 Bronwyn S. Bedrick
Bronwyn S. Bedrick Taylor P. Kohn2
Taylor P. Kohn2 Lydia H. Pecker
Lydia H. Pecker Mindy S. Christianson
Mindy S. Christianson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 24 October 2022
Sec. Pediatric Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.985525
This article is part of the Research Topic Fertility Preservation in the Pediatric Population View all 9 articles
Hemoglobinopathies are autosomal recessive disorders that occur when genetic mutations negatively impact the function of hemoglobin. Common hemoglobinopathies that are clinically significant include sickle cell disease, alpha thalassemia, and beta thalassemia. Advancements in disease-modifying and curative treatments for the common hemoglobinopathies over the past thirty years have led to improvements in patient quality of life and longevity for those who are affected. However, the diseases, their treatments and cures pose infertility risks, making fertility preservation counseling and treatment an important part of the contemporary comprehensive patient care. Sickle cell disease negatively impacts both male and female infertility, primarily by testicular failure and decreased ovarian reserve, respectively. Fertility in both males and females with beta thalassemia major are negatively impacted by iron deposition due to chronic blood transfusions. Hematopoietic stem cell transplant (HSCT) is currently the only curative treatment for SCD and transfusion dependent beta thalassemia. Many of the conditioning regimens for HSCT contain chemotherapeutic agents with known gonadotoxicity and whole-body radiation. Although most clinical studies on toxicity and impact of HSCT on long-term health do not evaluate fertility, gonadal failure is common. Male fertility preservation modalities that exist prior to gonadotoxic treatment include sperm banking for pubertal males and testicular cryopreservation for pre-pubertal boys. For female patients, fertility preservation options include oocyte cryopreservation and ovarian tissue cryopreservation. Oocyte cryopreservation requires controlled ovarian hyperstimulation (COH) with ten to fourteen days of intensive monitoring and medication administration. This is feasible once the patient has undergone menarche. Follicular growth is monitored via transvaginal or transabdominal ultrasound, and hormone levels are monitored through frequent blood work. Oocytes are then harvested via a minimally invasive approach under anesthesia. Complications of COH are more common in patients with hemoglobinopathies. Ovarian hyperstimulation syndrome creates a greater risk to patients with underlying vascular, pulmonary, and renal injury, as they may be less able to tolerate fluids shifts. Thus, it is critical to monitor patients undergoing COH closely with close collaboration between the hematology team and the reproductive endocrinology team. Counseling patients and families about future fertility must take into consideration the patient’s disease, treatment history, and planned treatment, acknowledging current knowledge gaps.
Hemoglobin is an oxygen-carrying protein comprised of four subunits: two alpha chains and two non-alpha globin chains. In a healthy adult, approximately 95-98% of hemoglobin is in the form of HbA1, which consists of two alpha and two beta chains; the remaining small percentage of hemoglobin is in the form of HbA2 (two alpha and two delta chains) and HbF (two alpha and two gamma chains) (1). Almost 2,000 hemoglobin gene variants have been described (2). However, most variants are not associated with clinically significant disease. Indeed, it is estimated that 24% of the world population carry at least one altered globin gene, but only 5% carry a clinically significant variant (3). Hemoglobinopathies are autosomal recessive disorders: sickle cell disease, alpha thalassemia, and beta thalassemia.
Advancements in disease-modifying and curative treatments for the common hemoglobinopathies over the past thirty years have led to improvements in patient quality of life and longevity for those who are affected. However, the diseases, their treatments and cures may pose infertility risks. Expanding opportunities to preserve fertility in childhood are thus relevant for children with these common genetic conditions.
In this article, we discuss the indications and complications of fertility preservation in pediatric patients with hemoglobinopathies, specifically sickle cell disease and beta thalassemia. We also review their etiologies and impact on fertility and summarize their main disease modifying treatment options, focusing on the use of hydroxyurea and hematopoietic stem cell transplant. Finally, we review healthcare and research disparities in this field.
Sickle cell disease (SCD) refers to a group of hemoglobinopathies characterized by two β-globin gene mutations or deletions, at least one of which is the point mutation that leads to the production of hemoglobin S (HbS) (Table 1). An adenine-to-thymine substitution in the sixth codon of the beta-globin chain results in HbS. This substitution creates an insoluble polymer that distorts the cellular membrane and promotes the characteristic red blood cell sickling in deoxygenated states. The inability of HbS to deform normally results in hemolysis, a shortened red blood cell lifespan, and a hypercoagulable state. Additionally, the sickled red blood cells may become entrapped within vessels, leading to vascular occlusion and ischemia that promotes further sickling. This is the mechanism responsible for vaso-occlusive pain crises (VOC), acute chest syndrome (ACS), stroke, splenic sequestration, neuropathy, osteonecrosis, and recurrent infections, among other severe complications of SCD (4).
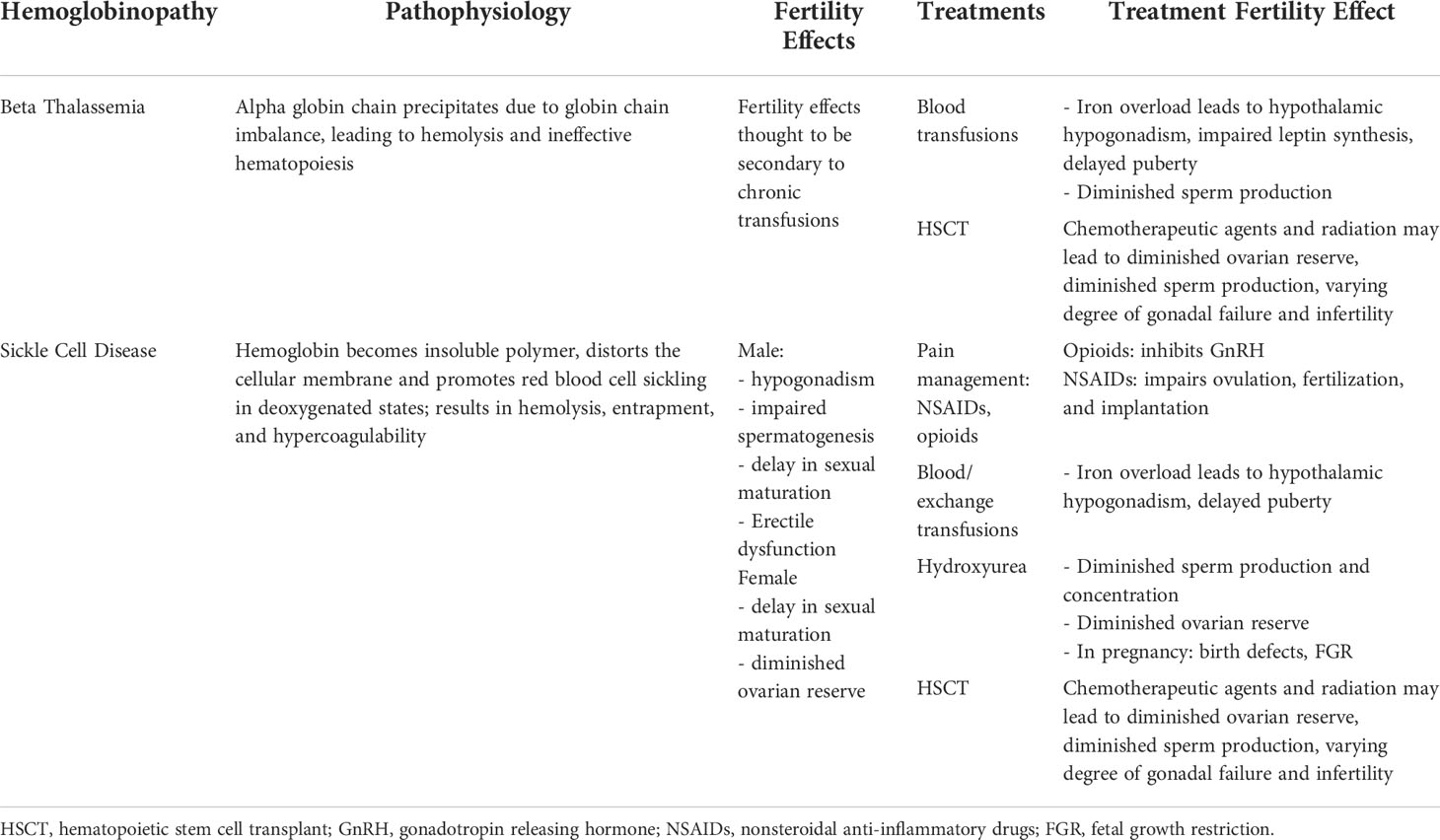
Table 1 Hemoglobinopathy pathophysiology, major treatment modalities, and impact on fertility.
SCD occurs when an individual is homozygous for HbS (i.e., HbSS, sickle cell anemia) or compound heterozygous with another beta globin gene mutation.
Thalassemia arises from globin chain imbalance due to mutations in one of the four alpha subunit genes or one of the two beta subunit genes. An imbalance in the production of alpha and beta globin chains produces unpaired globin chains that precipitate within red blood cells, resulting in hemolysis and ineffective hematopoiesis. Thalassemia severity depends on the type of genetic defect (i.e., missense versus full deletion) and on the number of genes affected.
Beta thalassemia is caused by mutations in the beta globin gene. Some mutations reduce expression of the beta subunit (β+), whereas others result in complete loss of expression from that allele (β0). Individuals with one functional beta globin (β/β+ or β/β0; beta thalassemia minor) are asymptomatic carriers. Patients with some normal beta globin production (β+/β+ or β+/β0; beta thalassemia intermedia) usually have mild to moderate anemia, although patients may require chronic transfusions. Beta thalassemia major (BTM) is characterized by severe anemia that results when both beta globin genes have deletions (β0/β0) or when a deletion is paired with another mutation that severely decreases beta globin expression (5).
When patients are dependent on transfusions for survival, regardless of genotype, they are said to have transfusion-dependent thalassemia (6). These patients usually have BTM and without treatment or cure, they are at risk of growth impairment, skeletal abnormalities, hepatosplenomegaly, and death within the first two decades of life (5). Life expectancy for individuals with BTM has significantly (7) improved over the years. In the 1970s, half of patients died before the age of 12 (8); however, many patients are now living into their 50s or 60s, making normal puberty important and parenthood viable (9).
Given the prevalence and severity of BTM and SCD, this review article will focus on these hemoglobinopathies. We note that a small, but growing number of people with alpha thalassemia major are surviving. These patients, like those with beta thalassemia major are at risk for iron overload and gonadotoxicity during HSCT. Given the lack of data, however, we do not focus on these patients (7, 10).
Studies suggest that males with SCD are at risk for infertility as a result of both hypergonadatropic hypogonadism from vaso-occlusive induced testicular ischemia as well as hypogonadotropic hypogonadism from chronic transfusion-induced iron deposition in the hypothalamus and pituitary (11–13). Indeed, studies have demonstrated that approximately 24% of adult men with SCD are hypogonadal from both hypergonadatropic hypogonadism as well as hypogonadotropic hypogonadism (13–16). As a result, adolescent males may experience a delay in sexual maturation by one to two years, and boys with more severe genotypes (HbSS, HbSβ0) experiencing greater delays (13, 17).
Infertility in males with SCD can also occur as a result of severe erectile dysfunction. Vaso-occlusion of the corpus cavernosum can result in priapism and repeated vasoocclusive episodes with priapism can result in high rates of erectile dysfunction, with some studies demonstrating as many as 48% of men will have impaired erectile function at an average age of 28 years old (18). Priapism is a true SCD emergency as the risk of erectile dysfunction increases with prolonged episodes of priapism (19, 20). Severe cases of erectile dysfunction can make spontaneous reproduction difficult and limit future fertility and may even require penile prosthesis to achieve an erection necessary for intercourse.
Even in eugonadal men with SCD, spermatogenesis is often affected; impaired semen parameters have been observed in men with normal FSH, LH and testosterone levels, possibly due to testicular infarction (21, 22). In one study, 91% of patients with SCD and taking no disease modifying therapy had an abnormality on semen analysis. The most common abnormality being impaired motility, though total motile counts were still on average around 32 million motile sperm (23). Despite a high published rates of abnormalities on semen analysis in men with sickle cell disease, in a large retrospective registry study of patients with sickle cell disease, Gordeuk et al. found that among 1018 men with sickle cell disease, 620 pregnancies conceptions had been reported for a rate of 0.61 per man (24).
Men with SCD can conceive an unassisted pregnancy with a partner, though no largescale study has assessed the frequency of infertility in this population, and further studies are needed to determine.
The majority of data on female sexual development in SCD is from the 1960s-1990s. These studies demonstrated that females with SCD achieve sexual development and undergo menarche at later ages than unaffected females (25), with more severe genotypes (HbSS, HbSβ0) having greater delays than those with less severe hemoglobinopathies (HbSβ+ or HbSC) (13, 26). The delay in menarche is thought to be constitutional (13, 26), and age of menarche is consistent with bone age (27). Once menarche is reached, however, patients can be expected to have regular menstruation (26). The effects of disease-modifying therapy on age of menarche remains poorly defined (28).
The extent to which fertility is impacted in female patients with SCD is also unclear. Historically, lower pregnancy rates among women with SCD was used as a surrogate for fertility (29), but this approach to estimating fertility is limited. Women with SCD have multiple risks for reduced ovarian reserve: chronic inflammation, oxidative stress, and ovarian ischemia and reperfusion injuries (30, 31). Three studies have demonstrated normal anti-müllerian hormone (AMH) levels in untreated adolescent and young adults with SCD (32–34). However, women with SCD experience a more rapid decline in ovarian reserve, with lower levels of AMH than age matched controls (33, 35, 36). Females with SCD develop diminished ovarian reserve (DOR) at younger ages (25-30 years) than age-matched women (33, 34). Yet, no studies to date define the definitive risk of infertility in this population (28). Interestingly, in 2021 Mamsen et al. evaluated ovarian health markers in adolescent females with hemoglobinopathies. They found no difference in ovarian follicular density, morphology, and expression of follicular and oocyte proteins between those with SCD and health age-matched controls, suggesting that the primordial follicle pool is normal in this population (37).
Delayed sexual development, menarche, and hypothalamic hypogonadism are common in adolescents with BTM (38) but are these thought to be secondary to iron deposition from chronic transfusions rather than a consequence of the disease itself (39). Impact of chronic transfusion on pubertal development and fertility and prevention options will be further discussed in the next section.
For both SCD and BTM, a patient’s disease and disease severity, may impact which fertility preservation treatment options are available to them and the success of their fertility treatment. When counseling patients and guardians about treatment options, it is important to consider how the individual’s unique disease presentation may impact success.
Chronic pain and opioid use are common sequelae of SCD. Additionally, almost 70% of patients with BTM report recent pain (40). Opioids have been shown to suppress the hypothalamic-pituitary-gonadal axis through inhibiting gonadotropin releasing hormone (41). Indeed, women taking opioids chronically have an approximately 50% reduction in estradiol and testosterone levels, a 30% reduction in gonadotropins (42), and may experience menstrual cycle disruption (42). Chronic opioid use also reduces testosterone levels in males (43) and leads to lower sperm motility and morphology (44) as well as increased DNA fragmentation (43).
The use of nonsteroidal anti-inflammatory drugs (NSAIDs) can impact fertility through inhibition of cyclooxygenase 2 (COX-2), leading to reduced prostaglandin synthesis and impairments in ovulation, fertilization, and implantation (45, 46). While small studies have linked impaired ovulation and infertility with NSAID use, the impact of chronic NSAID use on ovarian reserve and fertility is not well understood (30). Furthermore, polypharmacy may obfuscate associations between analgesic medications and fertility in patients with SCD or BTM.
Red blood cell transfusion or exchange is a cornerstone for symptomatic management and prevention in patients with SCD and BTM. For patients with SCD, these transfusions/exchanges dilute sickle cell hemoglobin and thus reduce the sequalae of sickling. For patients with BTM, transfusions supply normal red blood cells and inhibit ineffective erythropoiesis (47). These patients often undergo transfusions every three to four weeks (5).
Despite clear benefits, chronic transfusions often lead to iron overload, which has both direct and indirect impact on gonadal function. Interestingly, iron deposition appears to be a greater issue for individuals with BTM than those with SCD (48, 49), and effects from iron deposition are the major complications associated with BTM. For example, in a study of 73 patients with BTM and SCD who received chronic transfusions, 33% of BTM had gonadal failure compared to 0% of SCD patients (48). A similar 2006 study found that 40% of patients with BTM had hypogonadism, and they were eight times as likely to have hypogonadism as patients with SCD (49). Chelation therapy thus becomes essential for those receiving chronic blood transfusions and should often be initiated prior to puberty in order to encourage normal development (50). However, progressive deposition in the hypothalamus and pituitary will occur even in the setting of chelation therapy (51).
Hypothalamic hypogonadism may result from direct iron deposition in the hypothalamus, pituitary, and reproductive organs as well as free radial oxidative stress (39, 52–54). The anterior pituitary is particularly sensitive to iron deposition and demonstrates evidence of iron accumulation within the first decade of life. Damage to the anterior pituitary leads to disturbances in gonadotropic hormones and may lead to pubertal delays or arrest. In fact, hypothalamic hypogonadism is the most common endocrinopathy affecting individuals with BTM (38). It is estimated that 70% of boys with beta thalassemia intermedia or major will develop hypogonadotropic hypogonadism (55), and over 50% of females will not reach menarche spontaneously (38). In addition to iron deposition in the hypothalamus and pituitary, gonadal iron deposition may occur (56). However, ovarian function appears to be preserved as evidenced by an age-appropriate ovarian response to hyperstimulation (57, 58). Furthermore, ovarian tissue preserved for fertility preservation in females with BTM demonstrate normal follicular density and morphology (37).
Iron deposition also negatively impacts fertility in patients with BTM include impaired leptin synthesis and disruption of liver and pancreatic function, which are involved in hormone and antioxidant metabolism (39). It has been suggested that iron deposition in adipose tissue disrupts the production of leptin, a hormone now believed to be vital for the pubertal development. In a study of 101 adolescents with BTM, Perrone et al. found significantly lower leptin levels than expected for Tanner stage 1-4 males and stage 3-5 females (59). In a separate study, Dedoussis et al. found that leptin serum levels were significantly lower for BTM patients who received either sporadic or chronic transfusions than normal and that leptin level was negatively correlated with levels of transferrin reception for those who were transfusion dependent (60).
Males with transfusion dependent beta-thalassemia have high rates of oligospermia and azoospermia, but conception is still possible. In a study of 52 men, 60% were normospermic, 17% were oligospermic, and 23% were azoospermic (61). For men with impaired spermatogenesis, spermatogenesis can be induced with exogenous gonadotropin stimulation, with human chorionic gonadotropin (hCG) alone or combined with human menopausal gonadotropin, thus making paternity possible (9, 62–64) Indeed, in a survey of ten thalassemia centers, including 738 transfusion-dependent men over the age of 18, 75% of those married or living with a partner conceived a pregnancy within the first two years of the marriage. Of these pregnancies, 79% occurred via natural conception and 15% of men required exogenous gonadotropin stimulation (65).
Approved by the FDA in 1998 for use in adults with sickle cell anemia, hydroxyurea has dramatically improved patient quality of life and reduced disease complications. Patients taking hydroxyurea are less likely to be hospitalized or require transfusions. Studies have also found improvements in long-term survival and reduced risk of stroke (66–71).
Hydroxyurea inhibits ribonucleotide reductase and thus cell cycle specific DNA replication. Through unclear mechanisms, hydroxyurea shifts expression of the beta globin locus resulting in increased production of HbF and decreased production of HbS. As a result of decreased HbS concentration, hemoglobin is less prone to polymerization and sickling. Hydroxyurea also decreases circulating leukocytes and reticulocytes, increases red blood cell volume, and improves cellular deformability, thereby reducing painful events (72).
Given substantive improvements in patient symptoms and markers of disease control, the National Heart, Lung, and Blood Institute (NHLBI) recommends offering hydroxyurea in pediatric patients over 9 months of age, regardless of clinical severity (68). However, the optimal time to start hydroxyurea therapy has not been established, and other national guidelines recommend starting at later ages (73). Additionally, in patients not taking hydroxyurea, it may be recommended prior to bone marrow transplant to reduce the risk of rejection and improve chance of engraftment (74). While there have been some studies suggesting benefit in the use of hydroxyurea in patients with BTM (75), these results are not widespread and its use in this population is uncommon (5).
Hydroxyurea use may be lifelong for patients with SCD. While there is strong evidence as to the myelosuppressive effects of hydroxyurea, data on other long-term effects, such as on infertility, are conflicting. The National Toxicology Program (NTP) Center for the Evaluation of Risks to Human Reproduction (CERHR) gave hydroxyurea use in pregnancy its second highest concern level due to risk for birth defects and intrauterine growth restriction (76). CERHR also ranks use of hydroxyurea in post-pubertal males as “highly concerning.”
For men, hydroxyurea owes its rank of “highly concerning” to its impact on sperm parameters. In men, treatment with hydroxyurea has been shown to significantly impair total sperm concentration (pretreatment: 38.5 million sperm/mL; post treatment: 18.46 million sperm/mL) but forward motility remains similar (pretreatment: 28.6%; post treatment: 29.4%) (23). In another study of men with SCD on hydroxyurea therapy, 20% developed oligospermia and 10% developed azoospermia (77). The impact on spermatogenesis extends to prepubertal males as well. In one study, two young males initiated on hydroxyurea at ages 10 and 16 were found to have severe oligospermia eight years after treatment initiation; two others who began hydroxyurea at ages 8 and 11 were found to be azoospermic 15 and 12 years later, respectively (78).
Despite hydroxyurea’s deleterious impact on semen parameters, studies have shown normalization of semen parameters after discontinuing the medication. In one study, almost 75% of men who stopped hydroxyurea for three months had normalization of their semen parameters (77). In a recent study by Joseph et al., there was no difference in semen volume, sperm concentration, total sperm count, or spermatozoa motility, morphology, and vitality between men who received hydroxyurea prior to puberty and men who were hydroxyurea naïve. In this study, men who had a history of hydroxyurea use stopped hydroxyurea on average two and a half years prior to semen analysis (79). These studies suggest that while hydroxyurea may have more severe effects when started in the prepubertal period, the effects are potentially reversible. However, the reliability and duration to recovery of spermatogenesis after hydroxyurea has not been well elucidated, and so sperm banking or testicular tissue banking may be considered prior to initiating hydroxyurea therapy.
While hydroxyurea is considered ‘low risk’ for infertility in women, it is associated with diminished ovarian reserve in three small studies of people with sickle cell anemia (hemoglobin SS and hemoglobin S beta-null thalassemia). During the decade-long follow up of the Multicenter Study of Hydroxyurea (MSH) randomized control trial, AMH was lower for women who were on hydroxyurea than for those not taking it (33). In 2015, Elchuri et al. compared 10-21 year-old females with SCD who received supportive care, hydroxyurea, or underwent bone marrow transplantation. They found that 24% of patients treated with hydroxyurea had diminished ovarian reserve as defined by AMH <5% of expected for age-matched controls (32). Moreover, among patients taking hydroxyurea, those with DOR had been taking the medication for 2.8 years longer on average than those without DOR. No patient receiving hydroxyurea in this study met criteria for premature ovarian insufficiency as defined as FSH is > 40 IU/L (32). In another single center analysis of ovarian reserve in 26 women with sickle cell anemia, all (n=5) women diagnosed with DOR were taking hydroxyurea (34). Whether markers of ovarian reserve normalize when hydroxyurea is stopped and whether hydroxyurea impairs fertility, ovarian hyperstimulation, or oocyte quality is not established. Given the potential to affect oocyte quantity and quality, the potential risks associated with its use must be weighed against the potential benefit of reduced rejection and graft versus host disease risk.
Hematopoietic stem cell transplant (HSCT) is currently the only curative treatment for SCD and transfusion dependent beta thalassemia. It is recommended when symptoms are no longer controlled with supportive care and medical management or for those who with serious disease complications. For patients with SCD who underwent HLA-matched transplants, greater than 90% sustained engraftment and resolution of painful crises (80). Similar improvements are seen in patients with BTM. To date, more than 5,000 of HSCT have been performed for these disorders (81–83).
For patients with SCD, HLA matched sibling donors result in the highest event-free survival. However due to limited availability of HLA-matched siblings, haploidentical, matched unrelated, and mismatched unrelated donation are possible and mostly offered on an experimental basis (84). Age at transplantation impacts morbidity and mortality, which is an important consideration for fertility preservation. When HSCT is performed prior to 16 years of age, the 5-year survival is 95% compared to 81% at older ages (82), and children <10 years old have decreased mortality compared to those transplanted 10-21 years old (85). Similarly, for BTM HLA identical transplantation at younger ages (<14 years) improves outcomes and survival (83, 86) due to higher rates of disease complications with age; transplant related mortality is <5% if performed prior to 5 years old (83).
A variety of condition regimens for HSCT are described. The first successful regimens were myeloablative. However, given the intolerability of these regimens for those with severe disease, nonmyeloablative conditioning regimens have been developed. Many of the conditioning regimens contain chemotherapeutic agents with known gonadotoxicity, such as cyclophosphamide. Whole body radiation may also be included. The commonly used radiation dose (2-3Gy) is less than the effective sterilizing dose (>14 Gy in females; > 6 Gy in males) but falls within ranges known to cause significant gonadal damage (87–89). Despite the potential for significant deleterious gonadal damage, most clinical studies on toxicity and impact of HSCT on long-term health do not evaluate gonadal function or fertility. From the studies that do (Table 2), it is evident that gonadal failure is common, but the impact is variable and unpredictable (115). Additionally, younger age at transplant and male sex appear to reduces risk of gonadal failure (109).
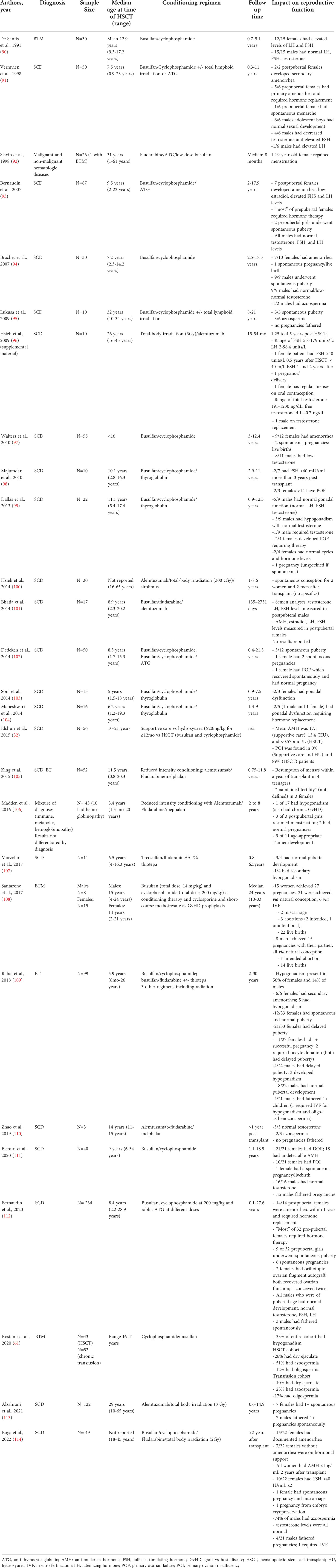
Table 2 Clinical studies of hematopoietic stem cell transplant in patients with sickle cell disease and beta thalassemia that evaluated reproductive function.
After transplantation, patients are often placed on additional immunosuppressive medications to reduce the risk for GvHD, such as cyclophosphamide or sirolimus (84, 116). Primary ovarian insufficiency is a common consequence of cyclophosphamide (117, 118), although ovarian reserve appears to be less affected if cyclophosphamide is given prior to menarche (119). Sirolimus has also been shown to significantly reduce sperm counts and fertility rates in male organ transplant recipients (120), lead to gonadal dysfunction and secondary infertility (121), and to negatively impact IVF outcomes (122).
In summary, counseling patients and families on future fertility is complicated by the wide range of conditioning regimens available and poor reporting on gonadal function post-transplant. Furthermore, impact on gonadal function may be highly variable even for the same conditioning regimen, with some patients experiencing rapid gonadal failure necessitating hormonal supplementation and others retaining full function. Long term follow-up and a greater understanding of the interplay between gonadotoxic treatment and gonadal function is needed.
Given the risk of gonadal failure after HSCT, fertility preservation before HSCT should be offered (123). Early counseling, no matter the patient age, is vital to provide patients with the optimal opportunity to protect their reproductive potential. Furthermore, fear of toxicity and sterility are important barriers to HSCT acceptance (124, 125). However, significant access to care remains including provider awareness, patient/family preferences, and financial barriers.
Fertility preservation is an important consideration for men and their provider to discuss prior to HSCT or even initiating hydroxyurea therapy (126). Male fertility preservation in many ways is more straight forward than for females - ejaculate or testicular tissue do not require stimulation cycles and can be collected nearly immediately, but counseling men about the risk of gonadotoxic agents is less frequently discussed with males than female cancer patients (127, 128).
For post pubertal males, sperm banking can readily be performed via ejaculation which allows sperm to be frozen until the patient desires fertility, at which time in vitro fertilization could be pursued (129). Given the impact of hydroxyurea on spermatogenesis, it is recommended that hydroxyurea be discontinued several months prior to sperm collection (123). This may allow for normalization of sperm parameters. However, discontinuing hydroxyurea for several months may be challenging for patients with severe SCD. While the process of sperm cryopreservation is relatively simple, informing patients of the risk of gonadotoxic agents is not always discussed or pursued (130). Studies have found low rates of fertility preservation counseling prior to gonadotoxic chemotherapy for cancer (128). However, these findings may not be applicable to adolescent males facing gonadotoxic treatment for their chronic lifelong disease and more research is needed about fertility preservation counseling in this population.
Discussion of fertility preservation and increasing state-mandated coverage of fertility preservation has resulted in an increase in rates of fertility preservation (128). In recent years, several online based companies have emerged that allow for in-home collection for sperm banking wherein the individual receives a kit, ejaculates in their own home into the provided container, and then ships the kit back for cryopreservation (131). With increasing insurance coverage of fertility preservation, awareness of fertility preservation options amongst providers, and online-based sperm preservation companies, hopefully the rates of sperm banking prior to gonadotoxic agents will increase.
Fertility preservation is more complex if the male has not yet gone through puberty. Indeed, rates of azoospermia are quite high in boys 13 years or younger (132). In these cases, surgery is required to harvest testicular tissue for cryopreservation (133). Only a small amount of testicular tissue is collected, and the procedure is well tolerated with minimal side effects (134). It is important however to note that there is no current ability to use this sperm for future fertility attempts; thus prepubescent testicular preservation is only offered in certain academic centers as part of a research protocol (135). Research into maturation of the testicular tissue and methods of reimplantation are ongoing. The field recently took a major step forward with the successful transplant of frozen rat spermatogonial stem cells into recipient mice, which produced differentiating germ cell types with production of spermatids (136). While this murine study is certainly promising for the future of reimplantation of testicular tissue, parents of boys undergoing gonadotoxic treatments should be aware that much research is still required and previously harvested testicular tissue may not be ready for spermatogenesis at the time when fertility may be desired. Currently, testicular tissue preservation is recommended for patients at significant risk for infertility, including patients with SCD and BTM (137). Operative considerations are discussed below.
The most important factors determining mode of fertility preservation in a female patient are whether she has undergone menarche and the urgency with which the gonadotoxic treatment is needed. Fertility preservation prior to HSCT for hemoglobinopathies is not usually urgent. This will allow for improved coordination and health optimization prior. In some cases, this may allow time for menarche to occur and thus permit the use of controlled ovarian hyperstimulation and oocyte cryopreservation. For patients who have not yet undergone menarche and for whom waiting until after menarche for transplantation is not feasible due to patient age and disease severity, ovarian tissue cryopreservation (OTC) is the only current option for fertility preservation. For females who have undergone menarche, OTC and oocyte cryopreservation are available. While embryo cryopreservation is another option, it is less likely in the pediatric and young adult population as it requires a sperm source and has greater ethical implications.
Controlled ovarian hyperstimulation (COH) requires ten to fourteen days of intensive monitoring and medication administration. Follicular growth is monitored via transvaginal or transabdominal ultrasound, and hormone levels are monitored through frequent blood work. Oocytes are then harvested via a minimally invasive approach under anesthesia.
Common complications of COH include headache, nausea, abdominal distention, and discomfort. Less commonly, ovarian hyperstimulation syndrome (OHSS) occurs, which may produce venous thromboembolism (VTE), ascites, and cardiopulmonary effusions (138). These consequences of COH are of even greater concern for patients with hemoglobinopathies, who may have altered pain perception and be less able to tolerate the discomfort of COH. Ovarian hyperstimulation syndrome also creates a greater risk to patients with underlying vascular, pulmonary, and renal injury, as they may be less able to tolerate fluids shifts (139). Indeed, COH increases the risk of VOC and ACS in patients with SCD. To date, there are 4 reported cases of acute pain crises during COH (Table 3).
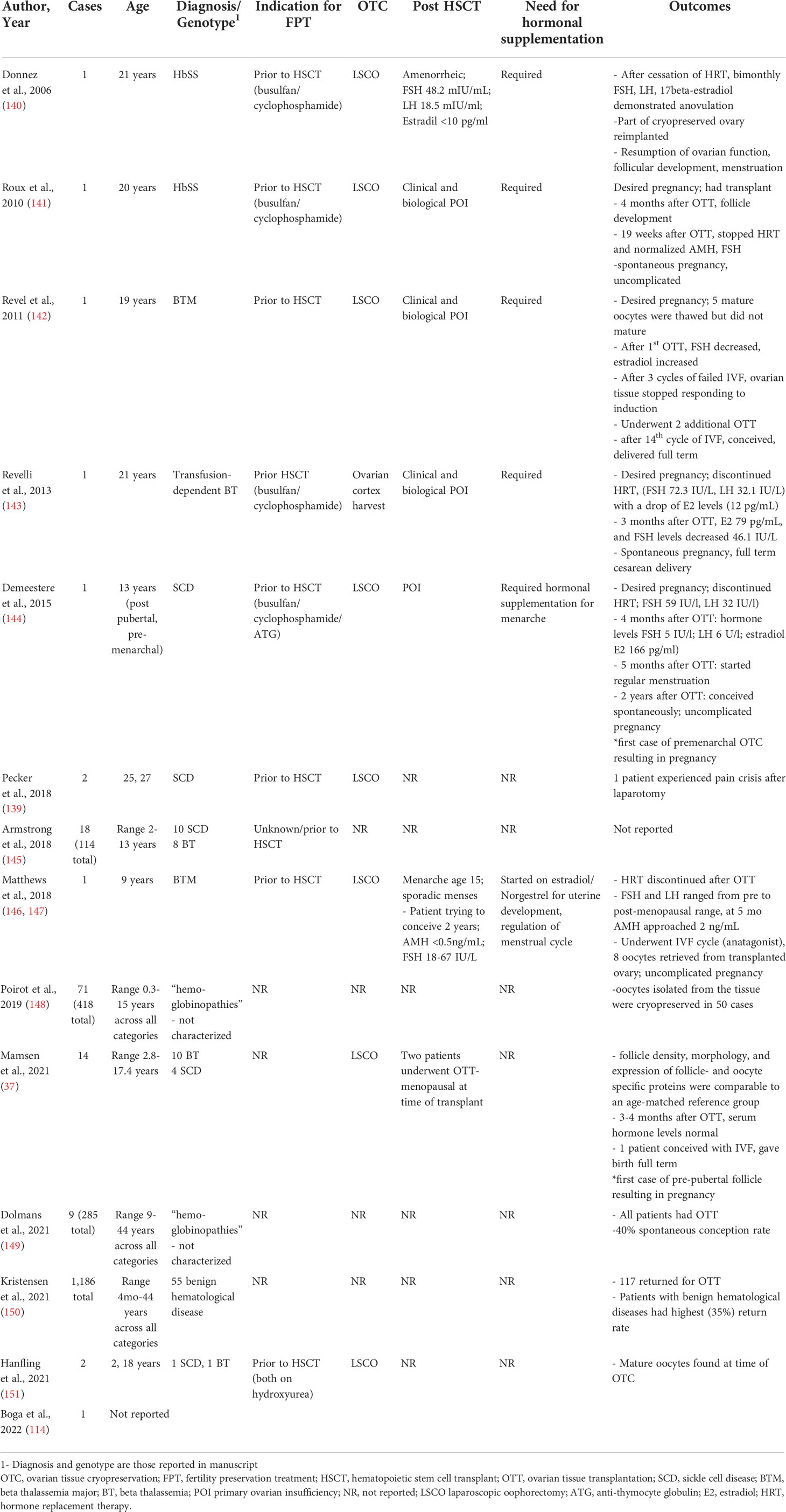
Table 3 Clinical reports of ovarian tissue cryopreservation for sickle cell disease and beta thalassemia.
Results of COH in patients with SCD are variable, with the number of oocytes retrieved ranging from 4 - 31. Fifteen is considered the minimum number of oocytes to harvest to optimize the change of pregnancy in one cycle (152). However, only 25% of the reported patients reached this goal. No patients in these cohorts underwent multiple cycles for fertility preservation, perhaps reflecting time and monetary constraints (139). There is also a scarcity of published data on ovarian stimulation protocols and outcomes in adolescent patients with and without hemoglobinopathies (153). It is generally recommended to use adult dosing regimens as a guide, adjusting for age, FSH level, and AFC. However, this may require frequent and significant dose adjustments. For example, in their cohort of eight teenage girls, Lavery et al., reported that dose adjustments were needed in 80% of cases (154).
To date, there have been several reports of successful and uncomplicated ovulation induction and IVF cycles for untransplanted patients with BTM. However, there have been no published COH protocols for BTM prior to fertility preservation (155, 156). These authors recommended discontinuing iron chelators prior to ovulation induction (156) as they are contraindicated in pregnancy, but this is not necessary for the purpose of fertility preservation.
Ovarian tissue cryopreservation (OTC) is an increasingly utilized method of FPT. As of 2019, OTC is no longer considered experimental by the American Society for Reproductive Medicine (ASRM), although it may be in other countries. As of 2017, there had been 130 live births from OTC (157), with estimates of greater than 200 births as of 2020 (158). Given the younger ages at which HSCT is recommended for patients with hemoglobinopathies, OTC may be the only mode of FPT available.
Ovarian tissue cryopreservation is commonly performed via an outpatient laparoscopic surgery in which an ovary, or portion of the ovary is removed. This tissue is then stored until future use at which point ovarian tissue transplantation (OTT) may occur. Since OTC enables preservation of a larger cohort of primordial follicles, ovarian endocrine function may be restored after OTT. Indeed, in a metanalysis of 309 cases of OTT, endocrine restoration, as defined by cyclic menstrual cycles, ovarian follicle growth on ultrasound, or pregnancy, was achieved in 64% of cases. Clinical pregnancy rate after OTT was 57.5% (159).
Importantly, ovarian endocrine function appears to be restored in the small number of reported post-OTT patients with hemoglobinopathies (Table 4). In 2006, Donnez et al. was the first to report restoration of ovarian function after orthotopic transplantation in a patient with HbSS. The patient underwent OTC prior to HSCT at age 21 years old. She required hormone supplementation after transplant and eventually had an OTT after which she had resumption of ovarian function, evidenced of follicular development, and regular menstruation (140). These findings have been replicated in several other reports of adolescent patients with hemoglobinopathies, including patients who were prepubertal (37).
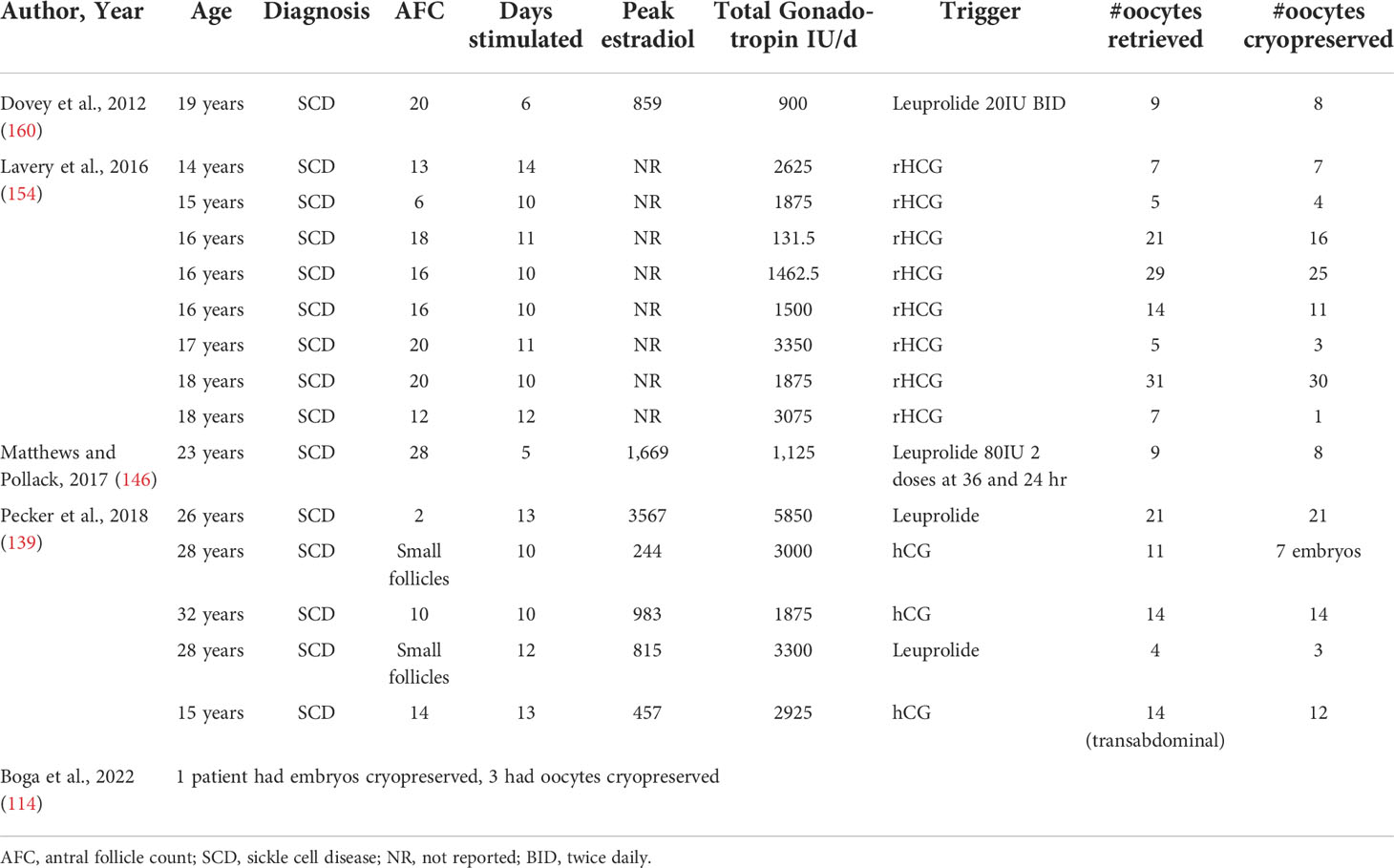
Table 4 Cases of controlled ovarian hyperstimulation in patients with hemoglobinopathies.
Spontaneous pregnancy may occur after OTC, although IVF is frequently required. In 2015, Demeestere et al., reported the first live birth from ovarian tissue cryopreserved from a pubertal female who was pre-menarchal. The OTC occurred at age 13. The patient, who had SCD, was confirmed to have primary ovarian insufficiency when she desired to conceive. Two years after ovarian transplantation, the patient spontaneously conceived (144). In 2021, Mamsen et al., reported the first case of pregnancy from OTC performed prior to puberty. The patient who had beta thalassemia underwent OTC prior to HSCT at 9 years old. She returned at 23 for OTT after which she had resumption of ovarian function and was able to conceive with IVF. In a separate case report, a patient required 14 cycles of IVF and 3 separate OTT to achieve a life birth (142). Unfortunately, most studies do not report on infertility rates among patients who have undergone OTT, nor do they report sufficient information on the indication for IVF or the number of cycles to draw conclusions about the chance of spontaneous conception after OTT. Furthermore, patients’ response to HSCT is variable; some patients appear to be completely cured after treatment while others have a less robust response. Individuals who are not cured likely differ in pregnancy outcomes given the higher rate of stillbirth and fetal growth restriction in untreated patients with SCD (161).
OTC has dramatically altered opportunities for fertility preservation in pediatric patients, especially those who are prepubertal. Despite the promise of OTC, outcomes should be viewed cautiously. Globally, few pregnancies have occurred for patients who had OTC prior to puberty. While spontaneous pregnancies occur, they should not be viewed as expected. Furthermore, ovarian tissue grafts have a finite life. It is estimated that ovarian grafts last approximately 2.25 years on average (159). Therefore, periodic OTT may be required throughout a female’s reproductive life.
Women with SCD and beta thalassemia are at increased risks for obstetric complications including maternal mortality, intrauterine fetal demise, preeclampsia, preterm delivery, and spontaneous miscarriage (162–167). These risks are partly due to high rate of comorbidities associated with these hemoglobinopathies, i.e., hypercoagulability. For women with significant comorbidities who wish to have biologic children, the option for surrogacy should be discussed along with appropriate preconception counseling with maternal-fetal medicine specialists.
Another important component of fertility treatment is the discussion of genetic testing. Individuals who carry mutations for hemoglobinopathies should be offered preimplantation genetic testing (PGT) to reduce the risk of an affected offspring. While patients may not be ready for parenthood soon after their fertility preservation, education on surrogacy and PGT may be helpful in informing patients and their families on the full scope of fertility options.
Both surgery and COH contribute to fluid shifts and hypercoagulability, which increase the risks for adverse outcomes among patients with SCD. It is estimated that 5% of pediatric patients experience postoperative VOC and ACS (168, 169), and moderate to severe OHSS occur in 1-5% of all COH cycles (138). To date, there have been six adverse outcomes reports from FPT: (Table 5): one episode of mild OHSS, four episodes of acute pain crises, and one episode of ACS requiring intubation and intensive care unit admission. All adverse events occurred in patients with SCD, and the more severe adverse events occurred in older patients with more comorbidities. Patients with BTM who have undergone a splenectomy are at increased risk of post-operative infections and those with hemosiderosis induced heart failure are more prone to fluid overload. However, these risks are less well described than for SCD. To date, no adverse events have been reported in pediatric SCD FPT or in any BTM patient undergoing FPT.
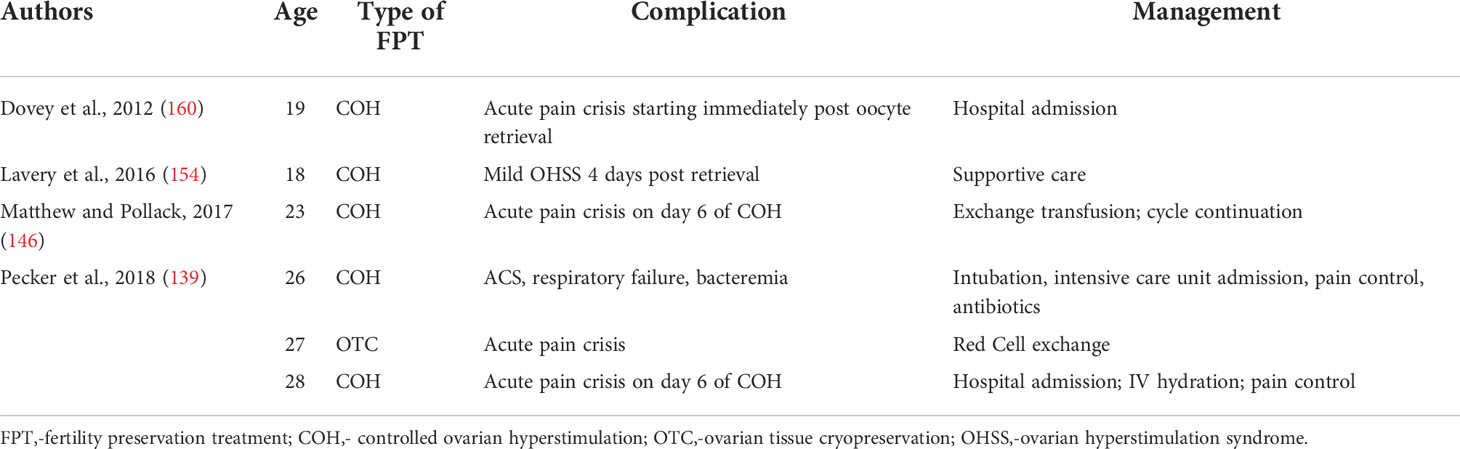
Table 5 Adverse outcomes associated with fertility preservation treatment in patients with sickle cell disease.
Preoperative and postoperative optimization are vital to reduce procedural complications and to reduce the risk of cancellation of high stakes cycles. While there are no standardized protocols for preoperative management, there are general principles which should be followed. Below, we discuss the available literature and include a protocol created by our center for management of COH in patients with SCD (Table 6).

Table 6 Perioperative considerations.
Prior to FPT, coordination with the patient’s hematologist is vital for procedural optimization and postprodcedural management. Universal preoperative anesthesia consult is not warranted (170) but should be considered based on patient’s medical comorbidities. Ensuring that children are up to date on disease-specific screening (i.e. transcranial Doppler ultrasound) is also recommended, although this is expected if the patient is in the process of undergoing HSCT. Patients may have a history of prior VTE, and thus an anticoagulation plan must be considered when planning for both surgery and COH. To reduce overall surgical risks, coordinating with other procedures, such as port placement, should be considered.
Creating a COH stimulation protocol that optimizes oocyte yield while minimizing risk of OHSS is vital. In our practice, we use either hCG or a gonadotropin-releasing hormone agonist for trigger to reduce the risk of OHSS (139). Given the unclear impact of hydroxyurea on ovarian reserve, oocyte quality and embryo development (171), we also recommend discussing medication discontinuation prior to FPT.
Data on the benefits of preoperative transfusion in patients with SCD is conflicting (172–176). Nevertheless, most experts recommend transfusion for a hemoglobin level ≥9-10 g/dL and exchange transfusions for hemoglobin S <30% (170, 177), and these are the benchmarks that our group has recommended for management of COH (139). For patients who receive regular transfusions or exchanges, these should be continued in the immediate preoperative period. If transfusion or exchange is planned, coordination with blood bank specialists who are familiar with the patient’s transfusion history and have knowledge of any red blood cell autoimmunization or alloimmunization is recommended (139). Decisions on if and when transfusions or exchanges are recommended should be discussed with the patient’s hematologist.
Triggers for sickling should be minimized in the perioperative setting, including dehydration, acidosis, hypoxia, and hypothermia. To prevent dehydration, it is recommended that patients avoid prolonged fasting, consume clear liquids up to two hours prior to their procedure, and receive IV hydration while fasting. Our group recommends administering IV hydration prior to anesthesia administration (139). Normothermia through use of body temperature monitoring systems, blankets, and ambient temperature control is recommended. Monitoring oxygen saturation is paramount, and supplemental oxygen used when indicated. Glucocorticoids, such as dexamethasone, should be avoided as they may precipitate pain crises (139, 178, 179).
Use of incentive spirometry, chest physiotherapy, and early ambulation in the postoperative period is widely recommended to reduce the risk of ACS (139, 170). Early pain control is another important facet to postoperative management. Patients with chronic pain, such as in SCD, may have altered pain perception and may already be taking daily narcotics. Preoperative discussion with the patient’s hematologist is absolutely critical when determining postoperative pain regimen. Narcotics are often first line agents, although patient-controlled analgesia may be warranted (170). Understanding the patient’s recent pain history may help to predict postoperative complications, as patients with recent hospitalizations for crises are more likely to have postoperative crises (180).
Hemoglobinopathies are the most commonly inherited monogenetic diseases, yet research and funding do not reflect the prevalence of the diseases (125, 181). For example, sickle cell related variables are not collected in large health outcomes databases and no robust dataset exists for hemoglobinopathies and fertility (28), thereby limiting providers’ ability to offer evidence based care.
No studies to date have evaluated access to FPT care for patients with hemoglobinopathies (125). However, fertility preservation is not commonly utilized prior to HSCT. For example, in a claims database study of over 400 adults who underwent HSCT, only 7% had claims for fertility preservation services before their transplant (182). A significant barrier to care, especially in the adolescent population is timely referrals. Pediatric providers may not be aware of infertility risks and may feel poorly equipped to discuss fertility preservation and uncomfortable discussing reproductive health with patients and their families (183–185), especially with rapidly evolving recommendations and practices.
For patients who do receive a referral for fertility preservation, the cost of FPT may be a significant barrier. The average cost of COH for fertility preservation is over $12,000 (186, 187), and the cost of laparoscopic oophorectomy is comparable (186). Storage fees for cryopreserved oocytes and ovarian tissue, which may be over twenty years, further adds to required costs. Whereas programs such as Livestrong exist to assist patients in fertility preservation prior to cancer treatment, no such national program exists for hemoglobinopathies. As of 2022, 12 states mandate coverage for fertility preservation prior to gonadotoxic treatment (188). However, mandates often do not cover government assistance such as Medicaid and Medicare, and some states such as Utah require that the patient has a cancer diagnosis, thereby disqualifying many patients with SCD or BTM. Some have argued for the need to change institutional programs (125) to provide coverage for patients with SCD. Ultimately, advocating for legislation change both on the local and national level is needed to expand coverage for this population.
Adoption and utilization of oncofertility patient navigators (189) may help to reduce some of these barriers to fertility preservation among patients with hemoglobinopathies. Patient navigators help guide patients and their families through fertility preservation, from identifying an in-network clinic, expediting fertility evaluation, providing education about different fertility options, and referring to different support groups (190). Through advocating for patients and their families, navigators may play a vital role in empowering them to make the right fertility preservation decisions for their circumstances and goals.
Sickle cell disease and beta thalassemia are the most common and morbid hemoglobinopathies. Disease modifying and curative treatments have improved quality of life and increased the chance of living into adulthood. However, many of these treatments negatively impact fertility and normal pubertal development. Fertility preservation should be discussed with all patients and families considering disease modifying and curative therapies. In very young children in which fertility preservation may be challenging, the risks and benefits to delaying HSCT for greater maturation should be discussed. Counseling patients and families about future fertility must take into consideration the patient’s disease, treatment history, and planned treatment, acknowledging current knowledge gaps. Preparing for fertility preservation must also include a multidisciplinary approach to optimize patient outcomes while reducing surgical and procedural risks. Further research and advocacy are needed to improve patient care and future fertility.
MC, BB, LP and TK contributed to conception and outline of the manuscript. BB wrote the first draft of the manuscript. TK, LP and MC wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. van Beers E, van Wijk R. Red blood cell biochemistry and physiology. In: Lazarus H, Schmaier A, editors. Concise guide to hematology, 2 ed. Cham, Switzerland: Springer (2019). p. 15–20.
2. Hardison RC, Chui DH, Riemer C, Giardine B, Lehvaslaiho H, Wajcman H, et al. Databases of human hemoglobin variants and other resources at the globin gene server. Hemoglobin (2001) 25(2):183–93. doi: 10.1081/HEM-100104027
3. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ (2008) 86(6):480–7. doi: 10.2471/BLT.06.036673
4. Pecker LH, Lanzkron S. Sickle cell disease. Ann Intern Med (2021) 174(1):ITC1–ITC16. doi: 10.7326/AITC202101190
5. Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet (2012) 379(9813):373–83. doi: 10.1016/S0140-6736(11)60283-3
6. Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica (2013) 98(6):833–44. doi: 10.3324/haematol.2012.066845
7. Songdej D, Babbs C, Higgs DR, Consortium BI. An international registry of survivors with hb bart's hydrops fetalis syndrome. Blood (2017) 129(10):1251–9. doi: 10.1182/blood-2016-08-697110
8. Borgna-Pignatti C, Marsella M, Zanforlin N. The natural history of thalassemia intermedia. Ann N Y Acad Sci (2010) 1202:214–20. doi: 10.1111/j.1749-6632.2010.05550.x
9. Berdoukas V, Modell B. Transfusion-dependent thalassaemia: a new era. Med J Aust (2008) 188(2):68–9. doi: 10.5694/j.1326-5377.2008.tb01523.x
10. Amid A, Barrowman N, Odame I, Kirby-Allen M. Optimizing transfusion therapy for survivors of haemoglobin bart's hydrops fetalis syndrome: Defining the targets for haemoglobin-h fraction and "functional" haemoglobin level. Br J Haematol (2022) 197(3):373–6. doi: 10.1111/bjh.18077
11. Osegbe DN, Akinyanju OO. Testicular dysfunction in men with sickle cell disease. Postgrad Med J (1987) 63(736):95–8. doi: 10.1136/pgmj.63.736.95
12. Parshad O, Stevens MC, Preece MA, Thomas PW, Serjeant GR. The mechanism of low testosterone levels in homozygous sickle-cell disease. West Indian Med J (1994) 43(1):12–4.
13. Platt OS, Rosenstock W, Espeland MA. Influence of sickle hemoglobinopathies on growth and development. N Engl J Med (1984) 311(1):7–12. doi: 10.1056/NEJM198407053110102
14. Abbasi AA, Prasad AS, Ortega J, Congco E, Oberleas D. Gonadal function abnormalities in sickle cell anemia. studies in adult male patients. Ann Intern Med (1976) 85(5):601–5. doi: 10.7326/0003-4819-85-5-601
15. Dada OA, Nduka EU. Endocrine function and haemoglobinopathies: relation between the sickle cell gene and circulating plasma levels of testosterone, luteinising hormone (LH) and follicle stimulating hormone (FSH) in adult males. Clin Chim Acta (1980) 105(2):269–73. doi: 10.1016/0009-8981(80)90469-6
16. Taddesse A, Woldie IL, Khana P, Swerdlow PS, Chu JW, Abrams J, et al. Hypogonadism in patients with sickle cell disease: central or peripheral? Acta Haematol (2012) 128(2):65–8. doi: 10.1159/000337344
17. Zemel BS, Kawchak DA, Ohene-Frempong K, Schall JI, Stallings VA. Effects of delayed pubertal development, nutritional status, and disease severity on longitudinal patterns of growth failure in children with sickle cell disease. Pediatr Res (2007) 61(5 Pt 1):607–13. doi: 10.1203/pdr.0b013e318045bdca
18. Anele UA, Burnett AL. Erectile dysfunction after sickle cell disease-associated recurrent ischemic priapism: profile and risk factors. J Sex Med (2015) 12(3):713–9. doi: 10.1111/jsm.12816
19. Bennett N, Mulhall J. Sickle cell disease status and outcomes of African-American men presenting with priapism. J Sex Med (2008) 5(5):1244–50. doi: 10.1111/j.1743-6109.2008.00770.x
20. Idris IM, Abba A, Galadanci JA, Mashi SA, Hussaini N, Gumel SA, et al. Men with sickle cell disease experience greater sexual dysfunction when compared with men without sickle cell disease. Blood Adv (2020) 4(14):3277–83. doi: 10.1182/bloodadvances.2020002062
21. Friedman G, Freeman R, Bookchin R, Boyar R, Murthy G, Hellman L. Testicular function in sickle cell disease. Fertil Steril (1974) 25(12):1018–21. doi: 10.1016/S0015-0282(16)40809-5
22. Li M, Fogarty J, Whitney KD, Stone P. Repeated testicular infarction in a patient with sickle cell disease: a possible mechanism for testicular failure. Urology (2003) 62(3):551. doi: 10.1016/S0090-4295(03)00482-5
23. Berthaut I, Guignedoux G, Kirsch-Noir F, de Larouziere V, Ravel C, Bachir D, et al. Influence of sickle cell disease and treatment with hydroxyurea on sperm parameters and fertility of human males. Haematologica (2008) 93(7):988–93. doi: 10.3324/haematol.11515
24. Gordeuk V, Kroner B, Pugh N, Hankins J, Kutlar A, King A, et al. Hydroxyurea use and outcomes of pregnancy in sickle cell disease. Blood (2020) 136:33. doi: 10.1182/blood-2020-143315
25. Alleyne SI, Rauseo RD, Serjeant GR. Sexual development and fertility of Jamaican female patients with homozygous sickle cell disease. Arch Intern Med (1981) 141(10):1295–7. doi: 10.1001/archinte.1981.00340100051014
26. Serjeant GR, Singhal A, Hambleton IR. Sickle cell disease and age at menarche in Jamaican girls: observations from a cohort study. Arch Dis Child (2001) 85(5):375–8. doi: 10.1136/adc.85.5.375
27. Luban NL, Leikin SL, August GA. Growth and development in sickle cell anemia. Preliminary Rep Am J Pediatr Hematol Oncol (1982) 4(1):61–5.
28. Pecker LH, Sharma D, Nero A, Paidas MJ, Ware RE, James AH, et al. Knowledge gaps in reproductive and sexual health in girls and women with sickle cell disease. Br J Haematol (2021) 194(6):970–9. doi: 10.1111/bjh.17658
29. Dunne GD, Joseph RR. Fertility in hemoglobin s-s and hemoglobin s-c disease. Fertil Steril (1970) 21(8):630–4. doi: 10.1016/S0015-0282(16)37688-9
30. Ghafuri DL, Stimpson SJ, Day ME, James A, DeBaun MR, Sharma D. Fertility challenges for women with sickle cell disease. Expert Rev Hematol (2017) 10(10):891–901. doi: 10.1080/17474086.2017.1367279
31. Chase AR, Howard J, Oteng-Ntim E. Ovarian sickling as a proposed mechanism for premature ovarian failure necessitating ovum donation. Menopause Int (2009) 15(2):70–1. doi: 10.1258/mi.2009.009015
32. Elchuri SV, Williamson RS, Clark Brown R, Haight AE, Spencer JB, Buchanan I, et al. The effects of hydroxyurea and bone marrow transplant on anti-mullerian hormone (AMH) levels in females with sickle cell anemia. Blood Cells Mol Dis (2015) 55(1):56–61. doi: 10.1016/j.bcmd.2015.03.012
33. Pecker LH, Hussain S, Christianson MS, Lanzkron S. Hydroxycarbamide exposure and ovarian reserve in women with sickle cell disease in the multicenter study of hydroxycarbamide. Br J Haematol (2020) 191(5):880–7. doi: 10.1111/bjh.16976
34. Pecker LH, Hussain S, Mahesh J, Varadhan R, Christianson MS, Lanzkron S. Diminished ovarian reserve in young women with sickle cell anemia. Blood (2022) 139(7):1111–5. doi: 10.1182/blood.2021012756
35. Kopeika J, Oyewo A, Punnialingam S, Reddy N, Khalaf Y, Howard J, et al. Ovarian reserve in women with sickle cell disease. PloS One (2019) 14(2):e0213024. doi: 10.1371/journal.pone.0213024
36. Garba SR, Makwe CC, Osunkalu VO, Kalejaiye OO, Soibi-Harry AP, Aliyu AU, et al. Ovarian reserve in nigerian women with sickle cell anaemia: a cross- sectional study. J Ovarian Res (2021) 14(1):174. doi: 10.1186/s13048-021-00927-5
37. Mamsen LS, Kristensen SG, Pors SE, Botkjaer JA, Ernst E, Macklon KT, et al. Consequences of beta-thalassemia or sickle cell disease for ovarian follicle number and morphology in girls who had ovarian tissue cryopreserved. Front Endocrinol (Lausanne) (2021) 11:593718. doi: 10.3389/fendo.2020.593718
38. Toumba M, Sergis A, Kanaris C, Skordis N. Endocrine complications in patients with thalassaemia major. Pediatr Endocrinol Rev (2007) 5(2):642–8.
39. Roussou P, Tsagarakis NJ, Kountouras D, Livadas S, Diamanti-Kandarakis E. Beta-thalassemia major and female fertility: the role of iron and iron-induced oxidative stress. Anemia (2013) 2013:617204. doi: 10.1155/2013/617204
40. Trachtenberg F, Foote D, Martin M, Carson S, Coates T, Beams O, et al. Pain as an emergent issue in thalassemia. Am J Hematol (2010) 85(5):367–70. doi: 10.1002/ajh.21670
41. Seyfried O, Hester J. Opioids and endocrine dysfunction. Br J Pain (2012) 6(1):17–24. doi: 10.1177/2049463712438299
42. Daniell HW. Opioid endocrinopathy in women consuming prescribed sustained-action opioids for control of nonmalignant pain. J Pain (2008) 9(1):28–36. doi: 10.1016/j.jpain.2007.08.005
43. Drobnis EZ, Nangia AK. Pain medications and Male reproduction. Adv Exp Med Biol (2017) 1034:39–57. doi: 10.1007/978-3-319-69535-8_6
44. Farag AGA, Basha MA, Amin SA, Elnaidany NF, Elhelbawy NG, Mostafa MMT, et al. Tramadol (opioid) abuse is associated with a dose- and time-dependent poor sperm quality and hyperprolactinaemia in young men. Andrologia (2018) 50(6):e13026. doi: 10.1111/and.13026
45. Stone S, Khamashta MA, Nelson-Piercy C. Nonsteroidal anti-inflammatory drugs and reversible female infertility: is there a link? Drug Saf (2002) 25(8):545–51. doi: 10.2165/00002018-200225080-00001
46. Gaytan M, Bellido C, Morales C, Sanchez-Criado JE, Gaytan F. Effects of selective inhibition of cyclooxygenase and lipooxygenase pathways in follicle rupture and ovulation in the rat. Reproduction (2006) 132(4):571–7. doi: 10.1530/rep.1.01236
47. Taher A, Mehio G, Isma'eel H, Cappellini MD. Stroke in thalassemia: a dilemma. Am J Hematol (2008) 83(4):343. doi: 10.1002/ajh.21117
48. Vichinsky E, Butensky E, Fung E, Hudes M, Theil E, Ferrell L, et al. Comparison of organ dysfunction in transfused patients with SCD or beta thalassemia. Am J Hematol (2005) 80(1):70–4. doi: 10.1002/ajh.20402
49. Fung EB, Harmatz PR, Lee PD, Milet M, Bellevue R, Jeng MR, et al. Increased prevalence of iron-overload associated endocrinopathy in thalassaemia versus sickle-cell disease. Br J Haematol (2006) 135(4):574–82. doi: 10.1111/j.1365-2141.2006.06332.x
50. Bronspiegel-Weintrob N, Olivieri NF, Tyler B, Andrews DF, Freedman MH, Holland FJ. Effect of age at the start of iron chelation therapy on gonadal function in beta-thalassemia major. N Engl J Med (1990) 323(11):713–9. doi: 10.1056/NEJM199009133231104
51. Skordis N, Gourni M, Kanaris C, Toumba M, Kleanthous M, Karatzia N, et al. The impact of iron overload and genotype on gonadal function in women with thalassaemia major. Pediatr Endocrinol Rev (2004) 2 Suppl 2:292–5.
52. Singer ST, Vichinsky EP, Gildengorin G, van Disseldorp J, Rosen M, Cedars MI. Reproductive capacity in iron overloaded women with thalassemia major. Blood (2011) 118(10):2878–81. doi: 10.1182/blood-2011-06-360271
53. Bergeron C, Kovacs K. Pituitary siderosis. a histologic, immunocytologic, and ultrastructural study. Am J Pathol (1978) 93(2):295–309.
54. Abdulzahra MS, Al-Hakeim HK, Ridha MM. Study of the effect of iron overload on the function of endocrine glands in male thalassemia patients. Asian J Transfus Sci (2011) 5(2):127–31. doi: 10.4103/0973-6247.83236
55. Chern JP, Lin KH, Tsai WY, Wang SC, Lu MY, Lin DT, et al. Hypogonadotropic hypogonadism and hematologic phenotype in patients with transfusion-dependent beta-thalassemia. J Pediatr Hematol Oncol (2003) 25(11):880–4. doi: 10.1097/00043426-200311000-00011
56. Chatterjee R, Katz M. Reversible hypogonadotrophic hypogonadism in sexually infantile male thalassaemic patients with transfusional iron overload. Clin Endocrinol (Oxf) (2000) 53(1):33–42. doi: 10.1046/j.1365-2265.2000.00962.x
57. De Sanctis V, Vullo C, Katz M, Wonke B, Tanas R, Bagni B. Gonadal function in patients with beta thalassaemia major. J Clin Pathol (1988) 41(2):133–7. doi: 10.1136/jcp.41.2.133
58. Skordis N, Petrikkos L, Toumba M, Hadjigavriel M, Sitarou M, Kolnakou A, et al. Update on fertility in thalassaemia major. Pediatr Endocrinol Rev (2004) 2 Suppl 2:296–302.
59. Perrone L, Perrotta S, Raimondo P, Mucerino J, De Rosa C, Siciliani MC, et al. Inappropriate leptin secretion in thalassemia: a potential cofactor of pubertal timing derangement. J Pediatr Endocrinol Metab (2003) 16(6):877–81. doi: 10.1515/JPEM.2003.16.6.877
60. Dedoussis GV, Kyrtsonis MC, Andrikopoulos NE, Voskaridou E, Loutradis A. Inverse correlation of plasma leptin and soluble transferrin receptor levels in beta-thalassemia patients. Ann Hematol (2002) 81(9):543–7. doi: 10.1007/s00277-002-0499-7
61. Rostami T, Mohammadifard MA, Ansari S, Kiumarsi A, Maleki N, Kasaeian A, et al. Indicators of male fertility potential in adult patients with beta-thalassemia major: a comparative study between patients undergone allogeneic stem cell transplantation and transfusion-dependent patients. Fertil Res Pract (2020) 6:4. doi: 10.1186/s40738-020-00071-6
62. Borgna-Pignatti C. The life of patients with thalassemia major. Haematologica (2010) 95(3):345–8. doi: 10.3324/haematol.2009.017228
63. De Sanctis V, Soliman AT, Canatan D, Di Maio S, Elsedfy H, Baioumi A, et al. Gonadotropin replacement in male thalassemia major patients with arrested puberty and acquired hypogonadotropic hypogonadism (AAH): preliminary results and potential factors affecting induction of spermatogenesis. Endocrine (2019) 63(1):167–70. doi: 10.1007/s12020-018-1772-4
64. De Sanctis V, Vullo C, Katz M, Wonke B, Nannetti C, Bagni B. Induction of spermatogenesis in thalassaemia. Fertil Steril (1988) 50(6):969–75. doi: 10.1016/S0015-0282(16)60382-5
65. De Sanctis V, Soliman AT, El-Hakim I, Christou S, Mariannis D, Karimi M, et al. Marital status and paternity in patients with transfusion- dependent thalassemia (TDT) and non transfusion-dependent thalassemia (NTDT): an ICET - a survey in different countries. Acta Biomed (2019) 90(3):225–37. doi: 10.23750/abm.v90i3.8586
66. Le PQ, Gulbis B, Dedeken L, Dupont S, Vanderfaeillie A, Heijmans C, et al. Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr Blood Cancer (2015) 62(11):1956–61. doi: 10.1002/pbc.25608
67. Lobo CL, Pinto JF, Nascimento EM, Moura PG, Cardoso GP, Hankins JS. The effect of hydroxcarbamide therapy on survival of children with sickle cell disease. Br J Haematol (2013) 161(6):852–60. doi: 10.1111/bjh.12323
68. National Heart L, and Blood Institute. Evidence-based management of sickle cell disease: Expert panel report, 2014. Bethesda, Maryland USA (2014).
69. Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA (2003) 289(13):1645–51. doi: 10.1001/jama.289.13.1645
70. Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol (2010) 85(6):403–8. doi: 10.1002/ajh.21699
72. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med (1995) 332(20):1317–22. doi: 10.1056/NEJM199505183322001
73. Elenga N, Kayemba-Kay's S, Nacher M, Archer N. A call to start hydroxyurea by 6 months of age and before the advent of sickle cell disease complications. Pediatr Blood Cancer (2022) 69(2):e29423. doi: 10.1002/pbc.29423
74. Brachet C, Azzi N, Demulder A, Devalck C, Gourdin A, Gulbis B, et al. Hydroxyurea treatment for sickle cell disease: impact on haematopoietic stem cell transplantation's outcome. Bone Marrow Transplant (2004) 33(8):799–803. doi: 10.1038/sj.bmt.1704443
75. Yavarian M, Karimi M, Bakker E, Harteveld CL, Giordano PC. Response to hydroxyurea treatment in Iranian transfusion-dependent beta-thalassemia patients. Haematologica (2004) 89(10):1172–8.
76. National Toxicology P. NTP-CERHR monograph on the potential human reproductive and developmental effects of hydroxyurea. NTP CERHR MON (2008) 21):vii–viii, v, ix-III1.
77. Sahoo LK, Kullu BK, Patel S, Patel NK, Rout P, Purohit P, et al. Study of seminal fluid parameters and fertility of Male sickle cell disease patients and potential impact of hydroxyurea treatment. J Assoc Physicians India (2017) 65(6):22–5.
78. Lukusa AK, Vermylen C. Use of hydroxyurea from childhood to adult age in sickle cell disease: semen analysis. Haematologica (2008) 93(11):e67. doi: 10.3324/haematol.13659
79. Joseph L, Jean C, Manceau S, Chalas C, Arnaud C, Kamdem A, et al. Effect of hydroxyurea exposure before puberty on sperm parameters in males with sickle cell disease. Blood (2021) 137(6):826–9. doi: 10.1182/blood.2020006270
80. Lucarelli G, Gaziev J, Isgro A, Sodani P, Paciaroni K, Alfieri C, et al. Allogeneic cellular gene therapy in hemoglobinopathies–evaluation of hematopoietic SCT in sickle cell anemia. Bone Marrow Transplant (2012) 47(2):227–30. doi: 10.1038/bmt.2011.79
81. Shenoy S, Angelucci E, Arnold SD, Baker KS, Bhatia M, Bresters D, et al. Current results and future research priorities in late effects after hematopoietic stem cell transplantation for children with sickle cell disease and thalassemia: A consensus statement from the second pediatric blood and marrow transplant consortium international conference on late effects after pediatric hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2017) 23(4):552–61. doi: 10.1016/j.bbmt.2017.01.009
82. Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood (2017) 129(11):1548–56. doi: 10.1182/blood-2016-10-745711
83. Baronciani D, Angelucci E, Potschger U, Gaziev J, Yesilipek A, Zecca M, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European society for blood and bone marrow transplantation hemoglobinopathy registry, 2000-2010. Bone Marrow Transplant (2016) 51(4):536–41. doi: 10.1038/bmt.2015.293
84. Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood (2012) 120(22):4285–91. doi: 10.1182/blood-2012-07-438408
85. Arnold SD, Brazauskas R, He N, Li Y, Aplenc R, Jin Z, et al. Clinical risks and healthcare utilization of hematopoietic cell transplantation for sickle cell disease in the USA using merged databases. Haematologica (2017) 102(11):1823–32. doi: 10.3324/haematol.2017.169581
86. Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, et al. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med (1993) 329(12):840–4. doi: 10.1056/NEJM199309163291204
87. Howell SJ, Shalet SM. Spermatogenesis after cancer treatment: damage and recovery. J Natl Cancer Inst Monogr (2005) 34):12–7. doi: 10.1093/jncimonographs/lgi003
88. Wallace WH, Shalet SM, Hendry JH, Morris-Jones PH, Gattamaneni HR. Ovarian failure following abdominal irradiation in childhood: the radiosensitivity of the human oocyte. Br J Radiol (1989) 62(743):995–8. doi: 10.1259/0007-1285-62-743-995
89. Wallace WH, Thomson AB, Saran F, Kelsey TW. Predicting age of ovarian failure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys (2005) 62(3):738–44. doi: 10.1016/j.ijrobp.2004.11.038
90. De Sanctis V, Galimberti M, Lucarelli G, Polchi P, Ruggiero L, Vullo C. Gonadal function after allogenic bone marrow transplantation for thalassaemia. Arch Dis Child (1991) 66(4):517–20. doi: 10.1136/adc.66.4.517
91. Vermylen C, Cornu G, Ferster A, Brichard B, Ninane J, Ferrant A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant (1998) 22(1):1–6. doi: 10.1038/sj.bmt.1701291
92. Slavin S, Nagler A, Naparstek E, Kapelushnik Y, Aker M, Cividalli G, et al. Nonmyeloablative stem cell transplantation and cell therapy as an alternative to conventional bone marrow transplantation with lethal cytoreduction for the treatment of malignant and nonmalignant hematologic diseases. Blood (1998) 91(3):756–63. doi: 10.1182/blood.V91.3.756
93. Bernaudin F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood (2007) 110(7):2749–56. doi: 10.1182/blood-2007-03-079665
94. Brachet C, Heinrichs C, Tenoutasse S, Devalck C, Azzi N, Ferster A. Children with sickle cell disease: growth and gonadal function after hematopoietic stem cell transplantation. J Pediatr Hematol Oncol (2007) 29(7):445–50. doi: 10.1097/MPH.0b013e31806451ac
95. Lukusa AK, Vermylen C, Vanabelle B, Curaba M, Brichard B, Chantrain C, et al. Bone marrow transplantation or hydroxyurea for sickle cell anemia: long-term effects on semen variables and hormone profiles. Pediatr Hematol Oncol (2009) 26(4):186–94. doi: 10.1080/07357900902892780
96. Hsieh MM, Kang EM, Fitzhugh CD, Link MB, Bolan CD, Kurlander R, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med (2009) 361(24):2309–17. doi: 10.1056/NEJMoa0904971
97. Walters MC, Sullivan KM. Stem-cell transplantation for sickle cell disease. N Engl J Med (2010) 362(10):955–6.
98. Majumdar S, Robertson Z, Robinson A, Starnes S, Iyer R, Megason G. Outcome of hematopoietic cell transplantation in children with sickle cell disease, a single center's experience. Bone Marrow Transplant (2010) 45(5):895–900. doi: 10.1038/bmt.2009.244
99. Dallas MH, Triplett B, Shook DR, Hartford C, Srinivasan A, Laver J, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant (2013) 19(5):820–30. doi: 10.1016/j.bbmt.2013.02.010
100. Hsieh MM, Fitzhugh CD, Weitzel RP, Link ME, Coles WA, Zhao X, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA (2014) 312(1):48–56. doi: 10.1001/jama.2014.7192
101. Bhatia M, Jin Z, Baker C, Geyer MB, Radhakrishnan K, Morris E, et al. Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant (2014) 49(7):913–20. doi: 10.1038/bmt.2014.84
102. Dedeken L, Le PQ, Azzi N, Brachet C, Heijmans C, Huybrechts S, et al. Haematopoietic stem cell transplantation for severe sickle cell disease in childhood: a single centre experience of 50 patients. Br J Haematol (2014) 165(3):402–8. doi: 10.1111/bjh.12737
103. Soni S, Gross TG, Rangarajan H, Baker KS, Sturm M, Rhodes M. Outcomes of matched sibling donor hematopoietic stem cell transplantation for severe sickle cell disease with myeloablative conditioning and intermediate-dose of rabbit anti-thymocyte globulin. Pediatr Blood Cancer (2014) 61(9):1685–9. doi: 10.1002/pbc.25059
104. Maheshwari S, Kassim A, Yeh RF, Domm J, Calder C, Evans M, et al. Targeted busulfan therapy with a steady-state concentration of 600-700 ng/mL in patients with sickle cell disease receiving HLA-identical sibling bone marrow transplant. Bone Marrow Transplant (2014) 49(3):366–9. doi: 10.1038/bmt.2013.188
105. King AA, Kamani N, Bunin N, Sahdev I, Brochstein J, Hayashi RJ, et al. Successful matched sibling donor marrow transplantation following reduced intensity conditioning in children with hemoglobinopathies. Am J Hematol (2015) 90(12):1093–8. doi: 10.1002/ajh.24183
106. Madden LM, Hayashi RJ, Chan KW, Pulsipher MA, Douglas D, Hale GA, et al. Long-term follow-up after reduced-intensity conditioning and stem cell transplantation for childhood nonmalignant disorders. Biol Blood Marrow Transplant (2016) 22(8):1467–72. doi: 10.1016/j.bbmt.2016.04.025
107. Marzollo A, Calore E, Tumino M, Pillon M, Gazzola MV, Destro R, et al. Treosulfan-based conditioning regimen in sibling and alternative donor hematopoietic stem cell transplantation for children with sickle cell disease. Mediterr J Hematol Infect Dis (2017) 9(1):e2017014. doi: 10.4084/mjhid.2017.014
108. Santarone S, Natale A, Olioso P, Onofrillo D, D'Incecco C, Parruti G, et al. Pregnancy outcome following hematopoietic cell transplantation for thalassemia major. Bone Marrow Transplant (2017) 52(3):388–93. doi: 10.1038/bmt.2016.287
109. Rahal I, Galambrun C, Bertrand Y, Garnier N, Paillard C, Frange P, et al. Late effects after hematopoietic stem cell transplantation for beta-thalassemia major: the French national experience. Haematologica (2018) 103(7):1143–9. doi: 10.3324/haematol.2017.183467
110. Zhao J, Beebe K, Magee K, Salzberg D, Stahlecker J, Miller HK, et al. Adolescent male fertility following reduced-intensity conditioning regimen for hematopoietic stem cell transplantation in non-malignant disorders. Pediatr Transplant (2019) 23(6):e13496. doi: 10.1111/petr.13496
111. Elchuri SV, Williamson Lewis R, Quarmyne MO, Haight AE, Cottrell HN, Meacham LR. Longitudinal description of gonadal function in sickle-cell patients treated with hematopoietic stem cell transplant using alkylator-based conditioning regimens. J Pediatr Hematol Oncol (2020) 42(7):e575–82. doi: 10.1097/MPH.0000000000001782
112. Bernaudin F, Dalle JH, Bories D, de Latour RP, Robin M, Bertrand Y, et al. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica (2020) 105(1):91–101. doi: 10.3324/haematol.2018.213207
113. Alzahrani M, Damlaj M, Jeffries N, Alahmari B, Singh A, Rondelli D, et al. Non-myeloablative human leukocyte antigen-matched related donor transplantation in sickle cell disease: outcomes from three independent centres. Br J Haematol (2021) 192(4):761–8. doi: 10.1111/bjh.17311
114. Boga C, Asma S, Ozer C, Bulgan Kilicdag E, Kozanoglu I, Yeral M, et al. Gonadal status and sexual function at long-term follow-up after allogeneic stem cell transplantation in adult patients with sickle cell disease. Exp Clin Transplant (2022). doi: 10.6002/ect.2021.0392
115. Guilcher GMT, Monagel DA, Nettel-Aguirre A, Truong TH, Desai SJ, Bruce A, et al. Nonmyeloablative matched sibling donor hematopoietic cell transplantation in children and adolescents with sickle cell disease. Biol Blood Marrow Transplant (2019) 25(6):1179–86. doi: 10.1016/j.bbmt.2019.02.011
116. Al-Homsi AS, Roy TS, Cole K, Feng Y, Duffner U. Post-transplant high-dose cyclophosphamide for the prevention of graft-versus-host disease. Biol Blood Marrow Transplant (2015) 21(4):604–11. doi: 10.1016/j.bbmt.2014.08.014
117. Manger K, Wildt L, Kalden JR, Manger B. Prevention of gonadal toxicity and preservation of gonadal function and fertility in young women with systemic lupus erythematosus treated by cyclophosphamide: the PREGO-study. Autoimmun Rev (2006) 5(4):269–72. doi: 10.1016/j.autrev.2005.10.001
118. Blumenfeld Z, Avivi I, Linn S, Epelbaum R, Ben-Shahar M, Haim N. Prevention of irreversible chemotherapy-induced ovarian damage in young women with lymphoma by a gonadotrophin-releasing hormone agonist in parallel to chemotherapy. Hum Reprod (1996) 11(8):1620–6. doi: 10.1093/oxfordjournals.humrep.a019457
119. Wallace WH, Shalet SM, Tetlow LJ, Morris-Jones PH. Ovarian function following the treatment of childhood acute lymphoblastic leukaemia. Med Pediatr Oncol (1993) 21(5):333–9. doi: 10.1002/mpo.2950210505
120. Zuber J, Anglicheau D, Elie C, Bererhi L, Timsit MO, Mamzer-Bruneel MF, et al. Sirolimus may reduce fertility in male renal transplant recipients. Am J Transplant (2008) 8(7):1471–9. doi: 10.1111/j.1600-6143.2008.02267.x
121. Boobes Y, Bernieh B, Saadi H, Raafat Al Hakim M, Abouchacra S. Gonadal dysfunction and infertility in kidney transplant patients receiving sirolimus. Int Urol Nephrol (2010) 42(2):493–8. doi: 10.1007/s11255-009-9644-8
122. Wald K, Cakmak H, Mok-Lin E, Cedars M, Rosen M, Letourneau J. Back-to-back random-start ovarian stimulation prior to chemotherapy to maximize oocyte yield. J Assist Reprod Genet (2019) 36(6):1161–8. doi: 10.1007/s10815-019-01462-5
123. Nickel RS, Maher JY, Hsieh MH, Davis MF, Hsieh MM, Pecker LH. Fertility after curative therapy for sickle cell disease: A comprehensive review to guide care. J Clin Med (2022) 11(9):1–21. doi: 10.3390/jcm11092318
124. Hansbury EN, Schultz WH, Ware RE, Aygun B. Bone marrow transplant options and preferences in a sickle cell anemia cohort on chronic transfusions. Pediatr Blood Cancer (2012) 58(4):611–5. doi: 10.1002/pbc.23304
125. Mishkin AD, Mapara MY, Reshef R. Iatrogenic infertility after curative stem cell transplantation in patients with sickle cell disease. Ann Intern Med (2018) 168(12):881–2. doi: 10.7326/M18-0185
126. Peterson AM, Singh M. Fertility preservation in benign and malignant conditions. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2022).
127. Flisser E. Identifying at-risk populations: are we simply not doing enough fertility preservation procedures? Fertil Steril (2018) 110(4):640–1. doi: 10.1016/j.fertnstert.2018.06.018
128. Patel P, Kohn TP, Cohen J, Shiff B, Kohn J, Ramasamy R. Evaluation of reported fertility preservation counseling before chemotherapy using the quality oncology practice initiative survey. JAMA Netw Open (2020) 3(7):e2010806. doi: 10.1001/jamanetworkopen.2020.10806
129. Softness K, Kohn TP, Perecman A, Carrasquillo R. Access to male fertility preservation information and referrals at national cancer institute cancer centers. Andrologia (2021) 53(5):e14020. doi: 10.1111/and.14020
130. Selter J, Huang Y, Williams SZ, Brady PC, Melamed A, Hershman DL, et al. Use of fertility preservation services in male reproductive-aged cancer patients. Gynecol Oncol Rep (2021) 36:100716. doi: 10.1016/j.gore.2021.100716
131. Rabinowitz M, Bowring M, Kohn T, Levy J, Herati A. Impact of environmental and socioeconomic factors on semen quality in the united states. J Sexual Med (2022) 19(4):S111–2. doi: 10.1016/j.jsxm.2022.01.235
132. Halpern JA, Thirumavalavan N, Kohn TP, Patel AS, Leong JY, Cervellione RM, et al. Distribution of semen parameters among adolescent males undergoing fertility preservation in a multicenter international cohort. Urology (2019) 127:119–23. doi: 10.1016/j.urology.2019.01.027
133. Burns KC, Hoefgen H, Strine A, Dasgupta R. Fertility preservation options in pediatric and adolescent patients with cancer. Cancer (2018) 124(9):1867–76. doi: 10.1002/cncr.31255
134. Ginsberg JP, Carlson CA, Lin K, Hobbie WL, Wigo E, Wu X, et al. An experimental protocol for fertility preservation in prepubertal boys recently diagnosed with cancer: a report of acceptability and safety. Hum Reprod (2010) 25(1):37–41. doi: 10.1093/humrep/dep371
135. Onofre J, Baert Y, Faes K, Goossens E. Cryopreservation of testicular tissue or testicular cell suspensions: a pivotal step in fertility preservation. Hum Reprod Update (2016) 22(6):744–61. doi: 10.1093/humupd/dmw029
136. Whelan EC, Yang F, Avarbock MR, Sullivan MC, Beiting DP, Brinster RL. Reestablishment of spermatogenesis after more than 20 years of cryopreservation of rat spermatogonial stem cells reveals an important impact in differentiation capacity. PloS Biol (2022) 20(5):e3001618. doi: 10.1371/journal.pbio.3001618
137. Delgouffe E, Braye A, Goossens E. Testicular tissue banking for fertility preservation in young boys: Which patients should be included? Front Endocrinol (Lausanne) (2022) 13:854186. doi: 10.3389/fendo.2022.854186
138. Brinsden PR, Wada I, Tan SL, Balen A, Jacobs HS. Diagnosis, prevention and management of ovarian hyperstimulation syndrome. Br J Obstet Gynaecol (1995) 102(10):767–72. doi: 10.1111/j.1471-0528.1995.tb10840.x
139. Pecker LH, Maher JY, Law JY, Beach MC, Lanzkron S, Christianson MS. Risks associated with fertility preservation for women with sickle cell anemia. Fertil Steril (2018) 110(4):720–31. doi: 10.1016/j.fertnstert.2018.05.016
140. Donnez J, Dolmans MM, Demylle D, Jadoul P, Pirard C, Squifflet J, et al. Restoration of ovarian function after orthotopic (intraovarian and periovarian) transplantation of cryopreserved ovarian tissue in a woman treated by bone marrow transplantation for sickle cell anaemia: case report. Hum Reprod (2006) 21(1):183–8. doi: 10.1093/humrep/dei268
141. Roux C, Amiot C, Agnani G, Aubard Y, Rohrlich PS, Piver P. Live birth after ovarian tissue autograft in a patient with sickle cell disease treated by allogeneic bone marrow transplantation. Fertil Steril (2010) 93(7):2413e2415–2419. doi: 10.1016/j.fertnstert.2009.12.022
142. Revel A, Laufer N, Ben Meir A, Lebovich M, Mitrani E. Micro-organ ovarian transplantation enables pregnancy: a case report. Hum Reprod (2011) 26(5):1097–103. doi: 10.1093/humrep/der063
143. Revelli A, Marchino G, Dolfin E, Molinari E, Delle Piane L, Salvagno F, et al. Live birth after orthotopic grafting of autologous cryopreserved ovarian tissue and spontaneous conception in Italy. Fertil Steril (2013) 99(1):227–30. doi: 10.1016/j.fertnstert.2012.09.029
144. Demeestere I, Simon P, Dedeken L, Moffa F, Tsepelidis S, Brachet C, et al. Live birth after autograft of ovarian tissue cryopreserved during childhood. Hum Reprod (2015) 30(9):2107–9. doi: 10.1093/humrep/dev128
145. Armstrong AG, Kimler BF, Smith BM, Woodruff TK, Pavone ME, Duncan FE. Ovarian tissue cryopreservation in young females through the oncofertility consortium's national physicians cooperative. Future Oncol (2018) 14(4):363–78. doi: 10.2217/fon-2017-0410
146. Matthews M, Pollack R. Acute pain crisis in a patient with sickle cell disease undergoing ovarian simulation for fertility preservation prior to curative stem cell transplantation: case report and literature review. J Assist Reprod Genet (2017) 34(11):1445–8. doi: 10.1007/s10815-017-1008-1
147. Matthews SJ, Picton H, Ernst E, Andersen CY. Successful pregnancy in a woman previously suffering from beta-thalassemia following transplantation of ovarian tissue cryopreserved before puberty. Minerva Ginecol (2018) 70(4):432–5. doi: 10.23736/S0026-4784.18.04240-5
148. Poirot C, Fortin A, Dhedin N, Brice P, Socie G, Lacorte JM, et al. Post-transplant outcome of ovarian tissue cryopreserved after chemotherapy in hematologic malignancies. Haematologica (2019) 104(8):e360–3. doi: 10.3324/haematol.2018.211094
149. Dolmans MM, von Wolff M, Poirot C, Diaz-Garcia C, Cacciottola L, Boissel N, et al. Transplantation of cryopreserved ovarian tissue in a series of 285 women: a review of five leading European centers. Fertil Steril (2021) 115(5):1102–15. doi: 10.1016/j.fertnstert.2021.03.008
150. Kristensen SG, Wakimoto Y, Colmorn LB, Dueholm M, Pors SE, Macklon KT, et al. Use of cryopreserved ovarian tissue in the Danish fertility preservation cohort. Fertil Steril (2021) 116(4):1098–106. doi: 10.1016/j.fertnstert.2021.05.096
151. Hanfling SN, Parikh T, Mayhew A, Robinson E, Graham J, Gomez-Lobo V, et al. Case report: two cases of mature oocytes found in prepubertal girls during ovarian tissue cryopreservation. F S Rep (2021) 2(3):296–9. doi: 10.1016/j.xfre.2021.03.007
152. Sunkara SK, Rittenberg V, Raine-Fenning N, Bhattacharya S, Zamora J, Coomarasamy A. Association between the number of eggs and live birth in IVF treatment: an analysis of 400 135 treatment cycles. Hum Reprod (2011) 26(7):1768–74. doi: 10.1093/humrep/der106
153. Manuel SL, Moravek MB, Confino R, Smith KN, Lawson AK, Klock SC, et al. Ovarian stimulation is a safe and effective fertility preservation option in the adolescent and young adult population. J Assist Reprod Genet (2020) 37(3):699–708. doi: 10.1007/s10815-019-01639-y
154. Lavery SA, Islam R, Hunt J, Carby A, Anderson RA. The medical and ethical challenges of fertility preservation in teenage girls: a case series of sickle cell anaemia patients prior to bone marrow transplant. Hum Reprod (2016) 31(7):1501–7. doi: 10.1093/humrep/dew084
155. Bajoria R, Chatterjee R. Current perspectives of fertility and pregnancy in thalassemia. Hemoglobin (2009) 33 Suppl 1:S131–135. doi: 10.3109/03630260903365023
156. Bajoria R, Chatterjee R. Hypogonadotrophic hypogonadism and diminished gonadal reserve accounts for dysfunctional gametogenesis in thalassaemia patients with iron overload presenting with infertility. Hemoglobin (2011) 35(5-6):636–42. doi: 10.3109/03630269.2011.623809
157. Donnez J, Dolmans MM. Fertility preservation in women. N Engl J Med (2017) 377(17):1657–65. doi: 10.1056/NEJMra1614676
158. Dolmans MM, Falcone T, Patrizio P. Importance of patient selection to analyze in vitro fertilization outcome with transplanted cryopreserved ovarian tissue. Fertil Steril (2020) 114(2):279–80. doi: 10.1016/j.fertnstert.2020.04.050
159. Pacheco F, Oktay K. Current success and efficiency of autologous ovarian transplantation: A meta-analysis. Reprod Sci (2017) 24(8):1111–20. doi: 10.1177/1933719117702251
160. Dovey S, Krishnamurti L, Sanfilippo J, Gunawardena S, McLendon P, Campbell M, et al. Oocyte cryopreservation in a patient with sickle cell disease prior to hematopoietic stem cell transplantation: first report. J Assist Reprod Genet (2012) 29(3):265–9. doi: 10.1007/s10815-011-9698-2
161. Boafor TK, Olayemi E, Galadanci N, Hayfron-Benjamin C, Dei-Adomakoh Y, Segbefia C, et al. Pregnancy outcomes in women with sickle-cell disease in low and high income countries: a systematic review and meta-analysis. BJOG (2016) 123(5):691–8. doi: 10.1111/1471-0528.13786
162. Lao TT. Obstetric care for women with thalassemia. Best Pract Res Clin Obstet Gynaecol (2017) 39:89–100. doi: 10.1016/j.bpobgyn.2016.09.002
163. Oteng-Ntim E, Ayensah B, Knight M, Howard J. Pregnancy outcome in patients with sickle cell disease in the UK–a national cohort study comparing sickle cell anaemia (HbSS) with HbSC disease. Br J Haematol (2015) 169(1):129–37. doi: 10.1111/bjh.13270
164. Oteng-Ntim E, Meeks D, Seed PT, Webster L, Howard J, Doyle P, et al. Adverse maternal and perinatal outcomes in pregnant women with sickle cell disease: systematic review and meta-analysis. Blood (2015) 125(21):3316–25. doi: 10.1182/blood-2014-11-607317
165. Savona-Ventura C, Bonello F. Beta-thalassemia syndromes and pregnancy. Obstet Gynecol Surv (1994) 49(2):129–37. doi: 10.1097/00006254-199402000-00025
166. Serjeant GR, Loy LL, Crowther M, Hambleton IR, Thame M. Outcome of pregnancy in homozygous sickle cell disease. Obstet Gynecol (2004) 103(6):1278–85. doi: 10.1097/01.AOG.0000127433.23611.54
167. Sun PM, Wilburn W, Raynor BD, Jamieson D. Sickle cell disease in pregnancy: twenty years of experience at Grady memorial hospital, Atlanta, Georgia. Am J Obstet Gynecol (2001) 184(6):1127–30. doi: 10.1067/mob.2001.115477
168. Gilbertson AA, Ball PA, Watson-Williams EJ. The management of anaesthesia in sickle cell states. Proc R Soc Med (1967) 60(7):631–6. doi: 10.1177/003591576706000704
169. Hyder O, Yaster M, Bateman BT, Firth PG. Surgical procedures and outcomes among children with sickle cell disease. Anesth Analg (2013) 117(5):1192–6. doi: 10.1213/ANE.0b013e3182a44d74
170. Schyrr F, Dolci M, Nydegger M, Canellini G, Andreu-Ullrich H, Joseph JM, et al. Perioperative care of children with sickle cell disease: A systematic review and clinical recommendations. Am J Hematol (2020) 95(1):78–96. doi: 10.1002/ajh.25626
171. Liebelt EL, Balk SJ, Faber W, Fisher JW, Hughes CL, Lanzkron SM, et al. NTP-CERHR expert panel report on the reproductive and developmental toxicity of hydroxyurea. Birth Defects Res B Dev Reprod Toxicol (2007) 80(4):259–366. doi: 10.1002/bdrb.20123
172. Haberkern CM, Neumayr LD, Orringer EP, Earles AN, Robertson SM, Black D, et al. Cholecystectomy in sickle cell anemia patients: perioperative outcome of 364 cases from the national preoperative transfusion study. preoperative transfusion in sickle cell disease study group. Blood (1997) 89(5):1533–42.
173. Howard J, Malfroy M, Llewelyn C, Choo L, Hodge R, Johnson T, et al. The transfusion alternatives preoperatively in sickle cell disease (TAPS) study: a randomised, controlled, multicentre clinical trial. Lancet (2013) 381(9870):930–8. doi: 10.1016/S0140-6736(12)61726-7
174. Koshy M, Weiner SJ, Miller ST, Sleeper LA, Vichinsky E, Brown AK, et al. Surgery and anesthesia in sickle cell disease. cooperative study of sickle cell diseases. Blood (1995) 86(10):3676–84.
175. Vichinsky EP, Neumayr LD, Haberkern C, Earles AN, Eckman J, Koshy M, et al. The perioperative complication rate of orthopedic surgery in sickle cell disease: report of the national sickle cell surgery study group. Am J Hematol (1999) 62(3):129–38. doi: 10.1002/(SICI)1096-8652(199911)62:3<129::AID-AJH1>3.0.CO;2-J
176. Waldron P, Pegelow C, Neumayr L, Haberkern C, Earles A, Wesman R, et al. Tonsillectomy, adenoidectomy, and myringotomy in sickle cell disease: perioperative morbidity. preoperative transfusion in sickle cell disease study group. J Pediatr Hematol Oncol (1999) 21(2):129–35. doi: 10.1097/00043426-199903000-00009
177. Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA (2014) 312(10):1033–48. doi: 10.1001/jama.2014.10517
178. Darbari DS, Fasano RS, Minniti CP, Castro OO, Gordeuk VR, Taylor JGt, et al. Severe vaso-occlusive episodes associated with use of systemic corticosteroids in patients with sickle cell disease. J Natl Med Assoc (2008) 100(8):948–51. doi: 10.1016/S0027-9684(15)31410-3
179. Darbari DS, Wang Z, Kwak M, Hildesheim M, Nichols J, Allen D, et al. Severe painful vaso-occlusive crises and mortality in a contemporary adult sickle cell anemia cohort study. PloS One (2013) 8(11):e79923. doi: 10.1371/journal.pone.0079923
180. Vichinsky EP, Haberkern CM, Neumayr L, Earles AN, Black D, Koshy M, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. the preoperative transfusion in sickle cell disease study group. N Engl J Med (1995) 333(4):206–13. doi: 10.1056/NEJM199507273330402
181. Smith LA, Oyeku SO, Homer C, Zuckerman B. Sickle cell disease: a question of equity and quality. Pediatrics (2006) 117(5):1763–70. doi: 10.1542/peds.2005-1611
182. Hwee T, Bergen K, Leppke S, Silver A, Loren A. Hematopoietic cell transplantation and utilization of fertility preservation services. Biol Blood Marrow Transplant (2019) 25(5):989–94. doi: 10.1016/j.bbmt.2019.01.005
183. Quinn GP, Vadaparampil ST, King L, Miree CA, Wilson C, Raj O, et al. Impact of physicians' personal discomfort and patient prognosis on discussion of fertility preservation with young cancer patients. Patient Educ Couns (2009) 77(3):338–43. doi: 10.1016/j.pec.2009.09.007
184. Quinn GP, Vadaparampil ST, Lee JH, Jacobsen PB, Bepler G, Lancaster J, et al. Physician referral for fertility preservation in oncology patients: a national study of practice behaviors. J Clin Oncol (2009) 27(35):5952–7. doi: 10.1200/JCO.2009.23.0250
185. Quinn GP, Vadaparampil ST, Fertility Preservation Research G. Fertility preservation and adolescent/young adult cancer patients: physician communication challenges. J Adolesc Health (2009) 44(4):394–400. doi: 10.1016/j.jadohealth.2008.08.014
186. Hirshfeld-Cytron J, van Loendersloot LL, Mol BW, Goddijn M, Grobman WA, Moolenaar LM, et al. Cost-effective analysis of oocyte cryopreservation: stunning similarities but differences remain. Hum Reprod (2012) 27(12):3639. doi: 10.1093/humrep/des339
187. Walter JR, Xu S. Woodruff TK. A call for fertility preservation coverage for breast cancer patients: The cost of consistency. J Natl Cancer Inst (2017) 109(5):1–5. doi: 10.1093/jnci/djx006
188. Preservation AfF. State Laws & Legislation. Available at: https://www.allianceforfertilitypreservation.org/state-legislation/ (2022) (Accessed May 20, 2022).
189. Scott-Trainer J. The role of a patient navigator in fertility preservation. Cancer Treat Res (2010) 156:469–70. doi: 10.1007/978-1-4419-6518-9_37
Keywords: fertility preservation, sickle cell disease, beta thalassemia, oocyte cryopreservation, ovarian tissue cryopreservation, sperm cryopreservation
Citation: Bedrick BS, Kohn TP, Pecker LH and Christianson MS (2022) Fertility preservation for pediatric patients with hemoglobinopathies: Multidisciplinary counseling needed to optimize outcomes. Front. Endocrinol. 13:985525. doi: 10.3389/fendo.2022.985525
Received: 04 July 2022; Accepted: 30 September 2022;
Published: 24 October 2022.
Edited by:
Asma Chattha, Mayo Clinic, United StatesReviewed by:
Maria T. Bourlon, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoCopyright © 2022 Bedrick, Kohn, Pecker and Christianson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mindy S. Christianson, bWNocmlzMjFAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.