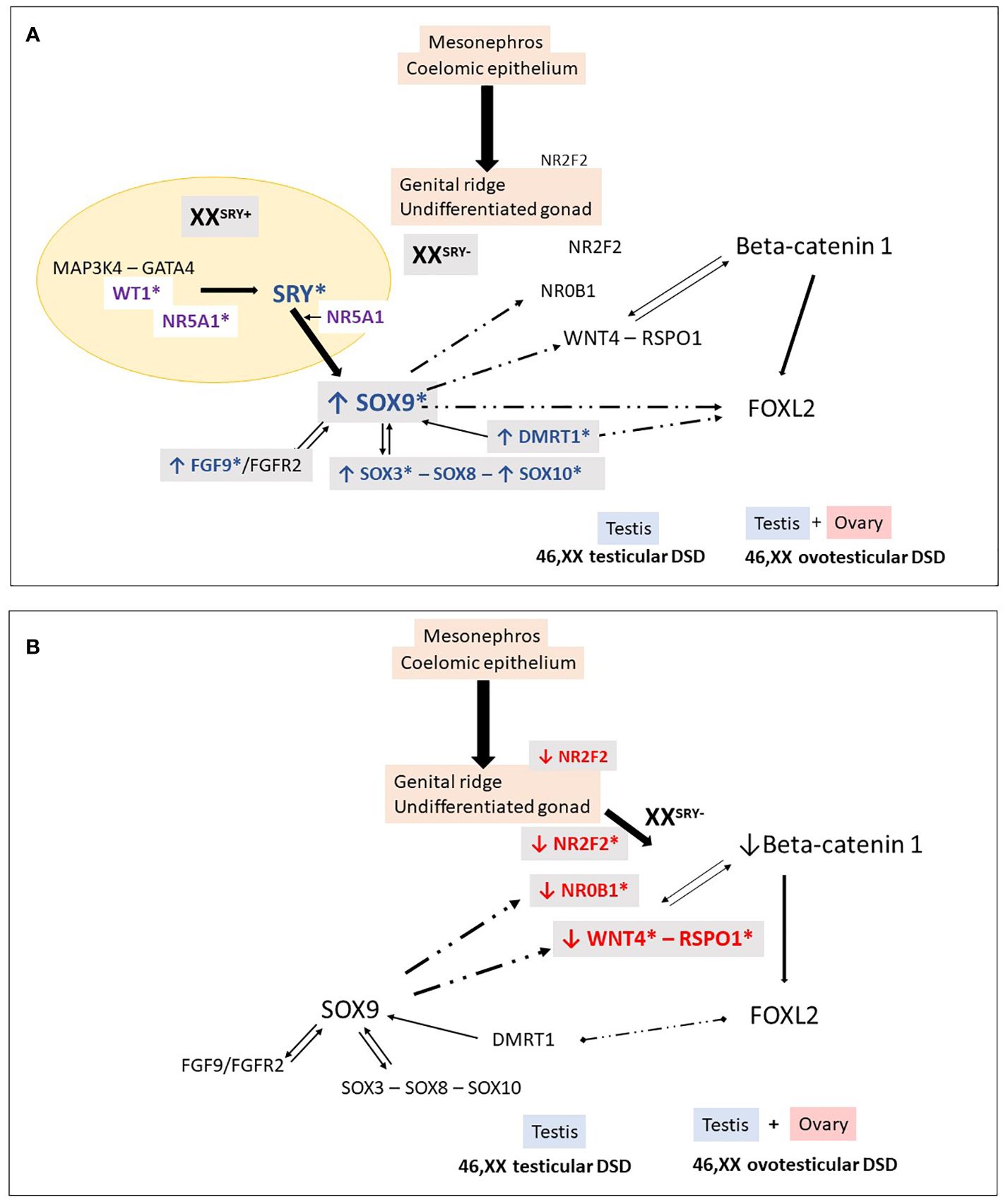
Figure 2 46,XX Testicular and ovotesticular DSD. Loss of the antagonistic balance of the RSPO1/WNT4/β-catenin pathway and the SRY/SOX9/FGF9 pathway can lead to the development of an abnormal gonad. (A) In XX individuals with Yp translocations and the presence of SRY, testicular differentiation can occur. In 46,XX SRY-negative individuals, testicular development may result from different conditions: overexpression of “pro-testicular” factors such as SOX9, SOX3, SOX10, FGF9, DMRT1, and (B) reduced expression of “pro-ovarian” factors such as RSPO1, WNT4, NR2F2. These changes in gene expression can be caused by an increase in the number of gene copies or their regulatory sequences. Additionally, in particular conditions, factors like WT1 and NR5A1 can also promote testicular development in 46,XX individuals. *Indicates genes associated with 46,XX testicular and ovotesticular DSD in humans.
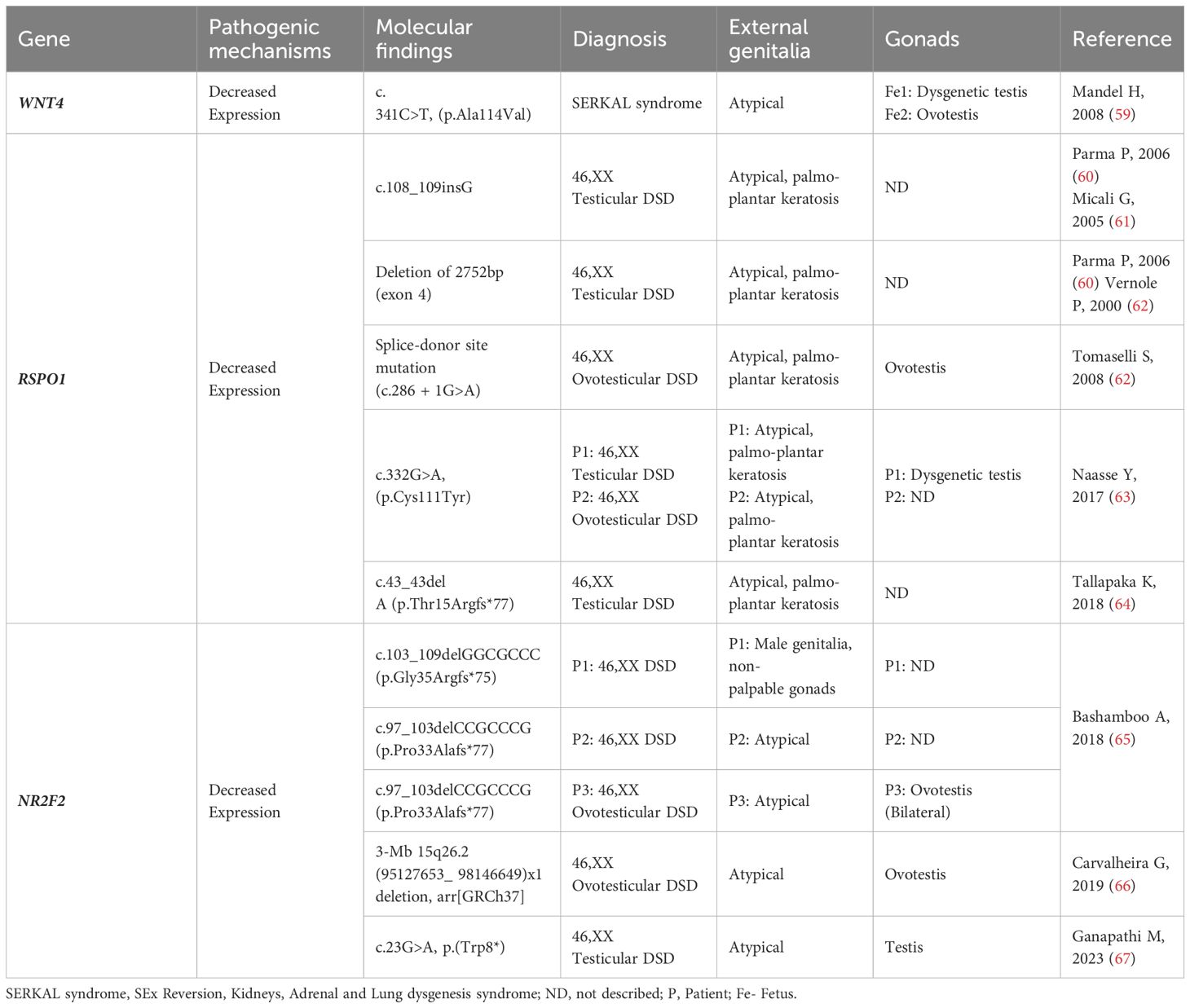
Table 2 WNT4, RSPO1 and NR2F2: Genotype and clinical and gonadal characteristics of patients with SRY-negative 46,XX Testicular and Ovotesticular DSD reported in the literature.
RSPO1 gene (1p34.3) encodes a secreted agonist protein of the canonical Wnt/β-catenin signaling pathway, that is widely expressed during fetal development (Table 1). RSPO1 plays a key role in gonad differentiation toward the ovary by synergizing the WNT4 to stabilize β-catenin in XX gonads (52, 59). In XX mice, the gonadal phenotype of the Rspo1 and the Wnt4 knockouts are strikingly similar: it ranges from small testes to ovotestes (26). RSPO1 is also expressed in fibroblasts and regulates the proliferation and differentiation of keratinocytes (69).
Homozygous deleterious RSOP1 variants have been identified in SRY-negative 46,XX DSD patients with atypical genitalia and palmoplantar hyperkeratosis and increased susceptibility to squamous cell carcinoma of the skin (60, 62, 69, 70) (Table 2). These variants are typically located in the cysteine-rich furin domains of RSPO1, which are important for stabilizing cytosolic β-catenin. Dysregulation of β-catenin might result in the inhibition of Sox9 degradation and contribute to testis development (Figure 2).
Histological examination of gonads of two affected individuals reveals testicular structures with Leydig cell hyperplasia and ovotestes with small residual ovarian tissue, respectively (63, 70). The absence of RSPO1 also affects the skin microenvironment and epidermal integrity, contributing to an elevated risk of squamous cell carcinoma in palmoplantar regions exposed to frictional stresses (71). Some patients may also present with congenital microphthalmia, cataracts, coloboma of the iris and choroid, onychodystrophy, laryngeal carcinoma, and hearing impairment (60–62, 70, 72).
The NR2F2 (Nuclear Receptor Subfamily 2 Group F Member 2) gene (15q26.2) encodes the chicken ovalbumin upstream promoter transcription factor 2 (COUP-TF2), which is an orphan nuclear receptor (Table 1). COUP-TFII plays important roles during embryogenesis, particularly in cell fate determination, organogenesis, angiogenesis, and metabolism (64, 73). It also plays a role in cell regeneration or dedifferentiation. High expression of COUP-TFII is observed in the mesenchymal component of various organs, including the heart, brain, kidney, adrenal cortex, genital tubercle, otocyst, periocular mesenchyme, optic stalk, and olfactory placode, during development and organogenesis (74). Knockout and heterozygous mice lacking COUP-TFII exhibit multiple vascular abnormalities, especially in the heart and brain. These abnormalities can lead to premature death, with embryonic mortality observed in COUP-TFII knockout mice and death occurring within the first few days of life in heterozygous mice (74).
In the gonadal ridges, COUP-TF2 acts as a “pro-ovary” and “anti-testis” factor (75). Previous studies suggest that the Nr2f2 repression is necessary for fetal Leydig cell differentiation (76).
Ferreira et al. demonstrated that the human NR2F2 is highly upregulated during bipotential gonad development, being detected in early somatic cells that precede the steroidogenic cell emergence in the undifferentiated gonad. The authors propose that COUP-TFII regulates cell fate during gonad development by modulating the WNT signaling pathway, Runx2 (RUNX family transcription factor 2) activity, and the expression of Pparg (Peroxisome Proliferator Activated Receptor Gamma) and Sox9. Impairment of its function might disrupt the transcriptional plasticity of early supporting gonadal cells. This disruption during early gonad development may cause early supporting gonadal cells to commit to the testicular pathway (77).
Less than 40 individuals with heterozygous pathogenic variants in NR2F2 have been reported (78). Congenital heart defects are the most well-known phenotypes associated with its pathogenic variants, according to the expression pattern of COUP-TF2 (67, 73). However, the clinical features associated with NR2F2 variants are variable. These features include intrauterine growth restriction (IUGR), congenital heart disease (CHD), congenital diaphragmatic hernia (CDH), blepharophimosis ptosis-epicanthus inversus syndrome (BPES), developmental delays, hypotonia, feeding difficulties, failure to thrive, congenital and acquired microcephaly, dysmorphic facial features (such as up-slanted or short palpebral fissures, micrognathia or retrognathia, low-set or dysplastic ears, hypertelorism, and full cheeks), renal failure, hearing loss, strabismus, asplenia, and vascular malformations. Genital anomalies and DSD have also been described (78).
The molecular mechanisms leading to testis development in some 46,XX patients with COUP-TFII loss-of-function have yet to be defined (77, 79). NR2F2 pathogenic variants/deletion were found to be associated with five patients who had a syndromic form of SRY-negative 46,XX T/OT DSD (Table 2) (65, 66, 78, 79).
These patients presented with atypical genitalia (4/5), congenital diaphragmatic hernia (CDH) (3/5), blepharophimosis ptosis-epicanthus inversus syndrome (BPES) (3/5), and congenital heart disease (CHD) (2/5). Three of the patients had frameshift variants affecting the N-terminal region of the protein, specifically, p.Gly35Argfs*75 and p.Pro33Alafs*77 and the fourth patient had a de novo nonsense variant, p.Trp8* (78, 79).
(Table 2). In the fifth patient, a CGH array assay identified a 3-Mb 15q26.2 [arr(GRCh37) 95127653_98146649] x1 deletion that encompassed the entire NR2F2 gene (65, 66).
Genotype-phenotype correlations cannot be identified, as individuals carrying identical NR2F2 variants may present with variable phenotypic manifestations. In the case of 46,XX patients, a single-copy genomic deletion that encompasses the entire NR2F2 gene may result in testicular tissue and atypical external genitalia in some cases, but in others, there may be no evidence of genital anomalies or DSD, despite the presence of other syndromic features (65, 66, 78, 80). These findings suggest that the phenotypic expression of NR2F2-related differences may be likely influenced by additional modifiers.
SRY initiates the formation of male gonadal tissue from bipotential gonadal primordia by stimulating a cascade of related genes, the SRY-related HMG box-containing genes (SOX) (Table 1). These genes play an essential role in the differentiation of Sertoli cells and the development of the testes (81).
The main cause of 46,XX T DSD patients is related to a chromosomal rearrangement during paternal meiosis that leads to the translocation of the SRY from the paternal Y chromosome to the X chromosome or an autosome. In such cases, patients typically exhibit external and internal male genitalia (82).
In such cases, the genetic etiological diagnosis of 46,XX T DSD can be established using the fluorescence in situ hybridization (FISH) technique, which identifies a fluorescent signal indicating the sequence of the SRY translocated onto the X chromosome or autosome. Alternatively, the polymerase chain reaction (PCR) can be used to identify the presence of the SRY in the individual’s DNA being evaluated. Microarray analysis is also used to detect the presence of SRY.
It is worth noting that the formation of the testis can occur in 46,XX individuals, even in the absence of SRY, particularly among those who have dosage variations in HMG-box transcription factors (Table 1).
The increased expression of genes associated with male gonadal determination is a well-established etiological cause of 46,XX T/OT DSD patients. Among these genes, members of the SOX family play a significant role in this process.
The SOX (SRY-related HMG box) family of proteins is a group of transcriptional regulators that contain a highly conserved high-mobility group domain (83, 84). The high-mobility group domain was first identified in the SRY gene, and several genes from the SOX family have been linked to the etiology of differences of gonadal developmental in mammals.
SOX9 (17q24.3) is a transcription factor that plays a significant role in various tissues, including chondrocytes and testes (84) (Table 1). Studies investigating the relationship between phenotype and genotype in humans and mice have demonstrated that SOX9 expression is a critical step in testis development, occurring downstream of SRY. SOX9 is responsible for the specification of Sertoli cells, which in turn initiates testicular differentiation and triggers the production of AMH (85, 86).
Overexpression of SOX9, often caused by gene duplications or copy number variations in the upstream promoter region, has been linked to testis determination in the absence of SRY (84, 87, 88) (Figure 2). In many cases of 46,XX T/OT DSD, SOX9 duplications have been identified as the most commonly observed genetic cause, second only to SRY translocation (Table 3).
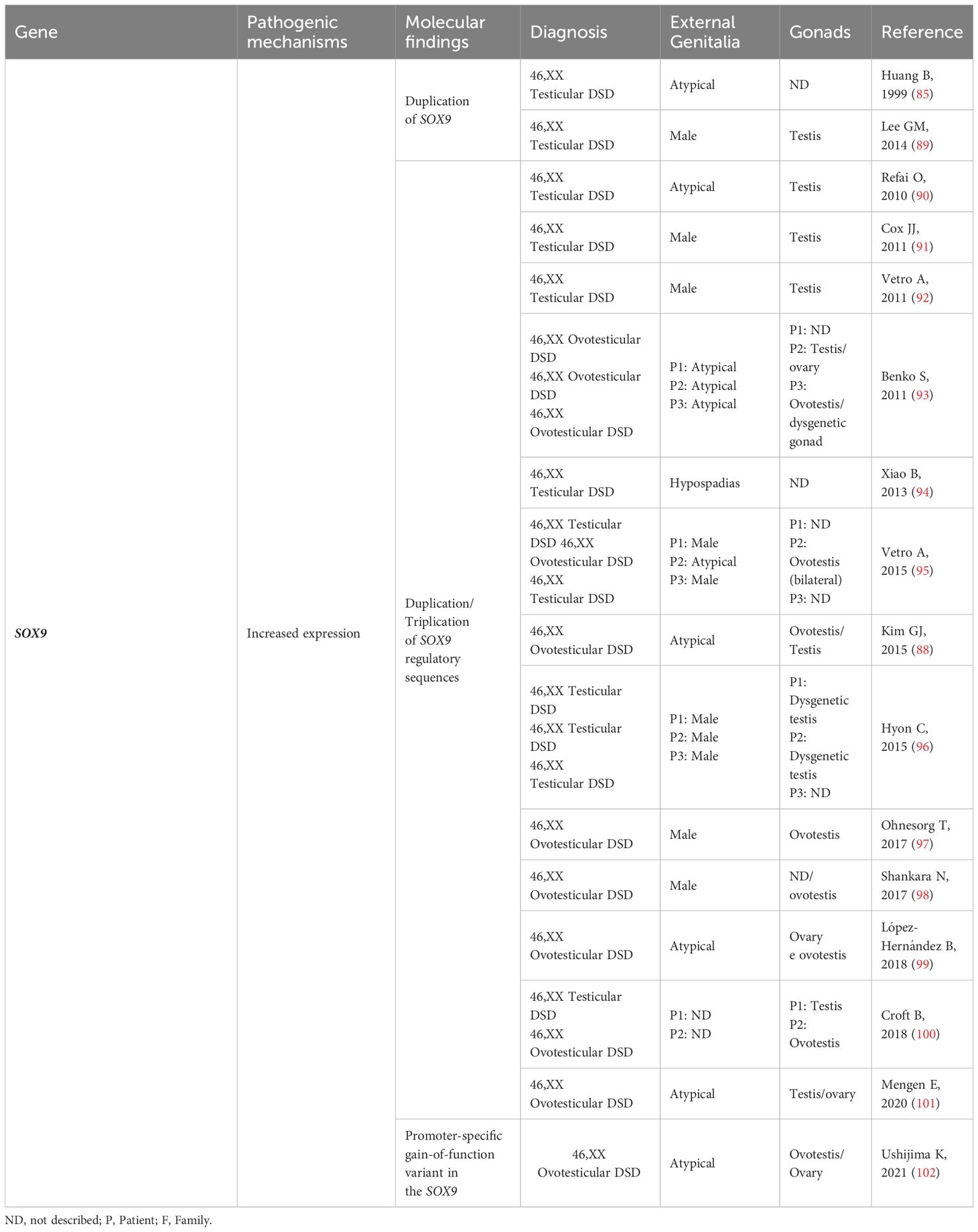
Table 3 SOX9: Genotype and clinical and gonadal characteristics of patients with SRY-negative 46,XX Testicular and Ovotesticular DSD reported in the literature.
Many of these duplications involve a noncoding region spanning at least 24 kb, known as the RevSex region, located approximately 0.5-0.6 Mb upstream of the SOX9 gene (89–100) (Table 3). This region is predicted to contain a human testis-specific enhancer, and the duplication of this enhancer drives the atypical expression of SOX9, leading to the activation of testicular differentiation (87, 92).
Ushijima et al. escribed an SRY-negative 46,XX OT DSD patient with a novel SOX9 missense variant (p.Glu50Lys) with promoter-specific gain-of-function (GoF) activity in in vitro studies. The authors demonstrated that E50K-SOX9 had (GoF) activity in the mTESCO-luciferase reporter, suggesting that it was due to change(s) in its bioactivity. GoF activity was observed in mTESCO-luc but not in mAmh-luc, thereby indicating that the acquisition of GoF activity was promoter-specific. To associate the promotor SOX9 variant with atypical expression of SOX9, and the beginning of testicular differentiation in the 46,XX OT DSD patient, mice carrying the Sox9 p.E50K were also generated and characterized. These mice, nevertheless, did not develop ovotestis (101). Such discordance of expressivity/phenotype among humans and mice are not limited to sox9/SOX9 (102) but are also described in other genes associated with DSD, including Nr5a1/NR5A1 (103) and Wt1/WT1 (104). The molecular mechanism of the promoter -specific GoF activity of E50K-SOX9 remains to be elucidated.
SOX3 (Xq27.1) is another member of the SOX gene family that is involved in gonadal development. It encodes a protein that is highly SRY-like, with an amino acid sequence similarity of 67% for the protein and 90% for the HMG DNA-binding domain (105) (Table 1). Studies in transgenic mice have shown that increased ectopic expression of Sox3 in undifferentiated gonads can lead to sex reversal in XX mice, with complete virilization of external genitalia observed in 77% of animals (106). These findings suggest that Sox3 hyperexpression acts as a counterpart of Sry, leading to increased expression of Sox9. Together with Nr5a1, Sox3 binds to the enhancer region of Sox9 (106).
Like the findings in mice, when increased expression of SOX3 is observed in humans, this gene acts in conjunction with NR5A1 to promote overexpression of SOX9. This phenomenon directs the gonads toward male determination (105) (Figure 2).
The duplication of the SOX3 in a patient with SRY-negative 46,XX OT DSD was initially identified by Sutton et al. (105). Several other 46,XX patients with testicular development (T and OT) and duplications of the SOX3 or in the regions located upstream of this gene have been reported (94, 107–114), supporting the importance of SOX3 in testis development (Table 3).
A heterozygous deletion downstream of SOX3 was also reported in an SRY-negative 46,XX infertility male. The authors of the study speculated that this deletion may play a role in the regulation of the SOX3, potentially resulting in increased expression of SOX3 (115).
SOX10 (22q13.1) is another gene closely correlated with SOX9 in humans (Table 1). Initially expressed in neural crest cells during the embryonic period, it plays a critical role in their development. SOX10 is also expressed in fetal gonads (116). In mice, the expression of Sox10 specifically in Sertoli cells strongly indicates its involvement in the testicular differentiation process and reinforces its role in the male pathway (117).
Studies in transgenic animal models have demonstrated that Sox10 overexpression causes sex reversal in XX mice (117). These studies demonstrated that the expression level of Sox10 is crucial in determining the gonadal phenotype. Complete testicular differentiation in all mice was observed in the lineage with higher levels of Sox10 expression, while the lineage expressing lower levels of the transgene showed only 30% of mice with complete sex reversal in the postnatal period. Interestingly, all fetuses from the second group (lower expression levels) were able to initiate Sertoli cell differentiation (presence of cells expressing Sox9 in XX transgenic gonads). In these gonads, cells committed to the female pathway, identified by the expression of Foxl2, were interspersed with Sox9-positive cells (117). This same pattern has been described in ovotestis in humans, as well as in mouse models of ovotestis development (118).
Similarly, gonadal, and reproductive system alterations have been reported in cases of partial duplication of chromosome 22q in 46,XX humans, a chromosomal region that contains SOX10 (Table 4). Rare patients diagnosed with 46,XX T/OT DSD, both syndromic and non-syndromic, have also been described in the literature with chromosome 22 aneuploidies (116, 119–121) (Table 4).
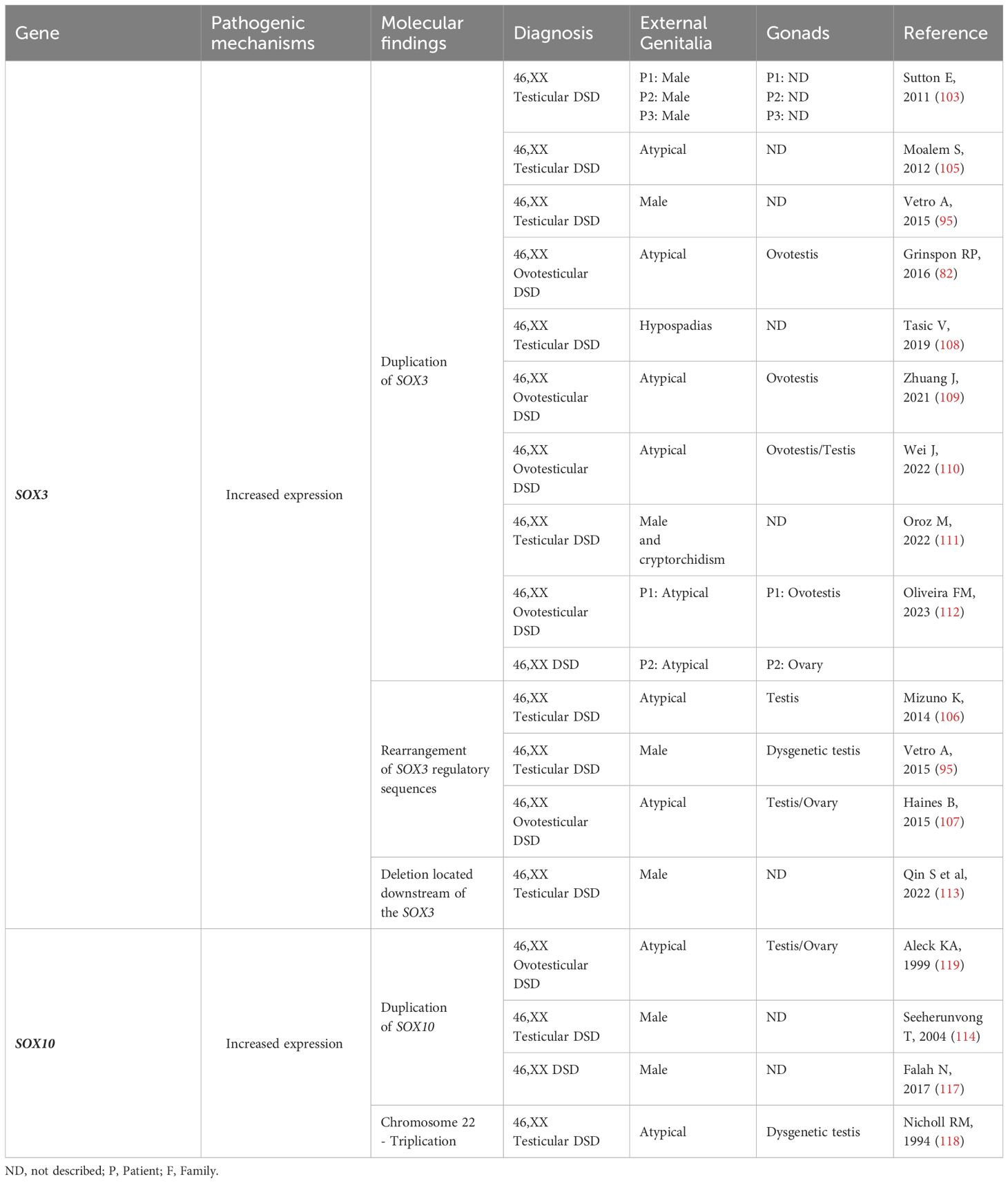
Table 4 SOX3 and SOX10: Genotype and clinical and gonadal characteristics of the patients with SRY-negative 46,XX Testicular and Ovotesticular DSD reported in the literature.
The DMRT1 gene (9p24.3) encodes a transcription factor that plays a crucial role in sex determination and gonadal development in various species (Table 1). It possesses a zinc-finger-like DNA binding domain known as the DM (doublesex/MAB-3) domain. This domain allows DMRT1 to bind to specific DNA sequences and regulate the expression of genes involved in sex differentiation (122). DMRT1 expression has been observed in the undifferentiated human XY gonadal primordium. During the early fetal period (gestational weeks 8-20), it is primarily expressed in Sertoli cells, which play a crucial role in testicular development. In the second gestational trimester, childhood, and post-puberty, DMRT1 expression becomes more abundant in spermatogonia (123) This dynamic pattern of expression suggests that DMRT1 plays a significant role in both the early and later stages of male gonadal development.
DMRT1 expression has indeed been detected in oogonia and oocytes during the early stages of ovarian development, up until gestational week 20. However, it is important to note that after the onset of meiotic germ cell division, DMRT1 expression becomes absent in these cells (123).
In contrast to DMRT1 homologs in other vertebrates, mammalian DMRT1 seems to not be involved in the initial sex determination but is instead required for maintaining male gonadal fate (124, 125). Studies in mice have demonstrated that the loss of expression of certain key genes in postnatal life can lead to the reprogramming of Sertoli cells into granulosa cells and vice versa. This suggests that there is a level of plasticity in gonadal fate even after the typical formation of a testis or ovary (31, 124).
Ectopic expression of DMRT1 has been shown to reprogram differentiated female granulosa cells into male Sertoli-like cells. DMRT1 functions in collaboration with other key male sex regulators like SOX9 to maintain and reprogram sexual cell fate. It acts as a singular transcription factor, by regulating gene expression and chromatin accessibility (126).
Bertini et al. reported a three-year-old boy who presented with a typical male phenotype and an SRY-negative 46,XX karyotype (127). The genetic study conducted showed a heterozygous de novo in tandem duplication of 50,221 bp on chromosome 9p. This duplication encompassed exons 2 and 3 of the DMRT1 and was detected using MPLA, CGH-array analysis, and Sanger sequencing. The breakpoints of the duplication were in the intronic regions, and it did not disrupt the coding frame of DMRT1. To investigate other potential genetic factors contributing to the phenotype, a custom NGS panel and whole genome sequencing were performed, but no additional pathogenic or uncertain variants were found in genes known to be involved in pro-testis/anti-ovary gene cascades.
The identified duplication might have allowed DMRT1 to escape the usual transcriptional repression that occurs in 46,XX fetal gonads, leading to the activation of the testicular determination cascade. Notably, no previous cases of SRY-negative 46,XX DSD associated with alterations in DMRT1 have been reported thus far.
The FGF9 gene (13q12.1) is a signaling peptide involved in the development of various organs, including limbs, lungs, the adenohypophysis, and the gonadal ridges (Table 1). FGFs are typically considered paracrine factors and play important roles in tissue patterning and organogenesis during embryogenesis. The FGF9 subfamily, which signals from epithelium to mesenchyme, stimulates mesenchymal proliferation. In Fgf9 knockout XY mice, gonadal development is severely impaired during embryonic and fetal life, leading to reproductive phenotypes ranging from different range of undervirilization to complete feminization of external genitalia (128).
In a study by Chiang et al., an SRY-negative 46,XX male with hypospadias and azoospermia was identified (129). Array-CGH analysis revealed duplicated regions on chromosomes 13q12.11 (21.143874–21.174184 Mb) and 13q31.1 (79.807500–79.813700 Mb). These duplicated regions encompassed the entire FGF9 and SPRY2 genes, respectively. The genomic gain of FGF9 was hypothesized to result in FGF9 overexpression, which could explain testicular development instead of ovarian development. Additionally, SPRY2 was previously related to a potential role in male sex organogenesis by controlling FGF9 gene-induced mesonephric cell migration to the developing testis (130). The higher amount of FGF9 would interfere with the expression of WNT4 in the embryo, thereby impeding ovarian development in SRY-negative 46,XX males.
WT1 gene (11p13) is a transcription factor that encodes a zinc-finger protein (Table 1). It is widely expressed in the condensing mesenchyme, genital ridge, fetal gonads, renal vesicle, developing podocytes of the fetal kidney, and mesothelium (131). The Wt1 and Lhx9 (Lim homeobox 9) genes act as direct activators of the Nr5a1 and play a critical role in the development of the undifferentiated gonad (132).
More than 30 protein isoforms originating from WT1 alternative splicing, alternative translation start sites, and different RNA editing are known. The alternative splice site in intron 9 allows WT1 isoforms with omission or inclusion of three amino acids [lysine-threonine-serine (KTS)] between the third and fourth zinc fingers. These isoforms regulate specific urogenital differentiation processes (133, 134).
Pathogenic WT1 variants are associated with several phenotypes, including 46,XY and 46,XX DSD (135).
WT1 also plays a crucial role in the differentiation and maintenance of Sertoli cells, and this function is positively related to the testicular abnormalities observed in XY patients with pathogenic WT1 variants (37).
The role of WT1 in ovarian development is not yet completely understood. In mice, Wt1 is essential for the maintenance of granulosa cells, and its inactivation leads to atypical ovary development, characterized by reduced ovary size and a fewer number of developing follicles (136, 137).
In SRY-negative 46,XX individuals with testicular and ovotesticular DSD, seven pathogenic variants of WT1 have been identified (Table 5) (147–150). These variants affect the fourth zinc finger, which is a highly conserved region of the WT1 protein. Testicular development in this condition may be influenced by the inappropriate interaction between the mutated WT1 protein and the main ovarian determinant, beta-catenin 1. Additionally, studies have shown that pathogenic variants in exon 10 increase the expression of genes such as SOX9, NR5A1, and DMRT1, which are involved in the development of Sertoli cells. It has been suggested that these alterations could promote the sequestration of beta-catenin 1, leading to the upregulation of pro-testicular pathways (148, 149).
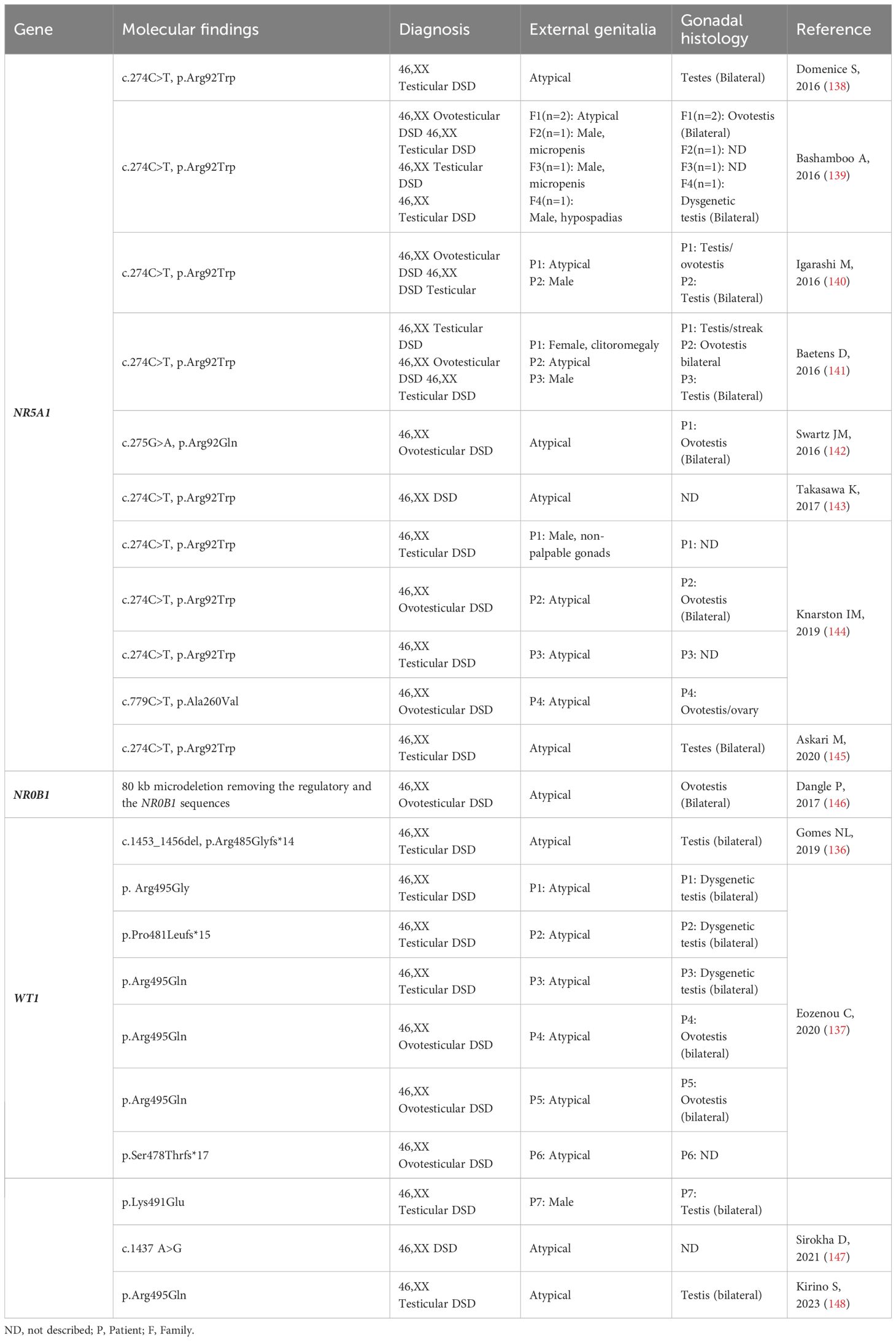
Table 5 NR5A1, NR0B1 and WT1: Genotype and clinical and gonadal characteristics of the patients with SRY-negative 46,XX Testicular and Ovotesticular DSD reported in the literature.
NR5A1 (9q33.3) encodes the steroidogenic factor 1 (SF-1), which is expressed in the developing urogenital ridge, hypothalamus, anterior pituitary gland, and steroidogenic tissues (Table 1). SF-1 plays a crucial role in controlling several steps of adrenal and gonadal development (138, 151). NR5A1 variants are associated with a wide phenotypic spectrum of 46,XX, and 46,XY DSD (139, 140).
A single and recurrent variant in the NR5A1 (c.C274T, p.Arg92Trp), present in a heterozygous state, was identified in several 46,XX OT/T DSD patients (Table 5) (140) (141–143, 145, 152). In the study by Askari et al. (152), the p.Arg92Trp variant was identified in a pair of siblings with 46,XX DSD (ovotesticular and testicular DSD patients), as well as in their father who had oligospermia. This further supports the notion that the NR5A1 variant can play a role in the development of different gonadal phenotypes (145). Another variant was identified in the Arg92 codon, just by changing the amino acid to Glutamine (c.G275A, p.Arg92Gln) in a 46,XX OT DSD patient (153). The arginine 92 residue is in a highly conserved region of NR5A1, which is crucial for its interaction with DNA. A third variant (c.C779T, p.Ala260Val) in the NR5A1 was identified in a single 46,XX OT DSD patient (144).
To date, 13 families consisting of 15 patients with 46,XX DSD, and deleterious NR5A1 variants have been reported (Table 5). These patients exhibit a variable range of virilization in the external genitalia, including isolated clitoromegaly, hypospadias, male genitalia with micropenis and cryptorchidism, or male genitalia and cryptorchidism. Likewise, the gonadal tissues also exhibit a diverse range, from streak/dysgenetic gonads to ovotestis or testis, depending on the specific case.
The mechanism which these three variants activate the testicular development in 46,XX OT/T DSD carriers remain elusive. It is suggested that they reduce the inhibition of the expression of male pathway genes, such as SOX9 and AMH (141, 143), by disrupting specific ovarian development signals, mainly in the WNT/β-catenin pathway (144, 153).
NR0B1 (Nuclear Receptor Subfamily 0 Group B Member 1) gene is located in the dosage-sensitive sex reversal (DSS) region at Xp21.2 (Table 1). It encodes an unusual orphan nuclear receptor that lacks the classic DNA-binding domain (154, 155). NR0B1/DAX1 is expressed in various tissues including the developing urogenital ridge, hypothalamus, anterior pituitary gland, adrenal glands, and gonads. It is known to have a role in both ovarian and testicular development, especially in spermatogenesis (156, 157) In mice, a coordinated expression of Nr0b1, Sry, and potentially other factors is necessary to upregulate Sox9 expression in precursor somatic cells. This coordinated expression is crucial for the development of Sertoli cells in the testes (158). These findings confirm an essential role for NR0B1 in both Sertoli and Leydig cell function (157, 159). However, the phenotype of male mice lacking Nr0b1 can vary depending on the strain due to the background-specific abundance of male-determining Sry gene transcripts. This means that the presence of different genetic backgrounds can lead to variability in the phenotypes of XY mice lacking Dax1 (Nr0b1) (160). Additionally, Nr0b1 can be upregulated by Wnt4 through the activation of the WNT/β-catenin pathway (161). Loss of function of NR0B1 causes X-linked primary adrenal insufficiency and hypogonadotropic hypogonadism (162, 163).
If normal levels of NR0B1 are crucial for testicular development and spermatogenesis, an excessive dosage of NR0B1 has been suggested to act as an anti-testicular factor (164) Xp21.1 duplications, which include NR0B1 and testis-specific MAGEB genes, have been identified in some XY patients with gonadal dysgenesis. These duplications contribute to abnormalities in gonadal development and function (146, 155, 165–168).
Dangle et al. (169) identified a copy number rearrangement in an SRY-negative 46,XX OT DSD patient using microarray analysis (Table 5). This rearrangement involved an 80 kb microdeletion and disrupted the Xp21.2 DSS critical region. The condition not only resulted in the removal of the regulatory sequences and the NR0B1 gene, but it also impacted the normal genomic organization. This disturbance led to modified gene expression patterns through a position effect (169).
Studies have indeed shown that epigenetic profiles undergo dynamic changes during mammalian development, serving as a critical mechanism in determining cell fate decisions and facilitating cellular differentiation (170). Although knowledge about the involvement of epigenetic regulators in human gonadal development remains limited, their role is unquestionable (171, 172).
Regarding the expression of miRNAs in fetal gonads, it is widely recognized that they play a role in the regulation of proteins that are critically involved in gonad development (173, 174). Moreover, it is observed that several miRNAs exhibit a sexually dimorphic expression pattern in fetal gonads, indicating their potential involvement in directing cell fate decisions and maintaining cellular states (174).
In the ovary, the role of miRNAs in follicle assembly, growth, differentiation, and ovulation has been identified (175). Real et al. (176) described miR-124 as a promising candidate gene for mice ovarian development. They found that miR-124 potentially targets several genes involved in sex determination, including Sox9, in their 3’-UTR regions. The authors also demonstrated that inhibiting miR-124 in XX gonadal cells resulted in the ectopic expression of Sox9, suggesting that this miRNA may down-regulate Sox9 in female gonads during the critical period of sex determination. Furthermore, miR-124 exhibited differential up-regulation in XX mice gonads during early stages of differentiation, but not in XY mice gonads (176). In humans, no report of miRNA abnormalities was related to 46,XX DSD etiology.
Various studies have also presented evidence suggesting the involvement of methylation patterns in the process of gonadal determination (171). However, there is currently no direct confirmation of a link between abnormal methylation patterns and the etiology of 46,XX DSD. It is known that DNA methylation and histone modifications are actively involved in the spatiotemporal expression of Sry by making the enhancers and the promoter accessible for the binding of multiple transcription factors (16, 171, 177). Furthermore, methylation of the promoter/regulatory region directly impacts the expression of the Sox9 gene in the testis and ovary of mammals. The adult testis exhibits strong Sox9 expression, while site-specific methylation in the adult ovary could play a crucial role in reducing Sox9 gene expression (178).
Certainly, innovative studies will play a crucial role in establishing the involvement of epigenetic mechanisms in the etiology of 46,XX DSD. These studies will contribute to expanding our understanding of gonads determination.
While our understanding of ovarian determination has significantly advanced, the process of testicular tissue development in an SRY-negative 46,XX gonad remains intriguing. It is worth noting that the majority of individuals with SRY-negative 46,XX testicular and ovotesticular DSD have not received a confirmed genetic diagnosis. This highlights the possibility of unknown genetic pathways or epigenetic mechanisms involved in these conditions. Further research and expansion of patient cohorts are needed to identify these other new members of the gonadal determination cascade.
MF: Writing – review & editing. ES: Writing – original draft, Writing – review & editing. MN: Writing – original draft, Writing – review & editing. RB: Writing – original draft, Writing – review & editing. BM:Funding acquisition, Writing – original draft, Writing – review & editing. SD: Funding acquisition, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) Grants No. 312543/2021-2 (to SD) and 307571/2021-1 (to BM), and by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) Grants No. 2019/26780-9 (to BM), and by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (to MF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nagahama Y, Chakraborty T, Paul-Prasanth B, Ohta K, Nakamura M. Sex determination, gonadal sex differentiation, and plasticity in vertebrate species. Physiol Rev. (2021) 101:1237–308. doi: 10.1152/physrev.00044.2019
2. Arboleda VA, Sandberg DE, Vilain E. DSDs: genetics, underlying pathologies and psychosexual differentiation. Nat Rev Endocrinol. (2014) 10:603–15. doi: 10.1038/nrendo.2014.130
3. Wu QY, Li N, Li WW, Li TF, Zhang C, Cui YX, et al. Clinical, molecular and cytogenetic analysis of 46, XX testicular disorder of sex development with SRY-positive. BMC Urol. (2014) 14:70. doi: 10.1186/1471-2490-14-70
4. Blackless M, Charuvastra A, Derryck A, Fausto-Sterling A, Lauzanne K, Lee E. How sexually dimorphic are we? Review and synthesis. Am J Hum Biol. (2000) 12:151–66. doi: 10.1002/(ISSN)1520-6300
6. Wiersma R. The clinical spectrum and treatment of ovotesticular disorder of sexual development. Adv Exp Med Biol. (2011) 707:101–3. doi: 10.1007/978-1-4419-8002-1_21
7. Mao Y, Chen S, Wang R, Wang X, Qin D, Tang Y. Evaluation and treatment for ovotesticular disorder of sex development (OT-DSD) - experience based on a Chinese series. BMC Urol. (2017) 17:21. doi: 10.1186/s12894-017-0212-8
8. Sircili MH, Denes FT, Costa EM, MaChado MG, Inacio M, Silva RB, et al. Long-term followup of a large cohort of patients with ovotesticular disorder of sex development. J Urol. (2014) 191:1532–6. doi: 10.1016/j.juro.2013.10.037
9. Ouhilal S, Turco J, Nangia A, Stotland M, Manganiello PD. True hermaphroditism presenting as bilateral gynecomastia in an adolescent phenotypic male. Fertil Steril. (2005) 83:1041. doi: 10.1016/j.fertnstert.2004.09.036
10. Matsui F, Shimada K, Matsumoto F, Itesako T, Nara K, Ida S, et al. Long-term outcome of ovotesticular disorder of sex development: a single center experience. Int J Urol. (2011) 18:231–6. doi: 10.1111/j.1442-2042.2010.02700.x
11. Hisamatsu E, Nakagawa Y, Sugita Y. Two cases of late-diagnosed ovotesticular disorder of sex development. APSP J Case Rep. (2013) 4:40.eCollection.
12. Maciel-Guerra AT, de Mello MP, Coeli FB, Ribeiro ML, Miranda ML, Marques-de-Faria AP, et al. XX Maleness and XX true hermaphroditism in SRY-negative monozygotic twins: additional evidence for a common origin. J Clin Endocrinol Metab. (2008) 93:339–43. doi: 10.1210/jc.2007-1115
13. Toublanc JE, Boucekkine C, Abbas N, Barama D, Vilain E, McElreavey K, et al. Hormonal and molecular genetic findings in 46,XX subjects with sexual ambiguity and testicular differentiation. Eur J Pediatr. (1993) 152 Suppl 2:S70–5. doi: 10.1007/BF02125443
14. Verkauskas G, Jaubert F, Lortat-Jacob S, Malan V, Thibaud E, Nihoul-Fékété C. The long-term followup of 33 cases of true hermaphroditism: a 40-year experience with conservative gonadal surgery. J Urol. (2007) 177:726–31. doi: 10.1016/j.juro.2006.10.003
15. Abbas NE, Toublanc JE, Boucekkine C, Toublanc M, Affara NA, Job JC, et al. A possible common origin of “Y-negative” human XX males and XX true hermaphrodites. Hum Genet. (1990) 84:356–60. doi: 10.1007/BF00196234
16. Stévant I, Nef S. Genetic control of gonadal sex determination and development. Trends Genet. (2019) 35:346–58. doi: 10.1016/j.tig.2019.02.004
17. MacLaughlin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med. (2004) 350:367–78. doi: 10.1056/NEJMra022784
18. Ostrer H, Huang HY, Masch RJ, Shapiro E. A cellular study of human testis development. Sex Dev. (2007) 1:286–92. doi: 10.1159/000108930
19. Nef S, Stévant I, Greenfield A. Characterizing the bipotential mammalian gonad. Curr Top Dev Biol. (2019) 134:167–94. doi: 10.1016/bs.ctdb.2019.01.002
20. Ross AJ, Capel B. Signaling at the crossroads of gonad development. Trends Endocrinol Metab. (2005) 16:19–25. doi: 10.1016/j.tem.2004.11.004
21. McLaren A. Somatic and germ-cell sex in mammals. Philos Trans R Soc Lond B Biol Sci. (1988) 322:3–9. doi: 10.1098/rstb.1988.0109
22. Richardson BE, Lehmann R. Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat Rev Mol Cell Biol. (2010) 11:37–49. doi: 10.1038/nrm2815
23. Xie Y, Wu C, Li Z, Wu Z, Hong L. Early gonadal development and sex determination in mammal. Int J Mol Sci. (2022) 23(14):7500. doi: 10.3390/ijms23147500
24. Ottolenghi C, Omari S, Garcia-Ortiz JE, Uda M, Crisponi L, Forabosco A, et al. Foxl2 is required for commitment to ovary differentiation. Hum Mol Genet. (2005) 14:2053–62. doi: 10.1093/hmg/ddi210
25. Chassot AA, Gillot I, Chaboissier MC. R-spondin1, WNT4, and the CTNNB1 signaling pathway: strict control over ovarian differentiation. Reproduction. (2014) 148:R97–110. doi: 10.1530/REP-14-0177
26. Chassot AA, Ranc F, Gregoire EP, Roepers-Gajadien HL, Taketo MM, Camerino G, et al. Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum Mol Genet. (2008) 17:1264–77. doi: 10.1093/hmg/ddn016
27. Bashamboo A, McElreavey K. Mechanism of sex determination in humans: insights from disorders of sex development. Sex Dev. (2016) 10:313–25. doi: 10.1159/000452637
28. Ohnesorg T, Vilain E, Sinclair AH. The genetics of disorders of sex development in humans. Sex Dev. (2014) 8:262–72. doi: 10.1159/000357956
29. Pannetier M, Chassot AA, Chaboissier MC, Pailhoux E. Involvement of FOXL2 and RSPO1 in ovarian determination, development, and maintenance in mammals. Sex Dev. (2016) 10:167–84. doi: 10.1159/000448667
30. Pannetier M, Fabre S, Batista F, Kocer A, Renault L, Jolivet G, et al. FOXL2 activates P450 aromatase gene transcription: towards a better characterization of the early steps of mammalian ovarian development. J Mol Endocrinol. (2006) 36:399–413. doi: 10.1677/jme.1.01947
31. Uhlenhaut NH, Jakob S, Anlag K, Eisenberger T, Sekido R, Kress J, et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. (2009) 139:1130–42. doi: 10.1016/j.cell.2009.11.021
32. Lin YT, Capel B. Cell fate commitment during mammalian sex determination. Curr Opin Genet Dev. (2015) 32:144–52. doi: 10.1016/j.gde.2015.03.003
33. Hacker A, Capel B, Goodfellow P, Lovell-Badge R. Expression of Sry, the mouse sex determining gene. Development. (1995) 121:1603–14. doi: 10.1242/dev.121.6.1603
34. Hossain A, Saunders GF. The human sex-determining gene SRY is a direct target of WT1. J Biol Chem. (2001) 276:16817–23. doi: 10.1074/jbc.M009056200
35. de Santa Barbara P, Méjean C, Moniot B, Malclès MH, Berta P, Boizet-Bonhoure B. Steroidogenic factor-1 contributes to the cyclic-adenosine monophosphate down-regulation of human SRY gene expression. Biol Reprod. (2001) 64:775–83. doi: 10.1095/biolreprod64.3.775
36. Katoh-Fukui Y, Miyabayashi K, Komatsu T, Owaki A, Baba T, Shima Y, et al. Cbx2, a polycomb group gene, is required for Sry gene expression in mice. Endocrinology. (2012) 153:913–24. doi: 10.1210/en.2011-1055
37. Miyamoto Y, Taniguchi H, Hamel F, Silversides DW, Viger RS. A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol Biol. (2008) 9:44. doi: 10.1186/1471-2199-9-44
38. Tevosian SG, Albrecht KH, Crispino JD, Fujiwara Y, Eicher EM, Orkin SH. Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2. Development. (2002) 129:4627–34. doi: 10.1242/dev.129.19.4627
39. Hanley NA, Hagan DM, Clement-Jones M, Ball SG, Strachan T, Salas-Cortés L, et al. SRY, SOX9, and DAX1 expression patterns during human sex determination and gonadal development. Mech Dev. (2000) 91:403–7. doi: 10.1016/S0925-4773(99)00307-X
40. Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. (2008) 453:930–4. doi: 10.1038/nature06944
41. Jameson SA, Lin YT, Capel B. Testis development requires the repression of Wnt4 by Fgf signaling. Dev Biol. (2012) 370:24–32. doi: 10.1016/j.ydbio.2012.06.009
42. Moniot B, Declosmenil F, Barrionuevo F, Scherer G, Aritake K, Malki S, et al. The PGD2 pathway, independently of FGF9, amplifies SOX9 activity in Sertoli cells during male sexual differentiation. Development. (2009) 136:1813–21. doi: 10.1242/dev.032631
43. Barrionuevo F, Bagheri-Fam S, Klattig J, Kist R, Taketo MM, Englert C, et al. Homozygous inactivation of Sox9 causes complete XY sex reversal in mice. Biol Reprod. (2006) 74:195–201. doi: 10.1095/biolreprod.105.045930
44. Chaboissier MC, Kobayashi A, Vidal VI, Lützkendorf S, van de Kant HJ, Wegner M, et al. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development. (2004) 131:1891–901. doi: 10.1242/dev.01087
45. Ono M, Harley VR. Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol. (2013) 9:79–91. doi: 10.1038/nrendo.2012.235
46. Eggers S, Ohnesorg T, Sinclair A. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol. (2014) 10:673–83. doi: 10.1038/nrendo.2014.163
47. Loke J, Pearlman A, Radi O, Zuffardi O, Giussani U, Pallotta R, et al. Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/β-catenin signaling. Hum Mol Genet. (2014) 23:1073–83. doi: 10.1093/hmg/ddt502
48. Pearlman A, Loke J, Le Caignec C, White S, Chin L, Friedman A, et al. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet. (2010) 87:898–904. doi: 10.1016/j.ajhg.2010.11.003
49. White S, Hewitt J, Turbitt E, van der Zwan Y, Hersmus R, Drop S, et al. A multi-exon deletion within WWOX is associated with a 46,XY disorder of sex development. Eur J Hum Genet. (2012) 20:348–51. doi: 10.1038/ejhg.2011.204
50. Ledig S, Hiort O, Scherer G, Hoffmann M, Wolff G, Morlot S, et al. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod. (2010) 25:2637–46. doi: 10.1093/humrep/deq167
51. da Silva TE, Gomes NL, Lerario AM, Keegan CE, Nishi MY, Carvalho FM, et al. Genetic evidence of the association of DEAH-box helicase 37 defects with 46,XY gonadal dysgenesis spectrum. J Clin Endocrinol Metab. (2019) 104:5923–34. doi: 10.1210/jc.2019-00984
52. Biason-Lauber A, Chaboissier MC. Ovarian development and disease: The known and the unexpected. Semin Cell Dev Biol. (2015) 45:59–67. doi: 10.1016/j.semcdb.2015.10.021
53. Carré GA, Greenfield A. Characterising novel pathways in testis determination using mouse genetics. Sex Dev. (2014) 8:199–207. doi: 10.1159/000358402
54. Vainio S, Heikkilä M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature. (1999) 397:405–9. doi: 10.1038/17068
55. Jordan BK, Mohammed M, Ching ST, Délot E, Chen XN, Dewing P, et al. Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet. (2001) 68:1102–9. doi: 10.1086/320125
56. Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46,XX woman. N Engl J Med. (2004) 351:792–8. doi: 10.1056/NEJMoa040533
57. Biason-Lauber A, De Filippo G, Konrad D, Scarano G, Nazzaro A, Schoenle EJ. WNT4 deficiency–a clinical phenotype distinct from the classic Mayer-Rokitansky-Kuster-Hauser syndrome: a case report. Hum Reprod. (2007) 22:224–9. doi: 10.1093/humrep/del360
58. Philibert P, Biason-Lauber A, Rouzier R, Pienkowski C, Paris F, Konrad D, et al. Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and müllerian duct abnormalities: a French collaborative study. J Clin Endocrinol Metab. (2008) 93:895–900. doi: 10.1210/jc.2007-2023
59. Tomizuka K, Horikoshi K, Kitada R, Sugawara Y, Iba Y, Kojima A, et al. R-spondin1 plays an essential role in ovarian development through positively regulating Wnt-4 signaling. Hum Mol Genet. (2008) 17:1278–91. doi: 10.1093/hmg/ddn036
60. Radi O, Parma P, Imbeaud S, Nasca MR, Uccellatore F, Maraschio P, et al. XX sex reversal, palmoplantar keratoderma, and predisposition to squamous cell carcinoma: genetic analysis in one family. Am J Med Genet A. (2005) 138a:241–6. doi: 10.1002/ajmg.a.30935
61. Micali G, Nasca MR, Innocenzi D, Frasin LA, Radi O, Parma P, et al. Association of palmoplantar keratoderma, cutaneous squamous cell carcinoma, dental anomalies, and hypogenitalism in four siblings with 46,XX karyotype: a new syndrome. J Am Acad Dermatol. (2005) 53:S234–9. doi: 10.1016/j.jaad.2005.02.033
62. Vernole P, Terrinoni A, Didona B, De Laurenzi V, Rossi P, Melino G, et al. An SRY-negative XX male with Huriez syndrome. Clin Genet. (2000) 57:61–6. doi: 10.1034/j.1399-0004.2000.570109.x
63. Naasse Y, Bakhchane A, Charoute H, Jennane F, Bignon-Topalovic J, Malki A, et al. A novel homozygous missense mutation in the FU-CRD2 domain of the R-spondin1 gene associated with familial 46,XX DSD. Sex Dev. (2017) 11:269–74. doi: 10.1159/000485393
64. Lin FJ, Qin J, Tang K, Tsai SY, Tsai MJ. Coup d’Etat: an orphan takes control. Endocr Rev. (2011) 32:404–21. doi: 10.1210/er.2010-0021
65. Carvalheira G, Malinverni AM, Moysés-Oliveira M, Ueta R, Cardili L, Monteagudo P, et al. The natural history of a man with ovotesticular 46,XX DSD caused by a novel 3-Mb 15q26.2 deletion containing NR2F2 gene. J Endocr Soc. (2019) 3:2107–13. doi: 10.1210/js.2019-00241
66. Carvalheira G, Malinverni AM, Moysés-Oliveira M, Ueta R, Cardili L, Monteagudo P, et al. CORRIGENDUM FOR “The natural history of a man with ovotesticular 46,XX DSD due to a Novel 3-Mb 15q26.2 deletion containing NR2F2 gene”. J Endocr Soc. (2020) 4:bvaa022. doi: 10.1210/jendso/bvaa022
67. Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. (2014) 94:574–85. doi: 10.1016/j.ajhg.2014.03.007
68. Mandel H, Shemer R, Borochowitz ZU, Okopnik M, Knopf C, Indelman M, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. (2008) 82:39–47. doi: 10.1016/j.ajhg.2007.08.005
69. Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, et al. R-spondin1 is essential in sex determination, skin differentiation and Malignancy. Nat Genet. (2006) 38:1304–9. doi: 10.1038/ng1907
70. Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat. (2008) 29:220–6. doi: 10.1002/humu.v29:2
71. Dellambra E, Cordisco S, Delle Monache F, Bondanza S, Teson M, Nicodemi EM, et al. RSPO1-mutated keratinocytes from palmoplantar keratoderma display impaired differentiation, alteration of cell-cell adhesion, EMT-like phenotype and invasiveness properties: implications for squamous cell carcinoma susceptibility in patients with 46XX disorder of sexual development. Orphanet J Rare Dis. (2022) 17:275. doi: 10.1186/s13023-022-02434-2
72. Tallapaka K, Venugopal V, Dalal A, Aggarwal S. Novel RSPO1 mutation causing 46,XX testicular disorder of sex development with palmoplantar keratoderma: A review of literature and expansion of clinical phenotype. Am J Med Genet A. (2018) 176:1006–10. doi: 10.1002/ajmg.a.38646
73. Polvani S, Pepe S, Milani S, Galli A. COUP-TFII in health and disease. Cells. (2019) 9(1):101. doi: 10.3390/cells9010101
74. Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. (1999) 13:1037–49. doi: 10.1101/gad.13.8.1037
75. Gomes NL, Chetty T, Jorgensen A, Mitchell RT. Disorders of sex development-novel regulators, impacts on fertility, and options for fertility preservation. Int J Mol Sci. (2020) 21(7):2282. doi: 10.3390/ijms21072282
76. van den Driesche S, Walker M, McKinnell C, Scott HM, Eddie SL, Mitchell RT, et al. Proposed role for COUP-TFII in regulating fetal Leydig cell steroidogenesis, perturbation of which leads to masculinization disorders in rodents. PloS One. (2012) 7:e37064. doi: 10.1371/journal.pone.0037064
77. Ferreira LGA, Kizys MML, Gama GAC, Pachernegg S, Robevska G, Sinclair AH, et al. COUP-TFII regulates early bipotential gonad signaling and commitment to ovarian progenitors. Cell Biosci. (2024) 14:3. doi: 10.1186/s13578-023-01182-5
78. Ganapathi M, Matsuoka LS, March M, Li D, Brokamp E, Benito-Sanz S, et al. Heterozygous rare variants in NR2F2 cause a recognizable multiple congenital anomaly syndrome with developmental delays. Eur J Hum Genet. (2023) 31:1117–24. doi: 10.1038/s41431-023-01434-5
79. Bashamboo A, Eozenou C, Jorgensen A, Bignon-Topalovic J, Siffroi JP, Hyon C, et al. Loss of function of the nuclear receptor NR2F2, encoding COUP-TF2, causes testis development and cardiac defects in 46,XX children. Am J Hum Genet. (2018) 102:487–93. doi: 10.1016/j.ajhg.2018.01.021
80. Brady PD, DeKoninck P, Fryns JP, Devriendt K, Deprest JA, Vermeesch JR. Identification of dosage-sensitive genes in fetuses referred with severe isolated congenital diaphragmatic hernia. Prenat Diagn. (2013) 33:1283–92. doi: 10.1002/pd.4244
81. Grinspon RP, Rey RA. Disorders of sex development with testicular differentiation in SRY-negative 46,XX individuals: clinical and genetic aspects. Sex Dev. (2016) 10:6089. doi: 10.1159/000445088
82. Grinspon RP, Rey RA. Molecular characterization of XX maleness. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20236089
83. Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. (2013) 140:4129–44. doi: 10.1242/dev.091793
84. Huang B, Wang S, Ning Y, Lamb AN, Bartley J. Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet. (1999) 87:349–53. doi: 10.1002/(ISSN)1096-8628
85. Lefebvre V, Dumitriu B, Penzo-Méndez A, Han Y, Pallavi B. Control of cell fate and differentiation by Sry-related high-mobility-group box (Sox) transcription factors. Int J Biochem Cell Biol. (2007) 39:2195–214. doi: 10.1016/j.biocel.2007.05.019
86. De Santa Barbara P, Bonneaud N, Boizet B, Desclozeaux M, Moniot B, Sudbeck P, et al. Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Müllerian hormone gene. Mol Cell Biol. (1998) 18:6653–65. doi: 10.1128/MCB.18.11.6653
87. Kim GJ, Sock E, Buchberger A, Just W, Denzer F, Hoepffner W, et al. Copy number variation of two separate regulatory regions upstream of SOX9 causes isolated 46,XY or 46,XX disorder of sex development. J Med Genet. (2015) 52:240–7. doi: 10.1136/jmedgenet-2014-102864
88. Lee GM, Ko JM, Shin CH, Yang SW. A Korean boy with 46,XX testicular disorder of sex development caused by SOX9 duplication. Ann Pediatr Endocrinol Metab. (2014) 19:108–12. doi: 10.6065/apem.2014.19.2.108
89. Refai O, Friedman A, Terry L, Jewett T, Pearlman A, Perle MA, et al. De novo 12;17 translocation upstream of SOX9 resulting in 46,XX testicular disorder of sex development. Am J Med Genet A. (2010) 152a:422–6. doi: 10.1002/ajmg.a.33201
90. Cox JJ, Willatt L, Homfray T, Woods CG. A SOX9 duplication and familial 46,XX developmental testicular disorder. N Engl J Med. (2011) 364:91–3. doi: 10.1056/NEJMc1010311
91. Vetro A, Ciccone R, Giorda R, Patricelli MG, Della Mina E, Forlino A, et al. XX males SRY negative: a confirmed cause of infertility. J Med Genet. (2011) 48:710–2. doi: 10.1136/jmedgenet-2011-100036
92. Ohnesorg T, van den Bergen JA, Belluoccio D, Shankara-Narayana N, Kean AM, Vasilaras A, et al. A duplication in a patient with 46,XX ovo-testicular disorder of sex development refines the SOX9 testis-specific regulatory region to 24 kb. Clin Genet. (2017) 92:347–9. doi: 10.1111/cge.12976
93. Xiao B, Ji X, Xing Y, Chen YW, Tao J. A rare case of 46, XX SRY-negative male with approximately 74-kb duplication in a region upstream of SOX9. Eur J Med Genet. (2013) 56:695–8. doi: 10.1016/j.ejmg.2013.10.001
94. Vetro A, Dehghani MR, Kraoua L, Giorda R, Beri S, Cardarelli L, et al. Testis development in the absence of SRY: chromosomal rearrangements at SOX9 and SOX3. Eur J Hum Genet. (2015) 23:1025–32. doi: 10.1038/ejhg.2014.237
95. Hyon C, Chantot-Bastaraud S, Harbuz R, Bhouri R, Perrot N, Peycelon M, et al. Refining the regulatory region upstream of SOX9 associated with 46,XX testicular disorders of Sex Development (DSD). Am J Med Genet A. (2015) 167a:1851–8. doi: 10.1002/ajmg.a.37101
96. Shankara Narayana N, Kean AM, Ewans L, Ohnesorg T, Ayers KL, Watson G, et al. Painful ovulation in a 46,XX SRY -ve adult male with SOX9 duplication. Endocrinol Diabetes Metab Case Rep. (2017) 2017:17–0045. doi: 10.1530/EDM-17-0045
97. Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, et al. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. (2011) 48:825–30. doi: 10.1136/jmedgenet-2011-100255
98. López-Hernández B, Méndez JP, Coral-Vázquez RM, Benítez-Granados J, Zenteno JC, Villegas-Ruiz V, et al. Duplication of SOX9 associated with 46,XX ovotesticular disorder of sex development. Reprod BioMed Online. (2018) 37:107–12. doi: 10.1016/j.rbmo.2018.03.017
99. Croft B, Ohnesorg T, Hewitt J, Bowles J, Quinn A, Tan J, et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat Commun. (2018) 9:5319. doi: 10.1038/s41467-018-07784-9
100. Mengen E, Kayhan G, Kocaay P, Uçaktürk SA. A duplication upstream of SOX9 associated with SRY negative 46,XX ovotesticular disorder of sex development: A case report. J Clin Res Pediatr Endocrinol. (2020) 12:308–14. doi: 10.4274/jcrpe
101. Ushijima K, Ogawa Y, Terao M, Asakura Y, Muroya K, Hayashi M, et al. Identification of the first promoter-specific gain-of-function SOX9 missense variant (p.E50K) in a patient with 46,XX ovotesticular disorder of sex development. Am J Med Genet A. (2021) 185:1067–75. doi: 10.1002/ajmg.a.62063
102. Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, et al. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A. (2001) 98:6698–703. doi: 10.1073/pnas.111092198
103. Miyado M, Inui M, Igarashi M, Katoh-Fukui Y, Takasawa K, Hakoda A, et al. The p.R92W variant of NR5A1/Nr5a1 induces testicular development of 46,XX gonads in humans, but not in mice: phenotypic comparison of human patients and mutation-induced mice. Biol Sex Differ. (2016) 7:56. doi: 10.1186/s13293-016-0114-6
104. Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, et al. WT-1 is required for early kidney development. Cell. (1993) 74:679–91. doi: 10.1016/0092-8674(93)90515-R
105. Sutton E, Hughes J, White S, Sekido R, Tan J, Arboleda V, et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J Clin Invest. (2011) 121:328–41. doi: 10.1172/JCI42580
106. Del Valle I, Buonocore F, Duncan AJ, Lin L, Barenco M, Parnaik R, et al. A genomic atlas of human adrenal and gonad development. Wellcome Open Res. (2017) 2:25. doi: 10.12688/wellcomeopenres
107. Moalem S, Babul-Hirji R, Stavropolous DJ, Wherrett D, Bägli DJ, Thomas P, et al. XX male sex reversal with genital abnormalities associated with a de novo SOX3 gene duplication. Am J Med Genet A. (2012) 158a:1759–64. doi: 10.1002/ajmg.a.35390
108. Mizuno K, Kojima Y, Kamisawa H, Moritoki Y, Nishio H, Nakane A, et al. Elucidation of distinctive genomic DNA structures in patients with 46,XX testicular disorders of sex development using genome wide analyses. J Urol. (2014) 192:535–41. doi: 10.1016/j.juro.2014.02.044
109. Haines B, Hughes J, Corbett M, Shaw M, Innes J, Patel L, et al. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J Clin Endocrinol Metab. (2015) 100:E815–20. doi: 10.1210/jc.2014-4383
110. Tasic V, Mitrotti A, Riepe FG, Kulle AE, Laban N, Polenakovic M, et al. Duplication of the SOX3 gene in an sry-negative 46,XX male with associated congenital anomalies of kidneys and the urinary tract: case report and review of the literature. Balkan J Med Genet. (2019) 22:81–8. doi: 10.2478/bjmg-2019-0006
111. Zhuang J, Chen C, Li J, Jiang Y, Wang J, Wang Y, et al. The 46, XX ovotesticular disorder of sex development with xq27.1q27.2 duplication involving the SOX3 gene: A rare case report and literature review. Front Pediatr. (2021) 9:682846. doi: 10.3389/fped.2021.682846
112. Wei J, Liu C, Zhang M, Liu S, Fu J, Lin P. Duplication of SOX3 in an SRY-negative 46,XX male with prostatic utricle: case report and literature review. BMC Med Genomics. (2022) 15:188. doi: 10.1186/s12920-022-01347-0
113. Oroz M, Vičić A, Požgaj Šepec M, Karnaš H, Stipančić G, Stipoljev F. The smallest dislocated microduplication of Xq27.1 harboring SOX3 gene associated with XX male phenotype. J Pediatr Endocrinol Metab. (2023) 36:86–90. doi: 10.1515/jpem-2022-0324
114. de Oliveira FM, Barros BA, Dos Santos AP, Campos NLV, Mazzola TN, Filho PL, et al. SOX3 duplication in a boy with 46,XX ovotesticular disorder of sex development and his 46,XX sister with atypical genitalia: Probable germline mosaicism. Am J Med Genet A. (2023) 191:592–8. doi: 10.1002/ajmg.a.63051
115. Qin S, Wang X, Wang J. Identification of an SRY-negative 46,XX infertility male with a heterozygous deletion downstream of SOX3 gene. Mol Cytogenet. (2022) 15:2. doi: 10.1186/s13039-022-00580-7
116. Seeherunvong T, Perera EM, Bao Y, Benke PJ, Benigno A, Donahue RP, et al. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am J Med Genet A. (2004) 127a:149–51. doi: 10.1002/ajmg.a.20630
117. Polanco JC, Wilhelm D, Davidson TL, Knight D, Koopman P. Sox10 gain-of-function causes XX sex reversal in mice: implications for human 22q-linked disorders of sex development. Hum Mol Genet. (2010) 19:506–16. doi: 10.1093/hmg/ddp520
118. Wilhelm D, Washburn LL, Truong V, Fellous M, Eicher EM, Koopman P. Antagonism of the testis- and ovary-determining pathways during ovotestis development in mice. Mech Dev. (2009) 126:324–36. doi: 10.1016/j.mod.2009.02.006
119. Falah N, Posey JE, Thorson W, Benke P, Tekin M, Tarshish B, et al. 22q11.2q13 duplication including SOX10 causes sex-reversal and peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease. Am J Med Genet A. (2017) 173:1066–70. doi: 10.1002/ajmg.a.38109
120. Nicholl RM, Grimsley L, Butler L, Palmer RW, Rees HC, Savage MO, et al. Trisomy 22 and intersex. Arch Dis Child Fetal Neonatal Ed. (1994) 71:F57–8. doi: 10.1136/fn.71.1.F57
121. Aleck KA, Argueso L, Stone J, Hackel JG, Erickson RP. True hermaphroditism with partial duplication of chromosome 22 and without SRY. Am J Med Genet. (1999) 85:2–4. doi: 10.1002/(ISSN)1096-8628
122. Raymond CS, Shamu CE, Shen MM, Seifert KJ, Hirsch B, Hodgkin J, et al. Evidence for evolutionary conservation of sex-determining genes. Nature. (1998) 391:691–5. doi: 10.1038/35618
123. Jørgensen A, Nielsen JE, Blomberg Jensen M, Græm N, Rajpert-De Meyts E. Analysis of meiosis regulators in human gonads: a sexually dimorphic spatio-temporal expression pattern suggests involvement of DMRT1 in meiotic entry. Mol Hum Reprod. (2012) 18:523–34. doi: 10.1093/molehr/gas030
124. Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature. (2011) 476:101–4. doi: 10.1038/nature10239
125. Matson CK, Zarkower D. Sex and the singular DM domain: insights into sexual regulation, evolution and plasticity. Nat Rev Genet. (2012) 13:163–74. doi: 10.1038/nrg3161
126. Zarkower D, Murphy MW. DMRT1: an ancient sexual regulator required for human gonadogenesis. Sex Dev. (2022) 16:112–25. doi: 10.1159/000518272
127. Bertini V, Baldinotti F, Parma P, Tyutyusheva N, Sepich M, Bertolucci G, et al. In tandem intragenic duplication of doublesex and mab-3-related transcription factor 1 (DMRT1) in an SRY-negative boy with a 46,XX disorder of sex development. Genes (Basel). (2023) 14:2067. doi: 10.3390/genes14112067
128. Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. (2001) 104:875–89. doi: 10.1016/S0092-8674(01)00284-7
129. Chiang HS, Wu YN, Wu CC, Hwang JL. Cytogenic and molecular analyses of 46,XX male syndrome with clinical comparison to other groups with testicular azoospermia of genetic origin. J Formos Med Assoc. (2013) 112:72–8. doi: 10.1016/j.jfma.2012.02.009
130. Chi L, Itäranta P, Zhang S, Vainio S. Sprouty2 is involved in male sex organogenesis by controlling fibroblast growth factor 9-induced mesonephric cell migration to the developing testis. Endocrinology. (2006) 147:3777–88. doi: 10.1210/en.2006-0299
131. Ambu R, Vinci L, Gerosa C, Fanni D, Obinu E, Faa A, et al. WT1 expression in the human fetus during development. Eur J Histochem. (2015) 59:2499. doi: 10.4081/ejh.2015.2499
132. Wilhelm D, Englert C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. (2002) 16:1839–51. doi: 10.1101/gad.220102
133. Haber DA, Sohn RL, Buckler AJ, Pelletier J, Call KM, Housman DE. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc Natl Acad Sci U S A. (1991) 88:9618–22. doi: 10.1073/pnas.88.21.9618
134. Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. (2001) 106:319–29. doi: 10.1016/S0092-8674(01)00453-6
135. Ferrari MTM, Elias FM, Gomes NLRA, Batista RL, Faria JAD, Nishi MY, et al. WT1: A single gene associated with multiple and severe phenotypes. Endocrine Metab Science. (2023) 13:100143. doi: 10.1016/j.endmts.2023.100143
136. Cen C, Chen M, Zhou J, Zhang L, Duo S, Jiang L, et al. Inactivation of Wt1 causes pre-granulosa cell to steroidogenic cell transformation and defect of ovary development†. Biol Reprod. (2020) 103:60–9. doi: 10.1093/biolre/ioaa042
137. Gao F, Zhang J, Wang X, Yang J, Chen D, Huff V, et al. Wt1 functions in ovarian follicle development by regulating granulosa cell differentiation. Hum Mol Genet. (2014) 23:333–41. doi: 10.1093/hmg/ddt423
138. Morohashi KI, Inoue M, Baba T. Coordination of multiple cellular processes by NR5A1/nr5a1. Endocrinol Metab (Seoul). (2020) 35:756–64. doi: 10.3803/EnM.2020.402
139. Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev. (2008) 2:200–9. doi: 10.1159/000152036
140. Domenice S, MaChado AZ, Ferreira FM, Ferraz-de-Souza B, Lerario AM, Lin L, et al. Wide spectrum of NR5A1-related phenotypes in 46,XY and 46,XX individuals. Birth Defects Res C Embryo Today. (2016) 108:309–20. doi: 10.1002/bdrc.21145
141. Igarashi M, Takasawa K, Hakoda A, Kanno J, Takada S, Miyado M, et al. Identical NR5A1 missense mutations in two unrelated 46,XX individuals with testicular tissues. Hum Mutat. (2017) 38:39–42. doi: 10.1002/humu.23116
142. Baetens D, Stoop H, Peelman F, Todeschini AL, Rosseel T, Coppieters F, et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet Med. (2017) 19:367–76. doi: 10.1038/gim.2016.118
143. Bashamboo A, Donohoue PA, Vilain E, Rojo S, Calvel P, Seneviratne SN, et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum Mol Genet. (2016) 25:3446–53. doi: 10.1093/hmg/ddw186
144. Knarston IM, Robevska G, van den Bergen JA, Eggers S, Croft B, Yates J, et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum Mutat. (2019) 40:207–16. doi: 10.1002/humu.23672
145. Takasawa K, Igarashi M, Ono M, Takemoto A, Takada S, Yamataka A, et al. Phenotypic variation in 46,XX disorders of sex development due to the NR5A1 p.R92W variant: a sibling case report and literature review. Sex Dev. (2017) 11:284–8. doi: 10.1159/000485868
146. García-Acero M, Molina M, Moreno O, Ramirez A, Forero C, Céspedes C, et al. Gene dosage of DAX-1, determining in sexual differentiation: duplication of DAX-1 in two sisters with gonadal dysgenesis. Mol Biol Rep. (2019) 46:2971–8. doi: 10.1007/s11033-019-04758-y
147. Gomes NL, de Paula LCP, Silva JM, Silva TE, Lerario AM, Nishi MY, et al. A 46,XX testicular disorder of sex development caused by a Wilms’ tumour Factor-1 (WT1) pathogenic variant. Clin Genet. (2019) 95:172–6. doi: 10.1111/cge.13459
148. Eozenou C, Gonen N, Touzon MS, Jorgensen A, Yatsenko SA, Fusee L, et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 (WT1) gene. Proc Natl Acad Sci U S A. (2020) 117:13680–8. doi: 10.1073/pnas.1921676117
149. Sirokha D, Gorodna O, Vitrenko Y, Zelinska N, Ploski R, Nef S, et al. A novel WT1 mutation identified in a 46,XX testicular/ovotesticular DSD patient results in the retention of intron 9. Biol (Basel). 30(12):1248. doi: 10.3390/biology10121248
150. Kirino S, Yogi A, Adachi E, Nakatani H, Gau M, Iemura R, et al. Phenotypic variation in 46,XX disorders of sex development due to the 4th zinc finger domain variant of WT1: A familial case report. Sex Dev. (2023) 17(1):51–5. doi: 10.1159/000529720
151. Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, et al. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. (2002) 57:19–36. doi: 10.1210/rp.57.1.19
152. Askari M, Rastari M, Seresht-Ahmadi M, McElreavey K, Bashamboo A, Razzaghy-Azar M, et al. A missense mutation in NR5A1 causing female to male sex reversal: A case report. Andrologia. (2020) 52:e13585. doi: 10.1111/and.13585
153. Swartz JM, Ciarlo R, Guo MH, Abrha A, Weaver B, Diamond DA, et al. A 46,XX ovotesticular disorder of sex development likely caused by a steroidogenic factor-1 (NR5A1) variant. Horm Res Paediatr. (2017) 87:191–5. doi: 10.1159/000452888
154. Zanaria E, Muscatelli F, Bardoni B, Strom TM, Guioli S, Guo W, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature. (1994) 372:635–41. doi: 10.1038/372635a0
155. Bardoni B, Zanaria E, Guioli S, Floridia G, Worley KC, Tonini G, et al. A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet. (1994) 7:497–501. doi: 10.1038/ng0894-497
156. Iyer AK, McCabe ER. Molecular mechanisms of DAX1 action. Mol Genet Metab. (2004) 83:60–73. doi: 10.1016/j.ymgme.2004.07.018
157. Meeks JJ, Weiss J, Jameson JL. Dax1 is required for testis determination. Nat Genet. (2003) 34:32–3. doi: 10.1038/ng1141
158. Bouma GJ, Albrecht KH, Washburn LL, Recknagel AK, Churchill GA, Eicher EM. Gonadal sex reversal in mutant Dax1 XY mice: a failure to upregulate Sox9 in pre-Sertoli cells. Development. (2005) 132:3045–54. doi: 10.1242/dev.01890
159. Meeks JJ, Crawford SE, Russell TA, Morohashi K, Weiss J, Jameson JL. Dax1 regulates testis cord organization during gonadal differentiation. Development. (2003) 130:1029–36. doi: 10.1242/dev.00316
160. Fernandes-Freitas I, Milona A, Murphy KG, Dhillo WS, Owen BM. Live birth in sex-reversed XY mice lacking the nuclear receptor dax1. Sci Rep. (2020) 10:1703. doi: 10.1038/s41598-020-58788-9
161. Mizusaki H, Kawabe K, Mukai T, Ariyoshi E, Kasahara M, Yoshioka H, et al. Dax-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1) gene transcription is regulated by wnt4 in the female developing gonad. Mol Endocrinol. (2003) 17:507–19. doi: 10.1210/me.2002-0362
162. Seminara SB, Achermann JC, Genel M, Jameson JL, Crowley WF Jr. X-linked adrenal hypoplasia congenita: a mutation in DAX1 expands the phenotypic spectrum in males and females. J Clin Endocrinol Metab. (1999) 84:4501–9. doi: 10.1210/jc.84.12.4501
163. El-Khairi R, Martinez-Aguayo A, Ferraz-de-Souza B, Lin L, Achermann JC. Role of DAX-1 (NR0B1) and steroidogenic factor-1 (NR5A1) in human adrenal function. Endocr Dev. (2011) 20:38–46. doi: 10.1159/000321213
165. Barbaro M, Oscarson M, Schoumans J, Staaf J, Ivarsson SA, Wedell A. Isolated 46,XY gonadal dysgenesis in two sisters caused by a Xp21.2 interstitial duplication containing the DAX1 gene. J Clin Endocrinol Metab. (2007) 92:3305–13. doi: 10.1210/jc.2007-0505
166. Igarashi M, Dung VC, Suzuki E, Ida S, Nakacho M, Nakabayashi K, et al. Cryptic genomic rearrangements in three patients with 46,XY disorders of sex development. PloS One. (2013) 8:e68194. doi: 10.1371/journal.pone.0068194
167. Norling A, Lindén Hirschberg A, Iwarsson E, Persson B, Wedell A, Barbaro M. Novel candidate genes for 46,XY gonadal dysgenesis identified by a customized 1 M array-CGH platform. Eur J Med Genet. (2013) 56:661–8. doi: 10.1016/j.ejmg.2013.09.003
168. Nishi MY, Faria Júnior JAD, Krepischi ACV, de Moraes DR, da Costa SS, Silva E, et al. A small supernumerary xp marker chromosome including genes NR0B1 and MAGEB causing partial gonadal dysgenesis and gonadoblastoma. Sex Dev. (2022) 16:55–63. doi: 10.1159/000517085
169. Dangle P, Touzon MS, Reyes-Múgica M, Witchel SF, Rajkovic A, Schneck FX, et al. Female-to-male sex reversal associated with unique Xp21.2 deletion disrupting genomic regulatory architecture of the dosage-sensitive sex reversal region. J Med Genet. (2017) 54:705–9. doi: 10.1136/jmedgenet-2016-104128
170. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. (2007) 447:425–32. doi: 10.1038/nature05918
171. Tachibana M. Epigenetics of sex determination in mammals. Reprod Med Biol. (2016) 15:59–67. doi: 10.1007/s12522-015-0223-7
172. Miyawaki S, Tachibana M. Role of epigenetic regulation in mammalian sex determination. Curr Top Dev Biol. (2019) 134:195–221. doi: 10.1016/bs.ctdb.2019.01.008
173. Knarston I, Ayers K, Sinclair A. Molecular mechanisms associated with 46,XX disorders of sex development. Clin Sci (Lond). (2016) 130:421–32. doi: 10.1042/CS20150579
174. Gunes SO, Metin Mahmutoglu A, Agarwal A. Genetic and epigenetic effects in sex determination. Birth Defects Res C Embryo Today. (2016) 108:321–36. doi: 10.1002/bdrc.21146
175. Grossman H, Shalgi R. A role of microRNAs in cell differentiation during gonad development. Results Probl Cell Differ. (2016) 58:309–36. doi: 10.1007/978-3-319-31973-5_12
176. Real FM, Sekido R, Lupiáñez DG, Lovell-Badge R, Jiménez R, Burgos M. A microRNA (mmu-miR-124) prevents Sox9 expression in developing mouse ovarian cells. Biol Reprod. (2013) 89:78. doi: 10.1095/biolreprod.113.110957
177. Tanaka SS, Nishinakamura R. Regulation of male sex determination: genital ridge formation and Sry activation in mice. Cell Mol Life Sci. (2014) 71:4781–802. doi: 10.1007/s00018-014-1703-3
Keywords: differences of sex development (DSD), 46, XX testicular DSD, 46, XX ovotesticular DSD, gonadal development, ovary
Citation: Ferrari MTM, Silva ESdN, Nishi MY, Batista RL, Mendonca BB and Domenice S (2024) Testicular differentiation in 46,XX DSD: an overview of genetic causes. Front. Endocrinol. 15:1385901. doi: 10.3389/fendo.2024.1385901
Received: 14 February 2024; Accepted: 18 March 2024;
Published: 24 April 2024.
Edited by:
Silvano Bertelloni, University of Pisa, ItalyReviewed by:
Laura Audí, Vall d’Hebron University Hospital, SpainCopyright © 2024 Ferrari, Silva, Nishi, Batista, Mendonca and Domenice. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sorahia Domenice, c29yYWhpYS5kQGhjLmZtLnVzcC5icg==
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.