 Xin Wang
Xin Wang Haotian Chen1†
Haotian Chen1† Bingding Huang
Bingding Huang- 1College of Big Data and Internet, Shenzhen Technology University, Shenzhen, China
- 2Chengdu NBbiolab. CO., LTD., SME Incubation Park, Chengdu, China
Introduction: Traditional methods for constructing synthetic nanobody libraries are labor-intensive and time-consuming. This study introduces a novel approach leveraging protein large language models (LLMs) to generate germline-specific nanobody sequences, enabling efficient library construction through statistical analysis.
Methods: We developed NanoAbLLaMA, a protein LLM based on LLaMA2, fine-tuned using low-rank adaptation (LoRA) on 120,000 curated nanobody sequences. The model generates sequences conditioned on germlines (IGHV3-301 and IGHV3S5301). Training involved dataset preparation from SAbDab and experimental data, alignment with IMGT germline references, and structural validation using ImmuneBuilder and Foldseek.
Results: NanoAbLLaMA achieved near-perfect germline generation accuracy (100% for IGHV3-301, 95.5% for IGHV3S5301). Structural evaluations demonstrated superior predicted Local Distance Difference Test (pLDDT) scores (90.32 ± 10.13) compared to IgLM (87.36 ± 11.17), with comparable TM-scores. Generated sequences exhibited fewer high-risk post-translational modification sites than IgLM. Statistical analysis of CDR regions confirmed diversity, particularly in CDR3, enabling the creation of synthetic libraries with high humanization (>99.9%) and low risk.
Discussion: This work establishes a paradigm shift in nanobody library construction by integrating LLMs, significantly reducing time and resource demands. While NanoAbLLaMA excels in germline-specific generation, limitations include restricted germline coverage and framework flexibility. Future efforts should expand germline diversity and incorporate druggability metrics for clinical relevance. The model’s code, data, and resources are publicly available to facilitate broader adoption.
1 Introduction
Nanobodies, derived from the heavy-chain antibodies of camelids, are single-domain antibody fragments that lack light chains (Hamers-Casterman et al., 1993). They possess unique characteristics, such as high stability and ease of production, which are of significant importance for diagnostics, therapeutics, and molecular research (Mullin et al., 2024). The antigen-binding site of nanobodies is composed of three complementarity-determining regions (CDR1, CDR2, and CDR3), which play a crucial role in antigen recognition and binding. CDR1 and CDR2 are relatively short and primarily provide auxiliary functions for antigen binding, while CDR3 is longer and highly diverse, serving as the key region for antigen binding. The rapid development of nanobodies highlights the importance of constructing diverse and high-quality nanobody libraries (Kunz et al., 2018).
Traditional methods for preparing synthetic nanobody libraries typically use a defined protein framework as a template, with artificial rules for designing the CDR3 region sequences. Although effective, these methods are usually time-consuming and restrictive, limiting the speed of discovery and development (Valdés-Tresanco et al., 2022). Nowadays, there are already some synthetic binding protein databases, such as the SYNBIP database. The nanobody libraries generated in this database are also potential synthetic protein binders (Li et al., 2024). With the continuous breakthroughs in the field of deep learning, especially the emergence of protein large language models (ProLLM), a new approach has been provided for the construction of nanobody libraries (Strokach and Kim, 2022).
Protein large language models, after training on a vast dataset of protein sequences and structural data, have the capability to generate new protein sequences with desired characteristics. These models use deep learning techniques to accurately understand protein sequences and predict protein folding to generate required protein sequences (Varadi et al., 2021; Ofer et al., 2021). By integrating these models into the construction process of nanobody libraries, researchers can significantly improve the efficiency, diversity, and specificity of library generation.
This paper explores the innovative application of protein large language models in constructing nanobody libraries. We trained the model on a nanobody dataset and evaluated its effectiveness in generating a diverse range of nanobody sequences. The model is capable of generating the required nanobody sequences conditioned on different germlines. By leveraging the predictive power of protein large language models, we aim to provide a new method for the preparation of nanobody libraries.
2 Methods
2.1 Nanobody sequence dataset
In the training of NanoAbLLaMA, we utilized ProLLaMA as the underlying model framework. ProLLaMA is a large language model for proteins that has been pre-trained on the LLaMA2 framework, specializing in protein language. Its superior scalability allows for the utilization of natural language to formulate user instructions and to create our dataset (Lv et al., 2024). We extracted nanobody sequences from SAbDab database and experimental datasets (Dunbar et al., 2013). The experimental dataset is the base sequence, since the final goal is to create a synthetic nanobody library for germlines IGHV3-
2.2 Model architecture and training framework
Large Language Model Meta AI 2 (LLaMA2) is the second-generation large language model developed by Meta, based on the classic Transformer architecture (Touvron et al., 2023). We train on the LLaMA2 framework using Low-Rank Adaptation (LoRA) (Hu et al., 2021) to reduce training costs. LoRA is an efficient method for fine-tuning large pre-trained models, aiming to reduce the computational resources and storage space required for fine-tuning. LoRA introduces low-rank matrix decomposition technology, decomposing the model’s weight matrix into two low-rank matrices, thereby greatly reducing the number of parameters that need to be updated. Conceptually, fine-tuning can be thought of as a process of finding parameter changes (Ding et al., 2023). Let W
Here,
We add LoRA to every decoder in LLaMA2, including

Figure 1. The overall architecture of NanoAbLLaMA. We added low-rank adapters (LoRA) to certain weights. During training, we freeze these weights and other parameters, focusing only on training LoRA.
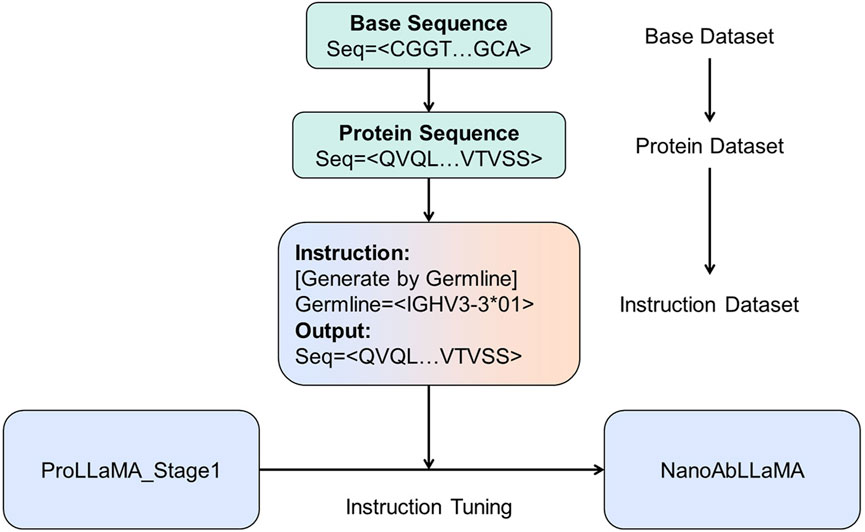
Figure 2. The training process of NanoAbLLaMA. The base data is translated from our own base data to obtain protein sequences, which are then made into instruction datasets for training.
2.3 Synthetic library
Aim for a synthetic library with high humanization and fewer risk sites, we independently generate CDR1, CDR2, and CDR3. To create a synthetic library, we need to generate a certain amount of data for statistical analysis and then create a synthetic library based on the statistical patterns. To ensure that the synthetic library can be applied in practice, we use frameworks with high humanization and fewer risk sites to generate our data. We obtained the required frameworks from the SAbDab database (Schneider et al., 2021). For example, in the germline IGHV3-
And in the germline IGHV3S
These two frameworks have a humanization rate as high as 99.9%, with fewer than 10 risk sites, making them very suitable for use as frameworks for generation. We generated 10,000 nanobody sequences for CDR1 and CDR2 regions using these two frameworks. Due to the lower diversity in CDR1 and CDR2 regions, the overall repetition rate is relatively high, but this also ensures the correctness of the statistical results. However, due to the higher diversity in the CDR3 region, we generated a total of 100,000 unique nanobody sequences. To ensure the correctness of the statistical results, for the germline IGHV3-
3 Results
3.1 NanoAbLLaMA
Our model NanoAbLLaMA was trained on 120,000 nanobody sequences for full-length nanobody sequence generation. NanoAbLLaMA can generate sequences conditioned on germline (IGHV3-
3.2 Controllable germline generation
To verify that the results generated by our model are valid, we generated 10,000 unique sequences for each of the two germlines (IGHV3-
The results shown in Table 1 indicate that NanoAbLLaMA can generate corresponding nanobody sequences based on instructions for the required germline, thus achieving controllable germline generation.

Table 1. Germline generation accuracy. Our NanoAbLLaMA achieved nearly 100% accuracy for the generation of both germlines.
3.3 Nanobody sequence generation
We compared our model with other state-of-the-art models in the field of antibody design, such as the Immunoglobulin Language Model (IgLM) (Shuai et al., 2023), which is a generative language model that uses bidirectional context to design antibody sequence spans of different lengths and is trained on a large-scale natural antibody dataset. IgLM can generate full-length antibody sequences conditioned on chain type and source species. To ensure the correctness of the calculation results, we selected the most abundant data for statistics, that is, sequences with lengths of 110 aa to 130 aa, and selected 10 sequences of each length for statistical averaging. We used ImmuneBuilder (Abanades et al., 2023) to predict the structure of the sequences. ImmuneBuilder is a set of deep learning models specifically for predicting the structures of antibodies, nanobodies, and T cell receptors, which is highly accurate and much faster than AlphaFold2. We calculated the predicted Local Distance Difference Test (pLDDT) (Varadi et al., 2021) based on the predicted structure, and pLDDT is used to measure whether the sequence is structurally reasonable. At the same time, we used Foldseek (van Kempen et al., 2024) to calculate the average template modeling score (TM-Score) (Zhang and Skolnick, 2004) and the root mean square deviation (RMSD), which reflect the degree of structural similarity. TM-score focuses more on the overall structure, while RMSD is more sensitive to the size and local changes of protein structures. The results are shown in Figure 3, our NanoAbLLaMA has a better pLDDT score, indicating that NanoAbLLaMA, through training on nanobody sequence data, can produce structurally reasonable nanobody sequences. The average value and standard deviation of pLDDT for the nanobody sequences generated by NanoAbLLaMA are 90.32 ± 10.13, while those for IgLM are 87.36 ± 11.17 Figure 4 displays the 3D structure of the generated sequences, and Table 2 shows the structural scores.
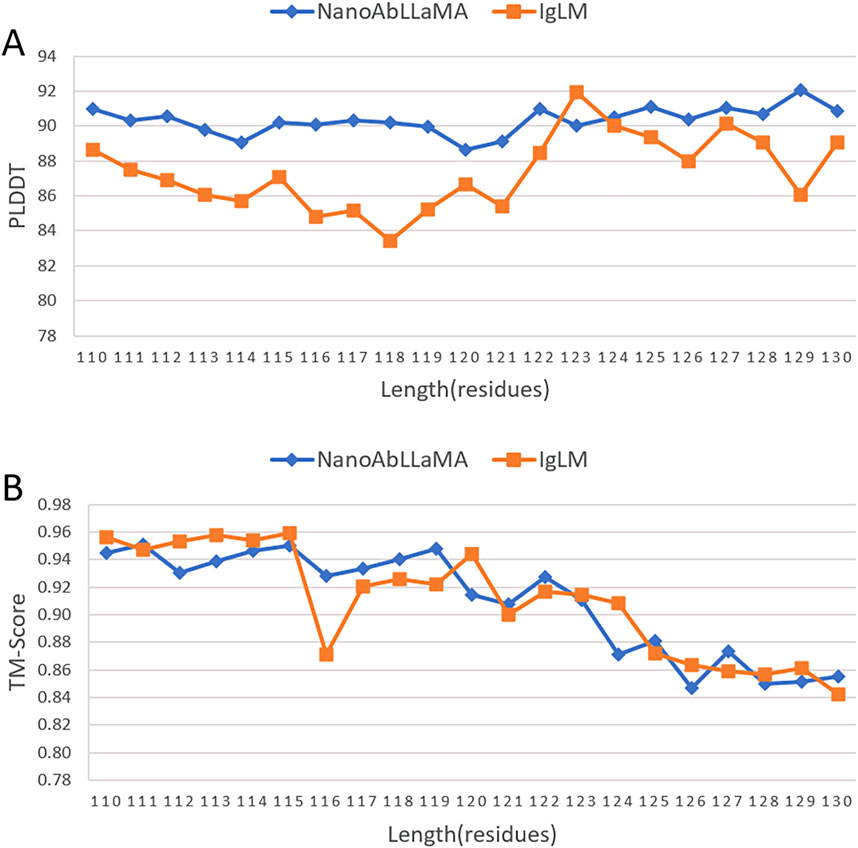
Figure 3. Structural experiments of NanoAbLLaMA. (A) Compared with IgLM, NanoAbLLaMA has a higher pLDDT score. (B) The TM-Score score is comparable to IgLM.
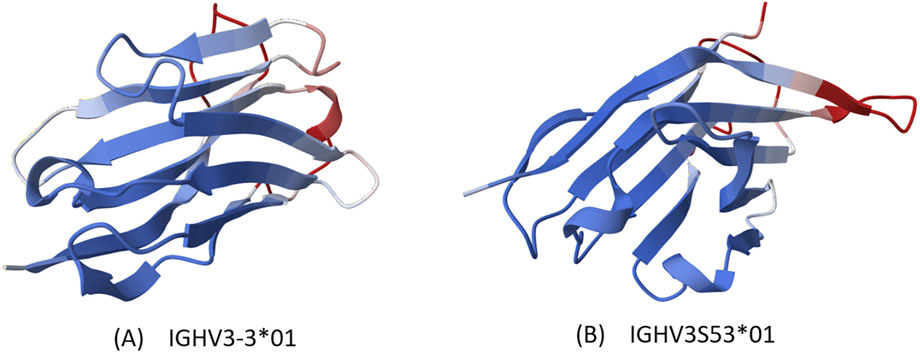
Figure 4. Protein visualization. Structures generated by AlphaFold based on sequences, with color representing credibility, blue being more credible. (A) IGHV3-3*01 (B) IGHV3S53*01.
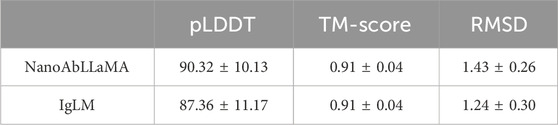
Table 2. Structural scoring table.
At the same time, the production of nanobody synthetic libraries also pays attention to post-translational modifications (PTM) (Ramazi and Zahiri, 2021) of proteins, which are common risks in biopharmaceutical development. Mainly including: Oxidation, Glycosylation, Hydrolysis, etc. Therefore, corresponding detection was also carried out. The results are shown in Figure 5, the number of high-risk sites in the sequences generated by our NanoAbLLaMA is less than that of IgLM.
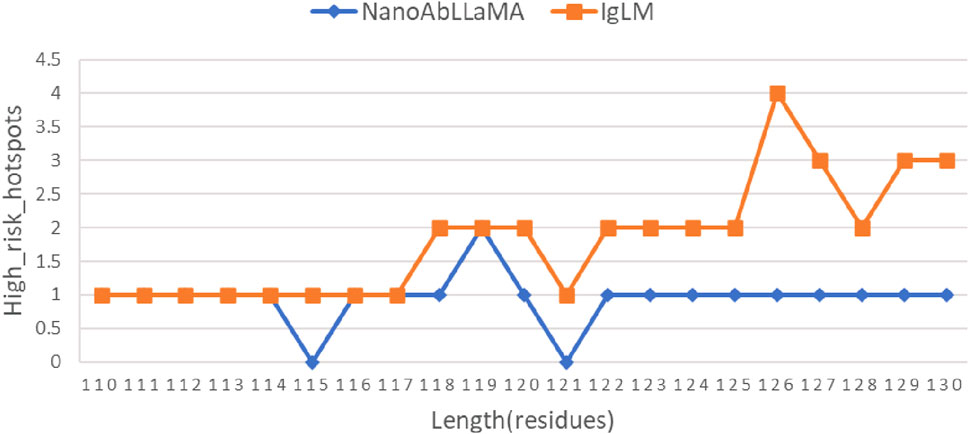
Figure 5. Comparison of the number of high-risk sites. The number of high-risk sites in the first half is quite similar, and the number of high-risk sites in our model is lower in the second half.
After generating sequences with NanoAbLLaMA, we still need to make a synthetic library, so we need to statistically determine the frequency of each amino acid site and make a synthetic library based on germline, framework, and amino acid frequency. Figure 6 is a SeqLogo created based on a portion of the data we generated.
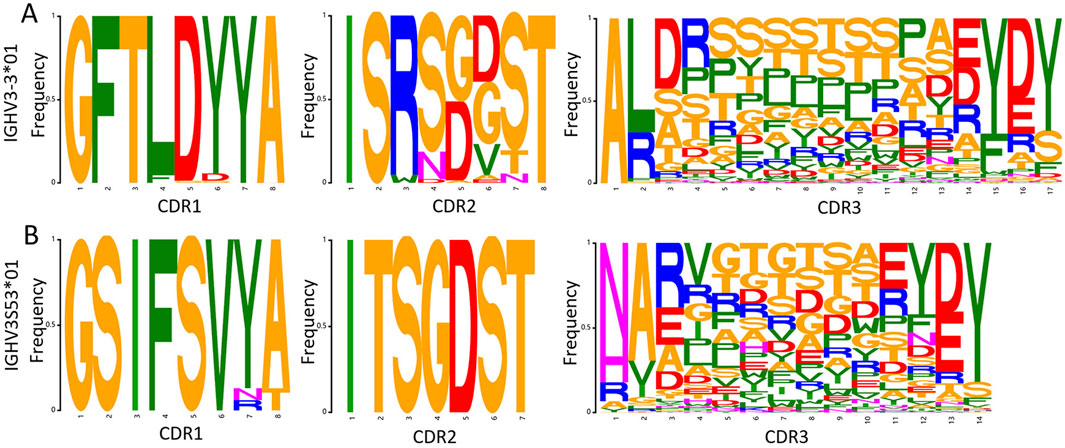
Figure 6. Amino acid frequency of each CDR region for the two germlines. (A) Amino acid frequency for germline IGHV3-
We utilized Discovery Studio to analyze the disulfide bond information of the generated sequences. The analysis revealed that all sequences contain a conserved disulfide bond connecting FR1 (C23) and FR3 (C104), which plays a crucial role in maintaining the structural stability of the protein. This conserved disulfide bond is a common feature in many proteins, contributing to their overall stability and functionality. However, no additional disulfide bonds were identified in the sequences.
4 Discussion
In this study, we propose an innovative approach to construct nanobody synthesis libraries using the nanobody large language model NanoAbLLaMA. This method not only improves the efficiency of library construction, but also provides new ideas for the design of nanobodies. The following is an in-depth discussion of our findings.
First of all, traditional nanobody library construction methods often rely on multi-step operations in the laboratory, involving many complex steps such as single-stranded antibody collection, vector preparation, and insertion of single-stranded antibody sequences. These methods are time-consuming and costly, limiting the widespread use of nanobodies. Our NanoAbLLaMA model leverages the powerful generation capabilities of large language models to rapidly generate high-quality nanobody sequences and generate libraries based on statistical analysis, significantly reducing the time and resource consumption of library construction.
Secondly, the existing antibody language models, such as AbLang (Olsen et al., 2022) and IgLM(Shuai et al., 2023), cannot meet our requirements for generating nanobody sequences based on germline, and most of these models use species and chain type as conditions to generate sequences. The NanoAbLLaMA model is trained using a low-rank adaptive technique, which allows it to be fine-tuned for specific germlines. The effectiveness of this strategy was validated in our experiments, where the model was able to generate the expected nanoantibody sequences and excelled in diversity and specificity. These results indicate that the combination of the flexibility of large language models and the advantages of targeted training can effectively improve the design efficiency and quality of nanobodies.
However, there are some limitations to this study. First, although NanoAbLLaMA has achieved good results in generating germline-specific nanoantibody sequences, it is unable to cover more germlines and cannot specify the framework to generate CDR sequences. Future research may consider expanding the training dataset to cover a wider range of germlines, or modifying the training mode of the model to improve the model’s capabilities.
In addition, factors such as druggability still need to be paid attention to in practical application. These factors are critical for the clinical application of nanobodies and therefore need to be explored in depth in follow-up studies.
5 Conclusion
Existing methods for making nanobody synthetic libraries are mature but laborious. In this work, we introduced a new way to make nanobody synthetic libraries by generating nanobody sequences with protein large language models and making nanobody synthetic libraries based on statistical results. We also developed NanoAbLLaMA, a ProLLM that can generate nanobody sequences based on germline. Experiments show that NanoAbLLaMA has excellent performance.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
XW: Conceptualization, Project administration, Resources, Supervision, Writing–original draft, Writing–review and editing. HC: Conceptualization, Data curation, Formal Analysis, Software, Visualization, Writing–original draft, Writing–review and editing. BC: Conceptualization, Resources, Writing–review and editing. LL: Resources, Writing–review and editing. FM: Supervision, Writing–review and editing. BH: Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Project of the Educational Commission of Guangdong Province of China (No. 2022ZDJS113).
Conflict of interest
Author BC was employed by Chengdu NBbiolab. CO., LTD.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1545136/full#supplementary-material
References
Abanades, B., Wong, W. K., Boyles, F., Georges, G., Bujotzek, A., and Deane, C. M. (2023). ImmuneBuilder: deep-Learning models for predicting the structures of immune proteins, Commun. Biol., 6, 575. doi:10.1038/s42003-023-04927-7
Aghajanyan, A., Zettlemoyer, L., and Gupta, S. (2020). Intrinsic dimensionality explains the effectiveness of language model fine-tuning. doi:10.48550/arXiv.2012.13255
Ding, N., Qin, Y., Yang, G., Wei, F., Yang, Z., Su, Y., et al. (2023). Parameter-efficient fine-tuning of large-scale pre-trained language models. Nat. Mach. Intell. 5, 220–235. doi:10.1038/s42256-023-00626-4
Dunbar, J., and Deane, C. M. (2016). ANARCI: antigen receptor numbering and receptor classification, , 32, 298–300. doi:10.1093/bioinformatics/btv552
Dunbar, J., Krawczyk, K., Leem, J., Baker, T., Fuchs, A., Georges, G., et al. (2013). SAbDab: the structural antibody database, Nucleic Acids Res., 42, D1140–D1146. doi:10.1093/nar/gkt1043Available at: https://academic.oup.com/nar/article-pdf/42/D1/D1140/3538157/gkt1043.pdf
Hamers-Casterman, C., Atarhouch, T., Muyldermans, S., Robinson, G., Hammers, C., Songa, E. B., et al. (1993). Naturally occurring antibodies devoid of light chains. Nat. Occur. antibodies devoid light chains 363, 446–448. doi:10.1038/363446a0
Hu, E. J., Shen, Y., Wallis, P., Allen-Zhu, Z., Li, Y., Wang, S., et al. (2021). LoRA: low-rank adaptation of large language models. doi:10.48550/arXiv.2106.09685
Kunz, P., Zinner, K., Mücke, N., Bartoschik, T., Muyldermans, S., and Hoheisel, J. D. (2018). The structural basis of nanobody unfolding reversibility and thermoresistance, Sci. Rep., 8, 7934. doi:10.1038/s41598-018-26338-z
Lefranc, M.-P. (2008). IMGT®, the international ImMunoGeneTics information System® for immunoinformatics: methods for querying IMGT® databases, tools, and web resources in the context of immunoinformatics, Mol. Biotechnol., 40, 101–111. doi:10.1007/s12033-008-9062-7
Lefranc, M.-P., Giudicelli, V., Busin, C., Bodmer, J., Müller, W., Bontrop, R., et al. (1998). IMGT, the international ImMunoGeneTics database, Nucleic Acids Res., 26, 297–303. doi:10.1093/nar/26.1.297
Li, Y., Li, F., Duan, Z., Liu, R., Jiao, W., Wu, H., et al. (2024). SYNBIP 2.0: epitopes mapping, sequence expansion and scaffolds discovery for synthetic binding protein innovation, 53, D595–D603. doi:10.1093/nar/gkae893._eprintAvailable at: https://academic.oup.com/nar/article-pdf/53/D1/D595/59812323/gkae893.pdf.
Lv, L., Lin, Z., Li, H., Liu, Y., Cui, J., Chen, C. Y.-C., et al. (2024). ProLLaMA: a protein language model for multi-task protein language processing
Mullin, M., McClory, J., Haynes, W., Grace, J., Robertson, N., and Van Heeke, G. (2024). Applications and challenges in designing VHH-based bispecific antibodies: leveraging machine learning solutions. MAbs 16, 2341443. doi:10.1080/19420862.2024.2341443
Ofer, D., Brandes, N., and Linial, M. (2021). The language of proteins: NLP, machine learning and protein sequences, Comput. Struct. Biotechnol. J., 19, 1750–1758. doi:10.1016/j.csbj.2021.03.022
Olsen, T. H., Moal, I. H., and Deane, C. M. (2022). AbLang: an antibody language model for completing antibody sequences. Bioinforma. Adv. 2, vbac046. doi:10.1093/bioadv/vbac046
Ramazi, S., and Zahiri, J. (2021). “Post-translational modifications in proteins: resources,” in Tools and prediction methods 2021. doi:10.1093/database/baab012baab012
Schneider, C., Raybould, M. I. J., and Deane, C. M. (2021). SAbDab in the age of biotherapeutics: updates including SAbDab-nano, the nanobody structure tracker. Nucleic Acids Res. 50, D1368–D1372. doi:10.1093/nar/gkab1050
Shuai, R. W., Ruffolo, J. A., and Gray, J. J. (2023). IgLM: infilling language modeling for antibody sequence design 14. Elsevier, 979–989.e4. doi:10.1016/j.cels.2023.10.001
Strokach, A., and Kim, P. M. (2022). Deep generative modeling for protein design, Curr. Opin. Struct. Biol., 72, 226–236. doi:10.1016/j.sbi.2021.11.008
Touvron, H., Martin, L., Stone, K., Albert, P., Almahairi, A., Babaei, Y., et al. (2023). Llama 2: open foundation and fine-tuned chat models
Valdés-Tresanco, M. S., Molina-Zapata, A., Pose, A. G., and Moreno, E. (2022). Structural insights into the design of synthetic nanobody libraries, Molecules, 27, 2198. doi:10.3390/molecules27072198
van Kempen, M., Kim, S. S., Tumescheit, C., Mirdita, M., Lee, J., Gilchrist, C. L. M., et al. (2024). Fast and accurate protein structure search with Foldseek, Nat. Biotechnol., 42, 243–246. doi:10.1038/s41587-023-01773-0
Varadi, M. e. a., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2021). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. doi:10.1093/nar/gkab1061
Keywords: reinforcement learning, generative AI, nanobodies, libraries, protein large language models
Citation: Wang X, Chen H, Chen B, Liang L, Mei F and Huang B (2025) NanoAbLLaMA: construction of nanobody libraries with protein large language models. Front. Chem. 13:1545136. doi: 10.3389/fchem.2025.1545136
Received: 14 December 2024; Accepted: 31 January 2025;
Published: 25 February 2025.
Edited by:
Rachelle J. Bienstock, RJB Computational Modeling LLC, United StatesReviewed by:
Weiwei Xue, Chongqing University, ChinaDapeng Wang, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Wang, Chen, Chen, Liang, Mei and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengcheng Mei, bWVpZmVuZ2NoZW5nQHN6dHUuZWR1LmNu; Bingding Huang, aHVhbmdiaW5nZGluZ0BzenR1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship