 Indranil Sinha
Indranil Sinha Dharaniya Sakthivel
Dharaniya Sakthivel David E. Varon
David E. Varon- Division of Plastic Surgery, Brigham and Women’s Hospital, Boston, MA, USA
Skeletal muscle maintenance is a dynamic process and undergoes constant repair and regeneration. However, skeletal muscle regenerative capacity declines in obesity. In this review, we focus on obesity-associated changes in inflammation, metabolism, and impaired insulin signaling, which are pathologically dysregulated and ultimately result in a loss of muscle mass and function. In addition, we examine the relationships between skeletal muscle, liver, and visceral adipose tissue in an obese state.
Introduction
Current estimates are that one-third of the United States population is obese, and this number is rapidly escalating (1). Many of these patients additionally suffer from preclinical or overt type 2 diabetes mellitus (T2DM) (1, 2). Multiple studies suggest that skeletal muscle wasting in these patients, especially those above the age of 60 years, can be severe (3–5). Diminished capacity for skeletal muscle regeneration likely contributes to the loss of lean muscle mass seen in diabetic patients (6). Obesity, a common precursor to T2DM, is also noted to have significant and independent negative effects on lean skeletal muscle mass (7). This is correlated with insulin resistance and reduced muscle performance (8). Overall, these patients suffer from a significant decline in muscle strength, as compared to age-matched controls, and a loss of functional independence (3–5). However, the effects of obesity on skeletal muscle regeneration remain largely unknown. Stimulation or preservation of skeletal muscle regeneration could possibly enable these patients to improve their strength and functional activity, as well as maintain skeletal muscle mass (8, 9).
Recent studies demonstrate that mice fed a high-fat diet (HFD) exhibit reduced hind limb muscle mass and form fewer and smaller fibers following skeletal muscle injury (10). Additionally, there exists a reduction in the total number of satellite cells, which are required for skeletal muscle regeneration (10, 11). Therefore, it is of significant clinical importance to understand how obesity impacts muscle regeneration and identify mechanisms that may be targeted for therapeutic benefit. Skeletal muscle mass in these patients is not only essential for ambulation but also necessary for glucose utilization and maintaining insulin sensitivity (12). Multiple factors affect muscle mass in patients with obesity including satellite cell function, inflammation, insulin signaling, and metabolic derangements. Furthermore, obesity-related increases in visceral adipose tissue (VAT) and fatty acid accumulation in the liver, as with non-alcoholic fatty liver disease (NAFLD), are intimately linked to the maintenance of muscle mass. When evaluating obesity and corollary loss of skeletal muscle mass, systemic mediators and their effect on muscle regeneration must be considered.
Satellite Cells
Satellite cells in skeletal muscle are located beneath the basal lamina of mature muscle fibers and are thought to be the major source of regeneration following muscle injury (13). It is now known that the satellite cell population is heterogeneous and contains both myogenic and non-myogenic cell populations. Using fluorescence-activated cell sorting (FACS), unique, myogenic stem cells, or skeletal muscle precursors (SMPs), within the satellite cell pool can be identified and isolated for further study (14). Early life obesity, induced by HFD, results in both a reduction in SMP cell frequency and impaired differentiation (10). Specifically, myogenic differentiation (MyoD), a critical factor in promoting skeletal muscle differentiation, is significantly reduced in satellite cells isolated in a diet-induced obesity (DIO) murine model (15).
In addition, satellite cell activation in a murine DIO model is impaired, which can be partially attributed to a loss in hepatocyte growth factor (HGF) signaling in skeletal muscle (16). Skeletal muscle-specific decrease in active HGF following injury limits activation of satellite cells from their quiescent state. HGF activation of SMPs requires AMP-activated protein kinase (AMPK), a protein essential to maintain satellite cell number and induce myotube formation. The active, or phosphorylated form of AMPK, promotes skeletal muscle glucose uptake and increases insulin sensitivity. Recent studies suggest that the satellite cells isolated from injured muscles of DIO mice demonstrate decreased AMPK activity and impaired regeneration (17). Fibrogenic/adipogenic precursors (FAPs) are a separate and distinct population in the satellite cell compartment (18). These cells are unable to directly form myofibers but can promote the differentiation of SMPs or form adipose tissue based on the local environment (18–20). A common observation in conditions associated with impaired skeletal muscle insulin sensitivity is an accumulation of ectopic lipids within (intracellular) and between (extracellular) skeletal muscle fibers (21), which is linked to reduced insulin sensitivity and diminished muscle function (22). The contribution of FAPs to these intramuscular lipid deposits remains unknown, as does their precise contribution to skeletal muscle regeneration in a model of obesity.
Inflammation
Obesity results in chronic, low-grade skeletal muscle inflammation (23). Recent studies further suggest DIO alone can reprogram both skeletal muscle and liver to increase the production of proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin 1-beta (IL-1β), and IL-6 (24). Increased IL-6 has been shown to limit skeletal muscle differentiation in vitro (25). In murine models of cachexia, both IL-6 or nuclear factor-kappa B (NF-κB) overexpression in skeletal muscle causes severe muscular atrophy (26, 27). Separately, in persistent inflammatory conditions, IL-6 actions are associated with increased muscle wasting (28). Despite multiple studies suggesting that skeletal muscle-specific upregulation of proinflammatory cytokines induces muscle wasting; this area warrants further research in regard to obesity. To date, no studies have shown that IL-6 or NF-κB inhibition, either systemically or in skeletal muscle, improves regeneration in an obesity model. In addition, it remains unclear whether local inflammation from skeletal muscle, increased cytokine release from liver or visceral fat, or a combination are required for impaired muscle regeneration and loss of muscle mass.
TNF-α, another proinflammatory cytokine, also has catabolic effects on muscles in chronic inflammatory state. Elevated TNF-α circulation in obese models can cause muscle wasting, inflammatory myopathies, and insulin resistance by regulating activation and secretion of other proinflammatory cytokines (29, 30). TNF-α supplementation additionally limits C2C12 muscle myoblast cell differentiation in vitro by repressing MyoD synthesis. The effects of TNF-α on skeletal muscle regeneration in an obese model remains unknown.
In addition, obesity further promotes deposition of macrophages in VAT, which contributes to inflammation, increased lipolysis, and subsequently ectopic fat deposition in skeletal muscles (31). In the early stages of obesity, an increase in these macrophages precedes T cell accumulation. T cells, in turn, are polarized into proinflammatory Th1 cells that cause myocyte inflammation through interferon secretion. The inhibition of ectopic macrophage accumulation in fat may reverse insulin resistance and thereby improve muscle function (32, 33). These points further highlight that muscle wasting in obesity is a systemic issue, instead of secondary to local changes in skeletal muscle alone.
Insulin Resistance
An array of growth factor signaling cascades, regulated by insulin, are required for the proper maintenance of skeletal muscle mass. Obesity-associated insulin resistance alters these pathways and can variably inhibit muscle regeneration. Insulin signaling is a highly complex pathway within skeletal muscle, mediated by insulin growth factor-1 (IGF-1) (34). Specifically, downstream of IGF-1, both the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathways are known to regulate skeletal muscle regeneration (35).
Mitogen-activated protein kinases are enzymes that become catalytically activated in response to diverse stimuli such as mitogens, osmotic stress, and proinflammatory cytokines. MAPK activity mediates the crosstalk between canonical and non-canonical transforming growth factor (TGF-β) in a DIO model (36). In skeletal muscle, TGF-β1 inhibits differentiation of fetal myoblasts (37). Separately, increased levels of TGF-β can cause muscle injury to heal with fibrosis, rather than regenerated skeletal muscle (38). Increased p38 MAPK and TGF-β activity within ectopic adipocytes may induce satellite cell senescence (39). Paradoxically, results from C2C12 studies, a murine myoblast model for skeletal muscle development, demonstrates a positive role for activated MAPK in cell migration (40). MAPK signaling and activity remain controversial with respect to skeletal muscle regeneration in obesity, and this topic warrants further research. Interestingly, follistatin supplementation improves muscle growth in circumstances with elevated TGF-β signaling (41).
In models of muscular dystrophy, an increase in PI3K activity can be beneficial for regeneration, as it increases Akt activity and downstream, promyogenic factors, which stimulate muscle growth. Akt activation also helps in preventing muscle atrophy by inducing the expression of mammalian target of rapamycin and ribosomal protein S6 kinase beta-1 (S6K1) (42). Specifically, in DIO models, an increase in Akt activity by phosphatase and tensin homolog (PTEN) inhibition restores skeletal muscle regeneration (11). The role of decreased insulin signaling with regard to skeletal muscle injury remains a topic of active research.
Metabolism
Obesity and chronic overnutrition are closely associated with increased mitochondrial-derived oxidative stress (43, 44). Skeletal muscle from obese or diabetic patients shows decreased mitochondrial content and a corollary loss of fatty acid oxidation (45, 46) associated with excess caloric consumption and non-inherent mitochondrial dysfunction (47). In patients with T2DM, targeted overexpression of catalase within mitochondria can protect skeletal muscle from ischemic injury, but the role of oxidative stress and mitochondrial dysfunction in obesity-related loss of skeletal muscle regeneration remains unknown (48, 49). In the context of obesity, skeletal muscle undergoes a protective shift to retain its functional capacity by converting to glycolytic, type II muscle fibers, mediated by Brg1/Brm-associated factor (Baf60c) (50–52). The Baf60c pathway increases Akt activation, which, as discussed previously, improves diet-based glucose tolerance and increases insulin sensitivity. Independently, muscle-specific Akt activation also leads to hypertrophy of type II muscle fibers with subsequent resolution of hepatic steatosis, decreased fat mass, and improved metabolic parameters (53). However, Baf60c signaling is decreased in obese rodent models, possibly due to the inhibitory effects of TNF-α (51, 52).
In contrast, many studies suggest that hypertrophy of oxidative muscle fibers (type I) can also promote metabolic homeostasis. Muscle-specific overexpression of peroxisome proliferator-activated receptor-delta (PPAR-δ) (54, 55) promotes higher levels of type I fibers relative to type II fibers, improved performance in endurance, exercise, and resistance to DIO. Conversely, mice that are deficient in peroxisome proliferator-activated receptor-gamma (PPAR-γ) coactivator 1-alpha (PGC-1α) display abnormal oxidative fiber growth and develop an increase in body fat (56). These studies suggest that increased energy expenditure in skeletal muscle mediated by hypertrophy can protect against weight gain and metabolic dysfunction. In addition, myostatin-deficient mice are resistant to DIO (57), but this metabolic effect may be due to either changes in type I or type II fibers, or from the direct action of myostatin on adipose tissue (58). Overall, it remains unclear whether or not a type I or type II fiber majority contributes to the improvement in metabolic parameters in DIO, but an increase in muscle mass, in general, appears to counteract the metabolic derangements seen in obesity.
In the absence of muscle hypertrophy, reactive oxygen species (ROS) accumulate in skeletal muscle. In obese conditions, increased ROS production is associated with contractile dysfunction, chronic oxidative stress followed by protein loss, and muscle atrophy (59). ROS is also capable of modulating the insulin signaling pathway, although the exact mechanism remains unclear. Studies suggest that ROS decreases insulin response and contributes to impaired mitochondrial activity (60). Sirtuin (SIRT), a NAD(+)-dependent histone deacetylase (HDAC) localized in mitochondria, has been found to regulate several mitochondrial genes and is important in muscle differentiation, activation of myogenesis, and skeletal muscle metabolism. Specifically, SIRT1 promotes glycolysis and inhibits adipogenesis, thereby attenuating obesity-related insulin resistance (61–63). Conversely, in T2DM, inhibition of SIRT1 alters mitochondrial metabolism and increases the production of ROS (64).
Histone deacetylases, in general, are a group of enzymes that regulate gene expression by altering chromatin structure. In obesity models, HDAC inhibition restores PPAR-γ function improving skeletal muscle glucose and fatty acid metabolism. HDAC inhibition also generates non-traditional effects such as reducing adipose tissue expansion, resistance to obesity, and improvement in insulin sensitivity (65, 66). HDAC inhibitors have proven their potency in hampering fibrosis and favorably encouraging therapeutic muscle regeneration (67). Evaluation of HDAC inhibitors for the treatment of obesity-related muscle wasting is underway (68).
In skeletal muscle, glucose transporter 4 (GLUT4) levels are directly associated with increased oxidative capacity (69). Increases in GLUT4 translocation to the plasma membrane promotes improved rates of satellite cell proliferation and differentiation (70). AMPK increases GLUT4 gene expression in human skeletal muscles (71). AMPK is also a widely recognized regulator of energy metabolism. Decreased AMPK activity is associated with metabolic disorders such as obesity and T2DM (18, 72). AMPK also plays a key role in upregulating the transcription levels of paired box protein 7 (Pax7), myogenic factor 5, myogenin, and MyoD, all of which are necessary for muscle growth. Although metabolic rate is stimulated through AMPK activity, ATP/AMP ratios for the AMPK activation pathways are not affected by obesity (18, 73).
Skeletal muscle isolated from patients with T2DM shows reduced levels of diacylglycerol kinase-delta (DGKδ), a key enzyme in triglyceride biosynthesis required for appropriate AMPK function. DGKs control the expression levels of diacylglycerol (DAG) by catalyzing its conversion to phosphatidic acid utilizing ATP (74). Elevated plasma free fatty acid (FFA) levels from enlarged adipose tissue in obese models force intramyocellular DAG accumulation (75). In an obese population, increased DAG accumulation, secondary to reduced DGK or increased, circulating FFA, results in inhibition of both glucose uptake and glycogen synthesis. This further exacerbates insulin resistance.
Liver and Fat
As previously noted, obesity-associated liver dysfunction can have a profound impact on skeletal muscle maintenance and regeneration. NAFLD commonly occurs in obesity and is correlated with sarcopenia, even in the absence of insulin resistance (76, 77). Loss of muscle mass reduces a key cellular target for insulin action, contributing to glucose intolerance and, in turn, further muscle depletion. In addition, NAFLD is associated with the production of multiple proinflammatory factors, including NF-κB, IL-6, and TNF-α, all of which are known to be protein catabolic (78). VAT also releases circulating FFA, leading to further liver damage (79). Independently, VAT can also result in higher levels of proinflammatory cytokines, similar to the liver (80). Thereby, liver damage and VAT accumulation work synergistically to impair skeletal muscle regeneration in obesity by increasing FFA circulation, proinflammatory cytokines, and limiting promyogenic insulin actions on muscle. These pathways are depicted in Figure 1.
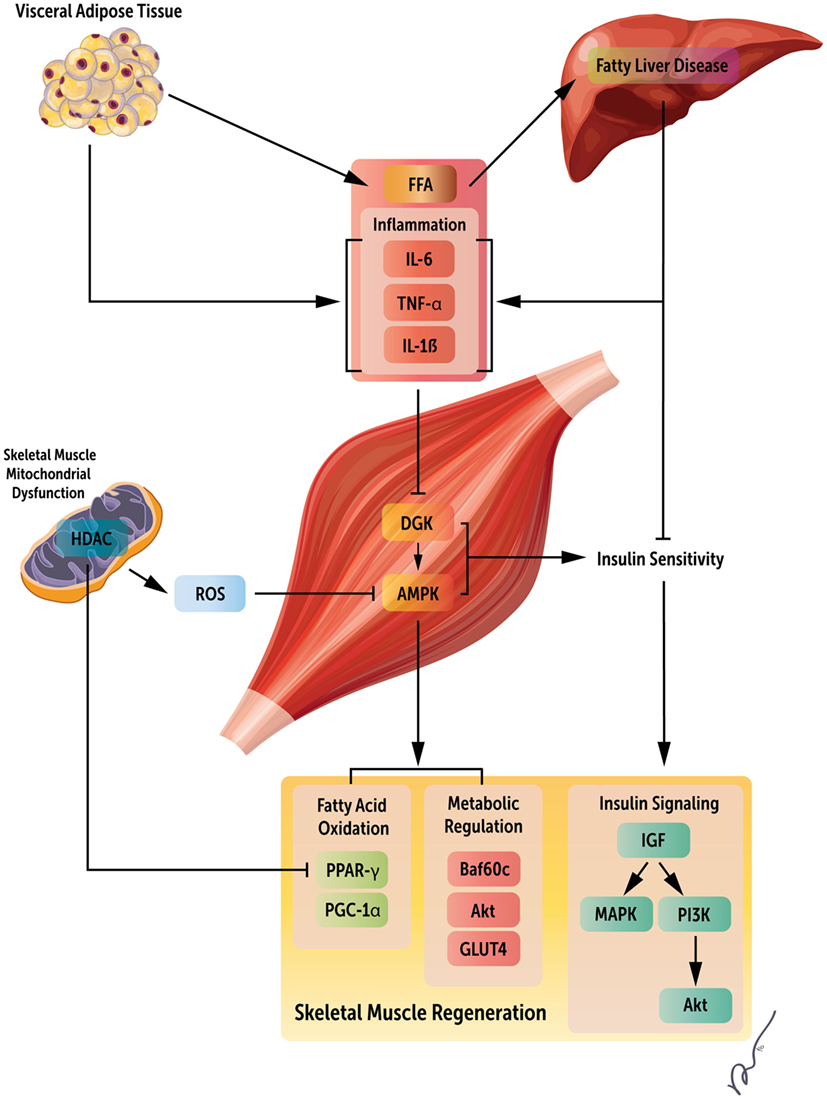
Figure 1. Systemic regulators of obesity mediated loss of skeletal muscle regeneration. Obesity results in both increased visceral adipose tissue and fatty acid accumulation in the liver. These changes manifest as increased circulating fatty acids, inflammatory mediators, and insulin resistance, leading to metabolic derangements within skeletal muscle, and ultimately, decreased skeletal muscle regeneration by the deregulation of multiple signaling pathways. This figure summarizes key factors limiting muscle regeneration in an obese state.
Conclusion and Perspectives
Obesity is accompanied by significant health concerns, including severe loss of skeletal muscle mass. The maintenance of skeletal muscle is necessary for ambulation, proper insulin signaling, and glucose homeostasis. Obesity-related loss of muscle mass perpetuates a cycle of increasing metabolic abnormality, associated liver dysfunction, and further muscle loss. Effective methods to target obesity-associated muscle wasting must account for multiple systemic changes that occur, including increased inflammatory mediators, circulating FFA, metabolic dysfunction, and insulin resistance. Further research is warranted to determine specific molecular mechanisms that limit skeletal muscle regeneration and induce atrophy in an obese state.
Author Contributions
IS and DS drafted the manuscript and DV developed the figure. All the authors contributed to its intellectual content and critical revisions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Guariguata L. By the numbers: new estimates from the IDF diabetes atlas update for 2012. Diabetes Res Clin Pract (2012) 98(3):524–5. doi:10.1016/j.diabres.2012.11.006
2. King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care (1998) 21(9):1414–31. doi:10.2337/diacare.21.9.1414
3. Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R, Kammerer C, et al. Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care (2007) 30(6):1507–12. doi:10.2337/dc06-2537
4. Kim TN, Park MS, Yang SJ, Yoo HJ, Kang HJ, Song W, et al. Prevalence and determinant factors of sarcopenia in patients with type 2 diabetes: the Korean Sarcopenic Obesity Study (KSOS). Diabetes Care (2010) 33(7):1497–9. doi:10.2337/dc09-2310
5. Srikanthan P, Hevener AL, Karlamangla AS. Sarcopenia exacerbates obesity-associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLoS One (2010) 5(5):e10805. doi:10.1371/journal.pone.0010805
6. Wang XH, Du J, Klein JD, Bailey JL, Mitch WE. Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int (2009) 76(7):751–9. doi:10.1038/ki.2009.260
7. Akhmedov D, Berdeaux R. The effects of obesity on skeletal muscle regeneration. Front Physiol (2013) 4:371. doi:10.3389/fphys.2013.00371
8. Goodpaster BH, Thaete FL, Kelley DE. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. Am J Clin Nutr (2000) 71(4):885–92.
9. Jang YC, Sinha M, Cerletti M, Dall’Osso C, Wagers AJ. Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function. Cold Spring Harb Symp Quant Biol (2011) 76:101–11. doi:10.1101/sqb.2011.76.010652
10. Woo M, Isganaitis E, Cerletti M, Fitzpatrick C, Wagers AJ, Jimenez-Chillaron J, et al. Early life nutrition modulates muscle stem cell number: implications for muscle mass and repair. Stem Cells Dev (2011) 20(10):1763–9. doi:10.1089/scd.2010.0349
11. Hu Z, Wang H, Lee IH, Modi S, Wang X, Du J, et al. PTEN inhibition improves muscle regeneration in mice fed a high-fat diet. Diabetes (2010) 59(6):1312–20. doi:10.2337/db09-1155
12. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell (2012) 148(5):852–71. doi:10.1016/j.cell.2012.02.017
13. Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol (1961) 9:493–5. doi:10.1083/jcb.9.2.493
14. Cerletti M, Jurga S, Witczak CA, Hirshman MF, Shadrach JL, Goodyear LJ, et al. Highly efficient, functional engraftment of skeletal muscle stem cells in dystrophic muscles. Cell (2008) 134(1):37–47. doi:10.1016/j.cell.2008.05.049
15. D’Souza DM, Trajcevski KE, Al-Sajee D, Wang DC, Thomas M, Anderson JE, et al. Diet-induced obesity impairs muscle satellite cell activation and muscle repair through alterations in hepatocyte growth factor signaling. Physiol Rep (2015) 3(8):e12506. doi:10.14814/phy2.12506
16. Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE. Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol Biol Cell (2002) 13(8):2909–18. doi:10.1091/mbc.E02-01-0062
17. Fu X, Zhu M, Zhang S, Foretz M, Viollet B, Du M. Obesity impairs skeletal muscle regeneration through inhibition of AMPK. Diabetes (2016) 65(1):188–200. doi:10.2337/db15-0647
18. Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol (2010) 12(2):153–63. doi:10.1038/ncb2015
19. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell (2013) 153(2):376–88. doi:10.1016/j.cell.2013.02.053
20. Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol (2010) 12(2):143–52. doi:10.1038/ncb2014
21. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med (2014) 371(12):1131–41. doi:10.1056/NEJMra1011035
22. Schrauwen-Hinderling VB, Hesselink MK, Schrauwen P, Kooi ME. Intramyocellular lipid content in human skeletal muscle. Obesity (Silver Spring) (2006) 14(3):357–67. doi:10.1038/oby.2006.47
23. Kristiansen OP, Mandrup-Poulsen T. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes (2005) 54(Suppl 2):S114–24. doi:10.2337/diabetes.54.suppl_2.S114
24. Ceddia RP, Lee D, Maulis MF, Carboneau BA, Threadgill DW, Poffenberger G, et al. The PGE2 EP3 receptor regulates diet-induced adiposity in male mice. Endocrinology (2016) 157(1):220–32. doi:10.1210/en.2015-1693
25. Cantini M, Massimino ML, Rapizzi E, Rossini K, Catani C, Dalla Libera L, et al. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem Biophys Res Commun (1995) 216(1):49–53. doi:10.1006/bbrc.1995.2590
26. Tsujinaka T, Fujita J, Ebisui C, Yano M, Kominami E, Suzuki K, et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J Clin Invest (1996) 97(1):244–9. doi:10.1172/JCI118398
27. Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell (2004) 119(2):285–98. doi:10.1016/j.cell.2004.09.027
28. Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J (2013) 280(17):4131–48. doi:10.1111/febs.12338
29. Chen SE, Jin B, Li YP. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am J Physiol Cell Physiol (2007) 292(5):C1660–71. doi:10.1152/ajpcell.00486.2006
30. Loell I, Lundberg IE. Can muscle regeneration fail in chronic inflammation: a weakness in inflammatory myopathies? J Intern Med (2011) 269(3):243–57. doi:10.1111/j.1365-2796.2010.02334.x
31. Kraakman MJ, Murphy AJ, Jandeleit-Dahm K, Kammoun HL. Macrophage polarization in obesity and type 2 diabetes: weighing down our understanding of macrophage function? Front Immunol (2014) 5:470. doi:10.3389/fimmu.2014.00470
32. Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J Obes (Lond) (2015) 39(11):1607–18. doi:10.1038/ijo.2015.104
33. Behan JW, Ehsanipour EA, Sheng X, Pramanik R, Wang X, Hsieh YT, et al. Activation of adipose tissue macrophages in obese mice does not require lymphocytes. Obesity (Silver Spring) (2013) 21(7):1380–8. doi:10.1002/oby.20159
34. Fuentes EN, Björnsson BT, Valdés JA, Einarsdottir IE, Lorca B, Alvarez M, et al. IGF-I/PI3K/Akt and IGF-I/MAPK/ERK pathways in vivo in skeletal muscle are regulated by nutrition and contribute to somatic growth in the fine flounder. Am J Physiol Regul Integr Comp Physiol (2011) 300(6):R1532–42. doi:10.1152/ajpregu.00535.2010
35. Ruiz-Alcaraz AJ, Lipina C, Petrie JR, Murphy MJ, Morris AD, Sutherland C, et al. Obesity-induced insulin resistance in human skeletal muscle is characterised by defective activation of p42/p44 MAP kinase. PLoS One (2013) 8(2):e56928. doi:10.1371/journal.pone.0056928
36. Tan CK, Leuenberger N, Tan MJ, Yan YW, Chen Y, Kambadur R, et al. Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes (2011) 60(2):464–76. doi:10.2337/db10-0801
37. Zhu S, Goldschmidt-Clermont PJ, Dong C. Transforming growth factor-beta-induced inhibition of myogenesis is mediated through Smad pathway and is modulated by microtubule dynamic stability. Circ Res (2004) 94(5):617–25. doi:10.1161/01.RES.0000118599.25944.D5
38. Burks TN, Cohn RD. Role of TGF-β signaling in inherited and acquired myopathies. Skelet Muscle (2011) 1(1):19. doi:10.1186/2044-5040-1-19
39. Farup J, Madaro L, Puri PL, Mikkelsen UR. Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis (2015) 6:e1830. doi:10.1038/cddis.2015.198
40. Dimchev GA, Al-Shanti N, Stewart CE. Phospho-tyrosine phosphatase inhibitor Bpv(Hopic) enhances C2C12 myoblast migration in vitro. Requirement of PI3K/AKT and MAPK/ERK pathways. J Muscle Res Cell Motil (2013) 34(2):125–36. doi:10.1007/s10974-013-9340-2
41. Zhu J, Li Y, Lu A, Gharaibeh B, Ma J, Kobayashi T, et al. Follistatin improves skeletal muscle healing after injury and disease through an interaction with muscle regeneration, angiogenesis, and fibrosis. Am J Pathol (2011) 179(2):915–30. doi:10.1016/j.ajpath.2011.04.008
42. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol (2001) 3(11):1014–9. doi:10.1038/ncb1101-1014
43. Anderson AS, Haynie KR, McMillan RP, Osterberg KL, Boutagy NE, Frisard MI, et al. Early skeletal muscle adaptations to short-term high-fat diet in humans before changes in insulin sensitivity. Obesity (Silver Spring) (2015) 23(4):720–4. doi:10.1002/oby.21031
44. Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest (2008) 118(2):789–800. doi:10.1172/JCI32601
45. Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab (2000) 279(5):E1039–44.
46. Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes (2002) 51(10):2944–50. doi:10.2337/diabetes.51.10.2944
47. Kraegen EW, Cooney GJ, Turner N. Muscle insulin resistance: a case of fat overconsumption, not mitochondrial dysfunction. Proc Natl Acad Sci U S A (2008) 105(22):7627–8. doi:10.1073/pnas.0803901105
48. Ryan TE, Schmidt CA, Green TD, Spangenburg EE, Neufer PD, Mcclung JM. Targeted expression of catalase to mitochondria protects against ischemic myopathy in high-fat diet-fed mice. Diabetes (2016) 65(9):2553–68. doi:10.2337/db16-0387
49. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet (2003) 34(3):267–73. doi:10.1038/ng1180
50. Meng ZX, Li S, Wang L, Ko HJ, Lee Y, Jung DY, et al. Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor-mediated Akt activation. Nat Med (2013) 19(5):640–5. doi:10.1038/nm.3144
51. Brown LA, Lee DE, Patton JF, Perry RA Jr, Brown JL, Baum JI, et al. Diet-induced obesity alters anabolic signalling in mice at the onset of skeletal muscle regeneration. Acta Physiol (Oxf) (2015) 215(1):46–57. doi:10.1111/apha.12537
52. Meng ZX, Wang L, Xiao Y, Lin JD. The Baf60c/Deptor pathway links skeletal muscle inflammation to glucose homeostasis in obesity. Diabetes (2014) 63(5):1533–45. doi:10.2337/db13-1061
53. Izumiya Y, Hopkins T, Morris C, Sato K, Zeng L, Viereck J, et al. Fast/glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab (2008) 7(2):159–72. doi:10.1016/j.cmet.2007.11.003
54. Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, et al. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J (2003) 17(15):2299–301. doi:10.1096/fj.03-0269fje
55. Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol (2004) 2(10):e294. doi:10.1371/journal.pbio.0020294
56. Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol (2005) 3(4):e101. doi:10.1371/journal.pbio.0030101
57. Mcpherron AC, Lee SJ. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest (2002) 109(5):595–601. doi:10.1172/JCI13562
58. Feldman BJ, Streeper RS, Farese RV, Yamamoto KR. Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc Natl Acad Sci U S A (2006) 103(42):15675–80. doi:10.1073/pnas.0607501103
59. Steinbacher P, Eckl P. Impact of oxidative stress on exercising skeletal muscle. Biomolecules (2015) 5(2):356–77. doi:10.3390/biom5020356
60. Martins AR, Nachbar RT, Gorjao R, Vinolo MA, Festuccia WT, Lambertucci RH, et al. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis (2012) 11:30. doi:10.1186/1476-511X-11-30
61. Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell (2015) 16(2):171–83. doi:10.1016/j.stem.2014.12.004
62. Lee C, Kim KH, Cohen P. MOTS-c: a novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic Biol Med (2016) 100:182–7. doi:10.1016/j.freeradbiomed.2016.05.015
63. Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab (2015) 21(3):443–54. doi:10.1016/j.cmet.2015.02.009
64. Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A (2011) 108(35):14608–13. doi:10.1073/pnas.1111308108
65. Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes (2013) 62(3):732–42. doi:10.2337/db12-0548
66. Ye J. Improving insulin sensitivity with HDAC inhibitor. Diabetes (2013) 62(3):685–7. doi:10.2337/db12-1354
67. Consalvi S, Saccone V, Mozzetta C. Histone deacetylase inhibitors: a potential epigenetic treatment for Duchenne muscular dystrophy. Epigenomics (2014) 6(5):547–60. doi:10.2217/epi.14.36
68. Christensen DP, Dahllöf M, Lundh M, Rasmussen DN, Nielsen MD, Billestrup N, et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med (2011) 17(5–6):378–90. doi:10.2119/molmed.2011.00021
69. Richter EA, Hargreaves M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev (2013) 93(3):993–1017. doi:10.1152/physrev.00038.2012
70. Mokbel N, Hoffman NJ, Girgis CM, Small L, Turner N, Daly RJ, et al. Grb10 deletion enhances muscle cell proliferation, differentiation and GLUT4 plasma membrane translocation. J Cell Physiol (2014) 229(11):1753–64. doi:10.1002/jcp.24628
71. McGee SL, Van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, et al. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes (2008) 57(4):860–7. doi:10.2337/db07-0843
72. Zarse K, Ristow M. A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell Metab (2015) 21(3):355–6. doi:10.1016/j.cmet.2015.02.013
73. Scheele C, Nielsen S, Kelly M, Broholm C, Nielsen AR, Taudorf S, et al. Satellite cells derived from obese humans with type 2 diabetes and differentiated into myocytes in vitro exhibit abnormal response to IL-6. PLoS One (2012) 7(6):e39657. doi:10.1371/journal.pone.0039657
74. Jiang LQ, De Castro Barbosa T, Massart J, Deshmukh AS, Löfgren L, Duque-Guimaraes DE, et al. Diacylglycerol kinase-δ regulates AMPK signaling, lipid metabolism, and skeletal muscle energetics. Am J Physiol Endocrinol Metab (2016) 310(1):E51–60. doi:10.1152/ajpendo.00209.2015
75. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am (2008) 37(3):635–46. doi:10.1016/j.ecl.2008.06.007
76. Hong HC, Hwang SY, Choi HY, Yoo HJ, Seo JA, Kim SG, et al. Relationship between sarcopenia and nonalcoholic fatty liver disease: the Korean Sarcopenic Obesity Study. Hepatology (2014) 59(5):1772–8. doi:10.1002/hep.26716
77. Milić S, Lulić D, Štimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol (2014) 20(28):9330–7. doi:10.3748/wjg.v20.i28.9330
78. Sakuma K, Yamaguchi A. Sarcopenia and cachexia: the adaptations of negative regulators of skeletal muscle mass. J Cachexia Sarcopenia Muscle (2012) 3(2):77–94. doi:10.1007/s13539-011-0052-4
79. Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology (2010) 51(2):679–89. doi:10.1002/hep.23280
Keywords: skeletal muscle, obesity, inflammation, metabolism, insulin
Citation: Sinha I, Sakthivel D and Varon DE (2017) Systemic Regulators of Skeletal Muscle Regeneration in Obesity. Front. Endocrinol. 8:29. doi: 10.3389/fendo.2017.00029
Received: 14 October 2016; Accepted: 01 February 2017;
Published: 16 February 2017
Edited by:
Sushil Kumar Mahata, University of California San Diego, USAReviewed by:
Sana Siddiqui, University of California San Francisco, USAJiyoung Park, Ulsan National Institute of Science and Technology, South Korea
Copyright: © 2017 Sinha, Sakthivel and Varon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Indranil Sinha, aXNpbmhhQHBhcnRuZXJzLm9yZw==