 Natalie Payne1*†
Natalie Payne1*† Leigh Combrink2†
Leigh Combrink2† Simona Kraberger3
Simona Kraberger3 Rafaela S. Fontenele3Kara Schmidlin3
Rafaela S. Fontenele3Kara Schmidlin3 Ivonne Cassaigne4Melanie Culver1,2,5
Ivonne Cassaigne4Melanie Culver1,2,5 Arvind Varsani3,6Koenraad Van Doorslaer1,7,8
Arvind Varsani3,6Koenraad Van Doorslaer1,7,8- 1Genetics Graduate Interdisciplinary Program, University of Arizona, Tucson, AZ, United States
- 2School of Natural Resources and the Environment, University of Arizona, Tucson, AZ, United States
- 3The Biodesign Center for Fundamental and Applied Microbiomics, Center for Evolution and Medicine, School of Life Sciences, Arizona State University, Tempe, AZ, United States
- 4Primero Conservation, Portal, AZ, United States
- 5U.S. Geological Survey, Arizona Cooperative Fish and Wildlife Research Unit, University of Arizona, Tucson, AZ, United States
- 6Structural Biology Research Unit, Department of Integrative Biomedical Sciences, University of Cape Town, Observatory, Cape Town, South Africa
- 7School of Animal and Comparative Biomedical Sciences, University of Arizona, Tucson, AZ, United States
- 8The BIO5 Institute, Department of Immunobiology, Cancer Biology Graduate Interdisciplinary Program, UA Cancer Center, University of Arizona, Tucson, AZ, United States
With viruses often having devastating effects on wildlife population fitness and wild mammals serving as pathogen reservoirs for potentially zoonotic diseases, determining the viral diversity present in wild mammals is both a conservation and One Health priority. Additionally, transmission from more abundant hosts could increase the extinction risk of threatened sympatric species. We leveraged an existing circular DNA enriched metagenomic dataset generated from bobcat (Lynx rufus, n = 9) and puma (Puma concolor, n = 13) scat samples non-invasively collected from Sonora, Mexico, to characterize fecal DNA viromes of each species and determine the extent that viruses are shared between them. Using the metaWRAP pipeline to co-assemble viral genomes for comparative metagenomic analysis, we observed diverse circular DNA viruses in both species, including circoviruses, genomoviruses, and anelloviruses. We found that differences in DNA virome composition were partly attributed to host species, although there was overlap between viruses in bobcats and pumas. Pumas exhibited greater levels of alpha diversity, possibly due to bioaccumulation of pathogens in apex predators. Shared viral taxa may reflect dietary overlap, shared environmental resources, or transmission through host interactions, although we cannot rule out species-specific host-virus coevolution for the taxa detected through co-assembly. However, our detection of integrated feline foamy virus (FFV) suggests Sonoran pumas may interact with domestic cats. Our results contribute to the growing baseline knowledge of wild felid viral diversity. Future research including samples from additional sources (e.g., prey items, tissues) may help to clarify host associations and determine the pathogenicity of detected viruses.
1. Introduction
Infectious diseases are playing a critical role in wildlife population conservation (Lewis et al., 2017). Through reducing the survival and reproduction of individuals (Woodroffe, 1999; Deem et al., 2001), parasites and infectious diseases can generate trophic cascades (Frainer et al., 2018; Baruzzi et al., 2022), contributing to significant wildlife population declines, affecting multiple species in a community (Pedersen et al., 2007). Furthermore, the transmission of generalist parasites or infectious agents to threatened species can increase the extinction risk of wild animals (Daszak et al., 1999; Woodroffe, 1999; Lafferty and Gerber, 2002; Pedersen et al., 2007). This is evident in carnivores (Woodroffe, 1999; Lafferty and Gerber, 2002; Pedersen et al., 2007), which appear to be particularly susceptible to long term impacts of epizootic diseases at the population-level (Malmberg et al., 2021).
Pathogen surveillance and understanding the viral diversity and potential disease threats associated with more abundant host species may reveal emerging infectious diseases and help prevent future outbreaks and subsequent potential loss of sympatric threatened species. More than just a conservation concern, an increased understanding of the viral diversity in wildlife and the potential for spillover to other species is essential for effectively managing future outbreaks (Olival et al., 2017; Carroll et al., 2018) and is a One Health priority (at the nexus of human, animal and environmental health; Mazzamuto et al., 2022).For example, a study on juvenile and adult red foxes (Vulpes vulpes) in peri-urban areas in Croatia noted the dominant presence of fox picobirnavirus and parvovirus in fecal samples, as well as a novel fox circovirus (Lojkić et al., 2016). With red foxes being the most abundant carnivore in the Northern Hemisphere and the novel fox circovirus being very similar to circoviruses isolated from diseased dogs in USA and Italy, it seems possible to posit that red foxes could serve as wildlife virus reservoirs (Lojkić et al., 2016). Such virome characterizations of carnivores are particularly relevant to advancing One Health priorities, with the order Carnivora being ranked among the top five mammalian orders as a source of zoonotic pathogens (Keesing and Ostfeld, 2021). Monitoring wildlife diseases (and particularly wildlife viromes) using non-invasive fecal sample collection is a nascent field (Pannoni et al., 2022; Mazzamuto et al., 2022; Schilling et al., 2022) and could bridge the gap between passive (e.g., voluntary disease reporting) and active wildlife disease surveillance (e.g., submission of samples from hunted game; Cardoso et al., 2022).
The sociality of a species can also affect the spread of infectious diseases (Sah et al., 2018). In group living or social species, group size was thought to influence disease transmission dynamics (Kappeler et al., 2015; Sah et al., 2018), with larger groups and animals living at higher population densities having higher parasite prevalence and burden (Patterson and Ruckstuhl, 2013; Albery et al., 2020). However, recent research suggests that animals can spatially organize their groups to minimize infections (Albery et al., 2020) and that the interactions within a social group and not only its size drive the spread of infectious diseases (Sah et al., 2018).
Social interactions are not limited to group living, as some relatively solitary species have been shown to exhibit complex social networks with a variety of social partners and interactions (Sah et al., 2018). Thus, despite many felids being solitary, the use of shared environmental resources, as well as occasional conflict or predation within and among felid species, creates opportunities for inter- and intra-specific pathogen transmission.
The Sonoran desert is a unique ecoregion home to four species of wild solitary felids: two being more common, bobcat (Lynx rufus) and puma (Puma concolor), and two listed as endangered, the ocelot (Leopardus pardalis) and jaguar (Panthera onca). Currently, little is known about the exact disease threats and viral diversity associated with these felids. As such, Sonoran desert felids provide both the conservation need and a unique opportunity to assess levels of viral diversity present within and shared between these populations of closely related sympatric host carnivore species. Bobcats and pumas, as the more abundant felids, are easier to sample, and surveys of viral diversity in these species may serve as a proxy for virome characterization or indication of potential viral spillover for the rarer felids.
In this paper, we leveraged a collection of fecal samples of wild bobcat and puma from Sonora, Mexico to determine (1) what DNA viruses are present in wild felids in Sonora and (2) the similarity of fecal DNA viromes between these sympatric species. These data will contribute to the growing field of wildlife viromics and to our understanding of the viral diversity present in wild mammalian species.
2. Methods
2.1. Sample collection and processing
Bobcat and puma scat samples were collected from Sonora, Mexico, between 2012 and 2014 (Figure 1). Scats possibly of felid origin, were collected only if determined to be fresh, based on color, moisture, smell, and texture. Host DNA was extracted using Qiagen’s DNeasy Blood and Tissue Kit, and species identification was performed through Sanger sequencing of a region of the mitochondrial cytochrome B gene (Verma and Singh, 2002; Naidu et al., 2011; Cassaigne et al., 2016), as previously described for these samples (Payne et al., 2020). Thirteen puma and nine bobcat scat samples were randomly selected for DNA virome analysis (Payne et al., 2020). Separate viral DNA extraction from scat cross-sections, circular viral DNA amplification, and library preparation followed the protocol in Payne et al. (2020). Sequencing libraries were generated using the TruSeq Nano DNA Sample Preparation kit and sequenced on an Illumina HiSeq 4,000 (2 × 100 bp) at Macrogen Inc. (Korea) in 2018.
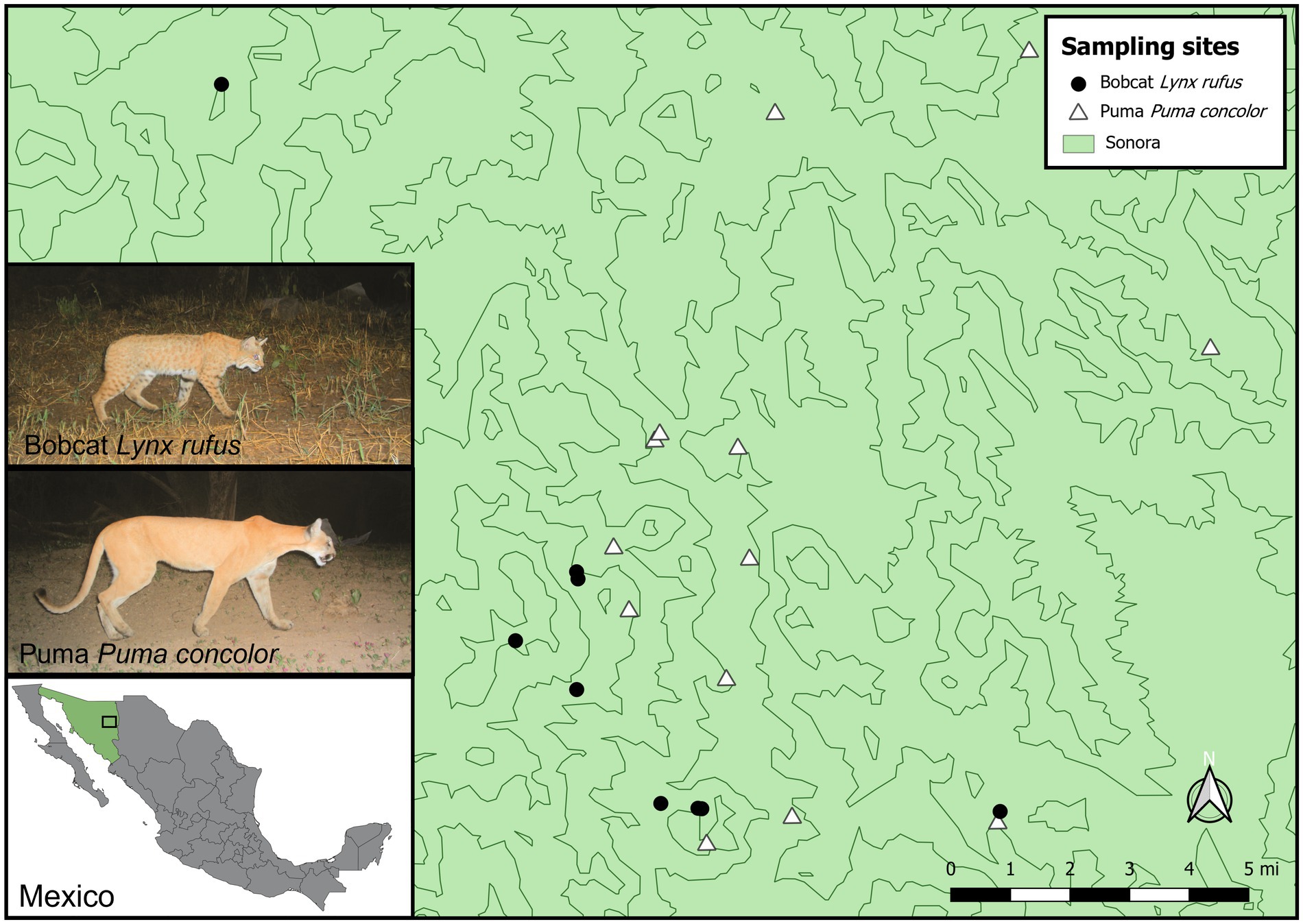
Figure 1. Map of bobcat and puma scat sampling sites in Sonora, Mexico. Camera trap photo insets show a bobcat and puma photographed at the same site in Sonora in 2011 (Photos: Primero Conservation and Jesús Moreno from the Management Unit for the Conservation of Wildlife (UMA) “Refugio Privado de Jaguares Silvestres”). Shapefiles: “States of Mexico”. Downloaded from http://tapiquen-sig.jimdo.com. Carlos Efraín Porto Tapiquén. Orogénesis Soluciones Geográficas. Porlamar, Venezuela 2015.
2.2. Bioinformatics and analyses
The metaWRAP pipeline v. 1.3.2 (Uritskiy et al., 2018) was used to process raw sequencing reads for comparative metagenomic analysis. Within the metaWRAP read_qc module, reads were trimmed using default parameters with Trim Galore v. 0.5.0 (Krueger, 2022) as a wrapper around Cutadapt v. 1.18 (Martin, 2011), human contamination was removed with BMTagger v. 3.101 (Rotmistrovsky and Agarwala, 2011) using the hg38 human genome assembly (GCA_000001405.15), and read quality was assessed with FastQC v. 0.11.8 (Andrews, 2010). Untrimmed reads with human contamination removed were deposited in the Sequence Read Archive. Reads from all samples were co-assembled using metaSPAdes v. 3.14.1 (Bankevich et al., 2012; Nurk et al., 2017) with default parameters outside of metaWRAP, and the scaffolds were then used in the metaWRAP assembly module, allowing for assembly of unused reads with MEGAHIT v. 1.1.3 (Li et al., 2015). We elected to perform one co-assembly across all samples to achieve better detection of rare taxa and allow for direct comparison of bobcat and puma virome composition. The metaWRAP Kraken 2 module was used to run Kraken 2 (Wood et al., 2019) and generate Krona plots (Ondov et al., 2011) to assess taxonomic composition of contigs in the final assembly, reads from individual samples (subset to 1,000,000 reads), and all pooled reads for each host species, using the premade Kraken 2 viral database (v. 9/8/2022 available at https://benlangmead.github.io/aws-indexes/k2). Contig abundances (in genome copies per million reads) were estimated with Salmon v. 0.13.1 (Patro et al., 2017) using the metaWRAP quant_bins module, and contig taxonomy was assigned using a megablast search against the NCBI nucleotide database (available at https://ftp.ncbi.nlm.nih.gov/blast/db/, downloaded 10/20/2022) using blast v. 2.13.0 (Altschul et al., 1990) outside of metaWRAP for v5 database compatibility, with the output further processed by the metaWRAP classify_bins module for pruning blast hits and assigning taxonomy with Taxator-tk (Dröge et al., 2015). CheckV v. 1.0.1 (Nayfach et al., 2020) was used to assess quality and completeness of viral contigs. Contigs assigned as viral (and not designated as phages) by classify_bins and which were determined to be complete or high-quality (>90% complete) viral genomes with CheckV were retained for community analyses in R. All downstream analyses were repeated with a second set of viral contigs (>66.7% genome completeness), referred to as our “lower-completeness” set (resulting figures in Supplementary Figures S31–S37). The CheckV quality summary, final taxnomic assignment, top blast hit, and abundance per sample of each contig in the high and lower completeness set can be found in Supplementary Table 1.
The R package vegan v. 2.5-7 (Oksanen et al., 2020) was used to conduct viral community ecology analyses on the contigs representing high-quality viral genomes. Alpha diversity metrics (species richness, Simpson Diversity Index, and Shannon Diversity Index) were calculated for each sample, and Wilcoxon rank sum tests were used to determine if significant differences in alpha diversity exist between bobcats and pumas. Beta diversity metrics were calculated among all pairs of samples, both considering contig abundances (Bray–Curtis Dissimilarity Index; abundances were in genome copies per million reads, as estimated by quant_bins) and considering contig presence/absence (Jaccard distance). Kruskal–Wallis rank sum tests were used to determine if beta diversity differed significantly between host species pairs, and Dunn’s test was used post hoc to determine which comparisons differed significantly using the FSA R package v. 0.9.3 (Ogle et al., 2022).
Vegan was also used to conduct ordination analyses non-metric multidimensional scaling (NMDS) and principal coordinates analysis (PCoA) with both Bray–Curtis and Jaccard distance matrices, and visualizations were generated using ggplot2 (Wickham, 2016) and ggord (Beck, 2022). To further assess differences in virome composition due to host species, permutational multivariate analyses of variance (PERMANOVA) and analyses of similarity (ANOSIM) were conducted using Bray–Curtis and Jaccard distance matrices with vegan’s adonis2 function and anosim function, respectively. To assess correlations between geographic Euclidean distance and beta diversity, separate Mantel tests were performed using Bray–Curtis and Jaccard distance matrices.
3. Results
3.1. MetaWRAP
We obtained 4,044 contigs of 1,000 to 164,510 nts in length after co-assembly (prior to identification of viral sequences), and Krona plots generated by the Kraken 2 module show the viral taxa represented in the assembly (Supplementary Figure S1), bobcat reads (Supplementary Figure S2), puma reads (Supplementary Figure S3), and individual sample reads (Supplementary Figures S4–S25). Of the portion of the final assembly matching the Kraken 2 viral database (with contig taxonomy weighted by contig length and coverage), 48% represented viruses within Monodnaviria, with 28% identified as belonging to circoviruses (Figure 2). Viruses within the family Anelloviridae comprised 16% of the viral portion of the assembly, and phages within the class Caudoviricetes comprised 33%, reflecting viruses likely associated with enteric bacteria of the felids. When reads from each sample of the same host species were pooled (Figure 2), 96% of bobcat viral reads matched to those of the viruses in the family Circoviridae, while puma viral reads largely represented viruses in the families Genomoviridae (51%), Retroviridae (felispumavirus, 14%), Anelloviridae (8%), and class Caudoviricetes (25%). The computational analysis identified 38 complete genomes and 16 additional high-quality viral contigs (using CheckV and classify_bins taxonomy results, not including phages), which were used for downstream analyses. We included an additional 34 medium-quality contigs in our “lower-completeness” set.
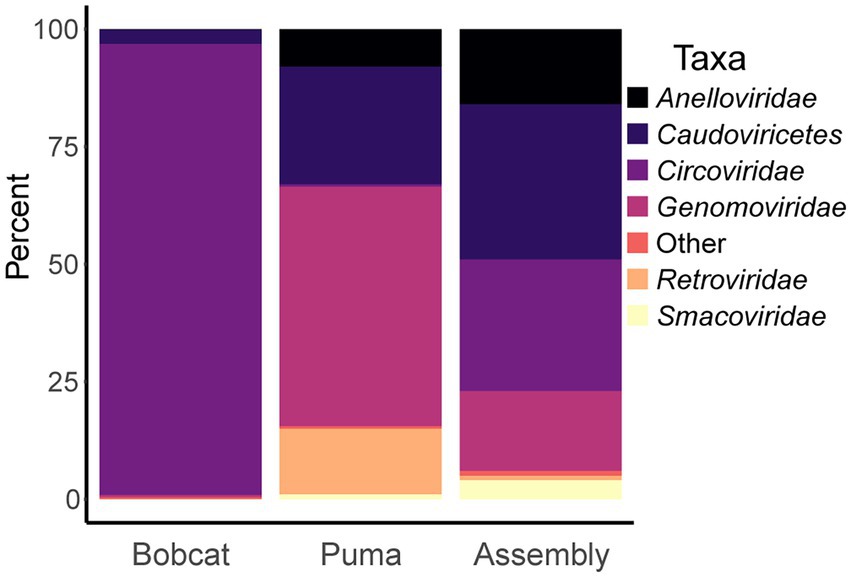
Figure 2. Bar plots showing taxonomy of reads matching viral database from bobcat and puma samples, as well as taxonomy of contigs matching viral database. Contigs are from the final assembly generated by metaSPAdes and MEGAHIT, and metaWRAP weights taxonomy results of contigs based on length and coverage.
3.2. Alpha and beta diversity
Species richness, Shannon Diversity Index, and Simpson Diversity Index were the alpha diversity metrics calculated for all samples. We observed a wider range of species richness values for pumas (Figure 3A), and pumas had higher median values for each of these metrics (Supplementary Figures S26, S27), although differences in alpha diversity metrics among bobcats and pumas were not significant (richness: p = 0.1677; Shannon: p = 0.1264; Simpson: p = 0.1264). However, using our “lower-completeness” set of viral contigs, we found richness differed significantly between pumas and bobcats (p < 0.05; Figure 3C). Median beta diversity values were greatest between pumas and bobcats and lowest among pumas (Figure 3B and Supplementary Figure S28), and both Bray–Curtis and Jaccard distances were significantly different among different host species pairings (p < 0.01 for both distances), with significant differences among puma–puma pairings and other host species pairings (p-adj < 0.05 and p-adj < 0.01 with Bray–Curtis and Jaccard distances, respectively, for puma–bobcat pairings and p-adj < 0.05 with both distances for bobcat–bobcat pairings). However, using our “lower-completeness” set of contigs and Jaccard distance (Figure 3D), significant differences among host species pairs were explained by puma–bobcat pairings having significantly higher beta diversity than puma–puma (p-adj < 0.01) and bobcat–bobcat pairings (p-adj < 0.05).
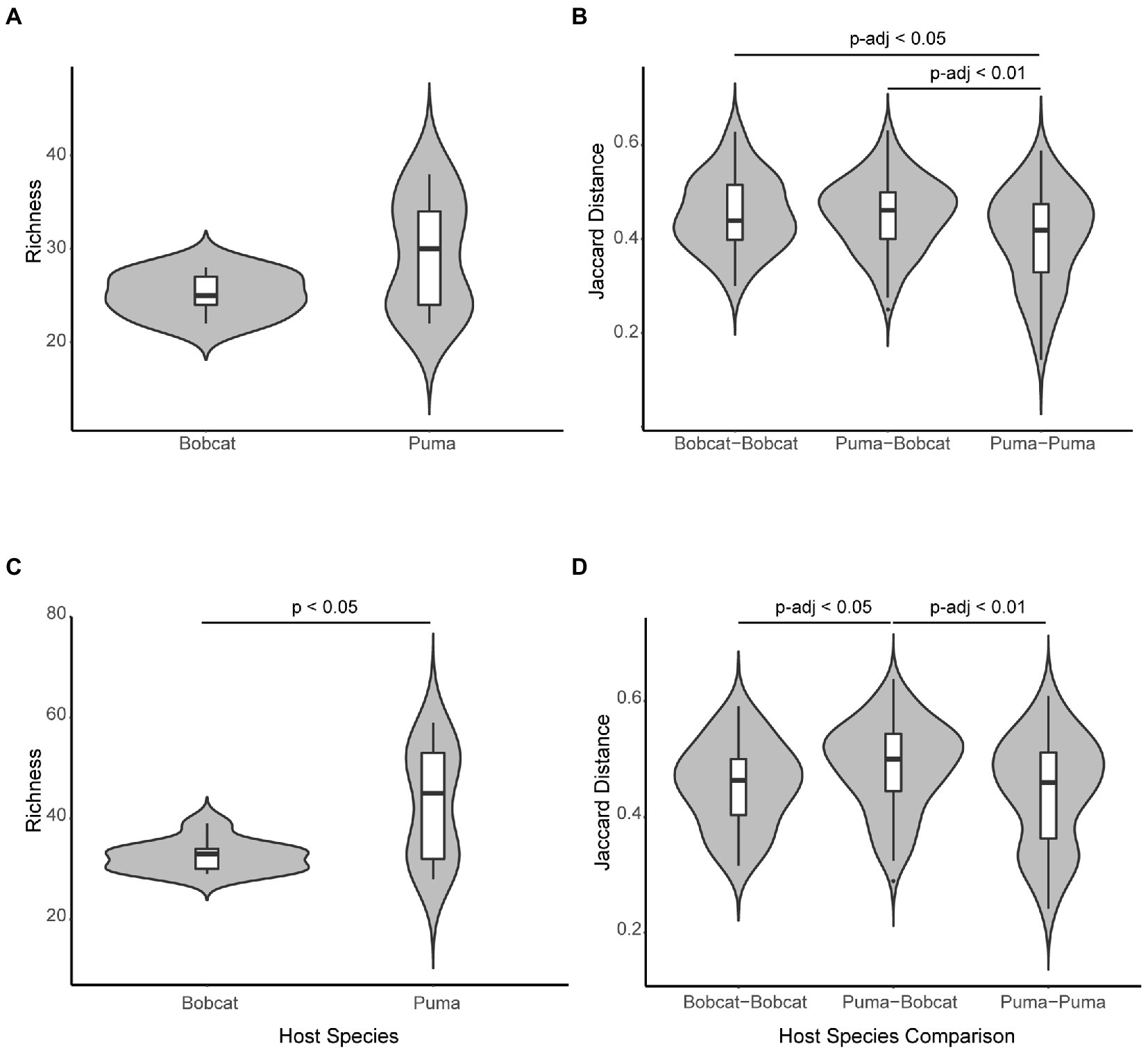
Figure 3. Violin plots showing alpha and beta diversity. (A) Species richness of bobcats and pumas, using contigs representing high-quality or complete viral genomes. (B) Jaccard distances between bobcats, between pumas and bobcats, and between pumas, using contigs representing high-quality or complete viral genomes. Mean Jaccard distance differed significantly between puma–puma pairings and other groups (p-adj < 0.01 for puma–bobcat pairings and p-adj < 0.05 for bobcat–bobcat pairings). (C) Species richness of bobcats and pumas, using viral contigs in the “lower-completeness” set. Mean species richness differed significantly between bobcats and pumas (p < 0.05). (D) Jaccard distance between bobcats, between pumas and bobcats, and between pumas, using viral contigs in the “lower-completeness” set. Mean Jaccard distance differed significantly between puma and bobcat pairings and other groups (p-adj < 0.01 for puma–puma pairings and p-adj < 0.05 for bobcat–bobcat pairings).
3.3. Effect of host species and geographic distance
The NMDS and PCoA plots reveal an extensive overlap between bobcat and puma viral communities (Figure 4 and Supplementary Figures S29, S30), although pumas and bobcats with the highest richness levels tended to cluster separately in the NMDS based on Bray–Curtis distances (stress = 0.155), and both PCoA and NMDS (stress = 0.211) based on Jaccard distances revealed puma samples outlying the region of overlap between the two species clusters. The PERMANOVAs revealed a significant effect of host species on community composition using Jaccard distances (p < 0.05, R2 = 0.09418), but not Bray–Curtis distances (p = 0.497, R2 = 0.04395). ANOSIM revealed significant differences between host species using both Jaccard (p < 0.05, R = 0.1901) and Bray–Curtis distances (p < 0.05, R = 0.1441). Differences between host species remained significant using the more complex “lower-completeness” set using Jaccard distances (PERMANOVA: p < 0.01, R2 = 0.12081; ANOSIM: p < 0.01, R = 0.2639) and ANOSIM with Bray–Curtis distances (p < 0.05, R = 0.1513; PERMANOVA: p = 0.649, R2 = 0.0398). Mantel tests did not reveal significant correlations between geographic distance and beta diversity for either Bray–Curtis (p = 0.813, r = −0.1011) or Jaccard distances (p = 0.165, r = 0.1191).
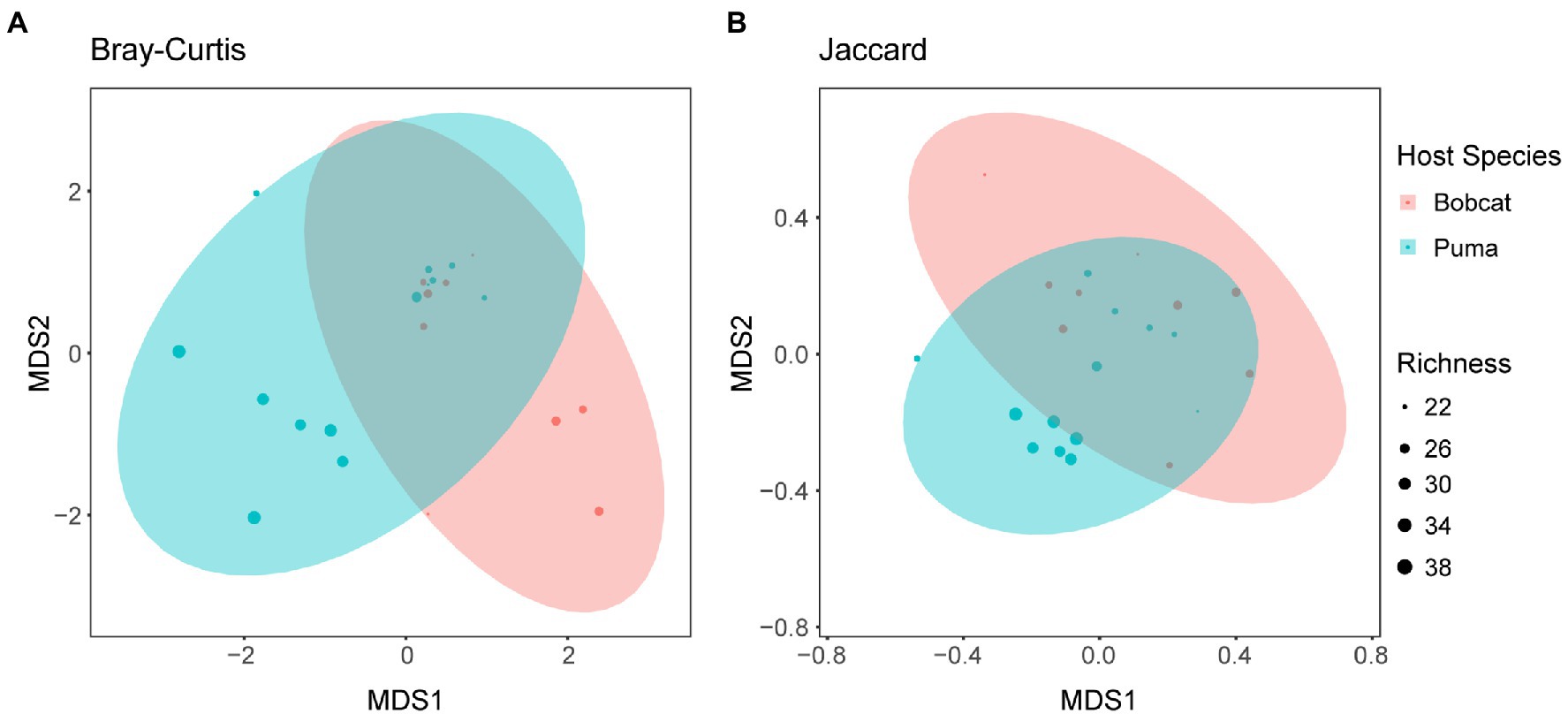
Figure 4. NMDS plots generated using (A) Bray–Curtis dissimilarity (stress = 0.155) and (B) Jaccard distance (stress = 0.211), using contigs representing high-quality or complete viral genomes. Each point represents the viral community composition within a specific sample. Points are colored by host species, and point size is proportional to species richness of each sample. Ellipses corresponding to the two host species groups are shown at the 95% confidence level. Axes (MDS1 and MDS2) correspond to the two axes of variation.
4. Discussion
Determining the viral diversity present in wildlife is essential for the management and control of emerging infectious diseases. Owing to the large potential for zoonotic spillover, characterizing mammalian viromes is vital to achieving One Health priorities. In non-invasively collected scat samples from wild pumas and bobcats, we observed diverse circular DNA viruses, including circoviruses, genomoviruses, and anelloviruses. Given that rolling circle amplification (RCA) was performed prior to sequencing, we expected to find a high proportion of circular DNA viruses present (although non-circular DNA is not excluded from sequencing). Specifically, one of the complete viral genomes, with particularly high prevalence in two bobcat samples, was identified as Sonfela circovirus 1, the genome of which was originally identified in these two samples (Payne et al., 2020). Additionally, the high proportion of contigs with homology to anelloviruses was also expected, as the diversity of anellovirus genomes isolated from these samples has been previously characterized (Kraberger et al., 2021).
Interestingly, we documented the presence of feline foamy virus (FFV) within our dataset as a 4.4 kb contig primarily derived from one puma sample (72% of the reads). This suggests that this is an integrated FFV and was detected through carry-over of host DNA. FFV, a contact-dependent, multi-host adapted retrovirus, is known to cause lifelong infection in both domestic and wild felid species (Linial, 2000; Dannemiller et al., 2020), including pumas and domestic cats (Felis catus; Kechejian et al., 2019; Kraberger et al., 2020). Most studies document prevalence in domestic cats, with comparatively few detecting the presence of FFV in wild felids (Dannemiller et al., 2020). Pumas have shown a high prevalence of FFV and a high frequency of intraspecies transmission in other studies (Kechejian et al., 2019; Dannemiller et al., 2020; Kraberger et al., 2020). Additionally, frequent cross-species spillover of FFV has been documented from domestic cats to pumas due to depredation events (Kraberger et al., 2020), and the presence of the virus in a Sonoran puma may indicate that interactions between wild and domestic felids have occurred. However, it is also possible that this was a result of social spillover from another puma. While FFV is generally considered apathogenic, clinically silent infection has been associated with histopathological changes in domestic cats (German et al., 2008; Ledesma-Feliciano et al., 2019), and further research is needed to clarify implications for feline health.
Our analyses also indicate extensive overlap between bobcat and puma DNA viral communities. The broader diversity of viruses observed in pumas may result from exposure to a wider variety of prey species. Pumas have been observed to predate a broad range of taxa, including ungulates, mesocarnivores, and small mammals (Cassaigne et al., 2016; Meyer et al., 2020), whereas bobcats are known to primarily specialize on rodents and lagomorphs (Hass, 2009; López-Vidal et al., 2014; Meyer et al., 2020). Furthermore, apex predators are known to experience greater bioaccumulation of viruses (Malmberg et al., 2021). Pumas are also known to occasionally prey upon smaller felids such as bobcats (Hass, 2009; Prude and Cain III, 2021), and previous studies have documented pathogen transmission from bobcat to puma, putatively through competitive contact or depredation (Franklin et al., 2007; Lee et al., 2017; Malmberg et al., 2021). While such interactions may facilitate viral transmission to Sonoran pumas, the presence of shared viral taxa in bobcats and pumas does not necessarily indicate cross-species transmission. As both pumas and bobcats are known to prey on small mammals, and bobcats have been known to predate deer on occasion (Leopold and Krausman, 1986; McKinney and Smith, 2007), shared viral taxa may instead reflect dietary overlap or shared environmental resources, such as water sources. For example, we identified complete viral genomes matching the rodent anelloviruses Neotofec virus NeonRodL2_5 and Neotofec virus NeonRodL2_6 in bobcats and a partial genome matching Dipodfec virus NeonRodF1_131 (within the phylum Cressdnaviricota) in pumas. These viruses were first isolated from white-throated woodrats (Neotoma albigula) and Merriam’s kangaroo rat (Dipodomys merriami), respectively, which could be suitable prey items for both felid species (Meyer et al., 2020). Alternatively, shared taxa that infect these felids may be co-evolved within each species. However, we were unable to determine the strains present within each sample (and species) since contigs were generated by co-assembly. Future research including samples from other sources (e.g., prey items, tissue samples) might help to clarify such host associations.
We found that geographic distance among scat samples did not have a significant effect on DNA virome composition at this spatial scale. Our results support previous findings of low levels of spatial autocorrelation of pathogen exposure in pumas and bobcats in Florida, Colorado, and California (Gilbertson et al., 2016). Instead, DNA virome composition appears to be shaped by a combination of host species dependent and independent factors, with extensive virome composition overlap observed between host species using ordination analyses, while PERMANOVA and ANOSIM revealed small yet significant effects of host species. Although pseudoreplication is not suspected, the possibility of some individuals being represented by more than one sample may contribute to the observed effect of host species on DNA virome composition.
Despite providing insight into the possible interactions between host and viral communities, further research is needed to clarify the implications of these results for Sonoran felid health. Fecal virome characterization of non-invasively collected scat samples includes many novel and known viruses derived from the scat depositors as well as prey species and environmental contacts, so the host associations and pathogenicity of each virus in the metagenome is unknown. Of the major viral taxa identified here, viruses in the family Circoviridae may be of most interest in terms of bobcat and puma health. Circoviruses are known as the smallest animal pathogens that replicate autonomously (Fisher et al., 2020). They are found in a number of species [freshwater fish, birds, bats, chimpanzees, minks, elk, and humans (Rosario et al., 2017; Fisher et al., 2020)], although their presence is often subclinical (Fisher et al., 2020). In some birds, circoviruses are considered potentially immunosuppressive (Todd, 2000), suggesting that concurrent co-infections could increase the symptoms and severity of disease (Fisher et al., 2020). Some circoviruses are known to cause clinical disease such as hemorrhagic gastroenteritis in dogs (Anderson et al., 2017; Kotsias et al., 2019), often fatal postweaning multisystemic wasting syndrome in pigs (Chae, 2005; Segalés et al., 2005) and virus (BFDV) in birds [beak and feather disease virus (Todd, 2000)]. The identification of potentially disease-causing circoviruses in bobcats and pumas is of concern for both wild felid population health and conservation. With their propensity for displaying tissue tropism (Todd, 2000), transmission of these circoviruses from more abundant/common species to threatened wild felids could result in catastrophic population declines, particularly if these felids are already immunocompromised from co-infections. Furthermore, the high abundance of Sonfela circovirus 1 reads in two bobcat samples may suggest an active infection. However, with these viruses being identified from non-invasively collected scat samples, further study is needed to clarify host associations and consequences for feline health. Although important feline pathogens present in the scats may have been missed through analysis of the circular viral DNA enriched dataset, these results contribute to the documentation of viral diversity in wild felids. Future studies coupling the characterization of broader virome composition and disease dynamics across sympatric populations of wild mammals could help with the identification of viral threats to wildlife, as well as potentially to humans and domestic animals.
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found at: NCBI; PRJNA922235.
Author contributions
NP: conceptualization, data analysis, writing, and manuscript preparation. LC: conceptualization, writing, manuscript preparation, and supervision. SK, RF, and KS: data collection. IC: sample collection. MC: supervision. AV and KD: data collection and supervision. All authors contributed to the article and approved the submitted version.
Funding
The high throughput sequencing work and viral molecular work was supported by a startup grant awarded to AV from Arizona State University.
Acknowledgments
The authors would like to acknowledge members of the Culver and Van Doorslaer labs for their support, and we acknowledge Melissa Bergeman for assistance in sample preparation. We also thank Bonnie Hurwitz and Karla Vargas for review and comments on previous versions of this manuscript. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Although this code has been processed successfully on a computer system at the U.S. Geological Survey (USGS), no warranty expressed or implied is made regarding the display or utility of the data for other purposes, nor on all computer systems, nor shall the act of distribution constitute any such warranty. The USGS or the U.S. Government shall not be held liable for improper or incorrect use of the data described and/or contained herein.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1126149/full#supplementary-material
References
Albery, G. F., Newman, C., Ross, J. B., MacDonald, D. W., Bansal, S., and Buesching, C. (2020). Negative density-dependent parasitism in a group-living carnivore. Proc. R. Soc. B 287:20202655. doi: 10.1098/rspb.2020.2655
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anderson, A., Hartmann, K., Leutenegger, C. M., Proksch, A. L., Mueller, R. S., and Unterer, S. (2017). Role of canine circovirus in dogs with acute haemorrhagic diarrhoea. Vet. Rec. 180:542. doi: 10.1136/vr.103926
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data [online]. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Baruzzi, C., Barton, B. T., and Lashley, M. A. (2022). Could parasite outbreaks be mediated by interspecific competition? The case of Entomophthora, blowflies, and vultures. Ecology 104:e3916. doi: 10.1002/ecy.3916
Beck, M. W. (2022). ggord: Ordination Plots with ggplot2. R package version 1.1.7. Available at: https://zenodo.org/badge/latestdoi/35334615
Cardoso, B., García-Bocanegra, I., Acevedo, P., Cáceres, G., Alves, P. C., and Gortázar, C. (2022). Stepping up from wildlife disease surveillance to integrated wildlife monitoring in Europe. Res. Vet. Sci. 144, 149–156. doi: 10.1016/j.rvsc.2021.11.003
Carroll, D., Daszak, P., Wolfe, N. D., Gao, G. F., Morel, C. M., Morzaria, S., et al. (2018). The global virome project. Science 359, 872–874. doi: 10.1126/science.aap7463
Cassaigne, I., Medellín, R. A., Thompson, R. W., Culver, M., Ochoa, A., Vargas, K., et al. (2016). Diet of pumas (Puma concolor) in Sonora, Mexico, as determined by GPS kill sites and molecular identified scat, with comments on jaguar (Panthera onca) diet. Southwest Nat. 61, 125–132. doi: 10.1894/0038-4909-61.2.125
Chae, C. (2005). A review of porcine circovirus 2-associated syndromes and diseases. Vet. J. 169, 326–336. doi: 10.1016/j.tvjl.2004.01.012
Dannemiller, N. G., Kechejian, S., Kraberger, S., Logan, K., Alldredge, M., Crooks, K. R., et al. (2020). Diagnostic uncertainty and the epidemiology of feline foamy virus in pumas (Puma concolor). Sci. Rep. 10, 1–7. doi: 10.1038/s41598-020-58350-7
Daszak, P., Berger, L., Cunningham, A. A., Hyatt, A. D., Green, D. E., and Speare, R. (1999). Emerging infectious diseases and amphibian population declines. Emerg. Infect. Dis. 5, 735–748. doi: 10.3201/eid0506.990601
Deem, S. L., Karesh, W. B., and Weisman, W. (2001). Putting theory into practice: wildlife health in conservation. Conserv. Biol. 15, 1224–1233. doi: 10.1046/j.1523-1739.2001.00336.x
Dröge, J., Gregor, I., and McHardy, A. C. (2015). Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 31, 817–824. doi: 10.1093/bioinformatics/btu745
Fisher, M., Harrison, T. M. R., Nebroski, M., Kruczkiewicz, P., Rothenburger, J. L., Ambagala, A., et al. (2020). Discovery and comparative genomic analysis of elk circovirus (ElkCV), a novel circovirus species and the first reported from a cervid host. Sci. Rep. 10:19548. doi: 10.1038/s41598-020-75577-6
Frainer, A., McKie, B. G., Amundsen, P.-A., Knudsen, R., and Lafferty, K. D. (2018). Parasitism and the biodiversity-functioning relationship. Trends Ecol. Evol. 33, 260–268. doi: 10.1016/j.tree.2018.01.011
Franklin, S. P., Troyer, J. L., Terwee, J. A., Lyren, L. M., Boyce, W. M., Riley, S. P. D., et al. (2007). Frequent transmission of immunodeficiency viruses among bobcats and pumas. J. Virol. 81, 10961–10969. doi: 10.1128/JVI.00997-07
German, A. C., Harbour, D. A., Helps, C. R., and Gruffydd-Jones, T. J. (2008). Is feline foamy virus really apathogenic? Vet. Immunol. Immunopathol. 123, 114–118. doi: 10.1016/j.vetimm.2008.01.035
Gilbertson, M. L. J., Carver, S., VandeWoude, S., Crooks, K. R., Lappin, M. R., and Craft, M. E. (2016). Is pathogen exposure spatially autocorrelated? Patterns of pathogens in puma (Puma concolor) and bobcat (Lynx Rufus). Ecosphere 7:e01558. doi: 10.1002/ecs2.1558
Hass, C. C. (2009). Competition and coexistence in sympatric bobcats and pumas. J. Zool. 278, 174–180. doi: 10.1111/j.1469-7998.2009.00565.x
Kappeler, P. M., Cremer, S., and Nunn, C. L. (2015). Sociality and health: impacts of sociality on disease susceptibility and transmission in animal and human societies. Philos. Trans. R. Soc. B Biol. Sci. 370:20140116. doi: 10.1098/rstb.2014.0116
Kechejian, S. R., Dannemiller, N., Kraberger, S., Ledesma-Feliciano, C., Malmberg, J., Parker, M. R., et al. (2019). Feline foamy virus is highly prevalent in free-ranging puma concolor from Colorado, Florida and southern California. Viruses 11, 1–9. doi: 10.3390/v11040359
Keesing, F., and Ostfeld, R. S. (2021). Impacts of biodiversity and biodiversity loss on zoonotic diseases. Proc. Natl. Acad. Sci. 118:e2023540118. doi: 10.1073/pnas.2023540118
Kotsias, F., Bucafusco, D., Nuñez, D. A., Lago Borisovsky, L. A., Rodriguez, M., and Bratanich, A. C. (2019). Genomic characterization of canine circovirus associated with fatal disease in dogs in South America. PLoS One 14:e0218735. doi: 10.1371/journal.pone.0218735
Kraberger, S., Fountain-Jones, N. M., Gagne, R. B., Malmberg, J., Dannemiller, N. G., Logan, K., et al. (2020). Frequent cross-species transmissions of foamy virus between domestic and wild felids. Virus Evol. 6:vez058. doi: 10.1093/ve/vez058
Kraberger, S., Serieys, L. E., Richet, C., Fountain-Jones, N. M., Baele, G., Bishop, J. M., et al. (2021). Complex evolutionary history of felid anelloviruses. Virology 562, 176–189. doi: 10.1016/j.virol.2021.07.013
Krueger, F. (2022). Trim Galore. Available at: https://github.com/FelixKrueger/TrimGalore (Accessed December 9, 2022).
Lafferty, K. D., and Gerber, L. R. (2002). Good medicine for conservation biology: the intersection of epidemiology and conservation theory. Conserv. Biol. 16, 593–604. doi: 10.1046/j.1523-1739.2002.00446.x
Ledesma-Feliciano, C., Troyer, R. M., Zheng, X., Miller, C., Cianciolo, R., Bordicchia, M., et al. (2019). Feline foamy virus infection: characterization of experimental infection and prevalence of natural infection in domestic cats with and without chronic kidney disease. Viruses 11:662. doi: 10.3390/v11070662
Lee, J., Malmberg, J. L., Wood, B. A., Hladky, S., Troyer, R., Roelke, M., et al. (2017). Feline immunodeficiency virus cross-species transmission: implications for emergence of new lentiviral infections. J. Virol. 91, e02134–e02116. doi: 10.1128/JVI.02134-16
Leopold, B. D., and Krausman, P. R. (1986). Diets of 3 predators in big bend National Park, Texas. J. Wildl. Manag. 50, 290–295. doi: 10.2307/3801915
Lewis, J. S., Logan, K. A., Alldredge, M. W., Carver, S., Bevins, S. N., Lappin, M., et al. (2017). The effects of demographic, social, and environmental characteristics on pathogen prevalence in wild felids across a gradient of urbanization. PLoS One 12:e0187035. doi: 10.1371/journal.pone.0187035
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Linial, M. (2000). Why aren’t foamy viruses pathogenic? Trends Microbiol. 8, 284–289. doi: 10.1016/S0966-842X(00)01763-7
Lojkić, I., Biđin, M., Prpić, J., Šimić, I., Krešić, N., and Bedeković, T. (2016). Faecal virome of red foxes from peri-urban areas. Comp. Immunol. Microbiol. Infect. Dis. 45, 10–15. doi: 10.1016/j.cimid.2016.01.005
López-Vidal, J. C., Elizalde-Arellano, C., Hernández, L., Laundré, J. W., González-Romero, A., and Cervantes, F. A. (2014). Foraging of the bobcat (Lynx Rufus) in the Chihuahuan Desert: generalist or specialist? Southwest Nat. 59, 157–166. doi: 10.1894/F01-CLG-59.1
Malmberg, J. L., White, L. A., and Vandewoude, S. (2021). Bioaccumulation of pathogen exposure in top predators. Trends Ecol. Evol. 36, 411–420. doi: 10.1016/j.tree.2021.01.008
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mazzamuto, M. V., Schilling, A.-K., and Romeo, C. (2022). Wildlife disease monitoring: methods and perspectives. Animals 12:3032. doi: 10.3390/ani12213032
McKinney, T., and Smith, T. W. (2007). Diets of sympatric bobcats and coyotes during years of varying rainfall in Central Arizona. West. North Am. Nat. 67, 8–15. doi: 10.3398/1527-0904(2007)67[8:dosbac]2.0.co;2
Meyer, J. M., Leempoel, K., Losapio, G., and Hadly, E. A. (2020). Molecular ecological network analyses: an effective conservation tool for the assessment of biodiversity, trophic interactions, and community structure. Front. Ecol. Evol. 8:588430. doi: 10.3389/fevo.2020.588430
Naidu, A., Smythe, L. A., Thompson, R. W., and Culver, M. (2011). Genetic analysis of scats reveals minimum number and sex of recently documented mountain lions. J. Fish Wildl. Manag. 2, 106–111. doi: 10.3996/042010-JFWM-008
Nayfach, S., Camargo, A. P., Schulz, F., Eloe-Fadrosh, E., Roux, S., and Kyrpides, N. C. (2020). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585. doi: 10.1038/s41587-020-00774-7
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Ogle, D. H., Doll, J. C., Wheeler, P., and Dinno, A. (2022). FSA: Fisheries stock analysis. Available at: https://github.com/fishR-Core-Team/FSA.
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2020). Vegan: Community ecology package. R package version 2. 5–7. Available at: https://cran.r-project.org/package=vegan.
Olival, K. J., Hosseini, P. R., Zambrana-Torrelio, C., Ross, N., Bogich, T. L., and Daszak, P. (2017). Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650. doi: 10.1038/nature22975
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a web browser. BMC Bioinf. 12:385. doi: 10.1186/1471-2105-12-385
Pannoni, S. B., Proffitt, K. M., and Holben, W. E. (2022). Non-invasive monitoring of multiple wildlife health factors by fecal microbiome analysis. Ecol. Evol. 12:e8564. doi: 10.1002/ece3.8564
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon: fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Patterson, J. E., and Ruckstuhl, K. E. (2013). Parasite infection and host group size: a meta-analytical review. Parasitology 140, 803–813. doi: 10.1017/S0031182012002259
Payne, N., Kraberger, S., Fontenele, R. S., Schmidlin, K., Bergeman, M. H., Cassaigne, I., et al. (2020). Novel circoviruses detected in feces of Sonoran felids. Viruses 12:1027. doi: 10.3390/v12091027
Pedersen, A. B., Jones, K. E., Nunn, C. L., and Altizer, S. (2007). Infectious diseases and extinction risk in wild mammals. Conserv. Biol. 21, 1269–1279. doi: 10.1111/j.1523-1739.2007.00776.x
Prude, C. H., and Cain, J. W. III (2021). Habitat diversity influences puma Puma concolor diet in the Chihuahuan Desert. Wildl. Biol. :wlb.00875. doi: 10.2981/wlb.00875
Rosario, K., Breitbart, M., Harrach, B., Segalés, J., Delwart, E., Biagini, P., et al. (2017). Revisiting the taxonomy of the family Circoviridae: establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 162, 1447–1463. doi: 10.1007/s00705-017-3247-y
Rotmistrovsky, K., and Agarwala, R. (2011). BMTagger: Best match tagger for removing human reads from metagenomics datasets. Available at: https://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/
Sah, P., Mann, J., and Bansal, S. (2018). Disease implications of animal social network structure: a synthesis across social systems. J. Anim. Ecol. 87, 546–558. doi: 10.1111/1365-2656.12786
Schilling, A.-K., Mazzamuto, M. V., and Romeo, C. (2022). A review of non-invasive sampling in wildlife disease and Health Research: What’s new? Animals 12:1719. doi: 10.3390/ani12131719
Segalés, J., Allan, G. M., and Domingo, M. (2005). Porcine circovirus diseases. Anim. Health Res. Rev. 6, 119–142. doi: 10.1079/ahr2005106
Todd, D. (2000). Circoviruses: immunosuppressive threats to avian species: a review. Avian Pathol. 29, 373–394. doi: 10.1080/030794500750047126
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158–113. doi: 10.1186/s40168-018-0541-1
Verma, S. K., and Singh, L. (2002). Novel universal primers establish identity of an enormous number of animal species for forensic application. Mol. Ecol. Notes 3, 28–31. doi: 10.1046/j.1471-8286.2003.00340.x
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
Keywords: viromes, comparative metagenomics, wildlife, bobcat (Lynx rufus), puma (Puma concolor), virus, Sonoran desert
Citation: Payne N, Combrink L, Kraberger S, Fontenele RS, Schmidlin K, Cassaigne I, Culver M, Varsani A and Van Doorslaer K (2023) DNA virome composition of two sympatric wild felids, bobcat (Lynx rufus) and puma (Puma concolor) in Sonora, Mexico. Front. Ecol. Evol. 11:1126149. doi: 10.3389/fevo.2023.1126149
Edited by:
Terri Maness, Louisiana Tech University, United StatesReviewed by:
Jill Pecon-Slattery, National Zoological Park (SI), United StatesMichael Tessler, St. Francis College, United States
Andrew Skidmore, University of New Mexico Health Sciences Center, United States
Copyright © 2023 Payne, Combrink, Kraberger, Fontenele, Schmidlin, Cassaigne, Culver, Varsani and Van Doorslaer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalie Payne, bmF0YWxpZXJtZXJjZXJAYXJpem9uYS5lZHU=
†These authors have contributed equally to this work