 Yen-Yu Tseng1Jyh-Ming Liou2Wei-Chieh Cheng1Jing-Ting Hsu2Tsui-Ling Hsu1Ming-Shiang Wu2*Chi-Huey Wong1,3*
Yen-Yu Tseng1Jyh-Ming Liou2Wei-Chieh Cheng1Jing-Ting Hsu2Tsui-Ling Hsu1Ming-Shiang Wu2*Chi-Huey Wong1,3*- 1Genomics Research Center, Academia Sinica, Taipei, Taiwan
- 2Division of Gastroenterology, Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan
- 3Department of Chemistry, The Scripps Research Institute, La Jolla, CA, United States
Current treatment of Helicobacter pylori involves a triple therapy comprising one proton pump inhibitor and two other antibiotics; however, the outcomes are limited due to the existence of antibiotic resistant strains. We previously reported that moenomycin A, a cell-wall transglycosylase inhibitor, is highly active against multidrug-resistant Helicobacter pylori. Herein we show that combination of moenomycin A with the protein synthesis inhibitor clarithromycin or metronidazole can synergistically achieve almost 95% eradication of multidrug-resistant Helicobacter pylori. We also found that the moenomycin A-non-susceptible strains of Helicobacter pylori with deletion of transglycosylase exhibit moenomycin A hyposensitivity, faster growth and impaired biofilm formation compared to the parental strain. Overall, the combination of moenomycin A and clarithromycin or metronidazole to achieve a synergistic effect on different targets is a promising treatment for multidrug-resistant Helicobacter pylori.
Introduction
Over half of the world’s population are infected with Helicobacter pylori (H. pylori), a microaerophilic Gram-negative bacterium. Separated from gastritis and peptic ulcer (Marshall et al., 1998), this bacterial infection was found to be the key risk factor of gastric cancer (Malfertheiner et al., 2012). Generally, the treatment of H. pylori infection involves a 1–2-week use of antibiotics including the combination of a proton pump inhibitor with two to three antibiotics such as clarithromycin, amoxicillin, metronidazole/tinidazole, tetracycline or levofloxacin to reduce acid production in the stomach (McColl, 2010; Malfertheiner et al., 2012; Liou et al., 2013a). However, about 20% of patients with H. pylori infection develop antibiotic resistance (Liou et al., 2013b; Megraud et al., 2013).
Moenomycin (Moe), known as flavomycin or bambermycin for fodder, is a mixture compounds (Moenomycins A, A12, C1, C3, and C4) which are separated from Streptomyces species (Wallhausser et al., 1965; Huber and Moenomycin, 1967; Huber and Nesemann, 1968). It was issued in the early 1960s as animal feedstock and was claimed to inhibit the transglycosylase (TGase) in Gram-positive bacteria to block the biosynthesis of peptidoglycan that forms bacterial cell wall (van Heijenoort et al., 1987; Chen et al., 2003; Halliday et al., 2006). Bacterial cell wall is mainly comprised of peptidoglycan which formed through two key reactions, transglycosylation and transpeptidation catalyzed respectively by the enzymes transglycosylase and transpeptidase which exist as separate domains of the penicillin binding protein (PBP). TGase catalyzes the transfer of the glycosyl moiety from a glycosyl donor to a hydroxyl group of lipid-II (van Heijenoort, 2007). TGase is an important antibiotic target since it is essential for bacteria and the polysaccharide backbone of the peptidoglycan is conserved in drug-resistant strains; therefore, antibiotics targeting the transglycosylation step may be difficult to develop resistance. On the other hand, the transpeptidase is extensively used for development of antibiotics such as β-lactam antibiotics and vancomycin. Due to the overuse or abuse of antibiotics that target the transpeptidase, some resistant strains have emerged.
There were reports on multi-drug resistant (MDR) H. pylori strains isolated from different gastrointestinal patients, but how these strains develop Moe A resistance was unknown (Orhan et al., 2005; Cheng et al., 2008; Salzberg et al., 2011; Tseng et al., 2014; Yuriy et al., 2014). So far, Moe A has been only used as animal feedstock and has never been developed for human use, probably due to its poor absorption. However, because of its extremely low absorption in the gastrointestinal tract with negligible toxicity, Moe A appears to be a suitable antibiotic for the treatment of patients infected with MDR H. pylori strains which are often found on the lining of the stomach. Based on our previous study in vitro (Tseng et al., 2014) and in vivo in animal models (data not shown), we found that MoeA has beneficial effect on multidrug-resistant H. pylori eradication in vitro but not in vivo. Hence, we tried to improve the therapeutic efficacy by combination therapy and we did see the synergistic effect in vitro. The in vitro method of detecting synergy is simple to manipulate, especially with Time-Kill, Checkerboard, and E test (White et al., 1996). Time-kill and Checkerboard methods are the most widely used techniques to analyze synergy. However, the Time-kill method of synergy test need to manipulate with the broth under microaerobic condition. Therefore, we selected convenient operation of Checkerboard methods with the agar under microaerobic condition to assess MoeA combination therapy for H. pylori eradication. Furthermore, using surface plasmon resonance analysis to compare the binding of moenomycin A with various truncated PBPs (Cheng et al., 2008), we previously found that the transmembrane domain of PBP is vital for moenomycin binding, particularly in the case of Gram-negative H. pylori. Hence, the purpose of this study was to study the efficacy of Moe A combination therapy on MDR H. pylori isolated from patients with gastrointestinal disease and to understand the role of PBP in Moe A-non-susceptible H. pylori strains.
Materials and methods
Chemicals
Moenomycin (>90%) was purified from FLAVOMYCIN 20 (Huvepharma, Antwerp, Belgium). Clarithromycin was purchased from K-mycin (Standard Chem and Pharm CO. Tainan, Taiwan) and metronidazole were obtained from FLAGYL (SHIONOGI and CO. Taipei, Taiwan).
Patients and isolation of H. pylori
H. pylori strains were obtained from the stomach of patients infected by H. pylori who were treatment-naïve or treated with eradication therapies at National Taiwan University Hospital (NTUH), Taipei, Taiwan. Patients with a history of partial gastric resection were excluded. The study protocol was authenticated by the Institutional Review Boards of NTUH (Becton Dickinson and Co., Rutherford, New Jersey, United States). Bacterial plates were incubated at 37°C under microaerophilic conditions of 5% O2, 10% CO2 and 85% N2 for 4 days. All strain cultures were stored at −80°C until the need for investigation.
Construction of the penicillin binding protein 1a knockout mutant
The gene encoding PBP1a with the upstream and downstream about 500 bp was amplified with the gDNA of the resistant strain by polymerase chain reaction (PCR). The primers of PBP1a KO were designed by Primer3Plus software: Upstream region (forward) CTCGAGTTCTTTCTTGCAAGATGTGCC; Upstream region (reverse) GTATATCCAGTGATTTTTTTCTCCATCTACATGGCTATAGGGACTTTAACA; Downstream region (forward) GGGCGGGGCGTAAATGTTTTCTAAATCTTTAGAAGCCCT; Downstream region (reverse) GGTACCTTTTAGGGGCGTTAAAGAGTTT. The PBP1a genes were truncated by multiplex PCR, and a chloramphenicol-resistant cassette about 660-bp codon was then cloned to replace the full length PBP1a gene. Furthermore, the DNA fragment was cloned into the pGEM-T easy vector (Promega, Madison, Wisconsin, United States). A chloramphenicol-resistant cassette with the upstream and downstream about 500 bp flanking region of PBP1a was cut off and ligated into the XhoI (New England Biolabs, Ipswich, Massachusett, United States) site and PknI (New England Biolabs, Ipswich, Massachusett, United States) of the shuttle vector pHel3 (plasmid pHel3 was a presentation from Rainer Haas, Universität München, Germany). This plasmid was electrotransformed into the resistant strain to generate truncating mutants. The DNA of the transformants was verified by PCR with external and internal primers and validated by DNA sequencing.
Electrotransformation of H. pylori
H. pylori cells were transformed with plasmids (pHel3-PBP1a-Cmr) by electroporation. Briefly, the H. pylori culture on plates was scraped and suspended with ice-cold ultrapure water. The cells were collected by centrifugation, and the pellet was washed using ice-cold 10% glycerol (Sigma-Aldrich, Saint Louis, Missouri, United States) twice and resuspended with 10% glycerol. Plasmid DNA was mixed with cell suspension. The mixture was poured into a cooled electroporation cuvette (Bio-Rad, Hercules, California), and shocked with a single-pulse electroporation (2.5 kV, 25 mF, 200 Ω). The sample was incubated on serum plate for 24 h at 37°C. Finally, the bacteria were inoculated onto chloramphenicol selective media for 4 days.
Checkerboard test
From the stock solutions, a serial dilution of each antibiotic was separated into each agar plate to obtain different concentrations of 0.125, 0.25, 0.5, 1, 2, 4, 8, 16, and 32 μg/ml for checkerboard analysis. The agar plates with one antibiotic and its combinations were placed by different concentrations. Thus, each of the agar plates presented a unique combination of concentrations of the two antibiotics. The plates were incubated for 3 days at 37°C. The fractional inhibitory concentration index (FIC I) was used to calculate the results. TheΣFractional inhibitory concentration (ΣFIC) values were counted as follows: ΣFIC = the fractional inhibitory concentration A (FIC A) + FIC B, where FIC A is the minimum inhibitory concentration (MIC) of antibiotic A in the combination/MIC of antibiotic A alone, and FIC B is the MIC of antibiotic B in the combination/MIC of antibiotic B alone. The combined effect is synergistic when ΣFIC I is ≤ 0.5. Additions was expressed by a FIC I from >0.5 to ≤1 and indifferent FIC I was >1 to ≤4 while antagonism is indicated as when the ΣFIC I is > 4 (Eliopoulos and Moellering, 1991; Salzberg et al., 2011).
Biofilm quantification
Bacteria were cultured based on the growth curves; however, biofilm ring assays were manipulated by 24-well tissue culture plates. Bacterial cultures were used to inoculate in liquid medium with each well to the optical density (OD) at 600 nm (OD600) of 0.1 optical density unit (ODU). Each well was sucked dry and washed two times with phosphate-buffered saline (PBS, Sigma-Aldrich, Saint Louis, Missouri, United States). Plates were dried for 5 min at 37°C, and then biofilm rings were fixed with methanol. Gram’s crystal violet (Sigma-Aldrich, Saint Louis, Missouri, United States) solution was added to each well. The plates were incubated, crystal violet was removed, and each well was washed three times with ultrapure water. The plates were desiccated. Then, differentiation solution was used to solubilize the dye. Plates were incubated with differentiation solution, solubilized crystal violet solution was transferred to a cuvette, and the absorbance at 590 nm (OD590) was read. The data was shown with three biologically independent experiments.
Scanning electron microscopy
Stock cultures were cultured in fresh Brain Heart Infusion Broth (BHI broth; Becton Dickinson and Co., Rutherford, New Jersey, United States) in plates. Biofilms were captured on coverslips and analyzed by scanning electron microscopy. Briefly, samples were fixed. After fixation, samples were washed three times with 0.05 M sodium cacodylate buffer. The samples were mounted onto carbon stubs, vacuumed sputter-coating with gold, observed with SEM. A Hitachi S-2400 N SEM (Hitachi High-Technologyies, Tokyo, Japan) was used to acquire all images.
Growth inhibition
Cultures of parental and knockout strains were diluted with fresh broth media. Moe A was added at 2-fold serial dilution from the highest concentration (0–16 µg/mL) directly to the culture tubes and the turbidity was monitored 4 days later by reading OD600.
Motility assay
Cultures of parental and knockout strains were diluted with fresh broth media, followed by incubation for 1–5 days and the turbidity was recorded by OD600.
Detection of PBPs from membrane
To detect PBPs with BOCILLIN FL (Thermo Fisher Scientific, Waltham, Massachusett, United States) labeling reagent, H. pylori cells were cultured on plates. The bacteria were collected by centrifugation, then washed with PBS, resuspended in the same buffer, and disrupted by sonication. The cell lysates were centrifuged. The supernatant fractions were harvested and centrifuged. The pellets were harvested, washed, and resuspended. The suspensions were utilized for membrane preparations and fluorescent binding assays with BOCILLIN FL. The protein concentrations on the membrane were quantified by using the Bradford protein assay (Bio-Rad Laboratories Hercules, California, United States). For detection of PBPs, each membrane preparation (≈300 μg of protein) was subjected to SDS-PAGE (Thermo Fisher Scientific, Waltham, Massachusett, United States) and proteins were blotted onto PVDF membrane (Merck Millipore Burlington, Massachusetts, United States). Non-specific binding was blocked in 5% blocking buffer for 60 min, and membranes were incubated with 10 μM BOCILLIN FL at 37°C for 60 min. To visualize the labeled PBPs, the PVDF was visualized under UV light (290 nm).
Results
Our previous report indicated that Moe A was effective against MDR H. pylori strains in vitro but the in vivo effect in animal models was not as significant as the in vitro effect. To further evaluate the efficacy of Moe A combination therapy, the effect of Moe A in combination with clarithromycin (CLR) was evaluated against MDR strains to determine the susceptibility. Table 1 shows the best combination of Moe A and CLR with FIC I. Synergism was found in 21 of 31 strains, additive effect was found in 9 of 31 strains and indifferent effect was displayed in 1 of 31 strains (FIC I = 1.063). Antagonism was not observed. The MICs of CLR for MDR strains decreased when tested in combination with Moe A with concentrations ranging from 0.001 to 11.167 μg/ml. Particularly, the best FIC I was observed for No. Three strain with MIC of 0.019 μg/ml, corresponding to a 66- and a 266-fold reduction of MIC for Moe A and CLR, respectively. In addition, the effect of Moe A in combination with metronidazole (MTZ) was evaluated against MDR strains to determine the susceptibility (Table 2). Table 2 shows the best combinations of Moe A and MTZ with FIC I. Synergism was observed in 21 of 32 strains, additive effect was observed in 10 of 32 strains and indifferent effect was displayed in 1 of 32 strains (FIC I = 1.063). Antagonism was not observed. The MICs of MTZ and Moe A combination against MDR strains were lower than the MICs of Moe A with concentrations ranging from 1.25 to 21.333 μg/ml. Especially, the best FIC I was observed for No. Two strain with MIC of 0.229 μg/ml, corresponding to a seven- and a 14-fold reduction of MIC for Moe A and MTZ, respectively. In addition, Figure 1 shows the checkerboard assays and the isobolograms obtained from different concentrations of Moe A and CLR against No. Two strain, the (MTZ + CLR)-resistant strain or from different concentrations of Moe A and MTZ against No. Three strain, the (MTZ + CLR)-resistant strain.
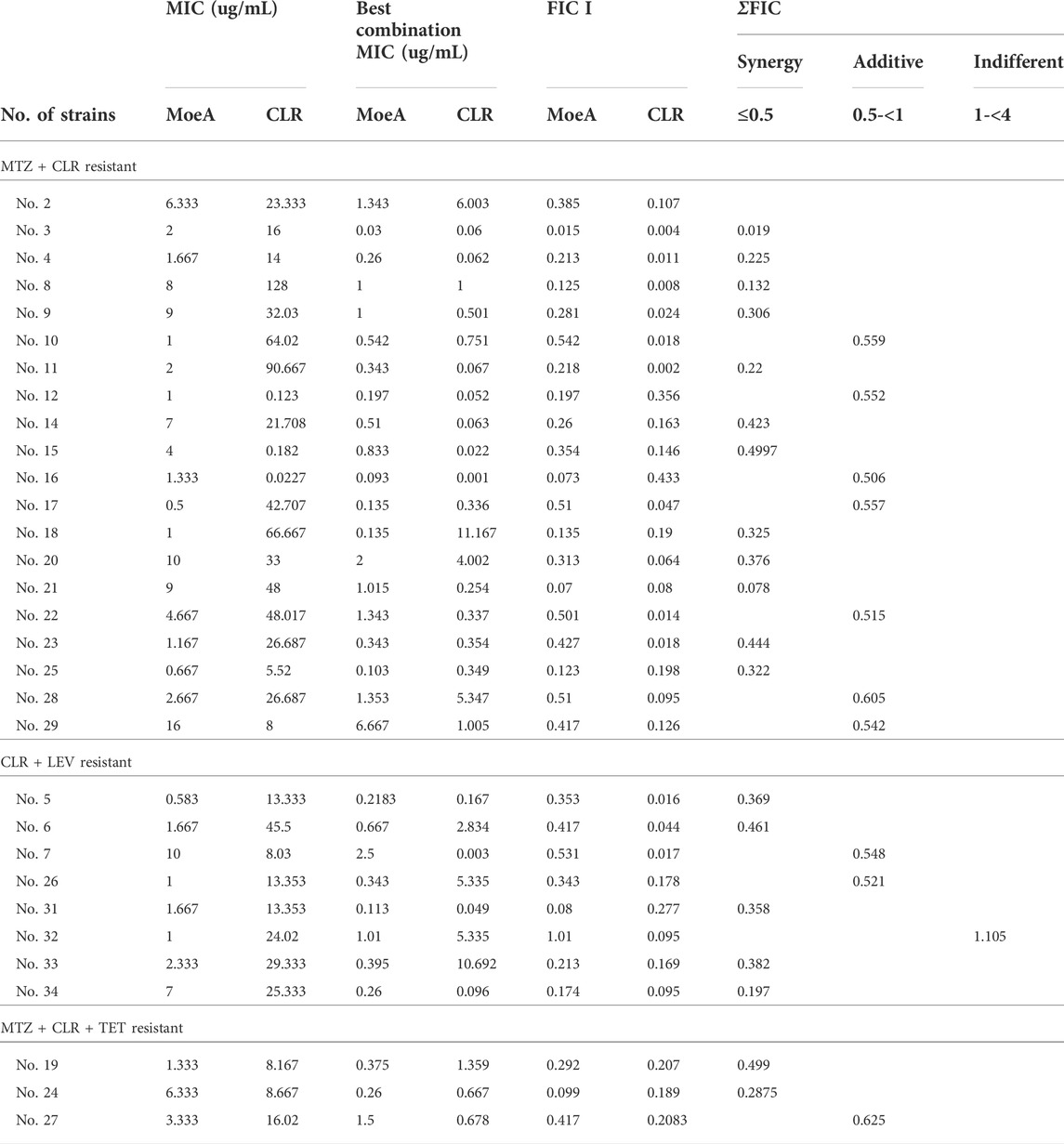
TABLE 1. FIC I of MoeA and CLR against multidrug resistant H. pylori strains.
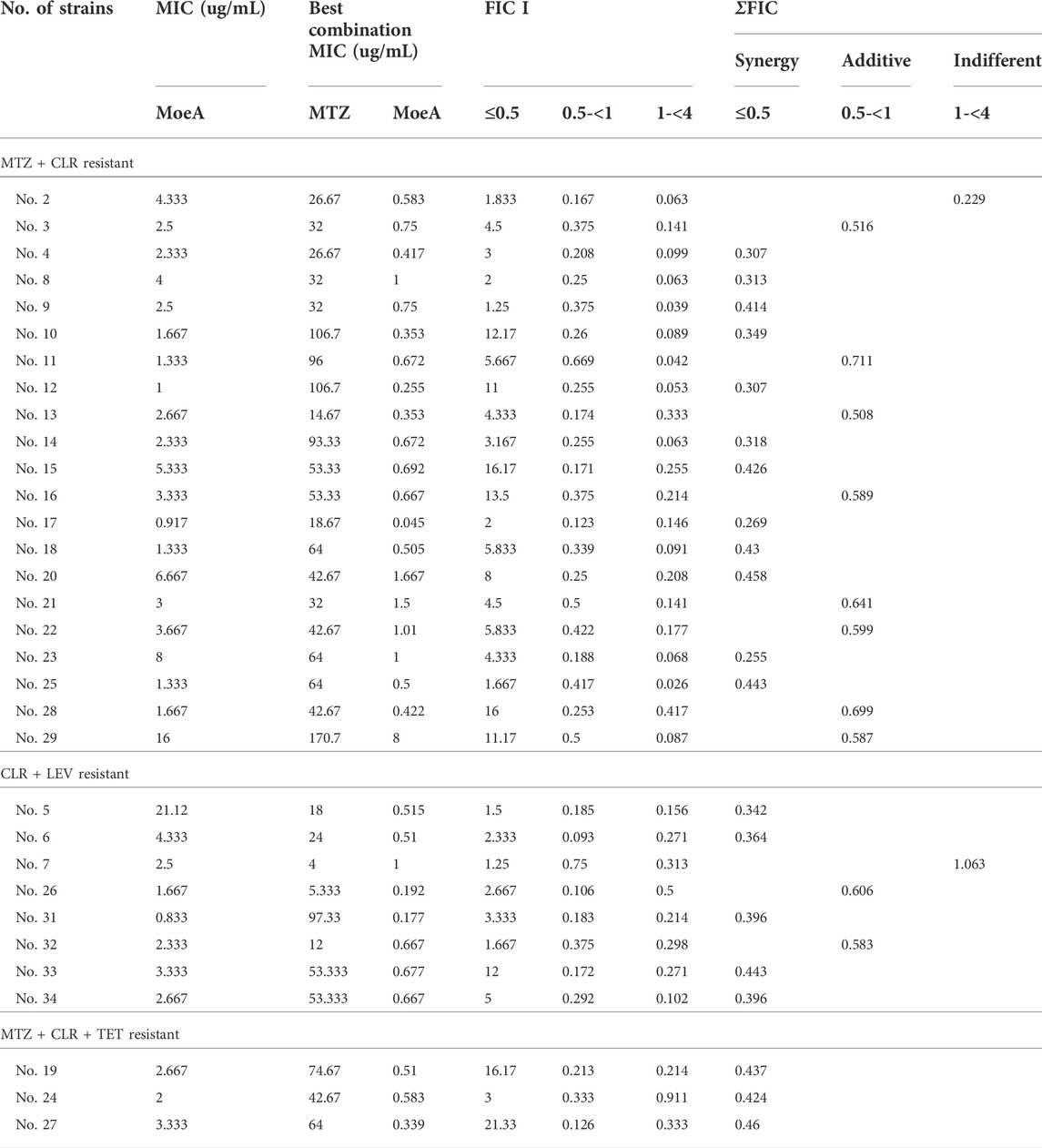
TABLE 2. FIC I of MoeA and MTZ against multidrug resistant H. pylori strains.
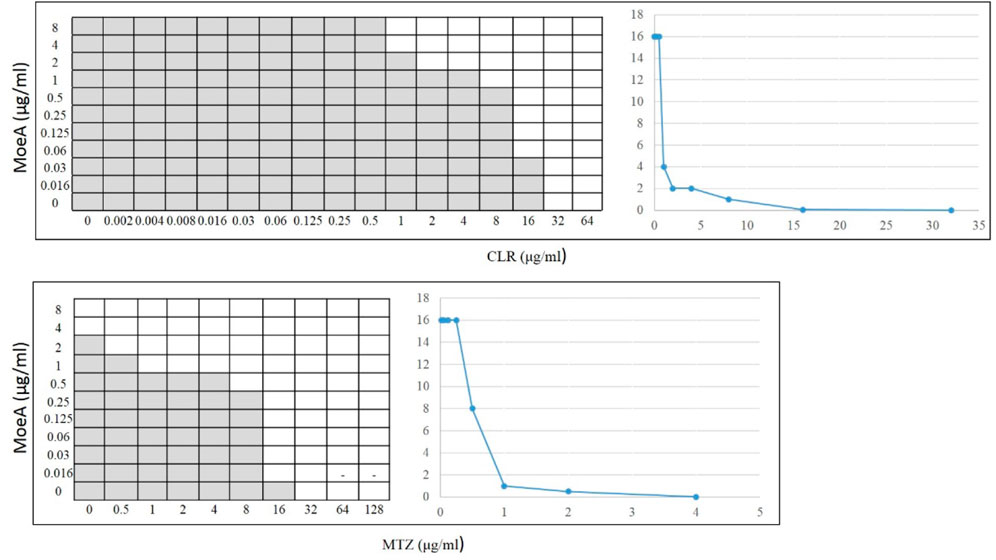
FIGURE 1. Checkerboard assays and isobolograms for assessment of synergism in the (MoeA + CLR) combination against No. 2 H. pylori strain or the (MoeA + MTZ) combination against No. 3 H. pylori strain. The interpretation of the best (MoeA + CLR) or (MoeA + MTZ) combination was based on synergism (FIC I ≤ 0.5), antagonism (FIC I ≥ 4.0), and additive effect (FIC I > 0.5–4.0). The grey zone represents bacterial viability, and the white zone represents bacterial mortality in the presence of (Moe A+ CLR) or (Moe A+ MTZ). On the right, the isobolograms showed the synergistic curve. The x axis of the isobologram is the dose of CLR or MTZ and the y axis is the dose of Moe A.
At present, the mechanism of Moe A resistance to H. pylori is not completely understood. Two Moe A-resistant H. pylori strains were isolated from patients of NTUH. The strains showed a high degree of similarity with ATCC43504 through PBP1a detection and identification. To clarify if PBP1a in these strains interacts with Moe A, PBP1a deletion mutants were constructed and confirmed with sequencing and functional assays as below and Cmr colonies were isolated (Figure 2A). The DNA was amplified with the primers P7/S1 and S2/P8 annealing to PBP1a of the parental strain to produce a 1,269 bp and a 1,953 bp sequence, separately (Figure 2B). A 660 bp product was amplified and sequenced from truncated strains. The results showed upstream and downstream genes of PBP1a as well as cmr gene instead of PBP1a, indicating success of the assembly (data not shown). To assess whether the PBP1a gene was involved in Moe A resistance, the PBP1a of Moe A-non-susceptible strain was knocked out. As shown in Figure 3, the PBP1a deletion strain grew in media containing 8 µg/ml of Moe A (MIC = 2 µg/ml). These results demonstrate that the genetic alteration of PBP1a gene may cause Moe A resistance. Because the parental strain showed a high-level Moe A resistance (MIC = 8 µg/ml) with genetic alteration in the PBP1a gene. This finding raised the question that the high-level Moe A resistance might be caused by PBP1a inactivation.
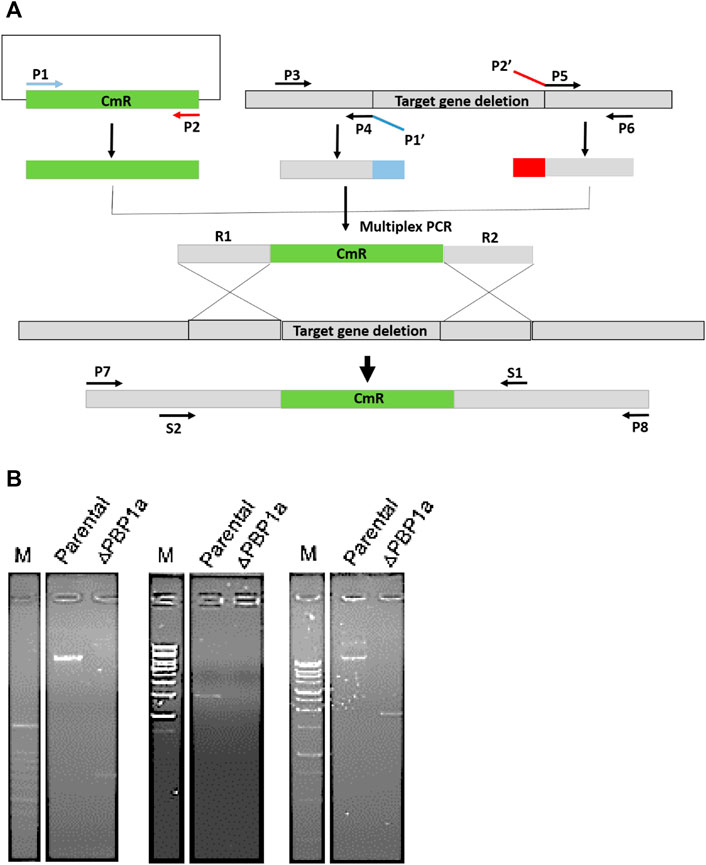
FIGURE 2. (A) The flowchart of gene truncation in H. pylori. The fragments of upstream and downstream were amplified with primers P3 and P4 and with P5 and P6, respectively. The CmR selective marker fragments was amplified with primers P1 and P2. Primers P1′ and P2′ were devised for generic linkers (shown in blue and red). The fragments of upstream and downstream were linked to the CmR fragments by using nested primers (S1 and S2). (B) Long range PCR analysis of genomic DNA from parental and ΔPBP1a before and after pHeL3 transfection was performed to confirm the integration (660 bp), the upstream flank (1,269 bp) and the downstream flank (1,953 bp), respectively.
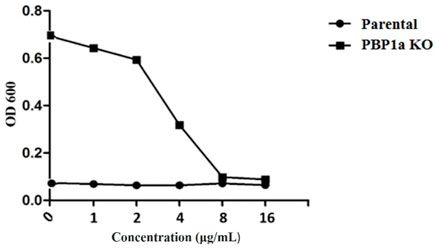
FIGURE 3. Comparison of sensitivity between parental and PBP1a KO strains for serial dilution antibiotic susceptibility testing. MIC of the parental -type and PBP1a KO strain in response to 0, 1, 2, 4, 8, 16 μg/ml MoeA. The results of at least three independent experiments are calculated.
In many bacteria, PBP contributes to biofilm forming. We evaluated the capability of H. pylori to form biofilm rings with an Improved Crystal Violet Assay, performed in 24-well polystyrene microtiter plates with measurements at 72 h. We observed that the Moe A-non-susceptible PBP1a deletion strain formed the weak biofilm ring (Figures 4A,B). In contrast, the parental strain formed moderate to the strong biofilm ring. Besides, we also applied SEM to gain more insight into the biofilm structure. We could observe mighty biofilm formation and countless adherent bacteria that contained mainly coccoid forms along with some bacilli. However, biofilm and adherent bacteria were less monitored for the PBP1a deletion strain (Figure 5). Interestingly, Rod-shaped cells were mostly visible in the PBP1a deletion strain. Despite these differences, the SEM data suggest that PBP1a is sophisticatedly involved in the biofilm formation of H. pylori.
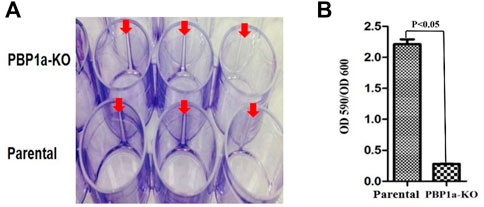
FIGURE 4. Effect of PBP1a KO on biofilm formation in a-non-susceptible clinical H. pylori strain. (A), Qualitative biofilm ring (arrow) in 24-well Corning Costar cell culture plates after crystal violet staining. Row A and B represent PBP1a KO strain and the parental strain respectively. (B), Quantitative biofilm formation in PBP1a KO strain and the parental strain. Standard deviation of optical densities (OD 590/OD 590) was from three independent experiments. Statistically significant difference (p < 0.05) was calculated by the t-test.

FIGURE 5. Effect of PBP1a deletion on the biofilm formation of a non-susceptible clinical H. pylori strain under SEM. Scanning electron micrographs were taken to show the structural differences in biofilm architecture of parental strain vs. PBP1a deletion strain. Bacteria were grown on cell culture-treated coverslips for 24 h (scale bars, 10 µm). SEM micrograph of the biofilm formation of parental strain (A) at magnification 3.5 k×, voltage 15.0 kV, working distance 4.4 mm, with 10 μm scale bar. (B) At magnification 3.7.0 k×, voltage 15.0 kV, working distance 4.4 mm, with 10 μm scale bar. (C) SEM micrograph of the biofilm formation of PBP1a KO strain at magnification 3.5 k×, voltage 15.0 kV, working distance 5.4 mm, with 10 μm scale bar. (D) At magnification 3.7 k×, voltage 15.0 kV, working distance 5.4 mm, with 10 μm scale.
The growth rate of parental strain was then analyzed by turbidimetry and compared to the PBP1a deletion strain. (Figure 6A), and it was shown that the PBP1a deletion strain exhibited a faster growth rate than the parental strain. The bacterial mass of the PBP1a deletion strain increased exponentially from day 2 to day 5 but the bacterial mass of Moe A-non-susceptible strains remained in a steady state until day 4. The differences in growth between the PBP1a deletion strain and the parental strain could be interpreted as a direct consequence of PBP1a deletion or mutation in elevating the MICs of Moe A. To determine whether other PBPs were involved in the growth rate, the PBP of parental strain was analyzed by BOCILLIN FL and compared to the PBP1a deletion strain. (Figure 6B). Two PBPs were identified in H. pylori. We refer to the PBP with 55–70 kDa as PBP 1a and the PBP with 32 kDa as PBP 4. The parental strain exhibited a significant loss of PBP four signal compared to the PBP1a deletion strain. The PBP4 difference between the deletion of PBP1a could result in the differences in growth.
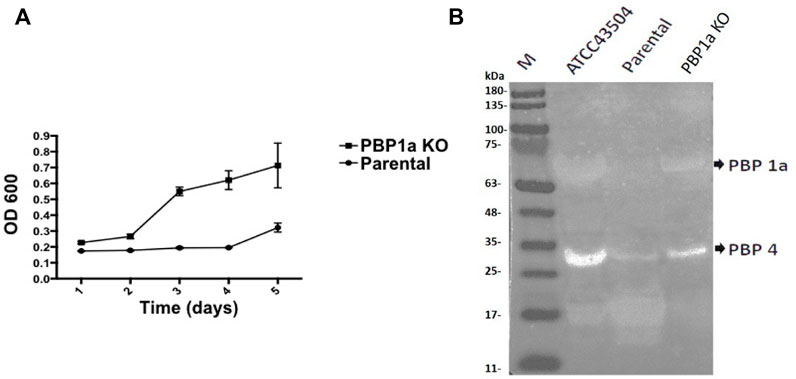
FIGURE 6. (A) Effect of PBP1a deletion on the growth rate of a non-susceptible clinical H. pylori strain. Growth curves of a non-susceptible clinical H. pylori strain (circles), and its PBP1a KO strain (squares). Each data point represents the mean and standard deviation (bars) of OD from triplicate wells. (B) PBP profiling of H. pylori. Extracted membrane labeled BOCILLIN FL were visualized under UV light (290 nm); lane 1, H. pylori ATCC 43504; lane 2, a non-susceptible clinical H. pylori strain; lane 3, PBP1a KO of the non-susceptible clinical H. pylori strain.
Discussion
Our findings in this study support the proposition that combination of Moe A with one of other antibiotics at present utilized for the treatment of H. pylori infection may eradicate H. pylori and its MDR strains. The eradication efficacy using the combination of Moe A and CLR was like the combination of Moe A and MTZ. Treatment of (MTZ + CLR)-resistant and (CLR + LEV)-resistant strains with Moe A in combination with CLR was shown to have more than 64% synergistic effect and 32% additive effect from the two antibiotics, and the combination effect of Moe A and CLR on (MTZ + CLR + TET)-resistant strains was 100% synergistic. If there is indeed a greater benefit for the Moe A combination therapy in these MDR strains, the synergistic effect is the mode of action (Caesar and Cech, 2019). Besides, the combination of Moe A and MTZ against the (MTZ + CLR)-resistant or the (CLR + LEV)-resistant strains was shown to have more than 62% synergistic effect and 34% additive effect. However, the Moe A and MTZ combination therapy against the (MTZ + CLR + TET)-resistant strains was also found to have 100% additive effect. Since CLR and TET are both protein synthesis inhibitors, the additive effect perhaps was more significant than the synergistic effect for the (MTZ + CLR + TET)-resistant strains (Gasparetto et al., 2012; Bollenbach, 2015).
Based on our previous data (Caesar and Cech, 2019), the incidence rate of Moe A-resistance to one, two, three, four, and five drugs were 16.7%, 27.3%, 29.4%, 20%, and 50%, respectively. Previous reports suggest that combination of antibiotics for the treatment of MDR H. pylori strains may be beneficial if the efficacy is ≥ 90% (Bergamaschi et al., 2007). Indeed, the treatment of H. pylori resistant to (MTZ + CLR), or (CLR + LEV), or (MTZ + CLR + TET) with (Moe A+ MTZ) or (Moe A+ CLR) was found to increase the effect of eradication (almost 90% vs. 27.3%) in this study. In our combination study, Moe A may disrupt the synthesis of peptidoglycan and affect cell-wall permeability, thus allowing CLR or MTZ to reach ribosomes or nucleus more easily and increasing their antibacterial activity by synergistic or additive effect (Figure 7).
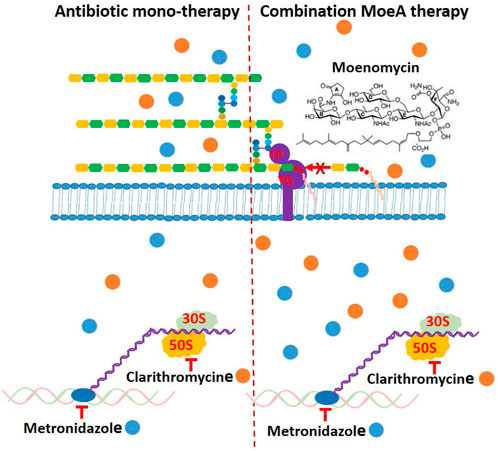
FIGURE 7. Schematic presentation of the mechanism of antibiotic monotherapy or combination Moe A therapy and possible binding of cytosolic proteins and genes, which might be a reason for enhancing antimicrobial activity.
Since PBP1a is a target for anti-bacterial drug design, we also investigated in this study the impact of PBP1a deletions on phenotypes, including susceptibility, cell morphology and biofilm formation. We sequenced the PBP1a of clinical strains and showed a high degree of similarity with ATCC 43504, and we reported for the first-time construction, verification, and characterization of PBP1a deletion strain using a pHel3 shuttle vector and found that deletion of PBP 1a had a high impact on bacterial growth, making cells to maintain in the rod forms. We believe this finding is significant and further research are needed to understand this interesting observation. The impaired biofilm formation with PBP1a deletion is consistent with the observation of PBP mutants in E. coli (Kumar et al., 2012), which exhibited significantly abated biofilm formation and motility. Biofilms are used to protect bacteria from antibiotic action and reduced antibiotic susceptibility (Stewart, 2002). In contrast, H. pylori with PBP1a KO are hyposensitive (Figure 3). Besides, the growth of H. pylori isolated from clinical patients has an elongating lag phase with maximum growth on day 5, while the standard strain (ATCC43504) with PBP1a deletion had maximum growth on day 4 (Figure 8). It appeared that the growth patterns of Moe A-non-susceptible strains isolated from clinical patients had longer lag phases compared to the standard strain. In E. coli, PBP1a mutants were shown that growth is more slowly, and viability also is lower than the wild-type and cause cells to become more sensitive to stress. Accordingly, AmpC, may aid resistance to β-lactam antibiotics (Pepper et al., 2006). However, based on our MIC experiments, the PBP1a deletion strain was less susceptible to the antibiotic than the parental strain. Although antibiotic sensitivity has been reported in E. coli PBP mutants, in P. aeruginosa, PBP mutants were antibiotic tolerant (Moya et al., 2009). Furthermore, inactivation of the E. coli dacB ortholog encoding PBP4 (Kishida et al., 2006; Ghosh et al., 2008) was demonstrated to be resistant to β-lactam antibiotics (Avison et al., 2001), and in general, the coccoid forms comprised high fatty acid and cholesterol showed antibiotic resistance (Kadkhodaei et al., 2020). Nevertheless, electron microscopy revealed that the cells with PBP1a deletion were rod-forms (Figures 5C,D), and this markedly altered susceptibility further emphasizes the importance of PBP1a in H. pylori physiology. Generally, the rod-form is viable, culturable and more susceptible compared with the coccoid form. In contrast to the current understanding, deletion of PBP1a induced a faster growth rate and produced more biomass, emphasizing the importance of PBP1a in antibiotic susceptibility. It is noted that PBP four of the PBP1a deletion strain appears to be overproduced compared to the parental strain (Figure 6B) and may contribute to the formation of the spiral form of H. pylori (Tuomanen, 1986). Moreover, it was also demonstrated that E. coli could grow at the biggest growth rate with fewer PBPs (Driehuis and Wouters, 1987; Guinote et al., 2011). BolA is a transcription factor to suppress the expression of the mreB gene. The alleviation of bacterial growth and survival can be considered to the overexpression of the bolA gene in E. coli, especially in the exponential growth phase. These data suggest that there is a significant regulation is between the peptidoglycan polymerization and cell growth. Overall, the effect of PBP1a on cell growth and cell shape could regulate the transcription of the gene involved in Moe A non-susceptibility.
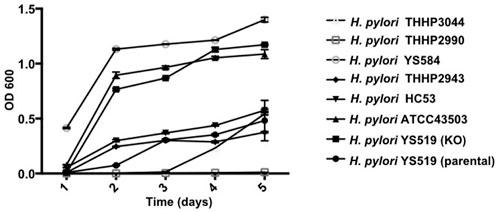
FIGURE 8. Growth curves of H. pylori ATCC43054 (Standard strain), H. pylori YS519 (parental), H. pylori YS519 (PBP1a KO), and other clinical Moe A-non-susceptible H. pylori strains. Growth curves were started at an OD600 of 0.05 in BHI medium, in triplicate.
Conclusion
In this research, our study provides an encouraging strategy that combination of Moe A with other antibiotics currently used in gastrointestinal therapy is potentially useful for the treatment of infection caused by MDR H. pylori strains. The study of PBP1a deletion on Moe A-non-susceptible strains also provided useful information for the future design of better therapies against Moe A-non-susceptible H. pylori infection.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Boards of National Taiwan University Hospital (NTUH). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Y-YT, J-ML, W-CC, and J-TH performed the experiments, collected and organized the data, and wrote the paper; W-CC and T-LH directed the research, M-SW and C-HW designed and directed the research and wrote paper, had full access to all the data in the study and final responsibility for the decision to submit for publication.
Funding
This work was supported by the Summit Program of Academia Sinica and the National Research Program for Biopharmaceuticals, Taiwan.
Acknowledgments
We thank Chia-Jung Lee for SEM analyses at College of Pharmacy, Taipei Medical University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Avison, M. B., Horton, R. E., Walsh, T. R., and Bennett, P. M. (2001). Escherichia coli CreBC is a global regulator of gene expression that responds to growth in minimal media. J. Biol. Chem. 276 (29), 26955–26961. doi:10.1074/jbc.m011186200
Bergamaschi, A., Magrini, A., and Pietroiusti, A. (2007). Recent advances in the treatment of Helicobacter pylori infection. Recent Pat. antiinfect. Drug Discov. 2 (3), 197–205. doi:10.2174/157489107782497281
Bollenbach, T. (2015). Antimicrobial interactions: Mechanisms and implications for drug discovery and resistance evolution. Curr. Opin. Microbiol. 27, 1–9. doi:10.1016/j.mib.2015.05.008
Caesar, L. K., and Cech, N. B. (2019). Synergy and antagonism in natural product extracts: When 1 + 1 does not equal 2. Nat. Prod. Rep. 36 (6), 869–888. doi:10.1039/c9np00011a
Chen, L., Walker, D., Sun, B., Hu, Y., Walker, S., and Kahne, D. (2003). Vancomycin analogues active against vanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc. Natl. Acad. Sci. U. S. A. 100 (10), 5658–5663. doi:10.1073/pnas.0931492100
Cheng, T. J., Sung, M. T., Liao, H. Y., Chang, Y. F., Chen, C. W., Huang, C. Y., et al. (2008). Domain requirement of moenomycin binding to bifunctional transglycosylases and development of high-throughput discovery of antibiotics. Proc. Natl. Acad. Sci. U. S. A. 105 (2), 431–436. doi:10.1073/pnas.0710868105
Driehuis, F., and Wouters, J. T. M. (1987). Effect of growth rate on the penicillin-binding proteins of Escherichia coli. FEMS Microbiol. Lett. 48 (1-2), 89–92. doi:10.1111/j.1574-6968.1987.tb02521.x
Eliopoulos, G. M., and Moellering, R. C. (1991). “Antimicrobial combination,” in Antibiotics Laboratory Medicine. 3rd Edn. Editors V. Lorian (Baltimore: the Williams & Wilkins Co), 432–492.
Gasparetto, M., Pescarin, M., and Guariso, G. (2012). Helicobacter pylori eradication therapy: Current availabilities. ISRN Gastroenterol. 2012, 186734–186742. doi:10.5402/2012/186734
Ghosh, A. S., Chowdhury, C., and Nelson, D. E. (2008). Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 16 (7), 309–317. doi:10.1016/j.tim.2008.04.006
Guinote, I. B., Matos, R. G., Freire, P., and Arraiano, C. M. (2011). BolA affects cell growth and binds to the promoters of penicillin-binding proteins 5 and 6 and regulates their expression. J. Microbiol. Biotechnol. 21 (3), 243–251. doi:10.4014/jmb.1009.09034
Halliday, J., McKeveney, D., Muldoon, C., Rajaratnam, P., and Meutermans, W. (2006). Targeting the forgotten transglycosylases. Biochem. Pharmacol. 71 (7), 957–967. doi:10.1016/j.bcp.2005.10.030
Huber, G., and Moenomycin, I. V. (1967). [Moenomycin, IV. Acid hydrolysis and characterisation of the cleavage product]. Justus Liebigs Ann. Chem. 707, 170–176. doi:10.1002/jlac.19677070124
Huber, G., and Nesemann, G. (1968). Glutamine synthetase deadenylylating enzyme. Biochem. Biophys. Res. Commun. 11, 32–37. doi:10.1016/0006-291x(68)90708-0
Kadkhodaei, S., Siavoshi, F., and Noghabi, K. A. (2020). Mucoid and coccoid Helicobacter pylori with fast growth and antibiotic resistance. Helicobacter 25 (2), e12678. doi:10.1111/hel.12678
Kishida, H., Unzai, S., Roper, D. I., Lloyd, A., Park, S. Y., and Tame, J. R. (2006). Crystal structure of penicillin binding protein 4 (dacB) from Escherichia coli, both in the native form and covalently linked to various antibiotics. Biochemistry 45 (3), 783–792. doi:10.1021/bi051533t
Kumar, A., Sarkar, S. K., Ghosh, D., and Ghosh, A. S. (2012). Deletion of penicillin-binding protein 1b impairs biofilm formation and motility in Escherichia coli. Res. Microbio 163 (4), 254–257. doi:10.1016/j.resmic.2012.01.006
Liou, J. M., Chen, C. C., Chang, C. Y., Chen, M. J., Fang, Y. J., Lee, J. Y., et al. (2013a). Efficacy of genotypic resistance-guided sequential therapy in the third-line treatment of refractory Helicobacter pylori infection: A multicentre clinical trial. J. Antimicrob. Chemother. 68 (2), 450–456. doi:10.1093/jac/dks407
Liou, J. M., Chen, C. C., Chen, M. J., Chen, C. C., Chang, C. Y., Fang, Y. J., et al. (2013b). Sequential versus triple therapy for the first-line treatment of Helicobacter pylori: A multicentre, open-label, randomised trial. Lancet 381 (9862), 205–213. doi:10.1016/s0140-6736(12)61579-7
Malfertheiner, P., Megraud, F., O’Morain, C. A., Atherton, J., Axon, A. T., Bazzoli, F., et al. (2012). Management of Helicobacter pylori infection--the maastricht IV/florence consensus report. Gut 61 (5), 646–664. doi:10.1136/gutjnl-2012-302084
Marshall, B. J., Goodwin, C. S., Warren, J. R., Murray, R., Blincow, E. D., Blackbourn, S. J., et al. (1998). Prospective double-blind trial of duodenal ulcer relapse after eradication of Campylobacter pylori. Lancet 2 (8626-8627), 1437–1442. doi:10.1016/s0140-6736(88)90929-4
McColl, K. E. (2010). Helicobacter pylori infection. N. Engl. J. Med. Overseas. Ed. 362 (17), 1597–1604. doi:10.1056/nejmcp1001110
Megraud, F., Coenen, S., Versporten, A., Kist, M., Lopez-Brea, M., Hirschl, A. M., et al. (2013). Helicobacter Pylori resistance to antibiotics in Europe and its relationship to antibiotic consumption. Gut 62 (1), 34–42. doi:10.1136/gutjnl-2012-302254
Moya, B., Dötsch, A., Juan, C., Blázquez, J., Zamorano, L., Haussler, S., et al. (2009). β-Lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 5 (3), e1000353. doi:10.1371/journal.ppat.1000353
Orhan, G., Bayram, A., Zer, Y., and Balci, I. (2005). Synergy tests by E test and checkerboard methods of antimicrobial combinations against Brucella melitensis. J. Clin. Microbiol. 43 (1), 140–143. doi:10.1128/jcm.43.1.140-143.2005
Pepper, E. D., Farrell, M. J., and Finkel, S. E. (2006). Role of penicillin-binding protein 1b in competitive stationary-phase survival of Escherichia coli. FEMS Microbiol. Lett. 263 (1), 61–67. doi:10.1111/j.1574-6968.2006.00418.x
Salzberg, L. I., Luo, Y., Hachmann, A. B., Mascher, T., and Helmann, J. D. (2011). The Bacillus subtilis GntR family repressor YtrA responds to cell wall antibiotics. J. Bacteriol. 193 (20), 5793–5801. doi:10.1128/jb.05862-11
Stewart, P. S. (2002). Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 292 (2), 107–113. doi:10.1078/1438-4221-00196
Tseng, Y. Y., Liou, J. M., Hsu, T. L., Cheng, W. C., Wu, M. S., and Wong, C. H. (2014). Development of bacterial transglycosylase inhibitors as new antibiotics: moenomycin A treatment for drug-resistant Helicobacter pylori. Bioorg. Med. Chem. Lett. 24 (11), 2412–2414. doi:10.1016/j.bmcl.2014.04.041
Tuomanen, E. (1986). Newly made enzymes determine ongoing cell wall synthesis and the antibacterial effects of cell wall synthesis inhibitors. J. Bacteriol. 167 (2), 535–543. doi:10.1128/jb.167.2.535-543.1986
van Heijenoort, J. (2007). Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 71 (4), 620–635. doi:10.1128/mmbr.00016-07
van Heijenoort, Y., Leduc, M., Singer, H., and van Heijenoort, J. (1987). Effects of moenomycin on Escherichia coli. Microbiology 133 (3), 667–674. doi:10.1099/00221287-133-3-667
Wallhausser, K. H., Nesemann, G., Prave, P., and Steigler, A. (1965). Moenomycin, a new antibiotic. I. fermentation and isolation. Antimicrob. Agents Chemother. 5, 734–736.
White, R. L., Burgess, D. S., Manduru, M., and Bosso, J. A. (1996). Comparison of three different in vitro methods of detecting synergy: Time-kill, checkerboard, and E test. Antimicrob. Agents Chemother. 40 (8), 1914–1918. doi:10.1128/aac.40.8.1914
Keywords: moenomycin, combination, Helicobacter, antibiotics, penicillin-binding protein knockout
Citation: Tseng Y-Y, Liou J-M, Cheng W-C, Hsu J-T, Hsu T-L, Wu M-S and Wong C-H (2022) Combating multidrug-resistant Helicobacter pylori with moenomycin A in combination with clarithromycin or metronidazole. Front. Chem. 10:897578. doi: 10.3389/fchem.2022.897578
Received: 16 March 2022; Accepted: 04 October 2022;
Published: 19 October 2022.
Edited by:
Nuno Manuel Xavier, University of Lisbon, PortugalReviewed by:
Enrique Domínguez-Álvarez, Institute of General Organic Chemistry (CSIC), SpainHao Wang, Minzu University of China, China
Copyright © 2022 Tseng, Liou, Cheng, Hsu, Hsu, Wu and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi-Huey Wong, chwong@gate.sinica.edu.tw; Ming-Shiang Wu, mingshiang@ntu.edu.tw