 William J. Netzer1*
William J. Netzer1* Anjana Sinha1*Mondana Ghias1
Anjana Sinha1*Mondana Ghias1 Emily Chang1Katherina Gindinova1Emily Mui1
Emily Chang1Katherina Gindinova1Emily Mui1 Ji-Seon Seo1
Ji-Seon Seo1 Subhash C. Sinha1,2*
Subhash C. Sinha1,2*- 1Laboratory of Molecular and Cellular Neuroscience, The Rockefeller University, New York, NY, United States
- 2Appel Alzheimer’s Disease Research Institute, Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY, United States
We previously showed that the anticancer drug imatinib mesylate (IMT, trade name: Gleevec) and a chemically distinct compound, DV2-103 (a kinase-inactive derivative of the potent Abl and Src kinase inhibitor, PD173955) lower Aβ levels at low micromolar concentrations primarily through a lysosome-dependent mechanism that renders APP less susceptible to proteolysis by BACE1 without directly inhibiting BACE1 enzymatic activity, or broadly inhibiting the processing of other BACE1 substrates. Additionally, IMT indirectly inhibits γ-secretase and stimulates autophagy, and thus may decrease Aβ levels through multiple pathways. In two recent studies we demonstrated similar effects on APP metabolism caused by derivatives of IMT and DV2-103. In the present study, we synthesized and tested radically altered IMT isomers (IMTi’s) that possess medium structural similarity to IMT. Independent of structural similarity, these isomers manifest widely differing potencies in altering APP metabolism. These will enable us to choose the most potent isomers for further derivatization.
1 Introduction
Neurotoxic β-amyloid peptides (Aβ) are major drivers of Alzheimer’s disease (AD) and are formed by sequential cleavage of the amyloid precursor protein (APP) by β-secretase (BACE1/2) and γ-secretase, respectively. Both β- and γ-secretases can be pharmacologically inhibited to reduce production of Aβ peptides. Indeed, there has been great interest in the development of inhibitors and modulators of the secretases as potential AD therapeutics (Miranda et al., 2021; Kumar et al., 2018; Zhao et al., 2020; Portelius et al., 2010; Hur, 2022; Panza et al., 2009; Golde et al., 2013; Rynearson et al., 2021) but at this time all clinical trials involving secretase inhibitors/modulators have failed. Reasons given have included timing of drug administration (too late in disease course for benefits to occur); non-specific inhibition of secretase substrates other than APP; lack of target engagement; toxicity; and even failure of the Amyloid hypothesis (Kim et al., 2022).
In our previous study, we have shown that the anticancer drug IMT, which is a potent Abl kinase inhibitor (Buchdunger et al., 1996) and PD173955 (Nagar et al., 2002), an Abl/Src kinase inhibitor, reduce Aβ production in cultured N2a695 cells, rat embryonic neurons, and in guinea pig brain in vivo by indirectly inhibiting γ-secretase processing of APP, while sparing γ-secretase processing of Notch1 in cellular assays (Netzer et al., 2003). In a recent study we further showed that a kinase inactive derivative of PD173955, DV2-103, as well as IMT, reduced Aβ levels in cells mainly by indirectly inhibiting BACE cleavage of APP (Netzer et al., 2017), adding to our earlier study suggesting that the Aβ-lowering effect of IMT and DV2-103 are not only Abl kinase-independent but also broadly kinase-independent and affect both γ-secretase and BACE processing of APP. IMT and DV2-103 decrease levels of APP-βCTF and sAPPβ, and raise levels of APP-αCTF, as well as a 141 amino acid APP-CTF (C141), and a 9 kDa APP-CTF (all consistent with reduced BACE processing of APP) in N2a695 cells (Netzer et al., 2017). Remarkably, this pattern of APP metabolites induced by IMT and DV2-103, and some of their analogs is observed when N2a695 cells are treated with a general, active-site-directed BACE inhibitor (Netzer et al., 2017; Sun et al., 2019; Sinha et al., 2019). We demonstrated that IMT does not inhibit BACE1 enzymatic activity in two in vitro BACE1 assays at concentrations up to 100 μM or inhibit processing of several non-APP BACE substrates in cells (Netzer et al., 2017) and that the inhibitory activities of IMT and DV2-103 require acidified lysosomes. We provided a model suggesting that the effects of these drugs on APP metabolism were a result of their effects on lysosomes, which caused APP to undergo increased trafficking to lysosomes and spend less time in the amyloidogenic pathway where Aβ and its direct precursor, the APP-βCTF, are formed (Netzer et al., 2017).
To understand how so many structurally different compounds reduce levels of secreted Aβ in cells and have a characteristic effect on APP metabolite levels, we designed novel IMT isomers, IMTi-1 – 3 (Figure 1A), and tested their effects on APP metabolism by measuring the Aβ levels in cell supernatants and APP metabolites in cell lysates. The design involved a large change in the structure of IMT to greatly alter the pharmacophore structurally but maintain IMT’s physical properties, in particular its property as a weak base, which is necessary for its sequestration in lysosomes through ion trapping (Kazmi et al., 2013; Burger et al., 2015). We further developed and evaluated 28 new analogs of the more potent IMT isomers, IMTi-1 and 2, to gain structure-activity relationship among the new analogs and to the previously described IMT analogs. The results of our studies described in this article support that the pharmacophores of IMT greatly affect APP processing and introduce IMTi-1 and 2 as new pharmacophores to further develop more potent analogs that may function similarly to IMT.
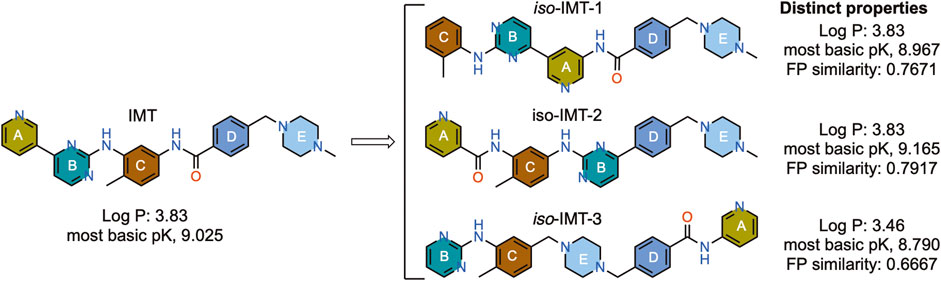
Figure 1. Structure and properties of IMT and the designed IMT isomers. Fingerprint (FP) similarity of IMTi-1 – 3 to parent IMT and their properties, including most basic PK, were calculated in silico.
2 Materials and methods
All commercial chemicals and solvents were reagent grade and used without further purification. All air-sensitive reactions were performed under argon protection. Column chromatography was performed using 230–400 mesh silica gel. Analytical thin layer chromatography was performed on 250 μM silica gel F254 plates. Preparative thin layer chromatography was performed on 1,000 μM silica gel F254 plates. All final compounds were purified using HPLC. The identity and purity of each product was determined using MS, HPLC, TLC, and NMR analyses. 1H NMR spectra were recorded on either a Bruker 400 or 600 MHz instrument. Chemical shifts are reported in δ values in ppm downfield from TMS as the internal standard. 1H data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constant (Hz), integration. Purity of target compounds has been determined to be >95% by LC/MS on a Waters purification system with PDA, MicroMass ZQ and ELSD detector and a reverse phase column (Waters X-Bridge C18, 4.6 × 150 mm, 5 µm) eluted with water/acetonitrile gradients, containing 0.1% TFA. All compounds tested in this study were prepared in house and their structures were confirmed using 1H NMR and MS analyses (Spectral data provided for new compounds only). Yields are from a single reaction and not optimized. All final compounds were obtained in >95% purity as judged by LCMS.
N2a695 were cultured in 1:1 OptiMem Reduced Serum Media (Life Technologies): Dulbecco’s Modified Eagle Medium ([+] 4.5 g/L D-glucose [+] L-Glutamine; [−] Sodium pyruvate (Life Technologies) supplemented with 5% fetal bovine serum, 0.4% Penstrep and 0.4% Geneticin and incubated at 37°C in 5% CO2. Antibodies were obtained from The Laboratory of Molecular and Cellular Neuroscience at The Rockefeller University. Human Aβ40 and Aβ42 ELISA plates (Life Technologies) and Plus MSD (Mesoscale Discovery) plates for Aβ Peptide (Aβ38, Aβ40 and Aβ42) Panel 1 (6E10) Kit (Catalog number K15200G) were obtained from Thermo Fisher, Life Technologies and Meso Scale Discovery.
2.1 Synthesis of IMTi-1 and analogs 1a-r
i) Intermediates 5a-b. To a solution of 4a (350 mg, 1.3 mmol) and o-toluidine (0.3 mL) in i-PrOH (3 mL) was added 1N HCl (1.5 mL), and the mixture was heated at 125°C using Microwave for 2 h. Solvents were removed under reduced pressure, residues treated with aqueous NaHCO3 to neutralize, and the resulting mixture extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous MgSO4, concentrated, and purified by Combi-Flash over Silica gel column using hexanes-EtOAc as eluents to afford intermediate 5a (300 mg, 68%). 1H NMR (600 MHz, CDCl3) of 5a: δ 9.16 (s, 1H), 8.85 (s, 1H), 8.51 (s, 2H), 8.03 (d, J = 7.92 Hz, 1H), 7.35–7.30 (m, 2H), 7.14–7.07 (m, 2H), 2.38 (s, 3H); HRMS: m/z 341.0391 and 343.0370 [M + H]+.
Similarly, intermediate 4b (270 mg, 1 mmol) was reacted with o-toluidine (0.25 mL) in i-PrOH (2 mL) and 1N HCl (1 mL) using the method described for 5a to afford intermediate 5b (245 mg, 72%). 1H NMR: (600 MHz, CDCl3) of 5b: δ 8.47 (d, J = 6.0 Hz, 1H), 8.22 (s, 1H), 8.09 (d, J = 6.0 Hz, 1H), 7.96 (d, J = 6.0 Hz, 1H), 7.62 (d, J = 6.0 Hz, 1H), 7.36 (t, J = 6.0 Hz, 1H), 7.29 (t, J = 6.0 Hz, 1H), 7.24 (d, J = 12 Hz, 1H), 7.11 (d, J = 6.0 Hz, 1H), 7.06 (t, J = 6.0 Hz, 1H), 6.95 (s, 1H), 2.37 (s, 3H); HRMS: m/z 340.0439 and 342.0418, [M + H]+.
ii) Compound IMTi-1 (General Buchwald coupling method). A solution of 5a (38 mg) and 6a (21 mg) in dioxane was degassed and charged with Pd2(dba)3 (4 mg), XanthPhos (7 mg), and Cs2CO3 (55 mg) and heated at 100°C temperature for 16 h. Solvents were removed and worked up using EtOAc and water. Combined organic layers were dried over anhydrous MgSO4 and concentrated under reduced pressure. The resulting residues were purified by preparative TLC (Silica gel, 1 mm plate; CH2Cl2:MeOH:Aq. NH3 (90:10:1)) to afford the target product IMTi-1 (35 mg, 63%). 1H NMR (400 MHz, CDCl3): δ 9.01 (d, J = 1.5 Hz, 1H), 8.92 (s, 1H), 8.88 (s, 1H), 8.47 (d, J = 5.12 Hz, 1H), 8.05 (d, J = 7.96 Hz, 1H), 7.88 (d, J = 7.96 Hz, 2H), 7.45 (d, J = 8.40 Hz, 1H), 7.28 (t, J = 4.28 Hz, 1H), 7.23 (d, J = 7.40 Hz, 1H), 7.15 (d, J = 5.16 Hz, 1H), 7.07–7.03 (m, 2H), 3.57 (s, 2H), 2.49 (br s, 8H), 2.35 (s, 3H), 2.30 (s, 3H); HRMS: m/z 494.2653 [M + H]+. Purity (HPLC): >98%.
iii) Compound 1a. Buchwald coupling of 5b (48 mg, 0.14 mmol) with 6a (33 mg, 0.14 mmol) in 1,4-Dioxane (3 mL) was performed by heating the mixture at 100°C in the presence of Pd2(dba)3 (5 mg), XanthPhos (8 mg), and Cs2CO3 (68 mg) for 16 h. Usual work-up and purification afforded 1a (33 mg, 48%). 1H NMR (400 MHz, CDCl3): δ 8.46 (d, J = 5.20 Hz, 1H), 8.36 (s, 1H), 8.12 (d, 8.04 Hz, 1H), 8.02 (s, 1H), 7.87–7.82 (m, 4H), 7.52–7.46 (m, 3H), 7.31–7.23 (m, 1H), 7.17 (d, J = 5.16 Hz, 1H), 7.06 (t, J = 3.6 Hz, 1H), 6.96 (s, 1H), 3.56 (s, 2H), 2.52 (br s, 8H), 2.37 and 2.33 (s, 3H each); HRMS: m/z 493.2671 [M + H]+.
iv) Compounds 1b-Boc and 1b. Buchwald coupling of 5a (160 mg, 0.47 mmol) with 6b (155 mg, 0.49 mmol) in 1,4-Dioxane (10 mL) was performed by heating the mixture at 100°C in the presence of Pd2(dba)3 (20 mg), XanthPhos (27 mg), and Cs2CO3 (155 mg) for 16 h. Usual work-up and purification afforded 1b-Boc (100 mg, 35%). MS of 1b-Boc: m/z 579.30.
Boc-deprotection. To a solution of 1b-Boc (100 mg) in EtOAc (3 mL) was added 4 M HCl in dioxane (1 mL) at room temperature (RT) and the mixture stirred overnight (16 h). Solvents were removed under reduced pressure and worked up using CH2Cl2 and Aq. NaHCO3 solution to afford compound 1b (79 mg, 95%). HRMS: m/z 480.2467 [M + H]+.
v) Compound 1c. Intermediate 5a (75 mg, 0.2 mmol) underwent Buchwald coupling with 6c (53 mg, 0.24 mmol) in the presence of Pd2(dba)3 (10 mg), Xanthphos (13 mg), and CS2CO3 (72 mg) in 1,4-Dioxane (5 mL) overnight at 95°C, as described above for IMTi-1 to give compound 1c (56 mg, 40%). 1H NMR (600 MHz, CDCl3): δ 9.04 (s, 1H), 8.94 (s, 1H), 8.86 (d, J = 6.0 Hz, 1H), 8.13 (dd, J = 12.0, 6.0 Hz, 1H), 7.87 (d, J = 6.0 Hz, 2H), 7.32 (t, J = 6.0 Hz, 2H), 7.265 (d, J = 6.0 Hz, 1H), 7.215 (d, J = 6.0 Hz, 1H), 7.08 (t, J = 6.0 Hz, 1H), 6.975 (d, J = 6.0 Hz, 2H), 3.40 (t, J = 6.0 Hz, 4H), 2.62 (t, J = 6.0 Hz, 4H), 2.40 and 2.39 (s, 3H each); MS: m/z 480.25 [M + H]+.
vi) Compound 1d. Intermediate 5b (145 mg, 0.42 mmol) underwent Buchwald coupling with 6c (93 mg, 0.42 mmol) in the presence of Pd2(dba)3 (19 mg), XanthPhos (25 mg), and Cs2CO3 (206 mg) in 1,4-Dioxane (10 mL) overnight at 100°C as described above and worked up and purified to give compound 1d (110 mg, 55%). 1H NMR (600 MHz, CDCl3) of 1k: δ 8.49 (d, J = 6.0 Hz, 1H), 8.36 (s, 1H), 8.17 (dd, J = 6.0, 12.0 Hz, 1H), 7.88–7.83 (m, 3H), 7.51 (t, J = 6.0 Hz, 1H), 7.33–7.29 (m, 2H), 7.27 (d, J = 6.0 Hz), 7.22 (d, J = 6.0 Hz, 1H), 7.09 (t, J = 6.0 Hz, 1H), 6.99 (d, J = 6.0 Hz, 2H), 3.41 (t, J = 6.0 Hz, 4H), 2.63 (br t, 4H), 2.41 and 2.40 (s, 3H each); MS: m/z 479.25 [M + H]+.
vii) Compound 1e. Intermediate 5a (35 mg, 0.1 mmol) underwent Buchwald coupling with amide 6d (21 mg, 0.11 mmol) in the presence of Pd2(dba)3 (4 mg), XanthPhos (6 mg), and Cs2CO3 (50 mg, 0.15 mmol) at 100°C overnight to give compound 1e (20 mg, 44%) after usual work-up using EtOAc and Aq. NH4Cl solution, and purification. 1H NMR (600 MHz, CDCl3): δ 9.03, 8.95 and 8.90 (s, 1H each), 8.50 (d, J = 6.0 Hz, 1H), 8.09 (d, J = 6.0 Hz, 1H), 7.87 (d, J = 6.0 Hz, 2H), 7.35 (d, J = 6.0 Hz, 2H), 7.32–7.29 (m, 1H), 7.25 (d, J = 6.0 Hz, 1H), 7.20 (d, J = 6.0 Hz, 1H), 7.08 (d, J = 6.0 Hz, 2H), 2.89 (t, J = 6.0 Hz, 2H), 2.59 (d, J = 6.0 Hz, 2H), 2.37 (s, 3H), 2.33 (s, 6H); HRMS: m/z 453.2358 [M + H]+.
viii) Compound 1f. Intermediate 5b (340 mg, 1 mmol) underwent Buchwald coupling with amide 6d (200 mg, 1 mmol) in the presence of Pd2(dba)3 (45 mg, 5 mol%), XanthPhos (60 mg, 10 mo%), and Cs2CO3 (500 mg, 1.5 mmol). Usual work-up after heating at 100°C for 16 h and purification gave compound 1f (270 mg, 60%). 1H NMR (600 MHz, CDCl3): δ 8.49 (d, J = 6.0 Hz, 1H), 8.38 (s, 1H), 8.15 (d, J = 6.0 Hz, 1H), 7.99 (br s, 1H), 7.88–7.83 (m, 4H), 7.68 (d, J = 6.0 Hz, 1H), 7.53 (t, J = 6.0 Hz, 1H), 7.42–7.38 (m, 2H), 7.33 (d, J = 12.0 Hz, 1H), 7.26 (d, J = 6.0 Hz, 1H), 7.21 (d, J = 6.0 Hz, 1H), 7.08 (t, J = 6.0 Hz, 1H), 6.97 (s, 1H), 2.29 (q, J = 6.0 Hz, 2H), 2.61 (q, J = 6.0 Hz, 2H), 2.40 (s, 3H), 2.38 (s, 6H); HRMS: m/z 452.2450 [M + H]+. Purity (HPLC): >98%.
ix) Compound 1g. Intermediate 5a (38 mg, 0.11 mmol) underwent Buchwald coupling with 6e (22 mg, 0.15 mmol) in the presence of Pd2(dba)3 (5 mg), XanthPhos (10 mg), and Cs2CO3 (73 mg mmol) in 1,4-Dioxane (2 mL) by heating overnight at 100°C. Sovents were removed under reduced pressure and the residues purified by preparative TLC to give compound 1 g (15 mg, 31%). HRMS: m/z 439.2243 [M + H]+.
x) Compounds 1h-Boc and 1h. Buchwald coupling of 5a (60 mg, 0.18 mmol) with 6f (56 mg, 0.18 mmol) in 1,4-Dioxane (3 mL) was performed by heating the mixture at 100°C in the presence of Pd2(dba)3 (8 mg), XanthPhos (10 mg), and Cs2CO3 (86 mg) for 16 h. Usual work-up and purification afforded 1h-Boc (71 mg, 70%). 1H NMR (400 MHz, CDCl3) of 1h-Boc: δ 9.04 (s, 1H), 8.43 (s, 1H), 8.85 (s, 1H), 8.49 (d, J = 5.04 Hz, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.89–7.87 (br, 2H), 7.27–7.17 (m, 6H), 7.07–7.03 (m, 2H), 4.58 (br, 2H), 3.16 (br s, 1H), 3.07 (br s, 1H), 2.36 (s, 3H), 1.50 and 1.34 (s, 6H and 3H); 0.97 (s, 9H); MS: m/z 581.32 [M + H]+.
Compound 1h-Boc (65 mg, 0.11 mmol) in EtOAc (3 mL) was stirred with 4 M HCl in dioxane (1 mL) at RT for 16 h. Solvents were removed under reduced pressure and worked-up using CH2Cl2 and Aq. NaHCO3 solution to afford compound 1h (42 mg, 77%). 1H NMR (400 MHz, CDCl3) of 1h: δ 9.06 (s, 1H), 8.94 (s, 1H), 8.85 (s, 1H), 8.50 (d, J = 5.04 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 8.0 Hz, 1H), 8.0 (s, 1H), 7.89 (d, J = 7.92 Hz, 2H), 7.52 (d, J = 7.80 Hz, 2H), 7.24–7.21 (m, 4H), 7.08 (t, J = 4.00 Hz, 1H), 7.00 (s, 1H), 3.92 (s, 2H), 2.38 (s, 2H), 2.37 (s, 3H), 0.95 (s, 9H); HRMS: m/z 481.2616 [M + H]+.
xi) Compounds 1i-Boc and 1i. Intermediate 5b (40 mg, 0.12 mmol) was reacted with 6f (38 mg, 0.12 mmol) under Buchwald coupling conditions using Pd2(dba)3 (2 mg), XanthPhos (4 mg), and Cs2CO3 (30 mg) in 1,4-Dioxane (2 mL) overnight as described above at 100°C to give the Boc-protected derivative, 1i-Boc (45 mg, 66%). MS of 1i-Boc: m/z 580.33 [M + H]+.
Compound 1i-Boc (40 mg, 0.07 mmol) was deprotected using 4 M HCl in dioxane (1 mL) at RT. Solvents were removed under reduced pressure and worked up using CH2Cl2 and Aq. NaHCO3 solution to afford compound 1f (30 mg, 90%). 1H NMR (400 MHz, CDCl3) of 1i: δ 8.47, 8.36 and 8.14 (s, 1H each), 7.88–7.83 (m, 4H), 7.50 (d, J = 7.40 Hz, 4H), 7.24–7.19 (m, 3H), 7.08–7.01 (m, 2H), 3.90 (s, 2H), 3.49 (s, 2H), 2.34 (s, 3H), 0.95 (m, 9H); HRMS: m/z 480.2776 [M + H]+.
xii) Compounds 1j-Boc and 1j. Intermediate 5a (19 mg) was reacted with 6g (20 mg) under Buchwald coupling conditions using Pd2(dba)3 (2 mg), XanthPhos (4 mg), and Cs2CO3 (30 mg) in 1,4-Dioxane (2 mL) overnight at 100°C to give 1j-Boc (15 mg, 46%). 1H NMR (400 MHz, CDCl3) of compound 1j-Boc: δ 9.04 (br s, 1H), 8.97 and 8.88 (s, 1H each), 8.72 (br, 1H), 8.47 (d, J = 4.28 Hz, 1H), 8.06 (d, J = 7.92 Hz, 4H), 7.94 (m, 2H), 7.44 7.36–7.17 (m, 5H), 7.06 (m, 2H), 4.42 (br s, 2H), 4.08 (m, 1H), 2.36 (s, 3H), 1.70–1.4 (m, 8H), 1.35–1.26 (m, (9H+2H); MS: m/z 593.32 [M + H]+.
Compound 1j-Boc (15 mg, 0.025 mmol) was deprotected using 4 M HCl in dioxane (0.5 mL) at RT. Solvents were removed under reduced pressure and worked up using CH2Cl2 and Aq. NaHCO3 solution to afford compound 1f (10 mg, 81%). 1H NMR (400 MHz, CDCl3) of compound 1j: δ 9.03 (br s, 1H), 8.94 (s, 1H), 8.85 (s, 1H), 8.48 (d, J = 4.84 Hz, 1H), 8.34 9 (br, 1H), 8.08 (d, J = 7.96 Hz, 1H), 7.88 (d, J = 7.80 Hz, 2H), 7.49 (d, J = 7.64 Hz, 2H), 7.30 (d, J = 7.56 Hz, 2H), 7.25–7.23 (m, 3H), 7.18 (d, J = 4.72 Hz, 1H), 7.09–7.05 (m, 2H), 3.94 (s, 2H), 2.57 (m, 1H), 2.37 (s, 3H), 1.98–1.45 (m, 8H), 1.29–1.18 (m, 2H); HRMS: m/z 493.2718 [M + H]+.
xiii) Compounds 1k-Boc and 1k. Intermediate 5b (46 mg, 0.14 mmol) reacted with 6g (45 mg, 0.14 mmol) under Buchwald coupling conditions using Pd2(dba)3 (5 mg), XanthPhos (8 mg), and Cs2CO3 (66 mg) in 1,4-Dioxane (2 mL) overnight at 100°C to give 1k-Boc (51 mg, 62%). MS of 1k-Boc: m/z 592.33 [M + H]+.
Compound 1k-Boc (40 mg, 0.068 mmol) was deprotected using 4 M HCl in dioxane/EtOAc (1:1, 2 mL) at RT. Solvents were removed under reduced pressure and worked up using CH2Cl2 and Aq. NaHCO3 solution to afford compound 1k (30 mg, 89%) after purification. 1H NMR (600 MHz, CDCl3) of 1k: δ 8.41 (d, J = 5.04 Hz, 1H), 8.31 (s, 1H), 8.04 (d, J = 7.96 Hz, 1H), 7.88 (d, J = 7.72 Hz, 3H), 7.78 (d, J = 7.64 Hz, 1H), 7.49–7.42 (m, 4H), 7.27–7.24 (m, 3H), 7.15 (d, J = 4.96 Hz, 1H), 7.03 (t, J = 7.24 Hz, 1H), 3.90 (s, 2H), 3.38 (m, 1H), 2.34 (s, 3H), 1–97–1.62 (m, 6H), 1.26–1.16 (m, 4H); HRMS: m/z 492.2772 [M + H]+.
xiv) Compound 1L. Intermediate 5a (100 mg, 0.29 mmol) underwent Buchwald coupling with 6h (98 mg, 0.29 mmol) in the presence of Pd2(dba)3 (14 mg), XanthPhos (20 mg), and Cs2CO3 (142 mg) in 1,4-Dioxane (10 mL) as described above for IMTi-1. After the reaction mixture was stirred at 100°C overnight, usual work up and purification gave the title product 1L-Boc (161 mg, 98%). 1HNMR (600 MHz, CDCl3) of 1L-Boc: δ 9.04 (s, 1H), 8.95 (s, 1H), 8.88 (s, 1H), 8.50 (s, 2H), 8.10 (d, J = 6.0 Hz, 1H), 7.91 (d, J = 6.0 Hz, 2H), 7.37 (br t, J =, 2H), 7.30 (d, J = 6.0 Hz, 1H), 7.26 (d, J = 6.0 Hz, 1H), 7.20 (m, 2H), 4.18 (m, 1H), 2.82 (br, 2H), 2.40 (s, 3H), 2.08 (m, 1H), 1.82 (m, 1H), 1.69 (m, 2H), 1.61 (s, 3H), 1.49 (s, 6H), 1.48 (m, 2H); MS (ESI) m/z 564.28 [M]+.
Intermediate 1L-Boc (90 mg, 0.16 mmol) in EtOAc (3 mL) was Boc-deprotected using 4 M HCl in Dioxane (2 mL) to give compound 1L (70 mg, 94%) after usual work up using CH2Cl2 and NaHCO3 and filtration over a short bed of Silica gel. HRMS of 1L: m/z 465.2401 [M + H]+.
xv) Compound 1m. Intermediate 5b (165 mg, 0.49 mmol) underwent Buchwald coupling with 6h (148 mg, 0.49 mmol) in the presence of Pd2(dba)3 (22 mg), XanthPhos (30 mg), and Cs2CO3 (235 mg) in 1,4-Dioxane (10 mL) as described above. After the reaction mixture was stirred at 100°C overnight, usual work up and purification gave the title product 1m-Boc (265 mg, 87%). 1HNMR (600 MHz, CDCl3) of 1m-Boc: δ 8.50 (d, J = 6.0 Hz, 1H), 8.37 (s, 1H), 8.16 (d, J = 12.0 Hz, 1H), 7.93 (s, 1H), 7.89–7.86 (m, 3H), 7.545 (t, J = 6.0 Hz, 1H), 7.42 (d, J = 6.0 Hz, 1H), 7.32 (t, J = 12.0 Hz, 2H), 7.27 (d, J = 6.0 Hz, 1H), 7.22 (d, J = 6.0 Hz, 1H), 7.08 (t, J = 6.0 Hz, 1H), 6.96 (s, 1H), 4.18 (m, 1H), 2.81 (br, 2H), 2.40 (s, 3H), 2.08 (m, 1H), 1.81 (m, 1H), 1.68 (m, 2H), 1.59 (s, 3H), 1.51 (s, 6H), 1.48 (m, 2H); MS: m/z 564.29 [M + H]+.
Intermediate 1m-Boc (95 mg, 0.17 mmol) was Boc-deprotected using 2 M HCl in Dioxane (2 mL) to give compound 1m (75 mg, 95%) after usual work up using CH2Cl2 and NaHCO3 and filtration over a short bed of Silica gel. HRMS of 1m: m/z 464.2459 [M + H]+.
xvi) Compound 1n. NaCNBH3 (25 mg) and AcOH (50 µL) were added sequentially to a solution of amine 1L.HCl (51 mg, 0.1 mmol) and paraformaldehyde (30 mg) in MeOH/2N Aq. KOH (10:1, 1.1 mL) at ice-water temperature and the reaction mixture was stirred at RT for another 8 h. Solvents were removed under reduced pressure, and the residue was suspended in CH2Cl2 and washed using water. Combined organic layers were dried using Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by Silica gel column to afford 1n (40 mg, 83%). 1H NMR (600 MHz, CD3OD + CDCl3): δ 8.99 (br s, 1H), 8.94 (s, 1H), 8.45 (s, 1H), 8.02 (d, J = 6.0 Hz, 2H), 7.74 (d, J = 6.0 Hz, 1H), 7.49 (d, J = 6.0 Hz, 2H), 7.30–7.24 (m, 3H), 7.10 (t, J = 6.0 Hz, 1H), 3.49 (m, 2H), 3.10 (t, J = 12.0 Hz, 1H), 2.95 (t, J = 12.0 Hz, 1H), 2.87 (m, 1H), 2.81 (s, 3H), 2.34 (s, 3H), 2.10–1.64 (m, 4H); HRMS: m/z 479.2560 [M + H]+.
xvii) Compound 1o. Reductive amination of amine 1m (50 mg, 0.1 mmol) with paraformaldehyde (30 mg), NaCNBH3 (30 mg), and AcOH (50 µL) in MeOH/2N Aq. KOH (10:1, 1.1 mL) as described for 1n afforded 1o (32 mg, 67%). 1HNMR (600 MHz, CDCl3) of 1o: δ 8.42 (d J = 6.0 Hz, 1H), 8.36 (s, 1H), 8.05 (d, J = 6.0 Hz, 1H), 7.87 (d, J = 6.0 Hz, 2H), 7.80 (d, J = 6.0 Hz, 1H), 7.47 (t, J = 6.0 Hz, 1H), 7.34 (d, J = 12.0 Hz, 2H), 7.265 (d, J = 6.0 Hz, 1H), 7.23 (d, J = 6.0 Hz, 1H), 7.16 (q, J = 6.0 Hz, 1H), 7.05 (t, J = 6.0 Hz, 1H), 3.29 (d, J = 12.0 Hz, 2H), 3.10 (t, J = 6.0 Hz, 1H), 2.66 (s, 3H), 2–63–2.54 (m, 2H), 2.34 (s, 3H), 2.03–1.92 (m, 3H), 1.65–1.61 (m, 1H); HRMS: m/z 478.2602 [M + H]+.
xviii) Intermediate 5c. Intermediate 4c (156 mg, 0.5 mmol) underwent Buchwald coupling with 3-aminopyridine (50 mg) in the presence of Pd2(dba)3 (18 mg), XanthPhos (20 mg), and Cs2CO3 (250 mg) in 1,4-Dioxane (3 mL) by reacting the reaction mixture overnight at 100°C. Usual work up using CH2Cl2 and water and purification of the concentrated organic layers using Combi Flash afforded 5c-Boc (160 mg, 86%). The latter product was treated with 4 M HCl in dioxane (2 mL) overnight, and solvents were removed to afford 5c as HCl salt. MS of 5c: m/z 264.12 [M + H]+.
xix) Compound 1p. Prepared by amide formation between 5c.HCl (30 mg, 0.1 mmol) and 6j (20 mg, 0.1 mmol) using PyBOP (78 mg, 0.15 mmol) and DIPEA (60 µL) in DMF (250 µL) to afford 1p (20 mg, 46%) after work up and purification by preparative TLC. 1H NMR (600 MHz, CDCl3): δ 8.49 (d, J = 6.0 Hz, 1H), 8.38 (s, 1H), 8.15 (d, J = 6.0 Hz, 1H), 7.99 (br s, 1H), 7.88–7.83 (m, 4H), 7.68 (d, J = 6.0 Hz, 1H), 7.53 (t, J = 6.0 Hz, 1H), 7.42–7.38 (m, 2H), 7.33 (d, J = 12.0 Hz, 1H), 7.26 (d, J = 6.0 Hz, 1H), 7.21 (d, J = 6.0 Hz, 1H), 7.08 (t, J = 6.0 Hz, 1H), 6.97 (s, 1H), 2.29 (q, J = 6.0 Hz, 2H), 2.61 (q, J = 6.0 Hz, 2H), 2.40 (s, 3H), 2.38 (s, 6H); HRMS: m/z 439.2256 [M + H]+. Purity (HPLC): >95%.
xx) Compound 1q (Prepared by Suzuki reaction). To a degassed solution of intermediate 5a (34 mg, 0.1 mmol) and boronic acid 7a (35 mg, 0.15 mmol) in DMF (2 mL) and 2 M aq. K2CO3 solution (2 M, 0.2 mL) in a microwave vial was added Pd(PPh3)4 (11 mg) and the mixture was heated at 100°C for 30 min using microwave. The reaction mixture was diluted using water, extracted using EtOAc, and the combined organic layers concentrated under reduced pressure and the residue was purified by preparative TLC to afford 1q (15 mg, 30%). 1H NMR (400 MHz, CDCl3): δ 9.18 (s, 1H), 8.92 (s, 1H), 8.56 (s, 1H), 8.51 (d, J = 5.04 Hz, 1H), 8.08 (d, J = 8.00 Hz, 1H), 7.63 (d, J = 7.88 Hz, 2H), 7.48 (d, J = 7.88 Hz, 2H), 7.25–7.21 (m, 4H), 7.09–7.05 (m, 1H), 3.49 (s, 2H), 2.37 (s, 3H), 2.32 (s, 6H); HRMS: m/z 396.2197 [M + H]+.
xxi) Compound 1r. As described above for 1q, Suzuki reaction of 5a (100 mg, 0.29 mmol) with 7b (84 mg, 0.38 mmol) in DMF (2 mL) and aq. K2CO3 solution (2 M, 0.3 mL) in a microwave vial in the presence of Pd(PPh3)4 (20 mg) and heating the reaction mixture in microwave at 110°C for 30 min afforded 1r (35 mg, 28%) after work up and purification using preparative TLC. 1H NMR (400 MHz, CDCl3) of 1r: δ 9.14 (s, 1H), 8.92 (s, 1H), 8.52 (d, J = 4.52 Hz, 2H), 8.13 (d, J = 6.0 Hz, 1H), 7.60 (d, J = 8.56 Hz, 2H), 7.48 (d, J = 8.56 Hz, 1H), 7.25–7.21 (m, 2H), 7.10–6.96 (m, 3H), 3.34–3.23 (m, 8H), 1H), 2.81 (s, 3H), 2.34 (s, 3H), 2.10–1.64 (m, 4H); HRMS: m/z 437.2449 [M + H]+.
2.2 Synthesis of IMTi-2 and analogs 2a-b and IMTi-3
i) IMTi-2. Buchwald coupling of intermediate 8 (54 mg, 0.24 mmol) with amine 9 (56 mg, 0.24 mmol) at 100°C overnight in the presence of Pd2(dba)3 (10 mg), XanthPhos (15 mg), and Cs2CO3 (140 mg) in 1,4-Dioxane (3 mL), as described above for IMTi-1, afforded intermediate 10 (41 mg, 42%). MS of 10: m/z 409.16 [M]+.
Intermediate 10 (41 mg, 0.1 mmol) underwent reductive amination with 4-methylpiperazine (25 µL) using NaCNBH3 (65 mg) in dichloroethane (DCE) (3 mL) and AcOH (0.1 mL) as described for 1o to give IMTi-2 (30 mg, 60%). 1H NMR (400 MHz, CDCl3+ CD3OD) of IMTi-2: δ 9.15 (s, 1H), 8.81 (s, 1H), 8.46 (d, J = 4.88 Hz, 1H), 8.35 (s, 1H), 8.27 (d, J = 6.88 Hz, 1H), 8.07 (d, J = 7.64 Hz, 2H), 7.85 (m, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.37 (m, 1H), 7.24 (d, J = 8.0 Hz, 1H), 7.15 (d, J = 4.96 Hz, 1H), 3.58 (s, 2H), 2.52 (br s, 8H), 2.33 (s, 6H); MS: m/z 494.2678 [M + H]+.
ii) Compounds 2a and 2b. Aldehyde 8 (116 mg, 0.53 mmol) underwent reductive amination with neopentyl amine (130 µL) using NaCNBH3 (324 mg) in DCE (2 mL) and AcOH (0.1 mL) over 2 h. Reaction mixture was extracted using CH2Cl2, concentrated, and the residues taken in acetonitrile was stirred with Boc2O (300 mg) overnight to afford intermediate 11a (165 mg, 80% in 2 steps). Subsequently, Amine 9 (27mg, 0.12 mmol) underwent Buchwald coupling with 11a (46 mg, 0.12 mmol) in the presence of Pd2(dba)3 (4 mg), XanthPhos (7 mg), and Cs2CO3 (58 mg) in 1,4-Dioxane (3 mL) to afford 2a-Boc (53 mg, 77%), and the latter product was deprotected using TFA in the presence of tri-isopropyl silane (TIPS) in CH2Cl2 giving 2a (38 mg, 90%) after filtration using a short bed of silica gel column. MS of 2a: m/z 481.27 [M + H]+.
Similarly, Aldehyde 8 (112 mg, 0.51 mmol) underwent reductive amination with cyclohexyl amine (100 µL) using NaCNBH3 (324 mg) in DCE (2 mL) and AcOH (0.1 mL) over 2 h, and the reaction mixture was extracted using CH2Cl2, concentrated, and the residues taken in acetonitrile was further reacted with Boc2O (300 mg) overnight to afford intermediate 11b (175 mg, 85% in 2 steps). Next, amine 9 (24mg, 0.09 mmol) underwent Buchwald coupling with 11b (36 mg, 0.09 mmol) in the presence of Pd2(dba)3 (4 mg), XanthPhos (6 mg), and Cs2CO3 (44 mg) in 1,4-Dioxane (2 mL) to afford 2b-Boc (36 mg, 68%), and the latter product was deprotected using TFA in the presence of TIPS in CH2Cl2 giving 2b (25 mg, 86%) after filtration using a short bed of silica gel column. MS of 2b: m/z 493.27 [M + H]+.
iii) Intermediate 13. To a solution of 3-aminopyridine (1 equiv.) and DIEA (3 equiv.) in dry THF (5 mL/mmol) was added 4-chloromethylbenzoyl chloride (1.2 equiv.) at room temperature and the resulting mixture was stirred for 16 h and evaporated under reduced pressure. The residues were worked up using CH2Cl2 and Aq. NaHCO3 and purified to afford intermediate 12. MS of 12: m/z 247.06/249.06 [M + H]+.
A solution of intermediate 12 (1 equiv.), N-Boc-piperidine (1 equiv.), and DIEA (3 equiv.) in dry THF (5 mL/mmol) was heated at 90°C for 16 h. Reaction mixture was worked-up using water and CH2Cl2, and the combined organic layers concentrated under reduced pressure and chromatographed over Silica gel using CH2Cl2-MeOH-aq. NH3 to afford 13-Boc. The latter underwent Boc deprotection using methanolic HCl to afford 13. MS: m/z 297.17 [M + H]+.
iv) Intermediate 14. 2-Aminopyrimidine (95 mg, 1.0 mmol) underwent Buchwald coupling with 3-bromo-4-methylbenzaldehyde (199 mg, 1.0 mmol) in the presence of Pd2(dba)3 (45 mg), XanthPhos (60 mg), and Cs2CO3 (489 mg) in 1,4-Dioxane (10 mL) overnight at 100°C to give intermediate 14 (206 mg, 96%). MS of 14: m/z 214.09 [M + H]+.
v) IMTi-3. A solution of 13 (40 mg, 0.13 mmol) and 14 (28 mg, 0.13 mmol) in dichloroethane (5 mL) and AcOH (0.2 mL) was added Na(OAc)3BH (100 mg, 0.47 mmol) in portions at 0°C. After the reaction mixture was stirred overnight at room temperature, usual work up using methylene chloride and Aq. NaHCO3 solution and purification over Silica gel gave IMTi-3 (34 mg, 50%). 1H NMR (400 MHz, CDCl3+ CD3OD) of IMTi-3: δ 8.68 (s, 1H), 8.37 (s, 1H, and d, J = 4.6 Hz, 2H), 8.30 (s, 2H), 7.83 (d, J = 7.13 Hz, 2H), 7.82 (s, 1H), 7.43 (d, J = 7.88 Hz, 2H), 7.31 (dd, J = 7.96, 4.6 Hz, 1H), 7.17 (d, J = 7.64 Hz, 1H), 7.01 (d, J = 7.48 Hz, 1H), 6.89 (s, 1H), 6.69 (t, J = 4.72 Hz, 1H), 3.57 (s, 4H), 3.53 (s, 4H), 2.29 (s, 3H); HRMS: m/z 494.2672 [M + H]+.
2.3 Screening and evaluation of IMTi’s and analogs
N2a695 cells were used to screen all new compounds and in the follow-up studies with compounds found active in the preliminary screen. In a typical experiment, 6-well tissue culture plates (Corning) were seeded at 4.0 × 105–4.5 × 105 N2a695 cells/mL, 2 mL/well for overnight incubation. When cells were >95% confluent, media were exchanged with fresh media containing 10 µM solutions of compounds or DMSO carrier alone and cells were incubated at 37°C in 5% CO2 for 5 h. Culture media were collected and soluble Aβ concentrations in the media were determined by ELISA or MSD plates for human Aβ peptides as per manufacturer instructions. Signals for Aβ were measured using Perkin Elmer Envision and SQ120 MSD ELISA reader. Follow-up studies with N2a695 cells were performed similarly.
2.4 Effects of IMT and IMTi’s on APP metabolism
N2a695 cells were treated with compounds for 5 h as described above, and media were aspirated out (or collected for determination of Aβ levels). Cells were scraped in cold Dulbecco’s PBS buffer (1 mL) containing mini EDTA-free protease inhibitor (Roche) and centrifuged for 1 min at 13,000 rpm at 4°C to form a cell pellet. The buffer was aspirated, and the cell pellets were lysed in 3% SDS plus protease inhibitor cocktail by sonication on ice for two rounds of 20 s on a low setting. Protein concentrations were measured using the Pierce BCA Protein Assay (Thermo Fisher) kit in accordance with the manufacturer’s instructions.
To perform WBs, N2a695 cell lysates from 1a and analogs-treated samples were run on a 10%–20% or a 16.5% Tris-Tricine gel (Criterion) and electro transferred to PVDF membranes (EMD Millipore) overnight at 30 V. PVDF membranes were incubated in PBS containing 0.25% glutaraldehyde (Sigma) for 30 min after electro transference, blocked for 30 min in milk PBST, incubated with primary antibody RU369 for 1 h at room temperature followed by washing and incubation with an HRP-linked secondary antibody and detected with enhanced chemiluminescence ECL reagents. WB images were analyzed using ImageJ to quantify the prominent bands.
To determine effects of compounds on BACE1 vs. GS inhibition, we used N2a cells transiently transfected with full length APP (APP-FL) or with APP99 (APP-βCTF) as described previously (Netzer et al., 2017; Sun et al., 2019). After 48 h, media were removed and fresh media containing compound 1a and analogs were added. Following 5 h of incubation, cell supernatants were collected, and analyzed using MSD-ELISA for Aβ and sAPPα and western blot for sAPPβ.
2.5 In vivo brain permeability and retention of IMT analogs
All procedures involving animals were approved by The Rockefeller University Institutional Animal Care and Use Committee and were in accordance with the National Institutes of Health guidelines. Mesylate salts of the isomeric IMT analogs (1 or 3 mg/mL in water, 125 μL, 50 mg/kg) were administered intraperitoneally (i.p.) or through oral gavage to 8 weeks old C57BL/6J WT mice (Sun et al., 2019). Mice were euthanized 4 h post drug administration and brain hemispheres and plasma were harvested and collected in pre-weighted tubes and snap-frozen in liquid nitrogen. To measure brain and plasma concentrations of the specific compounds, mouse brain tissue was homogenized and extracted using Ethanol, and plasma samples were extracted using Acetonitrile. Concentration of the drug and metabolites in brain and in plasma was determined by LC-MS/MS analysis.
2.6 Drug extraction from brain
After tubes were weighed to calculate brain weight and thawed to room temperature, 1 mL of EtOH (200 Proof) was added to the microcentrifuge tubes containing the harvested right brain hemispheres. 10 μL of 1 µM internal standard (ABG190, a synthetic analog of 1a) was added to each tube and samples were sonicated to homogeneity (∼2 min). Tubes were shaken at 40 min at room temperature (1K RPM) and centrifuged for 8 min at 13K RPM. The supernatant (0.9 mL) was transferred to a new collection tube and 0.5 mL EtOH was added to the pellet for a second round of extraction as described above. 600 μL of the supernatant was combined with the first collection before samples were submitted for LCMS-MS analysis (Sun et al., 2019).
2.7 Drug extraction from blood
300 μL of acetonitrile was added to collected blood samples. 10 μL of 1 µM internal standard (ABG190) was added to each tube and samples were sonicated to homogeneity (∼2 min). Tubes were contributed at 13 K RPM for 9 min 300 μL of the supernatant was collected and combined with 500 µL of 5 mM ammonium formate before samples were submitted for LCMS-MS analysis (Sun et al., 2019).
2.8 In vitro kinase activity assay
The assay was performed by Luceome Biotechnologies, LLC. Typically, 10 mM stock solutions of the compounds were diluted in DMSO to a concentration of 250 μM. Prior to initiating the assay, all test compounds were evaluated for false positive against split-luciferase (Jester et al., 2010). For kinase assays, each Cfluc-Kinase was translated along with Fos-Nfluc using a cell-free system (rabbit reticulocyte lysate) at 30°C for 90 min 24 μL aliquot of this lysate containing either 1 μL of DMSO (for no-inhibitor control) or compound solution in DMSO (10 μM final concentration) was incubated for 30 min at room temperature followed by 1 h in presence of a kinase specific probe. 80 μL of luciferin assay reagent was added to each solution and luminescence was immediately measured on a luminometer. The percent Inhibition was calculated using the following equation: % Inhibition = (ALUcontrol–ALUsamplex 100)/ALUcontrol.
3 Results
3.1 Chemistry
IMT isomers, IMTi-1 – 3, possess all five rings and the chemical functions that broadly match the parent compound (Figure 1). We designed these isomers by making one or two hypothetical fragmentations across C-N and C-C bonds and re-joining the resulting fragments through other ring(s) and keeping the functionalities similar to IMT, as outlined in Scheme 1A. Here, cleavage sites shown by “scissor” are evident in IMT at ‘a-c’ and the double arrows shown connect I and II; III, V and IV; and VI, II and III to give IMTi-1, IMTi-2, and IMTi-3, respectively. Similarly, we designed IMTi-1 and 2 analogs based on the previously described IMT analogs, in that ‘E’ ring has been modified with R = various alkyl and cycloalkyl amines and ‘A’ ring with phenyl and substituted phenyls besides pyridine (Scheme 1B) (Sun et al., 2019).
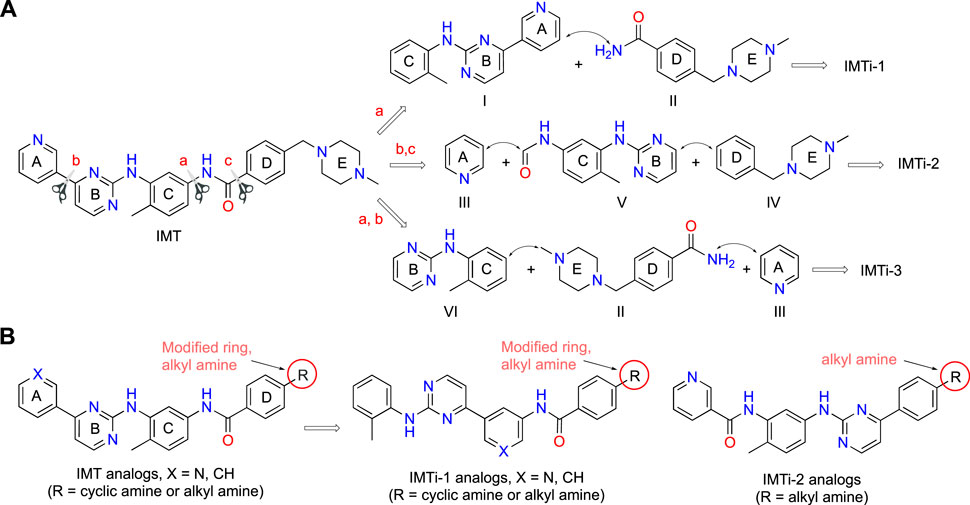
Scheme 1. Design of IMTi-1 – 3 and analogs. (A) Shown are hypothetical fragmentation of IMT involving (a) C-N bond or (b, c) C-C bond cleavage giving fragments I-VI, and re-assembly of these fragments to afford IMTi-1 – 3. Note: fragment II is common for both IMTi-1 and 3, and III for IMTi-2 and 3. Key: Scissor sign, site of C-C or C-N bond cleavage for fragmentation; double arrow, C-C or C-N bond connection for re-assembly of the molecules. (B) General structure of IMT analogs described previously (Sun et al., 2019) and of IMTi-1’s and 2’s designed here.
Synthesis of IMTi-1 – 3 and analogs. We prepared IMTi-1 and its analogs 1a-1r using the readily available intermediates, as outlined in Schemes 2, 3. First, to prepare IMTi-1, intermediate 4a was reacted with o-toluidine and the resulting product 5a underwent Buchwald coupling (Ruiz-Castillo and Buchwald, 2016) with amide 6a (Scheme 2). Similarly, intermediate 4b reacted with o-toluidine to give 5b, and both 5a and 5b underwent Buchwald coupling (Ruiz-Castillo and Buchwald, 2016) with various amides 6a-h giving products 1a-m, several after Boc deprotection as needed. Analogs 1n and 1o were prepared by reaction of 1l and 1m with formaldehyde under the reductive amination conditions using NaCNBH3. The analog 1p was obtained by reacting 4c with 3-aminopyridine, followed by Boc-deprotection giving amine 5c and reacting the latter with acid 6i (Scheme 3). Finally, Analog 1q and 1r were prepared by Suzuki coupling (Miyaura and Suzuki, 1995) of 5a with boronic acids, 7a and 7b (Scheme 3).
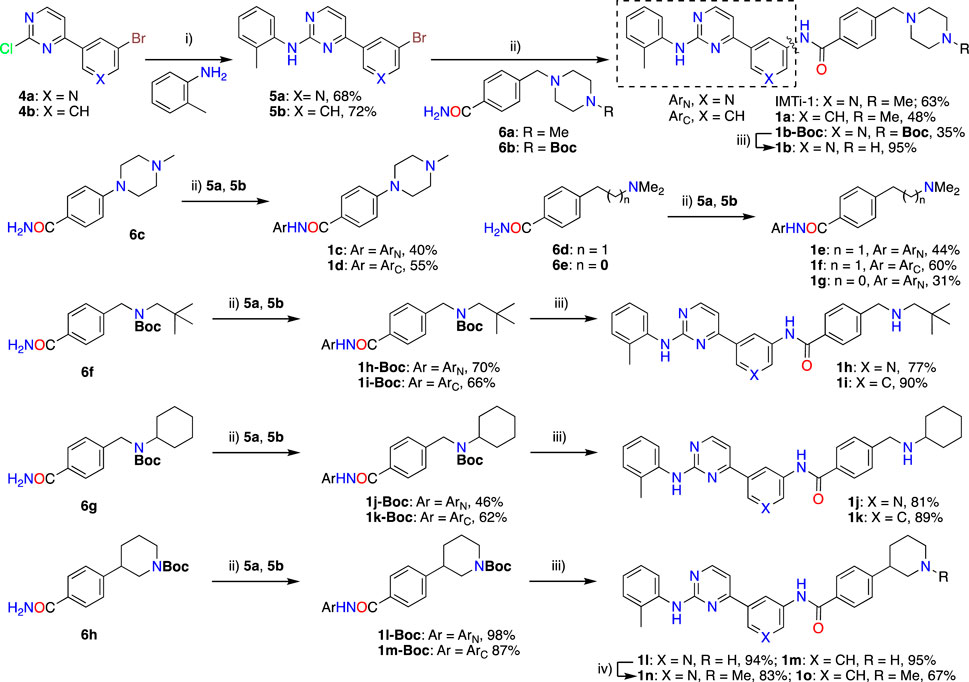
Scheme 2. Synthesis of IMTi-1 and analogs 1a-o Key: i) 3N HCl, Dioxane. microwave, 100°C, 2 h ii) Pd2(dba)3, XanthPhos, Cs2CO3, 1,4-Dioxane, microwave, 100°C. iii) 4M HCl in dioxane, EtOAc, RT, 2 h iv) CH2O, NaCNBH3, DCE, 0°C - RT, 16 h.
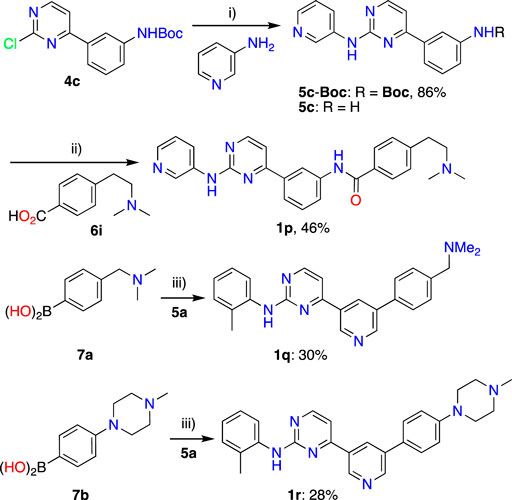
Scheme 3. Synthesis of IMTi-1 analogs 1p-r. Key: i) Pd2(dba)3, XanthPhos, Cs2CO3, 1,4-Dioxane, microwave, 100°C; 4M HCl in dioxane, EtOAc, RT, 2 h ii) PyBOP, DIPEA, DMF, RT, 16 h iii) Pd(PPh3)4, aq. K2CO3, 1,4-Dioxane, microwave, 100°C, 2 h.
Next, we prepared IMTi-2 and its analogs 2a-b using intermediates 8 and 9, as described in Scheme 4A. Intermediates 8 and 9 reacted together under the Buchwald coupling conditions affording 10, which underwent reductive amination with N-methylpiperazine to give IMTi-2. Alternatively, intermediate 8 underwent reductive amination with cyclohexyl amine and neopentyl amine and Boc-protection of the resulting amines to give intermediates 11a and 11b, which reacted with intermediate 9 under the Buchwald coupling conditions, followed by Boc-deprotection to give analogs 2a-b. Finally, to prepare IMTi-3, we prepared intermediate 13 by reacting 4-chloro-mthylbenzoyl chloride with 3-aminopyridine 3-amino-pyridine and the resulting product 12 with N-Boc-piperazine followed by N-deprotection, and intermediate 14 by reacting 3-bromo-4-methylbenzaldehyde with 2-amino-pyrimidine under Buchwald conditions. Subsequently, we coupled intermediates 13 and 14 together under the reductive amination conditions using NaCNBH4 to give the title IMTi-3 (Scheme 4B) (Afanasyev et al., 2019).
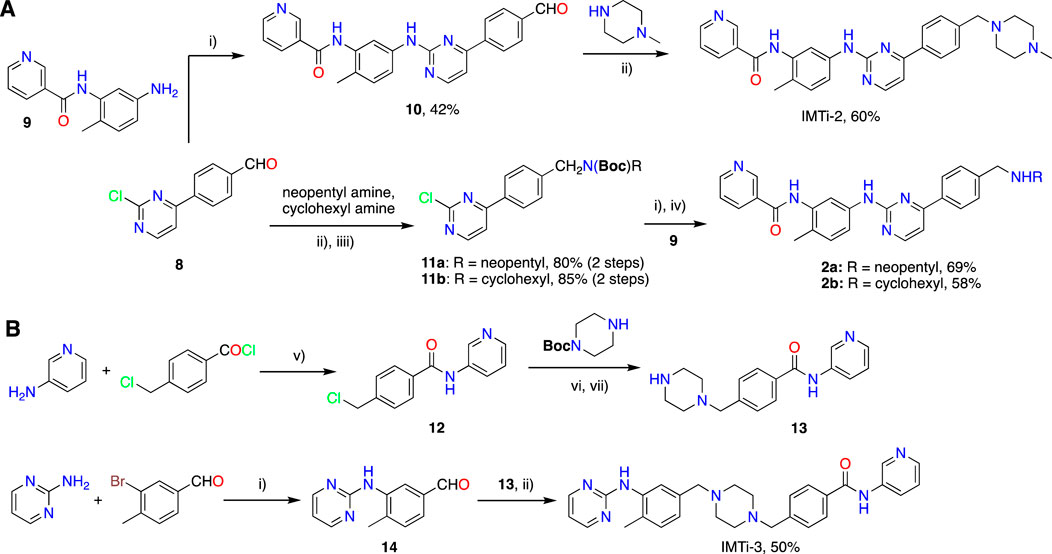
Scheme 4. Synthesis of (A) IMTi-2 analogs, and (B) IMTi-3. Key: i) Pd(dba)3, XanthPhos, Cs2CO3, 1,4-Dioxane, microwave, 100°C, 2 h ii) Na(OAc)3BH, DCE, AcOH. iii) Boc2O, ACN. iv) TFA, TIPS, CH2Cl2, 0°C-RT, 2 h v) DIEA, THF, RT, 3 h vi) DIEA, THF, 90°C, 2 h vii) 4M HCl in dioxane, EtOAc, RT, 2 h.
3.2 Structural diversity
As described above, many IMT analogs, in that either A ring changed to substituted benzene ring or E ring to cycloalkyl amines or alkyl amines, were prepared previously and evaluated (Sun et al., 2019). The majority of IMTi-1 analogs, including 1a-1p and both analogs of IMTi-2, i.e., 2a and 2b, differ from one-another in ring ‘A’ and/or in ‘E’ and possess fragments containing ‘D’ and ‘E’ rings previously prepared in IMT analogs (Sun et al., 2019). New IMTi-1 analogs contain piperazine ring, a cyclic amine or piperidine ring connected through C-C or C-N bond to ring D, while all other IMTi-1 and both IMTi-2 analogs possess a substituted alkylamine instead of the ring E. These modifications improved APP processing activity in IMT analogs (Sun et al., 2019). There was no additional difference between two analogs, 2a and 2b, of IMTi-2. Thus, all 18 analogs of IMTi-1 possess rings ‘A-D’ and their arrangement is similar with three exceptions. 1) Seven compounds possess 1,3-substituted benzene and the remaining 11 analogs contain 3,5-substituted pyridine (Py) as the middle ring ‘A’, 2) The first ring from the left (ring ‘C’) in 1 analog, 1p, is 3-aminopyridine instead of o-toluidine in all remaining 17 compounds. 3) Analogs 1q and 1r do not possess the ‘amide group’ that connects the middle ring ‘A’ to the 4th ring ‘D’.
3.3 Evaluation
Previously, we showed that two chemically distinct compounds, IMT and DV2-103 lower Aβ production primarily by reducing BACE processing of APP (Netzer et al., 2017). Similarly, numerous analogs of IMT also lowered Aβ production by reducing BACE processing of APP (Sun et al., 2019). In the present study, before evaluating new analogs, we further examined the effects of these compounds on γ-secretase catalyzed Aβ formation and compared these to the more radically isomeric analogs of IMT. Our results show that IMT, DV2-103, and IMTi-1 are γ-secretase modulators; i.e., these compounds favor production or inhibition of different lengths of Aβ peptides (differing in their C-termini). Specifically, we exposed N2a695 cells to increasing concentrations of each compound and measured the production of Aβ38, 40, and 42. IMT, DV2-103 and IMTi-1 (Figure 2A) inhibit the formation of Aβ38 least, compared to Aβ40 and 42, and even boost levels of Aβ38 above controls at a drug concentration of 5 μM. Remarkably, this occurs for all Aβ peptides tested shorter than 40 amino acids (Figure 2C). Moreover, for some compounds that modulate γ-secretase activity most conspicuously at 5 μM, this effect vanishes at 10 μM, relative to controls (Figure 2B, C).
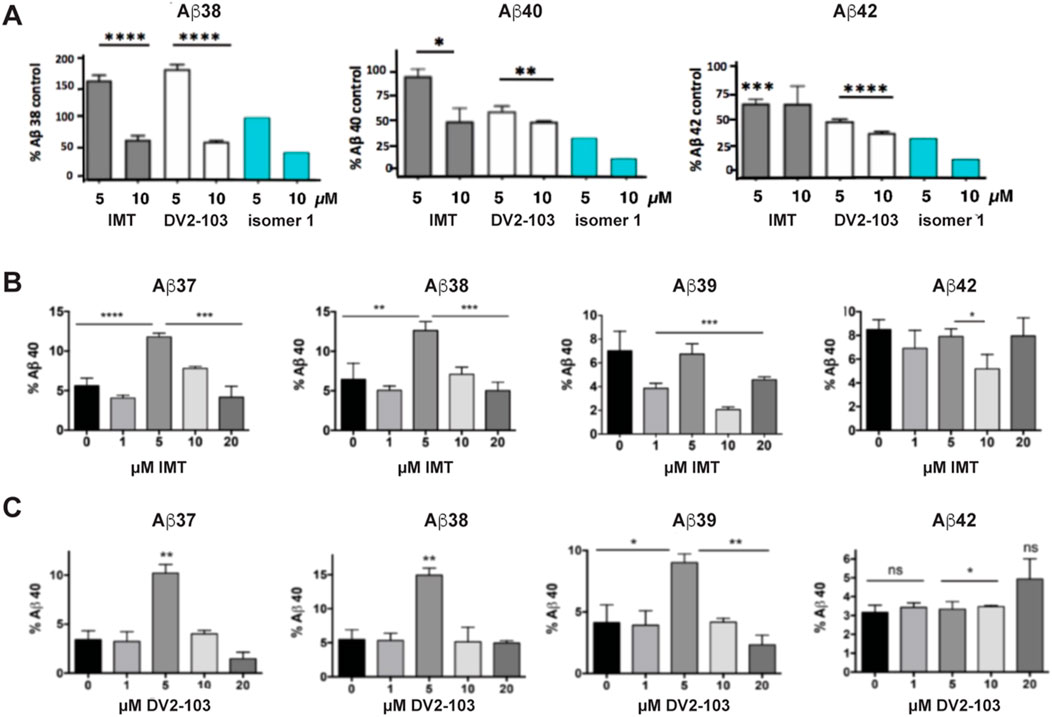
Figure 2. IMT, DV2-103, and IMT isomer 1 are γ-secretase modulators. (A) N2a695 cells incubated with IMT, DV2-103 and IMT isomer 1 lower levels of Aβ40 and 42 more than Aβ38, especially at a drug concentration of 5 μM, as measured by ELISA. Means differ significantly for IMT and DV2-103 compared to DMSO controls, N = 3 × 3. Data for Isomer 1 are from a representative sample. Differences between means for (A) are analyzed by One-way Anova for IMT and DV2-103 treated cells. Differences among means comparing 5 μM IMT and DMSO controls (B, C) are analyzed by Student’s T test (S.E.M.).
With new IMTi’s in hand, we first evaluated and compared the effects of IMT and IMTi-1 (Figure 3A) on APP processing in N2a695 cells using the methods described above and in our prior reports (Netzer et al., 2017; Sun et al., 2019). We used both compounds at 10 µM and analyzed cell supernatants using anti- Aβ40 ELISA to show that IMTi-1 reduced Aβ40 levels more strongly than IMT (Figure 3C). We further determine the sAPPβ levels in cell supernatants by performing western blotting (WB) experiments and probing the WB membranes using antibody RU anti-C-terminal sAPPβ (Figure 3B, bottom, and Figure 3D), and APP metabolites in cell lysates using antibody RU369 (anti-C terminal APP) (Figure 3B, upper). Similarly, we tested the effects of IMTi-2 and 3 on Aβ production in N2a695 cells to find that both IMTi-2 and IMTi-3 inhibited Aβ40 production weakly (Aβ40 levels: 68% for IMTi-2 and 93% for IMTi-3 at 10 µM concentration) compared to both IMT and IMTi-1.
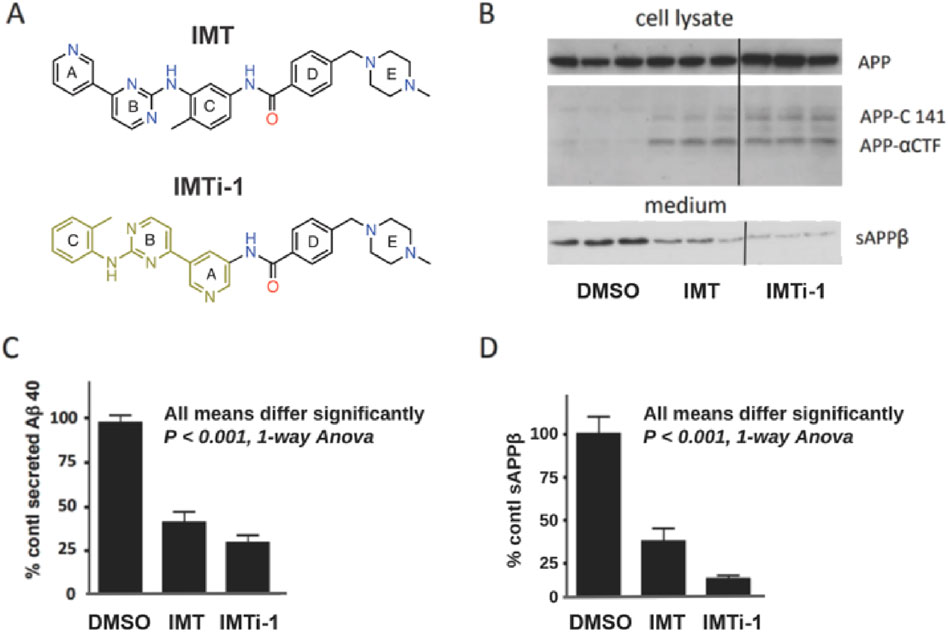
Figure 3. A Major structural change in IMT results in IMT isomer 1 (IMTi-1), which retains IMT’s Aβ-reducing effect and its reduction of BACE processing of APP in N2a 695 cells. (A) Structures of IMT and isomer-1, IMTi-1. (B) Western blots of N2a cell lysates (upper) and cell media (lower) from experiments using IMT and IMTi-1 probed with antibody RU369 (anti-C terminal APP) and RU anti-C-terminal sAPPβ (bottom), respectively. Each western blot panel shows lanes from a single gel. However, the three lanes at the right of each, which refer to incubation with IMTi-1, are from a different part of the same gel. (C) Quantification of secreted Aβ40 in N2a cells incubated with IMT or IMTi-1, One-way Anova, p < 0.001. (D) Quantification of sAPPβ levels. One-way Anova, p < 0.001.
Similarly, we evaluated all IMTi-1 and IMTi-2 analogs, including the Boc-protected compounds, using N2a695 cells. We found most Boc-protected compounds showed little or no inhibition of Aβ production (Supplementary Figure S1) under the above conditions. In fact, two Boc compounds, 1i-Boc and 1k-Boc, showed an increase in Aβ production, whereas several IMTi-1 analogs showed superior inhibitory effects compared to IMT on Aβ production (Supplementary Figure S1). To further examine the activities of the active analogs and whether IMTi’s has any bias on amyloidogenic vs. nonamyloidogenic cleavage of APP, we retested dozens of IMTi-1 and 2 analogs, including several found active in the screening assay and some tested for the first time using N2a695 cells as above, and performed MSD ELISA of the conditioned media to measure Aβ40, Aβ38, and Aβ42 peptides simultaneously. We found that most IMTi-1 analogs favored nonamyloidogenic cleavage of APP at both 10 and 5 µM concentrations and reduced production of Aβ40 and Aβ42 greater than Aβ38 peptide (Table 1). This indicates that IMTi-1 and analogs modulate γ-secretase cleavage of C-terminal APP since the differences in lengths of these peptides is determined by γ-secretase according to differences in utilization of the APP γ-secretase cleavage sites (Hur, 2022).
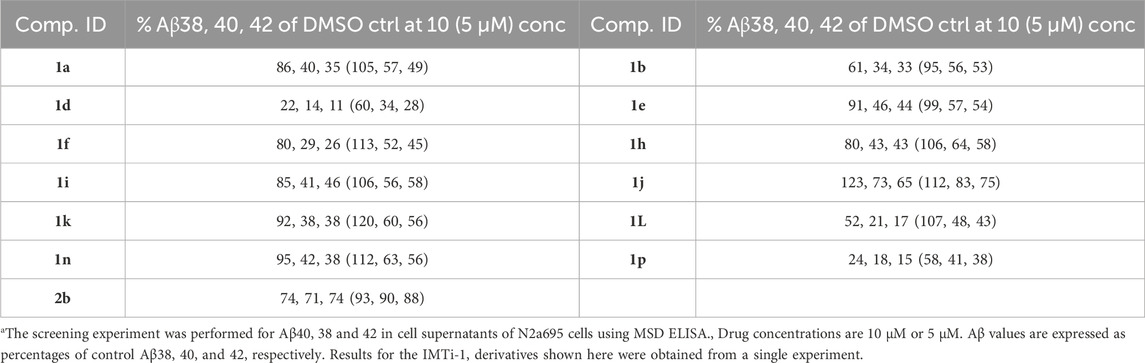
Table 1. Isomeric IMT analogs are γ-secretase modulatorsa.
We focused on IMTi-1 and its analogs 1d, 1f and 1p. All these analogs showed superior activities among all. To test whether these compounds lower Aβ levels by affecting the BACE and/or γ-secretase cleavages, we transfected wild-type (wt) cells with APP-FL and APP βCTF (C99), respectively, and incubated the cells with IMTi-1 (isomer) and analogs 1d, 1f and 1p. We used BACE inhibitor, MK8931, and γ-secretase inhibitor, DAPT, as controls and performed the experiments and processed the results as described previously (Sun et al., 2019). The results shown in Figures 4A, B revealed that all 4 compounds reduced β- and γ-cleavages of APP similarly to IMT. There were reductions in Aβ production in both cases, but more so in cells transfected with full-length APP indicating that these compounds, like IMT (Netzer et al., 2017), reduce both BACE and γ-secretase cleavages of APP but that attenuation of BACE processing accounted for the greater part of Aβ reduction (Figures 4A, B). None of these compounds showed any toxicity to N2a695 cells at 10 µM concentration (Figure 4C) under the experimental conditions used for the Aβ assay.
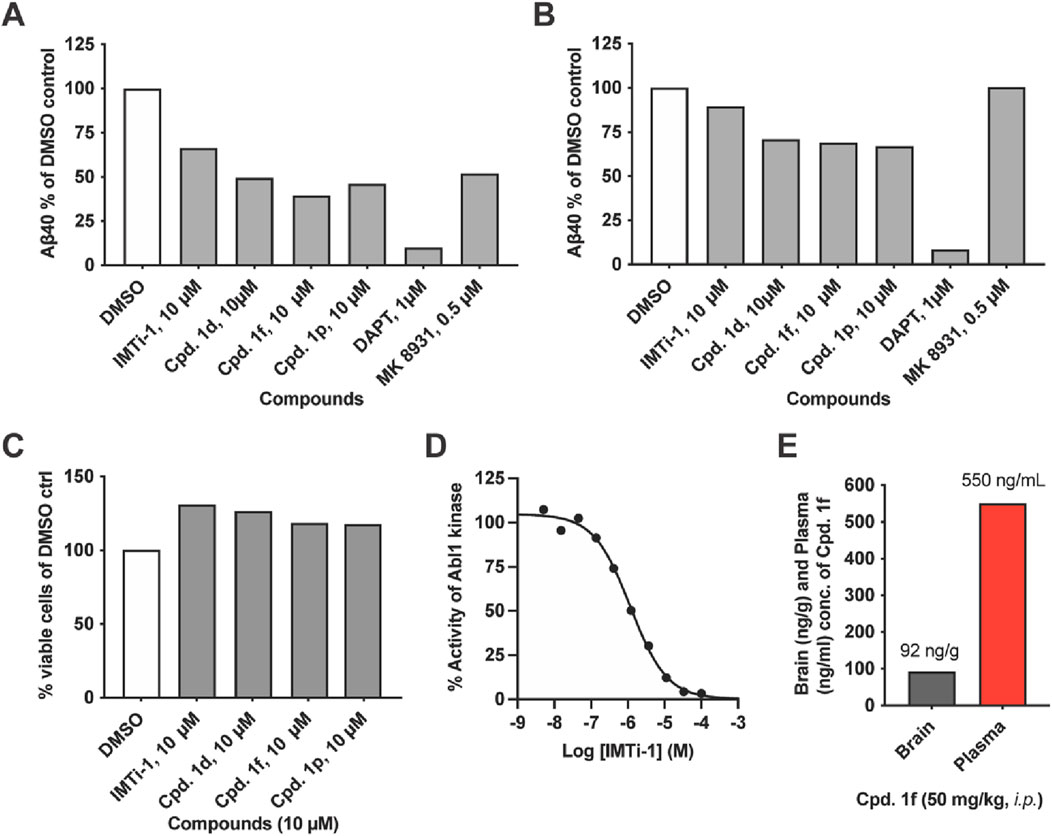
Figure 4. IMT isomer 1 (IMTi-1) and its analogs 1d, 1f and 1p lower BACE and γ-secretase cleavage of APP and lower levels of Aβ in N2a cells transiently transfected with APP 695 (left graph) or APP C99 (right graph): (A) full length APP695 (APP-FL) or (B) APP C99 (β-CTF). (C) Percentage of viable wild-type (WT) N2a cells upon treatment with IMT isomers 1, cpd. 1d, cpd. 1f and cpd. 1p compared to DMSO control under the same conditions used to test Aβ production. (D) Effects of IMTi-1 on Abl1 kinase in vitro. (E) Brain and plasma concentrations of IMTi-1 and cpd. 1f in 2 months old WT mice 4 h after i. p. injection of 50 mg/kg of each drug. Data for A-C are from representative samples.
IMT inhibits Abl1 kinase with low nanomolar affinity (Buchdunger et al., 1996). Earlier, we prepared and evaluated numerous IMT analogs to find that many of these analogs reduced Aβ levels in cells similarly to IMT, while inhibiting Abl kinase less potently, compared to IMT. In other words, there is not a good correlation between the Abl kinase inhibitory activity vs. the Aβ lowering effects in cells contacted with the IMT analogs. We have evaluated IMTi-1 to find that it inhibits Abl kinase less potently (IC50: 1.172 µM) (Figure 4D) than IMT IC50: 0.038 µM) (Buchdunger et al., 1996), while it reduced Aβ levels more potently than IMT (Figure 3C). This result further reinforces our prior observation that there is no or little connection between the Aβ-lowering activity of IMT and its inhibition of Abl1 kinase (Netzer et al., 2003). Finally, we tested the brain permeability of compound 1f by administering it to 2 months old mice. Plasma and brain tissue were collected 4 h post drug administration, and LC-MS/MS analysis of the acetonitrile and ethanol extracts was used to measure drug concentration. Compound 1f possesses similarity to IMT isomer-1a and is isomeric to an ABG-179 (Sun et al., 2019) analog that possessed a benzene instead of the pyridine (A) ring (see: Figure 1 for the ring numbering). Earlier, we have shown that ABG-179 possesses superior brain exposure compared to IMT and reduced both Aβ40 and 42 levels significantly in AD mice when delivered acutely for 5 days (Sun et al., 2019). Now, we have found that isomer 1f also possesses high brain exposure (Figure 4E) and that is comparable to ABG-179 based on the results of our prior studies (Sun et al., 2019).
4 Discussion
Based on the number and variety of chemically distinct compounds that produce the same biochemical effects on APP metabolism (Netzer et al., 2017) and that all active compounds are active at low micromolar concentration, we postulated that IMT, DV2-103 and their analogs are likely to produce their effects on APP metabolism by virtue of their physical rather than stereological properties. For example, physical properties would include acting as a weak base that would cause these molecules to be lysosomotropic. We came to this conclusion by showing that the effects of IMT and DV2-103 on APP metabolism are dependent on acidified lysosomes (Netzer et al., 2017), and with the knowledge that IMT is strongly lysosomotropic we hypothesized that IMT, DV2-103 and their active derivatives might bind to a polyspecific receptor where binding is less dependent on structural and electrostatic complementarity. However, our subsequent studies with IMT, IMTi-1 – 3 and the analogs of IMTi-1 provide a more complex picture.
In our current study, we designed IMT isomers, IMTi-1 – 3, each possessing a distinctly unique pharmacophore, yet maintaining IMT’s physical property as a weak base (Supplementary Table S1), determined through quantitative structure property relationship (QSPR) analysis (Li et al., 2018). The weakly basic property of IMT is necessary for its sequestration in lysosomes through ion trapping (Burger et al., 2015). In other words, IMT is lysosomotropic, as are IMTi-1 –3 (Netzer et al., 2017). However, IMTi-3 was found inactive in Aβ production assays, while IMT and IMTi-1 and 2 were active.
Interestingly, we found that IMT, IMTi-1, and DV2-103 show concentration dependent modulation of γ-secretase as tested in Aβ production assay in N2a695 cells (Figure 2). Moreover, by evaluating novel IMTi-1 and 2 analogs, it became evident that a subset of these analogs recapitulated IMT’s APP phenotype. Additionally, we showed that IMT, DV2-103, and the IMTi-1 isomers tested in this study are modulators of γ-secretase by virtue of the observation that their Aβ-lowering potency differentially affects Aβ peptide lengths depending on drug concentration. Remarkably, Aβ1-42 production is lowered at 5 μM drug concentrations, while Aβ1-38 production is inhibited least and, in some cases, raised. This is important because heightened production of Aβ38 has been considered benign, and more recently therapeutic (Cullen et al., 2022), while lowered production of Aβ42 is considered therapeutic; in either case, a decrease in Aβ peptide aggregation may occur.
IMTi-1 inhibited Abl kinase activity with over a 100-fold reduction in potency compared to previously published reports of IMT (Buchdunger et al., 1996). Although we had compared the relative effects of γ-secretase and BACE modulation of Aβ generation in cells, we could not rule out that the lowering of Aβ and sAPPβ was not a result of IMT’s effect of stimulating autophagy (Drullion et al., 2012), since autophagy was previously shown to accelerate lysosomal degradation of APP-βCTF and Aβ(Tian et al., 2011). Further examination of the structures and activities of IMTi-1 and analogs compared to IMT and similar analogs (depicted by ‘R’ in Scheme 1B) revealed that both classes of active compounds possessed similar ‘R’ groups. Yet, unlike IMT and its analogs, all active IMT-1 analogs behaved like g-secretase modulators, thereby providing a new pharmacophore for development of anti-Aβ therapy.
5 Conclusion
In summary, we suggest that the effect of IMT and related drugs on APP metabolism occurs through a mechanism that is, to a great extent, determined by physicochemical and structural properties of the drug molecules and is less dependent on similarities in stereochemical structure. Future studies may wish to focus on trafficking of full-length APP to determine whether these drugs affect APP trafficking by translocation of APP to lysosomes and away from amyloidogenic processing by BACE and γ-secretase. The fact that many of these compounds (structurally related or not) are γ-secretase modulators may also be consistent with a mechanism involving altered trafficking of APP that could affect the specificity of γ-secretase cleavage sites on APP during the formation of Aβ peptides.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
WN: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing–original draft, Writing–review and editing. AS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing–review and editing. MG: Methodology, Investigation, Writing–review and editing. EC: Methodology, Investigation, Writing–review and editing. KG: Methodology, Investigation, Writing–review and editing. EM: Methodology, Investigation, Writing–review and editing. J-SS: Formal Analysis, Visualization, Writing–review and editing. SS: Conceptualization, Data curation, Formal Analysis, Project administration, Supervision, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Funding support from JPB (#322 and #839 to SS) and Fisher Center for Alzheimer’s Research Foundation (PG) is duly acknowledged.
Acknowledgments
We are thankful to Paul Greengard (Deceased) of the Rockefeller University for his enthusiastic support to this work and Victor H. Bustos for helpful discussion. We also thank Proteomics Research Center for performing LC-MS/MS analysis of the brain and plasma extracts obtained from mice administered with compounds.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2024.1381205/full#supplementary-material
References
Afanasyev, O. I., Kuchuk, E., Usanov, D. L., and Chusov, D. (2019). Reductive amination in the synthesis of pharmaceuticals. Chem. Rev. 119, 11857–11911. doi:10.1021/acs.chemrev.9b00383
Buchdunger, E., Zimmermann, J., Mett, H., Meyer, T., Müller, M., Druker, B. J., et al. (1996). Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 56, 100–104.
Burger, H., den Dekker, A. T., Segeletz, S., Boersma, A. W., de Bruijn, P., Debiec-Rychter, M., et al. (2015). Lysosomal sequestration determines intracellular imatinib levels. Mol. Pharmacol. 88, 477–487. doi:10.1124/mol.114.097451
Drullion, C., Trégoat, C., Lagarde, V., Tan, S., Gioia, R., Priault, M., et al. (2012). Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell. Death and Dis. 3, e373–e73. doi:10.1038/cddis.2012.111
Golde, T. E., Koo, E. H., Felsenstein, K. M., Osborne, B. A., and Miele, L. (2013). γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 1828, 2898–2907. doi:10.1016/j.bbamem.2013.06.005
Hur, J.-Y. (2022). γ-Secretase in Alzheimer’s disease. Exp. and Mol. Med. 54, 433–446. doi:10.1038/s12276-022-00754-8
Jester, B. W., Cox, K. J., Gaj, A., Shomin, C. D., Porter, J. R., and Ghosh, I. (2010). A coiled-coil enabled split-luciferase three-hybrid system: applied toward profiling inhibitors of protein kinases. J. Am. Chem. Soc. 132, 11727–11735. doi:10.1021/ja104491h
Kazmi, F., Hensley, T., Pope, C., Funk, R. S., Loewen, G. J., Buckley, D. B., et al. (2013). Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. 41, 897–905. doi:10.1124/dmd.112.050054
Kim, C. K., Lee, Y. R., Ong, L., Gold, M., Kalali, A., and Sarkar, J. (2022). Alzheimer’s disease: Key insights from two decades of clinical trial failures. J. Alzheimers Dis. 87, 83–100. doi:10.3233/jad-215699
Kumar, D., Ganeshpurkar, A., Kumar, D., Modi, G., Gupta, S. K., and Singh, S. K. (2018). Secretase inhibitors for the treatment of Alzheimer's disease: long road ahead. Eur. J. Med. Chem. 148, 436–452. doi:10.1016/j.ejmech.2018.02.035
Li, M., Zhang, H., Liu, L., Chen, B., Guan, L., and Wu, Y. (2018). A quantitative structure-property relationship model based on chaos-enhanced accelerated particle swarm optimization algorithm and back propagation artificial neural network. Appl. Sci. 8, 1121. doi:10.3390/app8071121
Miranda, A., Montiel, E., Ulrich, H., and Paz, C. (2021). Selective secretase targeting for Alzheimer’s disease therapy. J. Alzheimer's Dis. 81, 1–17. doi:10.3233/jad-201027
Miyaura, N., and Suzuki, A. (1995). Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 95, 2457–2483. doi:10.1021/cr00039a007
Nagar, B., Bornmann, W. G., Pellicena, P., Schindler, T., Veach, D. R., Miller, W. T., et al. (2002). Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243.
Netzer, W. J., Bettayeb, K., Sinha, S. C., Flajolet, M., Greengard, P., and Bustos, V. (2017). Gleevec shifts APP processing from a β-cleavage to a nonamyloidogenic cleavage. Proc. Natl. Acad. Sci. U. S. A. 114, 1389–1394. doi:10.1073/pnas.1620963114
Netzer, W. J., Dou, F., Cai, D., Veach, D., Jean, S., Li, Y., et al. (2003). Gleevec inhibits β-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. U. S. A. 100, 12444–12449. doi:10.1073/pnas.1534745100
Panza, F., Solfrizzi, V., Frisardi, V., Capurso, C., D’Introno, A., Colacicco, A. M., et al. (2009). Disease-modifying approach to the treatment of Alzheimer’s disease. Drugs and Aging 26, 537–555. doi:10.2165/11315770-000000000-00000
Portelius, E., Van Broeck, B., Andreasson, U., Gustavsson, M. K., Mercken, M., Zetterberg, H., et al. (2010). Acute effect on the Aβ isoform pattern in CSF in response to γ-secretase modulator and inhibitor treatment in dogs. J. Alzheimers Dis. 21, 1005–1012. doi:10.3233/jad-2010-100573
Ruiz-Castillo, P., and Buchwald, S. L. (2016). Applications of palladium-catalyzed C–N cross-coupling reactions. Chem. Rev. 116, 12564–12649. doi:10.1021/acs.chemrev.6b00512
Rynearson, K. D., Ponnusamy, M., Prikhodko, O., Xie, Y., Zhang, C., Nguyen, P., et al. (2021). Preclinical validation of a potent γ-secretase modulator for Alzheimer’s disease prevention. J. Exp. Med. 218, e20202560. doi:10.1084/jem.20202560
Sinha, A., Gindinova, K., Mui, E., Netzer, W. J., and Sinha, S. C. (2019). Development of kinase inactive PD173955 analogues for reducing production of Aβ peptides. ACS Med. Chem. Lett. 10, 1430–1435. doi:10.1021/acsmedchemlett.9b00213
Sun, W., Netzer, W. J., Sinha, A., Gindinova, K., Chang, E., and Sinha, S. C. (2019). Development of Gleevec analogues for reducing production of β-amyloid peptides through shifting β-cleavage of amyloid precursor proteins. J. Med. Chem. 62, 3122–3134. doi:10.1021/acs.jmedchem.8b02007
Keywords: imatinib (IMT) or gleevec, DV2-103, gamma secretase, inhibitor, modulator, Aβ, isomer
Citation: Netzer WJ, Sinha A, Ghias M, Chang E, Gindinova K, Mui E, Seo J-S and Sinha SC (2024) Stretching the structural envelope of imatinib to reduce β-amyloid production by modulating both β- and γ-secretase cleavages of APP. Front. Chem. 12:1381205. doi: 10.3389/fchem.2024.1381205
Received: 03 February 2024; Accepted: 10 September 2024;
Published: 08 October 2024.
Edited by:
Abdul Sadiq, University of Malakand, PakistanCopyright © 2024 Netzer, Sinha, Ghias, Chang, Gindinova, Mui, Seo and Sinha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William J. Netzer, YmlsbG5ldHplckBnbWFpbC5jb20=; Anjana Sinha, c2luaGFhbmphbmE1ODE5QGdtYWlsLmNvbQ==; Subhash C. Sinha, c3VzMjA0NEBtZWQuY29ybmVsbC5lZHU=