 Jorge Lopez-Tello
Jorge Lopez-Tello
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 10 February 2023
Sec. Developmental Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1116770
This article is part of the Research TopicDevelopmental Biology and Endocrine Research for a Successful PregnancyView all 16 articles
Conditions such as small for gestational age (SGA), which is defined as birthweight less than 10th percentile for gestational age can predispose to neurodevelopmental abnormalities compared to babies with normal birthweight. Fetal growth and birthweight depend on placental function, as this organ transports substrates to the developing fetus and it acts as a source of endocrine factors, including steroids and prolactins that are required for fetal development and pregnancy maintenance. To advance our knowledge on the aetiology of fetal growth disorders, the vast majority of the research has been focused on studying the transport function of the placenta, leaving practically unexplored the contribution of placental hormones in the regulation of fetal growth. Here, using mice and natural variability in fetal growth within the litter, we compared fetuses that fell on or below the 10th percentile (classified as SGA) with those that had adequate weight for their gestational age (AGA). In particular, we compared placental endocrine metabolism and hormone production, as well as fetal brain weight and expression of developmental, growth and metabolic genes between SGA and AGA fetuses. We found that compared to AGA fetuses, SGA fetuses had lower placental efficiency and reduced capacity for placental production of hormones (e.g. steroidogenic gene Cyp17a1, prolactin Prl3a1, and pregnancy-specific glycoproteins Psg21). Brain weight was reduced in SGA fetuses, although this was proportional to the reduction in overall fetal size. The expression of glucose transporter 3 (Slc2a3) was reduced despite the abundance of AKT, FOXO and ERK proteins were similar. Developmental (Sv2b and Gabrg1) and microglia genes (Ier3), as well as the pregnancy-specific glycoprotein receptor (Cd9) were lower in the brain of SGA versus AGA fetuses. In this mouse model of SGA, our results therefore demonstrate that placental endocrine dysfunction is associated with changes in fetal growth and fetal brain development.
Abnormal birthweight is one of the most common complications of pregnancy that has immediate and long-term consequences for offspring wellbeing (1). Compared to babies that have an appropriate weight for their gestational age (AGA), babies born small (<10th percentile) or large for gestational age (>90th percentile) (SGA or LGA, respectively) are at higher risk of obstetric and neonatal complications (2, 3). Moreover, studies have shown that SGA or LGA increases the risk for metabolic diseases and cognitive and neurodevelopmental abnormalities compared to AGA (4–8). The identification and subsequent classification of babies with abnormal weight is challenging, as it requires segregation of babies that are constitutively small or large due to parental genetics versus babies that grew abnormally due to intrauterine problems (9–11). Therefore, work is required to understand the causes and mechanisms by which altered fetal growth may arise.
Fetal growth is regulated, in part, by the placenta which is a fetal organ responsible for the transport of gases and nutrients between the mother and the fetus. Aberrant placental function related to failure of substrate transport or deficits in vascular growth have been linked to fetal growth abnormalities, including fetal growth restriction, as well as stillbirth (12–14). However, the placenta is also a powerful endocrine organ that secretes an abundance of hormones and growth factors into the maternal and fetal circulation that allow for fetal development and pregnancy maintenance (15–20). Indeed, there is now growing evidence suggesting that placental metabolism and function impact on fetal brain development (21–27). For example, in mice, there is genomic linkage between placental and the fetal hypothalamic development (28). In mice, placental metabolism of serotonin is critical for fetal forebrain development. Others have found that placental allopregnanolone deficiency in mice alters cerebellar white matter development and programmes postnatal autistic-like behaviour in the offspring (24). Therefore, these studies suggest a potential link between placental endocrine function, fetal growth and fetal neurodevelopment, which could have long last effects on neurocognitive health outcomes.
In this scenario, animal models in which fetal and postnatal outcomes can be monitored are essential. Much of the investigations focused on fetoplacental development have been conducted in rodents, rabbits, sheep and pigs (29–36). However, the use of mice is still preferred over the aforementioned animal species due to multiple reasons. For example, the short gestational period and the relatively easy, lower cost maintenance of mice compared to large animal species is great for investigating fetal growth control. In mice, the sequence of events for brain maturation are largely similar to in humans (37). Moreover, a similar to in human, the mouse placenta is haemochorial in structure. An additional key advantage of the mouse is that its placenta is structurally divided in two functionally specialised regions; transport is carried out in the labyrinth zone, while hormone production is principally performed by the junctional zone (Jz). This characteristic is very helpful when studying placental function, as both layers can be easy separated (38). Nonetheless, the vast majority of published studies assessing the importance of the placenta for fetal growth have focused on placental transport function [e.g (31, 39–43).], leaving the significance of placental endocrine function practically unexplored.
Numerous animal models of pregnancy complications have been created, including those that mimic environmental and maternal conditions (11, 29, 36). However, models based on constructing fetal growth curves and percentile cut-offs, as applied in human obstetrics, are not commonplace for experimental research on placental function and fetal physiology (44). Here, using the mouse as an experimental animal model, we took advantage of the normal fetal weight variation observed within the litter to compare placental endocrine function and fetal brain development for fetuses that were classified as AGA or SGA using percentile cut-offs. We therefore hypothesize that abnormal placental endocrine output is an additional factor contributing to the development of SGA. Moreover, using fetal growth curves and percentile cut-offs in a model in which maternal health is preserved provides us with a valuable opportunity to study endocrine interaction between the placenta and fetus in a controlled intrauterine environment. Indeed, our work identifies that the production of prolactins and pregnancy specific glycoproteins (PSGs) by the placenta is compromised in conjunction with reduced fetal growth and brain development of SGA fetuses. Our work reinforces the idea of a placental-fetal brain axis that is controlled, in part, by placental hormones (25).
All animal work was performed under the UK Animals (Scientific Procedures) Act 1986. Experiments were conducted with a total of 11 C57BL6/J wild-type female mice (4 months old) housed at the University of Cambridge Animal Facility under a 12/12 dark-light system and fed ad libitum with a standard chow diet (RM3; Special Dietary Services). Females were time-mated with C57BL6/J males and the day a copulatory plug was detected was designated as gestational day (GD) 1 (term occurs ∼GD20). On GD16, pregnant females were killed by cervical dislocation, the uterus removed and fetuses cleaned from feto-placental membranes. Each fetus (and its corresponding placenta), was identified based on its position in the uterus, dried on tissue paper, weighed and immediately decapitated. Fetal brains were removed, weighed and immersed in liquid nitrogen for rapid freezing. The placental Jz was dissected from maternal decidua and placental labyrinth zone, and subsequently snap-frozen in liquid nitrogen. All frozen fetal and placental samples were stored at −80°C until tissues were powdered in individual pieces of foil using a hammer and dry ice for RNA/protein extraction (all samples were maintained in a frozen state, as far as possible). Only viable fetuses were used in the study and litter sizes ranged from 6 to 11 pups.
Placental Jz and fetal brain RNA (8-9 samples per group) was extracted using the RNeasy Plus Mini Kit (Qiagen) and the quantity of RNA obtained was determined using a NanoDrop spectrophotometer (NanoDrop Technologies) as previously described (45). RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to manufacturer’s instructions. Samples were analysed with a StepOne real-time PCR machine (Thermo Fisher Scientific) in duplicate using SYBR Green qPCR master mix (Applied Biosystems, Thermo Fisher Scientific) and primers described in Supplementary Table 1. Brain gene expression was normalized to the genomic mean of two housekeeping genes (Actb and Gapdh), while placental genes were normalized using Actb and Ywhaz as reference genes. These housekeeper genes remained stably expressed between the groups. Analysis was performed using the 2-ΔΔCt method (46).
Total protein was extracted from fetal brain and placental Jz (5 samples per group) using RIPA buffer (R0278-50M, Sigma Aldrich) supplemented with mini EDTA-free protease inhibitor cocktail mix (Roche), 1mM β-glycerophosphate (G-9891, Sigma Aldrich) and 1mM sodium orthovanadate (S65089891, Sigma Aldrich). Lysates were centrifuged at 2,500 rpm for 15 minutes at 4°C and protein concentration determined using the Pierce BCA protein assay kit (23225, Thermo Fisher Scientific). Samples were mixed with SDS gel loading buffer (L-4390, Sigma Aldrich) and protein denaturation was performed at 90°C for 5 minutes. After gel electrophoresis, proteins were transferred from the gel onto 0.45 µm nitrocellulose membranes (10600012, Amersham Protran). Membranes were then blocked either with 5% fetal bovine serum (A2153-100G, Sigma Aldrich) or semi-skimmed milk (Marvel) for 1 hour at room temperature and incubated overnight at 4°C with primary antibodies described in Table 1. The day after, membranes were washed with TBS-T and incubated for 1 hour at room temperature with secondary antibodies (1:10,000 NA934 or NA931, Amersham). Membranes were exposed to ECL substrate (SuperSignal West Femto, Thermo Fisher Scientific) and images were taken with an Invitrogen iBright imaging system (Thermo Fisher Scientific). Pixel intensity of protein bands was analysed with ImageJ software and normalization performed against beta-actin levels. Phosphorylated proteins were normalized to its corresponding total protein abundance.
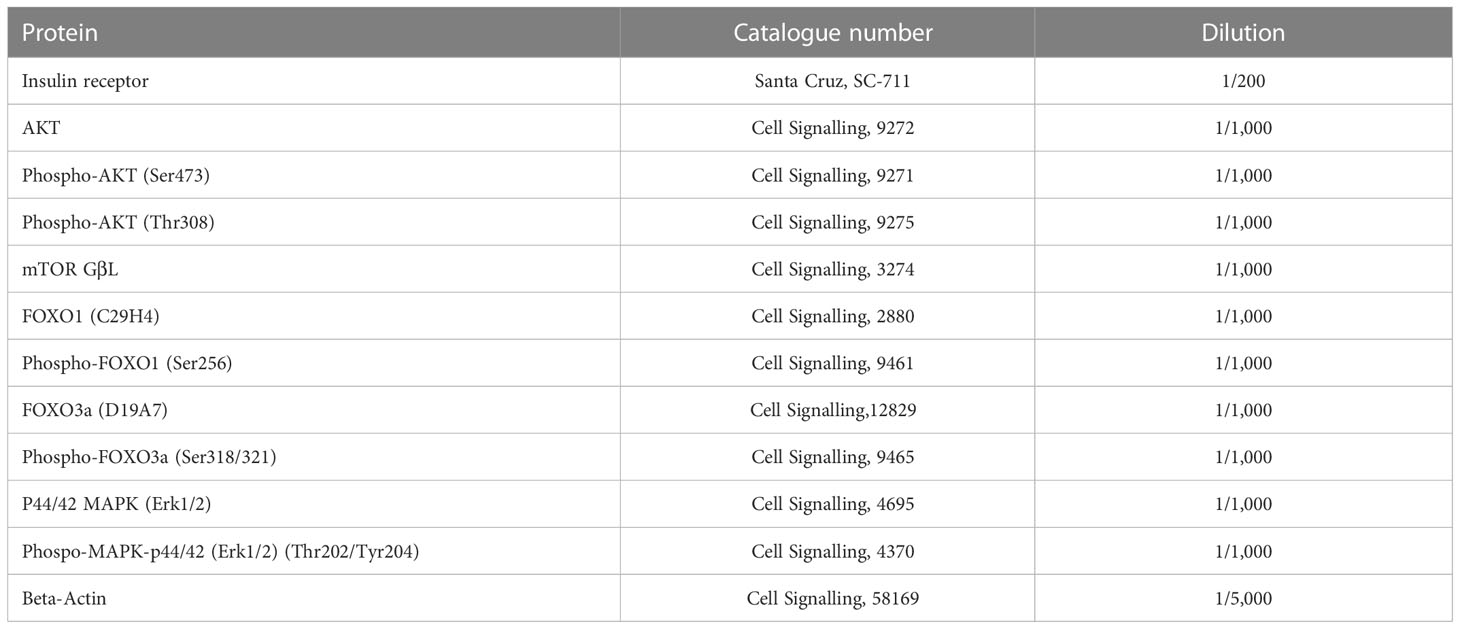
Table 1 List of antibodies used for western blotting.
No sample size calculation was conducted prior to undertaking this study. For the purpose of this paper, the fetus, and not the mother, was designated as the experimental unit. GraphPad Prism software (version 9) was used to determine statistical differences between groups. The distribution of fetal weights (graphed as a histogram) was constructed with a non-linear regression (Gaussian distribution) and percentiles calculated with the descriptive statistical analysis tool of GraphPad Prism software. Then, data in Excel were organized according to fetal weight and separated in three experimental groups based on the fetal weight threshold (SGA fetuses: 10th percentile ≤321 mg; AGA fetuses: 321-409 mg and LGA fetuses: 90th percentile≥410 mg). ROUT test was used to identify outlier values, which were then excluded for statistical purposes. Normality and homogeneity of variance of variables for western blotting and qPCR data were performed using the Shapiro-Wilks test in GraphPad Prism. For comparisons between two groups, the Student’s t-test or Mann-Whitney test were applied according to the normality of the variable. For data with more than two groups, one-way ANOVA coupled with Bonferroni post hoc test was employed. The relation between fetal intrauterine position as well as litter size with the presence of SGA, AGA and LGA was analysed by Chi square test using a contingency table in GraphPad Prism software. Pearson r correlations were performed with GraphPad Prism software and presented in a double gradient colour mapping graph that was subsequently modified with Adobe Illustrator to facilitate visualization. qPCR and western blotting data are expressed as individual data points and reported as mean ± SEM. P-values <0.05 were considered statistically significant.
The distribution of fetal weights across the 11 litters studied is shown in Figure 1A and litter sizes across the different litters used in this study can be found in Supplementary Table 2. A total of 9 fetuses were considered SGA (below the 10th percentile) and they were obtained from 5 different litters. On average, fetal weight was significantly lower by 19% in SGA fetuses when compared to AGA fetuses (Figures 1A, B). A total of 11 LGA fetuses (above the 90th percentile) were obtained from 3 different litters and these were on average, 12% heavier than the AGA group. All litters had at least 3 AGA fetuses. Analysis of placental weight did not show significant differences between the SGA, AGA and LGA fetuses (Figure 1C). However, the fetal weight to placental weight ratio, defined as grams of fetus produced by grams of placenta and also known as placental efficiency, was significantly reduced in the SGA fetuses compared to AGA and LGA fetuses. No differences in placental efficiency were observed between the AGA and LGA fetuses (Figure 1D).
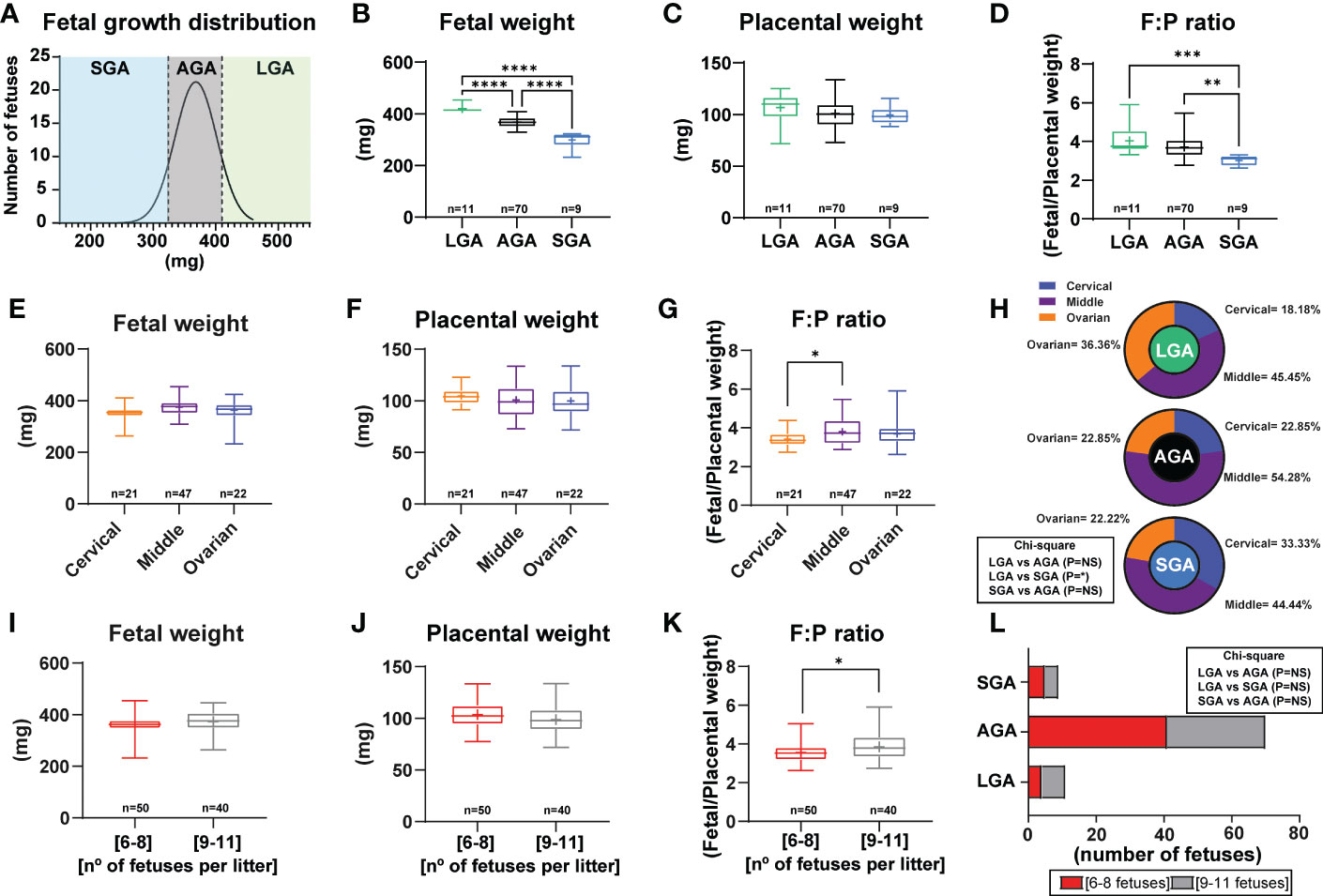
Figure 1 Fetal and placental weight in mice and establishment of a model to study small for gestational age. (A) Frequency distribution curve for fetal weights on GD16. Vertical line denotes the 10th (321mg) and 90th percentiles (410 mg). (B) Fetal weight for fetuses classified as SGA, AGA and LGA. (C) Placental weight classified as SGA, AGA and LGA. (D) Placental efficiency (defined as grams of fetus produced by grams of placenta) classified as SGA, AGA and LGA. (E–G) Feto-placental weights and placental efficiency based on their position in the uterus. (H) Distribution of LGA, AGA and SGA fetuses according to their position in the uterus. (I–K) Feto-placental weights and placental efficiency based on litter size. (L) Distribution of LGA, AGA and SGA fetuses according to litter size. Data are from 90 fetuses in total from 11 litters. Feto-placental weights are shown as box plots and whiskers. The rectangle shows the distribution, the line the median, the whiskers the maximum and minimum and the “+” is the mean of the group. Statistical analysis performed by one-way ANOVA, Student t-test, Mann-Whitney test and Chi-square test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AGA (appropriate for gestational age), LGA (large for gestational age), SGA (small for gestational age), n° (number).
Fetal and placental weights were not affected by the uterine position, as similar weights were found for fetuses positioned close to the ovary, cervix, and in between these two positions (Figures 1E, F). Nonetheless, placental efficiency was significantly lower by 10% for fetuses positioned close to the ovary compared to those in the middle of the uterus (Figure 1G). Individual group analysis demonstrated that there was not a clear pattern in the incidence of being SGA, AGA or LGA based on uterine position. Although a significant effect was observed between fetal position and being LGA or SGA (Figure 1H). Indeed, the number of SGA fetuses positioned close to the cervix was higher than for those proximal to the ovary (33.33% versus 22.22%; Figure 1H). This pattern was the opposite for LGA fetuses, with 22.22% located close to the cervix and 44.44% close to the ovary. AGA fetuses were primarily found in the middle part of the uterus (54.28%).
Finally, we divided our data according to the number of fetuses per litter to assess if litter size impacted the percentage of SGA, AGA and LGA (litters of 6 to 8 fetuses versus 9 to 11 fetuses; Figures 1I–L). This analysis revealed that there was no difference between fetal or placental weight, nor the proportion of fetuses classified as SGA, AGA or LGA between litters of 6 to 8 fetuses compared to those of 9 to 11 fetuses (Figure 1L). However, placental efficiency was significantly greater by 7.80% in litters with 9 to 11 fetuses compared to those with 6 to 8 (Figure 1K). Moreover, we then performed analysis of LGA and SGA fetuses by considering the litter size as a potential effect (comparison of LGA-SGA fetuses in litters of 6-8 fetuses versus LGA-SGA fetuses in litters of 9-11 fetuses). As displayed in Supplementary Table 3, we did not see differences in LGA or SGA fetuses in any of the aforementioned parameters.
To understand if the reduced placental efficiency for SGA fetuses was related to alterations in the endocrine function, key growth and metabolism proteins were quantified by western blotting in the placental Jz of SGA compared to AGA fetuses (Figures 2A, B). This revealed that the abundance of FOXO1, which functions in cell cycle and differentiation (47), was significantly reduced by 27% in the SGA compared to AGA group without affecting phosphorylated site at serine 256 (ratio of phosphorylated to total FOXO1; Figure 2B). However, the abundance the insulin receptor β, AKT (total and phosphorylated at threonine 308 and serine 473 sites), FOXO3 (total and phosphorylated at serine 318/321 sites), ERK (total and phosphorylated at threonine 202 and tyrosine 204 site) and mTOR- GβL were unaltered between SGA and AGA fetuses (Figures 2A, B).
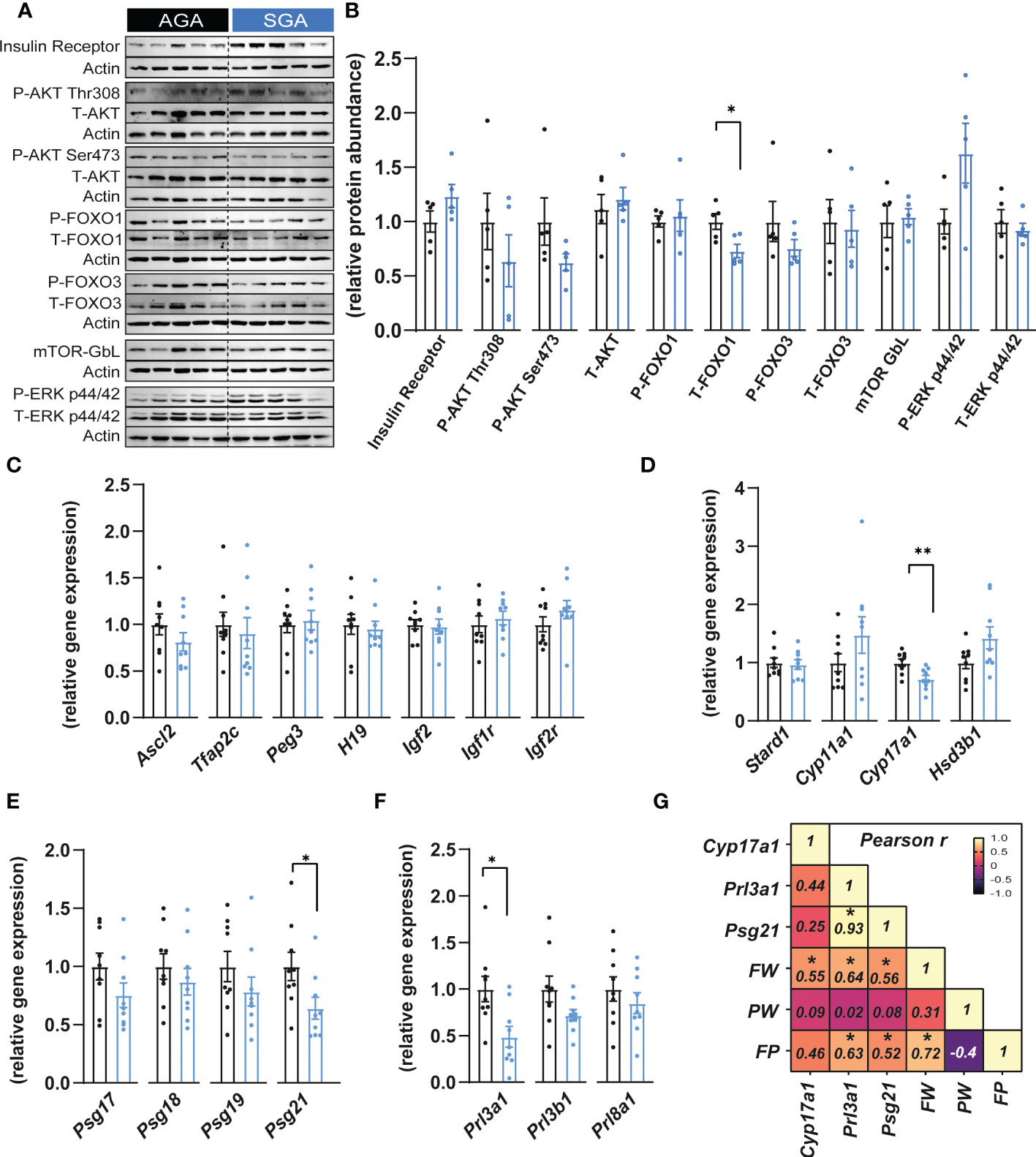
Figure 2 Small for gestational age is associated with altered expression of placental hormones. (A, B) Immunoblots and protein abundance of key growth and metabolic signalling proteins in dissected placental junctional zones (n=5 samples per group). (C) Expression of key genes involved in placental function and growth (n=9 samples per group). (D–F) Expression of steroidogenic pathway regulatory (D), pregnancy specific glycoprotein (E) and prolactin (F) genes in dissected placental junctional zones (n=9 samples per group). (G) Heatmap of Pearson’s correlations (number inside the box corresponds to the r value for the correlations that were significant) (n=18 samples). Data are shown as individual values and bars represent mean ± SEM. Statistical analysis performed by paired or unpaired t-test (Student t-test and Mann-Whitney test, respectively). *P<0.05, **P<0.01. AGA (appropriate for gestational age) and SGA (small for gestational age), FW (fetal weight), FP (feto-placental ratio, also known as placental efficiency), PW (placental weight), P (phosphorylated protein levels), T (total protein levels).
We next evaluated whether the expression of imprinted genes could be altered in placental Jz samples from SGA fetuses as previous studies have shown that, in mice, the imprinted loci Igf2-H19, Ascl2 and Peg3 are implicated in the control placental Jz formation and/or function (48–51). No changes were observed in any of the imprinted genes analysed in the Jz between SGA and AGA (Figure 2C). The Jz expression of Tfap2c, which has been shown to control the expression of certain imprinted genes such as H19 and Ascl2 (52), was also unaltered between the two experimental groups (Figure 2C). Moreover, mRNA levels of the IGF2 receptors, Igf1r and Igf2r remained similarly expressed in the Jz between SGA and AGA (Figure 2C).
We then assessed the placental capacity to produce hormones by measuring mRNA levels of genes involved in steroidogenesis (Stard1, Cyp11a1, Cyp17a1 and Hsd3b1), pregnancy specific glycoprotein genes (PSGs; Psg17, Psg18, Psg19 and Psg21) and prolactins (Prl3a1, Prl3b1 and Prl8a1) in the Jz of SGA and AGA fetuses (Figures 2D–F). We observed that Cyp17a1 expression was 28% lower in the placental Jz of the SGA group compared to AGA (Figure 2D). Moreover, Psg21 mRNA levels were reduced by 36% in the SGA versus AGA fetuses (Figure 2E). The expression of prolactin genes, Prl3a1 was also downregulated by 51% in the SGA group versus the AGA (Figure 2F). However, the Jz expression of other hormone-related genes quantified, namely Stard1, Cyp11a1, Hsd3b1, Psg17, Psg18, Psg19, Prl3a1, Prl3b1 and Prl8a1 were not different between the two experimental groups. To understand the contribution of changes in placental Jz Cyp17a1, Psg21 and Prl3a1 expression for feto-placental size and placental efficiency, we performed Pearson r correlations (Figure 2G). This revealed that Jz expression of all three genes correlated positively with fetal weight. Prl3a1 and Psg21 correlated positively with placental efficiency. Both, Prl3a1 and Psg21 correlated positively with each other. Lastly, placental efficiency, but not placental weight correlated positively with fetal weight (Figure 2G).
Short and long-term neurological impairments have been observed in offspring who were SGA (53). Therefore, we investigated if there could be any changes in fetal brain development in our mouse model of SGA. We found that SGA fetuses exhibited a 20% and 25% reduction in brain weight compared to the AGA and LGA groups, respectively (Figure 3A). In contrast, brain weight was similar between AGA and LGA fetuses (Figure 3A). Analysis of fetal brain weight relative to fetal body size revealed that the reduction in brain size was proportional to the reduced size of the SGA fetuses (brain to body weight ratio; not different between SGA, AGA and LGA fetuses; Figure 3B), which suggested that SGA fetuses were symmetrically smaller. We then determined the relationship between placental and fetal brain size by assessing the ratio of fetal brain weight to placental weight. This showed that fetal brain weight to placental weight ratio was significantly reduced by 18% in SGA compared to AGA fetuses (Figure 3C). However, this ratio was not different between LGA and AGA fetuses.
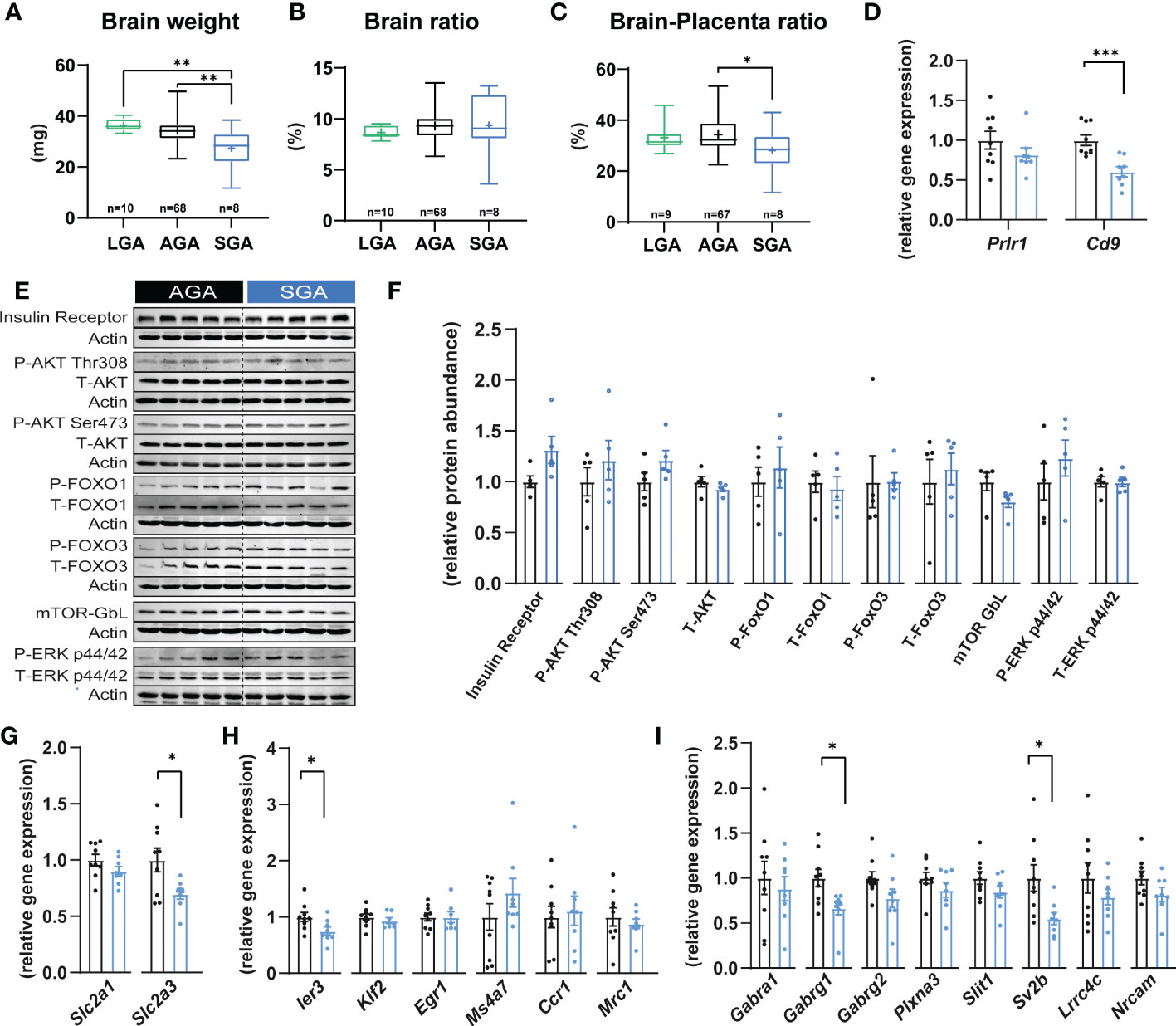
Figure 3 Small for gestational age and its impact on fetal brain. (A) Brain weight. (B) Brain weight expressed as a ratio of the brain weight divided by fetal weight. (C) Brain weight expressed as a ratio of brain weight divided by placental weight. (D) The expression of key prolactin and pregnancy-specific glycoprotein receptors in fetal brain (n=8-9 samples per group). (E, F) Immunoblots and protein abundance of key growth and metabolic signalling proteins in fetal brain (n=5 samples per group). (G–I) Expression levels of glucose transporters, microglia and axon developmental genes (n=8-9 samples per group). Data are from 90 fetuses in total from 11 litters (excluding outliers). Data are shown as individual values and bars represent mean ± SEM. Statistical analysis performed by one-way ANOVA followed by Bonferroni post hoc test (variables of three groups), paired or unpaired t-test (Student t-test and Mann-Whitney test, respectively). ROUT test was conducted to detect and eliminate outliers. *P<0.05, **P<0.01, ***P<0.001. AGA, (appropriate for gestational age); SGA, (small for gestational age); P, (phosphorylated protein levels); T, (total protein levels).
We also analysed the effect of uterine position and litter size on fetal brain development (Supplementary Figure 1). We did not see differences in the weight of the brain, although brain ratio was significantly increased in fetuses positioned closer to the ovaries compared to those in the middle position (Supplementary Figures 1A, B). No differences were found in the brain-placental ratio and litter size did not affect the weight of the brain, although brain ratio was significantly increased in litters of 6-8 fetuses compared to those of 9 to 11 fetuses (Supplementary Figure 1C–F). Analysis of LGA and SGA fetuses by considering the litter size as a potential effect (comparison of LGA-SGA fetuses in litters of 6-8 fetuses versus LGA-SGA fetuses in litters of 9-11 fetuses) showed that LGA fetuses in litters of 6-8 fetuses had reduced brain ratio and brain-placental ratio compared to those LGA in larger litters (Supplementary Table 3). These effects were not found in the SGA fetuses (Supplementary Table 3).
To further investigate the relationship between placental function and fetal brain we quantified the mRNA levels of receptors that mediate the action of prolactins and PSGs, Prlr1 and Cd9, respectively in SGA compared to AGA fetuses (54, 55). We found that the expression of Cd9 was 39.5% lower in the brain of SGA versus AGA fetuses (Figure 3D). However, the mRNA expression levels of Prlr1 in the brain were similar between SGA and AGA fetuses. To understand the potential molecular changes underlying brain weight differences in SGA versus AGA fetuses, we quantified the levels of key cellular signalling pathways involved in growth and metabolism and found that these remained similar in the brain of SGA and AGA fetuses (e.g. insulin receptor β, AKT, FOXO1-3, mTOR- GβL and ERK; Figures 3E, F). As the fetal brain is highly dependent on glucose metabolism for growth, we quantified the expression of genes encoding the main glucose transporters in the brain of SGA and AGA fetuses (56). Glucose transporter number 1 (Slc2a1) did not change between SGA and AGA fetuses; however, the mRNA levels Slc2a3 were significantly reduced by 30% in SGA compared to AGA fetuses (Figure 3G).
We then explored whether our model of SGA and placental malfunction may be linked to changes in the differentiation and maturation of microglia. We analysed mRNA levels of 6 microglia markers (Ier3, Klf2, Egr1, Ms4a7, Ccr1 and Mrc1) (57) and found that 5 out of 6 were stably expressed in the brain of SGA compared to AGA fetuses (Figure 3H). Interestingly, the expression of Ier3 was significantly reduced by 25% in SGA compared to AGA fetuses. We then quantified the expression of 8 genes involved in axonogenesis (58) (Plxna3, Slit1, Gabra1, Sv2b, Gabrg1, Gabrg2, Lrrc4c and Nrcam). We found that Sv2b and Gabrg1 were reduced by 45% and 12% in SGA compared to the AGA group, respectively (Figure 3I). The other axonogenesis genes analysed did not differ between SGA and AGA.
In this study we have shown that the defects in placental endocrine function are associated with reduced fetal growth and brain development in a mouse model of SGA. In particular, we found that the placental capacity to respond metabolically (FOXO1) and produce sex steroids (Cyp17a1), prolactins (Prl3a1) and PSGs (Psg21) were compromised in fetuses classified as SGA. Moreover, SGA fetuses have a proportional reduction in brain weight, low expression of glucose transporter 3 (Slc2a3), axogenesis (Sv2b and Gabrg1) and microglia (Ier3) genes along with reduced expression of the PSG receptor (Cd9) in the fetal brain of SGA fetuses compared to AGA. Finally, we found a significant link between the placenta and fetal brain development, as shown by the reduced brain-placental weight ratio in SGA fetuses. Together, our data reveal that placental hormones could be additional regulators of fetal growth with implications for fetal brain development. Furthermore, mis-communication of placental hormones with the developing fetal brain may have immediate and long-lasting implications for neurocognitive outcomes.
In the mouse, we have recently shown that the placental secretome comprises more than 1,000 proteins, including prolactins (17). In rodents, the prolactin family consists of 23 closely related genes and their expression varies spatially and temporally in the placenta (59). Although the role of prolactins in modulating maternal physiology is well-defined, for instance in metabolic adaptations during pregnancy (15, 60, 61) and in promoting maternal nurturing behaviour and lactation (62), their role in embryo and fetal development is not fully understood. In humans, prolactin can be found in the fetal circulation from mid-gestation and its receptors are expressed in fetal tissues by 7.5 weeks of gestation (63). In mice, prolactin receptors are widely expressed by the fetus, including its brain, with Prlr mRNA levels detectable as early as on GD8 (64). Prior work has speculated that prolactin in the fetus may be important for the growth of the adrenal cortex, production of pulmonary surfactant and in the control of the immune system (65, 66). However, further research is required to understand how the prolactin hormone family drives fetal growth, especially the growth and development of the fetal brain, given that placental production of PRL was decreased in our SGA mouse fetuses. Consistent with our findings, in humans, there is a down-regulation of the growth hormone/chorionic somatomammotropin (hGH/CSH) cluster in the SGA placenta (67). Moreover, other work has shown that mothers carrying SGA male fetuses display lower concentrations of prolactin in their circulation when compared to women carrying AGA male fetuses (68).
In our mouse model, the expression of Cyp17a1 was lower in the SGA versus AGA fetuses. Our findings are consistent with studies in humans that show polymorphisms in Cyp17a1 can influence the risk of SGA (69). In humans, the expression of Cyp17a1 is mainly localized in the syncytiotrophoblast, acting as a source of estrogen during pregnancy (70). In mice, placental Cyp17a1 expression in the placenta is modulated by Igf2 (48); however in the present study, Igf2 and other imprinted genes that regulate placental endocrine capacity were not differentially expressed between SGA and AGA fetuses. Other work has indicated that sex steroids interact with FOXO1 signalling (71). In addition, FOXO1 is important for embryonic development and placental differentiation (72). Thus, understanding the link between reduced FOXO1 abundance and decreased placental endocrine capacity in our SGA mouse model would be valuable.
The expression of PSGs was also altered in our mouse model of SGA, with lower Psg21 mRNA levels in the placental Jz and this was couple to decreased fetal brain expression of the PSG receptor, Cd9. In rodents, PSGs are synthesised by spongiotrophoblasts and giant cells, while in humans, PSGs are produced by the syncytiotrophoblast (73–75). PSGs belong to the carcinoembryonic antigen family (76) and although the functions of PSGs are not fully elucidated, they are proposed to modulate immune cells (77, 78) and angiogenesis (79). Moreover, low levels of PSGs are associated with the development of pregnancy complications, including early pregnancy loss, placental insufficiency, fetal growth restriction and fetal hypoxia (80–84). Reinforced by our findings that placental Prl3a1 and Psg21 expression are positively correlated with fetal weight, future work should evaluate circulating levels of these hormones in fetal plasma. The placental transport labyrinth region in the mouse expresses receptors for such hormones (49, 85). Thus, future studies should evaluate whether the relationship between placental hormone production and fetal growth may also be mediated by potential local changes in the formation and function of the placental labyrinth as a result of altered paracrine signalling.
An additional objective of our work was to characterize the potential changes occurring in the fetal brain in our mouse SGA model. We did see a symmetrical reduction in brain weight and decreased mRNA levels of the glucose transporter 3 (Slc2a3) and impaired axogenesis markers (as inferred by the low expression levels of Sv2b and Gabrg1) in SGA fetuses. Of note, we did not find any changes in PI3K-AKT-mTOR signalling, which is critical for biological processes like nutrient uptake, cell growth and migration (86), in the brain of SGA compared to AGA fetuses. Changes in the PI3K-AKT-mTOR signalling cascade have been linked to microcephaly (87), due to reductions in neuronal cell division (88). We also did not identify any changes in FOXO1-3 and ERK signalling, and microglia markers (57) were similarly expressed between SGA and AGA fetuses, aside from the reduced mRNA levels of Ier3. Microglia are important specialised cell types in the nervous system that influence brain development, including neuronal proliferation or synaptic remodelling (89). SGA is a condition that can be linked to inflammation, as SGA newborns can exhibit elevated interleukin (IL) IL-1β, IL-8 and tumor necrosis factor in their circulation during the first postnatal weeks (90). The gene Ier3 responds to changes in toll-like receptors (TLR3) (91) and its expression can be induced by different stimuli, including cytokines, infections and growth factors (92). In mice, low Ier3 levels results in abnormal immune regulation and inflammation (92). Therefore, future work should explore the contribution of differences in Ier3 in SGA fetus to the immune status, function and structural development of the developing brain. Regarding the changes in axogenesis markers, in the current study we employed whole brain lysates for our analyses and therefore cannot exclude the possibility of molecular changes in specific regions of the brain in the context of SGA. Nevertheless, in our SGA model, the expression of Sv2b, which encodes a synaptic vesicle protein and Gabrg1, which functions as a γ-Amino-butyric acid (GABA) receptor was reduced. Prior work has found that Sv2b is regulated by the maternal gut microbiota during mouse fetal brain development (58). This gene is broadly expressed in the central nervous system, with especially high expression in glutamatergic neurons (93, 94). Other investigations in mice, have also shown that intrauterine growth restriction is related to postnatal memory deficits that may be linked to abnormal proportions of maturing glutamatergic neurons at birth (95). Hence, study of the postnatal neurocognitive outcomes in our mouse model of SGA is warranted. In our study, the reduction in Sv2b and Gabrg1 was in line with decreased expression of Cd9 by the fetal brain in SGA fetuses, which may suggest a role for placental PSGs in shaping fetal axogenesis. Indeed, previous work in mice and humans has found CD9 is present in different cell types including astrocytes, sympathetic neurons and Schwann cells (96). Given that placental PSG production was also decreased in our SGA model, a greater understanding of how the placenta may influence fetal brain development through PSG signalling should be explored in future work.
A major limitation of the current study was that our study was not sufficiently powered to evaluate the influence of fetal sex on placental endocrine capacity and fetal brain development. This is important as previous work has demonstrated that fetal sex is an important determinant of placental endocrine capacity (48, 49). Furthermore, recent work in mice has revealed that placental steroid metabolism and metabolic signalling capacity differ depending on the size and the sex of the fetus (40). Moreover, the potential contribution of the labyrinth zone in the placental endocrine support of fetal development was not explored. This would be important in future work, as previous work has shown that the rodent labyrinth zone can assist in the production of steroids by the placenta (97). Moreover, steroids regulate glucose uptake through changes in Slc2a1 and Slc2a3 (98) and in our model we found reduced levels of Slc2a3 in the SGA fetal brain. Moreover, the mouse is a litter-bearing specie and despite that our analysis of fetal intrauterine position and litter size composition show that there are no significant differences in fetal-placental weights (including fetal brain weight), we observed that placental efficiency is affected by both, the intrauterine fetal position and the number of fetuses per litter. Previous work performed in rats have identified greater blood flow at the cervical and ovarian ends compared to middle of the uterus, which may partly explain our findings (99). Work in multiple species has also found that placental efficiency is greater in monotocous species with multiple gestations and in polytocous species with larger litters, which is not surprising given the higher fetal demand on maternal resources (100, 101). Therefore, the analysis in combination of both, endocrine and vascular/transport function of the placenta, will provide a more accurate and detailed angle to understand the causes and short/long-term consequences of SGA.
In the current study, we intentionally studied unmanipulated, wildtype litters using fetal growth curves and percentile cut-offs to preserve maternal health and precisely study the role of placental endocrine interactions with the fetus in a controlled intrauterine environment. However, in doing so, we likely limited our ability to detect important pathways and proteins involved in placental endocrine function. Nonetheless, our work provides new evidence about the importance of placental hormones in the regulation of fetal growth and development. Moreover, the low levels of Cd9 in the fetal brain suggest a direct link to changes in placental PSG production and sensitivity in the context of SGA. Further work is required to ascertain the specific contribution of the dysregulated placental hormones found in our SGA model (Cyp17a1, Prl3a1, Prl3b1 and Psg21) and brain development. Moreover, animal studies deploying in vivo approaches to specifically delete genes in mouse placenta endocrine cells would be highly useful for decoding the communication between placental hormones and fetal organogenesis (48, 49, 102). This work could be complemented by experiments using fetal brain explants or cerebral organoids that are cultured with different combinations of placental hormones. To sum up, our results identify placental hormones as key regulators of fetal growth and may have relevance for understanding the aetiology of fetal growth disorders, and the mechanistic basis of associations between poor fetal growth and the subsequent increased risk of poor neurocognitive outcomes.
The data presented in the study are deposited in the Gene Expression Omnibus repository, accession number GSE224682.
The animal study was reviewed and approved by the United Kingdom Home Office under the Animals (Scientific Procedures) Act 1986 and underwent review by the University of Cambridge AnimalWelfare and Ethical Review Body.
JL-T performed research. JL-T and AS-P wrote the paper and designed the study. All authors contributed to the article and approved the submitted version.
JL-T holds a Sir Henry Wellcome Postdoctoral Fellowship (220456/Z/20/Z). AS-P is supported by a MRC New Investigator Grant (MR/R022690/1/RG93186) and Lister Institute of Preventative Medicine Research Prize (RG93692).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1116770/full#supplementary-material
Supplementary Figure 1 | Brain phenotype based on the intrauterine position and litter size. Data are from 90 fetuses in total from 11 litters (excluding outliers). Data are shown as box plots and whiskers. The rectangle shows the distribution, the line the median, the whiskers the maximum and minimum and the “+” is the mean of the group. Statistical analysis performed by one-way ANOVA followed by Bonferroni post hoc test (variables of three groups), paired or unpaired t-test (Student t-test and Mann-Whitney test, respectively). *P<0.05.
1. Figueras F, Gratacos E. An integrated approach to fetal growth restriction. Best Pract Res Clin Obstetrics Gynaecol (2017) 38:48–58. doi: 10.1016/j.bpobgyn.2016.10.006
2. Sharma D, Shastri S, Sharma P. Intrauterine growth restriction: Antenatal and postnatal aspects. Clin Med Insights: Pediatr (2016) 10:CMPed.S40070. doi: 10.4137/CMPed.S40070
3. Ng S-K, Olog A, Spinks AB, Cameron CM, Searle J, McClure RJ. Risk factors and obstetric complications of large for gestational age births with adjustments for community effects: results from a new cohort study. BMC Public Health (2010) 10:460. doi: 10.1186/1471-2458-10-460
4. Padidela RNR, Bhat V. Neurobehavioral assessment of appropriate for gestational and small for gestational age babies. Indian Pediatr (2003) 40:1063–8.
5. Arcangeli T, Thilaganathan B, Hooper R, Khan KS, Bhide A. Neurodevelopmental delay in small babies at term: A systematic review. Ultrasound Obstet Gynecol (2012) 40:267–75. doi: 10.1002/uog.11112
6. Meher S, Hernandez-Andrade E, Basheer SN, Lees C. Impact of cerebral redistribution on neurodevelopmental outcome in small-for-gestational-age or growth-restricted babies: A systematic review. Ultrasound Obstet Gynecol (2015) 46:398–404. doi: 10.1002/uog.14818
7. Dong Y, Luo Z-C, Nuyt AM, Audibert F, Wei S-Q, Abenhaim HA, et al. Large-for-Gestational-Age may be associated with lower fetal insulin sensitivity and β-cell function linked to leptin. J Clin Endocrinol Metab (2018) 103:3837–44. doi: 10.1210/jc.2018-00917
8. Cho WK, Suh B-K. Catch-up growth and catch-up fat in children born small for gestational age. Korean J Pediatr (2016) 59:1–7. doi: 10.3345/kjp.2016.59.1.1
9. Burton GJ, Jauniaux E. Pathophysiology of placental-derived fetal growth restriction. Am J Obstetrics Gynecol (2018) 218:S745–61. doi: 10.1016/j.ajog.2017.11.577
10. Baschat AA, Hecher K. Fetal growth restriction due to placental disease. Semin Perinatol (2004) 28:67–80. doi: 10.1053/j.semperi.2003.10.014
11. Schröder HJ. Models of fetal growth restriction. Eur J Obstet Gynecol Reprod Biol (2003) 110 Suppl 1:S29–39. doi: 10.1016/S0301-2115(03)00170-2
12. MacDonald TM, Hui L, Tong S, Robinson AJ, Dane KM, Middleton AL, et al. Reduced growth velocity across the third trimester is associated with placental insufficiency in fetuses born at a normal birthweight: a prospective cohort study. BMC Med (2017) 15:164. doi: 10.1186/s12916-017-0928-z
13. Gibbins KJ, Pinar H, Reddy UM, Saade GR, Goldenberg RL, Dudley DJ, et al. Findings in stillbirths associated with placental disease. Am J Perinatol (2020) 37:708–15. doi: 10.1055/s-0039-1688472
14. Gaillard R, Steegers EAP, Tiemeier H, Hofman A, Jaddoe VWV. Placental vascular dysfunction, fetal and childhood growth, and cardiovascular development. Circulation (2013) 128:2202–10. doi: 10.1161/CIRCULATIONAHA.113.003881
15. Napso T, Yong HEJ, Lopez-Tello J, Sferruzzi-Perri AN. The role of placental hormones in mediating maternal adaptations to support pregnancy and lactation. Front Physiol (2018) 9:1091. doi: 10.3389/fphys.2018.01091
16. Lopez-Tello J, Jimenez-Martinez MA, Salazar-Petres E, Patel R, George AL, Kay RG, et al. Identification of structural and molecular signatures mediating adaptive changes in the mouse kidney in response to pregnancy. Int J Mol Sci (2022) 23. doi: 10.3390/ijms23116287
17. Napso T, Zhao X, Lligoña MI, Sandovici I, Kay RG, George AL, et al. Placental secretome characterization identifies candidates for pregnancy complications. Commun Biol (2021) 4:701. doi: 10.1038/s42003-021-02214-x
18. Freemark M. Placental hormones and the control of fetal growth. J Clin Endocrinol Metab (2010) 95:2054–7. doi: 10.1210/jc.2010-0517
19. Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev (2006) 27:141–69. doi: 10.1210/er.2005-0011
20. Handwerger S, Freemark M. The roles of placental growth hormone and placental lactogen in the regulation of human fetal growth and development. J Pediatr Endocrinol Metab (2000) 13:343–56. doi: 10.1515/jpem.2000.13.4.343
21. Strawn M, Samal A, Sarker MB, Dhakal P, Behura SK. Relevance of microRNAs to the regulation of the brain-placental axis in mice. Placenta (2021) 112:123–31. doi: 10.1016/j.placenta.2021.07.293
22. Guo H, Yang Y, Qiao Y, He J, Yao W, Zheng W. Heat stress affects fetal brain and intestinal function associated with the alterations of placental barrier in late pregnant mouse. Ecotoxicol Environ Saf (2021) 227:112916. doi: 10.1016/j.ecoenv.2021.112916
23. Devarshi PP, Grant RW, Ikonte CJ, Hazels Mitmesser S. Maternal omega-3 nutrition, placental transfer and fetal brain development in gestational diabetes and preeclampsia. Nutrients (2019) 11:E1107. doi: 10.3390/nu11051107
24. Bakalar D, O’Reilly JJ, Lacaille H, Salzbank J, Ellegood J, Lerch JP, et al. Lack of placental neurosteroid alters cortical development and female somatosensory function. Front Endocrinol (2022) 13:972033. doi: 10.3389/fendo.2022.972033
26. O’Donnell KJ, Meaney MJ. Fetal origins of mental health: The developmental origins of health and disease hypothesis. Am J Psychiatry (2017) 174:319–28. doi: 10.1176/appi.ajp.2016.16020138
27. Ursini G, Punzi G, Chen Q, Marenco S, Robinson JF, Porcelli A, et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med (2018) 24:792–801. doi: 10.1038/s41591-018-0021-y
28. Broad KD, Keverne EB. Placental protection of the fetal brain during short-term food deprivation. Proc Natl Acad Sci (2011) 108:15237–41. doi: 10.1073/pnas.1106022108
29. Lopez-Tello J, Arias-Alvarez M, Gonzalez-Bulnes A, Sferuzzi-Perri AN. Models of intrauterine growth restriction and fetal programming in rabbits. Mol Reprod Dev (2019) 86:1781–809. doi: 10.1002/mrd.23271
30. Gonzalez-Bulnes A, Parraguez VH, Berlinguer F, Barbero A, Garcia-Contreras C, Lopez-Tello J, et al. The impact of prenatal environment on postnatal life and performance: Future perspectives for prevention and treatment. Theriogenology (2020) 150:15–9. doi: 10.1016/j.theriogenology.2020.01.029
31. Lopez-Tello J, Jimenez-Martinez MA, Herrera EA, Krause BJ, Sferruzzi-Perri AN. Progressive uterine artery occlusion in the Guinea pig leads to defects in placental structure that relate to fetal growth. Placenta (2018) 72–73:36–40. doi: 10.1016/j.placenta.2018.10.003
32. Swanson AM, David AL. Animal models of fetal growth restriction: Considerations for translational medicine. Placenta (2015) 36:623–30. doi: 10.1016/j.placenta.2015.03.003
33. López-Tello J, Barbero A, González-Bulnes A, Astiz S, Rodríguez M, Formoso-Rafferty N, et al. Characterization of early changes in fetoplacental hemodynamics in a diet-induced rabbit model of IUGR. J Dev Orig Health Dis (2015) 6:454–61. doi: 10.1017/S2040174415001385
34. Sferruzzi-Perri AN, Camm EJ. The programming power of the placenta. Front Physiol (2016) 7:33. doi: 10.3389/fphys.2016.00033
35. Gonzalez-Bulnes A, Astiz S, Parraguez VH, Garcia-Contreras C, Vazquez-Gomez M. Empowering translational research in fetal growth restriction: Sheep and swine animal models. Curr Pharm Biotechnol (2016) 17:848–55. doi: 10.2174/1389201017666160519111529
36. Christoforou ER, Sferruzzi-Perri AN. Molecular mechanisms governing offspring metabolic programming in rodent models of in utero stress. Cell Mol Life Sci (2020) 77:4861–98. doi: 10.1007/s00018-020-03566-z
37. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol (2013) 0:1–16. doi: 10.1016/j.pneurobio.2013.04.001
38. Sandovici I, Reiterer M, Constância M, Branco CM. Protocol to isolate and culture primary mouse feto-placental endothelial cells. STAR Protoc (2022) 3:101721. doi: 10.1016/j.xpro.2022.101721
39. López-Tello J, Pérez-García V, Khaira J, Kusinski LC, Cooper WN, Andreani A, et al. Fetal and trophoblast PI3K p110α have distinct roles in regulating resource supply to the growing fetus in mice. Elife (2019) 8. doi: 10.7554/eLife.45282
40. Salazar-Petres E, Carvalho DP, Lopez-Tello J, Sferruzzi-Perri AN. Placental mitochondrial function, nutrient transporters, metabolic signalling and steroid metabolism relate to fetal size and sex in mice. Biology of Reproduction (2021) 106(6):1292–311. doi: 10.1101/2021.07.22.453249
41. Sferruzzi-Perri AN, López-Tello J, Fowden AL, Constancia M. Maternal and fetal genomes interplay through phosphoinositol 3-kinase(PI3K)-p110α signaling to modify placental resource allocation. Proc Natl Acad Sci (2016) 113:11255–60. doi: 10.1073/pnas.1602012113
42. Kusinski LC, Stanley JL, Dilworth MR, Hirt CJ, Andersson IJ, Renshall LJ, et al. eNOS knockout mouse as a model of fetal growth restriction with an impaired uterine artery function and placental transport phenotype. Am J Physiol Regul Integr Comp Physiol (2012) 303:R86–93. doi: 10.1152/ajpregu.00600.2011
43. Sandovici I, Georgopoulou A, Pérez-García V, Hufnagel A, López-Tello J, Lam BYH, et al. The imprinted Igf2-Igf2r axis is critical for matching placental microvasculature expansion to fetal growth. Developmental Cell (2021) 57(1):63–79. doi: 10.1101/520536
44. Dilworth MR, Kusinski LC, Baker BC, Renshall LJ, Greenwood SL, Sibley CP, et al. Defining fetal growth restriction in mice: A standardized and clinically relevant approach. Placenta (2011) 32:914–6. doi: 10.1016/j.placenta.2011.08.007
45. Lopez-Tello J, Schofield Z, Kiu R, Dalby MJ, van Sinderen D, Le Gall G, et al. Maternal gut microbiota bifidobacterium promotes placental morphogenesis, nutrient transport and fetal growth in mice. Cell Mol Life Sci (2022) 79:386. doi: 10.1007/s00018-022-04379-y
46. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001.1262
47. Polter A, Yang S, Zmijewska AA, van Groen T, J-H P, DePinho RA, et al. FoxO transcription factors in brain: Regulation and behavioral manifestation. Biol Psychiatry (2009) 65:150. doi: 10.1016/j.biopsych.2008.08.005
48. Aykroyd BRL, Tunster SJ, Sferruzzi-Perri AN. Igf2 deletion alters mouse placenta endocrine capacity in a sexually dimorphic manner. J Endocrinol (2020) 246:93–108. doi: 10.1530/JOE-20-0128
49. Aykroyd BRL, Tunster SJ, Sferruzzi-Perri AN. Loss of imprinting of the Igf2-H19 ICR1 enhances placental endocrine capacity via sex-specific alterations in signalling pathways in the mouse. Development (2022) 149:dev199811. doi: 10.1242/dev.199811
50. Tunster SJ, Boqué-Sastre R, McNamara GI, Hunter SM, Creeth HDJ, John RM. Peg3 deficiency results in sexually dimorphic losses and gains in the normal repertoire of placental hormones. Front Cell Dev Biol (2018) 6:123. doi: 10.3389/fcell.2018.00123
51. Tunster SJ, McNamara GI, Creeth HDJ, John RM. Increased dosage of the imprinted Ascl2 gene restrains two key endocrine lineages of the mouse placenta. Dev Biol (2016) 418:55–65. doi: 10.1016/j.ydbio.2016.08.014
52. Sharma N, Kubaczka C, Kaiser S, Nettersheim D, Mughal SS, Riesenberg S, et al. Tpbpa-cre-mediated deletion of TFAP2C leads to deregulation of Cdkn1a, Akt1 and the ERK pathway, causing placental growth arrest. Development (2016) 143:787–98. doi: 10.1242/dev.128553
53. Vollmer B, Edmonds CJ. School age neurological and cognitive outcomes of fetal growth retardation or small for gestational age birth weight. Front Endocrinol (Lausanne) (2019) 10:186. doi: 10.3389/fendo.2019.00186
54. Waterhouse R, Ha C, Dveksler GS. Murine CD9 is the receptor for pregnancy-specific glycoprotein 17. J Exp Med (2002) 195:277–82. doi: 10.1084/jem.20011741
55. Ha CT, Waterhouse R, Warren J, Zimmermann W, Dveksler GS. N-glycosylation is required for binding of murine pregnancy-specific glycoproteins 17 and 19 to the receptor CD9. Am J Reprod Immunol (2008) 59:251–8. doi: 10.1111/j.1600-0897.2007.00573.x
56. Ye X, Shin B-C, Baldauf C, Ganguly A, Ghosh S, Devaskar SU. Developing brain glucose transporters, serotonin, serotonin transporter and oxytocin receptor expression in response to early life hypocaloric, hypercaloric dietary and air pollutant exposures. Dev Neurosci (2021) 43:27–42. doi: 10.1159/000514709
57. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity (2019) 50:253–271.e6. doi: 10.1016/j.immuni.2018.11.004
58. Vuong HE, Pronovost GN, Williams DW, Coley EJL, Siegler EL, Qiu A, et al. The maternal microbiome modulates fetal neurodevelopment in mice. Nature (2020) 586:281–6. doi: 10.1038/s41586-020-2745-3
59. Simmons DG, Rawn S, Davies A, Hughes M, Cross JC. Spatial and temporal expression of the 23 murine Prolactin/Placental lactogen-related genes is not associated with their position in the locus. BMC Genomics (2008) 9:352. doi: 10.1186/1471-2164-9-352
60. Lopez-Vicchi F, De Winne C, Brie B, Sorianello E, Ladyman SR, Becu-Villalobos D. Metabolic functions of prolactin: Physiological and pathological aspects. J Neuroendocrinol (2020) 32:e12888. doi: 10.1111/jne.12888
61. Salazar-Petres ER, Sferruzzi-Perri AN. Pregnancy-induced changes in β-cell function: what are the key players? J Physiol (2002) 600(5):1089–117. doi: 10.1113/JP281082
62. Larsen CM, Grattan DR. Prolactin-induced mitogenesis in the subventricular zone of the maternal brain during early pregnancy is essential for normal postpartum behavioral responses in the mother. Endocrinology (2010) 151:3805–14. doi: 10.1210/en.2009-1385
63. Freemark M. Ontogenesis of prolactin receptors in the human fetus: roles in fetal development. Biochem Soc Trans (2001) 29:38–41. doi: 10.1042/0300-5127:0290038
64. Tzeng SJ, Linzer DI. Prolactin receptor expression in the developing mouse embryo. Mol Reprod Dev (1997) 48:45–52. doi: 10.1002/(SICI)1098-2795(199709)48:1<45::AID-MRD6>3.0.CO;2-P
66. Handwerger S, Freemark M. Role of placental lactogen and prolactin in human pregnancy. In: Mahesh VB, Dhindsa DS, Anderson E, Kalra SP, editors. Regulation of ovarian and testicular function. advances in experimental medicine and biology. Boston, MA: Springer US (1987). p. 399–420. doi: 10.1007/978-1-4684-5395-9_19
67. Männik J, Vaas P, Rull K, Teesalu P, Rebane T, Laan M. Differential expression profile of growth Hormone/Chorionic somatomammotropin genes in placenta of small- and Large-for-Gestational-Age newborns. J Clin Endocrinol Metab (2010) 95:2433–42. doi: 10.1210/jc.2010-0023
68. Ladyman SR, Larsen CM, Taylor RS, Grattan DR, McCowan LME. Case–control study of prolactin and placental lactogen in SGA pregnancies. Reprod Fertil (2021) 2:244–50. doi: 10.1530/RAF-21-0020
69. Yamada H, Sata F, Kato EH, Saijo Y, Kataoka S, Morikawa M, et al. A polymorphism in the CYP17 gene and intrauterine fetal growth restriction. Mol Hum Reprod (2004) 10:49–53. doi: 10.1093/molehr/gah005
70. Escobar JC, Patel SS, Beshay VE, Suzuki T, Carr BR. The human placenta expresses CYP17 and generates androgens de novo. J Clin Endocrinol Metab (2011) 96:1385–92. doi: 10.1210/jc.2010-2504
71. Labied S, Kajihara T, Madureira PA, Fusi L, Jones MC, Higham JM, et al. Progestins regulate the expression and activity of the forkhead transcription factor FOXO1 in differentiating human endometrium. Mol Endocrinol (2006) 20:35–44. doi: 10.1210/me.2005-0275
72. Ferdous A, Morris J, Abedin MJ, Collins S, Richardson JA, Hill JA. Forkhead factor FoxO1 is essential for placental morphogenesis in the developing embryo. Proc Natl Acad Sci U.S.A. (2011) 108:16307–12. doi: 10.1073/pnas.1107341108
73. Lei KJ, Sartwell AD, Pan CJ, Chou JY. Cloning and expression of genes encoding human pregnancy-specific glycoproteins. J Biol Chem (1992) 267:16371–8. doi: 10.1016/S0021-9258(18)42012-1
74. Rebstock S, Lucas K, Weiss M, Thompson J, Zimmermann W. Spatiotemporal expression of pregnancy-specific glycoprotein gene rnCGM1 in rat placenta. Dev Dyn (1993) 198:171–81. doi: 10.1002/aja.1001980303
75. Kromer B, Finkenzeller D, Wessels J, Dveksler G, Thompson J, Zimmermann W. Coordinate expression of splice variants of the murine pregnancy-specific glycoprotein (PSG) gene family during placental development. Eur J Biochem (1996) 242:280–7. doi: 10.1111/j.1432-1033.1996.0280r.x
76. McLellan AS, Fischer B, Dveksler G, Hori T, Wynne F, Ball M, et al. Structure and evolution of the mouse pregnancy-specific glycoprotein (Psg) gene locus. BMC Genomics (2005) 6:1–17. doi: 10.1186/1471-2164-6-4
77. Snyder SK, Wessner DH, Wessells JL, Waterhouse RM, Wahl LM, Zimmermann W, et al. Pregnancy-specific glycoproteins function as immunomodulators by inducing secretion of IL-10, IL-6 and TGF-beta1 by human monocytes. Am J Reprod Immunol (2001) 45:205–16. doi: 10.1111/j.8755-8920.2001.450403.x
78. Ha CT, Waterhouse R, Wessells J, Wu JA, Dveksler GS. Binding of pregnancy-specific glycoprotein 17 to CD9 on macrophages induces secretion of IL-10, IL-6, PGE2, and TGF-beta1. J Leukoc Biol (2005) 77:948–57. doi: 10.1189/jlb.0804453
79. McLellan AS, Fischer B, Dveksler G, Hori T, Wynne F, Ball M, et al. Structure and evolution of the mouse pregnancy-specific glycoprotein (Psg) gene locus. BMC Genomics (2005) 6:4. doi: 10.1186/1471-2164-6-4
80. Würz H, Geiger W, Künzig HJ, Jabs-Lehmann A, Bohn H, Lüben G. Radioimmunoassay of SP1 (pregnancy-specific beta1-glycoprotein) in maternal blood and in amniotic fluid normal and pathologic pregnancies. J Perinat Med (1981) 9:67–78. doi: 10.1515/jpme.1981.9.2.67
81. MacDonald DJ, Scott JM, Gemmell RS, Mack DS. A prospective study of three biochemical fetoplacental tests: serum human placental lactogen, pregnancy-specific beta 1-glycoprotein, and urinary estrogens, and their relationship to placental insufficiency. Am J Obstet Gynecol (1983) 147:430–6. doi: 10.1016/s0002-9378(16)32239-6
82. Masson GM, Anthony F, Wilson MS. Value of schwangerschaftsprotein 1 (SP1) and pregnancy-associated plasma protein-a (PAPP-a) in the clinical management of threatened abortion. Br J Obstet Gynaecol (1983) 90:146–9. doi: 10.1111/j.1471-0528.1983.tb08899.x
83. Tuzluoğlu S, Üstünyurt E, Karaşin SS, Karaşin ZT. Investigation of serum pregnancy-specific beta-1-Glycoprotein and relationship with fetal growth restriction. JBRA Assist Reprod (2022) 26:267–73. doi: 10.5935/1518-0557.20210068
84. Tamsen L, Johansson SG, Axelsson O. Pregnancy-specific beta 1-glycoprotein (SP1) in serum from women with pregnancies complicated by intrauterine growth retardation. J Perinat Med (1983) 11:19–25. doi: 10.1515/jpme.1983.11.1.19
85. Marsh B, Blelloch R. Single nuclei RNA-seq of mouse placental labyrinth development. Elife (2020) 9:e60266. doi: 10.7554/eLife.60266
86. Yu JSL, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development (2016) 143:3050–60. doi: 10.1242/dev.137075
87. Wang L, Zhou K, Fu Z, Yu D, Huang H, Zang X, et al. Brain development and akt signaling: the crossroads of signaling pathway and neurodevelopmental diseases. J Mol Neurosci (2017) 61:379–84. doi: 10.1007/s12031-016-0872-y
88. Cloëtta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, et al. Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci (2013) 33:7799–810. doi: 10.1523/JNEUROSCI.3294-12.2013
89. Harry GJ. Microglia during development and aging. Pharmacol Ther (2013) 139:313–26. doi: 10.1016/j.pharmthera.2013.04.013
90. Leviton A, Fichorova RN, O’Shea TM, Kuban K, Paneth N, Dammann O, et al. Two-hit model of brain damage in the very preterm newborn: small for gestational age and postnatal systemic inflammation. Pediatr Res (2013) 73:362–70. doi: 10.1038/pr.2012.188
91. Das A, Chai JC, Kim SH, Lee YS, Park KS, Jung KH, et al. Transcriptome sequencing of microglial cells stimulated with TLR3 and TLR4 ligands. BMC Genomics (2015) 16:517. doi: 10.1186/s12864-015-1728-5
92. Arlt A, Schäfer H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur J Cell Biol (2011) 90:545–52. doi: 10.1016/j.ejcb.2010.10.002
93. Crèvecœur J, Foerch P, Doupagne M, Thielen C, Vandenplas C, Moonen G, et al. Expression of SV2 isoforms during rodent brain development. BMC Neurosci (2013) 14:87. doi: 10.1186/1471-2202-14-87
94. Bartholome O, Van den Ackerveken P, Sánchez Gil J, de la Brassinne Bonardeaux O, Leprince P, Franzen R, et al. Puzzling out synaptic vesicle 2 family members functions. Front Mol Neurosci (2017) 10:148. doi: 10.3389/fnmol.2017.00148
95. Brown AS, Wieben M, Murdock S, Chang J, Dizon MLV, St. Pierre M, et al. Intrauterine growth restriction causes abnormal embryonic dentate gyrus neurogenesis in mouse offspring that leads to adult learning and memory deficits. eNeuro (2021) 8. doi: 10.1523/ENEURO.0062-21.2021
96. Doh-ura K, Mekada E, Ogomori K, Iwaki T. Enhanced CD9 expression in the mouse and human brains infected with transmissible spongiform encephalopathies. J Neuropathol Exp Neurol (2000) 59:774–85. doi: 10.1093/jnen/59.9.774
97. Matt DW, Macdonald GJ. Placental steroid production by the basal and labyrinth zones during the latter third of gestation in the rat. Biol Reprod (1985) 32:969–77. doi: 10.1095/biolreprod32.4.969
98. Hahn T, Barth S, Graf R, Engelmann M, Beslagic D, Reul JMHM, et al. Placental glucose transporter expression is regulated by Glucocorticoids1. J Clin Endocrinol Metab (1999) 84:1445–52. doi: 10.1210/jcem.84.4.5607
99. Even MD, Laughlin MH, Krause GF, Saal FSv. Differences in blood flow to uterine segments and placentae in relation to sex, intrauterine location and side in pregnant rats. Reproduction (1994) 102:245–52. doi: 10.1530/jrf.0.1020245
100. Fowden AL, Sferruzzi-Perri AN, Coan PM, Constancia M, Burton GJ. Placental efficiency and adaptation: endocrine regulation. J Physiol (Lond) (2009) 587:3459–72. doi: 10.1113/jphysiol.2009.173013
101. Sferruzzi-Perri AN, Lopez-Tello J, Salazar-Petres E. Placental adaptations supporting fetal growth during normal and adverse gestational environments. Exp Physiol (2022). doi: 10.1113/EP090442
Keywords: placenta, mouse, pregnancy, prolactin, pregnancy specific glycoprotein, fetal growth, animal models, endocrinology
Citation: Lopez-Tello J and Sferruzzi-Perri AN (2023) Characterization of placental endocrine function and fetal brain development in a mouse model of small for gestational age. Front. Endocrinol. 14:1116770. doi: 10.3389/fendo.2023.1116770
Received: 05 December 2022; Accepted: 27 January 2023;
Published: 10 February 2023.
Edited by:
Jayonta Bhattacharjee, Bangladesh Agricultural University, BangladeshReviewed by:
Shuhiba Mohammad, University of Calgary, CanadaCopyright © 2023 Lopez-Tello and Sferruzzi-Perri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge Lopez-Tello, jl898@cam.ac.uk; Amanda N. Sferruzzi-Perri, ans48@cam.ac.uk
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.