
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 15 November 2022
Sec. Reproduction
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.1017468
This article is part of the Research Topic Reviews in Receptor-Receptor Interactions as Novel Targets for Drug Development View all 6 articles
 Rebecca H. K. Emanuel1
Rebecca H. K. Emanuel1 Josh Roberts1
Josh Roberts1 Paul D. Docherty1,2*
Paul D. Docherty1,2* Helen Lunt3,4
Helen Lunt3,4 Rebecca E. Campbell5
Rebecca E. Campbell5 Knut Möller2
Knut Möller2Polycystic ovary syndrome (PCOS) affects up to 20% of women but remains poorly understood. It is a heterogeneous condition with many potential comorbidities. This review offers an overview of the dysregulation of the reproductive and metabolic systems associated with PCOS. Review of the literature informed the development of a comprehensive summarizing ‘wiring’ diagram of PCOS-related features. This review provides a justification for each diagram aspect from the relevant academic literature, and explores the interactions between the hypothalamus, ovarian follicles, adipose tissue, reproductive hormones and other organ systems. The diagram will provide an efficient and useful tool for those researching and treating PCOS to understand the current state of knowledge on the complexity and variability of PCOS.
Polycystic ovary syndrome (PCOS) is a heterogeneous condition that is reported to affect between 8% to 20% of women (1, 2). Currently, PCOS is diagnosed with the Rotterdam diagnostic criteria (3). To diagnose PCOS with the Rotterdam criteria, two of the following criteria must be observed: evidence of clinical and/or biochemical hyperandrogenism, evidence of oligo-ovulation and/or anovulation, or evidence of polycystic ovarian morphology through ultrasound (1). PCOS has a number of significant comorbidities associated with it, including type 2 diabetes (T2DM) (4, 5), cardiovascular disease (6), insulin resistance (7), obesity (8), infertility (6), pregnancy complications (9, 10), sleep disturbances (11), hypothyroidism (12), decreased mental health (13), and non-alcoholic fatty liver disease (NAFLD) (14).
PCOS is a common metabolic and reproductive syndrome with heterogenous clinical presentations (‘syndromes’) that have no simple, single diagnostic or clinical management pathway. In addition there are multiple, poorly understood etiological factors that contribute to the heterogenous clinical expression of PCOS and these include genetic, environmental and hormonal factors. This paper focuses on a review of hormonal factors for the following reasons: There is an extensive body of research in this area but previous research has tended to focus on one hormonal sub-system, rather than consider the detailed interactions present in the hormonal system as a whole. Having a better understanding of the interactions between various parts of these hormonal sub-systems may ultimately lead to better interpretation of diagnostic tests and also help with the development therapeutics that allow for individualized treatment plans. The aim of this paper is to describe each hormonal element of PCOS in detail, then unify these descriptions into a single codified ‘wiring’ diagram. The unifying diagram is presented at the end of this paper, but a simplified version is shown in Figure 1. This simplified version will be used as a basis to construct a more complex representation of the hormonal pathways affected by PCOS upon.
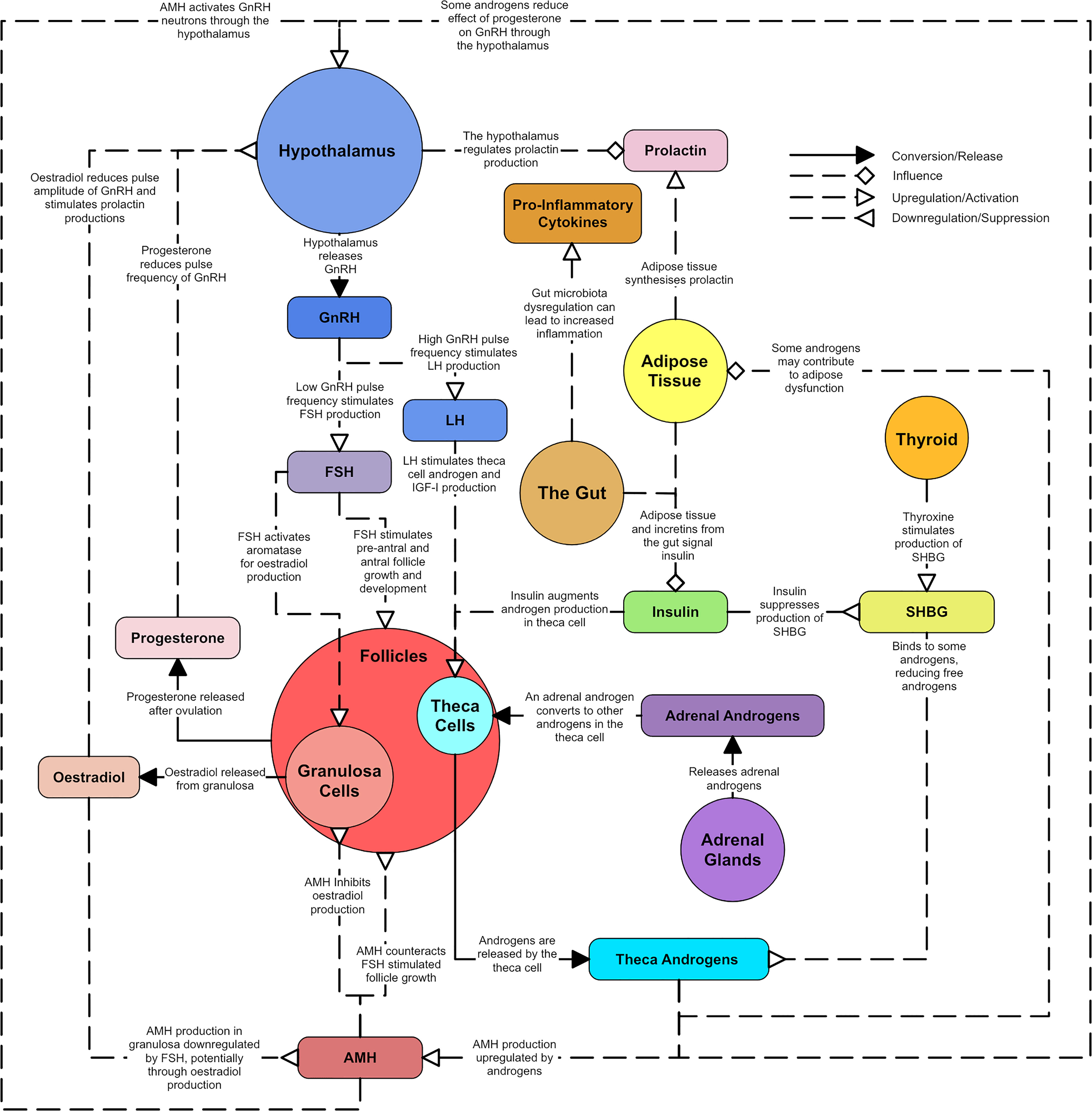
Figure 1 A simplified overview of the master diagram showing the key relationships and dysregulations of endocrine components in PCOS. Solid line arrows between features are used to indicated conversion or release. Dashed lines with diamond arrowhead connections are used to indicate influence. Dashed lines with arrowheads are used to indicate upregulation and dashed lines with reverse arrowheads are used to represent downregulation.
Original research articles were located through topic searches for the relationships between the hormones or biological regions known to be altered in PCOS (Figure 1). Clinical research involving humans was prioritized over pre-clinical research using animal models, with animal studies serving as support for human based evidence or suggestions of mechanisms that may underpin the evidence identified in humans. An exhaustive search was not possible due to the volume of research and complexity of the subject. However, effort was made to explore each primary facet of PCOS with evidence from several sources. The ‘wiring’ diagram outcome of this review is intended to capture the major endocrine dysfunctions in PCOS. Specific mechanistic hypotheses are noted where relevant but full coverage of endocrine mechanisms is outside the scope of this review.
Hyperandrogenism is a key component of PCOS. Approximately 60-80% of women with PCOS present with biochemical evidence of hyperandrogenism using androgen plasma levels and 60% present with clinical evidence of hyperandrogenism such as hirsutism, acne and androgenic alopecia (15, 16). The androgens in women considered to be present in the highest concentrations, in order of most to least concentrated, are dehydroepiandrosterone-sulphate (DHEA-S), dehydroepiandrosterone (DHEA), androstenedione (A4), testosterone and dihydrotestosterone (DHT) (17). DHEA is considered a “weak” androgen that converts to the more potent testosterone and DHT (17). DHEA can be sulphated into DHEA-S, another “weak” androgen with a longer half-life (17). Androgen production in the ovaries is regulated by upstream signals from the hypothalamic region of the brain. Neurons in the hypothalamus secrete pulsatile gonadotropin releasing hormone (GnRH) into the portal vasculature of the pituitary gland (18). GnRH is responsible for stimulating luteinizing hormone (LH) and follicle stimulating hormone (FSH) production and release (17). LH secretion mirrors the pulsatile release of GnRH and stimulates ovarian theca cells to synthesize A4 and testosterone from cholesterol (17). In the ovarian granulosa cell, A4 and testosterone are converted to estrogens through aromatization (17). DHT cannot be converted to estrogen, as it is said to be non-aromatizable, and is the most potent androgen isoform (17). In addition to the ovaries, androgens are synthesized in the adrenal glands, which mainly produce DHEA and A4 (17). About 10-25% of testosterone is produced in the adrenal cortex but this is regarded by some as a negligible contribution to the total concentration of testosterone in a woman (19). Figure 2 shows a diagram of the primary androgens and their significant relationships.
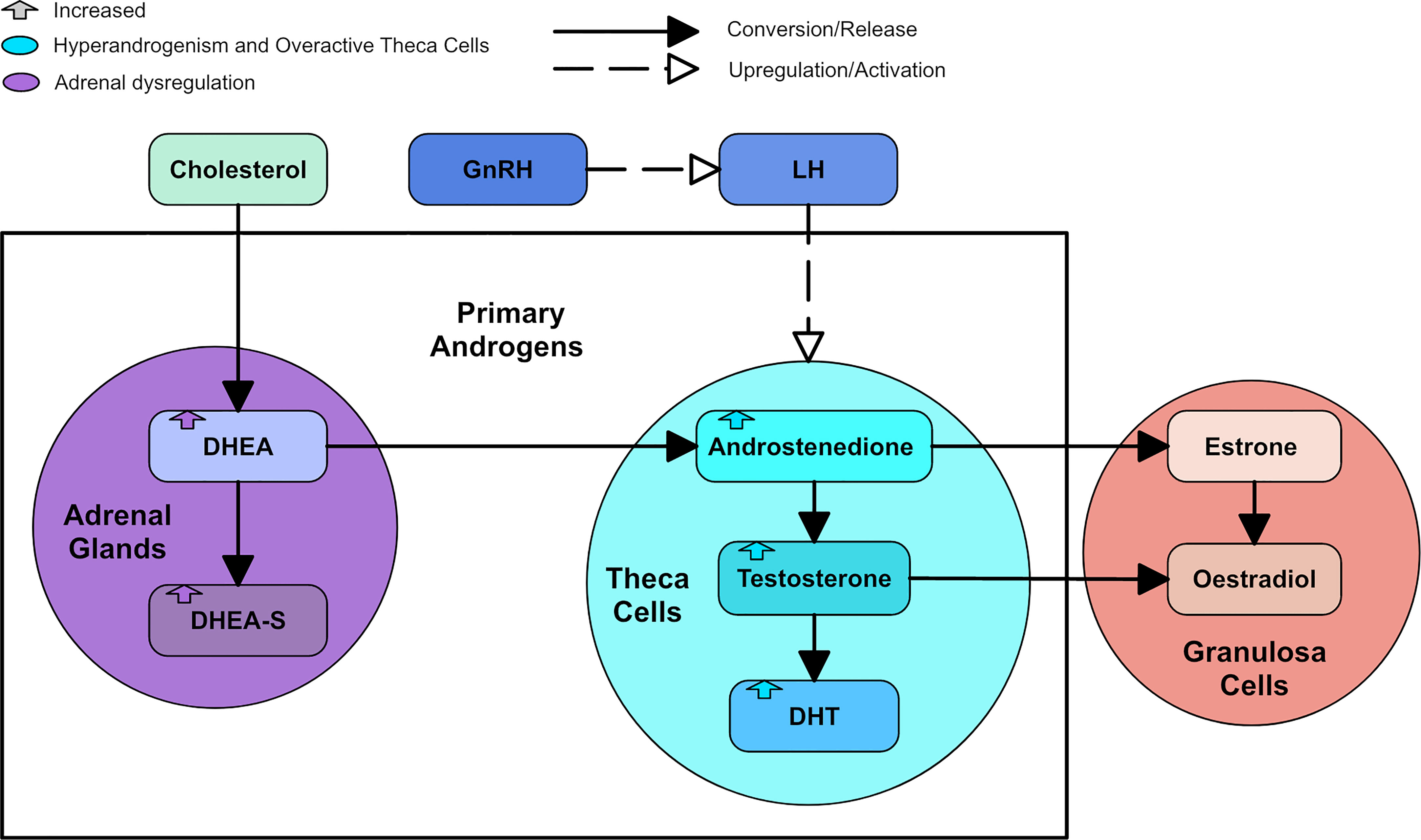
Figure 2 A diagram of the primary androgens, with colored arrows to represent how the androgens are changed by types of dysregulation.
Androgen excess (hyperandrogenism) is a key feature in PCOS and detrimentally affects ovarian function. Androgens are thought to stimulate pre-antral and small antral follicle growth through the androgen receptor (20). In women with PCOS, the androgen receptor may experience increased activity in the hypothalamus, ovary, skeletal muscle or adipose cells (21). High androgens in PCOS at least partially contribute to an increase in GnRH/LH pulse frequency (1, 22) and vice versa, generating a cycle of hormonal dysregulation (23). Functional ovarian hyperandrogenism can be directly or indirectly identified in the vast majority of PCOS patients (1). Functional adrenal hyperandrogenism is also present in a portion of PCOS patients, with a small percentage of this group not presenting with functional ovarian hyperandrogenism (1). A minor percentage of mostly obese PCOS patients present with neither functional ovarian nor functional adrenal hyperandrogenism. This small subgroup may present an endocrine state that appears to be a phenotype of PCOS but has an etiology related to obesity (excess adipose tissue) (1, 24).
Hyperandrogenism may also influence endometrial function (10). People with PCOS have an altered endometrium (25). This altered endometrium, along with other abnormalities commonly associated with PCOS, may lead to endometrial dysfunction, which can lead to pregnancy complications (25). Reduced reproductive potential in PCOS is often associated with anovulation and oligo-ovulation, but endometrial dysfunction may also contribute (26).
PCOS theca cells appear to have a gene expression profile that is distinct from normal theca cells (27). There is strong evidence that PCOS theca cells have an intrinsic abnormality that causes an overexpression of most steroidogenic enzymes, including LH receptors (1, 28–30). This overexpression leads to the increased androgen production that is ubiquitous with PCOS theca cells (31). Additionally, this increased androgen production appears independent to ovulation status (32).
A follicle in the ovary is composed of the oocyte, granulosa cells, and theca cells (33). Follicles start as primordial follicles which contain an oocyte surrounded by a single layer of granulosa cells and a basal lamina (34). Hyperandrogenism may have a negative effect on oocyte development and quality through the increase of reactive oxygen species levels (26). However, oocyte competence and quality appear equivalent in people with and without PCOS, so whether the oocyte contributes to decreased reproductive potential in PCOS is unclear (26). There are no theca cells until 2-3 layers of granulosa cells are acquired (34). There are large antral (later stage) and small pre-antral (earlier stage) follicles in the ovary. The density of pre-antral and small antral follicles in a person with Polycystic ovarian morphology (PCOM), one of the three diagnostic criteria of PCOS, has been shown to be six times greater than in a normal ovary (35).
While PCOM may occur in PCOS, PCOM can also occur in women who do not fully meet the diagnostic criteria for PCOS. In such cases, PCOM generally has a granulosa cell abnormality that is similar to PCOS but not as severe (36). Normal ovarian morphology and PCOS seem to exist on a spectrum with PCOM sitting between them (1). PCOM groups have been shown to have LH levels similar to non-PCOS and non-PCOM controls, but their anti-Mullerian hormone (AMH) levels were between that of controls and PCOS patients (36). Approximately half the women with PCOM have subclinical evidence of PCOS-related dysregulation whereas the other half have no apparent relation to PCOS, but most women with PCOM appear to be ovulatory (1). Figure 3 shows a simple diagram of follicle growth.
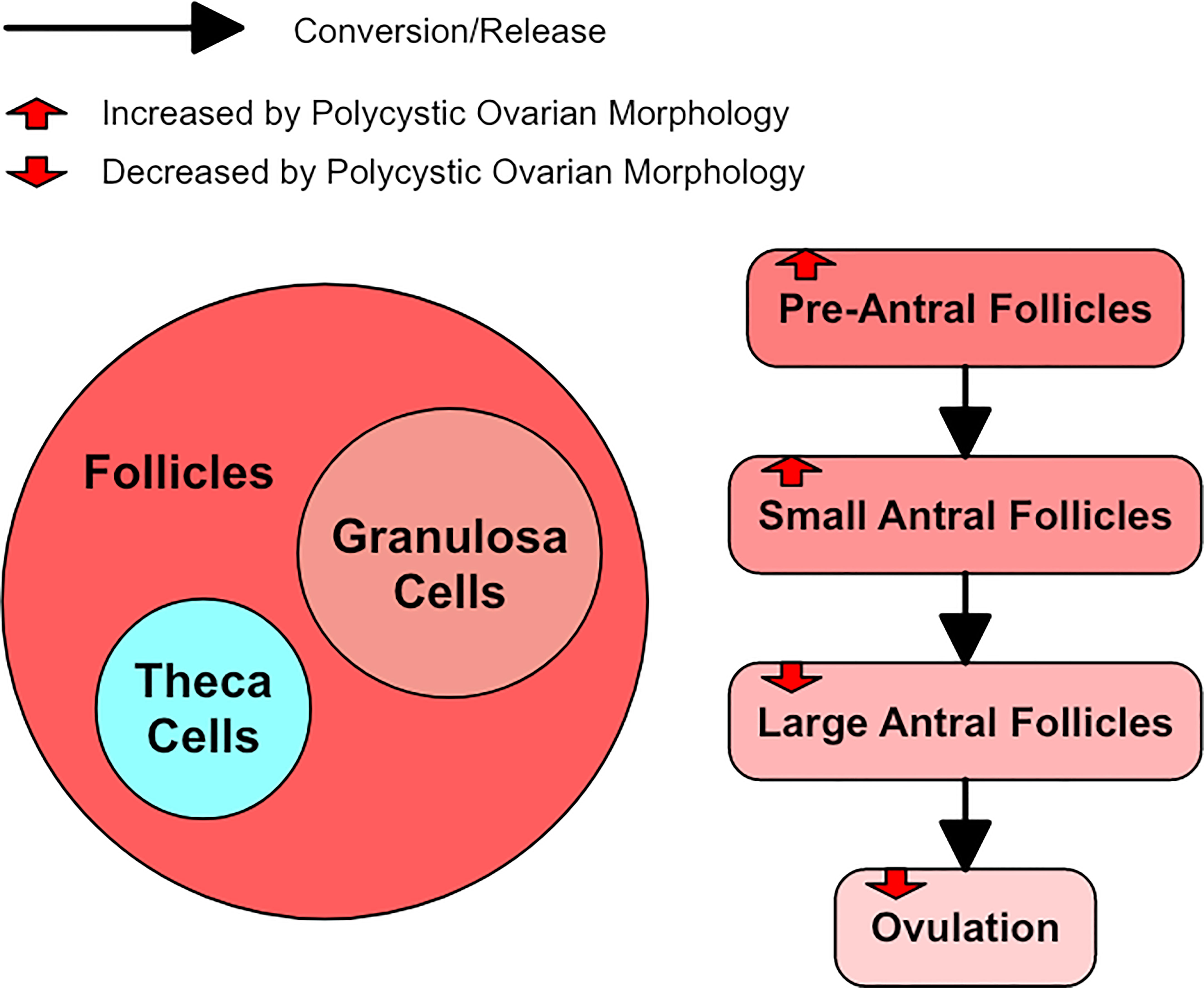
Figure 3 A diagram of follicle growth, with red arrows to represent how polycystic ovarian morphology alters it.
Serum LH concentrations have been found to be significantly increased in women with PCOS (37, 38). In contrast, FSH concentrations have been significantly lower in women with PCOS (39). Consequently, elevated LH/FSH ratios are generally reported (37, 38, 40). One study of 192 women showed that LH and FSH appear lower in women with PCOS who are also overweight or obese when compared to women with PCOS that were in the healthy BMI range (41). Among women in the healthy BMI range, those with PCOS had significantly higher FSH concentrations than controls (41). However, the women in the overweight or obese BMI categories had significantly lower FSH concentrations than controls (41).
Increased LH secretion is thought to have several consequences. LH stimulation of thecal cells in the ovary drives the synthesis of testosterone. Thus, the androgen synthesis in ovarian thecal cells is promoted, and can lead to hyperandrogenism. Elevated serum LH concentration is closely associated with a reduced chance of conception and an increased risk of miscarriage (42). However, it is unclear how substantially excessive LH in PCOS contributes to ovarian dysfunction (23).
The A4 and testosterone formed by the thecal cells in response to LH stimulation is converted in the granulosa cell to estradiol, but the activation of the enzyme aromatase necessary for this estradiol synthesis is dependent on FSH (43). FSH also promotes pre-antral follicle growth in synergy with theca cell-derived androgen. In PCOS, both these functions become dysregulated (44). Serum concentration and follicular fluid concentrations of FSH are generally lower in PCOS, with some studies reporting significantly decreased levels (45), and others reporting lower levels that are still within normal limits (46). However, these lower FSH levels do not seem to be enough to account for the disturbance of antral follicle growth and estradiol synthesis (47). Figure 4 shows the consequences of dysregulated LH and FSH.
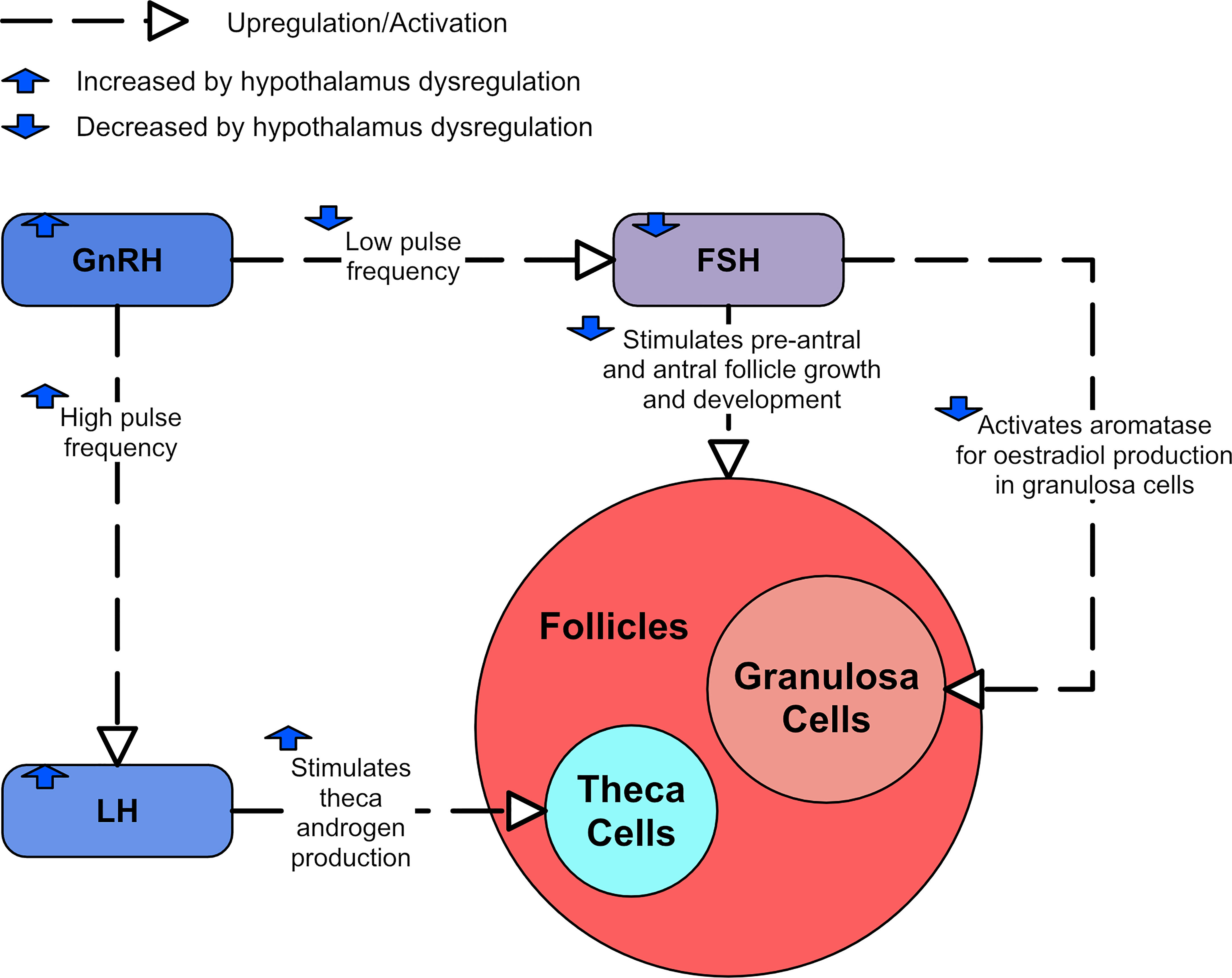
Figure 4 A diagram showing the effects of LH and FSH dysregulation in PCOS.
LH release is a result of pulsatile GnRH transfer from the hypothalamus to the pituitary (48). GnRH neurons in the hypothalamus are controlled by gonadal steroid feedback and release patterned pulsatile GnRH peptide to maintain pituitary function. A high pulse frequency of GnRH secretion favors release of LH while a low frequency of GnRH secretion favors greater FSH release. Thus, increased GnRH pulse frequency promotes LH synthesis over FSH synthesis (38, 49). Increasing levels of estradiol produced during the majority of the follicular phase cause a switch to positive feedback, triggering ovulation with an increase in LH (50). Ovulation triggers the increase in progesterone during the luteal phase, and low levels of progesterone are used as a clinical indicator that ovulation has not occurred (51). During the luteal phase, progesterone and estradiol produced by the corpus luteum signal the lowering of the GnRH pulses via negative feedback (50, 52) and by increased progesterone levels in the presence of estradiol (53). Evidence in the ewe suggests that estradiol alone inhibits GnRH and LH pulse amplitude, whereas progesterone alone inhibits GnRH and LH pulse frequency (54). Androgens have also been shown to contribute to increasing GnRH pulse frequency (55).
In PCOS, the sensitivity of the GnRH pulse generator to progesterone suppression is impaired (56, 57). Thus, ovarian steroid negative feedback that tightly regulates the Hypothalamic-Pituitary-Gonadal (HPG) axis is impaired (58), leading to hyperactive GnRH/LH secretion and disruptions to the neuroendocrine regulation of the reproductive system. Mechanistic studies in rodents demonstrate that DHT impairs the ability of progesterone to reduce the firing frequency of GnRH neurons from the HPG axis (59).
The mechanisms underpinning increased GnRH activity are not completely understood but have been explored in animal models. Figure 5 displays a summary of the likely hypothalamic dysregulation. One potential contributor is the GABAergic network. γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the brain, binds to two primary receptors: the GABAA receptor, and the GABAB receptor (60). While GABAergic signaling is predominantly inhibitory in the adult brain, GABA has a predominantly stimulatory effect on GnRH neurons through the GABAA receptor (50, 60), but an inhibitory effect through the GABAB receptor can also be observed (60). Increased GABA signaling to GnRH neurons has been identified in preclinical models of PCOS (61, 62) and cerebrospinal fluid GABA levels are higher in women with PCOS (63). There is a strong suggestion that this increased GABA signaling plays a role in driving the hyperactive GnRH release associated with PCOS as the GABAergic system is also thought to play a role in progesterone negative feedback (59). Hyperandrogenic mouse models of PCOS were shown to have significantly less progesterone receptors in a population of GABA neurons and exhibited impaired progesterone negative feedback (64, 65). This decrease in progesterone receptors likely contributes to the impaired progesterone inhibition of GnRH secretion found in some women with PCOS (66). Flutamide, an androgen receptor blocker, restores normal GABA innervation and neurotransmission in hyperandrogenic mice models (61, 62) and sensitivity to progesterone in women with PCOS (67). Mice models have also been used to demonstrate that DHT may increase the pulse frequency via the GABAergic system (55).
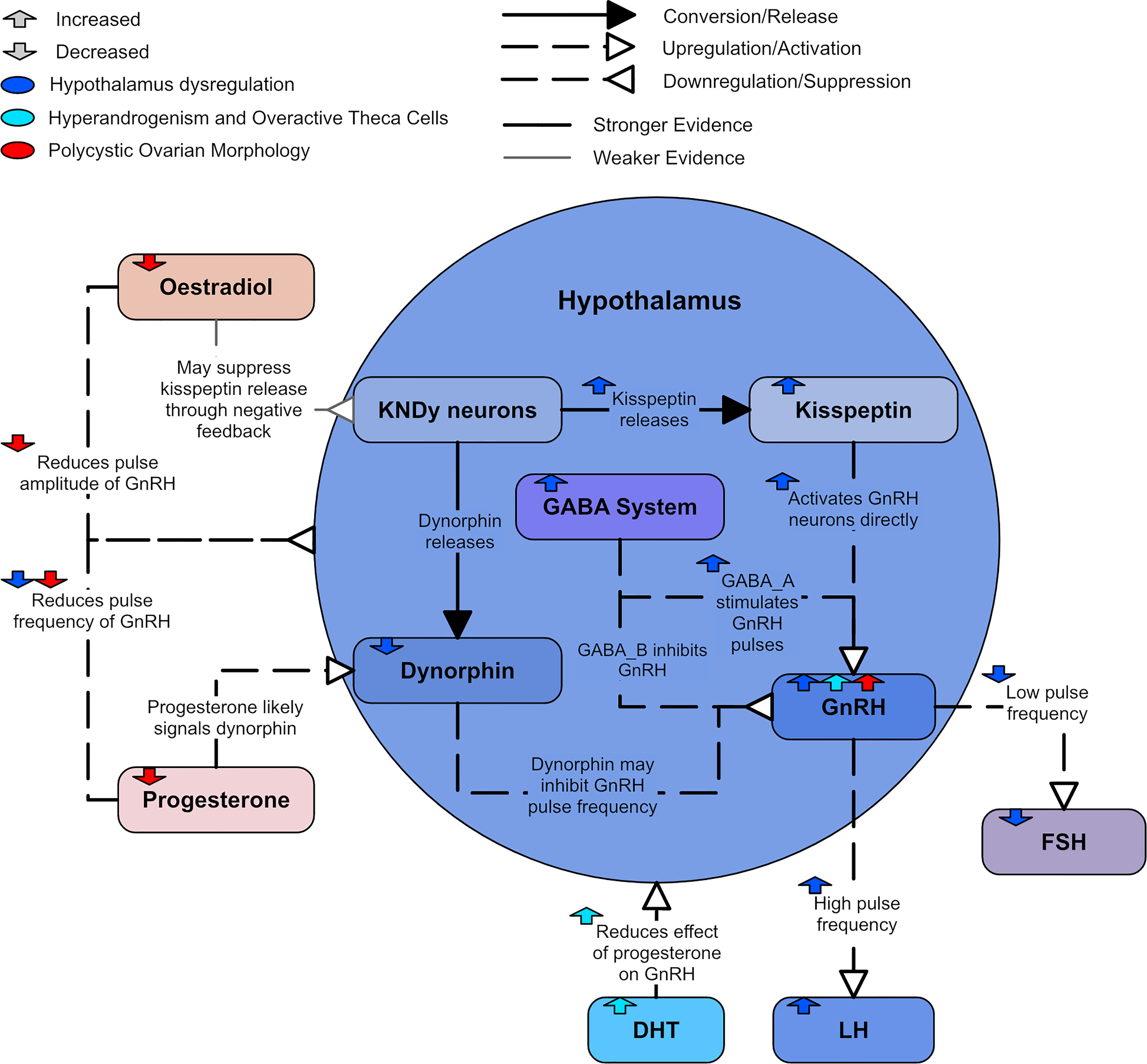
Figure 5 A diagram of hypothalamic dysregulation in PCOS.
Kisspeptin, neurokinin B, and dynorphin are three distinct neuropeptides that are found co-localize in a single subpopulation in the hypothalamus of several mammalian species. They are referred to as kisspeptin-/neurokinin B-/dynorphin-expressing or KNDy neurons. There is strong evidence that KNDy neurons have an important role in mediating the negative feedback of GnRH secretion by ovarian steroids (22, 68). It is possible that estradiol inhibits GnRH pulse amplitude by suppressing kisspeptin release from KNDy neurons (69). The kisspeptin neuropeptides that release from KNDy neurons have been identified as a primary GnRH pulse generator (50).
There is evidence that kisspeptin may contribute to GnRH neuron hyperactivity (22). Kisspeptin potently and directly activates GnRH neurons and drives GnRH/LH secretion (70, 71). Mice models have suggested that the presence of estradiol leads to kisspeptin indirectly activating GnRH neurons as well (70). It has been found that women with PCOS had increased kisspeptin levels, suggesting that kisspeptins play an important role in regulating LH levels (72, 73). It is unclear if this elevated kisspeptin is related to increased kisspeptin signaling in the brain. Animal models suggest not all PCOS phenotypes present with high kisspeptin levels (73). However, the models imply kisspeptin is elevated in PCOS with higher LH levels and normal body weight (73).
There is also considerable evidence that dynorphin is an important mediator of the inhibitory feedback control of progesterone on GnRH secretion (54). Evidence in the ewe shows a very high percentage of dynorphin neurons contain progesterone receptors; as such, dynorphin is likely to inhibit GnRH pulse frequency (74). Dynorphin expression has been found to be reduced in ewes prenatally exposed with androgen (75), which is in agreement with the theory that PCOS may be caused by prenatal exposure to excess androgens (76, 77).
The formation of pre-antral and small antral follicles is thought to be accelerated by the hyperandrogenism present in PCOS (78). Granulosa cells are considered the only source of AMH in the ovary (79). Mice models have shown that AMH is released throughout the development of a primordial follicle into a small antral follicle but AMH release wanes as the follicle develops into a pre-ovulatory stage (78). As there are often more pre-antral and small follicles in PCOS ovaries, more AMH is generally produced than in normal ovaries (47). Furthermore, each individual follicle in PCOS has been shown to produce more AMH than normal (47). However, this increased AMH may not be solely due to PCOM. In particular, serum AMH has been positively correlated to high androgen levels, and women with high androgens and PCOM seemed to have the highest AMH levels of women with PCOS (79). Therefore, high androgens may also contribute to increased AMH levels.
FSH promotes the development of small antral follicles through to an ovulatory stage (47). AMH has been shown to both inhibit FSH-induced aromatase activity and counteract FSH growth-promoting effects on granulosa cells (44, 80), consequently deterring estradiol production (79). In PCOS, antral follicle growth can be disturbed by high AMH levels inhibiting FSH effects (80). Thus, FSH-stimulated pre-antral follicle growth is attenuated (81). This suggests that increased AMH levels likely play a role in the causation of anovulation in PCOS (47). AMH has also been shown to contribute to GnRH hyperactivity in animal models, as it can directly activate GnRH neurons (82). As there is a negative correlation between estradiol and AMH in small antral follicles, AMH is thought to be down-regulated by FSH, potentially via estradiol synthesis (83).
Serum AMH concentrations are higher in PCOS patients and there is evidence to suggest that more severe PCOS phenotypes display higher AMH levels (46, 84, 85). One study of 104 women found that PCOS patients with amenorrhea had higher AMH serum concentrations than those with oligomenorrhoea (86). Similarly, another study of 215 women found that oligo/anovulatory women with PCOS had higher AMH levels than ovulatory women with PCOM (36). Figure 6 shows how high AMH relates to the dysregulation in PCOS.
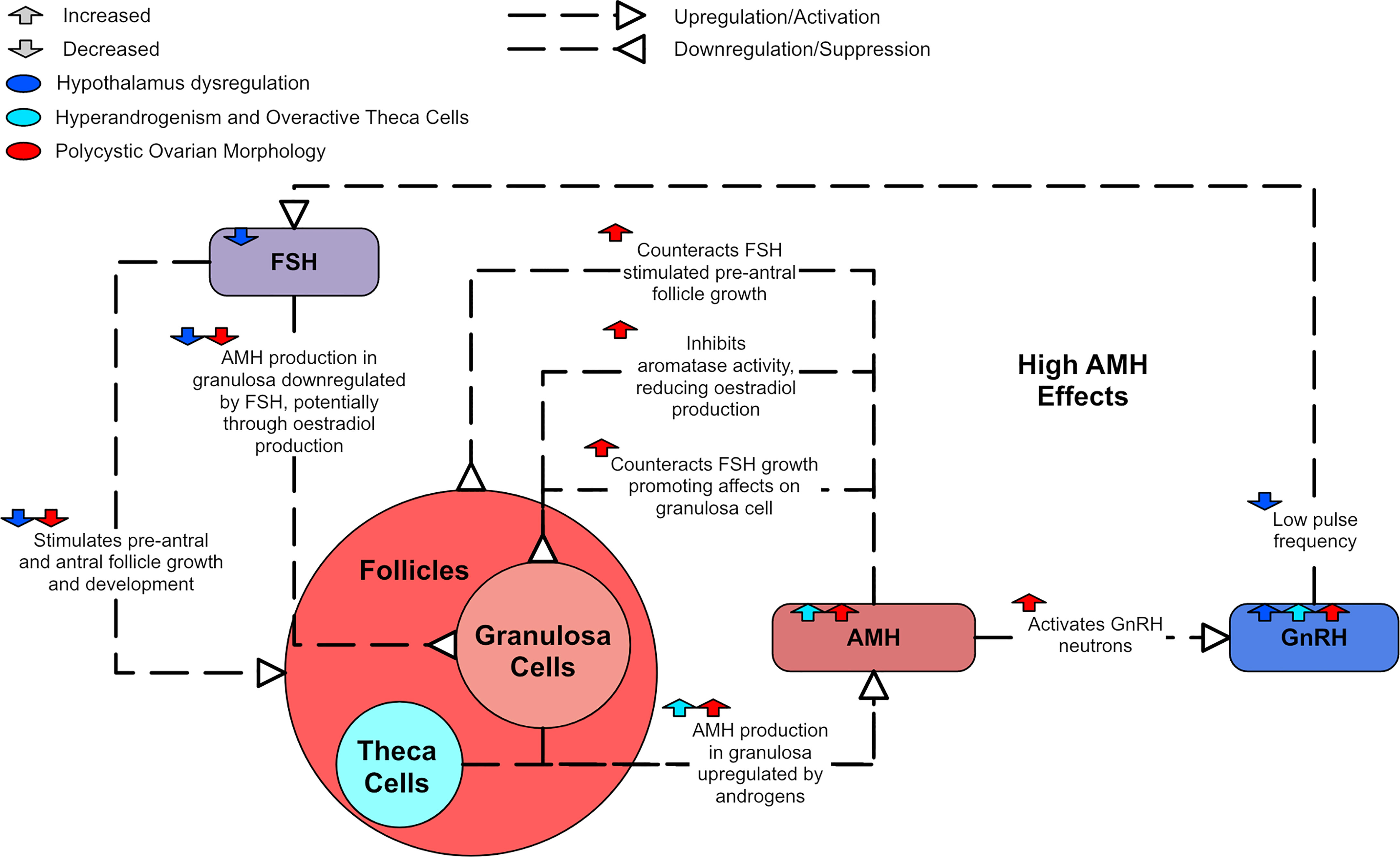
Figure 6 A diagram of the effects of AMH in PCOS.
In humans, almost all A4 is produced from DHEA (19), which is produced in the adrenal gland. It is commonly believed that DHEA and DHEA-S can freely convert between each other, with DHEA-S acting as a reservoir for DHEA due to its long half-life (17). However, this belief has been challenged by a study finding no evidence of a rise in DHEA-S upon DHEA administration in men (87). The study also noted that although pregnant women have been shown to convert DHEA-S to DHEA, they were unable to find direct evidence of continuous conversion between DHEA and DHEA-S in women in the literature (87).
Since DHEA-S is almost solely produced by the adrenal cortices, increased DHEA-S is often used as an indicator for overactive adrenal cortex production of androgens. Approximately 50-60% of women with PCOS exhibit adrenal originating androgen excess by increased DHEA-S (88). Currently, it is thought the ovary has a limited to negligible effect on the adrenocortical function (89). DHEA-S has been found to positively correlate with total testosterone, A4, and free androgen index (FAI) (90), as well as 17-hydroxyprogesterone (91). DHEA-S has also been found to decline with age (89, 92).
Insulin and insulin resistance will be discussed directly in section 3. However, there are complex links between DHEA-S and insulin that will be discussed in this section. Some studies negatively correlate DHEA-S and homeostasis model assessment-estimated insulin resistance (HOMA-IR) (89, 91). Other studies report no association (90), or a positive correlation between higher adrenal precursor androgen levels and insulin resistance (89). However, the HOMA-IR metric is imprecise and can fail to identify insulin resistance in woman with PCOS when compared to the gold standard of insulin resistance detection (93). There may also be ethnic differences, with one study finding that those they classified as white patients displayed a negative correlation between DHEA-S and HOMA-IR while those they classified as black patients did not (89). However, the number of patients in the ‘black’ cohort was too low to allow strong conclusions.
There is evidence to suggest communication between the adrenals and the hypothalamus. DHEA-S levels have been observed to decrease after administration of a GnRH antagonist in women with PCOS and high DHEA-S (94). Sullivan and Moenter reported that DHEA-S decreases GnRH neuron excitation through modulating the GABAA receptors in mice (95). It was also found that asymptomatic ovulatory women with PCOS had the highest DHEA-S levels when compared to other women with PCOS and those without PCOS (1). Figure 7 shows a diagram of the effects of an overactive adrenal cortex.
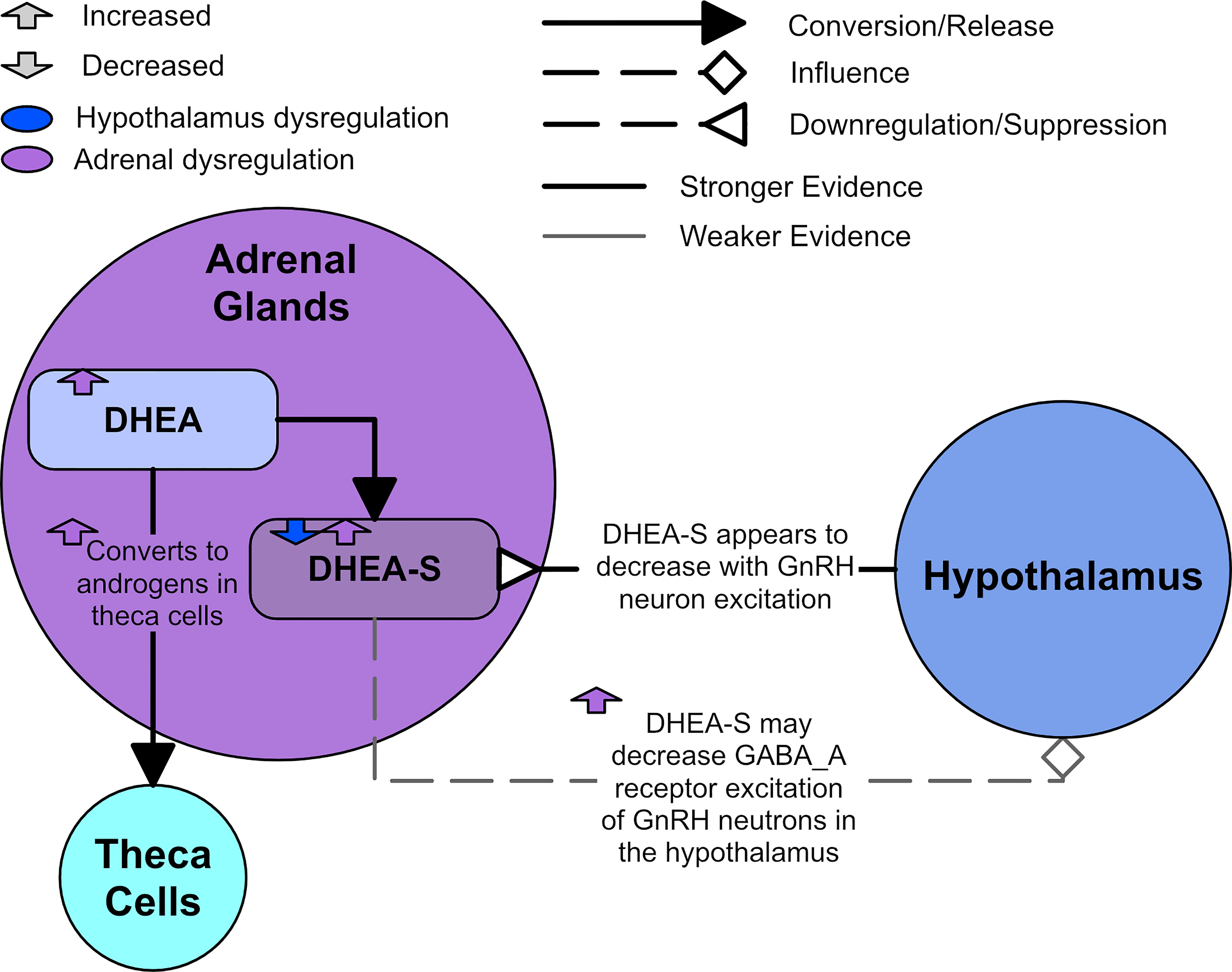
Figure 7 Adrenal dysregulation in PCOS.
PCOS is strongly associated with insulin resistance – around 75% of women diagnosed with PCOS also have impaired insulin sensitivity (93). Furthermore, lean women with PCOS have equivalent peripheral insulin resistance to obese women with PCOS (96). Thus, women with PCOS are vulnerable to developing metabolic syndrome and its associated dysfunction, which include hyperglycemia, central obesity, hypertension, and dyslipidemia. Metabolic syndrome is typically caused by interactions between insulin resistance and obesity. PCOS-linked insulin resistance is more severe in hyperandrogenic PCOS than non-hyperandrogenic PCOS (97). The pathogenesis of metabolic dysfunction in women with PCOS is not fully understood, but there is suggestion that hyperandrogenism influences the metabolic facets of PCOS (98, 99).
Insulin resistance induces hyperinsulinemia that can exacerbate PCOS dysfunction. Insulin and testosterone levels appear highly correlated and hyperinsulinemia has been suggested as the primary cause of increased testosterone (100). Insulin resistance also significantly increases the risk of T2DM (4, 5). Furthermore, obstructive sleep apnea is a PCOS comorbidity and is known to exacerbate insulin resistance (1). As with typical insulin resistance, PCOS-related insulin resistance is characterized by reduced sensitivity and responsiveness to insulin-mediated glucose utilization primarily in skeletal muscle and adipose tissue (101). Of interest, insulin sensitivity is not diminished in the ovaries, pituitary gland, or the adrenal gland in women with PCOS.
Insulin is thought to augment LH-stimulated testosterone production through activation of its receptor (102, 103). Rat studies have shown hyperinsulinemia upregulates LH-binding sites, thus augmenting LH-induced testosterone production in theca cells (34, 104, 105). Insulin in rats also augments the GnRH-stimulated production of LH, which has been shown to be glucose dependent (103). Insulin seems to augment the production of estradiol and progesterone in human granulosa cells through its own receptor (106). Therefore, elevated insulin can promote and exacerbate hyperandrogenism and PCOS symptoms (Figure 8).
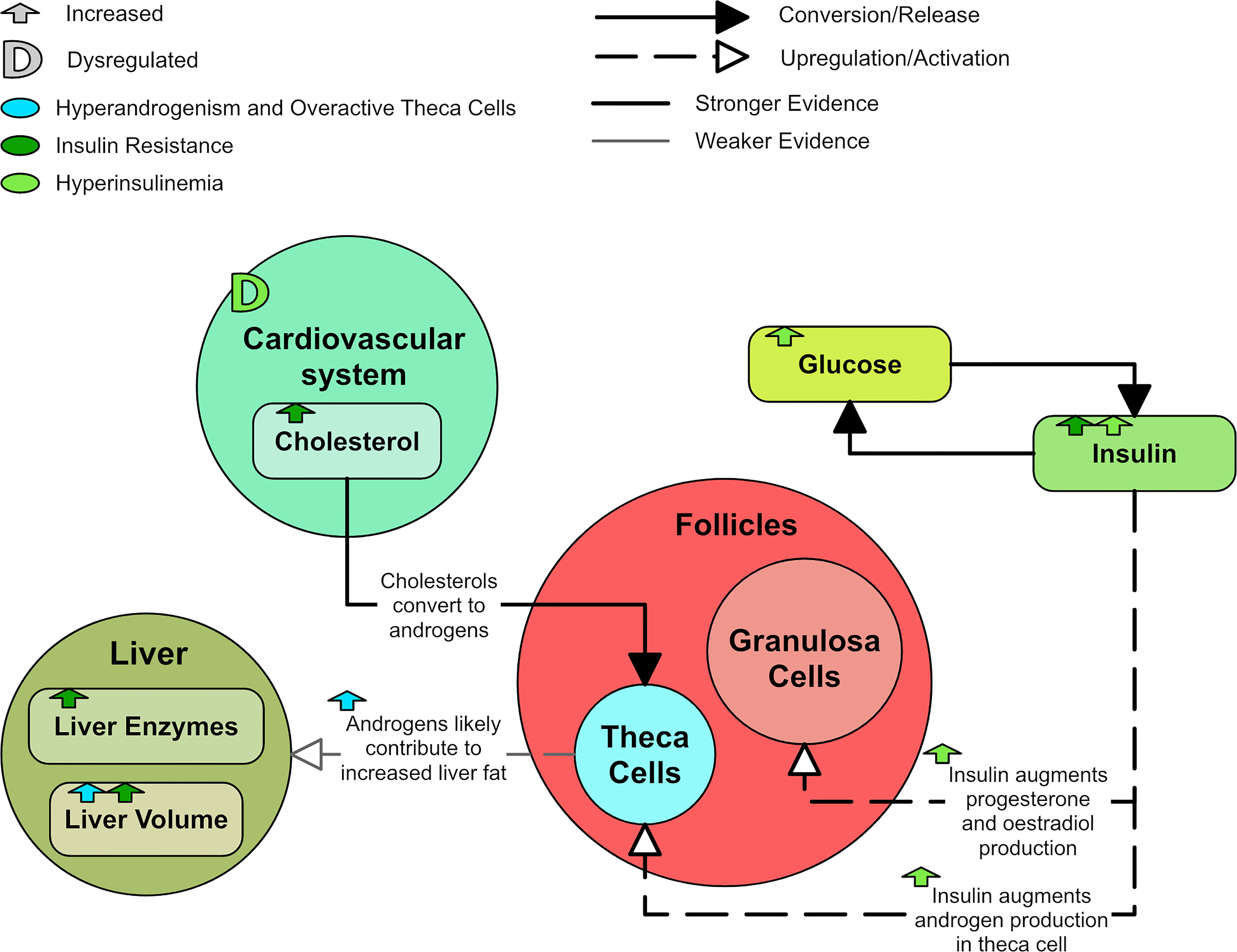
Figure 8 A diagram of the effects of insulin resistance and hyperinsulinemia in PCOS.
A relationship between insulin resistance, hyperinsulinemia and hypertension has been observed, leading to the postulation that hyperinsulinemia contributes to cardiovascular disease through direct mechanisms such as increasing sympathetic activity (107). Hyperinsulinemia may also contribute to cardiovascular disease by inducing abnormalities in endothelial function and vascular reactivity (108). In a study of over 2,000 PCOS patients, total cholesterol, triglycerides and low-density lipoprotein (LDL) cholesterol were significantly higher in PCOS patients than in controls even after correcting for body mass index (BMI) (39).
PCOS and its comorbidities are known to affect the liver. Insulin resistance (109) and PCOS (14) are compounding risk factors for NAFLD. In particular, PCOS doubles the risk factor for NAFLD in women (14). The liver enzymes aspartate transaminase (AST) and alanine transaminase (ALT) are significantly higher in PCOS groups (39) and are known markers of liver disease. Androgen excess has also been shown to contribute to the occurrence of NAFLD in PCOS rat models (110). It has been demonstrated that hyperandrogenic women with PCOS have increased liver fat compared to non-hyperandrogenic women with PCOS and non-PCOS controls, even after correction for BMI, adipose tissue volume, and HOMA-IR (98, 109). There is suggestion that an androgen-dependent proapoptotic PCOS environment contributes to NAFLD (111).
Insulin-like growth factor I (IGF-I) is produced in theca cells (33) and has been shown to stimulate testosterone production (112), likely by increasing LH binding affinity with theca cells (104). IGF-I may also augment FSH stimulated production of estradiol (113). Insulin amplifies these effects as it suppresses insulin-like growth factor binding protein I (IGFBP-I) production (114). This suppression of IGFBP-I enhances IGF-I bioactivity (114). LH is also thought to promote ovarian secretion of IGF-I (104) and DHEA-S has been found to positively correlate with IGF-I (91).
A meta-analysis found that women with PCOS appear to have lower levels of IGFBP-I than controls (115). However, adjustments for BMI suggested that decreased IGFBP-I may be the result of obesity and not have a role in the pathogenesis of PCOS (115). Thus, the relationship between IGF-I, IGFBP-I and PCOS remains unclear. IGF-I, IGF-II, and insulin can each augment LH-induced A4 production in the theca cell. Thus, it is possible for insulin resistance to have a role in hyperandrogenism, even in the absence of hyperinsulinemia (116). IGF-I has been shown in vitro to stimulate follicle growth in normal human ovaries but not polycystic ovaries (117). Figure 9 shows the dysregulation of IGF-I and IGFBP-I.
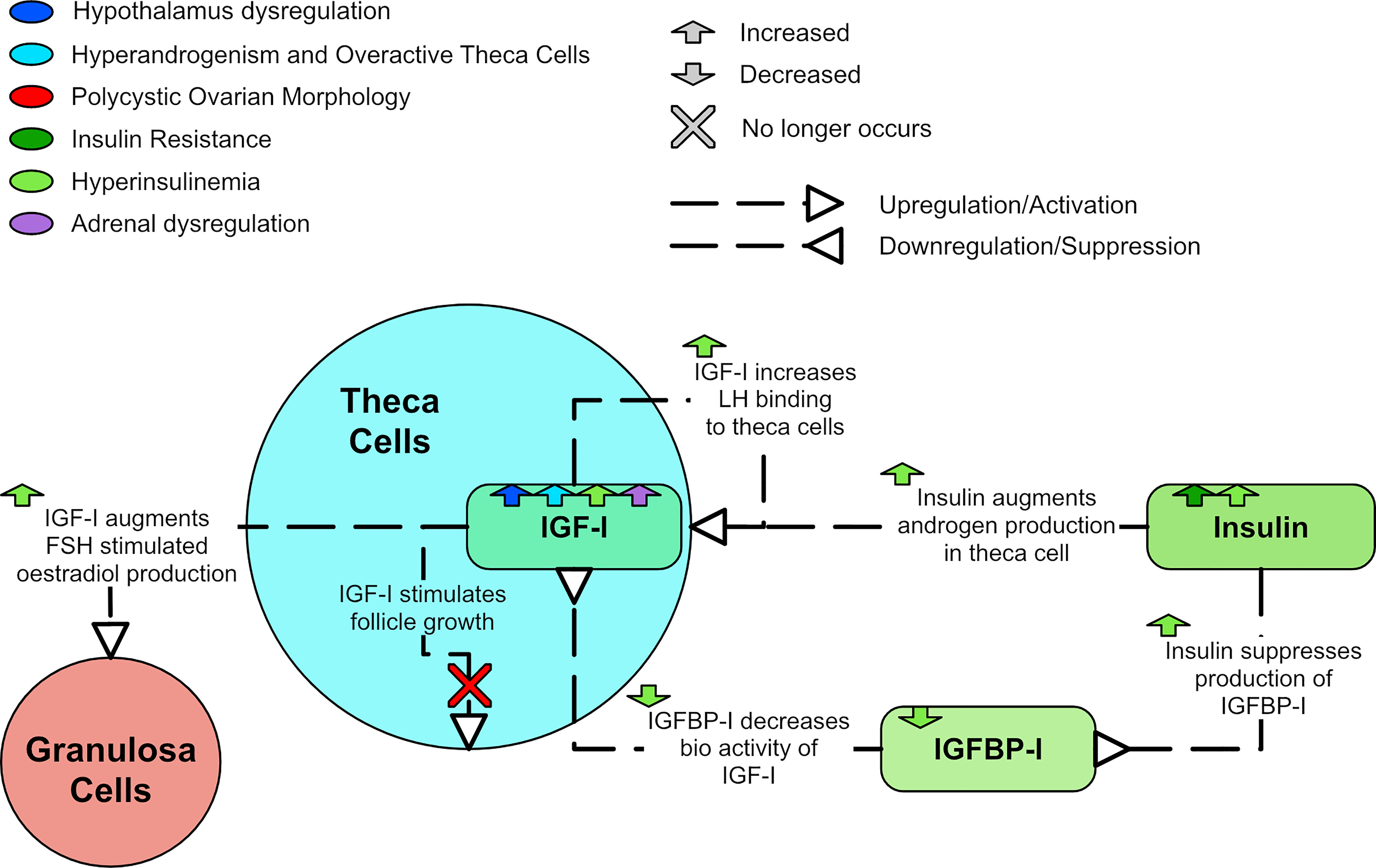
Figure 9 A diagram of the relationships of IGF-I and IGFBP-I in PCOS.
Low concentrations of sex hormone binding globulin (SHBG) are prevalent in T2DM, impaired glucose tolerance, insulin resistance, and obesity (118). Most circulating testosterone and estrogen can bind to SHBG, preventing the hormones from entering cells and binding to their receptors (17). Insulin and insulin resistance suppresses SHBG production (102, 119), resulting in increased circulating free testosterone levels and enhanced hyperandrogenism. SHBG levels have been shown to be raised by estrogen and possibly suppressed by androgens (120). However, testosterone has also been shown in vitro to increase SHBG levels to the same degree that estradiol does (119). A meta-analysis determined that SHBG levels are decreased in women with PCOS and appear to increase with treatments that improve their endo-metabolic profile (118). The meta-analysis also suggested that women with PCOS and lower SHBG levels were more likely to have hyperandrogenism, metabolic issues and infertility (118). SHBG levels are especially lower in those who also possess a high BMI (121–123). Figure 10 shows how SHBG is dysregulated in PCOS.
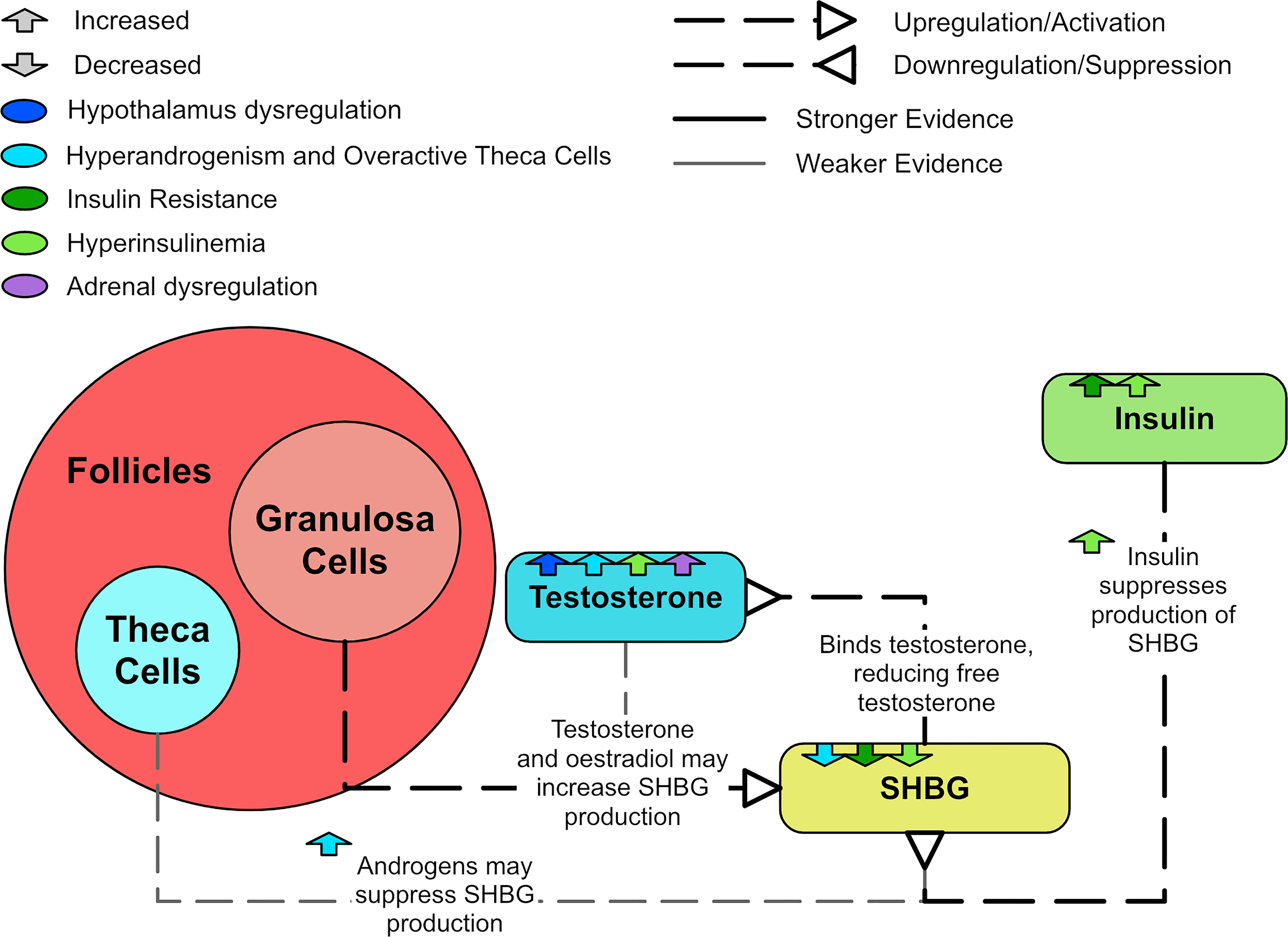
Figure 10 A diagram explaining SHBG and PCOS.
Thyroid disorders, especially the autoimmune disorder Hashimoto’s thyroiditis (HT), are observed significantly more frequently in PCOS patients than in the general population (12). Patients with both PCOS and HT have more severe metabolic symptoms than patients with either condition in isolation (12). A study of 125 women found that women with PCOS had increased thyroid volume compared to controls and the volume was highest in the insulin resistant PCOS group (124). In a study of 800 women, levels of thyroid stimulating hormone (TSH) were found to be higher in women with PCOS and appeared to be associated with hyperandrogenism (125). High TSH is a marker for an underactive thyroid and underproduction of the thyroid hormone, thyroxine. In a study of 103 women with PCOS, associations were found between high TSH values and high BMI, increased fasting insulin, high HOMA-IR indices, high testosterone, high FAI, and low SHBG (126). A study of 164 women found that those with PCOS and HT had lower SHBG than those with PCOS alone and normally functioning thyroids (12). This reduced SHBG in HT could occur because thyroxine has been shown to have a stimulatory effect on SHBG in vivo (119). Figure 11 shows the major aspects of dysregulation of the thyroid observed in PCOS.
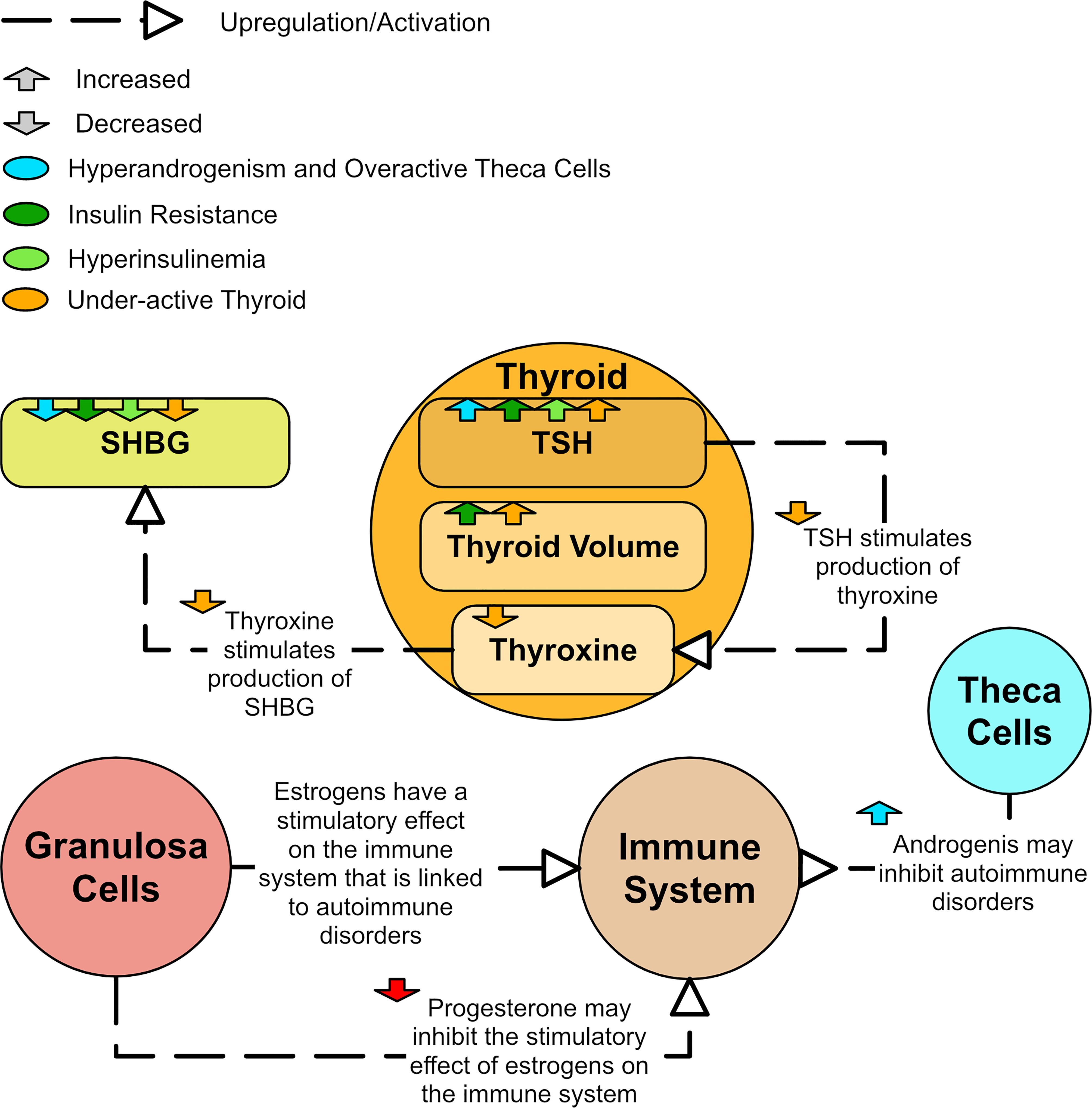
Figure 11 A diagram of how the thyroid and immune system could be dysregulated in PCOS.
The link between Hashimoto’s and PCOS is well established, but there also appears to be a link between PCOS and autoimmune disorders in general (127–129). Certain rheumatic diseases, which are autoimmune and inflammatory diseases, are more prevalent in PCOS (129). Some even claim that PCOS could be classed as an autoimmune disorder (128). Excess estrogen has been linked to different autoimmune diseases (128). The stimulatory effect of estrogen on the immune system may be inhibited by progesterone (127). As such, low levels of progesterone could lead to an overstimulated immune system (128) even though estradiol is not necessarily elevated in PCOS. However, high levels of androgens in PCOS appear to have a protective role against development of autoimmune disorders (127). Therefore, it is likely that the association of PCOS with autoimmune disorders differs by phenotype.
While prolactin is primarily known for regulating breast development and lactation, it has many other functions and is closely associated with metabolism. Prolactin secretion from the pituitary is stimulated by estradiol and regulated by the hypothalamus (130). Prolactin synthesis can also occur elsewhere in lower quantities, including from adipose tissue and the uterus (131). An in vivo study involving rats and humans found that high glucose and inflammation may stimulate the synthesis of prolactin in adipose tissue (132).
Hyperprolactinemia has been shown to induce an insulin resistant state in non-PCOS cohorts (133, 134). Prolactin also appears to inhibit SHBG production (119) and positively correlate with TSH (39). Due to the possibility that hyperprolactinemia may mimic PCOS, it is often one of the exclusion criteria in PCOS diagnosis (135). However, approximately 20% of women with PCOS also have hyperprolactinemia (135, 136). Women with coincident PCOS and hyperprolactinemia appear more insulin resistant than women with PCOS and normal prolactin levels (137). Some believe hyperprolactinemia is not more frequent in women with PCOS and that any association between them is casual (136). Others believe that hyperprolactinemia may be an integral part of PCOS (135) and PCOS with hyperprolactinemia may be a specific phenotype of PCOS.
While a subset of women with PCOS have high prolactin levels, a study of 2,052 PCOS patients and 9,696 controls found that levels of prolactin are significantly lower in women with PCOS when compared to controls before and after BMI adjustment (39). Due to the heterogeneous nature of PCOS, it is possible that both hyperprolactinemia and low prolactin are associated with different phenotypes of PCOS (Figure 12). Low prolactin seems to correlate with high BMI (138) and this effect appeared more significant in women with PCOS (41). In women with PCOS, prolactin appears to negatively correlate with total cholesterol, triglycerides and LDL cholesterol (39).
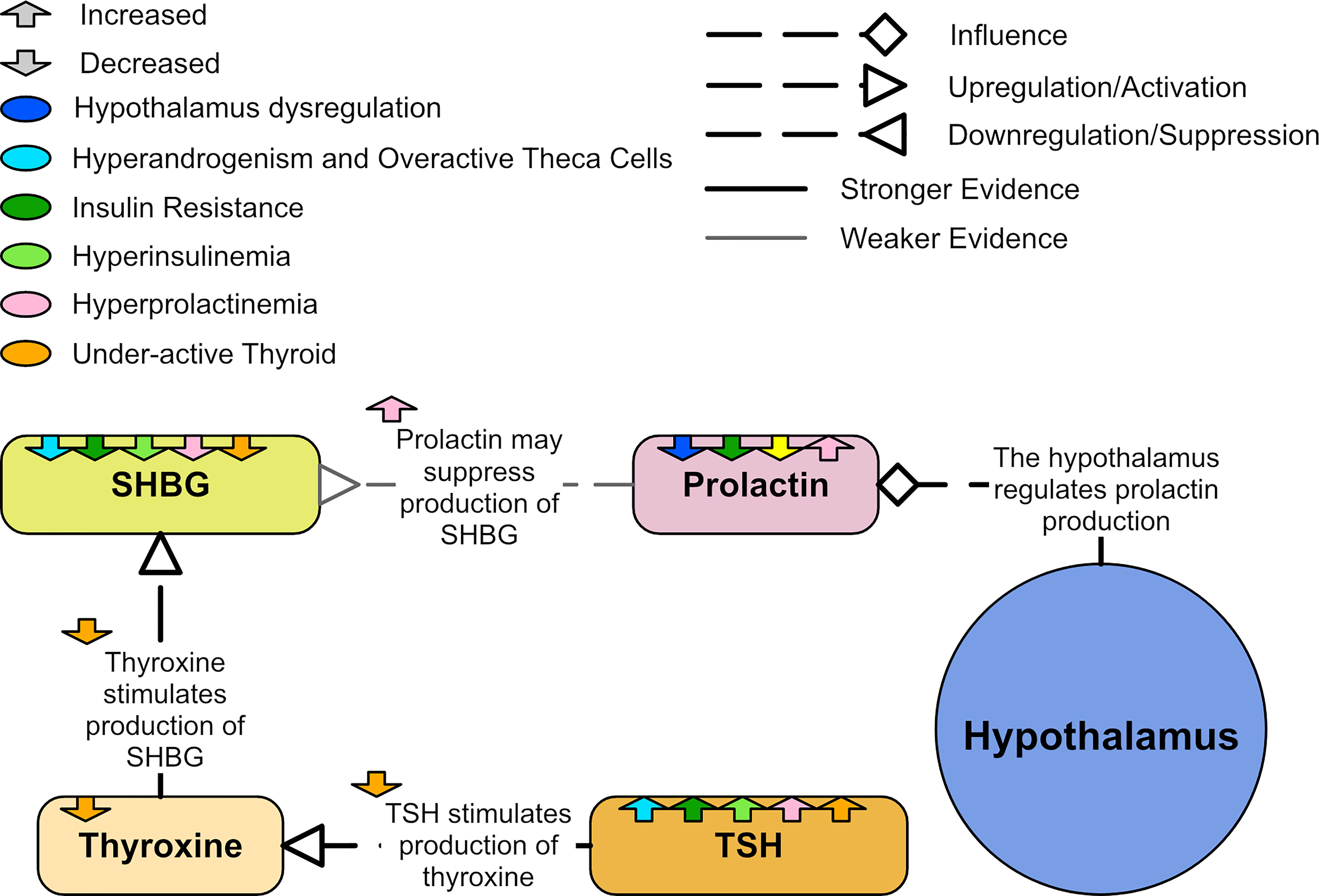
Figure 12 A diagram of showing the relationships to prolactin in PCOS.
Low prolactin seems to be an effective marker of a poor metabolic spectrum and high cardiovascular risk (39). Low prolactin levels are associated with metabolic syndrome and T2DM, while higher prolactin levels within the normal range appear to improve insulin sensitivity (139, 140). Prolactin negatively correlates with AST and ALT, suggesting that low prolactin may damage liver cells (39). Prolactin also plays a role in suppressing stress and anxiety (130). There are strong associations between PCOS and poor mental health. One meta-analysis found that women with PCOS are three times more likely to have depressive symptoms and five times more likely to have anxiety symptoms than controls (13).
Adipose tissue of women with PCOS is characterized by hypertrophic adipocytes and impairments in lipolysis and insulin action (99). The expression and secretion of a wide variety of adipokines implicated in insulin resistance are altered in PCOS (99). In particular, adiponectin, an adipose-specific protein, is downregulated in obesity and lower adiponectin levels are associated with insulin resistance (141–144). Women with PCOS are reported to have lower adiponectin levels compared with BMI-matched controls (141). Women with PCOS also appear to have larger adipocytes than BMI matched controls. These large adipocytes are also more prevalent in people with a genetic predisposition to T2DM (145). Large adipocytes are strongly correlated with insulin resistance (142). Adipose tissue dysregulation and insulin resistance seems to correlate more strongly with enlarged adipocytes rather than obesity itself (145). Adipocyte size is also inversely correlated with adiponectin levels and Glucose transporter type 4 (GLUT-4) expression (145).
GLUT-4 content in adipocyte membranes is independently decreased by obesity and by PCOS (146). Decreased expression of GLUT-4 leads to decreased insulin sensitivity and responsiveness of the adipocyte to insulin (145). Since diminished adipocyte insulin responsiveness in PCOS is associated with decreased GLUT-4 abundance (146), the problem may compound itself, and lead to divergence from healthy glucose homeostasis.
Leptin is another adipokine that can also be produced from granulosa cells (147). Leptin plays a key role in regulating appetite and energy expenditure (99). Leptin also plays a role in reproductive and immune function (99), as well as insulin action and lipid metabolism (148). Leptin may have an inhibitory effect on IGF-I augmentation of FSH-stimulated granulosa cell production of estradiol and LH-stimulated theca cell production of A4 (149). High leptin may also interfere with oocyte development and contribute to infertility in PCOS (150). A meta-analysis of leptin levels in people with PCOS found that leptin levels were significantly higher in individuals with PCOS when compared to controls (148). However, when separating obese and non-obese groups, the obese PCOS group still had significantly higher leptin levels than the obese controls but the non-obese PCOS group compared to non-obese controls did not (148). The meta-analysis also found a strong positive correlation between leptin and HOMA-IR, and a weaker positive correlation between leptin and BMI (148). Therefore, the increased leptin in PCOS may be secondary to obesity and hyperinsulinemia (151). There was also a negative correlation between leptin and testosterone, possibly because testosterone may suppress leptin synthesis (148). It is also possible that leptin positively correlates with A4 in non-obese individuals with PCOS (152). Additionally, there appears to be a relationship between leptin and prolactin as increased prolactin may influence increased leptin levels (131) and leptin may raise prolactin levels (138).
There may be relationships between PCOS adipose tissue and the reproductive hormones. In PCOS, there is a decrease in LH pulse amplitude with increasing BMI (1). In a study of 192 women, estradiol levels were lower in overweight and obese women with PCOS than in normal weight women with or without PCOS (41). In a study of 105 women, women with PCOS appeared to have a higher waist-to-hip ratio than BMI matched controls (142). No other differences in anthropometric variables or abdominal adipose tissue volume and distribution were significant (142). It is therefore unlikely that insulin resistance in PCOS is strongly associated with increased visceral and abdominal fat (142).
It is possible that many of the abnormalities related to adipose tissue in women with PCOS could be secondary to hyperandrogenism (99). An in vitro study of human pre-adipocytes found that testosterone caused a time and concentration dependent 50% reduction in lipolysis in subcutaneous fat cells (153). Subcutaneous adipose cells pre-treated with testosterone showed significantly impaired glucose uptake and insulin response in vitro (154). Hyperandrogenism has been linked to a decrease in adipose LDL receptor mRNA expression (111). Prolactin may induce changes in adipose tissue, inhibit lipid activity in adipose tissue and decrease adiponectin serum concentration (131). Figure 13 shows how adipose tissue can be dysregulated in PCOS.
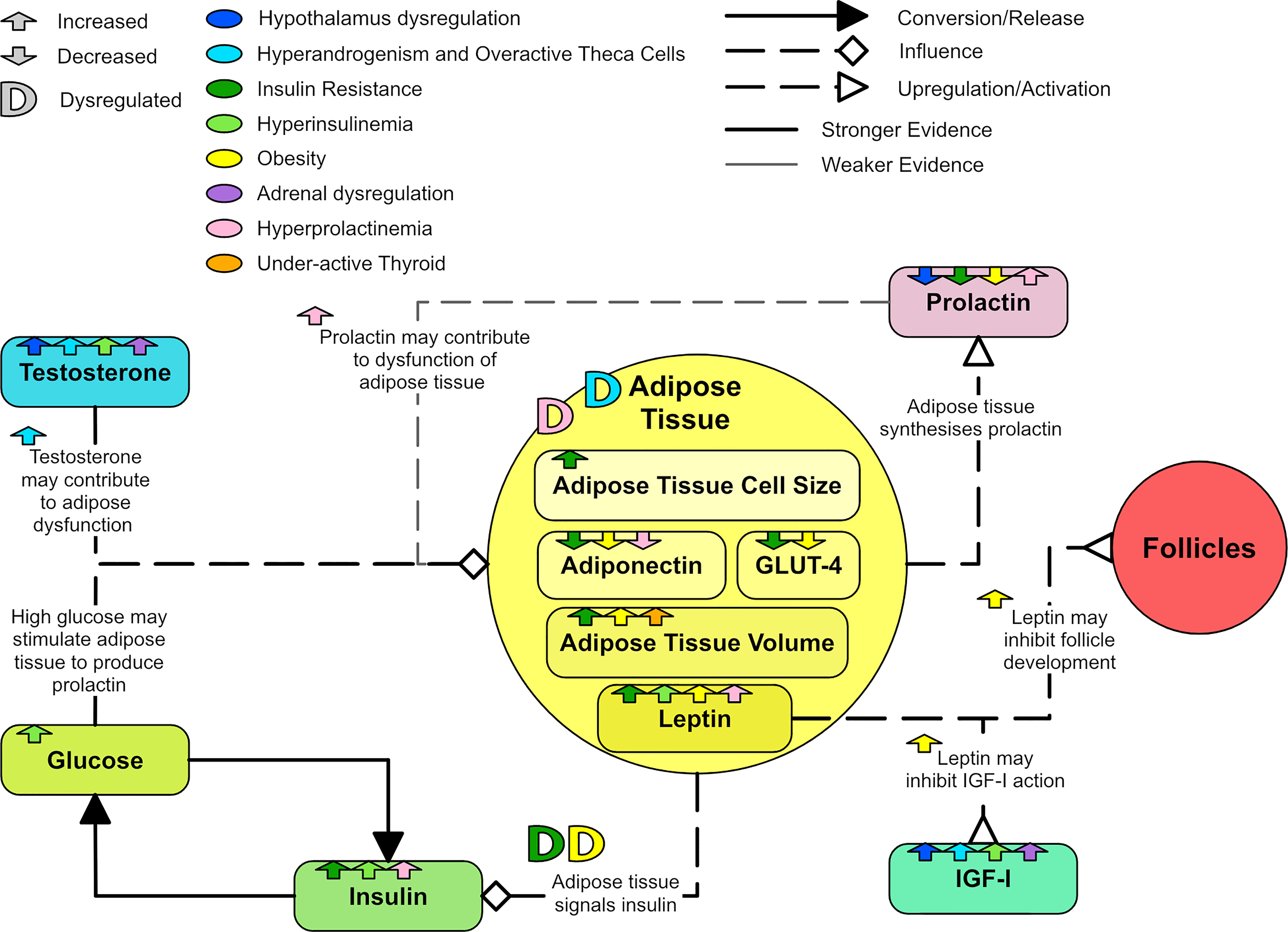
Figure 13 A diagram showing the dysregulation of adipose tissue in PCOS.
The gut is the largest endocrine organ in the body, producing multiple hormones that have important signaling roles in multiple metabolic pathways (155). The gut contains the largest number of bacteria and the greatest number of species compared to other areas of the body (156). Gut microbiota (GM) also have many important signaling and metabolic functions (156). Unsurprisingly, the gut is emerging as an important organ in the hormonal signaling pathways associated with PCOS.
Gastric inhibitory polypeptide (GIP) and glucagon-like peptide 1 (GLP-1) are gut hormones known as incretins. Incretins influence insulin secretion from the pancreas in response to ingested food (157). In healthy individuals, GIP and GLP-1 account for 60-75% of insulin secretion following glucose ingestion (158). Fasting GIP appears elevated in individuals with PCOS compared to controls (159, 160). Suppressing GIP has been shown to alleviate insulin resistance (161). GLP-1 increases insulin sensitivity, cognitive function and satiety (161). During a glucose tolerance test, late phase active GLP-1 levels have been shown to be decreased in lean individuals with PCOS compared with controls (160). One study of obese individuals with PCOS found that GLP-1 was lower in prediabetic participants compared to those with normal glucose tolerance (162). They also found correlations between decreased GLP-1 response, increased visceral adipose tissue and decreased insulin sensitivity as measured by the oral glucose insulin sensitivity index (162). Treatment with metformin appears to increase GIP and GLP-1 (163). Exploration of using GLP-1 receptor antagonists as a treatment for PCOS has shown that GLP-1 receptor antagonists appear effective in weight reduction and decreasing HOMA-IR (157).
The human gut holds many different communities of GM. There are four prime phylum, with Firmicutes and Bacteroidetes making up approximately 90%, and Actinobacteria and Proteobacterium making up approximately 10%, of all GM (164). Many factors can lead to changes in GM, including age, antibiotics and diet, with diet being one of the most influential factors (165). GM have a range of functions and researchers have often found relationships between different GM and obesity, diabetes, and liver function (166). Some GM are also associated with increased androgens and decreased estrogens (166). More recently, the relationship between PCOS and GM is being explored, with most researchers agreeing that people with PCOS have different GM communities when compared to healthy controls (167). However, at this early stage, the specific differences in the GM of an individual with PCOS are difficult to conclude, with many studies finding contradictory results.
Many studies report a decrease in alpha-diversity (168–171) and/or beta-diversity (169, 172) in PCOS groups when compared to control groups (167, 173, 174). Diversity is often negatively correlated with obesity (169, 172), androgens (169–171, 175) and markers of metabolic issues (169). There is an acknowledged relationship between decreased GM diversity and metabolic dysfunction (167, 170). Some studies also reported a decreased richness in GM in PCOS groups compared to controls, with the obese PCOS group being the least enriched (176, 177).
One of the most consistent findings throughout the literature was an increase in the Bacteroides genus (belonging to the Bacteroidetes phylum) within PCOS groups (169, 170, 176–179). Bacteroides are pro-inflammatory bacteria that can reduce activation of the gut-brain axis control of insulin through a reduction of GLP-1 (178). Increased Bacteroides can lead to an increase in branched chain amino acids (BCAAs) (179) and a reduction in bile acids (166). Increased BCAAs are associated with T2DM (165) and are a predictor in IR and diabetes (180). Bile acids emulsify fats, promote digestion, increase the absorption rate of fat-soluble substances, affect lipid metabolism, regulate glucose metabolism and enhance insulin sensitivity (165, 180). Bacteroides also produce lipopolysaccharide (LPS) that have links to chronic inflammation, obesity and insulin resistance (177). LPS is linked to signaling that promotes tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6) (167), two pro-inflammatory cytokines. Both may be increased in PCOS and higher in IR PCOS (151, 178), leading to a chronic inflammatory state and IR (165). TNF-α increase can also lead to increased intestinal permeability (167). One study found that the increase in Bacteroides were greater in IR PCOS when compared to non-IR PCOS (178). Another found the increase was greater in obese PCOS when compared to non-obese PCOS (177). A positive correlation between a Bacteroides species and LH levels has also been reported (176).
The Gammaproteobacteria class (of the Proteobacteria phylum) has been consistently observed to be elevated in individuals with PCOS compared to controls (175–177, 179). At a genus level, an increase in Escherichia (176, 177, 179) and/or Shigella (177, 179) were often mentioned. Proteobacteria have been found to be higher in those with T2DM, metabolic syndrome and inflammatory bowel disease, while Gammaproteobacteria is specifically higher in those individuals with NAFLD (175). One study found that the species of Escherichia they found increased in PCOS also positively correlated with insulin and negatively correlated with good cholesterol (176).
Some studies found that Actinobacteria were increased in obese or PCOS groups when compared to healthy controls (171, 172, 177). At a genus level Atopobium (172), Scardovia (172), Collinsella (177) and Slackia (177) were increased in obese groups when compared to non-obese groups, while Rothia was found increased in an obese PCOS group when compared to controls (177). Results regarding the various species and families in the Firmicutes phylum were inconsistent (171, 174, 177, 179).
There are many potential pathways between dysregulated GM and PCOS (Figure 14). GM are involved in the production of short chain fatty acids (SCFAs), which protect intestinal barrier integrity, promote insulin secretion and improve metabolism (180). There are many mediators of the brain-gut axis, such as serotonin, ghrelin, and peptide YY (PYY). Some studies reported a decrease in ghrelin and PYY in PCOS groups compared to controls (177, 179). Ghrelin and PPY negatively correlate with testosterone and liver enzymes and are lower in more severe PCOS phenotypes (177). PYY generally appears lower in obese individuals (161) and SCFAs are known to stimulate PYY release (165). PYY also promotes energy absorption in the intestinal tract (165). Ghrelin may inhibit estradiol and progesterone production (181). Levels of serotonin, which appears to be involved in appetite regulation and psychological wellbeing in PCOS, appears significantly lower in PCOS groups and obese controls compared to non-obese controls (177). Serotonin may also inhibit GnRH and LH secretion (182).
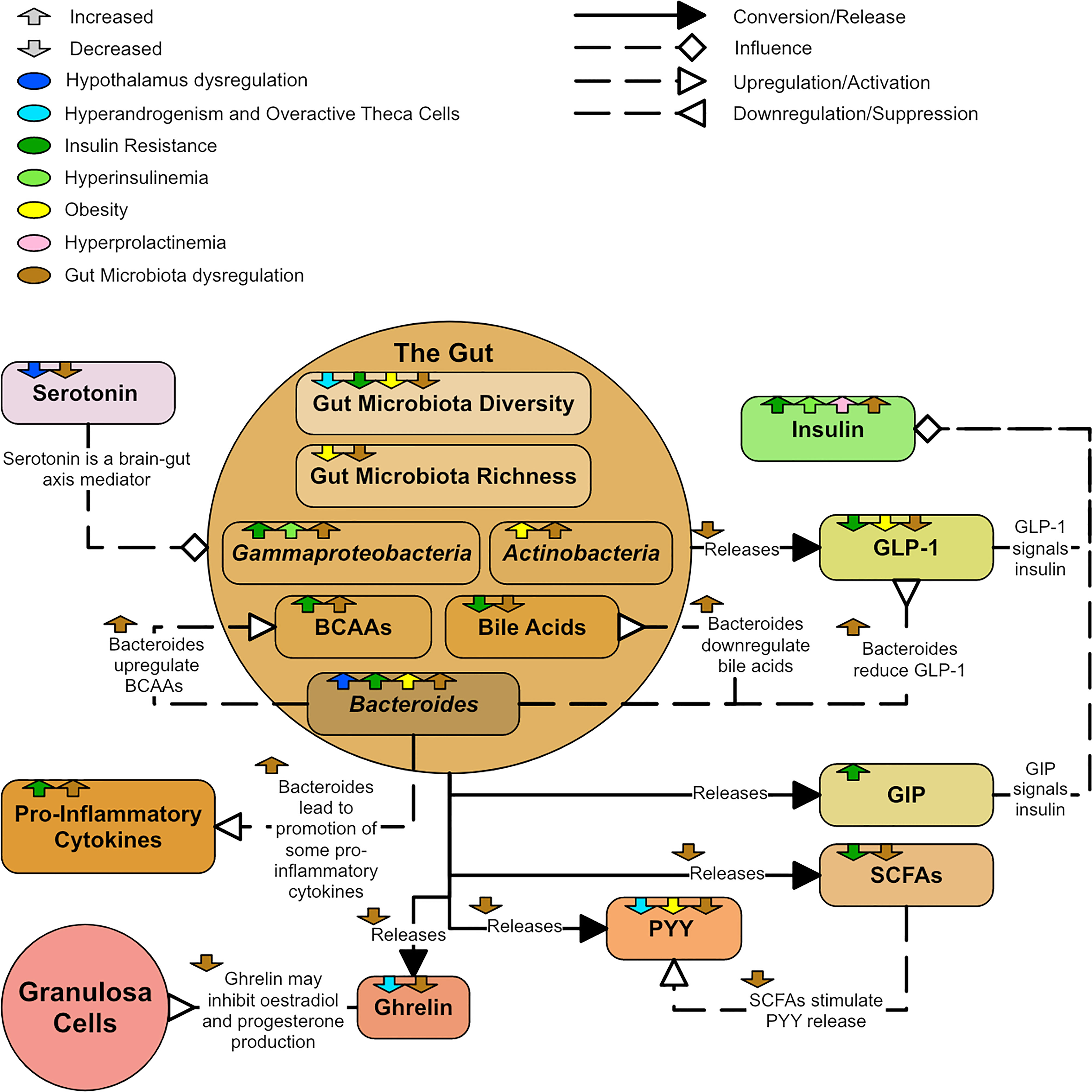
Figure 14 A diagram showing the dysregulation of the gut in PCOS.
A diagram was created to describe the major hormonal signaling pathways associated with endocrine and metabolic dysregulation seen in PCOS (Figure 15). The diagram aims to capture the different aspects of PCOS and suggest why PCOS may present with distinct phenotypes. In particular, colored arrows have been included to show how hormones and processes may increase or decrease, with different colors representing potentially different etiologies or phenotypes. They were also used to show known correlations between hormones. While many hormones can be produced and circulate in many different parts of the body only the most significant or pertinent hormone interactions and pathways are noted within the diagram. The coloring of the text boxes and circles are for aesthetic purposes.
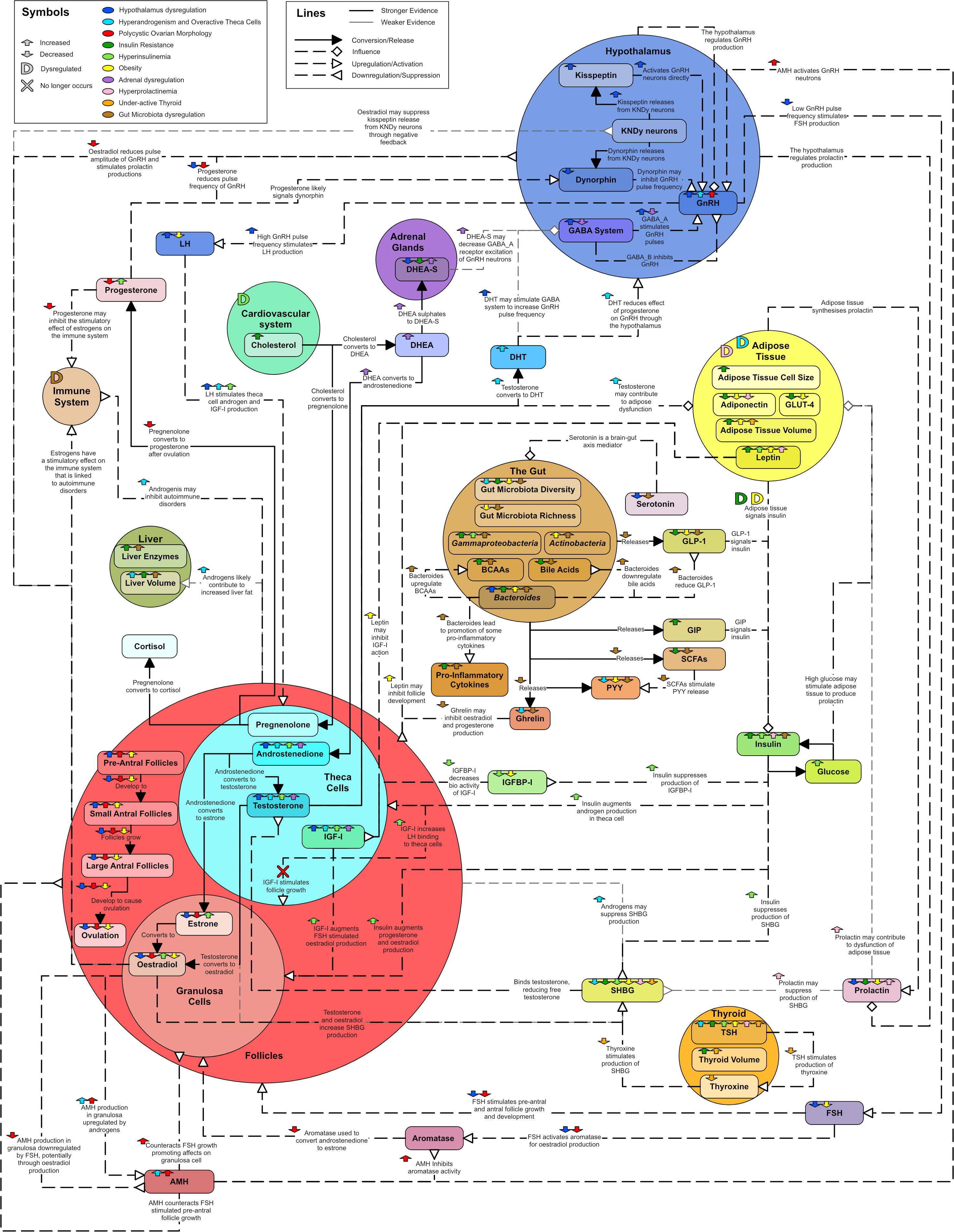
Figure 15 Unifying diagram of PCOS reproductive and metabolic dysregulation. Colored arrows are used to show how various features of the diagram are increased or decreased by possible types of dysregulation or correlations between features. A colored ‘D’ is used to indicate when a feature is dysregulated by something. A colored ‘X’ indicates a relationship that no longer occurs due to a dysregulation. The colors used to represent types of dysregulation are as follows: dark blue for dysregulation in the hypothalamus, light blue for overactive theca cells and hyperandrogenism, red for PCOM, dark green for insulin resistance, light green for hyperinsulinemia, yellow for obesity, purple for adrenal dysregulation, pink for hyperprolactinemia, orange for an underactive thyroid and brown for gut microbiota dysregulation. Solid line arrows between features are used to indicated conversion or release. Dashed lines with diamond arrowhead connections are used to indicate influence. Dashed lines with arrowheads are used to indicate upregulation and dashed lines with reverse arrowheads are used to represent downregulation. Thin grey lines indicate weaker evidence.
PCOS is an overlapping collection of phenotypes, associated with a complex hormonal dysregulation, which is not fully understood. In Figure 15 it can be observed that any of the featured dysregulation types can cause PCOS like symptoms due to the interconnectivity of the endocrine system. With specific blood test results to inform the process, Figure 15 could be used as a basis to enable patient, or phenotype specific models of PCOS. Such models may enable estimation of the likely efficacy of hormone treatments on individuals.
Due to the heterogeneous nature of PCOS, it is important for researchers to specify the pathological features of each PCOS group they are studying. Without this specification, PCOS studies can appear contradictory. Currently, most researchers use phenotypes that can be derived from the Rotterdam criteria (Table 1) to group women with PCOS. However, the Rotterdam phenotypes do not directly consider the metabolic dysfunctions underlying PCOS. Other researchers sometimes classify a subset of PCOS called ‘lean’ PCOS in an attempt to capture women who do not present with metabolic effects. However, 75% of lean women with PCOS are still insulin resistant (7) and a large literature review on lean PCOS indicated that the clinical presentation is comparable to that of overweight/obese PCOS, but that evidence is limited (183). Some research suggests splitting PCOS based on the presence of reproductive issues and/or metabolic issues (184, 185) and possibly even renaming the metabolic side as a different disorder (185). However, as Figure 15 implies, the reproductive and metabolic aspects of PCOS are interwoven.

Table 1 Rotterdam phenotypes (1).
Many clinicians stress the importance of tailored (individualized) clinical care when treating PCOS (186). However, patient-specific treatment requires an ability to describe the phenotype of the patient, in as much detail as possible. With such an approach, rather than treating the most prominent symptoms, the treatment of PCOS phenotypes may be best considered as managing both the symptoms and the most prominent underlying dysregulations. It is possible that Figure 15 could elucidate the underlying phenotype of PCOS patients by augmenting the interpretation of laboratory test results. This ability to capture the patient phenotype in a more holistic way (dual focus on both hormonal pathophysiology and presenting symptoms), may reduce the clinical and personal burden of treatment for this complex condition.
JR and PD conceived the idea. RE and JR researched and wrote the first draft. RE made the diagrams. RE, PD, HL, RC, JR, and KM reviewed and edited. All authors contributed to the article and approved the submitted version.
This study was supported by the UC Accelerator Scholarship, University of Canterbury and the Canterbury Medical Research Foundation Summer Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rosenfield RL, Ehrmann DA. The pathogenesis of polycystic ovary syndrome (PCOS): The hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocrine Rev (2016) 37(5):467–520. doi: 10.1210/er.2015-1104
2. Coyle C, Campbell RE. Pathological pulses in PCOS. Mol Cell Endocrinol (2019) 498:110561. doi: 10.1016/j.mce.2019.110561
3. Teede HJ, Misso ML, Costello MF, Dokras A, Laven J, Moran L, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Clin Endocrinol (2018) 89(3):251–68. doi: 10.1111/cen.13795
4. Ovalle F, Azziz R. Insulin resistance, polycystic ovary syndrome, and type 2 diabetes mellitus. Fertility Sterility (2002) 77(6):1095–105. doi: 10.1016/S0015-0282(02)03111-4
5. Venkatesan AM, Dunaif A, Corbould A. Insulin resistance in polycystic ovary syndrome: Progress and paradoxes. Recent Prog Hormone Res (2001) 56:295–308. doi: 10.1210/rp.56.1.295
6. Hart R, Doherty DA. The potential implications of a PCOS diagnosis on a woman’s long-term health using data linkage. J Clin Endocrinol Metab (2015) 100(3):911–9. doi: 10.1210/jc.2014-3886
7. Stepto NK, Cassar S, Joham AE, Hutchison SK, Harrison CL, Goldstein RF, et al. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic–hyperinsulaemic clamp. Hum Reprod (2013) 28(3):777–84. doi: 10.1093/humrep/des463
8. Kakoly NS, Khomami MB, Joham AE, Cooray SD, Misso ML, Norman RJ, et al. Ethnicity, obesity and the prevalence of impaired glucose tolerance and type 2 diabetes in PCOS: A systematic review and meta-regression. Hum Reprod Update (2018) 24(4):455–67. doi: 10.1093/humupd/dmy007
9. Palomba S, deWilde MA, Falbo A, Koster MPH, La Sala GB, Fauser BCJM. Reply: Polycystic ovary syndrome and pregnancy. Hum Reprod Update (2015) 22(2):278–9. doi: 10.1093/humupd/dmv052
10. Palomba S. Is fertility reduced in ovulatory women with polycystic ovary syndrome? an opinion paper. Hum Reprod (2021) 36(9):2421–8. doi: 10.1093/humrep/deab181
11. Fernandez RC, Moore VM, Van Ryswyk EM, Varcoe TJ, Rodgers RJ, March WA, et al. Sleep disturbances in women with polycystic ovary syndrome: Prevalence, pathophysiology, impact and management strategies. Nat Sci Sleep (2018) 10:45–64. doi: 10.2147/nss.S127475
12. Zhao H, Zhang Y, Ye J, Wei H, Huang Z, Ning X, et al. A comparative study on insulin secretion, insulin resistance and thyroid function in patients with polycystic ovary syndrome with and without hashimoto's thyroiditis. Diabetes Metab syndrome Obes (2021) 14:1817–21. doi: 10.2147/DMSO.S300015
13. Cooney LG, Lee I, Sammel MD, Dokras A. High prevalence of moderate and severe depressive and anxiety symptoms in polycystic ovary syndrome: A systematic review and meta-analysis. Hum Reprod (2017) 32(5):1075–91. doi: 10.1093/humrep/dex044
14. Sarkar M, Terrault N, Chan W, Cedars MI, Huddleston HG, Duwaerts CC, et al. Polycystic ovary syndrome (PCOS) is associated with NASH severity and advanced fibrosis. Liver Int (2020) 40(2):355–9. doi: 10.1111/liv.14279
15. Chang WY, Knochenhauer ES, Bartolucci AA, Azziz R. Phenotypic spectrum of polycystic ovary syndrome: Clinical and biochemical characterization of the three major clinical subgroups. Fertil Steril (2005) 83(6):1717–23. doi: 10.1016/j.fertnstert.2005.01.096
16. Conway GS, Honour JW, Jacobs HS. Heterogeneity of the polycystic ovary syndrome: Clinical, endocrine and ultrasound features in 556 patients. Clin Endocrinol (Oxf) (1989) 30(4):459–70. doi: 10.1111/j.1365-2265.1989.tb00446.x
17. Bienenfeld A, Azarchi S, Lo Sicco K, Marchbein S, Shapiro J, Nagler AR. Androgens in women: Androgen-mediated skin disease and patient evaluation. J Am Acad Dermatol (2019) 80(6):1497–506. doi: 10.1016/j.jaad.2018.08.062
18. Campbell RE, Coolen LM, Hoffman GE, Hrabovszky E. Highlights of neuroanatomical discoveries of the mammalian gonadotropin-releasing hormone system. J Neuroendocrinol (2022) 34(5):e13115. doi: 10.1111/jne.13115
19. Longcope C. Adrenal and gonadal androgen secretion in normal females. Clinics Endocrinol Metab (1986) 15(2):213. doi: 10.1016/S0300-595X(86)80021-4
20. Walters K. Role of androgens in normal and pathological ovarian function. Reproduction (2015) 149(4):R193–218. doi: 10.1530/REP-14-0517
21. Baculescu N. The role of androgen receptor activity mediated by the CAG repeat polymorphism in the pathogenesis of PCOS. J Med Life (2013) 6(1):18–25.
22. Coutinho EA, Kauffman AS. The role of the brain in the pathogenesis and physiology of polycystic ovary syndrome (PCOS). Med Sci (2019) 7(8):84. doi: 10.3390/medsci7080084
23. McCartney CR, Campbell RE, Marshall JC, Moenter SM. The role of gonadotropin-releasing hormone neurons in polycystic ovary syndrome. J Neuroendocrinol (2022) 34(5):e13093. doi: 10.1111/jne.13093
24. Witchel Selma F, Teede HJ, Peña AS. Curtailing PCOS. Pediatr Res (2020) 87(2):353–61. doi: 10.1038/s41390-019-0615-1
25. Palomba S, Piltonen TT, Giudice LC. Endometrial function in women with polycystic ovary syndrome: A comprehensive review. Hum Reprod Update (2020) 27(3):584–618. doi: 10.1093/humupd/dmaa051
26. Palomba S, Daolio J, La Sala GB. Oocyte competence in women with polycystic ovary syndrome. Trends Endocrinol Metab (2017) 28(3):186–98. doi: 10.1016/j.tem.2016.11.008
27. Wood JR, Nelson VL, Ho C, Jansen E, Wang CY, Urbanek M, et al. The molecular phenotype of polycystic ovary syndrome (PCOS) theca cells and new candidate PCOS genes defined by microarray analysis. J Biol Chem (2003) 278(29):26380–90. doi: 10.1074/jbc.M300688200
28. Jakimiuk AJ, Weitsman SR, Navab A, Magoffin DA. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J Clin Endocrinol Metab (2001) 86(3):1318–23. doi: 10.1210/jcem.86.3.7318
29. Gilling-Smith C, Story H, Rogers V, Franks S. Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol (Oxf) (1997) 47(1):93–9. doi: 10.1046/j.1365-2265.1997.2321049.x
30. Nelson VL, Qin K-n, Rosenfield RL, Wood JR, Penning TM, Legro RS, et al. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab (2001) 86(12):5925–33. doi: 10.1210/jcem.86.12.8088
31. Nelson VL, Legro RS, Strauss JF III, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol (1999) 13(6):946–57. doi: 10.1210/mend.13.6.0311
32. Gilling-Smith C, Willis DS, Beard RW, Franks S. Hypersecretion of androstenedione by isolated thecal cells from polycystic ovaries. J Clin Endocrinol Metab (1994) 79(4):1158–65. doi: 10.1210/jcem.79.4.7962289
33. Skinner MK, Schmidt M, Savenkova MI, Sadler-Riggleman I, Nilsson EE. Regulation of granulosa and theca cell transcriptomes during ovarian antral follicle development. Mol Reprod Dev (2008) 75(9):1457–72. doi: 10.1002/mrd.20883
34. Magoffin DA, Weitsman SR. Insulin-like growth factor-I regulation of luteinizing hormone (LH) receptor messenger ribonucleic acid expression and LH-stimulated signal transduction in rat ovarian theca-interstitial cells. Biol Reprod (1994) 51(4):766–75. doi: 10.1095/biolreprod51.4.766
35. Webber L, Stubbs S, Stark J, Trew G, Margara R, Hardy K, et al. Formation and early development of follicles in the polycystic ovary. Lancet (2003) 362(9389):1017–21. doi: 10.1016/S0140-6736(03)14410-8
36. Homburg R, Ray A, Bhide P, Gudi A, Shah A, Timms P, et al. The relationship of serum anti-mullerian hormone with polycystic ovarian morphology and polycystic ovary syndrome: A prospective cohort study. Hum Reprod (2013) 28(4):1077–83. doi: 10.1093/humrep/det015
37. Fauser BCJM, Pache TD, Lamberts SW, Hop WC, de Jong FH, Dahl KD. Serum bioactive and immunoreactive luteinizing hormone and follicle-stimulating hormone levels in women with cycle abnormalities, with or without polycystic ovarian disease. J Clin Endocrinol Metab (1991) 73(4):811–7. doi: 10.1210/jcem-73-4-811
38. Taylor AE, McCourt B, Martin KA, Anderson EJ, Adams JM, Schoenfeld D, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab (1997) 82(7):2248–56. doi: 10.1210/jcem.82.7.4105
39. Yang H, Di J, Pan J, Yu R, Teng Y, Cai Z, et al. The association between prolactin and metabolic parameters in PCOS women: A retrospective analysis. Front Endocrinol (Lausanne) (2020) 11:263. doi: 10.3389/fendo.2020.00263
40. Waldstreicher J, Santoro NF, Hall JE, Filicori M, Crowley JR WF. Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: Indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab (1988) 66(1):165–72. doi: 10.1210/jcem-66-1-165
41. Franik G, Madej P, Guz-Lem M, Owczarek A, Chudek J, Olszanecka-Glinianowicz M. Daytime decrease of prolactin levels is associated with PCOS regardless to nutritional status and other hormones levels. Gynecological Endocrinol (2017) 33(5):336–41. doi: 10.1080/09513590.2016.1276555
42. Balen AH, Tan SL, Jacobs HS. Hypersecretion of luteinising hormone: A significant cause of infertility and miscarriage. BJOG: Int J Obstetrics Gynaecol (1993) 100(12):1082–9. doi: 10.1111/j.1471-0528.1993.tb15170.x
43. Couse JF, Yates MM, Deroo BJ, Korach KS. Estrogen receptor-β is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology (2005) 146(8):3247–62. doi: 10.1210/en.2005-0213
44. Dewailly D, Robin G, Peigne M, Decanter C, Pigny P, Catteau-Jonard S. Interactions between androgens, FSH, anti-müllerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum Reprod Update (2016) 22(6):709–24. doi: 10.1093/humupd/dmw027
45. Desforges-Bullet V, Gallo C, Lefebvre C, Pigny P, Dewailly D, Catteau-Jonard S. Increased anti-müllerian hormone and decreased FSH levels in follicular fluid obtained in women with polycystic ovaries at the time of follicle puncture for in vitro fertilization. Fertility Sterility (2010) 94(1):198–204. doi: 10.1016/j.fertnstert.2009.03.004
46. Laven JS, Mulders AG, Visser JA, Themmen AP, De Jong FH, Fauser BC. Anti-mullerian hormone serum concentrations in normoovulatory and anovulatory women of reproductive age. J Clin Endocrinol Metab (2004) 89(1):318–23. doi: 10.1210/jc.2003-030932
47. Homburg R, Crawford G. The role of AMH in anovulation associated with PCOS: A hypothesis. Hum Reprod (2014) 29(6):1117–21. doi: 10.1093/humrep/deu076
48. Evans JJ, Wilkinson TM, Wall DJN. A two-pathway mathematical model of the LH response to GnRH that predicts self-priming. Int J Endocrinol (2013) 2013:410348. doi: 10.1155/2013/410348
49. Doi SAR, Towers PA, Scott CJ, Al-Shoumer KAS. PCOS: An ovarian disorder that leads to dysregulation in the hypothalamic–Pituitary–Adrenal axis? Eur J Obstetrics Gynecology Reprod Biol (2005) 118(1):4–16. doi: 10.1016/j.ejogrb.2004.06.024
50. Ruddenklau A, Campbell RE. Neuroendocrine impairments of polycystic ovary syndrome. Endocrinology (2019) 160(10):2230–42. doi: 10.1210/en.2019-00428
51. McCartney CR, Marshall JC. Polycystic ovary syndrome. New Engl J Med (2016) 375(1):54–64. doi: 10.1056/NEJMcp1514916
52. Karsch FJ. Central actions of ovarian steroids in the feedback regulation of pulsatile secretion of luteinizing hormone. Annu Rev Physiol (1987) 49(1):365–82. doi: 10.1146/annurev.ph.49.030187.002053
53. Soules MR, Steiner RA, Clifton DK, Cohen NL, Aksel S, Bremner WJ. Progesterone modulation of pulsatile luteinizing hormone secretion in normal women. J Clin Endocrinol Metab (1984) 58(2):378–83. doi: 10.1210/jcem-58-2-378
54. Foradori CD, Goodman RL, Adams VL, Valent M, Lehman MN. Progesterone increases dynorphin a concentrations in cerebrospinal fluid and preprodynorphin messenger ribonucleic acid levels in a subset of dynorphin neurons in the sheep. Endocrinology (2005) 146(4):1835–42. doi: 10.1210/en.2004-1326
55. Sullivan SD, Moenter SM. GABAergic integration of progesterone and androgen feedback to gonadotropin-releasing hormone neurons. Biol Reprod (2005) 72(1):33–41. doi: 10.1095/biolreprod.104.033126
56. Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: Evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and Progesterone1. J Clin Endocrinol Metab (1998) 83(2):582–90. doi: 10.1210/jcem.83.2.4604
57. Blank SK, McCartney CR, Helm KD, Marshall JC. Neuroendocrine effects of androgens in adult polycystic ovary syndrome and female puberty. Semin Reprod Med (2007) 25(5):352–9. doi: 10.1055/s-2007-984741
58. Daniels TL, Berga SL. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J Clin Endocrinol Metab (1997) 82(12):4179–83. doi: 10.1210/jc.82.12.4179
59. Pielecka J, Quaynor SD, Moenter SM. Androgens increase gonadotropin-releasing hormone neuron firing activity in females and interfere with progesterone negative feedback. Endocrinology (2006) 147(3):1474–9. doi: 10.1210/en.2005-1029
60. Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABAA receptor activation on gonadotrophin-releasing hormone neurones: Towards an emerging consensus. J Neuroendocrinol (2011) 23(7):557–69. doi: 10.1111/j.1365-2826.2011.02145.x
61. Sullivan SD, Moenter SM. Prenatal androgens alter GABAergic drive to gonadotropin-releasing hormone neurons: Implications for a common fertility disorder. Proc Natl Acad Sci (2004) 101(18):7129–34. doi: 10.1073/pnas.0308058101
62. Berg T, Silveira MA, Moenter SM. Prepubertal development of GABAergic transmission to gonadotropin-releasing hormone (GnRH) neurons and postsynaptic response are altered by prenatal androgenization. J Neurosci (2018) 39(8):2304–17. doi: 10.1523/jneurosci.2304-17.2018
63. Kawwass JF, Sanders KM, Loucks TL, Rohan LC, Berga SL. Increased cerebrospinal fluid levels of GABA, testosterone and estradiol in women with polycystic ovary syndrome. Hum Reprod (2017) 32(7):1450–6. doi: 10.1093/humrep/dex086
64. Moore AM, Prescott M, Campbell RE. Estradiol negative and positive feedback in a prenatal androgen-induced mouse model of polycystic ovarian syndrome. Endocrinology (2013) 154(2):796–806. doi: 10.1210/en.2012-1954
65. Moore AM, Prescott M, Marshall CJ, Yip SH, Campbell RE. Enhancement of a robust arcuate GABAergic input to gonadotropin-releasing hormone neurons in a model of polycystic ovarian syndrome. Proc Natl Acad Sci (2015) 112(2):596–601. doi: 10.1073/pnas.1415038112
66. Chhabra S, McCartney CR, Yoo RY, Eagleson CA, Chang RJ, Marshall JC. Progesterone inhibition of the hypothalamic gonadotropin-releasing hormone pulse generator: Evidence for varied effects in hyperandrogenemic adolescent girls. J Clin Endocrinol Metab (2005) 90(5):2810–5. doi: 10.1210/jc.2004-2359
67. Eagleson CA, Gingrich MB, Pastor CL, Arora TK, Burt CM, Evans WS, et al. Polycystic ovarian syndrome: Evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab (2000) 85(11):4047–52. doi: 10.1210/jcem.85.11.6992
68. Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocrine Rev (2009) 30(6):713–43. doi: 10.1210/er.2009-0005
69. Lehman MN, Coolen LM, Goodman RL. Minireview: Kisspeptin/Neurokinin B/Dynorphin (KNDy) cells of the arcuate nucleus: A central node in the control of gonadotropin-releasing hormone secretion. Endocrinology (2010) 151(8):3479–89. doi: 10.1210/en.2010-0022
70. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology (2008) 149(4):1979–86. doi: 10.1210/en.2007-1365
71. Prague JK, Dhillo WS. Potential clinical use of kisspeptin. Neuroendocrinology (2015) 102(3):238–45. doi: 10.1159/000439133
72. Wang T, Han S, Tian W, Zhao M, Zhang H. Effects of kisspeptin on pathogenesis and energy metabolism in polycystic ovarian syndrome (PCOS). Gynecological Endocrinol (2019) 35(9):807–10. doi: 10.1080/09513590.2019.1597343
73. Tang R, Ding X, Zhu J. Kisspeptin and polycystic ovary syndrome. Front Endocrinol (2019) 10:298(298). doi: 10.3389/fendo.2019.00298
74. Goodman RL, Coolen LM, Anderson GM, Hardy SL, Valent M, Connors JM, et al. Evidence that dynorphin plays a major role in mediating progesterone negative feedback on gonadotropin-releasing hormone neurons in sheep. Endocrinology (2004) 145(6):2959–67. doi: 10.1210/en.2003-1305
75. Cheng G, Coolen LM, Padmanabhan V, Goodman RL, Lehman MN. The Kisspeptin/Neurokinin B/Dynorphin (KNDy) cell population of the arcuate nucleus: Sex differences and effects of prenatal testosterone in sheep. Endocrinology (2010) 151(1):301–11. doi: 10.1210/en.2009-0541
76. Filippou P, Homburg R. Is foetal hyperexposure to androgens a cause of PCOS? Hum Reprod Update (2017) 23(4):421–32. doi: 10.1093/humupd/dmx013
77. Stener-Victorin E, Padmanabhan V, Walters KA, Campbell RE, Benrick A, Giacobini P, et al. Animal models to understand the etiology and pathophysiology of polycystic ovary syndrome. Endocrine Rev (2020) 41(4):1–39. doi: 10.1210/endrev/bnaa010
78. Durlinger AL, Gruijters MJ, Kramer P, Karels B, Ingraham HA, Nachtigal MW, et al. Anti-mullerian hormone inhibits initiation of primordial follicle growth in the mouse ovary. Endocrinology (2002) 143(3):1076–84. doi: 10.1210/endo.143.3.8691
79. Pellatt L, Rice S, Mason HD. Anti-müllerian hormone and polycystic ovary syndrome: A mountain too high? Reproduction (2010) 139(5):825–33. doi: 10.1530/REP-09-0415
80. Pellatt L, Rice S, Dilaver N, Heshri A, Galea R, Brincat M, et al. Anti-müllerian hormone reduces follicle sensitivity to follicle-stimulating hormone in human granulosa cells. Fertility sterility (2011) 96(5):1246–51.e1. doi: 10.1016/j.fertnstert.2011.08.015
81. Durlinger A, Gruijters M, Kramer P, Karels B, Kumar TR, Matzuk M, et al. Anti-müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology (2001) 142:4891–9. doi: 10.1210/endo.142.11.8486
82. Cimino I, Casoni F, Liu X, Messina A, Parkash J, Jamin SP, et al. Novel role for anti-müllerian hormone in the regulation of GnRH neuron excitability and hormone secretion. Nat Commun (2016) 7(1):1–12. doi: 10.1038/ncomms10055
83. Andersen CY, Byskov AG. Estradiol and regulation of anti-mullerian hormone, inhibin-A, and inhibin-B secretion: Analysis of small antral and preovulatory human follicles’ fluid. J Clin Endocrinol Metab (2006) 91(10):4064–9. doi: 10.1210/jc.2006-1066
84. Cook CL, Siow Y, Brenner AG, Fallat ME. Relationship between serum müllerian-inhibiting substance and other reproductive hormones in untreated women with polycystic ovary syndrome and normal women. Fertility Sterility (2002) 77(1):141–6. doi: 10.1016/S0015-0282(01)02944-2
85. Pigny P, Jonard S, Robert Y, Dewailly D. Serum anti-mullerian hormone as a surrogate for antral follicle count for definition of the polycystic ovary syndrome. J Clin Endocrinol Metab (2006) 91(3):941–5. doi: 10.1210/jc.2005-2076
86. Pigny P, Merlen E, Robert Y, Cortet-Rudelli C, Decanter C, Jonard S, et al. Elevated serum level of anti-mullerian hormone in patients with polycystic ovary syndrome: Relationship to the ovarian follicle excess and to the follicular arrest. J Clin Endocrinol Metab (2003) 88(12):5957–62. doi: 10.1210/jc.2003-030727
87. Hammer F, Subtil S, Lux P, Maser-Gluth C, Stewart PM, Allolio B, et al. No evidence for hepatic conversion of dehydroepiandrosterone (DHEA) sulfate to DHEA: In vivo and in vitro studies. J Clin Endocrinol Metab (2005) 90(6):3600–5. doi: 10.1210/jc.2004-2386
88. Gonzalez F. Adrenal involvement in polycystic ovary syndrome. Semin Reprod Endocrinol (1997) 15(2):137–57. doi: 10.1055/s-2007-1016296
89. Goodarzi MO, Carmina E, Azziz R. DHEA, DHEAS and PCOS. J Steroid Biochem Mol Biol (2015) 145:213–25. doi: 10.1016/j.jsbmb.2014.06.003
90. Paschou SA, Palioura E, Ioannidis D, Anagnostis P, Panagiotakou A, Loi V, et al. Adrenal hyperandrogenism does not deteriorate insulin resistance and lipid profile in women with PCOS. Endocr Connect (2017) 6(8):601–6. doi: 10.1530/EC-17-0239
91. Doi SAR, Al-Zaid M, Towers PA, Scott CJ, Al-Shoumer KAS. Steroidogenic alterations and adrenal androgen excess in PCOS. Steroids (2006) 71(9):751–9. doi: 10.1016/j.steroids.2006.05.005
92. Kumar A, Woods KS, Bartolucci AA, Azziz R. Prevalence of adrenal androgen excess in patients with the polycystic ovary syndrome (PCOS). Clin Endocrinol (2005) 62(6):644–9. doi: 10.1111/j.1365-2265.2005.02256.x
93. Tosi F, Bonora E, Moghetti P. Insulin resistance in a Large cohort of women with polycystic ovary syndrome: A comparison between euglycaemic-hyperinsulinaemic clamp and surrogate indexes. Hum Reprod (2017) 32(12):2515–21. doi: 10.1093/humrep/dex308
94. Gonzalez F, Hatala DA, Speroff L. Adrenal and ovarian steroid hormone responses to gonadotropin-releasing hormone agonist treatment in polycystic ovary syndrome. Am J Obstetrics Gynecology (1991) 165(3):535–45. doi: 10.1016/0002-9378(91)90280-5
95. Sullivan SD, Moenter SM. Neurosteroids alter Γ-aminobutyric acid postsynaptic currents in gonadotropin-releasing hormone neurons: A possible mechanism for direct steroidal control. Endocrinology (2003) 144(10):4366–75. doi: 10.1210/en.2003-0634
96. Marsden PJ, Murdoch AP, Taylor R. Tissue insulin sensitivity and body weight in polycystic ovary syndrome. Clin Endocrinol (Oxf) (2001) 55(2):191–9. doi: 10.1046/j.1365-2265.2001.01303.x
97. Broekmans F, Knauff E, Valkenburg O, Laven J, Eijkemans M, Fauser B. PCOS according to the Rotterdam consensus criteria: Change in prevalence among WHO-II anovulation and association with metabolic factors. BJOG: Int J Obstetrics Gynaecology (2006) 113(10):1210–7. doi: 10.1111/j.1471-0528.2006.01008.x
98. Jones H, Sprung VS, Pugh CJA, Daousi C, Irwin A, Aziz N, et al. Polycystic ovary syndrome with hyperandrogenism is characterized by an increased risk of hepatic steatosis compared to nonhyperandrogenic PCOS phenotypes and healthy controls, independent of obesity and insulin resistance. J Clin Endocrinol Metab (2012) 97(10):3709–16. doi: 10.1210/jc.2012-1382
99. Villa J, Pratley RE. Adipose tissue dysfunction in polycystic ovary syndrome. Curr Diabetes Rep (2011) 11(3):179. doi: 10.1007/s11892-011-0189-8
100. Pateguana NB, Janes A. The contribution of hyperinsulinemia to the hyperandrogenism of polycystic ovary syndrome. J Resistance (2019) 4(1):1–3. doi: 10.4102/jir.v4i1.50
101. Ciaraldi TP, Aroda V, Mudaliar S, Chang RJ, Henry RR. Polycystic ovary syndrome is associated with tissue-specific differences in insulin resistance. J Clin Endocrinol Metab (2009) 94(1):157–63. doi: 10.1210/jc.2008-1492
102. Nestler JE, Jakubowicz DJ, Falcon de Vargas A, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab (1998) 83(6):2001–5. doi: 10.1210/jcem.83.6.4886
103. Adashi EY, Hsueh AJ, Yen SS. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology (1981) 108(4):1441–9. doi: 10.1210/endo-108-4-1441
104. Cara JF, Fan J, Azzarello J, Rosenfield RL. Insulin-like growth factor-I enhances luteinizing hormone binding to rat ovarian theca-interstitial cells. J Clin Invest (1990) 86(2):560–5. doi: 10.1172/JCI114745
105. Cara JF, Rosenfield RL. Insulin-like growth factor I and insulin potentiate luteinizing hormone-induced androgen synthesis by rat ovarian thecal-interstitial cells. Endocrinology (1988) 123(2):733–9. doi: 10.1210/endo-123-2-733
106. Willis D, Franks S. Insulin action in human granulosa cells from normal and polycystic ovaries is mediated by the insulin receptor and not the type-I insulin-like growth factor receptor. J Clin Endocrinol Metab (1995) 80(12):3788–90. doi: 10.1210/jcem.80.12.8530637
107. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities — the role of insulin resistance and the sympathoadrenal system. New Engl J Med (1996) 334(6):374–82. doi: 10.1056/nejm199602083340607
108. Westerbacka J, Vehkavaara S, Bergholm R, Wilkinson I, Cockcroft J, Yki-Järvinen H. Marked resistance of the ability of insulin to decrease arterial stiffness characterizes human obesity. Diabetes (1999) 48(4):821–7. doi: 10.2337/diabetes.48.4.821
109. Petta S, Ciresi A, Bianco J, Geraci V, Boemi R, Galvano L, et al. Insulin resistance and hyperandrogenism drive steatosis and fibrosis risk in young females with PCOS. PloS One (2017) 12(11):e0186136. doi: 10.1371/journal.pone.0186136
110. Cui P, Hu W, Ma T, Hu M, Tong X, Zhang F, et al. Long-term androgen excess induces insulin resistance and non-alcoholic fatty liver disease in PCOS-like rats. J Steroid Biochem Mol Biol (2021) 208:105829. doi: 10.1016/j.jsbmb.2021.105829
111. Baranova A, Tran TP, Afendy A, Wang L, Shamsaddini A, Mehta R, et al. Molecular signature of adipose tissue in patients with both non-alcoholic fatty liver disease (NAFLD) and polycystic ovarian syndrome (PCOS). J Trans Med (2013) 11(1):133. doi: 10.1186/1479-5876-11-133
112. Cara JF. Insulin-like growth factors, insulin-like growth factor binding proteins and ovarian androgen production. Hormone Res Paediatrics (1994) 42(1-2):49–54. doi: 10.1159/000184145
113. Erickson GF, Garzo VG, Magoffin DA. Insulin-like growth factor-I regulates aromatase activity in human granulosa and granulosa luteal cells. J Clin Endocrinol Metab (1989) 69(4):716–24. doi: 10.1210/jcem-69-4-716
114. Ehrman DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocrine Rev (1995) 16(3):322–53. doi: 10.1210/edrv-16-3-322
115. Kelly CJ, Stenton SR, Lashen H. Insulin-like growth factor binding protein-1 in PCOS: A systematic review and meta-analysis. Hum Reprod Update (2010) 17(1):4–16. doi: 10.1093/humupd/dmq027
116. Nahum R, Thong K, Hillier S. Metabolic regulation of androgen production by human thecal cells in vitro. Hum Reprod (1995) 10(1):75–81. doi: 10.1093/humrep/10.1.75
117. Stubbs SA, Webber LJ, Stark J, Rice S, Margara R, Lavery S, et al. Role of insulin-like growth factors in initiation of follicle growth in normal and polycystic human ovaries. J Clin Endocrinol Metab (2013) 98(8):3298–305. doi: 10.1210/jc.2013-1378
118. Deswal R, Yadav A, Dang AS. Sex hormone binding globulin - an important biomarker for predicting PCOS risk: A systematic review and meta-analysis. Syst Biol Reprod Med (2018) 64(1):12–24. doi: 10.1080/19396368.2017.1410591
119. Plymate SR, Matej LA, Jones RE, Friedl KE. Inhibition of sex hormone-binding globulin production in the human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin Endocrinol Metab (1988) 67(3):460–4. doi: 10.1210/jcem-67-3-460
120. Pugeat M, Nader N, Hogeveen K, Raverot G, Déchaud H, Grenot C. Sex hormone-binding globulin gene expression in the liver: Drugs and the metabolic syndrome. Mol Cell Endocrinol (2010) 316(1):53–9. doi: 10.1016/j.mce.2009.09.020
121. Neubronner SA, Indran IR, Chan YH, Thu AWP, Yong E-L. Effect of body mass index (BMI) on phenotypic features of polycystic ovary syndrome (PCOS) in Singapore women: A prospective cross-sectional study. BMC Women's Health (2021) 21(1):135. doi: 10.1186/s12905-021-01277-6
122. Cupisti S, Dittrich R, Binder H, Kajaia N, Hoffmann I, Maltaris T, et al. Influence of body mass index on measured and calculated androgen parameters in adult women with hirsutism and PCOS. Exp Clin Endocrinol Diabetes (2007) 115(6):380. doi: 10.1055/s-2007-970163
123. Simons P, Valkenburg O, Bons JAP, Stehouwer CDA, Brouwers M. The relationships of sex hormone-binding globulin, total testosterone, androstenedione and free testosterone with metabolic and reproductive features of polycystic ovary syndrome. Endocrinol Diabetes Metab (2021) 4(3):e00267. doi: 10.1002/edm2.267
124. Wright CS, Craddock A, Weinheimer-Haus EM, Lim E, Conley TB, Janle EM, et al. Thyroid status, insulin sensitivity and glucose tolerance in overweight and obese adults before and after 36 weeks of whey protein supplementation and exercise training. Endocrine Res (2016) 41(2):103–9. doi: 10.3109/07435800.2015.1094083
125. Cai J, Zhang Y, Wang Y, Li S, Wang L, Zheng J, et al. High thyroid stimulating hormone level is associated with hyperandrogenism in euthyroid polycystic ovary syndrome (PCOS) women, independent of age, BMI, and thyroid autoimmunity: A cross-sectional analysis. Front Endocrinol (Lausanne) (2019) 10:222. doi: 10.3389/fendo.2019.00222
126. Dittrich R, Kajaia N, Cupisti S, Hoffmann I, Beckmann MW, Mueller A. Association of thyroid-stimulating hormone with insulin resistance and androgen parameters in women with PCOS. Reprod BioMedicine Online (Reproductive Healthcare Limited) (2009) 19(3):319–25. doi: 10.1016/S1472-6483(10)60165-4
127. Petríková J, Lazúrová I, Yehuda S. Polycystic ovary syndrome and autoimmunity. Eur J Internal Med (2010) 21(5):369–71. doi: 10.1016/j.ejim.2010.06.008
128. Mobeen H, Afzal N, Kashif M. Polycystic ovary syndrome may be an autoimmune disorder. Scientifica (2016) 2016:4071735–. doi: 10.1155/2016/4071735
129. Sharmeen S, Nomani H, Taub E, Carlson H, Yao Q. Polycystic ovary syndrome: Epidemiologic assessment of prevalence of systemic rheumatic and autoimmune diseases. Clin Rheumatol (2021) 40(12):4837–43. doi: 10.1007/s10067-021-05850-0
130. Grattan DR, Kokay IC. Prolactin: A pleiotropic neuroendocrine hormone. J Neuroendocrinol (2008) 20(6):752–63. doi: 10.1111/j.1365-2826.2008.01736.x
131. Carré N, Binart N. Prolactin and adipose tissue. Biochimie (2014) 97:16–21. doi: 10.1016/j.biochi.2013.09.023
132. Bouckenooghe T, Sisino G, Aurientis S, Chinetti-Gbaguidi G, Kerr-Conte J, Staels B, et al. Adipose tissue macrophages (ATM) of obese patients are releasing increased levels of prolactin during an inflammatory challenge: A role for prolactin in diabesity? Biochim Biophys Acta (BBA) - Mol Basis Dis (2014) 1842(4):584–93. doi: 10.1016/j.bbadis.2013.12.005
133. Gustafson AB, Banasiak MF, Kalkhoff RK, Hagen TC, Kim HJ. Correlation of hyperprolactinemia with altered plasma insulin and gluCAGon: Similarity to effects of late human pregnancy. J Clin Endocrinol Metab (1980) 51(2):242. doi: 10.1210/jcem-51-2-242
134. Serri O, Li L, Mamputu J-C, Beauchamp M-C, Maingrette F, Renier G. The influences of hyperprolactinemia and obesity on cardiovascular risk markers: Effects of cabergoline therapy. Clin Endocrinol (2006) 64(4):366–70. doi: 10.1111/j.1365-2265.2006.02469.x
135. Kyritsi EM, Dimitriadis GK, Kyrou I, Kaltsas G, Randeva HS. PCOS remains a diagnosis of exclusion: A concise review of key endocrinopathies to exclude. Clin Endocrinol (2017) 86(1):1–6. doi: 10.1111/cen.13245
136. Szosland K, Pawlowicz P, Lewiński A. Prolactin secretion in polycystic ovary syndrome (PCOS). Neuro-endocrinology Lett (2015) 36(1):53.
137. Bahceci M, Tuzcu A, Bahceci S, Tuzcu S. Is hyperprolactinemia associated with insulin resistance in non-obese patients with polycystic ovary syndrome? J Endocrinological Invest (2003) 26(7):655–9. doi: 10.1007/BF03347025
138. Roelfsema F, Pijl H, Keenan DM, Veldhuis JD. Prolactin secretion in healthy adults is determined by gender, age and body mass index. PloS One (2012) 7(2):e31305. doi: 10.1371/journal.pone.0031305
139. Balbach L, Wallaschofski H, Völzke H, Nauck M, Dörr M, Haring R. Serum prolactin concentrations as risk factor of metabolic syndrome or type 2 diabetes? BMC endocrine Disord (2013) 13(1):12–. doi: 10.1186/1472-6823-13-12
140. Wang T, Lu J, Xu YP, Li M, Sun J, Zhang J, et al. Circulating prolactin associates with diabetes and impaired glucose regulation: A population-based study. Diabetes Care (2013) 36(7):1974–80. doi: 10.2337/dc12-1893
141. Toulis KA, Goulis D, Farmakiotis D, Georgopoulos N, Katsikis I, Tarlatzis B, et al. Adiponectin levels in women with polycystic ovary syndrome: A systematic review and a meta-analysis. Hum Reprod Update (2009) 15(3):297–307. doi: 10.1093/humupd/dmp006
142. Mannerås-Holm L, Leonhardt H, Kullberg J, Jennische E, Odén A, Holm G, et al. Adipose tissue has aberrant morphology and function in PCOS: Enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab (2011) 96(2):E304–E11. doi: 10.1210/jc.2010-1290
143. Yilmaz M, Bukan N, Demırcı H, Öztürk Ç, Kan E, Ayvaz G, et al. Serum resistin and adiponectin levels in women with polycystic ovary syndrome. Gynecological Endocrinol (2009) 25(4):246–52. doi: 10.1080/09513590802653833
144. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: Close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab (2001) 86(5):1930–5. doi: 10.1210/jcem.86.5.7463
145. Hammarstedt A, Graham TE, Kahn BB. Adipose tissue dysregulation and reduced insulin sensitivity in non-obese individuals with enlarged abdominal adipose cells. Diabetol Metab Syndrome (2012) 4(1):42. doi: 10.1186/1758-5996-4-42
146. Rosenbaum D, Haber RS, Dunaif A. Insulin resistance in polycystic ovary syndrome: Decreased expression of GLUT-4 glucose transporters in adipocytes. Am J Physiology-Endocrinology And Metab (1993) 264(2):E197–202. doi: 10.1152/ajpendo.1993.264.2.E197
147. Cioffi JA, Van Blerkom J, Antczak M, Shafer A, Wittmer S, Snodgrass HR. The expression of leptin and its receptors in pre-ovulatory human follicles. Mol Hum Reprod (1997) 3(6):467–72. doi: 10.1093/molehr/3.6.467
148. Zheng S-H, Du D-F, Li X-L. Leptin levels in women with polycystic ovary syndrome: A systematic review and a meta-analysis. Reprod Sci (2017) 24(5):656–70. doi: 10.1177/1933719116670265
149. Agarwal SK, Vogel K, Weitsman SR, Magoffin DA. Leptin antagonizes the insulin-like growth factor-I augmentation of steroidogenesis in granulosa and theca cells of the human ovary. J Clin Endocrinol Metab (1999) 84(3):1072–6. doi: 10.1210/jcem.84.3.5543
150. Peng Y, Yang H, Song J, Feng D, Na Z, Jiang H, et al. Elevated serum leptin levels as a predictive marker for polycystic ovary syndrome. Front Endocrinol (Lausanne) (2022) 13:845165. doi: 10.3389/fendo.2022.845165
151. Wang J, Gong P, Li C, Pan M, Ding Z, Ge X, et al. Correlation between leptin and IFN-Γ involved in granulosa cell apoptosis in PCOS. Gynecological Endocrinol (2020) 36(12):1051–6. doi: 10.1080/09513590.2020.1760817
152. Kikuchi N, Andoh K, Mizunuma H, Minegishi T. Relationships between circulating leptin concentrations and other hormonal parameters in obese and non-obese women with polycystic ovary syndrome. Reprod Med Biol (2002) 1(2):49–54. doi: 10.1046/j.1445-5781.2002.00008.x
153. Dicker A, Rydén M, Näslund E, Muehlen IE, Wirén M, Lafontan M, et al. Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia (2004) 47(3):420–8. doi: 10.1007/s00125-003-1324-0
154. Corbould A. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol (2007) 192(3):585. doi: 10.1677/joe.1.07070
155. Rehfeld JF. The endocrine gut. In: Belfiore A, LeRoith D, editors. Principles of endocrinology and hormone action, Cham: Springer International Publishing (2016). p. 1–15.
157. Lamos EM, Malek R, Davis SN. GLP-1 receptor agonists in the treatment of polycystic ovary syndrome. Expert Rev Clin Pharmacol (2017) 10(4):401–8. doi: 10.1080/17512433.2017.1292125
158. Gasbjerg LS, Bergmann NC, Stensen S, Christensen MB, Rosenkilde MM, Holst JJ, et al. Evaluation of the incretin effect in humans using GIP and GLP-1 receptor antagonists. Peptides (2020) 125:170183. doi: 10.1016/j.peptides.2019.170183
159. Chang CL, Huang SY, Soong YK, Cheng PJ, Wang CJ, Liang IT. Circulating irisin and glucose-dependent insulinotropic peptide are associated with the development of polycystic ovary syndrome. J Clin Endocrinol Metab (2014) 99(12):E2539–48. doi: 10.1210/jc.2014-1180
160. Vrbikova J, Hill M, Bendlova B, Grimmichova T, Dvorakova K, Vondra K, et al. Incretin levels in polycystic ovary syndrome. Eur J Endocrinol (2008) 159(2):121–7. doi: 10.1530/EJE-08-0097
161. Moffett RC, Naughton V. Emerging role of GIP and related gut hormones in fertility and PCOS. Peptides (2020) 125:170233. doi: 10.1016/j.peptides.2019.170233
162. Ferjan S, Jensterle M, Oblak T, Zitnik IP, Marc J, Goricar K, et al. An impaired gluCAGon-like peptide-1 response is associated with prediabetes in polycystic ovary syndrome with obesity. J Int Med Res (2019) 47(10):4691–700. doi: 10.1177/0300060519865351
163. Svendsen PF, Nilas L, Madsbad S, Holst JJ. Incretin hormone secretion in women with polycystic ovary syndrome: Roles of obesity, insulin sensitivity, and treatment with metformin. Metabolism (2009) 58(5):586–93. doi: 10.1016/j.metabol.2008.11.009
164. Angelakis E, Armougom F, Million M, Raoult D. The relationship between gut microbiota and weight gain in humans. Future Microbiol (2012) 7(1):91–109. doi: 10.2217/fmb.11.142
165. Zhao X, Jiang Y, Xi H, Chen L, Feng X. Exploration of the relationship between gut microbiota and polycystic ovary syndrome (PCOS): A review. Geburtshilfe Frauenheilkd (2020) 80(2):161–71. doi: 10.1055/a-1081-2036
166. Wang L, Zhou J, Gober H-J, Leung WT, Huang Z, Pan X, et al. Alterations in the intestinal microbiome associated with PCOS affect the clinical phenotype. Biomedicine Pharmacotherapy (2021) 133:110958. doi: 10.1016/j.biopha.2020.110958
167. Guo J, Shao J, Yang Y, Niu X, Liao J, Zhao Q, et al. Gut microbiota in patients with polycystic ovary syndrome: A systematic review. Reprod Sci (2022) 29(1):69–83. doi: 10.1007/s43032-020-00430-0
168. Lindheim L, Bashir M, Münzker J, Trummer C, Zachhuber V, Leber B, et al. Alterations in gut microbiome composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome (PCOS): A pilot study. PloS One (2017) 12(1):e0168390. doi: 10.1371/journal.pone.0168390
169. Lüll K, Arffman RK, Sola-Leyva A, Molina NM, Aasmets O, Herzig K-H, et al. The gut microbiome in polycystic ovary syndrome and its association with metabolic traits. J Clin Endocrinol Metab (2020) 106(3):858–71. doi: 10.1210/clinem/dgaa848
170. Torres PJ, Siakowska M, Banaszewska B, Pawelczyk L, Duleba AJ, Kelley ST, et al. Gut microbial diversity in women with polycystic ovary syndrome correlates with hyperandrogenism. J Clin Endocrinol Metab (2018) 103(4):1502–11. doi: 10.1210/jc.2017-02153
171. Jobira B, Frank DN, Pyle L, Silveira LJ, Kelsey MM, Garcia-Reyes Y, et al. Obese adolescents with PCOS have altered biodiversity and relative abundance in gastrointestinal microbiota. J Clin Endocrinol Metab (2020) 105(6):e2134–44. doi: 10.1210/clinem/dgz263
172. Insenser M, Murri M, del Campo R, Martínez-García MÁ, Fernández-Durán E, Escobar-Morreale HF. Gut microbiota and the polycystic ovary syndrome: Influence of sex, sex hormones, and obesity. J Clin Endocrinol Metab (2018) 103(7):2552–62. doi: 10.1210/jc.2017-02799
173. Gu Y, Zhou G, Zhou F, Li Y, Wu Q, He H, et al. Gut and vaginal microbiomes in PCOS: Implications for women's health. Front Endocrinol (Lausanne) (2022) 13:808508. doi: 10.3389/fendo.2022.808508
174. Zhou L, Ni Z, Cheng W, Yu J, Sun S, Zhai D, et al. Characteristic gut microbiota and predicted metabolic functions in women with PCOS. Endocr Connect (2020) 9(1):63–73. doi: 10.1530/ec-19-0522
175. Mammadova G, Ozkul C, Yilmaz Isikhan S, Acikgoz A, Yildiz BO. Characterization of gut microbiota in polycystic ovary syndrome: Findings from a lean population. Eur J Clin Invest (2021) 51(4):e13417. doi: 10.1111/eci.13417
176. Liang Z, Di N, Li L, Yang D. Gut microbiota alterations reveal potential gut–brain axis changes in polycystic ovary syndrome. J Endocrinological Invest (2021) 44(8):1727–37. doi: 10.1007/s40618-020-01481-5
177. Liu R, Zhang C, Shi Y, Zhang F, Li L, Wang X, et al. Dysbiosis of gut microbiota associated with clinical parameters in polycystic ovary syndrome. Front Microbiol (2017) 8:324. doi: 10.3389/fmicb.2017.00324
178. Zeng B, Lai Z, Sun L, Zhang Z, Yang J, Li Z, et al. Structural and functional profiles of the gut microbial community in polycystic ovary syndrome with insulin resistance (IR-PCOS): A pilot study. Res Microbiol (2019) 170(1):43–52. doi: 10.1016/j.resmic.2018.09.002
179. Zhang J, Sun Z, Jiang S, Bai X, Ma C, Peng Q, et al. Probiotic bifidobacterium lactis V9 regulates the secretion of sex hormones in polycystic ovary syndrome patients through the gut-brain axis. mSystems (2019) 4(2):e00017-19. doi: 10.1128/mSystems.00017-19
180. He FF, Li YM. Role of gut microbiota in the development of insulin resistance and the mechanism underlying polycystic ovary syndrome: A review. J Ovarian Res (2020) 13(1):73. doi: 10.1186/s13048-020-00670-3
181. Viani I, Vottero A, Tassi F, Cremonini G, Sartori C, Bernasconi S, et al. Ghrelin inhibits steroid biosynthesis by cultured granulosa-lutein cells. J Clin Endocrinol Metab (2008) 93(4):1476–81. doi: 10.1210/jc.2007-2063
182. Chaudhari N, Dawalbhakta M, Nampoothiri L. GnRH dysregulation in polycystic ovarian syndrome (PCOS) is a manifestation of an altered neurotransmitter profile. Reprod Biol Endocrinol (2018) 16(1):37. doi: 10.1186/s12958-018-0354-x
183. Toosy S, Sodi R, Pappachan JM. Lean polycystic ovary syndrome (PCOS): An evidence-based practical approach. J Diabetes Metab Disord (2018) 17(2):277–85. doi: 10.1007/s40200-018-0371-5
184. Dapas M, Lin FTJ, Nadkarni GN, Sisk R, Legro RS, Urbanek M, et al. Distinct subtypes of polycystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis. PloS Med (2020) 17(6):e1003132. doi: 10.1371/journal.pmed.1003132
185. Dunaif A, Fauser BCJM. Renaming PCOS–a two-state solution. J Clin Endocrinol Metab (2013) 98(11):4325–8. doi: 10.1210/jc.2013-2040
186. Copp T, Hersch J, Doust J, McCaffery K, Dokras A, Dokrasl A, et al. 95 challenges and uncertainties regarding polycystic ovary syndrome (PCOS) and the potential for overdiagnosis: Clinicians’ views and experiences. BMJ Evidence-Based Med (2018) 23(Suppl 2):A45. doi: 10.1136/bmjebm-2018-111070.95
Keywords: PCOS, hormones, endocrine, reproductive, metabolic
Citation: Emanuel RHK, Roberts J, Docherty PD, Lunt H, Campbell RE and Möller K (2022) A review of the hormones involved in the endocrine dysfunctions of polycystic ovary syndrome and their interactions. Front. Endocrinol. 13:1017468. doi: 10.3389/fendo.2022.1017468
Received: 12 August 2022; Accepted: 27 October 2022;
Published: 15 November 2022.
Edited by:
Kok-Min Seow, Shin Kong Wu Ho-Su Memorial Hospital, TaiwanReviewed by:
Stefano Palomba, Magna Græcia University, ItalyCopyright © 2022 Emanuel, Roberts, Docherty, Lunt, Campbell and Möller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul D. Docherty, cGF1bC5kb2NoZXJ0eUBjYW50ZXJidXJ5LmFjLm56
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.