 Radosław Michalski1
Radosław Michalski1 Renata Smulik-Izydorczyk1
Renata Smulik-Izydorczyk1 Jakub Pięta1
Jakub Pięta1 Monika Rola1
Monika Rola1 Angelika Artelska1Karolina Pierzchała1
Angelika Artelska1Karolina Pierzchała1 Jacek Zielonka2
Jacek Zielonka2 Balaraman Kalyanaraman2
Balaraman Kalyanaraman2 Adam Bartłomiej Sikora1*
Adam Bartłomiej Sikora1*- 1Institute of Applied Radiation Chemistry, Lodz University of Technology, Lodz, Poland
- 2Department of Biophysics, Medical College of Wisconsin, Milwaukee, WI, United States
Azanone (HNO, also known as nitroxyl) is the protonated form of the product of one-electron reduction of nitric oxide (•NO), and an elusive electrophilic reactive nitrogen species of increasing pharmacological significance. Over the past 20 years, the interest in the biological chemistry of HNO has increased significantly due to the numerous beneficial pharmacological effects of its donors. Increased availability of various HNO donors was accompanied by great progress in the understanding of HNO chemistry and chemical biology. This review is focused on the chemistry of HNO, with emphasis on reaction kinetics and mechanisms in aqueous solutions.
1 Introduction
The discovery of endogenous enzymatic production of nitric oxide (•NO) and its important physiological functions in mammals as a signalling molecule, playing an important role as a vasorelaxant agent, an immune response effector, and a regulator of central nervous system (Ignarro et al., 1987) has established a new paradigm in physiology and medicine (Kerwin et al., 1995). Subsequently, over the past 3 decades, several other small, two or three atomic molecules have been identified as signaling agents. This class of low molecular weight signaling molecules includes •NO, carbon monoxide (CO), hydrogen sulfide (H2S) and azanone (HNO) (Wang, 2012; Bianco et al., 2017). Formally, HNO is the protonated product of one-electron reduction of •NO and a highly reactive electrophilic species of growing pharmacological significance (Kemp-Harper et al., 2016; Felker et al., 2019; Fukuto, 2019; Kemp-Harper et al., 2021). Interest in azanone donors and in the biological chemistry of HNO has increased significantly during the last 2 decades, due to the beneficial pharmacological effects of its donors in the treatment of heart failure. Currently, an HNO donor cimlanod (BMS-986231, CXL-1427) is in clinical trials for heart failure treatments (Felker et al., 2019; Felker et al., 2021; Greenberg and Urey, 2021; Lang et al., 2021). This review focuses on the chemistry of aqueous solutions of HNO, with emphasis on the reaction kinetics and mechanisms. The cited HNO rate constants are summarized in Tables 1 and 2.
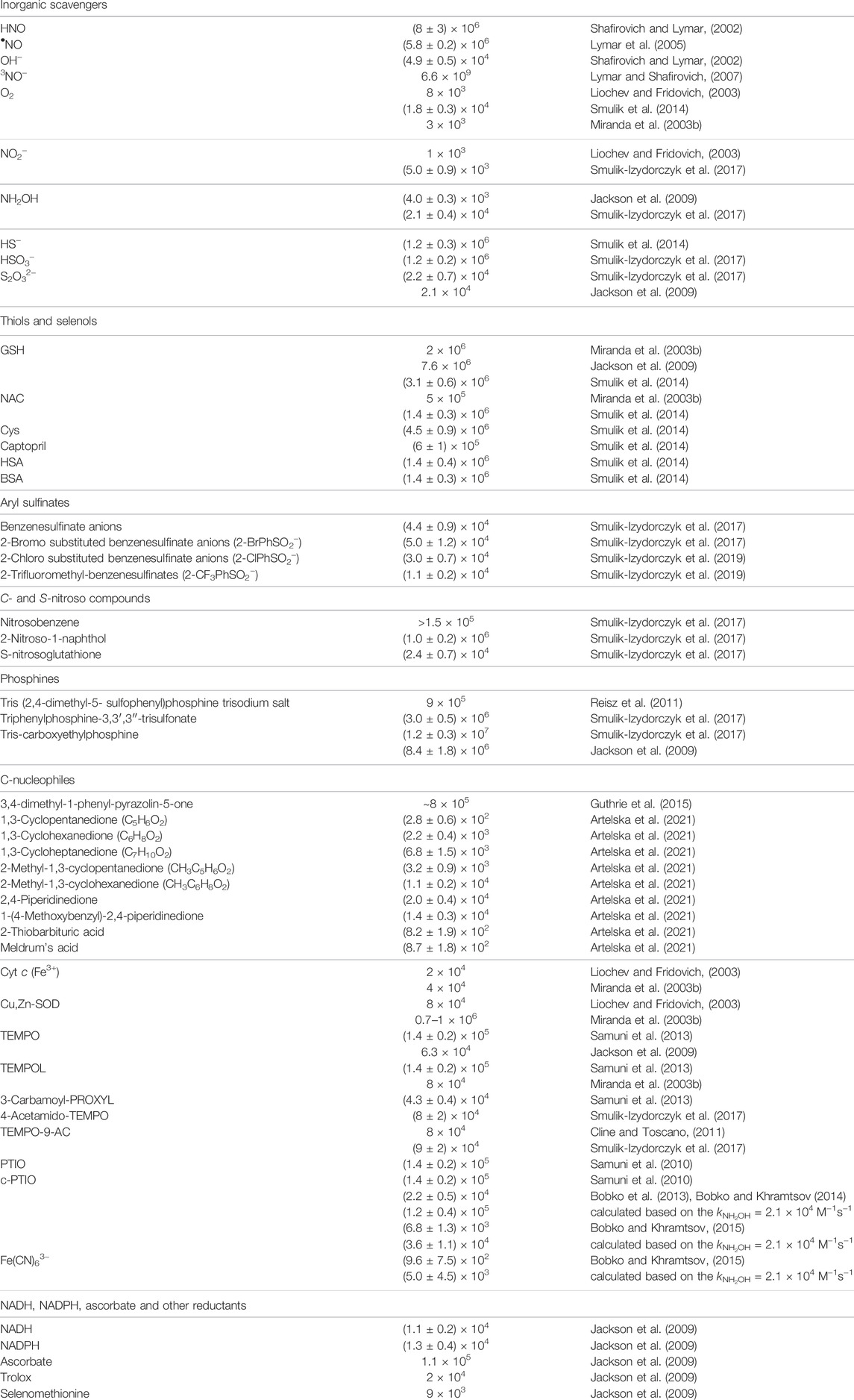
TABLE 1. Second-order rate constants for reactions of HNO with its scavengers.
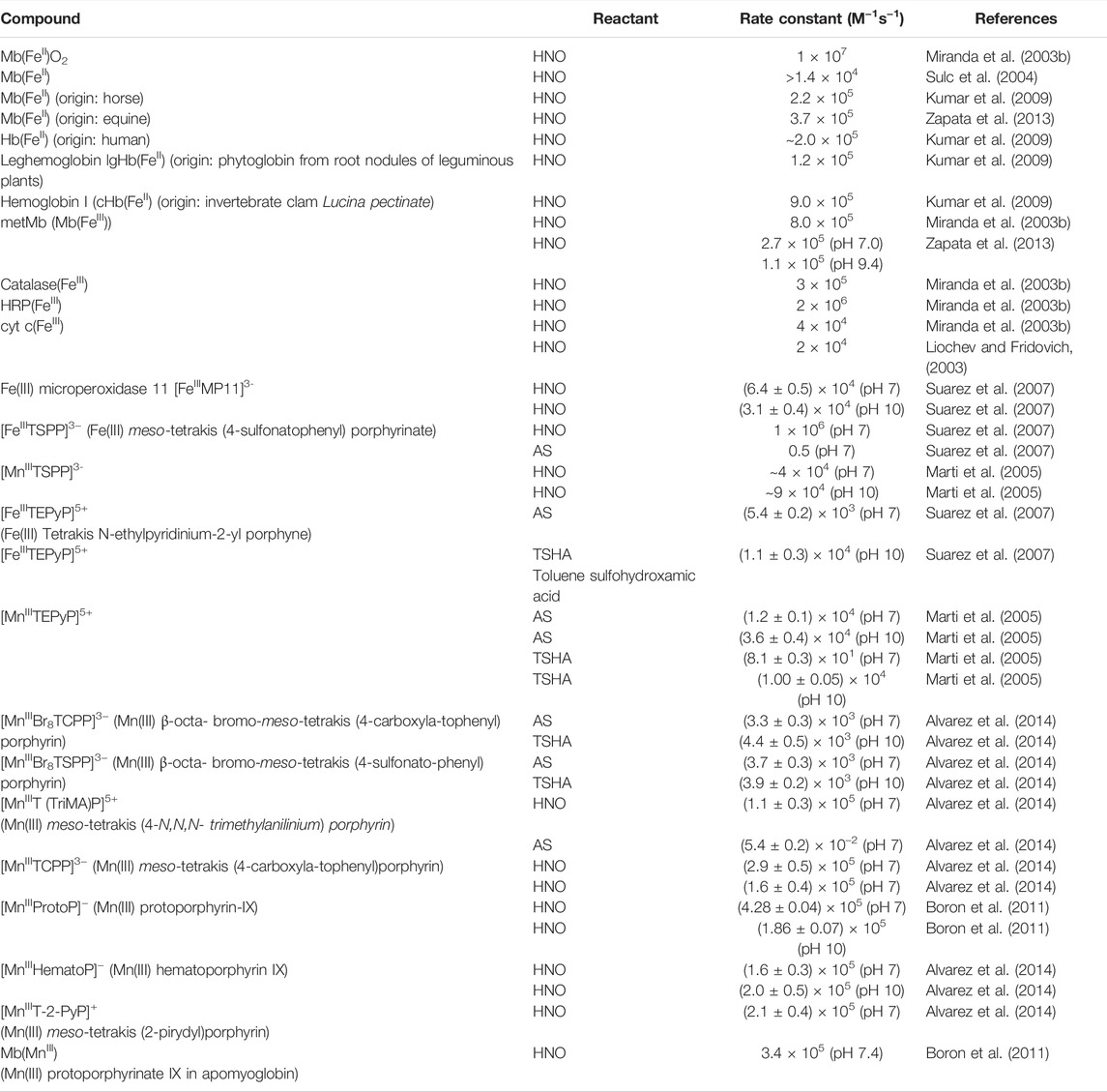
TABLE 2. Second-order rate constants for reactions of porphyrins with HNO and HNO donors.
2 HNO Chemistry
2.1 Nomenclature
Prior to describing HNO chemistry, it is important to briefly discuss the recommended and the used nomenclature. The IUPAC recommended name of HNO is “azanone” or “hydridooxidonitrogen” (which name is based on additive nomenclature). The commonly used name is “nitroxyl.” It has also been used to describe azanone anion, NO− [which IUPAC recommended name is “oxidonitrate (1−)”]. The term “nitroxyl” may be misleading as aminoxyl radicals, of the general formula R2N-O•, are known as “nitroxyl radicals.” Therefore, the reader must be aware that a literature search with the keyword “nitroxyl” may yield references to aminoxyl radicals R2N-O•. In some of the scientific literature names “nitroso hydrogen,” “monomeric hyponitrous acid,” “nitrosyl hydride” or “hydrogen oxonitrate,” were also used. For the compliance with the IUPAC recommendations and to avoid ambiguity, we suggest the use of the name “azanone” for HNO and “azanone anion” for NO−.
2.2 Structure, Spin States, Acidity
HNO is a simple triatomic molecule and its structure is described by three geometrical parameters: the N-O bond length rN-O, the N-H bond length rN-H and the H-N-O angle θH-N-O. The qualitative discussion of the electronic structure of HNO has been given by Orgel (1953) in relation to the properties of the isoelectronic and paramagnetic molecular oxygen and diamagnetic nitroso compounds. He has predicted, that the HNO ground state is singlet. The bent structure of HNO has also been proposed (Orgel, 1953). For the very first time, the molecular geometry of HNO has been experimentally determined by Dalby, based on the constants obtained from the rotational analysis (Dalby, 1958). For the HNO singlet ground state rN-O = 1.212 Å, rN-H = 1.063 Å, and θH-N-O = 108.6°. HNO structure and NO bond vibrational frequency υNO has been the subject of numerous theoretical studies [they have been recently reviewed (Zhang, 2013)]. Deprotonation of a singlet 1HNO molecule leads to the formation of a NO− anion, which is isoelectronic to molecular oxygen (O2) and has a triplet ground state. Due to different spin states of 1HNO and 3NO−, deprotonation of 1HNO and the protonation of 3NO− in aqueous solution, are spin-forbidden and slow reactions.
The acid-base equilibrium of HNO has been a subject of a long-standing debate. In a pulse radiolysis study on the nitric oxide reduction to NO− a pKa value of 4.7 for HNO was reported in 1970 (Grätzel et al., 1970). This value was reevaluated in 2001, based on the theoretical calculations, to 7.2 ± 1.0 (Bartberger et al., 2001). Further theoretical reevaluation led to pKa(1HNO/3NO−) value of 11.4 (Shafirovich and Lymar, 2002), and 11.6 ± 3.4 (Bartberger et al., 2002). Recently, a pKa value of 11.5 was reported, based on the advanced kinetic analysis of Piloty’s acid reaction with aquacobalamin (Polaczek et al., 2021). Based on the agreement between theoretical and experimental values, the use of the value of 11.4 for the pKa(1HNO/3NO−) is recommended. Shafirovich and Lymar have also estimated the pKa values for other HNO acid-base equilibria: pKa(1HNO/1NO−) ∼ 23, pKa(3HNO/3NO−) = −1.8 (Shafirovich and Lymar, 2002), suggesting that when formed, 1NO− will behave as a strong base, while 3HNO will behave as a strong acid.
Electron affinity of •NO, due to its electronic structure, has very low value — Eea = 0.026 ± 0.005 eV (Siegel et al., 1972; Travers et al., 1989). •NO reduction to 1HNO/3NO− in aqueous solutions has long been considered as nonrelevant in biological systems. It is important to note that the reported values of the reduction potential of •NO in aqueous solutions, discussed below, are results of estimation, since this value was not measured directly, due to several experimental limitations, including the instability of HNO. In 2002, it was postulated, that the standard reduction potential for •NO/3NO− redox pair is highly negative E°(•NO/3NO−) = −0.81 V, and practically outside of the biologically compatible range (Bartberger et al., 2002; Shafirovich and Lymar, 2002). At physiological pH, however, •NO reduction leads to the formation of HNO. Taking into account the postulated pKa(1HNO/3NO−) value of 11.4, Lymar and co-workers, estimated standard redox potential for •NO/HNO redox pair to be equal to −0.14 V (E°(•NO/HNO) = −0.14 V, and E°’(•NO/HNO) = −0.55 V for pH 7) (Shafirovich and Lymar, 2002). Those values have been recently revised to E°(•NO/HNO) = 0.27 V, and E°’(•NO/HNO) = −0.14 V for pH 7, based on the computational reevaluation of HNO solvation free energy (Venâncio et al., 2017). Such value, close to the one-electron reduction potential of molecular oxygen (O2/O2•– redox couple), would suggest the feasibility of •NO reduction to HNO by a variety of cellular enzymatic redox centers. The •NO/HNO interconversion under physiological conditions has been recently discussed in detail (Suarez et al., 2021), and is out of the scope of this article.
2.3 Stability of HNO and its Reactivity Towards Small Inorganic Scavengers
It should be noted, that direct experimental determination of the chemical reactivity of HNO is hampered by its rapid dimerization to hyponitrous acid H2N2O2 (Reaction 1a, k1a = (8 ± 3) × 106 M−1s−1) (Shafirovich and Lymar, 2002). This rate constant translates to the half-life time of HNO of ∼0.6 ms, when present at 100 µM concentration in aqueous solution. The acid-base equilibria of H2N2O2 have been reported with pKa (H2N2O2/HN2O2−) = 7.0 and pKa (HN2O2−/N2O22–) = 10.9. Therefore, at physiological pH (pH > 7), H2N2O2 exists predominantly in its mono-deprotonated form (HN2O2−). This form is responsible for the instability of H2N2O2 in aqueous solutions and for the formation of N2O (Reaction 1b, k1b = 5 × 10–4 s−1, t1/2 = 23 min).
Due to the instability of HNO in aqueous solutions, in chemical studies on HNO reactivity it is necessary to use HNO donors, that release HNO in a controlled manner, or produce azanone in situ, e.g., with the use of radiation techniques (e.g., pulse radiolysis) involving one-electron reduction of •NO by solvated electrons or by hydrogen atoms (H•), or photochemical techniques (e.g., laser flash photolysis), including photolysis of alkaline aqueous solutions of trioxodinitrate (N2O32−, anion of Angeli’s salt). Below, the radiation chemistry of aqueous solutions of •NO and the laser flash photolysis of aqueous solutions of N2O32− will be described, as those methods greatly contributed to our understanding of azanone chemistry and to the determination of the rate constants of the reactions involved. Further development of such methodologies for rapid in situ formation of HNO/NO−and for monitoring their reactions may overcome current obstacles in the studies of HNO/NO−chemistry and result in improved knowledge of the azanone chemistry and its biological fates.
2.3.1 Radiolysis of Aqueous Solutions of Nitric Oxide
Early works from the 1960s showed that the radiolysis of aqueous solutions of •NO leads to the formation of nitrite (NO2−) and N2O. The primary transient reactive species produced within approximately 10–8 s after the exposition of water to ionizing radiation are hydroxyl radical (•OH), hydrated electron (eaq−), and hydrogen atom (H•) [the more detailed description of water radiolysis can be found elsewhere (Bobrowski, 2017)]. The formation of N2O and NO2− in the irradiated aqueous solutions of nitric oxide is explicable by the mechanism presented below (Reactions 2–8) (Lymar et al., 2005):
Nitric oxide, being a radical, rapidly reacts with all three primary radical species from water radiolysis (•OH, eaq− and H•). Fast reaction with •OH radical results in the nitrite formation (Reaction 4, k4 = (1–2) × 1010 M−1s−1) (Treinin and Hayon, 1970; Seddon et al., 1973; Strehlow and Wagner, 1982). Reactions with hydrogen atom (Reaction 3, k3 = 1.1 × 1010 M−1s−1) (Knight and Sutton, 1967) and hydrated electron (Reaction 2, k2 = (2.1–3.1) × 1010 M−1s−1) (Gordon et al., 1963; Seddon and Young, 1970; Lymar et al., 2005) lead to 1HNO and 3NO−, respectively. Both, 1HNO and its anion 3NO− react subsequently with •NO with the formation of hyponitrite radical anion (N2O2•−) (Reactions 5, 6, k5 = (3.0 ± 0.8) × 109 M−1s−1, k6 = (5.8 ± 0.2) × 106 M−1s−1, pKa (HN2O2•) = 5.6 ± 0.3) (Poskrebyshev et al., 2004; Lymar et al., 2005; Poskrebyshev et al., 2008). Further reaction of N2O2•− with •NO generates N3O3− anion (Reaction 7, k7 = (5.4 ± 1.4) × 109 M−1s−1, pKa (HN3O3) = 3.1), that decomposes to N2O and NO2− (Reaction 8, k8 ∼ 3 × 102 s−1) (Lymar et al., 2005). The stoichiometric analyses indicate that for 1 mol of N2O generated, 2 mol of NO2− are produced, and 4 mol of •NO are consumed. Addition of aliphatic alcohols to the solution, leads to scavenging of •OH by the alcohols with the formation of highly reactive ketyl radicals (Reaction 9), capable to reduce •NO to HNO (Reaction 10), thus resulting in an increase in N2O yield (Woodward and Sutton, 1966).
Based on the above described reactions, it can be expected that when HNO is produced by •NO reduction in the presence of the excess of •NO, the transient generation of N2O2•− and N3O3− anions will occur resulting in the formation of N2O and NO2− in 1:1 M ratio, as it was observed in several studies (Suarez et al., 2015; Suarez et al., 2017). It has been shown that N2O2•− is quite stable both kinetically and thermodynamically with respect to its dissociation to •NO and 3NO− (Poskrebyshev et al., 2004). N2O2•− decay, observed in pulse radiolysis experiments, could be reasonably well described by a second-order kinetics. In the first and rate-determining step, two N2O2•− molecules disproportionate with the formation of N2O22− and generation of two •NO (Reaction 11, 2k11 = (8.2 ± 0.5) × 107 M−1s−1).
Subsequently, •NO rapidly adds to another N2O2•− molecule, yielding the N3O3− anion (Reaction 7), which slowly decomposes to the final products: N2O and NO2− (Reaction 8) (Poskrebyshev et al., 2008). The standard reduction potentials of N2O2•− and its protonated form, HN2O2•, were estimated–E°(N2O2•−/N2O22−) = 0.96 V and E°(HN2O2•, H+/H2N2O2) = 1.75 V (Poskrebyshev et al., 2004), suggesting that both species are strong one-electron oxidants.
2.3.2 Photolysis of Trioxodinitrate Solutions
Laser flash photolysis of sodium trioxodinitrate (Angeli’s salt, Na2N2O3) solutions has been used to study the kinetics of 1HNO/3NO− reactions (Shafirovich and Lymar, 2002, 2003; Lymar et al., 2005). Angeli’s salt anion in strongly alkaline solutions exists exclusively in fully deprotonated form (N2O32−) and is stable for hours. It has been shown, that steady state UV photolysis of alkaline Angeli’s salt solutions saturated with N2 results in the formation of nitrite and N2O, whereas the photolysis of solutions equilibrated with air, or saturated with oxygen, leads to the formation of ONOO− (Donald et al., 1986; Shafirovich and Lymar, 2002). The kinetics of ONOO− formation has been studied with the use of laser flash photolysis technique. It has been determined that the rate of ONOO− formation at fixed pH is independent of oxygen concentration and it has been proposed, that the rate-determining step is the 1HNO deprotonation in the reaction with OH−. The proposed mechanism assumes that ONOO−is formed only via the reaction between 3NO−and O2, and can be described by reaction sequence presented below (Reactions 12–15). The first step is the heterolytic photochemical cleavage of N2O32−, resulting in the formation of NO− in its singlet state and NO2− (Reaction 12). Azanone anion in its singlet state is an extremely strong base (pKa (1HNO/1NO−) ∼ 23) and is protonated by water instantaneously (Reaction 13). The next and rate-determining step is deprotonation of 1HNO to 3NO− (pKa (1HNO/3NO−) ∼ 11.4) (Reaction 14). The process of ONOO− generation is completed by a rapid and spin-allowed reaction of 3NO− and 3O2 (Reaction 15). The results obtained in those experiments were used to determine the second order rate constants of (Reaction 14 and Reaction 1a) (k14 = (4.9 ± 0.5) × 104 M−1s−1 and k1a = (8 ± 3) × 106 M−1s−1) (Shafirovich and Lymar, 2002).
The second order rate constant of 3NO− reaction with 3O2 (Reaction 15) was estimated to be equal to (2.7 ± 0.2) × 109 M−1s−1 (Shafirovich and Lymar, 2002). The photolysis of Angeli’s salt solutions at neutral pH, where HN2O3− is the main acid-base form, also resulted in the formation of ONOO−. The estimated yield of ONOO− formation at neutral pH was close to 0.25, and based on that observation it was proposed, that photolysis of aqueous solutions of HN2O3− leads to the formation of 3HNO and 1HNO in a 1:3 ratio (Reactions 16, 17). As mentioned earlier, 3HNO is a strong acid (pKa (3HNO/3NO−) = −1.8), and should dissociate rapidly to 3NO− in buffered solutions (Reaction 18) followed by rapid reaction with O2 (Reaction 15).
The distribution of the primary photolysis products of Angeli’s salt aqueous solution at neutral pH (22% 3NO−, 58% 1HNO, 20% •NO), was determined in a follow-up study (Lymar and Shafirovich, 2007). It has also been determined that the photolysis of aqueous solutions of amine-based diazeniumdiolates results in the generation of •NO (the distribution of the primary photolysis products of DTPA NONOate: 3% 3NO−, 12% 1HNO, 85% •NO). Laser flash photolysis experiments also allowed the determination of the second-order rate constant of the 1HNO reaction with 3NO− (Reaction 19, k19 = 6.6 × 109 M−1s−1) (Lymar and Shafirovich, 2007).
2.3.3 Reaction of Azanone With Molecular Oxygen
One of the most intriguing aspects of 1HNO chemistry is its reactivity toward molecular oxygen. As discussed above, the rapid reaction between triplet azanone anion (3NO−) and O2 leads to the formation of a strong biological oxidizing and nitrating agent, ONOO− (Reaction 15, k15 = (2.7 ± 0.2) × 109 M−1s−1) (Shafirovich and Lymar, 2002). The formation of ONOO−, as a transient product, was postulated in the oxidation reaction of potassium hydroxylamine-N-sulfonate (KO3SNHOH) (Belt and Baenziger, 1957) in oxygenated alkaline aqueous solutions (Ackermann and Powell, 1967). It has been proposed earlier, that this compound is an HNO donor (Ackermann and Powell, 1966). Formation of ONOO− was also confirmed for decomposition of Angeli’s salt or 2-bromo derivative of Piloty’s acid in alkaline solutions (Yagil and Anbar, 1964; Kirsch and de Groot, 2002; Smulik-Izydorczyk et al., 2019). ONOO− was also shown to be formed in oxygenated aqueous alkaline solutions of hydroxylamine, with azanone anion NO− proposed as a transient product reacting with O2 to form ONOO− (Yagil and Anbar, 1964; Hughes and Nicklin, 1971; Seel and Kaschuba, 1975).
In the case of 1HNO, the product of its reaction with O2 for many years has not been fully identified. It has been shown, that during the decomposition of Angeli’s salt in aerated aqueous solutions at neutral pH an oxidant is formed in the reaction of HNO with O2, capable of oxidizing dihydrorhodamine 123 (DHR) or dichlorodihydrofluorescein (DCFH) to their fluorescent products, rhodamine 123 (Rh) and dichlorofluorescein (DCF), respectively (Liochev and Fridovich, 2001; Miranda et al., 2001; Kirsch and de Groot, 2002). The yield of Rh was similar for both oxidants–Angeli’s salt and authentic ONOO−. Similar was also the effect of CO2, known to increase the yield of peroxynitrite-derived oxidation products (Miranda et al., 2001). It has also been shown, that the yield of DHR and NADH oxidation was similar for Angeli’s salt and SIN-1, a slow donor of ONOO− under aerobic conditions via co-generation of superoxide radical anion (O2•−) and •NO (Kirsch and de Groot, 2002). The yield of nitrate was also similar in the case of aerated aqueous solutions of Angeli’s salt and SIN-1 (Kirsch and de Groot, 2002). All those observations suggested that ONOO− is the product of the reaction of 1HNO with O2 (Liochev and Fridovich, 2001; Miranda et al., 2001; Kirsch and de Groot, 2002). There were, however, also some significant differences in the effects observed with Angeli’s salt and authentic ONOO−. It has been shown that Angeli’s salt, unlike ONOO−, is unable to oxidize phenols to their dimeric fluorescent products. This observation led to the conclusion that ONOO− is not formed in the reaction of HNO with O2 (Miranda et al., 2001; Miranda et al., 2002). As a result, the possibility of ONOO− formation from the reaction between 1HNO and O2 has been a matter of a long-term controversy. This was, in part, due to the lack of molecular probes that unambiguously identify and quantify ONOO−under the studied conditions.
The development of boronate-based probes for ONOO− provided an opportunity for quantitative measurement of ONOO− formation at neutral pH. Boronic acids and their esters react directly and rapidly with ONOO− (k ∼ 105–106 M−1s−1), with the formation of corresponding phenols as major products (Sikora et al., 2009; Zielonka et al., 2010; Zielonka et al., 2012; Sikora et al., 2013; Dębowska et al., 2016; Rios et al., 2016). It has been shown, that the reaction of boronates with ONOO− yields unique minor peroxynitrite-specific products (Sikora et al., 2009; Sikora et al., 2011; Sikora et al., 2013; Zielonka et al., 2015; Zielonka et al., 2016b; Zielonka et al., 2021). It has been proposed, that the reaction between ONOO− and boronic acid leads to the formation of ONOO− adduct, followed by the heterolytic (major pathway) or homolytic (minor pathway) O-O bond cleavage, resulting in the formation of phenols (major pathway) and minor radical-derived products (Sikora et al., 2011). It has been proposed, that the formation of the products of both major and minor pathways in a specific ratio can be used as a “peroxynitrite fingerprint” (Sikora et al., 2011; Zielonka et al., 2015; Zielonka et al., 2016a; Szala et al., 2020; Grzelakowska et al., 2021; Zielonka et al., 2021; Grzelakowska et al., 2022). This strategy has been applied to unambiguously determine the identity of the product of HNO reaction with O2 (Smulik et al., 2014). The formation of ONOO− in the reaction of HNO with O2 in aqueous solutions at neutral pH was confirmed based on the oxidation of the boronate probes in the solutions of Angeli’s salt, and the formation of ONOO−-specific products (Smulik et al., 2014). The second order rate constant for 1HNO reaction with O2 at pH 7.4 (Reaction 20) (k20 = (1.8 ± 0.3) × 104 M−1s−1) has been determined with the use of a set of HNO scavengers of known reactivity and the competition kinetics approach (Smulik et al., 2014). It should be noted that this value is significantly higher than previously reported 8 × 103 M−1s−1 (Liochev and Fridovich, 2003) or 3 × 103 M−1s−1 (Miranda et al., 2003b) - see Table 1.
2.3.4 Reaction of Azanone With Nitrite
The estimation of k20 reported by Liochev and Fridovich was based on the effect of NO2− on the aerobic decomposition of Angeli’s salt. The decomposition of Angeli’s salt anion in aqueous solutions at neutral pH leads to the formation of HNO and NO2− (Reaction 21). Due to the occurrence of the opposite reaction – HNO scavenging by NO2− (Reaction 22), the observed rate of Angeli’s salt decomposition is significantly slower in solutions containing millimolar concentrations of NO2−. Based on the observed effect of NO2− on the Angeli’s salt decomposition in aerated solutions, the second-order rate constant of HNO reaction with NO2− (Reaction 22) was estimated to be equal to 1 × 103 M−1s−1 (Liochev and Fridovich, 2003).
The use of boronate fluorogenic/colorimetric probes, in combination with a competition kinetics approach was proposed as a novel approach to study the kinetics of HNO reactions (Smulik et al., 2014). Over the last 7 years, the usefulness of this methodology was demonstrated in the studies of HNO reactivity toward a number of its scavengers (Smulik et al., 2014; Smulik-Izydorczyk et al., 2017; Smulik-Izydorczyk et al., 2019; Artelska et al., 2021; Smulik-Izydorczyk et al., 2021). The principle of this method is based on the competition between O2 and the scavenger of interest for their reaction with HNO released from Angeli’s salt or other HNO donor. Reaction of HNO with O2 results in the formation of ONOO− (Reaction 20), which oxidizes boronate probe (ArB(OH)2, e.g., coumarin 7-boronic acid (CBA), resorufin boronate derivative (PC1), fluorescein boronate derivative (FlBA)—see Scheme 1) to the corresponding phenolic product (ArOH, e.g., umbelliferone, resorufin or fluorescein), that can be easily monitored by UV-Vis absorption or fluorescence spectroscopy.
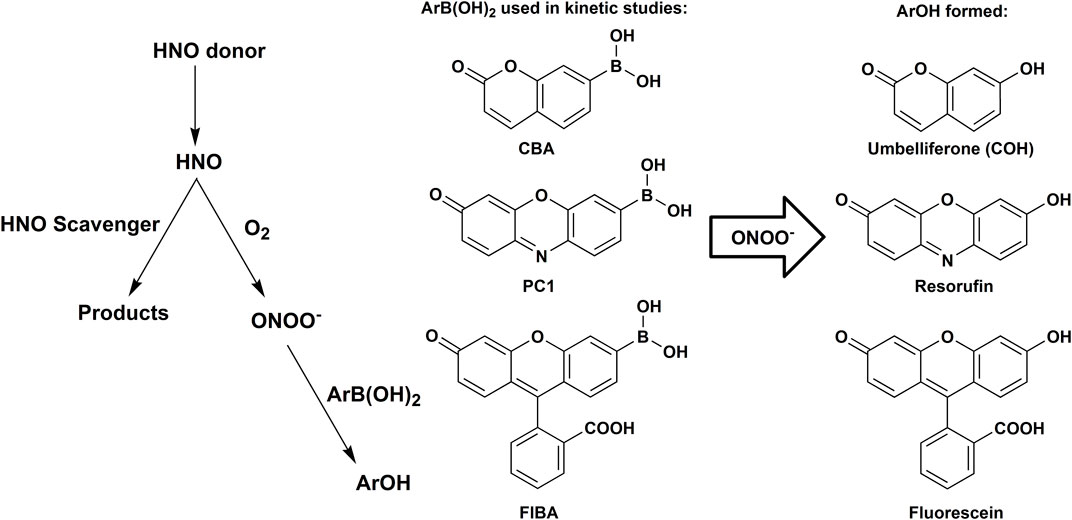
SCHEME 1. Reaction model used to determine the rate constants of the reactions between HNO and its scavengers with the use of boronate probes, the competition kinetic approach and HNO reaction with O2 as a reference reaction.
The rate of accumulation of the phenolic reporter (ArOH) over time can be expressed by Eq. 1,
where
where kdonor is the rate constant of HNO donor decomposition, and kscavenger and koxygen are the rate constants of azanone reaction with its scavenger and O2, respectively. To solve Eqs 2, 3, a steady state approximation was made (Eqs 4, 5).
The solution leads to Eqs 6, 7.
The rate of ArOH formation vi in the presence of azanone scavenger is expressed by Eq. 8, whereas, in the absence of that scavenger, it is expressed by Eq. 9.
Comparison of Eqs 8, 9 results in Eq. 10 describing the competition for HNO between O2 and the scavenger studied.
It should be noted that this model is based on several assumptions, including: 1) the HNO scavenger of interest does not affect the rate of HNO release from the donor; 2) the scavenger does not react with ONOO−, or such reaction is outcompeted by the reaction between ONOO−and the boronate probe; 3) the scavenger does not react with or interferes with the detection of the product of oxidation of the boronic probe; and 4) the amount of HNO released is significantly lower than the amount of the scavenger and of O2 in the solution.
With the use of this approach and a boronate derivative of resorufin, the rate constant of the reaction of HNO with NO2− was determined to be equal to (5.0 ± 0.9) × 103 M−1s−1 (Smulik-Izydorczyk et al., 2017), 5-fold higher than reported previously (Liochev and Fridovich, 2003).
2.3.5 Reaction of Azanone With Hydroxylamine
It has been shown, that the reaction of azanone with hydroxylamine (NH2OH) results in the formation of molecular nitrogen (N2) and water (Reaction 23) (Bonner et al., 1978). Jackson et al. (2009) estimated the rate constant for this reaction as k23 = (4.0 ± 0.3) × 103 M−1s−1. Recent analysis of the competition between O2 and NH2OH for HNO using the boronate reporter for ONOO−, resulted in the rate constant equal to (2.1 ± 0.4) × 104 M−1s−1 (Smulik-Izydorczyk et al., 2017).
2.3.6 Reaction of Azanone With HS−, Thiosulfate, and Sulfite
Published theoretical study predicted that HNO does not react with hard nucleophiles – water or alcohols (Bartberger et al., 2001). However, experimental data show, that it is highly reactive, towards soft nucleophiles. Using the above-described competition kinetics approach, it was shown that HNO reacts with HS−, HSO3− and S2O32− anions (Smulik et al., 2014; Smulik-Izydorczyk et al., 2017). The products of those reactions were not identified, but the corresponding adducts can be expected as primary reaction products (Reactions 24–26).
The second-order rate constants were determined to be equal to: k24 = (1.2 ± 0.3) × 106 M−1s−1, k25 = (1.2 ± 0.2) × 106 M−1s−1 and k26 = (2.2 ± 0.7) × 104 M−1s−1, for HS−, HSO3−, and S2O32−, respectively (Smulik et al., 2014; Smulik-Izydorczyk et al., 2017). The rate constant for HNO reaction with S2O32− (Reaction 26) has also been reported by Jackson to be equal to 2.1 × 104 M−1s−1 (Jackson et al., 2009) in perfect agreement with the competition method described above. In case of the reaction between HS− and HNO (Reaction 24) the product formed, HSNHOH, may react with HS− to form HS2− and release NH2OH (Reaction 27). Similarly, the reaction of −O3SSNHOH adduct (the product of Reaction 26) with S2O32− could result in the formation of NH2OH and S4O62− anion (Reaction 28). The stoichiometric analyses of such reaction should also take into account the possibility of the reaction between HNO and released NH2OH (Reaction 23). The recent study of Zarenkiewicz et al. (2021) on HNO reaction with H2S shows that this reaction leads to the formation of hydrogen polysulfides (H2Sn) or sulfur S8, depending on the relative concentrations of H2S and HNO. The reactivity of HNO towards selected inorganic scavengers is summarized in Scheme 2 and Table 1.
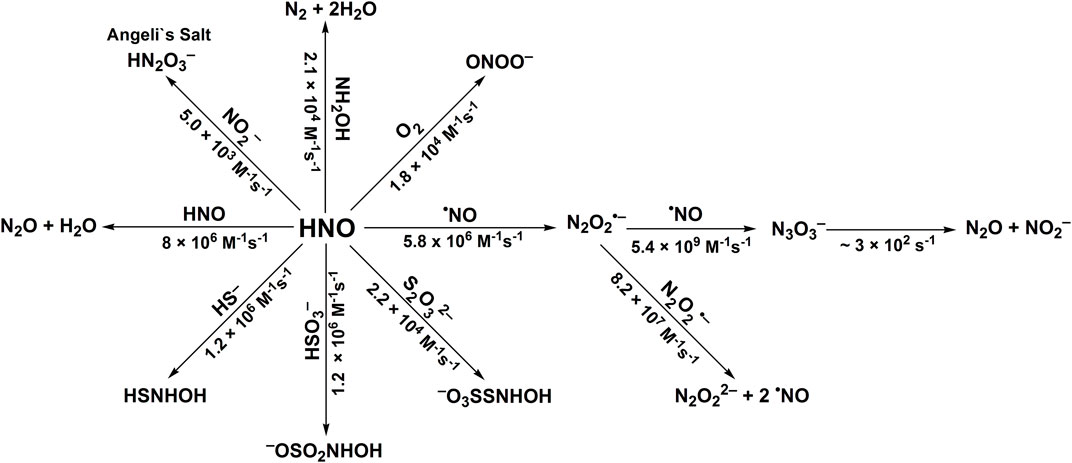
SCHEME 2. Reactivity of azanone towards selected inorganic scavengers.
2.4 Reactivity of HNO Towards Organic Nucleophiles
2.4.1 Reaction of Azanone With Thiols, Selenols and Hydropersulfides
One of the earliest reports describing the oxidation of thiols in solutions of HNO donors was by Doyle et al. (1988). The authors have shown that in Angeli’s salt solution in 40% aqueous acetonitrile at pH 7.8, thiophenol is oxidized to phenyl disulfide, with concomitant formation of NO2− and NH2OH (Reaction 29).
The proposed reaction mechanism involves HNO generation and its subsequent reaction with thiophenol resulting in the formation of C6H5SNHOH as a transient product (Reaction 30). The reaction of latter with C6H5SH was proposed to explain the formation of disulfide and NH2OH (Reaction 31). NO2− was the byproduct of the decomposition of Angeli’s salt (Reaction 21).
Subsequently, it was shown that high concentrations of L-cysteine inhibit completely the vasorelaxant response of isolated rat aortic rings to HNO generated from Angeli’s salt (Pino and Feelisch, 1994). Based on the known reaction of thiols with nitrosoarenes (Kazanis and McClelland, 1992) (Reactions 32, 33), it has been proposed and shown, that HNO reacts with thiols to form N-hydroxysulfenamides (Reaction 34), that can rearrange to generate a sulfinamides (Reaction 35), as stable products (Shoeman and Nagasawa, 1998; Wong et al., 1998; Shen and English, 2005).
To the best of our knowledge, due to an apparent instability of thiols N-hydroxysulfenamides, all attempts to chemically characterize those primary products of HNO reaction with thiols were thus far unsuccessful.
In 2016 Bianco et al. (2016) reported, that selenols are also highly reactive towards HNO and demonstrated that reactions of organoselenols and HNO result only in diselenide formation. It has been proposed, that such product’s profile is unique to selenols and selenoproteins (Scheme 3B).
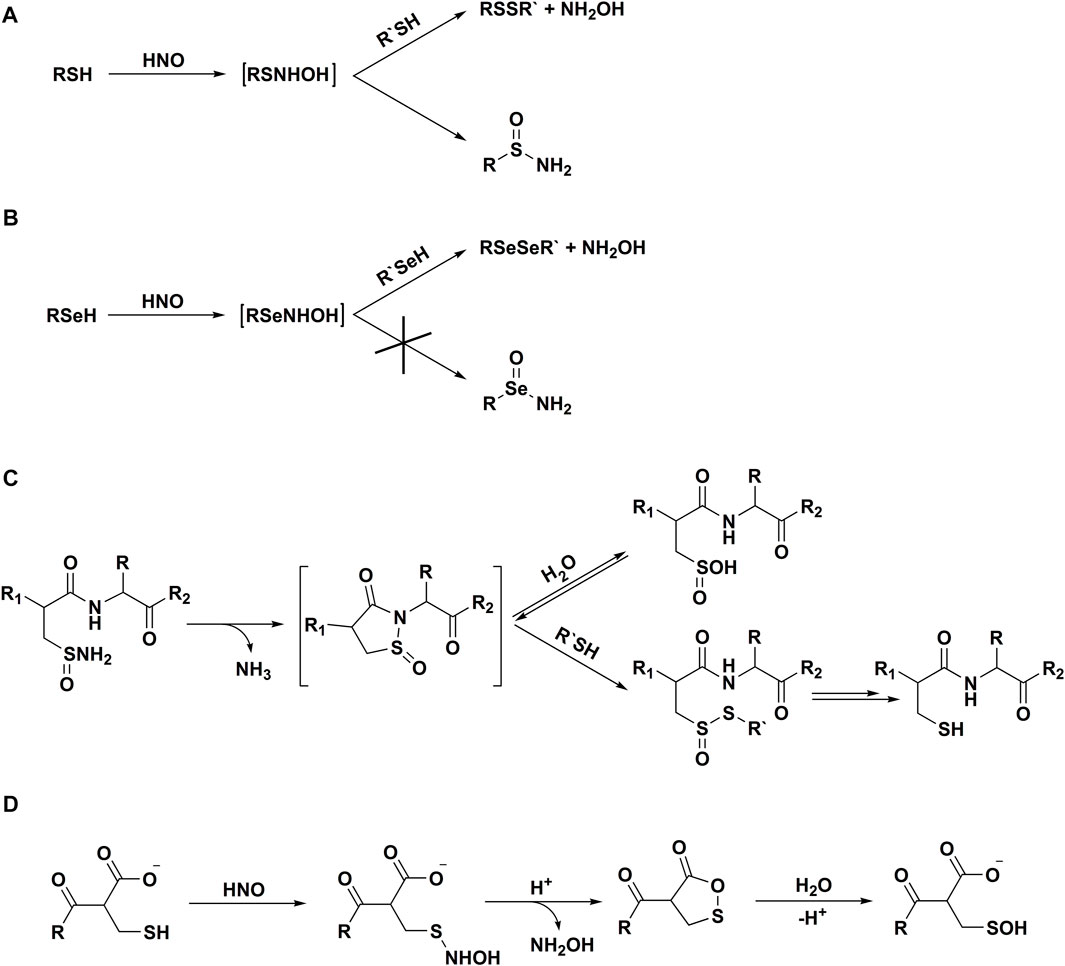
SCHEME 3. Reactivity of azanone towards thiols and selenols. Reaction of HNO with thiols (A) and selenols (B); (C) chemical transformations of sulfinamides, (D) reaction of HNO with C-terminal cysteines.
Exposure of reduced glutathione (GSH) to HNO was shown to lead to the formation of both the sulfinamide derivative (GS(O)NH2) and the glutathione disulfide (oxidized glutathione, GSSG) (Donzelli et al., 2006). Higher GSH concentrations favored a higher [GSSG]/[GS(O)NH2] ratio, suggesting that both GSSG and GS(O)NH2 are formed from a single transient intermediate, supporting the hypothesis of GSNHOH formation (Scheme 3A). It has also been shown, that formation of GS(O)NH2 is not observed with other reactive nitrogen species (RNS): •NO, nitrogen dioxide (•NO2), ONOO−, or N2O3, making GS(O)NH2 a unique and HNO-specific product. GS(O)NH2 was, therefore, used as a marker for HNO generation from several biologically-relevant pathways: thiol-mediated decomposition of S-nitrosothiols, peroxidase-dependent NH2OH and N-hydroxy-L-arginine oxidation (Donzelli et al., 2006).
It has been long assumed that HNO-driven oxidative transformation of thiols to sulfinamides is irreversible. Keceli and Toscano (2012) have examined the stability of HNO-derived sulfinamides in several systems, including small organic molecules, peptides, and a protein and observed that sulfinamides can be reduced back to the corresponding thiols in the presence of thiol excess. It was also shown that upon prolonged incubation of sulfinamides in water, sulfinic acid (RS(O)OH) is formed. However, in the presence of thiols, the reduction of sulfinamides is effectively outcompeting the hydrolysis process. The proposed mechanism of peptide sulfinamide reduction involving a cyclic transient product is presented in Scheme 3C.
The same authors have also shown that in the case of C-terminal cysteines the nature of HNO-derived modifications is highly affected by the C-terminal carboxylate (Keceli and Toscano, 2014). In this case, the formation of sulfinamides is not observed. It was proposed, that the N-hydroxysulfenamide formed in HNO reaction with the thiol undergoes intramolecular cyclization to the transient cyclic product, that is hydrolyzed to sulfenic acid (RSOH) (Scheme 3D).
Several groups studied the kinetics of the reaction of HNO with GSH. Based on the effect of GSH on the rate of Angeli’s salt decomposition in aqueous solution at pH 7.4 in the presence of NO2− and O2, it was concluded that the rate constant for the reaction of GSH with NO−/HNO is significantly higher than 1 × 105 M−1s−1 (Liochev and Fridovich, 2003). In an independent study, the second order rate constants for the reactions of HNO with GSH and N-acetyl cysteine (NAC) were determined to be equal to 2 × 106 M−1s−1 and 5 × 105 M−1s−1, respectively (Miranda et al., 2003b). The rate constant for HNO reaction with GSH has also been reported to be equal to 7.6 × 106 M−1s−1 (Jackson et al., 2009). More recent reexamination of the reactivity of selected thiols, based on their competition with O2 for HNO (using boronate probes for ONOO−), yielded the following values of the rate constants in aqueous solutions at pH 7.4: cysteine (kCys = (4.5 ± 0.9) × 106 M−1s−1, pKa = 8.3), glutathione (kGSH = (3.1 ± 0.6) × 106 M−1s−1, pKa = 8.8), N-acetylcysteine (kNAC = (1.4 ± 0.3) × 106 M−1s−1, pKa = 9.5) and captopril (kCap = (6 ± 1) × 105 M−1s−1, pKa = 9.8) (Smulik et al., 2014). It was shown, that for those small molecular weight thiols, there is an inverse correlation between the determined rate constants and pKa values of SH groups: the higher was the value of thiol’s pKa, the lower was the reaction rate constant. This strongly suggests that the deprotonated SH group (i.e., thiolate anion) is the active form reacting with HNO. The second order rate constants for HNO reaction with protein-bound thiols were also determined in that study for human and bovine serum albumins (pKa ∼ 8.5 for Cys34) (kHSA = (1.4 ± 0.4) × 106 M−1s−1, kBSA = (1.4 ± 0.3) × 106 M−1s−1). Such a high reactivity of thiols towards HNO, together with high concentrations of GSH and albumin in cells and plasma, respectively, suggest that thiols may be the potential targets of HNO in biological systems.
The pH-dependent kinetics of HNO reactions with selected thiols–glutathione, N-acetylcysteine, bovine serum albumin and human serum albumin was also recently reported (Smulik-Izydorczyk et al., 2021). It was shown, that the observed rate constant (kobs) can be expressed as a function of pH, which depends on thiol pKa, and the rate constants of azanone reaction with RS− (kthiolate) and with RSH (kthiol) (Eq. 11):
The results of that study showed, that the reaction of HNO with thiolate (RS−) is favored (kthiolate ∼ 107 M−1s−1) over reactions with the thiol (RSH, kthiol = 105–106 M−1s−1). Such observation is consistent with the observed inverse correlation between the reaction rate constant and the pKa value of the thiol.
Another class of thiol-related, biologically important sulfur nucleophiles are hydropersulfides (RSSH) (Saund et al., 2015; Fukuto and Hobbs, 2021). At physiological pH hydropersulfides (RSSH) are significantly more nucleophilic than thiols (RSH), due to their lower pKa [e.g., pKa (GSSH) = 5.45 ± 0.03, 3.5 units below the pKa of GSH (Benchoam et al., 2020)] and increased nucleophilicity of their anions RSS−. One may expect, that RSSH/RSS− are highly reactive towards HNO. As hydropersulfides have been detected in biological systems at high micromolar concentrations, they could be among important biological targets of azanone. The recent study of Zarenkiewicz et al. (2021) on HNO reaction with hydropersulfides shows that RSSH are more potent traps for HNO, than RSH. They also reported that HNO reaction with small molecule RSSH leads to the formation of various RSSnSR and RSS-NH-SnR species (Zarenkiewicz et al., 2021).
2.4.2 Reaction of Azanone With Aryl Sulfinates
Similar to sulfites, organic sulfinates are nucleophiles capable of HNO scavenging. The corresponding Piloty acid analogs (used as thermal HNO donors) would be expected to be the products of those reactions (Reaction 36). The rate constant for the HNO reaction of benzenesulfinate anion (C6H5SO2−), the decomposition product of Piloty’s acid, has been determined as kbenzenesulfinate = (4.4 ± 0.9) × 104 M−1s−1 (Smulik-Izydorczyk et al., 2017).
The rate constants of HNO reactions with 2-bromo-, 2-chloro- and 2-trifluoromethylbenzenesulfinates (2-BrPhSO2−, 2-ClPhSO2− and 2-CF3PhSO2−) were also determined and are equal to (5.0 ± 1.2) × 104 M−1s−1, (3.0 ± 0.7) × 104 M−1s−1 and (1.1 ± 0.2) × 104 M−1s−1, respectively (Smulik-Izydorczyk et al., 2017; Smulik-Izydorczyk et al., 2019). Compared to the rate constant for the reaction of HNO with nitrite ions (the products of decomposition of Angeli’s salt, k ∼ 103 M−1s−1), the rate constant for the reaction between HNO and benzenesulfinate (the products of the decomposition of Piloty’s salt) is one order of magnitude higher.
2.4.3 Reaction of Azanone With C- and S-Nitrosocompounds
It has been shown that the reaction of nitrosobenzene with HNO results in the formation of stable C-diazeniumdiolate product, cupferron (Reaction 37) (Shoeman and Nagasawa, 1998). The second-order rate constant of that reaction was recently estimated to be higher than 1.5 × 105 M−1s−1 (Smulik-Izydorczyk et al., 2017). In the same study, the reactivity of 2-nitroso-1-naphthol toward HNO was also studied, and the rate constant was determined to be equal to (1.0 ± 0.2) × 106 M−1s−1 (Smulik-Izydorczyk et al., 2017).
In 1998, the study on the reaction of HNO (generated from Angeli’s salt) with S-nitrosoglutathione (GSNO) was reported (Wong et al., 1998). It was shown that the incubation of GSNO with Angeli’s salt results in S-nitrosothiol decomposition and the generation of •NO. The proposed reaction mechanism included the HNO attack at the nitrogen atom of the -SNO group resulting in the formation of S-diazeniumdiolate, which subsequently decomposes with the generation of •NO and GSH.
The formation of S-diazeniumdiolate transient product has also been recently proposed for the reaction of thiols with •NO, resulting in the formation of HNO (Reaction 38) (Neuman et al., 2021).
The second-order rate constant of the reaction of HNO with GSNO has been determined to be equal to (2.4 ± 0.7) × 104 M−1s−1 (Smulik-Izydorczyk et al., 2017).
2.4.4 Reaction of Azanone With Phosphines
In 2009, it was reported that HNO reacts rapidly with organic phosphines to produce corresponding phosphine oxides and aza-ylides (Scheme 4A) (Reisz et al., 2009). This report was based on the previously reported reactions of organic phosphines with C- and S-nitroso compounds resulting in the formation of phosphine oxides and aza-ylides (Haake, 1972; Wang and Xian, 2008; Zhang et al., 2009). In aqueous solutions aza-ylides hydrolyze spontaneously to yield the corresponding amine and phosphine oxide, but this unstable transient product can also be converted to a stable covalent adduct. It has been shown that an appropriately situated electrophilic trap within the phosphine structure, such as an ester functional group, can capture the nucleophilic aza-ylide in the intramolecular cyclization reaction, producing a stable amide (Scheme 4B) (Saxon and Bertozzi, 2000). Applying this strategy, Reisz et al. (2009) have shown that azanone can be effectively trapped by the methyl ester of 2-(diphenylphosphino)benzoic acid to form the corresponding amide as an HNO-specific product. Further exploration of this azanone detection strategy resulted in the development of the first phosphine-based colorimetric probe for the detection of HNO (Reisz et al., 2011). Using the competition kinetics approach and HNO reaction with GSH as a reference (kGSH = 2 × 106 M−1s−1), the rate constant for the reaction of HNO with a water-soluble phosphine, tris(2,4-dimethyl-5- sulfophenyl)phosphine trisodium salt, was estimated at 37°C and pH 7.4 to be equal to 9 × 105 M−1s−1 (Reisz et al., 2011). Subsequently, Kawai et al. (2013) reported the design, synthesis and characterization of the first phosphine-based fluorogenic probe for the detection of HNO (Scheme 4C). Those seminal works were followed by a large number of reports of various arylphosphine-based fluorogenic probes for the detection of HNO, as reviewed elsewhere (Smulik-Izydorczyk et al., 2018). The detection mechanism used is based on the reaction of esters of fluorescent dyes and 2-(diphenylphosphino)benzoic acid as HNO-sensing moiety, the reaction of which with HNO leads to the release of the parent dye and fluorescence “turn on.” The reaction of HNO with a triphenylphosphine sensing site results in the formation of the aza-ylide, its subsequent intramolecular nucleophilic attack on the carbonyl carbon of the ester, leading to the liberation of the fluorescent dye and the formation of the P-oxide of the amide of 2-(diphenylphosphino)benzoic acid. The proposed mechanisms of HNO reaction with phosphines are summarized in Scheme 4.
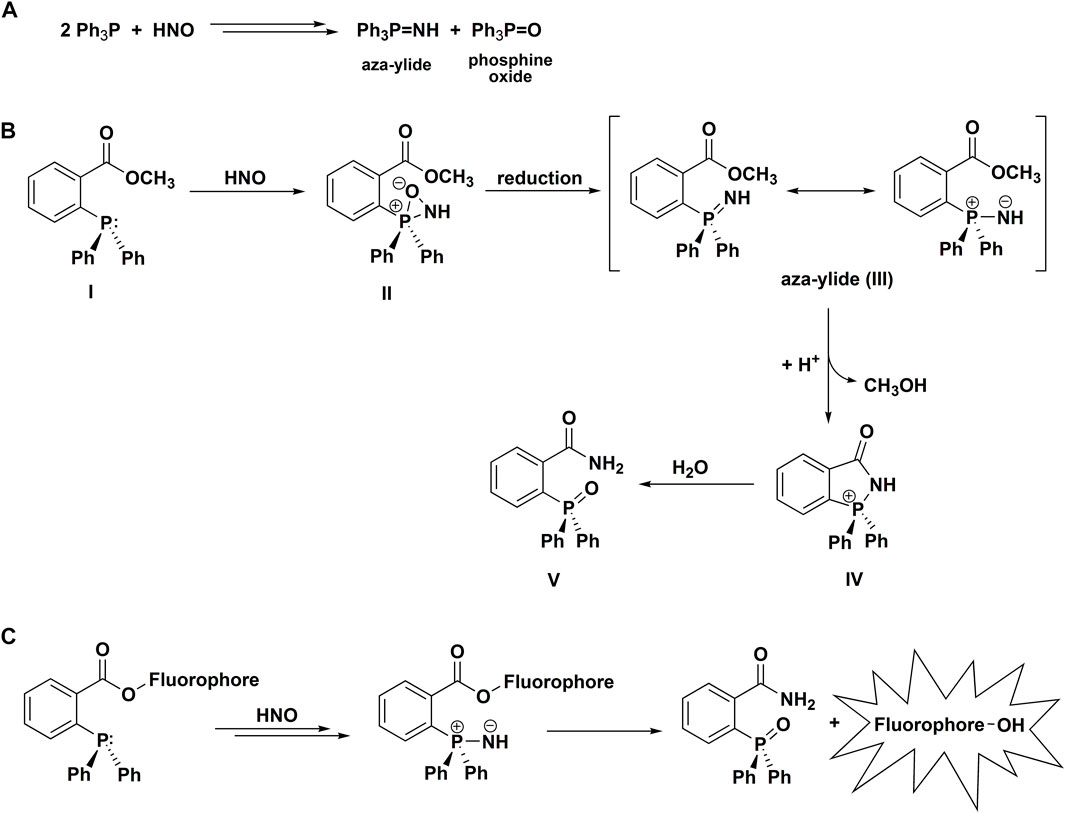
SCHEME 4. Reactivity of azanone towards phosphines (A), (B) The Staudinger ligation of HNO-derived aza-ylide. (C) The mechanism of HNO detection with the use of phosphine-based fluorogenic probes.
The rate constants for the reactions of HNO with triphenylphosphine-3,3′,3″-trisulfonate and tris-carboxyethylphosphine (TCEP) were recently re-evaluated and the values determined at pH 7.4 and 25°C are equal to (3.0 ± 0.5) × 106 M−1s−1 and (1.2 ± 0.3) × 107 M−1s−1 (Smulik-Izydorczyk et al., 2017). The value for TCEP is in good agreement with the previously reported value of 8.4 × 106 M−1s−1 (Jackson et al., 2009).
2.4.5 Reaction of Azanone With C-Nucleophiles
In 2015 Guthrie et al. (2015) reported, that HNO reacts with variety of pyrazolones with the formation of corresponding HNO adducts (pyrazolone substituted hydroxylamines). They developed a general protocol for the synthesis of new and versatile class of HNO donors (hydroxylamino-pyrazolones–HAPY) from pyrazolones and Angeli’s salt, as a HNO donor. The kinetic study of HNO scavenging by pyrazolones showed that this reaction is fast and the second order rate constant at pH 7.4 and 37°C can reach value 8 × 105 M−1s−1.
Recently, it was shown that C-nucleophiles can act as efficient scavengers of HNO (Artelska et al., 2021). There was a strong dependence of the reactivity of C-nucleophiles toward HNO on nucleophile structure (Figure 1), while due to the low pKa values all those nucleophiles are present in the solution in their deprotonated, anionic forms in aqueous solution at pH 7.4.
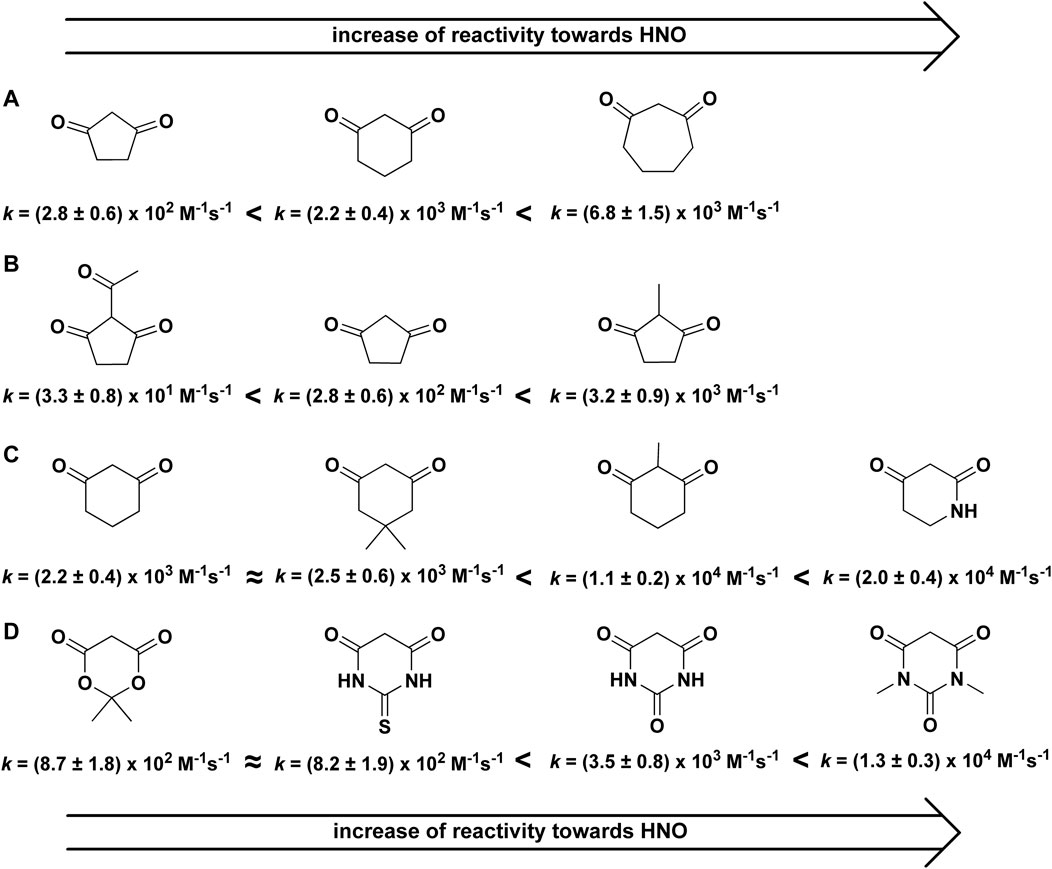
FIGURE 1. Reactivity of HNO towards cyclic C-nucleophiles.
The reactivity of 1,3-cyclopentanedione (C5H6O2), 1,3-cyclohexanedione (C6H8O2), and 1,3-cycloheptanedione (C7H10O2) was studied and the obtained second order rate constants were equal to 2.8 × 102 M−1s−1, 2.2 × 103 M−1s−1 and 6.8 × 103 M−1s−1, respectively. This indicates that the reactivity of cyclic C-nucleophiles toward HNO significantly increases with an increase in their ring size (Figure 1A). Methylation of α-carbon also resulted in a marked increase in the reactivity of C-nucleophiles (Figures 1B,C). The acylation of the same position has the opposite effect to methylation and results in decreased rate constant (Figure 1B).
The substitution of one or more carbon atoms in the ring of 1,3-cyclohexanedione with nitrogen resulted in an increase in C-nucleophile reactivity toward HNO (Figure 1C). In the case of 2,4-piperidinedione and 1-(4-methoxybenzyl)-2,4-piperidinedione the rate constants show an increase in nucleophile reactivity toward HNO, compared to 1,3-cyclohexanedione: k1,3-cyclohexanedione = 2.2 × 103 M−1s−1, k2,4-piperidinedione = 2.0 × 104 M−1s−1 and k1-(4-methoxybenzyl)-2,4-piperidinedione = 1.4 × 104 M−1s−1.
The reactivity of barbituric acid, 1,3-dimethylbarbituric acid, 2-thiobarbituric acid and Meldrum’s acid (all those cyclic C-nucleophiles contain two heteroatoms in the ring) towards azanone (Figure 1D) was also reported. Barbituric acid had similar reactivity toward HNO to 1,3-cyclohexanedione. In the case of 1,3-dimethylbarbituric acid a slight increase in the second order rate constant was observed. The reactivity of 2-thiobarbituric acid and Meldrum’s acid toward HNO was, however, rather low: k2-thiobarbituric acid = 8.2 × 102 M−1s−1 and kMeldrum’s acid = 8.7 × 102 M−1s−1.
The reactivity of HNO towards different classes of organic nucleophiles has been summarized in Scheme 5.
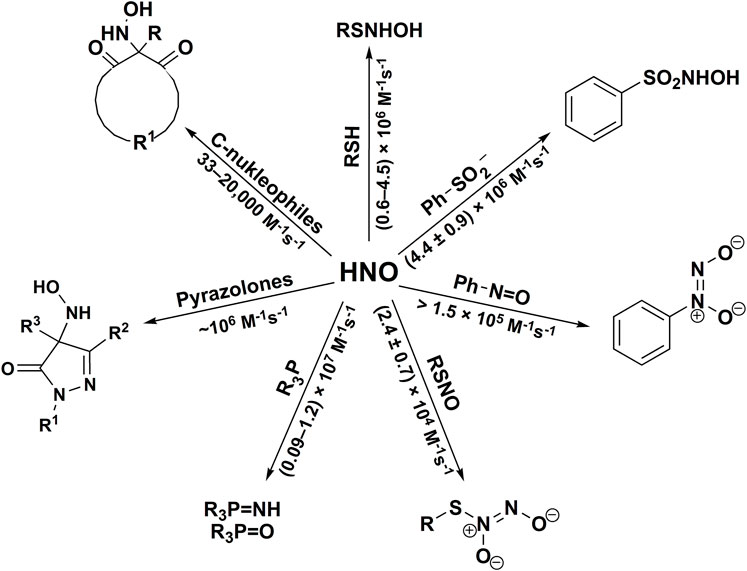
SCHEME 5. The reactivity of HNO towards organic nucleophiles.
2.5 Oxidation of HNO to •NO.
From the recent estimations of the one-electron reduction potential of •NO (E°’ (•NO/1HNO) = −0.16 V for pH 7) (Venâncio et al., 2017), it could be expected that HNO can be oxidized to •NO even by relatively weak one-electron oxidants. In fact, it has been shown that HNO is able to reduce cytochrome c (cyt c (Fe3+)) (E°’ = 0.256 V (Koller and Hawkridge, 1985)), Cu,Zn-SOD (E°’ = 0.403 V (St. Clair et al., 1992)), aminoxyl radicals: TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl, E°’ = 0.204 V (Kato et al., 1995)), TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl, E°’ = 0.226 V (Kato et al., 1995)) and 3-carbamoyl-PROXYL (3-carbamoyl-2,2,5,5 -tetramethyl-1-pyrrolidineoxyl, E°’ = 0.102 V (Kato et al., 1995)) and nitronyl nitroxide radicals: PTIO (2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, E°’ = 0.270 V (Goldstein et al., 2003)) and c-PTIO (2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, E°’ = 0.270 V (Goldstein et al., 2003)).
In 1988 it was reported that the treatment of cyt c(Fe3+) with an excess of Angeli’s salt in phosphate buffer at pH 7.0 at 25°C leads to the formation of ferrocytochrome c (cyt c(Fe2+)) (Doyle et al., 1988). The initial rate of cyt c(Fe3+) reduction was nearly zero order in the concentration of cyt c(Fe3+) and first order in the concentration of HN2O3−, suggesting that the decomposition of Angeli’s salt was controlling the rate of the reaction, consistent with HNO acting as the reductant of cyt c (Fe3+) (Reaction 39). The reported yield of cytochrome c reduction at low [HN2O3−]0/[cyt c (Fe3+)]0 ratio was close to 50%.
Two close values of the second-order rate constant of cyt c(Fe3+) reduction by HNO have been reported in the literature: k39 = 2 × 104 M−1s−1, (Liochev and Fridovich, 2003), and k39 = 4 × 104 M−1s−1 (Miranda et al., 2003b). It is worth to note that the reduction of cyt c(Fe3+) in aqueous solutions of Angeli’s salt was not observed in the presence of O2 (Liochev and Fridovich, 2001) further supporting the involvement of HNO in the reduction process. Moreover, it was observed that cyt c (Fe2+) is oxidized to cyt c(Fe3+) in aerated solutions of Angeli’s salt (Liochev and Fridovich, 2002). Those observations can be explained, taking into account the formation of ONOO− in the reaction of HNO and O2 (Reaction 20) and subsequent reoxidation of cyt c(Fe2+) by ONOO− (Reaction 40, k40 = 1.3 × 104 M−1s−1 at pH 7.4 and 25°C, (Thomson et al., 1995)) or ONOO−-derived one-electron oxidants, the explanation already proposed by the authors (Liochev and Fridovich, 2002).
Addition of Angeli’s salt to a buffered solution of Cu,Zn-SOD resulted in Cu2+ reduction to Cu+ in active site of the enzyme, which was attributed to the formation and oxidation of HNO (Reaction 41) (Liochev and Fridovich, 2001; 2002).
Two values differing by one order of magnitude for the second-order rate constant of the HNO-dependent Cu2+, Zn-SOD reduction have been reported: 8 × 104 M−1s−1, (Liochev and Fridovich, 2003), and ∼1 × 106 M−1s−1 (Miranda et al., 2003b). Interestingly, the occurrence of the reverse reaction, conversion of •NO into HNO by the reduced form of Cu, Zn-SOD has been also proposed (Murphy and Sies, 1991). Clearly, further research on the reaction kinetics, thermodynamics and mechanism is warranted.
Other mild one-electron oxidants reported to oxidize HNO to •NO are aminoxyl radicals (also known as nitroxide radicals or nitroxyl radicals). In an elegant kinetic study on the reactivity of nitroxides toward HNO with the use of EPR spectrometry, the reduction of cyclic stable aminoxyl radicals (RN-O•) by HNO to the corresponding hydroxylamines (RN-OH) (Reaction 42) was reported. (Samuni et al., 2013).
The second-order rate constants of HNO reactions with RN-O• radicals have been determined with the use of the competition kinetics against the reference reaction of HNO with ferric myoglobin (MbFeIII). In the presence of HNO donor, MbFeIII undergoes reductive nitrosylation with the formation of relatively stable ferrous nitrosyl complex, MbFeIINO, which can be followed by UV-Vis absorption spectroscopy. Using kMbFe(III) = 8 × 105 M−1s−1 (Miranda et al., 2003b) as a reference value, the rate constant values for HNO-derived reduction were determined for TEMPO (kTEMPO = (1.4 ± 0.2) × 105 M−1s−1), TEMPOL (kTEMPOL = (1.4 ± 0.2) × 105 M−1s−1) and 3-carbamoyl-PROXYL (k3-CP = (4.3 ± 0.4) × 104 M−1s−1) (Samuni et al., 2013). Those values were close to previously reported rate constants for TEMPOL (kTEMPOL = 8 × 104 M−1s−1) (Miranda et al., 2003b) and TEMPO (kTEMPO = 6.3 × 104 M−1s−1) (Jackson et al., 2009) and a most recent report for 4-acetamido-TEMPO (k = (8 ± 2) × 104 M−1s−1) (Smulik-Izydorczyk et al., 2017). Nitroxides are known to efficiently quench the excited singlet states of molecules, and when they are covalently linked to a fluorophore, the intramolecular quenching of the fluorescence signal is observed (Matsuoka et al., 2016). This property, in combination with the ability of HNO to reduce the nitroxides, enabled the design of fluorogenic probes, that can be used for the detection of HNO. In 2011, a fluorogenic probe 4-((9-acridinecarbonyl)amino)-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO-9-AC), built by covalent linking of a stable nitroxide to acridine fluorophore, for the fluorescent detection of HNO in aqueous solution was reported (Cline and Toscano, 2011). The probe reacts with HNO to produce the highly fluorescent hydroxylamine derivative, with concomitant oxidation of HNO to •NO. The probe was proposed as a chemical tool to differentiate HNO from •NO. The second-order rate constant for the probe reduction by HNO was reported as 8 × 104 M−1s−1 (Cline and Toscano, 2011), a value confirmed in an independent study (kTEMPO-9-AC = (9 ± 2) × 104 M−1s−1) (Smulik-Izydorczyk et al., 2017). Due to the high reactivity of aminoxyl (nitroxide) radicals towards various biological reductants (e.g., ascorbate), the nitroxide-based fluorogenic probes can be used for HNO detection only in the relatively simple chemical systems in the absence of reductants capable of reducing the probe.
In case of continuous generation of HNO, it can be expected that the generated •NO will react with HNO to form the N2O2•− radical anion (Reaction 6), which may react with another •NO to form N2O3− (Reaction 7), eventually producing N2O and NO2− (Reaction 8).
The other class of stable free radicals are nitronyl nitroxides. Those compounds, described in the scientific literature for the first time in 1968 (Osiecki and Ullman, 1968), similarly to aminoxyl radicals are sufficiently stable for isolation and characterization in the presence of O2. Nitronyl nitroxides, such as 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), are widely used in redox biology studies as scavengers of •NO (Akaike et al., 1993; Joseph et al., 1993). PTIO and its analogs were shown to react with •NO to form the corresponding imino nitroxides (e.g., PTI) of characteristic EPR spectrum and •NO2 (Akaike et al., 1993; Joseph et al., 1993) (Scheme 6). The reported values of the second order rate constants of nitronyl nitroxides reaction with •NO are in the range (0.4–16) × 104 M−1s−1 (Akaike et al., 1993; Woldman et al., 1994; Goldstein et al., 2003; Rosen et al., 2003). In 2010 it was demonstrated that nitronyl nitroxides react with HNO to form the corresponding hydroxylamines (Scheme 6) (Samuni et al., 2010).
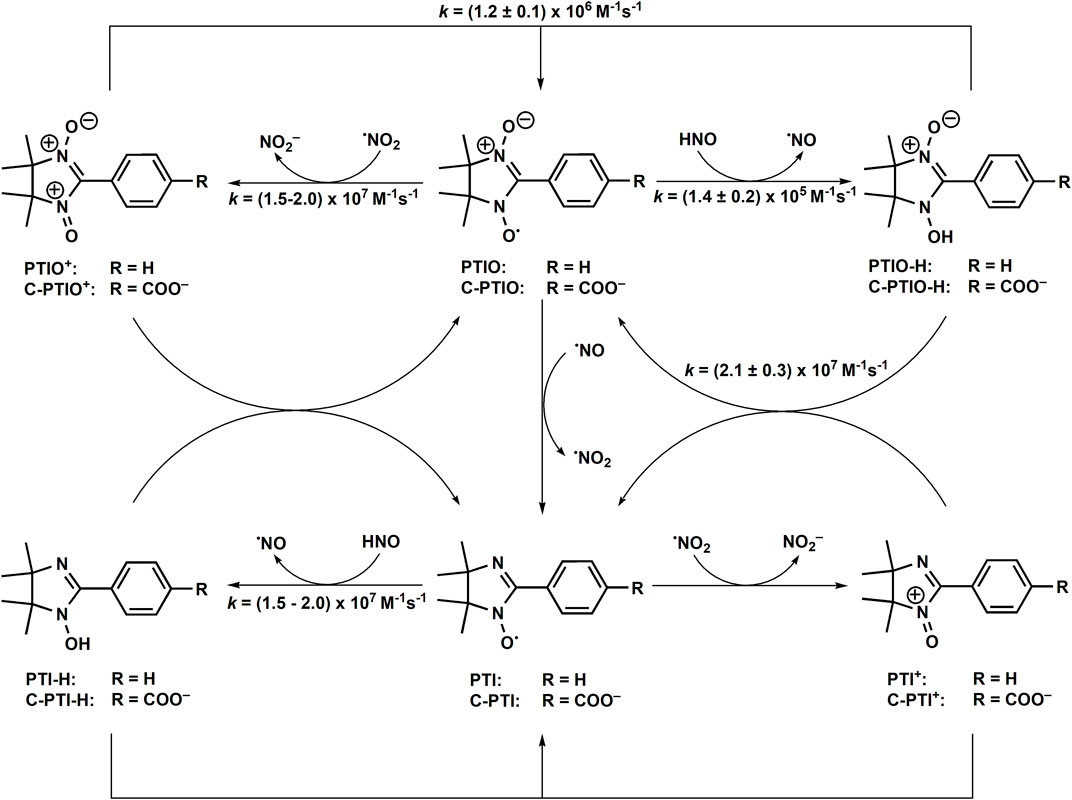
SCHEME 6. Redox reactions in HNO/nitronyl nitroxide system.
Due to the subsequent reactions of nitronyl nitroxide and imino nitroxide radicals with •NO, resulting in the formation of highly oxidizing •NO2 radicals, the chemistry of such a system is rather complex. The second-order rate constants of the HNO reaction with nitronyl nitroxides have been determined with the use of competition kinetics against MbFeIII using kMbFe(III) = 8 × 105 M−1s−1 (Miranda et al., 2003b) as a reference value. The determined rate constant values for PTIO and c-PTIO reduction by HNO at pH 7 were identical: kPTIO = kc-PTIO = (1.4 ± 0.2) × 105 M−1s−1. Independently, the kinetics of c-PTIO reaction with HNO was determined using NH2OH as a competing agent and the kC-PTIO/k23 ratio of 5.5 ± 0.9 was obtained (Bobko et al., 2013). Using k23 = (4.0 ± 0.3) × 103 M−1s−1 (Jackson et al., 2009) or k23 = (2.1 ± 0.4) × 104 M−1s−1 (Smulik-Izydorczyk et al., 2017) one can calculate kC-PTIO = (2.2 ± 0.5) × 104 M−1s−1 (Bobko et al., 2013; Bobko and Khramtsov, 2014) or kC-PTIO = (1.2 ± 0.4) × 105 M−1s−1. It has been also shown, that •NO2 radicals formed in such a system can oxidize Angeli’s salt anion to transient radical product, that decomposes to NO2− and •NO (Reaction 43, k43 = 7.7 × 106 M−1s−1) (Bobko and Khramtsov, 2014).
The kinetics of HNO reaction with nitronyl nitroxide radicals was further reevaluated by the competition kinetics approach, using hydroxylamine as an HNO scavenger and Fe(CN)64− anion as a •NO2 radical scavenger (Bobko and Khramtsov, 2015). Such a system is still relatively complex as Fe(CN)64− and Fe(CN)63− anions are in redox equilibrium with nitronyl nitroxide radicals and Fe(CN)63− anion is an oxidant of HNO. Based on the established competition, the kc-PTIO/k23 ratio of 1.7 ± 0.2 and
2.6 Reactivity of HNO With Metal-Porphyrins
The reaction of HNO with heme-proteins has been studied extensively for nearly 40 years. Among the studied heme-enzymes are myoglobin (Mb), hemoglobin (Hb), horseradish peroxidase (HRP), catalase (cat), cytochrome c (cyt c), soluble guanylate cyclase (sGC), and cytochromes P450 (CYP450) (Reisz et al., 2010). The reactions between HNO and Mb as well as HNO and Hb are well documented. The oxygenated ferrous heme of myoglobin (oxymyoglobin, Mb (FeII)O2) reacts with HNO forming ferric heme and possibly •NO and H2O2 (reaction 44) (Doyle and Mahapatro, 1984; Doyle et al., 1988) and the rate constant for this reaction was determined to be k44 = 1 × 107 M−1s−1 (Miranda et al., 2003b). •NO formed upon HNO oxidation reacts with another Mb (FeII)O2 molecule with rate constant k45 = 3–5 × 107 M−1s−1 producing ferric heme and NO3− (Reaction 45) (Doyle and Hoekstra, 1981; Doyle et al., 1983; Herold et al., 2001). Identical mechanism including similar reaction rate constants can be proposed for Hb(FeII)O2 (Herold et al., 2001; Reisz et al., 2010). Overall, it can be stated that both Mb (FeII)O2 and Hb(FeII)O2 convert HNO to •NO rapidly. It is worth to emphasize that in the literature an alternative mechanism for the heme (FeII)O2/HNO reaction was also proposed. In that mechanism, two ferric hemes, hydroxylamine and O2 are the final products (Miranda, 2005).
Deoxymyoglobin (Mb (FeII)) reacts with HNO forming coordination complex Mb (FeII)HNO (Reaction 46). Originally the rate constant of this reaction was estimated to be equal to 1.4 × 104 M−1s−1 (Sulc et al., 2004). The re-evaluation of reaction kinetics using a singular value decomposition (SVD) method provided a corrected value of 3.7 × 105 M−1s−1 (Zapata et al., 2013; Zapata et al., 2017). The reaction of Mb (FeII) with HNO is followed by subsequent reaction of Mb (FeII)HNO with HNO yielding Mb (FeIII)NO, with the rate constant estimated as 1.67 × 104 M−1s−1 (reaction 47) (Zapata et al., 2013; Zapata et al., 2017). For deoxyhemoglobin the analogical mechanism can be proposed and the rate constant of HNO reaction with Hb(FeII) is equal to ca. 2.0 × 105 M−1s−1 (Kumar et al., 2009). For other ferrous globins the rate constants of the reaction with HNO are in the range 1.2–9.0 × 105 M−1s−1 (Kumar et al., 2009).
The ferric hemes of metmyoglobin and methemoglobin undergo reductive nitrosylation in the presence of HNO producing the ferrous-nitrosyl complexes, Mb (FeII)NO and Hb(FeII)NO, respectively (reaction 48) (Miranda et al., 2003b; Miranda, 2005; Reisz et al., 2010). This reaction proceeds via the direct FeIII–HNO complex formation, followed by an electron transfer (Reisz et al., 2010). The rate constant for the reductive nitrosylation of metMb by HNO was originally estimated to be 8 × 105 M−1s−1 (Miranda et al., 2003b). Recent re-evaluation using SVD method led to the conclusion that the rate constant is about three times lower (k48 = 2.75 × 105 M−1s−1) (Zapata et al., 2013; Zapata et al., 2017).
The rate constant of reductive nitrosylation of metHb is difficult to determine due to the side reaction of HNO with β-93 cysteines of hemoglobin (Doyle et al., 1988). Nonetheless, the observed rate of HbNO formation for the chemically modified hemoglobin containing thioacetamide derivative instead of β-93 cysteines was in agreement with the rate of MbNO formation (Doyle et al., 1988).
Catalase and HRP, similarly to metMb and metHb, are 5-coordinate ferric heme proteins and undergo reductive nitrosylation upon exposure to HNO, with estimated rate constants equal to 3 × 105 M−1s−1 and 2 × 106 M−1s−1 (Reaction 48) (Miranda et al., 2003b). The heme-FeIINO adducts formed in case of both enzymes are stable under anaerobic conditions, but in the presence of O2 the ferric hemes are regenerated and NO3− is formed (Reaction 49) (Huang et al., 2004; King, 2005).
The reaction of cyt c(FeIII) with HNO results in the reduction of iron heme and the formation of •NO, as already discussed in Section 2.5. (Oxidation of HNO to NO) (Liochev and Fridovich, 2003; Miranda et al., 2003b). It has been proposed that iron-nitrosyl complex is an unstable intermediate of this reaction (Doyle et al., 1988).
In the case of sGC there is no estimated rate constant for its reaction with HNO. Miller and coworkers proposed that activation of sGC proceeds directly through HNO and the ferrous heme reaction without conversion of HNO to •NO (Miller et al., 2009). The expected product is sGC(FeII)HNO complex (Reaction 46) but not sGC(FeII)NO. sGC(FeIII) is unreactive toward HNO or •NO. However, a contrary report was published, where the HNO-dependent activation of sGC was excluded (Zeller et al., 2009). That report suggested that the secondary reaction between sGC(FeII)HNO and HNO that follows to the formation of sGC(FeII)NO (reaction 47) is responsible for the activation of sGC (Zapata et al., 2013).
It was shown that the ferric heme of cytochrome P450 is reduced by HNO to heme-FeII(NO) complex similarly to other heme enzymes (Reaction 48) (Miranda et al., 2003a). The interaction with HNO and the formation of the ferrous nitroxyl complex leads to the inactivation of CYP450.
On the basis of the described examples of HNO reactions with heme proteins the following conclusions can be drawn. Oxy-heme centers are the fastest scavengers of HNO, contrary to ferrous- and ferric-hemes centers, for which the values of the rate constant are at least an order of magnitude lower. The ferric forms of heme-proteins react with HNO producing appropriate ferrous heme–NO complexes with rate constants in the range between 4 × 104 M−1s−1 and 2 × 106 M−1s−1 (Reaction 48, Table 2). The two orders of magnitude span of the rate constants indicates that there are additional factors controlling the rate of this reaction. It has been suggested that in the case of cyt c, for which rate constant of reductive nitrosylation is the lowest one, the very likely reason of this phenomenon is the axially bounded histidine-18 and methionine-80 to the heme iron (Miranda, 2005). Thus, to bind to iron, HNO molecule needs to displace the axial ligand during the reaction. Another parameter that may control the kinetics of the HNO binding to the ferric center is the lability of the water ligand (Suarez et al., 2007; Alvarez et al., 2014). It is very likely that ferric hemes of proteins and ferric-porphyrins are hexacoordinated centers with axially bounded water molecules or hydroxide ions and the rate limiting step of the HNO binding is the release of such ligands (Suarez et al., 2007; Alvarez et al., 2014). For comparison, in ferrous hemes the water ligand is weakly bound, which correlates with higher rate constants of •NO binding to the ferrous hemes of Hb or Mb (∼107 M−1s−1) (Fukuto et al., 2013).
Based on the determined rate constants, it can be expected that heme proteins constitute one of the biological targets of HNO. Porphyrins are used as model compounds of heme-proteins that enable structure/reactivity studies with HNO without unnecessary interactions of HNO with a protein matrix. Microperoxidase-11 is an example of a pentacoordinate porphyrin-iron complex with axially bound histidine that enabled the evaluation of the influence of such ligand on the kinetics of HNO/porphyrin reaction (Suarez et al., 2007). The measured rate constant for this reaction (6.4 × 104 M−1s−1) (Suarez et al., 2007) is in reasonable agreement with the rate constants of HNO reactions with other metal-porphyrins (Table 2) indicating that axial histidine does not interfere with the HNO/iron reaction.
The kinetics of HNO reaction with other porphyrins containing iron and manganese were also studied and the appropriate rate constants are compiled in Table 2 (Suarez et al., 2007; Alvarez et al., 2014). Such reactivity is important not only from the point of view of the biological relevance of such complexes as heme models, especially in the case of iron porphyrins, but also due to the potential application for detection and stabilization of HNO. The formation of nitrosyl adducts by Fe- or Mn-porphyrins is accompanied by the shift in the Soret band that can be followed by UV-Vis absorption spectroscopy (Alvarez et al., 2014). Comparing the rate constants for Fe(III)- and Mn(III)-porphyrins (Table 2) it can be seen that the reactivity of Mn(III)-porphyrins toward HNO is similar to Fe(III)-porphyrins (Alvarez et al., 2014). Taking into account the fact that in the case of Mn(III)-porphyrins the formation of nitrosyl adduct is accompanied by 60–100 nm shift of the Soret band relative to around 5 nm for Fe-porphyrins (Doctorovich et al., 2014; Vásquez et al., 2017), the use of Mn(III)-porphyrins for the detection of HNO seems to be a reasonable choice.
The reactivity studies of HNO and porphyrins revealed another interesting chemistry related to the HNO donor/porphyrin interactions. It was shown that the reaction of HNO/metal-porphyrin complexes proceeds according to two alternative mechanisms. In the first mechanism, HNO is released by the donor molecule and a free HNO molecule reacts with metal-porphyrin leading to the appropriate nitrosyl adduct (Reaction 48). In the second mechanism, the donor interacts directly with a metal center via the possible metal porphyrin-donor complex (Reactions 50a, 50b) (Alvarez et al., 2014; Vásquez et al., 2017). The measured rate constants of HNO donor binding to metal-porphyrins are in the range of 1 × 103–1 × 104 M−1s−1 (Table 2). Such direct decomposition of HNO donor was observed for the oxidizing metal-porphyrin complexes. In the case of reducing complexes, HNO must be first released by the donor, followed by reductive nitrosylation. This finding implies the possibility of a direct interaction of HNO donors with heme-proteins. In fact, the decomposition of Angeli’s salt by cytochrome P450 was reported (Shibata et al., 1997).
The reactivity of cobalt-complexes based on porphyrins, corroles, and corrins toward HNO was also reported (Vásquez et al., 2017; Gallego et al., 2021). Cobalamin (vitamin B12, Cbl (III)) is an important example, since it occurs naturally and is an essential mammalian coenzyme. It was shown that aquacobalamin (H2O-Cbl+) and hydroxycobalamin (HO-Cbl) in the presence of Angeli’s salt are converted to the nitroxyl cobalamin (NO−-CoIIICbl) (Subedi et al., 2014). The mechanism of the reaction was dependent on the pH. At high pH over 10.8 the rate determining step was the spontaneous decomposition of Angeli’s salt to HNO and nitrite, followed by the rapid reaction of HNO with the complex. At lower pH (≤ 9.9) a direct reaction between Angeli’s salt anion and H2O-Cbl+ occurs and the complex was observed. The rate constant for the AS/H2O-Cbl+ reaction at acidic pH was determined to be 122.6 ± 5.3 M−1s−1 (Subedi et al., 2014). Recent study on the reaction between H2O-Cbl+ and Piloty’s acid (PA) has shown that PA also reacts directly with H2O-Cbl+ at neutral pH (28.4 ± 0.4 M−1s−1) (Gallego et al., 2021). In turn, in basic solutions, it was shown that HO-Cbl reacts with HNO released from PA, but the rate constant of this reaction remains unknown (Gallego et al., 2021). The mechanistic and kinetic studies on the reaction of reduced cobalamins, cob (II)alamin (Cbl (II)) and cob(I)alamin (Cbl(I)) were also performed (Subedi and Brasch, 2015; 2016). The reduced derivatives of cobalamins react with HNO as well, but mechanisms of these reactions are complex. Nonetheless, the final observed product for Cbl (II) and Cbl(I) is (NO−-CoIIICbl) (Subedi and Brasch, 2015; 2016).
To summarize, the reaction scheme of HNO with heme-proteins is very complex and still remains an area that requires further studies and analyses. In addition, an interesting aspect relates to the direct reactions of HNO-donors with metal-porphyrins, of potential pharmacological relevance.
2.7 Reactivity of HNO Towards NADH, NADPH, Ascorbate, Indoles and Other Reductants
2.7.1 HNO-Dependent NADH, and NADPH, Oxidation
The reactivity of HNO towards NADH and NADPH was a subject of several studies. The mechanism of HNO-dependent NADH and NADPH oxidation, however, is still not completely understood (Wink et al., 1998; Liochev and Fridovich, 2001; Reif et al., 2001; Kirsch and de Groot, 2002; Liochev and Fridovich, 2002; Jackson et al., 2009). In 1998, it was shown that NADPH is oxidized in aerated and deaerated Angeli’s salt solutions (Wink et al., 1998). Aerobic oxidation of NADPH was partially inhibited by TEMPOL and hydroxylamine. At low concentrations of Angeli’s salt in aerated solutions, 1 mol of NADPH was oxidized per about 4 mol of HN2O3−, however the exact stoichiometry of this reaction with respect to HNO is unclear (Reif et al., 2001). NADPH oxidation in such solutions was inhibitable by Cu,Zn-SOD. In an independent study, it was shown that NADPH consumption accounted for ∼50% of decomposed Angeli’s salt (Liochev and Fridovich, 2001). Both Cu,Zn-SOD and Mn-SOD were able to inhibit the oxidation of NADPH by HNO. When Angeli’s salt was aerobically oxidizing NADPH in the presence of cyt c(Fe3+) (10–40 μM), the Cu,Zn-SOD-inhibitable reduction of cyt c(Fe3+) was observed. The Cu,Zn-SOD concentration was selected to be sufficient to compete with cyt c(Fe3+) for O2•–, but too low to detectably inhibit NADPH oxidation. That experiment strongly suggested the generation of O2•– in HNO/O2/NADPH system. The authors proposed that NADPH is oxidized in such a solution to NADP• radicals, which subsequently reduce O2 to O2•− with the formation of NADP+. At higher concentration (∼2 μM), Cu,Zn-SOD inhibited by ∼50% the oxidation of NADPH (Liochev and Fridovich, 2001). Similar effect of Cu,Zn-SOD on the NADH oxidation in aerated Angeli’s salt solution was reported subsequently (Kirsch and de Groot, 2002). In that paper it was also demonstrated that NADH oxidation in Angeli’s salt solution can be significantly inhibited by the addition of a •NO donor DEA-NONOate, when [•NO]/[HNO] ratio is closed to 2. Under those conditions, HNO reaction with •NO leading to the formation of N2O2•− and its subsequent reactions with •NO were favored (Reactions 6, 7). The second-order rate constant of the reactions between NADH and HNO equal to k51 = (1.1 ± 0.2) × 104 M−1s−1 was reported (Jackson et al., 2009). Similar value k52 = (1.3 ± 0.4) × 104 M−1s−1 was also determined for NADPH. In the direct reaction of HNO with NADH or NADPH in deaerated Angeli’s salt solution, one may expect the formation of hydroxylamine as an HNO reduction product (Reactions 51, 52). However, experimental data did not support that hypothesis (Jackson et al., 2009). Clearly, further research on this type of HNO reactions is needed.
2.7.2 Ascorbate, Trolox and Selenomethionine
Using the competition kinetic approach and HNO reaction with NADH as a reference, the rate constants for the reaction of HNO with ascorbate, trolox and selenomethionine were determined to be equal to 1.1 × 105, 2 × 104 and 9 × 103 M−1s−1, respectively (Jackson et al., 2009). The mechanisms of those reactions remain to be established.
2.7.3 Indoles
The results of the studies on the reaction of various indolic compounds (e.g., N-acetyl-L-tryptophan, indol-3-acetic acid and melatonin) with Angeli’s salt in aerobic buffered aqueous solutions (pH 7.4) has been presented in several scientific reports (Suzuki et al., 2004; Peyrot and Ducrocq, 2007; Keceli et al., 2014). All these studies show that this reaction leads to the formation of N-nitroso derivative as the main product. The yield of its formation is rather low (below 20%) and depends on the indolic compound: Angeli’s salt concentrations ratio (Peyrot et al., 2006; Keceli et al., 2014). It was shown that the yield of N-nitroso derivative is significantly higher in the presence of bicarbonate (Suzuki et al., 2004), which may suggest the participation of peroxynitrite in the N-nitrosation mechanism. The recent study of Keceli et al. (2014) on the reaction of HNO with N-acetyl-L-tryptophan and small peptides containing either tryptophan or both tryptophan and cysteine residues, has shown that in the presence of cysteine, excess HNO is required for efficient TrpNO formation, clearly indicating, that tryptophan residues are significantly less reactive towards HNO, than cysteine residues.
3 Conclusion
HNO show a different chemical reactivity than •NO, and may be expected to exhibit unique physiological activity. The experimental limitations due to its instability under physiological conditions provide a significant hindrance in study design to further explore HNO chemistry. The development of new donors and rapid generation methods for time-resolved kinetic studies may overcome some of those obstacles. In addition, the combination of the experimental data with theoretical calculations may help with drawing reliable mechanistic conclusions. Finally, as a note of caution, it has to be emphasized that many rate constants of HNO reactions listed in this review were determined by competition kinetics, via kinetic simulations or other indirect approaches, and are based on very few values determined directly. Therefore, it can be expected that with the development of new experimental methods, the rate constants may need to be re-evaluated and the mechanistic conclusions adjusted.
Author Contributions
All authors contributed to the review concept, design, and bibliographic research. RS-I, RM, JP, JZ, and AS prepared the first version of the manuscript. AA, KP, RS-I, and RM prepared the tables and MR, RM, and JP prepared the schemes and figures. AS, JZ, and BK critically reviewed and prepared the final version of the manuscript. All authors accepted the manuscript in its final form.
Funding
AS was supported by Polish National Science Center within the SONATA BIS program (Grant no. 2015/18/E/ST4/00235). RM was supported by a grant from the Polish National Science Center SONATA program (Grant no. 2018/31/D/ST4/03494).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ackermann, M. N., and Powell, R. E. (1967). Air Oxidation of Hydroxylamine-N-Sulfonate. Inorg. Chem. 6 (9), 1718–1720. doi:10.1021/ic50055a023
Ackermann, M. N., and Powell, R. E. (1966). Alkaline Hydrolysis of Hydroxylamine-N-Sulfonate. Inorg. Chem. 5 (8), 1334–1337. doi:10.1021/ic50042a008
Akaike, T., Yoshida, M., Miyamoto, Y., Sato, K., Kohno, M., Sasamoto, K., et al. (1993). Antagonistic Action of Imidazolineoxyl N-Oxides against Endothelium-Derived Relaxing factor/.NO through a Radical Reaction. Biochemistry 32 (3), 827–832. doi:10.1021/bi00054a013
Alvarez, L., Suarez, S. A., Bikiel, D. E., Reboucas, J. S., Batinic-Haberle, I., Marti, M. A., et al. (2014). Redox Potential Determines the Reaction Mechanism of HNO Donors with Mn and Fe Porphyrins: Defining the Better Traps. Inorg. Chem. 53 (14), 7351–7360. doi:10.1021/ic5007082
Artelska, A., Rola, M., Rostkowski, M., Pięta, M., Pięta, J., Michalski, R., et al. (2021). Kinetic Study on the Reactivity of Azanone (HNO) toward Cyclic C-Nucleophiles. Int. J. Mol. Sci. 22 (23). doi:10.3390/ijms222312982
Bartberger, M. D., Fukuto, J. M., and Houk, K. N. (2001). On the Acidity and Reactivity of HNO in Aqueous Solution and Biological Systems. Proc. Natl. Acad. Sci. U. S. A. 98 (5), 2194–2198. doi:10.1073/pnas.041481598
Bartberger, M. D., Liu, W., Ford, E., Miranda, K. M., Switzer, C., Fukuto, J. M., et al. (2002). The Reduction Potential of Nitric Oxide (NO) and its Importance to NO Biochemistry. Proc. Natl. Acad. Sci. U. S. A. 99 (17), 10958–10963. doi:10.1073/pnas.162095599
Belt, R. F., and Baenziger, N. C. (1957). The Crystal Structure of Potassium Hydroxylamine-N-Sulfonate1. J. Am. Chem. Soc. 79 (2), 316–318. doi:10.1021/ja01559a019
Benchoam, D., Semelak, J. A., Cuevasanta, E., Mastrogiovanni, M., Grassano, J. S., Ferrer-Sueta, G., et al. (2020). Acidity and Nucleophilic Reactivity of Glutathione Persulfide. J. Biol. Chem. 295 (46), 15466–15481. doi:10.1074/jbc.RA120.014728
Bianco, C. L., Moore, C. D., Fukuto, J. M., and Toscano, J. P. (2016). Selenols Are Resistant to Irreversible Modification by HNO. Free Radic. Biol. Med. 99, 71–78. doi:10.1016/j.freeradbiomed.2016.07.008
Bianco, C. L., Toscano, J. P., Bartberger, M. D., and Fukuto, J. M. (2017). The Chemical Biology of HNO Signaling. Arch. Biochem. Biophys. 617, 129–136. doi:10.1016/j.abb.2016.08.014
Bobko, A. A., Ivanov, A., and Khramtsov, V. V. (2013). Discriminative EPR Detection of NO and HNO by Encapsulated Nitronyl Nitroxides. Free Radic. Res. 47 (2), 74–81. doi:10.3109/10715762.2012.746460
Bobko, A. A., and Khramtsov, V. V. (2014). Mechanistic Studies of Oxidative Decomposition of Angeli's Salt and PAPA NONOate. Nitric Oxide 40, 92–98. doi:10.1016/j.niox.2014.05.013
Bobko, A. A., and Khramtsov, V. V. (2015). Redox Properties of the Nitronyl Nitroxide Antioxidants Studied via Their Reactions with Nitroxyl and Ferrocyanide. Free Radic. Res. 49 (8), 919–926. doi:10.3109/10715762.2015.1013951
Bobrowski, K. (2017). “Radiation Chemistry of Liquid Systems,” in Applications of Ionizing Radiation in Materials Processing (Warszawa: Institute of Nuclear Chemistry and Technology), 81–116.
Bonner, F. T., Dzelzkalns, L. S., and Bonucci, J. A. (1978). Properties of Nitroxyl as Intermediate in the Nitric Oxide-Hydroxylamine Reaction and in Trioxodinitrate Decomposition. Inorg. Chem. 17 (9), 2487–2494. doi:10.1021/ic50187a030
Boron, I., Suarez, S. A., Doctorovich, F., Marti, M. A., and Bari, S. E. (2011). A Protective Protein Matrix Improves the Discrimination of Nitroxyl from Nitric Oxide by Mn-III Protoporphyrinate IX in Aerobic Media. J. Inorg. Biochem. 105 (8), 1044–1049. doi:10.1016/j.jinorgbio.2011.05.002
Cline, M. R., and Toscano, J. P. (2011). Detection of Nitroxyl (HNO) by a Prefluorescent Probe. J. Phys. Org. Chem. 24 (10), 993–998. doi:10.1002/poc.1871
Dalby, F. W. (1958). THE SPECTRUM AND STRUCTURE OF THE HNO MOLECULE. Can. J. Phys. 36 (10), 1336–1371. doi:10.1139/p58-138
Dębowska, K., Dębski, D., Michałowski, B., Dybala-Defratyka, A., Wójcik, T., Michalski, R., et al. (2016). Characterization of Fluorescein-Based Monoboronate Probe and its Application to the Detection of Peroxynitrite in Endothelial Cells Treated with Doxorubicin. Chem. Res. Toxicol. 29 (5), 735–746. doi:10.1021/acs.chemrestox.5b00431
Doctorovich, F., Bikiel, D. E., Pellegrino, J., Suarez, S. A., and Marti, M. A. (2014). Reactions of HNO with Metal Porphyrins: Underscoring the Biological Relevance of HNO. Accounts Chem. Res. 47 (10), 2907–2916. doi:10.1021/ar500153c
Donald, C. E., Hughes, M. N., Thompson, J. M., and Bonner, F. T. (1986). Photolysis of the Nitrogen-Nitrogen Double Bond in Trioxodinitrate: Reaction between Triplet Oxonitrate(1-) and Molecular Oxygen to Form Peroxonitrite. Inorg. Chem. 25 (16), 2676–2677. doi:10.1021/ic00236a004
Donzelli, S., Espey, M. G., Thomas, D. D., Mancardi, D., Tocchetti, C. G., Ridnour, L. A., et al. (2006). Discriminating Formation of HNO from Other Reactive Nitrogen Oxide Species. Free Radic. Biol. Med. 40 (6), 1056–1066. doi:10.1016/j.freeradbiomed.2005.10.058
Doyle, M. P., and Hoekstra, J. W. (1981). Oxidation of Nitrogen-Oxides by Bound Dioxygen in Hemoproteins. J. Inorg. Biochem. 14 (4), 351–358. doi:10.1016/S0162-0134(00)80291-3
Doyle, M. P., Mahapatro, S. N., Broene, R. D., and Guy, J. K. (1988). Oxidation and Reduction of Hemoproteins by Trioxodinitrate(Ii) - the Role of Nitrosyl Hydride and Nitrite. J. Am. Chem. Soc. 110 (2), 593–599. doi:10.1021/ja00210a047
Doyle, M. P., and Mahapatro, S. N. (1984). Nitric-Oxide Dissociation from Trioxodinitrate(Ii) in Aqueous-Solution. J. Am. Chem. Soc. 106 (12), 3678–3679. doi:10.1021/ja00324a049
Doyle, M. P., Pickering, R. A., and Cook, B. R. (1983). Oxidation of Oxymyoglobin by Nitric-Oxide through Dissociation from Cobalt Nitrosyls. J. Inorg. Biochem. 19 (4), 329–338. doi:10.1016/0162-0134(83)80006-3
Felker, G. M., Borentain, M., Cleland, J. G., DeSouza, M. M., Kessler, P. D., O'Connor, C. M., et al. (2019). Rationale and Design for the Development of a Novel Nitroxyl Donor in Patients with Acute Heart Failure. Eur. J. Heart Fail 21 (8), 1022–1031. doi:10.1002/ejhf.1504
Felker, G. M., McMurray, J. J. V., Cleland, J. G., O'Connor, C. M., Teerlink, J. R., Voors, A. A., et al. (2021). Effects of a Novel Nitroxyl Donor in Acute Heart Failure: The STAND-UP AHF Study. JACC Heart Fail 9 (2), 146–157. doi:10.1016/j.jchf.2020.10.012
Fukuto, J. M. (2019). A Recent History of Nitroxyl Chemistry, Pharmacology and Therapeutic Potential. Br. J. Pharmacol. 176 (2), 135–146. doi:10.1111/bph.14384
Fukuto, J. M., Cisneros, C. J., and Kinkade, R. L. (2013). A Comparison of the Chemistry Associated with the Biological Signaling and Actions of Nitroxyl (HNO) and Nitric Oxide (NO). J. Inorg. Biochem. 118, 201–208. doi:10.1016/j.jinorgbio.2012.08.027
Fukuto, J. M., and Hobbs, A. J. (2021). A Comparison of the Chemical Biology of Hydropersulfides (RSSH) with Other Protective Biological Antioxidants and Nucleophiles. Nitric Oxide 107, 46–57. doi:10.1016/j.niox.2020.11.004
Gallego, C. M., Mazzeo, A., Gaviglio, C., Pellegrino, J., and Doctorovich, F. (2021). Structure and Reactivity of NO/NO+/NO- Pincer and Porphyrin Complexes. Eur. J. Inorg. Chem. 2021 (46), 4712–4730. doi:10.1002/ejic.202100682
Goldstein, S., Russo, A., and Samuni, A. (2003). Reactions of PTIO and Carboxy-PTIO with *NO, *NO2, and O2-. J. Biol. Chem. 278 (51), 50949–50955. doi:10.1074/jbc.M308317200
Gordon, S., Hart, E. J., Matheson, M. S., Rabani, J., and Thomas, J. K. (1963). Reactions of the Hydrated Electron. Discuss. Faraday Soc. 36 (0), 193–205. doi:10.1039/DF9633600193
Grätzel, M., Taniguchi, S., and Henglein, A. (1970). Pulsradiolytische Untersuchung kurzlebiger Zwischenprodukte der NO-Reduktion in wäßriger Lösung. Berichte Bunsenges. für Phys. Chem. 74 (10), 1003–1010. doi:10.1002/bbpc.19700741012
Greenberg, B., and Urey, M. A. (2021). Stand [Up] and Stand by for New Strategies for Treating Acute Heart Failure. JACC Heart Fail 9 (2), 158–160. doi:10.1016/j.jchf.2020.11.001
Grzelakowska, A., Modrzejewska, J., Kolińska, J., Szala, M., Zielonka, M., Dębowska, K., et al. (2022). Water-soluble Cationic Boronate Probe Based on Coumarin Imidazolium Scaffold: Synthesis, Characterization, and Application to Cellular Peroxynitrite Detection. Free Radic. Biol. Med. 179, 34–46. doi:10.1016/j.freeradbiomed.2021.12.260
Grzelakowska, A., Zielonka, M., Dębowska, K., Modrzejewska, J., Szala, M., Sikora, A., et al. (2021). Two-photon Fluorescent Probe for Cellular Peroxynitrite: Fluorescence Detection, Imaging, and Identification of Peroxynitrite-specific Products. Free Radic. Biol. Med. 169, 24–35. doi:10.1016/j.freeradbiomed.2021.04.011
Guthrie, D. A., Ho, A., Takahashi, C. G., Collins, A., Morris, M., and Toscano, J. P. (2015). Catch-and-Release” of HNO with Pyrazolones. J. Org. Chem. 80 (3), 1338–1348. doi:10.1021/jo502330w
Haake, M. (1972). Zur desoxygenierung von tritylthionitrit. Tetrahedron Lett. 13 (33), 3405–3408. doi:10.1016/S0040-4039(01)94056-0
Herold, S., Exner, M., and Nauser, T. (2001). Kinetic and Mechanistic Studies of the NO Center Dot-Mediated Oxidation of Oxymyoglobin and Oxyhemoglobin. Biochemistry 40 (11), 3385–3395. doi:10.1021/bi002407m
Huang, J. M., Kim-Shapiro, D. B., and King, S. B. (2004). Catalase-mediated Nitric Oxide Formation from Hydroxyurea. J. Med. Chem. 47 (14), 3495–3501. doi:10.1021/jm030547z
Hughes, M. N., and Nicklin, H. G. (1971). Autoxidation of Hydroxylamine in Alkaline Solutions. J. Chem. Soc. A Inorg. Phys. Theor. 1971, 164–168. doi:10.1039/J19710000164
Ignarro, L. J., Byrns, R. E., Buga, G. M., and Wood, K. S. (1987). Endothelium-derived Relaxing Factor from Pulmonary Artery and Vein Possesses Pharmacologic and Chemical Properties Identical to Those of Nitric Oxide Radical. Circ. Res. 61 (6), 866–879. doi:10.1161/01.res.61.6.866
Jackson, M. I., Han, T. H., Serbulea, L., Dutton, A., Ford, E., Miranda, K. M., et al. (2009). Kinetic Feasibility of Nitroxyl Reduction by Physiological Reductants and Biological Implications. Free Radic. Biol. Med. 47 (8), 1130–1139. doi:10.1016/j.freeradbiomed.2009.06.034
Joseph, J., Kalyanaraman, B., and Hyde, J. S. (1993). Trapping of Nitric Oxide by Nitronyl Nitroxides: an Electron Spin Resonance Investigation. Biochem. Biophys. Res. Commun. 192 (2), 926–934. doi:10.1006/bbrc.1993.1504
Kato, Y., Shimizu, Y., Yijing, L., Unoura, K., Utsumi, H., and Ogata, T. (1995). Reversible Half-Wave Potentials of Reduction Processes on Nitroxide Radicals. Electrochimica Acta 40 (17), 2799–2802. doi:10.1016/0013-4686(95)00261-C
Kawai, K., Ieda, N., Aizawa, K., Suzuki, T., Miyata, N., and Nakagawa, H. (2013). A Reductant-Resistant and Metal-free Fluorescent Probe for Nitroxyl Applicable to Living Cells. J. Am. Chem. Soc. 135 (34), 12690–12696. doi:10.1021/ja404757s
Kazanis, S., and McClelland, R. A. (1992). Electrophilic Intermediate in the Reaction of Glutathione and Nitroso Arenes. J. Am. Chem. Soc. 114 (8), 3052–3059. doi:10.1021/ja00034a043
Keceli, G., Moore, C. D., and Toscano, J. P. (2014). Comparison of HNO Reactivity with Tryptophan and Cysteine in Small Peptides. Bioorg Med. Chem. Lett. 24 (16), 3710–3713. doi:10.1016/j.bmcl.2014.07.014
Keceli, G., and Toscano, J. P. (2014). Reactivity of C-Terminal Cysteines with HNO. Biochemistry 53 (22), 3689–3698. doi:10.1021/bi500360x
Keceli, G., and Toscano, J. P. (2012). Reactivity of Nitroxyl-Derived Sulfinamides. Biochemistry 51 (20), 4206–4216. doi:10.1021/bi300015u
Kemp-Harper, B. K., Horowitz, J. D., and Ritchie, R. H. (2016). Therapeutic Potential of Nitroxyl (HNO) Donors in the Management of Acute Decompensated Heart Failure. Drugs 76 (14), 1337–1348. doi:10.1007/s40265-016-0631-y
Kemp-Harper, B. K., Velagic, A., Paolocci, N., Horowitz, J. D., and Ritchie, R. H. (2021). Cardiovascular Therapeutic Potential of the Redox Siblings, Nitric Oxide (NO•) and Nitroxyl (HNO), in the Setting of Reactive Oxygen Species Dysregulation. Handb. Exp. Pharmacol. 264, 311–337. doi:10.1007/164_2020_389
Kerwin, J. F., Lancaster, J. R., and Feldman, P. L. (1995). Nitric Oxide: a New Paradigm for Second Messengers. J. Med. Chem. 38 (22), 4343–4362. doi:10.1021/jm00022a001
King, S. B. (2005). N-hydroxyurea and Acyl Nitroso Compounds as Nitroxyl (HNO) and Nitric Oxide (NO) Donors. Curr. Top. Med. Chem. 5 (7), 665–673. doi:10.2174/1568026054679362
Kirsch, M., and de Groot, H. (2002). Formation of Peroxynitrite from Reaction of Nitroxyl Anion with Molecular Oxygen. J. Biol. Chem. 277 (16), 13379–13388. doi:10.1074/jbc.M108079200
Knight, R., and Sutton, H. (1967). Radiolysis of Aqueous Solutions of Nitric Oxide. Trans. Faraday Soc. 63, 2628–2639. doi:10.1039/tf9676302628
Koller, K. B., and Hawkridge, F. M. (1985). Temperature and Electrolyte Effects on the Electron-Transfer Reactions of Cytochrome C. J. Am. Chem. Soc. 107 (25), 7412–7417. doi:10.1021/ja00311a032
Kumar, M. R., Pervitsky, D., Chen, L., Poulos, T., Kundu, S., Hargrove, M. S., et al. (2009). Nitrosyl Hydride (HNO) as an O-2 Analogue: Long-Lived HNO Adducts of Ferrous Globins. Biochemistry 48 (22), 5018–5025. doi:10.1021/bi900122r
Lang, N. N., Ahmad, F. A., Cleland, J. G., O'Connor, C. M., Teerlink, J. R., Voors, A. A., et al. (2021). Haemodynamic Effects of the Nitroxyl Donor Cimlanod (BMS‐986231) in Chronic Heart Failure: a Randomized Trial. Eur. J. heart Fail. 23 (7), 1147–1155. doi:10.1002/ejhf.2138
Liochev, S. I., and Fridovich, I. (2001). Copper,zinc Superoxide Dismutase as a Univalent NO(-) Oxidoreductase and as a Dichlorofluorescin Peroxidase. J. Biol. Chem. 276 (38), 35253–35257. doi:10.1074/jbc.M104237200
Liochev, S. I., and Fridovich, I. (2002). Nitroxyl (NO-): a Substrate for Superoxide Dismutase. Arch. Biochem. Biophys. 402 (2), 166–171. doi:10.1016/s0003-9861(02)00074-7
Liochev, S. I., and Fridovich, I. (2003). The Mode of Decomposition of Angeli's Salt (Na2N2O3) and the Effects Thereon of Oxygen, Nitrite, Superoxide Dismutase, and Glutathione. Free Radic. Biol. Med. 34 (11), 1399–1404. doi:10.1016/s0891-5849(03)00111-4
Lymar, S. V., and Shafirovich, V. (2007). Photoinduced Release of Nitroxyl and Nitric Oxide from Diazeniumdiolates. J. Phys. Chem. B 111 (24), 6861–6867. doi:10.1021/jp070959+
Lymar, S. V., Shafirovich, V., and Poskrebyshev, G. A. (2005). One-electron Reduction of Aqueous Nitric Oxide: a Mechanistic Revision. Inorg. Chem. 44 (15), 5212–5221. doi:10.1021/ic0501317
Marti, M. A., Bari, S. E., Estrin, D. A., and Doctorovich, F. (2005). Discrimination of Nitroxyl and Nitric Oxide by Water-Soluble Mn(III) Porphyrins. J. Am. Chem. Soc. 127 (13), 4680–4684. doi:10.1021/ja044632n
Matsuoka, Y., Yamato, M., and Yamada, K. (2016). Fluorescence Probe for the Convenient and Sensitive Detection of Ascorbic Acid. J. Clin. Biochem. Nutr. 58 (1), 16–22. doi:10.3164/jcbn.15-105
Miller, T. W., Cherney, M. M., Lee, A. J., Francoleon, N. E., Farmer, P. J., King, S. B., et al. (2009). The Effects of Nitroxyl (HNO) on Soluble Guanylate Cyclase Activity INTERACTIONS at FERROUS HEME and CYSTEINE THIOLS. J. Biol. Chem. 284 (33), 21788–21796. doi:10.1074/jbc.M109.014282
Miranda, K. M., Espey, M. G., Yamada, K., Krishna, M., Ludwick, N., Kim, S., et al. (2001). Unique Oxidative Mechanisms for the Reactive Nitrogen Oxide Species, Nitroxyl Anion. J. Biol. Chem. 276 (3), 1720–1727. doi:10.1074/jbc.M006174200
Miranda, K. M., Nims, R. W., Thomasa, D. D., Espey, M. G., Citrin, D., Bartberger, M. D., et al. (2003a). Comparison of the Reactivity of Nitric Oxide and Nitroxyl with Heme Proteins - A Chemical Discussion of the Differential Biological Effects of These Redox Related Products of NOS. J. Inorg. Biochem. 93 (1-2), 52–60. doi:10.1016/s0162-0134(02)00498-1
Miranda, K. M., Paolocci, N., Katori, T., Thomas, D. D., Ford, E., Bartberger, M. D., et al. (2003b). A Biochemical Rationale for the Discrete Behavior of Nitroxyl and Nitric Oxide in the Cardiovascular System. Proc. Natl. Acad. Sci. U. S. A. 100 (16), 9196–9201. doi:10.1073/pnas.1430507100
Miranda, K. M. (2005). The Chemistry of Nitroxyl (HNO) and Implications in Biology. Coord. Chem. Rev. 249 (3-4), 433–455. doi:10.1016/j.ccr.2004.08.010
Miranda, K. M., Yamada, K., Espey, M. G., Thomas, D. D., DeGraff, W., Mitchell, J. B., et al. (2002). Further Evidence for Distinct Reactive Intermediates from Nitroxyl and Peroxynitrite: Effects of Buffer Composition on the Chemistry of Angeli's Salt and Synthetic Peroxynitrite. Arch. Biochem. Biophys. 401 (2), 134–144. doi:10.1016/s0003-9861(02)00031-0
Murphy, M. E., and Sies, H. (1991). Reversible Conversion of Nitroxyl Anion to Nitric Oxide by Superoxide Dismutase. Proc. Natl. Acad. Sci. U. S. A. 88 (23), 10860–10864. doi:10.1073/pnas.88.23.10860
Neuman, N. I., Venâncio, M. F., Rocha, W. R., Bikiel, D. E., Suárez, S. A., and Doctorovich, F. (2021). Nitric Oxide Reacts Very Fast with Hydrogen Sulfide, Alcohols, and Thiols to Produce HNO: Revised Rate Constants. Inorg. Chem. 60 (21), 15997–16007. doi:10.1021/acs.inorgchem.1c01061
Orgel, L. E. (1953). The Electronic Structures and Spectra of Some Molecules Related to Oxygen. J. Chem. Soc. (Resumed) 255, 1276–1278. doi:10.1039/JR9530001276
Osiecki, J. H., and Ullman, E. F. (1968). Studies of Free Radicals. I. .alpha.-Nitronyl Nitroxides, a New Class of Stable Radicals. J. Am. Chem. Soc. 90 (4), 1078–1079. doi:10.1021/ja01006a053
Peyrot, F., and Ducrocq, C. (2007). Nitrosation of N-Terminally Blocked Tryptophan and Tryptophan-Containing Peptides by Peroxynitrite. Chembiochem 8 (2), 217–223. doi:10.1002/cbic.200600385
Peyrot, F., Fernandez, B. O., Bryan, N. S., Feelisch, M., and Ducrocq, C. (2006). N-nitroso Products from the Reaction of Indoles with Angeli's Salt. Chem. Res. Toxicol. 19 (1), 58–67. doi:10.1021/tx050253b
Pino, R. Z., and Feelisch, M. (1994). Bioassay Discrimination between Nitric Oxide (NO.) and Nitroxyl (NO-) Using L-Cysteine. Biochem. Biophys. Res. Commun. 201 (1), 54–62. doi:10.1006/bbrc.1994.1668
Polaczek, J., Subedi, H., Orzeł, Ł., Lisboa, L. S., Cink, R. B., Stochel, G., et al. (2021). Mechanistic Studies on the Reaction between Aquacobalamin and the HNO Donor Piloty's Acid over a Wide pH Range in Aqueous Solution. Inorg. Chem. 60 (5), 2964–2975. doi:10.1021/acs.inorgchem.0c02968
Poskrebyshev, G. A., Shafirovich, V., and Lymar, S. V. (2008). Disproportionation Pathways of Aqueous Hyponitrite Radicals (HN2O2•/N2O2•−). J. Phys. Chem. A 112 (36), 8295–8302. doi:10.1021/jp803230c
Poskrebyshev, G. A., Shafirovich, V., and Lymar, S. V. (2004). Hyponitrite Radical, a Stable Adduct of Nitric Oxide and Nitroxyl. J. Am. Chem. Soc. 126 (3), 891–899. doi:10.1021/ja038042l
Reif, A., Zecca, L., Riederer, P., Feelisch, M., and Schmidt, H. H. (2001). Nitroxyl Oxidizes NADPH in a Superoxide Dismutase Inhibitable Manner. Free Radic. Biol. Med. 30 (7), 803–808. doi:10.1016/s0891-5849(01)00477-4
Reisz, J. A., Bechtold, E., and King, S. B. (2010). Oxidative Heme Protein-Mediated Nitroxyl (HNO) Generation. Dalton Trans. 39 (22), 5203–5212. doi:10.1039/c000980f
Reisz, J. A., Klorig, E. B., Wright, M. W., and King, S. B. (2009). Reductive Phosphine-Mediated Ligation of Nitroxyl (HNO). Org. Lett. 11 (13), 2719–2721. doi:10.1021/ol900914s
Reisz, J. A., Zink, C. N., and King, S. B. (2011). Rapid and Selective Nitroxyl (HNO) Trapping by Phosphines: Kinetics and New Aqueous Ligations for HNO Detection and Quantitation. J. Am. Chem. Soc. 133 (30), 11675–11685. doi:10.1021/ja203652z
Rios, N., Piacenza, L., Trujillo, M., Martínez, A., Demicheli, V., Prolo, C., et al. (2016). Sensitive Detection and Estimation of Cell-Derived Peroxynitrite Fluxes Using Fluorescein-Boronate. Free Radic. Biol. Med. 101, 284–295. doi:10.1016/j.freeradbiomed.2016.08.033
Rosen, G. M., Porasuphatana, S., Tsai, P., Ambulos, N. P., Galtsev, V. E., Ichikawa, K., et al. (2003). Dendrimeric-Containing Nitronyl Nitroxides as Spin Traps for Nitric Oxide: Synthesis, Kinetic, and Stability Studies. Macromolecules 36 (4), 1021–1027. doi:10.1021/ma021292m
Samuni, U., Samuni, Y., and Goldstein, S. (2010). On the Distinction between Nitroxyl and Nitric Oxide Using Nitronyl Nitroxides. J. Am. Chem. Soc. 132 (24), 8428–8432. doi:10.1021/ja101945j
Samuni, Y., Samuni, U., and Goldstein, S. (2013). The Use of Cyclic Nitroxide Radicals as HNO Scavengers. J. Inorg. Biochem. 118, 155–161. doi:10.1016/j.jinorgbio.2012.10.002
Saund, S. S., Sosa, V., Henriquez, S., Nguyen, Q. N., Bianco, C. L., Soeda, S., et al. (2015). The Chemical Biology of Hydropersulfides (RSSH): Chemical Stability, Reactivity and Redox Roles. Arch. Biochem. Biophys. 588, 15–24. doi:10.1016/j.abb.2015.10.016
Saxon, E., and Bertozzi, C. R. (2000). Cell Surface Engineering by a Modified Staudinger Reaction. Science 287 (5460), 2007–2010. doi:10.1126/science.287.5460.2007
Seddon, W. A., and Young, M. J. (1970). Pulse Radiolysis of Nitric Oxide in Aqueous Solution. Can. J. Chem. 48 (2), 393–394. doi:10.1139/v70-063
Seddon, W., Fletcher, J., and Sopchyshyn, F. (1973). Pulse Radiolysis of Nitric Oxide in Aqueous Solution. Can. J. Chem. 51 (7), 1123–1130. doi:10.1139/v73-166
Seel, F., and Kaschuba, J. (1975). Autoxydation von Hydroxylamin-N-monosulfonat in alkalischer lösung. Z. für Anorg. Allg. Chem. 414 (1), 56–61. doi:10.1002/zaac.19754140107
Shafirovich, V., and Lymar, S. V. (2002). Nitroxyl and its Anion in Aqueous Solutions: Spin States, Protic Equilibria, and Reactivities toward Oxygen and Nitric Oxide. Proc. Natl. Acad. Sci. U. S. A. 99 (11), 7340–7345. doi:10.1073/pnas.112202099
Shafirovich, V., and Lymar, S. V. (2003). Spin-forbidden Deprotonation of Aqueous Nitroxyl (HNO). J. Am. Chem. Soc. 125 (21), 6547–6552. doi:10.1021/ja034378j
Shen, B., and English, A. M. (2005). Mass Spectrometric Analysis of Nitroxyl-Mediated Protein Modification: Comparison of Products Formed with Free and Protein-Based Cysteines. Biochemistry 44 (42), 14030–14044. doi:10.1021/bi0507478
Shibata, Y., Sato, H., Sagami, I., and Shimizu, T. (1997). Interaction of Angeli's Salt with Cytochrome P450 1A2 Distal Mutants: an Optical Absorption Spectral Study. Biochimica Biophysica Acta-Protein Struct. Mol. Enzym. 1343 (1), 67–75. doi:10.1016/S0167-4838(97)00104-0
Shoeman, D. W., and Nagasawa, H. T. (1998). The Reaction of Nitroxyl (HNO) with Nitrosobenzene Gives Cupferron (N-Nitrosophenylhydroxylamine). Nitric Oxide 2 (1), 66–72. doi:10.1006/niox.1998.0166
Siegel, M. W., Celotta, R. J., Hall, J. L., Levine, J., and Bennett, R. A. (1972). Molecular Photodetachment Spectrometry. I. The Electron Affinity of Nitric Oxide and the Molecular Constants of N${\Mathrm{O}}^{\ensuremath{-}}$. Phys. Rev. A 6(2), 607–631. doi:10.1103/PhysRevA.6.607
Sikora, A., Zielonka, J., Adamus, J., Debski, D., Dybala-Defratyka, A., Michalowski, B., et al. (2013). Reaction between Peroxynitrite and Triphenylphosphonium-Substituted Arylboronic Acid Isomers: Identification of Diagnostic Marker Products and Biological Implications. Chem. Res. Toxicol. 26 (6), 856–867. doi:10.1021/tx300499c
Sikora, A., Zielonka, J., Lopez, M., Dybala-Defratyka, A., Joseph, J., Marcinek, A., et al. (2011). Reaction between Peroxynitrite and Boronates: EPR Spin-Trapping, HPLC Analyses, and Quantum Mechanical Study of the Free Radical Pathway. Chem. Res. Toxicol. 24 (5), 687–697. doi:10.1021/tx100439a
Sikora, A., Zielonka, J., Lopez, M., Joseph, J., and Kalyanaraman, B. (2009). Direct Oxidation of Boronates by Peroxynitrite: Mechanism and Implications in Fluorescence Imaging of Peroxynitrite. Free Radic. Biol. Med. 47 (10), 1401–1407. doi:10.1016/j.freeradbiomed.2009.08.006
Smulik, R., Dębski, D., Zielonka, J., Michałowski, B., Adamus, J., Marcinek, A., et al. (2014). Nitroxyl (HNO) Reacts with Molecular Oxygen and Forms Peroxynitrite at Physiological pH. Biological Implications. J. Biol. Chem. 289 (51), 35570–35581. doi:10.1074/jbc.M114.597740
Smulik-Izydorczyk, R., Dębowska, K., Pięta, J., Michalski, R., Marcinek, A., and Sikora, A. (2018). Fluorescent Probes for the Detection of Nitroxyl (HNO). Free Radic. Biol. Med. 128, 69–83. doi:10.1016/j.freeradbiomed.2018.04.564
Smulik-Izydorczyk, R., Dębowska, K., Rostkowski, M., Adamus, J., Michalski, R., and Sikora, A. (2021). Kinetics of Azanone (HNO) Reactions with Thiols: Effect of pH. Cell Biochem. Biophys. 79 (4), 845–856. doi:10.1007/s12013-021-00986-x
Smulik-Izydorczyk, R., Mesjasz, A., Gerbich, A., Adamus, J., Michalski, R., and Sikora, A. (2017). A Kinetic Study on the Reactivity of Azanone (HNO) toward its Selected Scavengers: Insight into its Chemistry and Detection. Nitric Oxide 69, 61–68. doi:10.1016/j.niox.2017.05.003
Smulik-Izydorczyk, R., Rostkowski, M., Gerbich, A., Jarmoc, D., Adamus, J., Leszczyńska, A., et al. (2019). Decomposition of Piloty's Acid Derivatives - toward the Understanding of Factors Controlling HNO Release. Arch. Biochem. Biophys. 661, 132–144. doi:10.1016/j.abb.2018.11.012
St. Clair, C. S., Gray, H. B., and Valentine, J. S. (1992). Spectroelectrochemistry of Copper-Zinc Superoxide Dismutase. Inorg. Chem. 31 (5), 925–927. doi:10.1021/ic00031a041
Strehlow, H., and Wagner, I. (1982). Flash Photolysis in Aqueous Nitrite Solutions. Z. für Phys. Chem. 132 (2), 151–160. doi:10.1524/zpch.1982.132.2.151
Suarez, S. A., Marti, M. A., De Biase, P. M., Estrin, D. A., Bari, S. E., and Doctorovich, F. (2007). HNO Trapping and Assisted Decomposition of Nitroxyl Donors by Ferric Hemes. Polyhedron 26 (16), 4673–4679. doi:10.1016/j.poly.2007.05.040
Suarez, S. A., Muñoz, M., Alvarez, L., Venâncio, M. F., Rocha, W. R., Bikiel, D. E., et al. (2017). HNO Is Produced by the Reaction of NO with Thiols. J. Am. Chem. Soc. 139 (41), 14483–14487. doi:10.1021/jacs.7b06968
Suarez, S. A., Neuman, N. I., Muñoz, M., Álvarez, L., Bikiel, D. E., Brondino, C. D., et al. (2015). Nitric Oxide Is Reduced to HNO by Proton-Coupled Nucleophilic Attack by Ascorbate, Tyrosine, and Other Alcohols. A New Route to HNO in Biological Media? J. Am. Chem. Soc. 137 (14), 4720–4727. doi:10.1021/ja512343w
Suarez, S. A., Vargas, P., and Doctorovich, F. A. (2021). Updating NO(•)/HNO Interconversion under Physiological Conditions: A Biological Implication Overview. J. Inorg. Biochem. 216, 111333. doi:10.1016/j.jinorgbio.2020.111333
Subedi, H., and Brasch, N. E. (2016). Mechanistic Studies of the Reactions of the Reduced Vitamin B-12 Derivatives with the HNO Donor Piloty's Acid: Further Evidence for Oxidation of cob(I)alamin by (H)NO. Dalton Trans. 45 (1), 352–360. doi:10.1039/C5dt03459k
Subedi, H., and Brasch, N. E. (2015). Studies on the Reaction of Reduced Vitamin B-12 Derivatives with the Nitrosyl Hydride (HNO) Donor Angeli's Salt: HNO Oxidizes the Transition-Metal Center of Cob(I)alamin. Eur. J. Inorg. Chem. 2015, 3825–3834. doi:10.1002/ejic.201500442
Subedi, H., Hassanin, H. A., and Brasch, N. E. (2014). Kinetic and Mechanistic Studies on the Reaction of the Vitamin B-12 Complex Aquacobalamin with the HNO Donor Angeli's Salt: Angeli's Salt and HNO React with Aquacobalamin. Inorg. Chem. 53 (3), 1570–1577. doi:10.1021/ic402613z
Sulc, F., Immoos, C. E., Pervitsky, D., and Farmer, P. J. (2004). Efficient Trapping of HNO by Deoxymyoglobin. J. Am. Chem. Soc. 126 (4), 1096–1101. doi:10.1021/ja0376184
Suzuki, T., Mower, H. F., Friesen, M. D., Gilibert, I., Sawa, T., and Ohshima, H. (2004). Nitration and Nitrosation of N-Acetyl-L-Tryptophan and Tryptophan Residues in Proteins by Various Reactive Nitrogen Species. Free Radic. Biol. Med. 37 (5), 671–681. doi:10.1016/j.freeradbiomed.2004.05.030
Szala, M., Grzelakowska, A., Modrzejewska, J., Siarkiewicz, P., Słowiński, D., Świerczyńska, M., et al. (2020). Characterization of the Reactivity of Luciferin Boronate - A Probe for Inflammatory Oxidants with Improved Stability. Dyes Pigments 183, 108693. doi:10.1016/j.dyepig.2020.108693
Thomson, L., Trujillo, M., Telleri, R., and Radi, R. (1995). Kinetics of Cytochrome C2+ Oxidation by Peroxynitrite: Implications for Superoxide Measurements in Nitric Oxide-Producing Biological Systems. Arch. Biochem. Biophys. 319 (2), 491–497. doi:10.1006/abbi.1995.1321
Travers, M. J., Cowles, D. C., and Ellison, G. B. (1989). Reinvestigation of the Electron Affinities of O2 and NO. Chem. Phys. Lett. 164 (5), 449–455. doi:10.1016/0009-2614(89)85237-6
Treinin, A., and Hayon, E. (1970). Absorption Spectra and Reaction Kinetics of NO2, N2O3, and N2O4 in Aqueous Solution. J. Am. Chem. Soc. 92 (20), 5821–5828. doi:10.1021/ja00723a001
Vásquez, M. A. M., Pellegrino, J., Álvarez, L., Neuman, N. I., Doctorovich, F., and Martí, M. A. (2017). “9 - Interactions of HNO with Metallated Porphyrins, Corroles, and Corrines,” in The Chemistry and Biology of Nitroxyl (HNO). Editors F. Doctorovich, P. J. Farmer, and M. A. Marti (Boston: Elsevier), 193–205.
Venâncio, M. F., Doctorovich, F., and Rocha, W. R. (2017). Solvation and Proton-Coupled Electron Transfer Reduction Potential of (2)NO(•) to (1)HNO in Aqueous Solution: A Theoretical Investigation. J. Phys. Chem. B 121 (27), 6618–6625. doi:10.1021/acs.jpcb.7b03552
Wang, H., and Xian, M. (2008). Fast Reductive Ligation of S-Nitrosothiols. Angew. Chem. Int. Ed. Engl. 47 (35), 6598–6601. doi:10.1002/anie.200801654
Wang, R. (2012). Shared Signaling Pathways Among Gasotransmitters. Proc. Natl. Acad. Sci. U. S. A. 109 (23), 8801–8802. doi:10.1073/pnas.1206646109
Wink, D. A., Feelisch, M., Fukuto, J., Chistodoulou, D., Jourd'heuil, D., Grisham, M. B., et al. (1998). The Cytotoxicity of Nitroxyl: Possible Implications for the Pathophysiological Role of NO. Arch. Biochem. Biophys. 351 (1), 66–74. doi:10.1006/abbi.1997.0565
Woldman, Y., Khramtsov, V. V., Grigor'ev, I. A., Kiriljuk, I. A., and Utepbergenov, D. I. (1994). Spin Trapping of Nitric Oxide by Nitronylnitroxides: Measurement of the Activity of No Synthase from Rat Cerebellum. Biochem. Biophys. Res. Commun. 202 (1), 195–203. doi:10.1006/bbrc.1994.1912
Wong, P. S., Hyun, J., Fukuto, J. M., Shirota, F. N., DeMaster, E. G., Shoeman, D. W., et al. (1998). Reaction between S-Nitrosothiols and Thiols: Generation of Nitroxyl (HNO) and Subsequent Chemistry. Biochemistry 37 (16), 5362–5371. doi:10.1021/bi973153g
Woodward, T., and Sutton, H. (1966). Radiolysis of Aqueous Solutions Containing Nitric Oxide and Aliphatic Alcohols. Trans. Faraday Soc. 62, 70–80. doi:10.1039/tf9666200070
Yagil, G., and Anbar, M. (1964). The Formation of Peroxynitrite by Oxidation of Chloramine, Hydroxylamine and Nitrohydroxamate. J. Inorg. Nucl. Chem. 26 (3), 453–460. doi:10.1016/0022-1902(64)90010-7
Zapata, A. L., Kumar, M. R., and Farmer, P. J. (2017). “15 - Global Kinetic Analysis and Singular Value Decomposition Methods Applied to Complex Multicomponent Reactions of HNO,” in The Chemistry and Biology of Nitroxyl (HNO). Editors F. Doctorovich, P. J. Farmer, and M. A. Marti (Boston: Elsevier), 287–303. doi:10.1016/b978-0-12-800934-5.00015-3
Zapata, A. L., Kumar, M. R., Pervitsky, D., and Farmer, P. J. (2013). A Singular Value Decomposition Approach for Kinetic Analysis of Reactions of HNO with Myoglobin. J. Inorg. Biochem. 118, 171–178. doi:10.1016/j.jinorgbio.2012.10.005
Zarenkiewicz, J., Khodade, V. S., and Toscano, J. P. (2021). Reaction of Nitroxyl (HNO) with Hydrogen Sulfide and Hydropersulfides. J. Org. Chem. 86 (1), 868–877. doi:10.1021/acs.joc.0c02412
Zeller, A., Wenzl, M. V., Beretta, M., Stessel, H., Russwurm, M., Koesling, D., et al. (2009). Mechanisms Underlying Activation of Soluble Guanylate Cyclase by the Nitroxyl Donor Angeli's Salt. Mol. Pharmacol. 76 (5), 1115–1122. doi:10.1124/mol.109.059915
Zhang, J., Wang, H., and Xian, M. (2009). An Unexpected Bis-Ligation of S-Nitrosothiols. J. Am. Chem. Soc. 131 (11), 3854–3855. doi:10.1021/ja900370y
Zhang, Y. (2013). Computational Investigations of HNO in Biology. J. Inorg. Biochem. 118, 191–200. doi:10.1016/j.jinorgbio.2012.09.023
Zielonka, J., Podsiadły, R., Zielonka, M., Hardy, M., and Kalyanaraman, B. (2016a). On the Use of Peroxy-Caged Luciferin (PCL-1) Probe for Bioluminescent Detection of Inflammatory Oxidants In Vitro and In Vivo - Identification of Reaction Intermediates and Oxidant-specific Minor Products. Free Radic. Biol. Med. 99, 32–42. doi:10.1016/j.freeradbiomed.2016.07.023
Zielonka, J., Sikora, A., Adamus, J., and Kalyanaraman, B. (2015). Detection and Differentiation between Peroxynitrite and Hydroperoxides Using Mitochondria-Targeted Arylboronic Acid. Methods Mol. Biol. 1264, 171–181. doi:10.1007/978-1-4939-2257-4_16
Zielonka, J., Sikora, A., Joseph, J., and Kalyanaraman, B. (2010). Peroxynitrite Is the Major Species Formed from Different Flux Ratios of Co-generated Nitric Oxide and Superoxide: Direct Reaction with Boronate-Based Fluorescent Probe. J. Biol. Chem. 285 (19), 14210–14216. doi:10.1074/jbc.M110.110080
Zielonka, J., Sikora, A., Podsiadly, R., Hardy, M., and Kalyanaraman, B. (2021). Identification of Peroxynitrite by Profiling Oxidation and Nitration Products from Mitochondria-Targeted Arylboronic Acid. Methods Mol. Biol. 2275, 315–327. doi:10.1007/978-1-0716-1262-0_20
Zielonka, J., Zielonka, M., Sikora, A., Adamus, J., Joseph, J., Hardy, M., et al. (2012). Global Profiling of Reactive Oxygen and Nitrogen Species in Biological Systems: High-Throughput Real-Time Analyses. J. Biol. Chem. 287 (5), 2984–2995. doi:10.1074/jbc.M111.309062
Keywords: azanone, nitroxyl, HNO, chemical kinetics, reaction mechanism
Citation: Michalski R, Smulik-Izydorczyk R, Pięta J, Rola M, Artelska A, Pierzchała K, Zielonka J, Kalyanaraman B and Sikora AB (2022) The Chemistry of HNO: Mechanisms and Reaction Kinetics. Front. Chem. 10:930657. doi: 10.3389/fchem.2022.930657
Received: 28 April 2022; Accepted: 06 June 2022;
Published: 05 July 2022.
Edited by:
Loredana Serpe, University of Turin, ItalyReviewed by:
Gizem Keceli, Johns Hopkins Medicine, United StatesMateus Venancio, Federal University of Bahia, Brazil
Copyright © 2022 Michalski, Smulik-Izydorczyk, Pięta, Rola, Artelska, Pierzchała, Zielonka, Kalyanaraman and Sikora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adam Bartłomiej Sikora, YWRhbS5zaWtvcmFAcC5sb2R6LnBs