 Deepika Watts
Deepika Watts Mangesh T. Jaykar†
Mangesh T. Jaykar† Ben Wielockx
Ben Wielockx- Institute of Clinical Chemistry and Laboratory Medicine, Technische Universität Dresden, Dresden, Germany
Adequate oxygen levels are essential for the functioning and maintenance of biological processes in virtually every cell, albeit based on specific need. Thus, any change in oxygen pressure leads to modulated activation of the hypoxia pathway, which affects numerous physiological and pathological processes, including hematopoiesis, inflammation, and tumor development. The Hypoxia Inducible Factors (HIFs) are essential transcription factors and the driving force of the hypoxia pathway; whereas, their inhibitors, HIF prolyl hydroxylase domain (PHDs) proteins are the true oxygen sensors that critically regulate this response. Recently, we and others have described the central role of the PHD/HIF axis in various compartments of the adrenal gland and its potential influence in associated tumors, including pheochromocytomas and paragangliomas. Here, we provide an overview of the most recent findings on the hypoxia signaling pathway in vivo, including its role in the endocrine system, especially in adrenal tumors.
Hypoxia inducible factors: Overview of structure, function, and regulation
Oxygen is essential for the functioning and survival of cells and tissues because it is required for cellular energy production and as a cofactor/substrate for various enzymes; thus, the absence of sufficient oxygen pressure results in hypoxia. However, the term hypoxia is relative and should be used in the correct context as normal oxygen levels vary among tissues, e.g., from ~13% in arterial blood to ~4% in the brain (1). At the molecular level, mechanisms of cellular adaptation to hypoxia involve the hypoxia-inducible factors (HIF-1α, HIF-2α and HIF-3α), which are transcription factors that regulate the expression of hundreds of genes involved in adaptation processes and cell survival. ChIP-seq analysis and genome-wide chromatin immunoprecipitation, combined with DNA microarrays (ChIP-on-chip), have revealed that more than 800 genes regulated by HIFs are involved in various biological functions (2, 3). Furthermore, HIFs also regulate many microRNAs (4) and chromatin-modifying enzymes (5).
Structurally, each functional HIF transcription factor is a heterodimer of two subunits, an oxygen- sensitive HIF-α subunit and a constitutively expressed HIF-1β subunit, and both these subunits belong to the basic helix–loop-helix (HLH)-PER-ARNT-SIM (bHLH-PAS) protein family (6, 7). The β subunit is also called the aryl hydrocarbon receptor nuclear translocator, ARNT. In the presence of adequate oxygen, HIF prolyl hydroxylases (PHDs) hydroxylate the HIF-α subunit at conserved proline residues located in its oxygen-dependent degradation domain (8, 9). Subsequently, an E3 ubiquitin ligase, von Hippel Lindau (VHL), binds to the hydroxylated HIF-α, leading to its ubiquitination and proteasomal degradation (Figure 1) (10, 11). The PHD enzymes belong to the 2-oxoglutarate-dependent oxygenase superfamily and are dependent on oxygen, iron, and ascorbate for their activity (12); hence, in hypoxic cells, neither HIF-1α nor HIF-2α is hydroxylated by the PHDs, which halts HIFs degradation. This results in the accumulation of HIF-1α or HIF-2α in the cell and their consequent binding to the HIF-1β subunit, leading to the formation of the functional HIF heterodimer that then translocates to the nucleus along with its co-activators (p300 and CBP) to form the transcriptional complex. Specifically, the HIF transcription factor binds to hypoxia-responsive elements (A/GCGTG consensus motif) in the promoter region of several genes that regulate various processes such as erythropoiesis, angiogenesis, metabolism, apoptosis, cellular differentiation, and metastasis (Figure 1) (13–15).
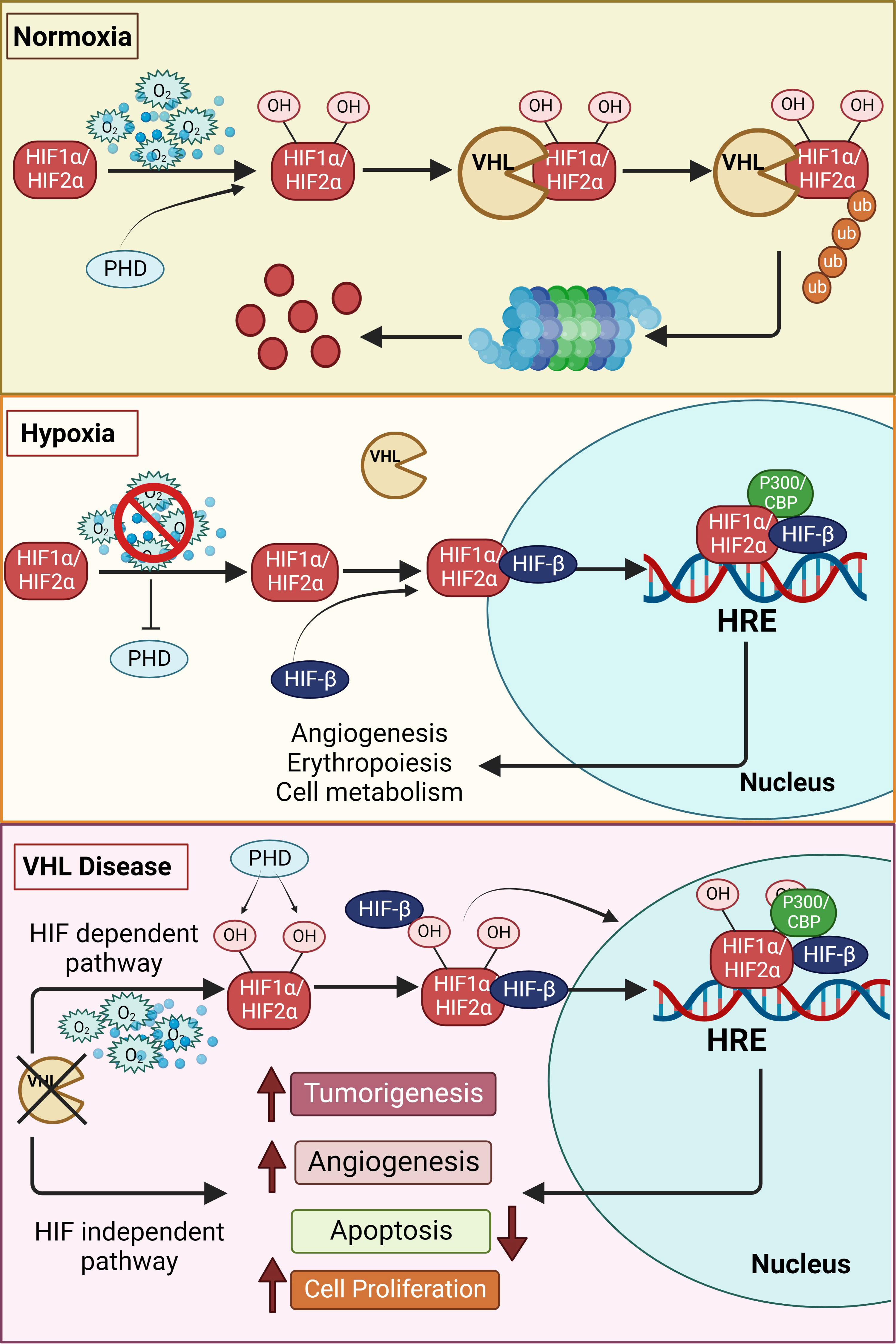
Figure 1 Schematic overview of the hypoxia pathway in normoxia, hypoxia and VHL-disease. In the presence of oxygen, the hypoxia-inducible factors (HIF)-α subunits are synthesized, hydroxylated by HIF prolyl hydroxylase domain (PHDs) proteins in the presence of oxygen, leading to the subsequent binding to von Hippel–Lindau (VHL) tumor suppressor protein. VHL mediated ubiquitination of hydroxylated HIFs, results in proteasomal degradation under normoxia. However, the hydroxylation of the alpha subunits is repressed under hypoxia due to inactivation of PHDs, thus stabilizing the HIF-α subunits. The stabilized alpha subunits bind to the beta subunit and translocate to the nucleus. The HIFs, together with other cofactors, bind to the hypoxia response elements (HREs), promoting the transcription of genes essentially involved in erythropoiesis, angiogenesis and other genes regulated by hypoxia pathway proteins. However, in the VHL disease, even in the presence of oxygen, HIFs are stabilized due to inactivity of the VHL resulting in increased tumorigenesis. This occurs directly by upregulation of tumor-associated genes or is facilitated by suppression of apoptosis and increase in angiogenesis. VHL mediated tumor development can also occur in an HIF-independent manner by upregulation of tumorigenesis. Additional information can be found in the text.
Another oxygen-dependent mechanism of HIF regulation involves Factor inhibiting HIF-1 (FIH1), an asparaginyl hydroxylase, which hydroxylates the HIF-α subunit at the asparagine in the C-terminal activation domain (N-803 in human HIF-1α) under normoxia and mild hypoxia. Such hydroxylation prevents activation of HIF as it inhibits interactions between HIF-1α and its co-activators p300/CBP (16). FIH1 also acts as a safety net because it is less sensitive to a reduction in oxygen pressure compared to PHDs and remains active even under mild hypoxia to block HIF-α activation that has escaped PHD-mediated degradation (17).
While the above pertained to oxygen-dependent regulation of HIFs, various tumor suppressor and oncogenic pathways (such as MAPK/ERK and PI3K/AKT) non-specifically regulate HIFs in an oxygen-independent manner. Tumor suppressors such as p53 and GSK3β decrease HIF-1α stability or transcriptional activity and thereby interfere with HIF function (18); in contrast, PI3K/AKT pathway activation has been shown to increase HIF-1α mRNA translation and production (19, 20). Another important mechanism of regulation is phosphorylation, which affects both HIF-1α stability and its transcriptional activity. Here, the tumor suppressor GSK3β phosphorylates HIF-1α at three serine residues within the human HIF-1α N-terminal transactivation domain (21, 22), which results in the Fbw7 and USP28-mediated HIF-1α ubiquitination and VHL-independent proteasomal degradation (23). Similarly, HIF-1α is also destabilized by PLK3, which phosphorylates two serine residues (24). Contrarily, HIF-1α phosphorylation can also lead to stabilization because it has been shown that HIF-1α stability increases upon phosphorylation at Ser-696 by ataxia-telangiectasia mutated (ATM) protein kinase (25). Further, ERK1 phosphorylates HIF-1α in its C-terminal activation domain, increasing its transcriptional activity, but not its stability (26).
Notably, even though both HIF-1α and HIF-2α contain similar binding and dimerization domains, they have different transactivation domains (27). Therefore, despite the presence of several common target genes, they can also individually modulate a unique sets of genes (28). Interestingly, these differentially regulated unique target genes can have opposing effects, as recently demonstrated in endothelial cells (29).
Hypoxia and HIFs in cancers and tumors
Solid tumors are characterized by hypoxic or even anoxic regions within the tumor mass because existing blood vessels fail to meet the oxygen requirements of the rapidly proliferating cancer cells (30). The consequent up-regulation of angiogenic factors, i.e., in response to tumor hypoxia, leads to the formation of non-functional blood vessels with structural and functional abnormalities (31) and this aberrant tumor vasculature severely restrains oxygen supply in the tumor microenvironment resulting in acute hypoxia (32). The involvement of tumor hypoxia in chemo- and radio-resistance is well established (33, 34), and new data suggests its involvement in resistance to immunotherapy as well (35). As the cellular response to hypoxia is controlled by HIF transcription factors that regulate several genes related to adaptation and progression of cancer cells, tumor hypoxia and HIFs govern many attributes of cancer cells, such as proliferation, metabolism, apoptosis, genomic instabilities, vascularization, immune responses, and invasion and metastasis. Typically, sustained hypoxia activates cell apoptosis; however, in tumors, it directs the selection of tumor cells that are resistant to apoptosis, and thereby, contributes to the malignant phenotype (36–38). Understandably, anticancer agents that target rapidly dividing cells are less effective against hypoxic cells that are more distant from vasculature and have reduced rates of proliferation. Moreover, cancer stem-like cells (CSCs) are a rare population of tumor cells that have self-renewal capacity and contribute to treatment resistance. As CSCs reside in the more hypoxic niches of the tumor, they escape chemo- and radiotherapy-induced DNA damage and thereby not only survive treatment but also repopulate the tumor with their progeny (39).
Hypoxia signaling pathway in endocrine tumors
Endocrine tumors can originate in any of the hormone-producing endocrine organs— thyroid, pituitary glands, pancreas, and adrenal gland, and their nomenclature reflects both origin and location. Thyroid cancer is considered the most common malignancy among all endocrine tumors and anaplastic thyroid carcinomas (ATCs) are predominantly aggressive tumors with an average survival of 3–4 months (40, 41). On the other hand, pheochromocytoma (PCC) and non-secretory pancreatic islet cell cancers are caused by mutations in the VHL tumor suppressor and are characterized by marked interfamilial variations in frequency, significant morbidity and, sometimes, even mortality.
Processes and factors such as signaling pathways, metabolic reprogramming, extracellular matrix remodeling, and epigenetic changes regulate the development and propagation of endocrine tumors (42, 43). As endocrine cancers, like most solid tumors, frequently exhibit major hypoxic areas, hypoxia signaling pathway genes have been associated with endocrine tumor development as well (14, 44–47). Further, given that the hypoxia pathway is one of the major drivers of endocrine tumors, identifying the exact molecular mechanisms of these dysregulated processes could help in the discovery of key therapeutic targets (Figure 1).
Pheochromocytoma and paraganglioma
Pheochromocytomas (PCCs) and paragangliomas (PGLs; together termed as PPGLs) are unique neuroendocrine tumors that make up less than 1% of all endocrine neoplasia. PCCs arise from chromaffin cells of the adrenal medulla whereas PGLs are extra-adrenal, neural crest-derived, neuroendocrine tumors (NETs) of the sympathetic and parasympathetic ganglia (48, 49). PGLs arising from parasympathetic paraganglia are mostly found in the head and neck region, including in the carotid body, due to the presence of neuroendocrine chief cells in the vagus and the glossopharyngeal nerves (50). PCCs and PGLs of sympathetic origin often secrete catecholamines, leading to systemic cardiometabolic effects (51) such as palpitations, tachycardia, hypertension, headaches, diaphoresis, heat intolerance, and anxiety. Even though PCCs and PGLs are typically benign, malignancy can occur in 10–15% of the cases with metastasis to bone, liver, lungs, and lymph nodes (52).
Mutations associated with PCCs and PGLs
Approximately 35–40% of PCCs and PGLs have a hereditary predisposition that is attributable to germline pathogenic variants (PVs) in over twenty susceptibility genes (53–55). Rates of predisposition to such germline PVs range between 25–30% in PCC, up to 40% in PGL, and about 50% in metastatic disease (56). Tumors with germline PVs are broadly categorized in two clusters, viz., Cluster 1 (pseudohypoxia) and Cluster 2 (kinase signaling). Cluster 1 includes PV mutations in SDH, VHL, fumarate hydratase (FH) and EPAS1, whereas cluster 2 PPGLs bears mutations in NF1, rearranged during transfection (RET) proto-oncogene, TMEM127, and MAX. While cluster 1 PPGLs are characterized by an immature catecholamine phenotype (noradrenergic phenotype) and higher aggressiveness, cluster 2 PPGLs have more mature phenotype (adrenergic phenotype) and are mostly non-metastatic (57, 58). However, in recent years, mRNA expression analysis for The Cancer Genome Atlas (TCGA) has found an additional cluster for PCC/PGL, namely, the WNT-altered cluster 3, which is associated with increased expression of genes in the WNT signaling pathway (58). Many WNT tumors are driven by novel somatic alterations in CSDE1 (Cold Shock Domain Containing E1) and recurrent fusions involving MAML3 and a cortical admixture subtype (58). CSDE1 is a tumor suppressor gene that encodes the CSDE1 factor, which is involved in development, messenger RNA stability, internal initiation of translation, cell-type-specificapoptosis, and neuronal differentiation (59). It has been found that the CSDE1 is significantly mutated in PCCs/PGLs and that these mutations result in the downregulation of the apoptosis protease activator protein 1 (APAF1), which is required for controlling apoptosis in PCC cells (60, 61).
Germline and somatic mutations in major susceptibility genes associated with hypoxia signaling involved in the PCC development, include the tumor suppressors such as VHL1, the SDH complex (genes encoding the four subunits, A,B,C,D) and occasionally, the egl-9 family hypoxia-inducible factor 1/Prolyl hydroxylase domain 2 protein (EGLN1/PHD2) (62, 63). Recently, ten new genes have been added to this list and those associated with the hypoxia pathway include HIF-2α (endothelial PAS domain containing protein 1, EPAS1) (64, 65), FH (66), and PHD1 (egl nine homolog 2, EGLN2) (67), suggesting that a mutation in any of these major genes involved in the VHL-HIF axis can lead to PCC or PGL development.
Von Hippel-Lindau disease
Von Hippel-Lindau (VHL) disease is an autosomal dominant neoplastic disorder that is characterized by multiple benign and malignant tumors, including cysts, that develop in the central nervous system and visceral organs (68). Various mutations in VHL have been found to cause diverse clinical symptoms; sometimes even the same mutation yields different phenotypes (69, 70). As pVHL has multiple functional domains, one of the potential explanations for this phenomenon is that a specific mutation causes a particular dysfunction. Specifically, mutations in the VHL gene on chromosome 3 affect the functionality of pVHL, i.e., as pVHL is incapable of recognizing hydroxylated HIFs, their greater stability leads to HIF-mediated transcription of genes and consequent development of VHL disease (Figure 1) (71, 72). The role of VHL in disease development has been described in detail elsewhere by Hudler and colleagues (71).
PCC is a hallmark of VHL disease and its absence or presence defines phenotypic classification as VHL type - 1 (protein-truncating mutations) or type - 2, which is linked to missense mutations (68). About 20% of patients with VHL disease will develop either unilateral or bilateral intra-adrenal PCC; however, the incidence of extra adrenal PGLs is rare and they occur only in type 2 disease with about 5% of the cases showing metastasis (73, 74). Type 2 disease is further divided into 3 subgroups - 2A, 2B, which correspond to low and high-risk clear cell renal cell carcinoma (ccRCC), respectively, and 2C with only PGL. Interestingly, there is a correlation between degree of HIF dysregulation and mutant VHL alleles in types 1, 2A, 2B, and 2C; specifically, highest HIF dysregulation is seen in type 1 while lowest is seen in type 2C (75, 76). Additionally, existing literature points towards Chuvash polycythemia being a type 3 VHL disease (77, 78). Nevertheless, type 2 VHL disease related PCCs/PGLs show overexpression of several genes involved in angiogenesis, glucose metabolism, and cell proliferation, with the hypoxic pathway particularly associated with stabilization of HIF-2α (Figure 1), i.e., the main isoform expressed in catecholamine-producing cells (79, 80). As PCC/PGL-linked biochemical testing is correlated with norepinephrine/normetanephrine predominance, as established from plasma levels, this phenotype is caused by the silencing of the norepinephrine-to-epinephrine converting enzyme Phenylethanolamine-N-Methyl-Transferase (PNMT) in the adrenal medulla (45, 81–83). Interestingly, this characteristic is similar to the immature phenotype seen in tumors with VHL mutations, i.e., a phenomenon that is associated with HIF-2α overexpression and stabilization. HIF-2α accumulation, rather than HIF-1α, is a major phenomenon in VHL tumors and it results in the overexpression of hypoxia-induced angiogenic genes, such as vascular endothelial growth factor (VEGF), erythropoietin (EPO) and EPO-receptor, cyclin D1 (CCND1), and other genes involved in extracellular matrix reorganization that facilitate tumor development (45, 84). Nonetheless, other HIF-independent proteins and pathways can also be dysregulated in VHL-mutated tumors, e.g., the developmental neuronal apoptosis pathway, p53-related networks, and glucose metabolism. Likewise, the HIF-independent defective apoptosis pathway (for example in type 2C VHL disease) cannot induce apoptosis in chromaffin cells due to greater stability of p53 (85), and some of the type 2C mutations interfere with the regulation of transcription factor AP-1 (JUN)-induced apoptosis due to a VHL-mediated reduction in Jun-B and EGLN3/PHD3 levels (71, 86, 87).
SDH gene mutations
PCC/PGL development is not only directly associated with mutations in the hypoxia pathway but also correlated to modifications in other genes responsible for HIF stabilization, included in cluster 1. One such example is the succinate dehydrogenase (SDHx) family comprising of 4 SDH subunits (A, B, C, D) (47, 88). Mutation in one of the SDHx genes promotes accumulation of the oncometabolite succinate, which inhibits the 2-oxogluarate-dependent PHDs and subsequently leading to stabilization and accumulation of HIF-2α (Figure 2). Additionally, metastatic SDHx-related PCC/PGL overexpress heat shock protein 90 (HSP90), a molecular chaperone that facilitates binding to HIF-2α by promoting its stability and preventing ubiquitination and proteasomal degradation (89–91).
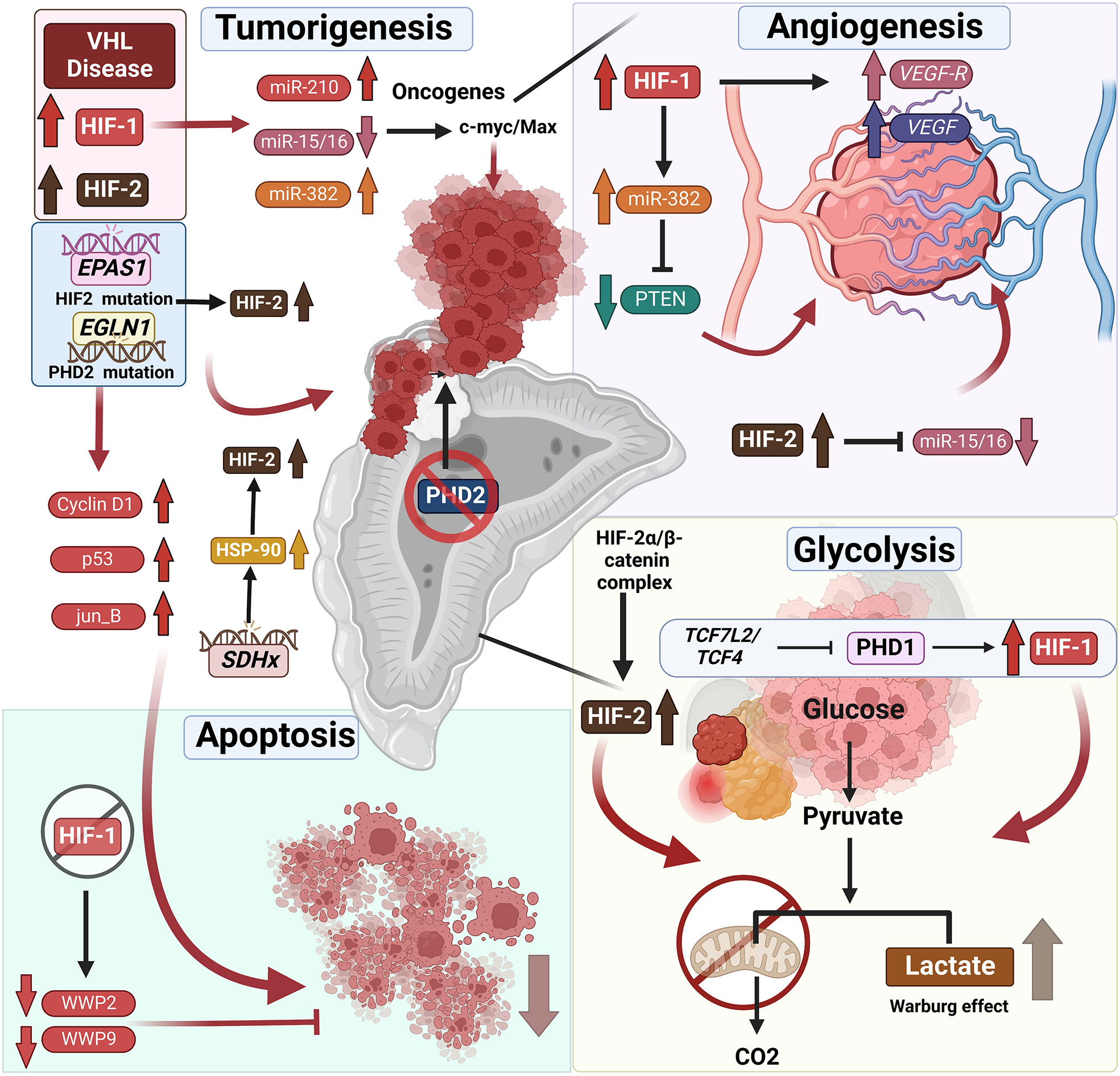
Figure 2 Mutations associated with hypoxia pathway proteins and their role in endocrine tumor development. VHL disease leads to the stabilization of HIFs, which is frequently associated with endocrine tumors – specifically pheochromocytomas (PCCs) and paragangliomas (PGLs). Not only does HIF upregulation result in tumorigenesis directly, it also results in reduced apoptosis of endocrine tumor cells. It was shown that HIF-1α repression induces apoptosis by downregulation of the expression of WW domain containing E3 ubiquitin protein ligase (WWP9, WWP2), promoting tumor cell apoptosis. Furthermore, HIF-1α and HIF-2α stabilization leads to changes in tumor-associated microRNAs. The HIF-associated increase or decrease in micro-RNAs correlated with upregulation of oncogenes and thus tumorigenesis. Likewise, the mutations in the genes such as EPAS1 or EGLN1 directly affects the stabilization of the HIFs resulting in tumor development and reduced apoptosis of endocrine tumor cells. SDHx mutations in PCCs and PGLs result in the overexpression of the heat shock protein 90 (HSP-90) resulting in the stabilization of HIF-2α. TH : Cre mediated medullar deletion of PHD2/EGLN1 in the mice resulted in the growth of aberrant structures from the adrenal medulla. Next to the direct effect of HIF stabilization on the tumor development, it also affects angiogenesis by upregulation of VEGF and VEGFR or miR-15/16 repression initiating angiogenesis. Furthermore, stabilization of HIFs, results in a shift from oxidative phosphorylation to glycolysis, converting pyruvate to lactate in the TCA cycle often mentioned as Warburg effect in the tumors, including endocrine tumors. Additional information can be found in the text.
SDHx-mediated HIF stability is one of the major drivers of PCC/PGL development as it activates genes associated with pseudohypoxia, angiogenesis, protein transport, energy metabolism regulation, and proliferation. Moreover, we and others have previously demonstrated that increased stabilization of HIF-2α is directly associated with a neuroendocrine to mesenchymal transition in these cells, contributing to a more pro-metastatic state of these cluster 1 PPGLs (57, 92). SDHx-associated PCCs/PGLs also have a norepinephrine/normetanephrine predominant profile (81) and, like VHL-associated tumors, are associated with PNMT gene silencing. Specifically, SDHB mutations are predominantly associated with multiple tumors, and although heterozygous SDHA pathogenic variants account for less than 1% of all PCC/PGL, SDHB mutations account for most common mutation associated with malignant PCCs (93, 94). SDHA pathogenic carriers can develop PCC/PGL at any location in the body, including head and neck PGLs and patients who develop SDHA mutation related PCC/PGL report to have high rates of metastatic disease (12%) (89, 95). Further, PCC patients with SDHB or SDHD mutations present overexpression of HIF-2α and its transcriptional target VEGF.
PHD mutations
The PHD family consists of PHD1, PHD2, and PHD3; and PHD2, encoded by EGLN1, is a crucial oxygen sensor that regulates HIFα levels (96). PHD2 dysregulation in the hypoxia pathway results in HIF-2α stabilization and consequent accumulation, leading to a pseudohypoxic state that may underlie the pathologic conditions encountered in PCC/PGL (96). Various mutations in EGLN1 have been associated with HIF-2 stabilization, e.g., a heterozygous germline mutation at H374 predisposes to instability and loss of PHD2 activity, leading to upregulation of HIF-2α due to greater stability (96). The H374R mutation is also associated with recurrent PGL, suggesting a crucial role for PHD2/EGLN1 as a tumor suppressor gene that is similar to VHL missense mutations seen in type 2 VHL disease, i.e., high risk of PCC/PGL (97). Moreover, Yang and colleagues have demonstrated that a germline mutation in PHD1 and a novel germline PHD2 mutation are associated with PCC/PGL and polycythemia (67). Additionally, a unique association between polycythemia and mildly elevated erythropoietin (EPO) levels has been observed in patients, which was linked to inappropriate hypersensitivity of erythroid progenitors to EPO, indicating increased EPOR expression/activity (67). Contrastingly, Provenzano and colleagues have recently described a novel germline EGLN1 gene variant in a patient with metastatic PCC and chronic myeloid leukemia (CML) in the absence of polycythemia (98). Recently, Eckardt and colleagues have reported that inactivation of PHD2 in the adrenal medulla, using a (Tyrosine Hydroxylase) TH-restricted Cre mouse line, resembles a combination of pseudohypoxic PGL and a PNMT negative noradrenergic phenotype (99). This TH:cre specific deletion of PHD2 was associated with morphological abnormalities in adrenal development, including ectopic TH+ cells (Figure 2) (99). Thus, these findings collectively establish that mutations in PHDs are associated with susceptibility to pheochromocytomas and paragangliomas (PPGLs), with or without polycythemia. However, compared to VHL, mutations in PHDs are relatively rare with an incidence of less than 2% in patients with PPGLs (58, 100, 101).
HIF stabilization
As mentioned above, upregulation and stabilization of HIFs is widely associated with angiogenesis, tumor progression, and immune evasion in various tumors (46, 102–104), and HIFs are crucial for mediating the tumorigenic effects of mutated VHL, SDHx, and EGLN1 in PCC and PGL (105). The role of HIFs in catecholamine synthesis has been extensively studied, and tyrosine hydroxylase (TH), the rate-limiting enzyme in this process that is responsible for the conversion of tyrosine to L-dihydroxyphenylalanine (L-DOPA), can be induced by hypoxia (106). Importantly, both HIFs can bind to the TH promoter and thereby increase TH expression (107). While HIF-2α knockdown has no effect on Th mRNA expression in a rodent adrenomedullary chromaffin cell line (108), it seems to be more important for the proper development of differentiated adrenal chromaffin cells. Further, HIF-2α upregulation and accumulation is associated with an immature chromaffin cell phenotype due to reduced PNMT expression and epinephrine synthesis (109). Thus, a noradrenergic phenotype with no epinephrine production is seen upon HIF accumulation during aggressive PGL development (84, 110, 111).
The role of direct mutations in HIFs leading to abnormal stabilization has also been studied in the development of PCC/PGLs, and recently, a transgenic mouse line with whole body HIF-2α gain-of-function mutation showed reduced PNMT in the adrenal glands (64). Moreover, compared to HIF-1α in VHL-related PCC and PGL, HIF-2α stabilization and accumulation is a major phenomenon, and multiple studies have described the oncogenic role of somatic mutations in EPAS1 in PCC/PGL (105, 112, 113). Consequently, HIF-2α/EPAS1 has been added to the pool of genes associated with PCC/PGL in the past few years (Figure 2) (114). Zhang and colleagues have reported two gain-of-function somatic mutations in exon 12 of HIF2α (c.1588G>A, p.Ala530Thr and c.1589C>T, p.Ala530Val) that result in PGL and polycythemia, respectively (112, 115). Furthermore, two other somatic mutations in HIF2α (c.1595A>G p.Y532C and c.1586T>C p.L529P) in patients with congenital polycythemia, multiple recurrent PPGLs, or somatostatinoma have also been reported (65).
Germline mutations in HIF-2α have also been reported to be associated with PCC/PGL development; nonetheless, as certain germline mutations in HIF-2α only lead to polycythemias and not tumors (115, 116), it is thought that such gain-of-function mutations alone are not sufficient for tumorigenesis, and that, presumably, simultaneous loss-of-function or somatic mutations in other genes may also be necessary (115–117). Another scenario involves gain-of-function mutations in HIF-2α (c.1589C>T) leading to concurrent PPGL and polycythemia (118), as seen in the case of the germline mutation in exon 9 (c.1121T>A, p.F374Y), which predisposes patients to polycythemia and PGL development (119). Further, tumors have been associated with EPAS1 mutations, either in the absence or presence of polycythemia, and polycythemia alone is seen in cases with mosaic mutations in EPAS1. Likewise, Buffet and colleagues have reported the presence of mosaic mutations in two patients with HIF-2α-related polycythemia/PGL syndrome (120). In summary, PHD2 mutations result in a norepinephrine/normetanephrine immature cell phenotype that is caused by silencing of the norepinephrine-to-epinephrine converting enzyme (PNMT) in the chromaffin cells of the adrenal medulla and is related to aggressive PCC development in the presence of mosaic mutations (114, 120, 121).
Adrenal cortical tumors
Adrenocortical carcinoma (ACC) represent a rare, aggressive, and heterogeneous tumors that arises from the cortex of the adrenal gland. The 5-year survival rate for ACC ranges from 16% to 47% (122). As mentioned above, hypoxia and HIFs are a common feature of many endocrine tumors because these lesions are characterized by rapid proliferation that is associated with metastasis, immune evasion, resistance to therapy, and increased mortality (123). Even though advances over the past few decades in several biomarkers associated with metastasis, prognosis, and survival in ACC patients have led to a better understanding of its molecular genetics, the specific effects of HIF-1 activity in ACC and hypoxia signatures for predicting ACC prognosis have not been established (124). However, a case study of a very rare erythropoietin-producing adrenocortical carcinoma accompanied by lung and liver metastases has been reported (125). Tumor associated hypoxia could be one of the players of erythropoietin production in tumor cells. Recently, a bioinformatic study has reported that a hypoxia-related gene signature could predict prognosis and reflect the immune microenvironment in ACC (126). Specifically, based on hypoxia-related gene expression, ACC patients in the TCGA database were divided into three molecular subtypes (C1, C2, and C3), each with different clinical outcomes with C3 having reported of shortest survival (127).
On the other hand, aldosterone producing adenomas (APAs) are benign aldosterone producing tumors associated with Primary aldosteronism (PA) causing secondary hypertension (128). These are characterized by autonomous production of aldosterone from the adrenal glands leading to low-renin levels and thus hypertension. Most APAs (~ 90%) harbor somatic pathogenic variants in genes encoding ion channels or transporters such as KCNJ5, ATP1A1, ATP2B3, CACNA1D, CACNA1H, CLCN2, and CTNNB1 (128, 129). However, no correlation to hypoxia signaling has been described in the aldosterone producing adenomas until now.
Mixed corticomedullary tumor
Mixed corticomedullary tumor (MCT), an extremely rare condition with unclear tumorigenesis, is an adrenal tumor with cortical and medullary cells. Adrenal MCT was first described by Mathison and Water-House in 1969 and only 30 cases have been reported to date. Adrenal tumors, including MCTs, exhibit different stemness expression (130), e.g., a patient with MCT displayed typical Cushing’s syndrome and hypertension, and hence, MCT tumorigenesis is thought to involve the two-hit hypothesis. Pathway enrichment analysis from exosome sequencing of MCT has identified enriched pathways, including the hypoxia-inducible factor-1 (HIF-1) signaling pathway (hsa04066; 1.3%), and some of the germline mutations were involved in stemness regulation, the first hit of which may drive adrenocortical adenoma (ACA) and PCC formation. Additional mutations affecting different pathways, including the HIF-1 signaling pathway, may accelerate tumor growth and intimately mix ACA and PCC (131).
Additional endocrine tumors
Thyroid cancer
As with many types of solid tumors, hypoxia due to inadequate vascularization is a prominent micro-environmental component in thyroid cancer (133). Differentiated thyroid cancer (DTC), which includes papillary cancer (PTC) and follicular cancer (FTC), accounts for >90% of all thyroid cancer cases (132). As angiogenesis is an important factor in the development, growth, and metastasis of cancers, the proangiogenic factor VEGF is a significant mediator of angiogenesis in the thyroid gland. Notably, this makes HIF-1 mediated regulation of VEGF a key factor in the development of thyroid tumors (44, 133, 134). Several studies have reported greater expression of HIF-1α and HIF-2α in thyroid cancer compared to normal thyroid tissue or benign lesions (135–137), and a strong correlation has been observed between HIF-2α expression and tumor size. Specifically, tumors with more intense HIF-1α and HIF-2α staining had a higher TNM stage (137), and overexpression of both HIF-1α and HIF-2α was associated with capsular invasion and lymph node metastasis.
Hyperactive PI3K signaling leads to stabilization of HIF-1α not only in normoxia but also in many cancers, including thyroid cancer and is predominantly involved in the metastasis of PTC and FTC (138, 139). Moreover, Ding and colleagues have reported that silencing of HIF-1α represses cell invasion and induces apoptosis by downregulating the expression of WW domain containing E3 ubiquitin protein ligase (WWP9, WWP2), VEGF, and VEGFR2 in thyroid cancer (44) as depicted in Figure 2. Thus, HIF-1α represents a potential therapeutic target for the treatment of thyroid cancer (140).
Pancreatic neuroendocrine tumors
Pancreatic cancer is a highly fatal malignancy and the lesion is predominantly characterized by severe hypoxic regions because its median partial oxygen pressure (pO2) is 0–5.3 mmHg (0–0.7%); in contrast, the pO2 of the adjacent normal pancreatic tissue is 24.3–92.7 mmHg (3.2–12.3%) (141). Thus, pancreatic cancer cells have developed effective adaptive metabolic responses to satisfy oxygen demand for biosynthesis and energy. For example, a major adaptation is the metabolic shift from oxidative phosphorylation to glycolysis, i.e., converting pyruvate to lactate instead of oxidation through the tricarboxylic acid (TCA) cycle (142). More significantly, 5–10% of VHL patients develop pancreatic tumors, which are most commonly non-secretory islet cell tumors known as pancreatic neuro-endocrine tumors (pNET) (143). Patients with missense mutations (type 2 VHL) exhibit a higher prevalence of pancreatic tumors and a hotspot on codons 161/67 in exon 3 is associated with higher risk of metastases compared to truncating mutations or large deletions (type 1 VHL) (144).
In addition to the above, Wnt signaling plays a crucial role in pancreatic tumor development and also alters cell metabolic plasticity to support immediate requirements (145). The transcription factor 7-like2/transcription factor 4 (TCF7L2/TCF4) plays a vital role in the Wnt/β-catenin signaling pathway, is responsible for PHD1 (EGLN2) silencing, leads to upregulation of HIF-1α, and affects glycolysis reprogramming (146). There is positive-feedback regulation between HIF-2α and β-catenin in that the HIF-2α/β-catenin complex can not only upregulate the activity of β-catenin but also stabilize and increase transcriptional activity of HIF-2α, which then promotes the metabolic shift to aerobic glycolysis in PC cells (147). VEGF stimulation is also known to promote angiogenesis and it can enhance glycolysis in pancreatic cancer by upregulating HIF-1α (148). Targeting the hypoxic tumor environment and the hypoxia pathway in pancreatic tumors has been described in detail elsewhere by Tao et al. (142) (Figure 2).
Micro-RNAs/HIF axis in endocrine tumors
MicroRNAs (MiR) are noncoding, about 20–22 nucleotides in length, and regulate gene expression by binding to 3′ UTRs of their related parental mRNAs. MiRNAs have been shown to control many physiological and pathophysiological processes, such as proliferation, differentiation, metabolism, and apoptosis by modulating target gene expression. Altered miRNA expression has been identified in several endocrine diseases, including tumors. For example, the 14q32 miRNA cluster is frequently dysregulated in human diseases and has been implicated in tumorigenesis of multiple endocrine glands (149). These 14q32 miRNAs may be oncogenic or tumor suppressing depending on cell type and are associated with downregulation of almost all miRNAs encoded by the 14q32 cluster in PCC (149). miR-382, a 14q32.2 miRNA cluster member, is upregulated in PPGLs associated with VHL and SDHB (150), is reported to be an angiogenic miRNA that is upregulated by HIF-1α, and acts as an angiogenic oncogene by repressing PTEN (151).
Several miRNA expression profiling studies have been performed on different endocrine tumors including PCC/PGL, and repression of 2 miRs, namely, miR-15a and miR-16, has been reported in pituitary adenomas and prostate cancer (152). Furthermore, several groups have also studied miRNA expression signatures in benign and metastatic PCC and have reported that miR-15a and miR-16 are indeed under-expressed, and that miR-483-5p, miR-183, and miR-101 are overexpressed in malignant PCC (152). For instance, pre-miR-15a and pre-miR-16 induce cell death and inhibit proliferation in rat PC12 cells but under-expression of miR-15a and miR-16 is associated with malignant tumors rather than benign PGLs. Thus, these miRNAs represent diagnostic and prognostic markers for malignant PCC (152). HIF-2α-induced repression of miR-15 and miR-16 enhances the stability of the c-Myc/Max heterodimer and thereby enhances tumor angiogenesis and metastasis (153). Dysregulation of miR-193b/365 (on chromosome 16p13.12) and miR-183/96 (on chromosome 7q32.2) has been associated with all PCC/PGL and SDHB-mutated tumors, respectively (150). Increased miR-21-3p is associated with a general increase in the expression of mesenchymal markers, whereas miR-183-5p decreases the expression of neuroendocrine genes (154). Moreover, compared to benign tumors, miR-101 expression is higher in SDHD-mutant malignant PCC tissues (155), and miR-375 is emerging as a new epigenetic alteration that is involved in neuroendocrine tumorigenesis because its overexpression in MTC has been demonstrated both by Hudson et al., and Manso et al. (156, 157).
MiR-210, a recent discovery in the field of hypoxia, is the most consistently and predominantly upregulated miRNA in response to hypoxia. Functional studies have demonstrated that miR-210 is a central gene that regulates many aspects of the hypoxia pathway, both under physiological and malignant conditions (158). MiR-210 is upregulated during hypoxia by HIF-1α (159); hence, it may promote tumorigenesis by activating important oncogenic genes linked to the hypoxia pathway (Figure 2) (159). Additionally, Tsang and colleagues have observed that miR-210 is overexpressed in PPGLs associated with VHL and SDHB germline mutations (159). However, miR-210 overexpression in head and neck PGLs has been reported to be independent of SDHx germline mutations, indicating that the HIF-1α/miR-210/SDHB axis may play a role in the pathogenesis of PGLs (160). Indeed, further studies are required to better understand the functional interplay between HIFα and miR-210, and its significance in the pathogenesis of PCC/PGLs (Figure 2). The role of microRNAs as potential biomarkers and therapeutic targets in PCC/PGL has been described elsewhere by Turai and colleagues (161) (Figure 2).
HIFs inhibitors in cancers/endocrine tumors
Targeting tumor hypoxia and HIFs is an extremely interesting therapeutic approach as tumor hypoxia mediates the aggressive, the metastatic, and the resistant phenotypes. Different approaches have been developed to target hypoxic cancer cells, such as gene therapy, hypoxia-activated prodrugs, recombinant anaerobic bacteria, pathways important in hypoxic cells like mTOR and UPR, and specific targeting of HIFs (162–165). However, HIF targeting is extremely difficult due to the complexity of the HIF pathway and the interconnected signaling cascades involved. Therefore, HIF transcription factors were considered un-druggable; nevertheless, recently, two HIF-2α-inhibitors, PT2385 and PT2399 have been successfully discovered based on the structure of HIF-2α (166–168). These compounds inhibit the growth of ccRCC, both in vitro and in vivo (169) and a phase I clinical trial yielded complete response, partial response, and stable disease in 2%, 12%, and 52% of the patients, respectively (170).
HIF-2α inhibitors are thought to possess great prospects for the treatment of advanced PPGL (171) and these promising initial results could potentially lead to preclinical and clinical studies to evaluate their efficiency in other types of tumors. Indeed, compound PT2385 is in Phase II clinical trials to assess its effectiveness in advanced cancers with VHL germline mutations. Another inhibitor of HIF-2α, PT2977, was found to have increased potency and better pharmacokinetic profile than PT2385 and PT1977 (172). Previous studies have shown that, compared to HIF-1α, HIF-2α is overexpressed in pseudohypoxic PPGLs (92, 173) and contribute to a more aggressive phenotype in these tumors (174). Thus, HIF-2α inhibitors are potentially more promising compared to HIF-1α inhibitors for the treatment of endocrine tumors, especially PCCs and PGLs.
Summary
Taken together, this review underlines that the hypoxia pathway proteins are essential contributors in the development and progression of endocrine tumors. Even though several mutations such as VHL, SDHx, EPAS1 and EGLN1 are associated with endocrine tumor initiation, many of these mutations directly or indirectly lead to stabilization of HIFs and consequently triggering the hypoxia pathway. This contributes to tumor development either by upregulation of oncogenes or genes facilitating tumor progression such as angiogenesis, skewed glycolysis or suppression of apoptosis in tumor cells. Therefore, considering the essential role of hypoxia signaling in endocrine tumors, the development of HIF inhibitors as therapeutic agents would be a potential anti-tumor strategy. Considering the upregulation of HIF-2α in many PCC/PGLs, HIF-2α inhibitors are potentially most promising, although more research is certainly warranted.
Author contributions
DW and MJ, wrote the manuscript. NB and BW contributed to the discussion and edited the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
This work was supported by grants from the DFG (German Research Foundation) within the CRC/Transregio 205/1, Project No. 314061271 - TRR205, “The Adrenal: Central Relay in Health and Disease” (A02) to BW and (A12) to NB; DFG grants WI3291/5-1 and 12-1 to BW. This work was supported by a grant from the DFG priority program µBONE 2084 to BW; BW was further supported by the DFG Heisenberg program (WI3291/13-1). We thank Dr. Vasuprada Iyengar for English Language and content editing. Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci (2012) 37:364–72. doi: 10.1016/j.tibs.2012.06.004
2. Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U.S.A. (2009) 106:4260–5. doi: 10.1073/pnas.0810067106
3. Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood (2011) 117:e207–17. doi: 10.1182/blood-2010-10-314427
4. Kulshreshtha R, Davuluri RV, Calin GA, Ivan M. A microRNA component of the hypoxic response. Cell Death Differ (2008) 15:667–71. doi: 10.1038/sj.cdd.4402310
5. Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell (2011) 43:811–22. doi: 10.1016/j.molcel.2011.07.012
6. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U.S.A. (1995) 92:5510–4. doi: 10.1073/pnas.92.12.5510
7. Bersten DC, Sullivan AE, Peet DJ, Whitelaw ML. bHLH-PAS proteins in cancer. Nat Rev Cancer (2013) 13:827–41. doi: 10.1038/nrc3621
8. Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem (2003) 278:30772–80. doi: 10.1074/jbc.M304982200
9. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J (2003) 22:4082–90. doi: 10.1093/emboj/cdg392
10. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (1999) 399:271–5. doi: 10.1038/20459
11. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science (2001) 292:464–8. doi: 10.1126/science.1059817
12. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5:343–54. doi: 10.1038/nrm1366
13. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer (2003) 3:721–32. doi: 10.1038/nrc1187
14. Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2011) 12:9–22. doi: 10.1038/nrc3183
15. Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005 (2005) 2005(306):re12. doi: 10.1126/stke.3062005re12
16. Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res (2006) 66:3688–98. doi: 10.1158/0008-5472.CAN-05-4564
17. Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell (2008) 30:393–402. doi: 10.1016/j.molcel.2008.04.009
18. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell (2010) 40:294–309. doi: 10.1016/j.molcel.2010.09.022
19. Bárdos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta (2005) 1755:107–20. doi: 10.1016/j.bbcan.2005.05.001
20. Déry MA, Michaud MD, Richard DE. Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int J Biochem Cell Biol (2005) 37:535–40. doi: 10.1016/j.biocel.2004.08.012
21. Flügel D, Görlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol (2007) 27:3253–65. doi: 10.1128/MCB.00015-07
22. Mottet D, Dumont V, Deccache Y, Demazy C, Ninane N, Raes M, et al. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem (2003) 278:31277–85. doi: 10.1074/jbc.M300763200
23. Flügel D, Görlach A, Kietzmann T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood (2012) 119:1292–301. doi: 10.1182/blood-2011-08-375014
24. Xu D, Yao Y, Lu L, Costa M, Dai W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J Biol Chem (2010) 285:38944–50. doi: 10.1074/jbc.M110.160325
25. Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol Cell (2010) 40:509–20. doi: 10.1016/j.molcel.2010.10.030
26. Minet E, Arnould T, Michel G, Roland I, Mottet D, Raes M, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett (2000) 468:53–8. doi: 10.1016/S0014-5793(00)01181-9
27. Infantino V, Santarsiero A, Convertini P, Todisco S, Iacobazzi V. Cancer cell metabolism in hypoxia: Role of HIF-1 as key regulator and therapeutic target. Int J Mol Sci (2021) 22(11). doi: 10.3390/ijms22115703
28. Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW. Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer (2007) 96:1284–92. doi: 10.1038/sj.bjc.6603675
29. Downes NL, Laham-Karam N, Kaikkonen MU, Ylä-Herttuala S. Differential but complementary HIF1α and HIF2α transcriptional regulation. Mol Ther (2018) 26:1735–45. doi: 10.1016/j.ymthe.2018.05.004
30. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26:225–39. doi: 10.1007/s10555-007-9055-1
31. Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res (2001) 61:8903–8.
32. Matsumoto S, Yasui H, Mitchell JB, Krishna MC. Imaging cycling tumor hypoxia. Cancer Res (2010) 70:10019–23. doi: 10.1158/0008-5472.CAN-10-2821
33. McAleese CE, Choudhury C, Butcher NJ, Minchin RF. Hypoxia-mediated drug resistance in breast cancers. Cancer Lett (2021) 502:189–99. doi: 10.1016/j.canlet.2020.11.045
34. Chédeville AL, Madureira PA. The role of hypoxia in glioblastoma radiotherapy resistance. Cancers (Basel) (2021) 13(3). doi: 10.3390/cancers13030542
35. Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las Rivas J, et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Update (2021) 59:100787. doi: 10.1016/j.drup.2021.100787
36. Giaccia AJ. Hypoxic stress proteins: Survival of the fittest. Semin Radiat Oncol (1996) 6:46–58. doi: 10.1016/S1053-4296(96)80035-X
37. Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature (1996) 379:88–91. doi: 10.1038/379088a0
38. Luis R, Brito C, Pojo M. Melanoma metabolism: Cell survival and resistance to therapy. Adv Exp Med Biol (2020) 1219:203–23. doi: 10.1007/978-3-030-34025-4_11
39. Wigerup C, Pahlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther (2016) 164:152–69. doi: 10.1016/j.pharmthera.2016.04.009
40. Lin B, Ma H, Ma M, Zhang Z, Sun Z, Hsieh IY, et al. The incidence and survival analysis for anaplastic thyroid cancer: a SEER database analysis. Am J Transl Res (2019) 11:5888–96.
41. Coelho RG, Fortunato RS, Carvalho DP. Metabolic reprogramming in thyroid carcinoma. Front Oncol (2018) 8:82. doi: 10.3389/fonc.2018.00082
42. Igaz P. [Genetics of neuroendocrine tumours, hereditary tumour syndromes]. Orv Hetil (2013) 154:1541–8. doi: 10.1556/OH.2013.29706
43. Jimenez C, Armaiz-Pena G, Dahia PLM, Lu Y, Toledo RA, Varghese J, et al. Endocrine and neuroendocrine tumors special issue-checkpoint inhibitors for adrenocortical carcinoma and metastatic pheochromocytoma and paraganglioma: Do they work? Cancers (Basel) (2022) 14(3). doi: 10.3390/cancers14030467
44. Ding ZY, Huang YJ, Tang JD, Li G, Jiang PQ, Wu HT. Silencing of hypoxia-inducible factor-1alpha promotes thyroid cancer cell apoptosis and inhibits invasion by downregulating WWP2, WWP9, VEGF and VEGFR2. Exp Ther Med (2016) 12:3735–41. doi: 10.3892/etm.2016.3826
45. Amorim-Pires D, Peixoto J, Lima J. Hypoxia pathway mutations in pheochromocytomas and paragangliomas. Cytogenet Genome Res (2016) 150:227–41. doi: 10.1159/000457479
46. Sormendi S, Wielockx B. Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.00040
47. Gaete D, Rodriguez D, Watts D, Sormendi S, Chavakis T, Wielockx B. HIF-prolyl hydroxylase domain proteins (PHDs) in cancer-potential targets for anti-tumor therapy? Cancers (Basel) (2021) 13(5). doi: 10.3390/cancers13050988
48. Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (Who) of Endocrine Tumours. Endocr Pathol (2017) 28(3):213–27. doi: 10.1007/s12022-017-9484-5
49. Whalen RK, Althausen AF, Daniels GH. Extra-adrenal pheochromocytoma. J Urol (1992) 147:1–10. doi: 10.1016/S0022-5347(17)37119-7
50. Merlo A, de Quirós SB, de Santa-María IS, Pitiot AS, Balbín M, Astudillo A, et al. Identification of somatic VHL gene mutations in sporadic head and neck paragangliomas in association with activation of the HIF-1α/miR-210 signaling pathway. J Clin Endocrinol Metab (2013) 98:E1661–6. doi: 10.1210/jc.2013-1636
51. Wachtel H, Fishbein L. Genetics of pheochromocytoma and paraganglioma. Curr Opin Endocrinol Diabetes Obes (2021) 28:283–90. doi: 10.1097/MED.0000000000000634
52. Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer (2014) 14:108–19. doi: 10.1038/nrc3648
53. Buffet A, Burnichon N, Favier J, Gimenez-Roqueplo A-P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab (2020) 34:101416. doi: 10.1016/j.beem.2020.101416
54. Sarkadi B, Saskoi E, Butz H, Patocs A. Genetics of pheochromocytomas and paragangliomas determine the therapeutical approach. Int J Mol Sci (2022) 23(3). doi: 10.3390/ijms23031450
55. Dariane C, Goncalves J, Timsit MO, Favier J. An update on adult forms of hereditary pheochromocytomas and paragangliomas. Curr Opin Oncol (2021) 33:23–32. doi: 10.1097/CCO.0000000000000694
56. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol (2015) 11:101–11. doi: 10.1038/nrendo.2014.188
57. Bechmann N, Moskopp ML, Ullrich M, Calsina B, Wallace PW, Richter S, et al. HIF2α supports pro-metastatic behavior in pheochromocytomas/paragangliomas. Endocr Relat Cancer (2020) 27:625–40. doi: 10.1530/ERC-20-0205
58. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001
59. Mihailovich M, Militti C, Gabaldón T, Gebauer F. Eukaryotic cold shock domain proteins: highly versatile regulators of gene expression. Bioessays (2010) 32:109–18. doi: 10.1002/bies.200900122
60. Dormoy-Raclet V, Markovits J, Malato Y, Huet S, Lagarde P, Montaudon D, et al. Unr, a cytoplasmic RNA-binding protein with cold-shock domains, is involved in control of apoptosis in ES and HuH7 cells. Oncogene (2007) 26:2595–605. doi: 10.1038/sj.onc.1210068
61. Alrezk R, Suarez A, Tena I, Pacak K. Update of pheochromocytoma syndromes: Genetics, biochemical evaluation, and imaging. Front Endocrinol (Lausanne) (2018) 9:515. doi: 10.3389/fendo.2018.00515
62. Fishbein L. Pheochromocytoma/Paraganglioma: Is this a genetic disorder? Curr Cardiol Rep (2019) 21:104. doi: 10.1007/s11886-019-1184-y
63. Castro-Teles J, Sousa-Pinto B, Rebelo S, Pignatelli D. Pheochromocytomas and paragangliomas in von hippel-lindau disease: not a needle in a haystack. Endocr Connect (2021) 10:R293–304. doi: 10.1530/EC-21-0294
64. Wang H, Cui J, Yang C, Rosenblum JS, Zhang Q, Song Q, et al. A transgenic mouse model of pacak(-)Zhuang syndrome with an Epas1 gain-of-Function mutation. Cancers (Basel) (2019) 11(5). doi: 10.3390/cancers11050667
65. Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, et al. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol (2013) 31:1690–8. doi: 10.1200/JCO.2012.47.1912
66. Castro-Vega LJ, Buffet A, De Cubas AA, Cascón A, Menara M, Khalifa E, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet (2014) 23:2440–6. doi: 10.1093/hmg/ddt639
67. Yang C, Zhuang Z, Fliedner SM, Shankavaram U, Sun MG, Bullova P, et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med (2015) 93:93–104. doi: 10.1007/s00109-014-1205-7
68. Chittiboina P, Lonser RR. Chapter 10 - Von hippel–lindau disease. In: Islam MP, Roach ES, editors. Handbook of clinical neurology. Elsevier (2015). p. 139–56.
69. Hong B, Ma K, Zhou J, Zhang J, Wang J, Liu S, et al. Frequent mutations of VHL gene and the clinical phenotypes in the largest Chinese cohort with Von hippel-lindau disease. Front Genet (2019) 10:867. doi: 10.3389/fgene.2019.00867
70. Liu Q, Yuan G, Tong D, Liu G, Yi Y, Zhang J, et al. Novel genotype-phenotype correlations in five Chinese families with Von hippel-lindau disease. Endocr Connect (2018) 7:870–8. doi: 10.1530/EC-18-0167
71. Hudler P, Urbancic M. The role of VHL in the development of von hippel-lindau disease and erythrocytosis. Genes (Basel) (2022) 13(2). doi: 10.3390/genes13020362
72. Chou A, Toon C, Pickett J, Gill AJ. Von hippel-lindau syndrome. Front Horm Res (2013) 41:30–49. doi: 10.1159/000345668
73. Delman KA, Shapiro SE, Jonasch EW, Lee JE, Curley SA, Evans DB, et al. Abdominal visceral lesions in von hippel-lindau disease: Incidence and clinical behavior of pancreatic and adrenal lesions at a single center. World J Surg (2006) 30:665–9. doi: 10.1007/s00268-005-0359-4
74. Maher ER, Neumann HPH, Richard S. Von hippel–lindau disease: A clinical and scientific review. Eur J Hum Genet (2011) 19:617–23. doi: 10.1038/ejhg.2010.175
75. Kaelin WG Jr. Von Hippel-lindau disease: insights into oxygen sensing, protein degradation, and cancer. J Clin Invest (2022) 132(18). doi: 10.1172/JCI162480
76. Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG Jr. Von hippel-lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet (2001) 10:1019–27. doi: 10.1093/hmg/10.10.1019
77. Coco D, Leanza S. Von Hippel-lindau syndrome: Medical syndrome or surgical syndrome? a surgical perspective. J Kidney Cancer VHL (2022) 9:27–32. doi: 10.15586/jkcvhl.v9i1.206
78. Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, et al. Disruption of oxygen homeostasis underlies congenital chuvash polycythemia. Nat Genet (2002) 32:614–21. doi: 10.1038/ng1019
79. Szabó PM, Pintér M, Szabó DR, Zsippai A, Patócs A, Falus A, et al. Integrative analysis of neuroblastoma and pheochromocytoma genomics data. BMC Med Genomics (2012) 5:48. doi: 10.1186/1755-8794-5-48
80. Jimenez C, Fazeli S, Roman-Gonzalez A. Antiangiogenic therapies for pheochromocytoma and paraganglioma. Endocr Relat Cancer (2020) 27:R239–54. doi: 10.1530/ERC-20-0043
81. Eisenhofer G, Lenders JWM, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem (2011) 57:411–20. doi: 10.1373/clinchem.2010.153320
82. Eisenhofer G, Peitzsch M, Bechmann N, Huebner A. Biochemical diagnosis of catecholamine-producing tumors of childhood: Neuroblastoma, pheochromocytoma and paraganglioma. Front Endocrinol (Lausanne) (2022) 13:901760. doi: 10.3389/fendo.2022.901760
83. Vogel TW, Brouwers FM, Lubensky IA, Vortmeyer AO, Weil RJ, Walther MM, et al. Differential expression of erythropoietin and its receptor in von hippel-lindau-associated and multiple endocrine neoplasia type 2-associated pheochromocytomas. J Clin Endocrinol Metab (2005) 90:3747–51. doi: 10.1210/jc.2004-1899
84. Qin N, de Cubas AA, Garcia-Martin R, Richter S, Peitzsch M, Menschikowski M, et al. Opposing effects of HIF1alpha and HIF2alpha on chromaffin cell phenotypic features and tumor cell proliferation: Insights from MYC-associated factor X. Int J Cancer (2014) 135:2054–64. doi: 10.1002/ijc.28868
85. Dong Y, Huang Y, Fan C, Wang L, Zhang R, Li W, et al. GIPC2 is an endocrine-specific tumor suppressor gene for both sporadic and hereditary tumors of RET- and SDHB-, but not VHL-associated clusters of pheochromocytoma/paraganglioma. Cell Death Dis (2021) 12:444. doi: 10.1038/s41419-021-03731-7
86. Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell (2005) 8:155–67. doi: 10.1016/j.ccr.2005.06.015
87. Sorrell AD, Lee S, Stolle C, Ellenhorn J, Grix A, Kaelin WG Jr., et al. Clinical and functional properties of novel VHL mutation (X214L) consistent with type 2A phenotype and low risk of renal cell carcinoma. Clin Genet (2011) 79:539–45. doi: 10.1111/j.1399-0004.2010.01464.x
88. Shankavaram U, Fliedner SM, Elkahloun AG, Barb JJ, Munson PJ, Huynh TT, et al. Genotype and tumor locus determine expression profile of pseudohypoxic pheochromocytomas and paragangliomas. Neoplasia (2013) 15:435–47. doi: 10.1593/neo.122132
89. van der Tuin K, Mensenkamp AR, Tops CMJ, Corssmit EPM, Dinjens WN, van de Horst-Schrivers AN, et al. Clinical aspects of SDHA-related pheochromocytoma and paraganglioma: A nationwide study. J Clin Endocrinol Metab (2017) 103:438–45. doi: 10.1210/jc.2017-01762
90. Moog S, Lussey-Lepoutre C, Favier J. Epigenetic and metabolic reprogramming of SDH-deficient paragangliomas. Endocr Relat Cancer 27 (2020) 27(12):R451–r463. doi: 10.1530/ERC-20-0346
91. Eniafe J, Jiang S. The functional roles of TCA cycle metabolites in cancer. Oncogene (2021) 40:3351–63. doi: 10.1038/s41388-020-01639-8
92. Morin A, Goncalves J, Moog S, Castro-Vega LJ, Job S, Buffet A, et al. TET-mediated hypermethylation primes SDH-deficient cells for HIF2α-driven mesenchymal transition. Cell Rep (2020) 30:4551–4566.e7. doi: 10.1016/j.celrep.2020.03.022
93. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Kerlan V, Plouin PF, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab (2002) 87:4771–4. doi: 10.1210/jc.2002-020525
94. Welander J, Söderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer (2011) 18:R253–76. doi: 10.1530/ERC-11-0170
95. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol (2017) 3:1204–12. doi: 10.1001/jamaoncol.2017.0223
96. Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. New Engl J Med (2008) 359:2685–92. doi: 10.1056/NEJMoa0806277
97. Ladroue C, Hoogewijs D, Gad S, Carcenac R, Storti F, Barrois M, et al. Distinct deregulation of the hypoxia inducible factor by PHD2 mutants identified in germline DNA of patients with polycythemia. Haematologica (2012) 97:9–14. doi: 10.3324/haematol.2011.044644
98. Provenzano A, Chetta M, De Filpo G, Cantini G, La Barbera A, Nesi G, et al. Novel germline PHD2 variant in a metastatic pheochromocytoma and chronic myeloid leukemia, but in the absence of polycythemia. Medicina (Kaunas) (2022) 58(8). doi: 10.3390/medicina58081113
99. Eckardt L, Prange-Barczynska M, Hodson EJ, Fielding JW, Cheng X, Lima J, et al. Developmental role of PHD2 in the pathogenesis of pseudohypoxic pheochromocytoma. Endocr Relat Cancer (2021) 28:757–72. doi: 10.1530/ERC-21-0211
100. Peng S, Zhang J, Tan X, Huang Y, Xu J, Silk N, et al. The VHL/HIF axis in the development and treatment of Pheochromocytoma/Paraganglioma. Front Endocrinol (Lausanne) (2020) 11:586857. doi: 10.3389/fendo.2020.586857
101. Majewska A, Budny B, Ziemnicka K, Ruchala M, Wierzbicka M. Head and neck paragangliomas-a genetic overview. Int J Mol Sci (2020) 41(3):1622–43. doi: 10.3390/ijms21207669
102. You L, Wu W, Wang X, Fang L, Adam V, Nepovimova E, et al. The role of hypoxia-inducible factor 1 in tumor immune evasion. Med Res Rev (2021) 41:1622–43. doi: 10.1002/med.21771
103. Lv C, Wang S, Lin L, Wang C, Zeng K, Meng Y, et al. USP14 maintains HIF1-α stabilization via its deubiquitination activity in hepatocellular carcinoma. Cell Death Dis (2021) 12:803. doi: 10.1038/s41419-021-04089-6
104. Gladek I, Ferdin J, Horvat S, Calin GA, Kunej T. HIF1A gene polymorphisms and human diseases: Graphical review of 97 association studies. Genes Chromosomes Cancer (2017) 56:439–52. doi: 10.1002/gcc.22449
105. Comino-Mendez I, de Cubas AA, Bernal C, Alvarez-Escola C, Sanchez-Malo C, Ramirez-Tortosa CL, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet (2013) 22:2169–76. doi: 10.1093/hmg/ddt069
106. Bechmann N, Poser I, Seifert V, Greunke C, Ullrich M, Qin N, et al. Impact of extrinsic and intrinsic hypoxia on catecholamine biosynthesis in absence or presence of Hif2alpha in pheochromocytoma cells. Cancers (Basel) (2019) 11(5). doi: 10.3390/cancers11050594
107. Schnell PO, Ignacak ML, Bauer AL, Striet JB, Paulding WR, Czyzyk-Krzeska MF. Regulation of tyrosine hydroxylase promoter activity by the von hippel-lindau tumor suppressor protein and hypoxia-inducible transcription factors. J Neurochem (2003) 85:483–91. doi: 10.1046/j.1471-4159.2003.01696.x
108. Brown ST, Kelly KF, Daniel JM, Nurse CA. Hypoxia inducible factor (HIF)-2α is required for the development of the catecholaminergic phenotype of sympathoadrenal cells. J Neurochem (2009) 110:622–30. doi: 10.1111/j.1471-4159.2009.06153.x
109. Watts D, Bechmann N, Meneses A, Poutakidou IK, Kaden D, Conrad C, et al. HIF2alpha regulates the synthesis and release of epinephrine in the adrenal medulla. J Mol Med (2021) 99:1655–66. doi: 10.1007/s00109-021-02121-y
110. Pietras A, Johnsson AS, Påhlman S. The HIF-2α-driven pseudo-hypoxic phenotype in tumor aggressiveness, differentiation, and vascularization. Curr Top Microbiol Immunol (2010) 345:1–20. doi: 10.1007/82_2010_72
111. Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, Nilsson H, et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell (2006) 10:413–23. doi: 10.1016/j.ccr.2006.08.026
112. Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med (2012) 367:922–30. doi: 10.1056/NEJMoa1205119
113. Favier J, Buffet A, Gimenez-Roqueplo AP. HIF2A mutations in paraganglioma with polycythemia. N Engl J Med (2012) 367:2161. doi: 10.1056/NEJMc1211953
114. Welander J, Andreasson A, Brauckhoff M, Bäckdahl M, Larsson C, Gimm O, et al. Frequent EPAS1/HIF2α exons 9 and 12 mutations in non-familial pheochromocytoma. Endocr Relat Cancer (2014) 21:495–504. doi: 10.1530/ERC-13-0384
115. Percy MJ, Beer PA, Campbell G, Dekker AW, Green AR, Oscier D, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood (2008) 111:5400–2. doi: 10.1182/blood-2008-02-137703
116. Percy MJ, Furlow PW, Lucas GS, Li X, Lappin TR, McMullin MF, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med (2008) 358:162–8. doi: 10.1056/NEJMoa073123
117. Liu Q, Tong D, Liu G, Yi Y, Zhang D, Zhang J, et al. HIF2A germline-mutation-induced polycythemia in a patient with VHL-associated renal-cell carcinoma. Cancer Biol Ther (2017) 18:944–7. doi: 10.1080/15384047.2017.1394553
118. Liu Q, Wang Y, Tong D, Liu G, Yuan W, Zhang J, et al. A somatic HIF2α mutation-induced multiple and recurrent Pheochromocytoma/Paraganglioma with polycythemia: Clinical study with literature review. Endocr Pathol (2017) 28:75–82. doi: 10.1007/s12022-017-9469-4
119. Lorenzo FR, Yang C, Ng Tang Fui M, Vankayalapati H, Zhuang Z, Huynh T, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (2013) 91:507–12. doi: 10.1007/s00109-012-0967-z
120. Buffet A, Smati S, Mansuy L, Menara M, Lebras M, Heymann MF, et al. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab (2014) 99:E369–73. doi: 10.1210/jc.2013-2600
121. Eisenhofer G, Huynh TT, Pacak K, Brouwers FM, Walther MM, Linehan WM, et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von hippel-lindau syndrome. Endocr Relat Cancer (2004) 11:897–911. doi: 10.1677/erc.1.00838
122. Puglisi S, Calabrese A, Basile V, Pia A, Reimondo G, Perotti P, et al. New perspectives for mitotane treatment of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab (2020) 34:101415. doi: 10.1016/j.beem.2020.101415
123. Rankin EB, Nam J-M, Giaccia AJ. Hypoxia: Signaling the metastatic cascade. Trends Cancer (2016) 2:295–304. doi: 10.1016/j.trecan.2016.05.006
124. Andrysik Z, Bender H, Galbraith MD, Espinosa JM. Multi-omics analysis reveals contextual tumor suppressive and oncogenic gene modules within the acute hypoxic response. Nat Commun (2021) 12:1375. doi: 10.1038/s41467-021-21687-2
125. Oka T, Onoe K, Nishimura K, Tsujimura A, Sugao H, Takaha M, et al. Erythropoietin-producing adrenocortical carcinoma. Urologia Internationalis (1996) 56:246–9. doi: 10.1159/000282852
126. Chen X, Yan L, Lu Y, Jiang F, Zeng N, Yang S, et al. And tumor immune microenvironment in adrenocortical carcinoma. J Oncol (2021) 2021:2298973. doi: 10.1155/2021/2298973
127. Zhang T, Song X, Qiao J, Zhu R, Ren Y, Shan PF. A novel predictive model for adrenocortical carcinoma based on hypoxia- and ferroptosis-related gene expression. Front Med (Lausanne) (2022) 9:856606. doi: 10.3389/fmed.2022.856606
128. Pitsava G, Faucz FR, Stratakis CA, Hannah-Shmouni F. Update on the genetics of primary aldosteronism and aldosterone-producing adenomas. Curr Cardiol Rep (2022) 24:1189–95. doi: 10.1007/s11886-022-01735-z
129. Rege J, Turcu AF, Rainey WE. Primary aldosteronism diagnostics: KCNJ5 mutations and hybrid steroid synthesis in aldosterone-producing adenomas. Gland Surg (2020) 9:3–13. doi: 10.21037/gs.2019.10.22
130. Donatini G, Van Slycke S, Aubert S, Carnaille B. Corticomedullary mixed tumor of the adrenal gland–a clinical and pathological chameleon: case report and review of literature. Updates Surg (2013) 65:161–4. doi: 10.1007/s13304-011-0132-1
131. Chiou HC, Jiang HJ, Yang SF, Kuo KK, Lin PC, Hsiao PJ. Stemness regulation of the adrenal mixed corticomedullary tumorigenesis-a case-control study. Neoplasia (2020) 22:263–71. doi: 10.1016/j.neo.2020.04.003
133. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncol 69(suppl (2005) 69(Suppl. 3):4–10. doi: 10.1159/000088478
134. Ceric S, Ceric T, Pojskic N, Bilalovic N, Musanovic J, Kucukalic-Selimovic E. Immunohistochemical expression and prognostic significance of VEGF-c in well-differentiated thyroid cancer. Acta Endocrinol (Buchar) (2020) 16:409–16. doi: 10.4183/aeb.2020.409
135. Wang N, Luo Hj Fau - Yin G-B, Yin Gb Fau - Dong C-R, Dong Cr Fau - Xu M, Xu M Fau - Chen GG, Chen Gg Fau - Liu Z-M, et al. Overexpression of HIF-2α, TWIST, and CXCR4 is associated with lymph node metastasis in papillary thyroid carcinoma. (2013) (1740–2530 (Electronic)).
136. Koperek O, Akin E, Asari R, Niederle B, Neuhold N. Expression of hypoxia-inducible factor 1 alpha in papillary thyroid carcinoma is associated with desmoplastic stromal reaction and lymph node metastasis. Virchows Archiv (2013) 463(6):795–802. doi: 10.1007/s00428-013-1484-3.
137. Liu YM, Ying SP, Huang YR, Pan Y, Chen WJ, Ni LQ, et al. Expression of HIF-1alpha and HIF-2alpha correlates to biological and clinical significance in papillary thyroid carcinoma. World J Surg Oncol (2016) 14:30. doi: 10.1186/s12957-016-0785-9
138. Burrows N, Resch J, Cowen RL, von Wasielewski R, Hoang-Vu C, West CM, et al. Expression of hypoxia-inducible factor 1 alpha in thyroid carcinomas. Endocr Relat Cancer (2010) 17:61–72. doi: 10.1677/ERC-08-0251
139. Burrows N, Babur M, Resch J, Williams KJ, Brabant G. Hypoxia-inducible factor in thyroid carcinoma. J Thyroid Res (2011) 2011:762905. doi: 10.4061/2011/762905
140. Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
141. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
142. Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo W, et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J Hematol Oncol (2021) 14:14. doi: 10.1186/s13045-020-01030-w
143. Geurts JL. Inherited syndromes involving pancreatic neuroendocrine tumors. J Gastrointest Oncol (2020) 11:559–66. doi: 10.21037/jgo.2020.03.09
144. Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and endocrine gland tumors: Noteworthy and (Not so) rare associations. Front Endocrinol (Lausanne) (2021) 12:678869. doi: 10.3389/fendo.2021.678869
145. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science (2008) 321:1801–6. doi: 10.1126/science.1164368
146. Xiang J, Hu Q, Qin Y, Ji S, Xu W, Liu W, et al. TCF7L2 positively regulates aerobic glycolysis via the EGLN2/HIF-1alpha axis and indicates prognosis in pancreatic cancer. Cell Death Dis (2018) 9:321. doi: 10.1038/s41419-018-0367-6
147. Zhang Q, Lou Y, Zhang J, Fu Q, Wei T, Sun X, et al. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with wnt/β-catenin signaling in pancreatic cancer. Mol Cancer (2017) 16:119. doi: 10.1186/s12943-017-0689-5
148. Shi S, Xu J, Zhang B, Ji S, Xu W, Liu J, et al. VEGF promotes glycolysis in pancreatic cancer via HIF1α up-regulation. Curr Mol Med (2016) 16:394–403. doi: 10.2174/1566524016666160316153623
149. Krokker L, Patócs A, Butz H. Essential role of the 14q32 encoded miRNAs in endocrine tumors. Genes (2021) 12(5). doi: 10.3390/genes12050698
150. de Cubas AA, Leandro-Garcia LJ, Schiavi F, Mancikova V, Comino-Mendez I, Inglada-Perez L, et al. Integrative analysis of miRNA and mRNA expression profiles in pheochromocytoma and paraganglioma identifies genotype-specific markers and potentially regulated pathways. Endocr Relat Cancer (2013) 20:477–93. doi: 10.1530/ERC-12-0183
151. Seok JK, Lee SH, Kim MJ, Lee YM. MicroRNA-382 induced by HIF-1α is an angiogenic miR targeting the tumor suppressor phosphatase and tensin homolog. Nucleic Acids Res (2014) 42:8062–72. doi: 10.1093/nar/gku515
152. Meyer-Rochow GY, Jackson NE, Conaglen JV, Whittle DE, Kunnimalaiyaan M, Chen H, et al. MicroRNA profiling of benign and malignant pheochromocytomas identifies novel diagnostic and therapeutic targets. Endocrine-Related Cancer (2010) 17:835–46. doi: 10.1677/ERC-10-0142
153. Xue G, Yan HL, Zhang Y, Hao LQ, Zhu XT, Mei Q, et al. C-myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2α and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene (2015) 34:1393–406. doi: 10.1038/onc.2014.82
154. Calsina B, Castro-Vega LJ, Torres-Perez R, Inglada-Perez L, Curras-Freixes M, Roldan-Romero JM, et al. Integrative multi-omics analysis identifies a prognostic miRNA signature and a targetable miR-21-3p/TSC2/mTOR axis in metastatic pheochromocytoma/paraganglioma. Theranostics (2019) 9:4946–58. doi: 10.7150/thno.35458
155. Zong L, Meng L, Shi R. Role of miR-101 in pheochromocytoma patients with SDHD mutation. Int J Clin Exp Pathol (2015) 8:1545–54.
156. Manso J, Bertazza L, Barollo S, Mondin A, Censi S, Carducci S, et al. Overexpression of miR-375 and l-type amino acid transporter 1 in pheochromocytoma and their molecular and functional implications. Int J Mol Sci (2022) 23(5). doi: 10.3390/ijms23052413
157. Censi S, Bertazza L, Piva I, Manso J, Benna C, Iacobone M, et al. Serum miR-375 for diagnostic and prognostic purposes in medullary thyroid carcinoma. Front Endocrinol (Lausanne) (2021) 12:647369. doi: 10.3389/fendo.2021.647369
158. Huang X, Le QT, Giaccia AJ. Mir-210–Micromanager of the Hypoxia Pathway. Trends Mol Med (2010) 16(5):230–7. doi: 10.1016/j.molmed.2010.03.004.
159. Tsang VH, Dwight T, Benn DE, Meyer-Rochow GY, Gill AJ, Sywak M, et al. Overexpression of miR-210 is associated with SDH-related pheochromocytomas, paragangliomas, and gastrointestinal stromal tumours. Endocr Relat Cancer (2014) 21:415–26. doi: 10.1530/ERC-13-0519
160. Merlo A, de Quiros SB, Secades P, Zambrano I, Balbín M, Astudillo A, et al. Identification of a signaling axis HIF-1α/microRNA-210/ISCU independent of SDH mutation that defines a subgroup of head and neck paragangliomas. J Clin Endocrinol Metab (2012) 97:E2194–200. doi: 10.1210/jc.2012-2410
161. Turai PI, Nyiro G, Butz H, Patocs A, Igaz P. MicroRNAs, long non-coding RNAs, and circular RNAs: Potential biomarkers and therapeutic targets in Pheochromocytoma/Paraganglioma. Cancers (Basel) (2021) 13(7). doi: 10.3390/cancers13071522
162. Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer Metastasis Rev (2007) 26:341–52. doi: 10.1007/s10555-007-9059-x
163. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer (2011) 11:393–410. doi: 10.1038/nrc3064
164. Baran N, Konopleva M. Molecular pathways: Hypoxia-activated prodrugs in cancer therapy. Clin Cancer Res (2017) 23:2382–90. doi: 10.1158/1078-0432.CCR-16-0895
165. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33:207–14. doi: 10.1016/j.tips.2012.01.005
166. Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, et al. A small-molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res (2016) 76:5491–500. doi: 10.1158/0008-5472.CAN-16-0473
167. Erbel PJ, Card PB, Karakuzu O, Bruick RK, Gardner KH. Structural basis for PAS domain heterodimerization in the basic helix–loop–helix-PAS transcription factor hypoxia-inducible factor. Proc Natl Acad Sci U.S.A. (2003) 100:15504–9. doi: 10.1073/pnas.2533374100
168. Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature (2016) 539:107–11. doi: 10.1038/nature19795
169. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) 539:112–7. doi: 10.1038/nature19796
170. Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I dose-escalation trial of PT2385, a first-in-Class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol (2018) 36:867–74. doi: 10.1200/JCO.2017.74.2627
171. Toledo RA. New HIF2α inhibitors: potential implications as therapeutics for advanced pheochromocytomas and paragangliomas. Endocr Relat Cancer (2017) 24:C9–c19. doi: 10.1530/ERC-16-0479
172. Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell renal cell carcinoma. J Medicinal Chem (2019) 62:6876–93. doi: 10.1021/acs.jmedchem.9b00719
173. Favier J, Brière J-J, Burnichon N, Rivière J, Vescovo L, Benit P, et al. The warburg effect is genetically determined in inherited pheochromocytomas. PLos One (2009) 4:e7094. doi: 10.1371/journal.pone.0007094
Keywords: hypoxia, endocrinology, tumor, pheochromocytoma, therapy
Citation: Watts D, Jaykar MT, Bechmann N and Wielockx B (2023) Hypoxia signaling pathway: A central mediator in endocrine tumors. Front. Endocrinol. 13:1103075. doi: 10.3389/fendo.2022.1103075
Received: 19 November 2022; Accepted: 21 December 2022;
Published: 09 January 2023.
Edited by:
Ralf Jockers, Université de Paris, FranceReviewed by:
Leonardo Rossi, University of Pisa, ItalyKarim Abid, Centre Hospitalier Universitaire Vaudois (CHUV), Switzerland
Copyright © 2023 Watts, Jaykar, Bechmann and Wielockx. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Wielockx, QmVuLldpZWxvY2t4QHR1LWRyZXNkZW4uZGU=
†These authors have contributed equally to this work