 Mengyun Wang1Sai Jiang1
Mengyun Wang1Sai Jiang1 Nusrat Hussain2Salman Zafar3Qingling Xie1Feibing Huang1Linxi Mao1Bin Li1Yuqing Jian1*
Nusrat Hussain2Salman Zafar3Qingling Xie1Feibing Huang1Linxi Mao1Bin Li1Yuqing Jian1* Wei Wang1*
Wei Wang1*- 1TCM and Ethnomedicine Innovation & Development International Laboratory, Innovative Material Medical Research Institute, School of Pharmacy, Hunan University of Chinese Medicine, Changsha, China
- 2Department of Chemistry, University of Baltistan Skardu, Skardu, Pakistan
- 3Institute of Chemical Sciences, University of Peshawar, Peshawar, Pakistan
A pair of 3,4-seco-cycloartane triterpenoid isomers with a rare peroxy bridge, namely, xuetonins A and B (1 and 2), four new lignans xuetonlignans A–D (3–6), a new sesquiterpene xuetonpene (7), and a new natural product xuetonin C (8), along with 43 known compounds, were obtained from the leaves of Tujia ethnomedicine, Kadsura heteroclita. Their structures and configurations were determined with the help of a combination of 1D- and 2D-NMR, HRESIMS spectra, electronic circular dichroism (ECD), and X-ray diffraction data. Compounds 2, 10, 13–15, and 17–19 showed moderate-to-potent activity against rheumatoid arthritis fibroblast-like synoviocytes (RAFLS) with IC50 values of 19.81 ± 0.26, 12.73 ± 0.29, 5.70 ± 0.24, 9.25 ± 0.79, 5.66 ± 0.52, 11.91 ± 0.44, 13.22 ± 0.27, and 15.94 ± 0.36 μM, respectively. Furthermore, compounds 22, 25, and 31 exhibited significant hepatoprotective effects against N-acetyl-p-aminophenol (APAP)–induced toxicity in HepG2 cells at 10 μM, and the cell viability increased by 12.93, 25.23, and 13.91%, respectively, compared with that in the model group (cf. bicyclol, 12.60%).
1 Introduction
Kadsura heteroclita (Roxb.) Craib (Schizandraceae) is an important ingredient of traditional Chinese medicine (TCM), which was widely distributed in the southwest part of China (Cao et al., 2019b). The plant is locally called “Xuetong” in Tujia ethnomedicine to treat rheumatoid arthritis (RA) and hepatitis (Cao et al., 2019a; Cao et al., 2019c; Wang et al., 2020). Previous phytochemical investigations have indicated that the main bioactive chemical constituents of K. heteroclita are dibenzocyclooctadienes and spirobenzofuranoid dibenzocyclooctadienes lignans, lanostanes, and cycloartane triterpenoids exhibiting various bioactivities such as anti-RA, anti-inflammation and analgesic, hepatoprotection, anti-HIV, anticancer, and anti-HBV (Liu Y. B et al., 2018; Wang et al., 2020). Previous reports from our research group on the stem of K. heteroclita describe the isolation of a series of triterpenoids and lignans (Wang et al., 2006b; Cao et al., 2019b).
The stem of the plant has always been used for medicinal purposes. Moreover, studies have also been carried out on its chemical constituents and pharmacological potential over the years (Wang et al., 2020). However, there is no specific literature on the phytochemistry and bioactivities of the leaves of K. heteroclita. Thus, in order to comprehend and understand the importance of the plant, the leaves of the plant were studied in this research endeavor, leading to the isolation of seven new compounds (1–7), one natural product (8) (Figure 1), and 43 known compounds. Furthermore, these secondary metabolites were tested for their anti-RAFLS effect and hepatoprotective potential. Compounds 2, 10, 13–15, and 17–19 exhibited a moderate-to-potent anti-RAFLS activity. Furthermore, compounds 22, 25, and 31 exhibited significant hepatoprotective effects against APAP-induced toxicity in HepG2 cells. Herein, the isolation, identification, bioactivity evaluation, and molecular docking studies of these isolated compounds are presented.
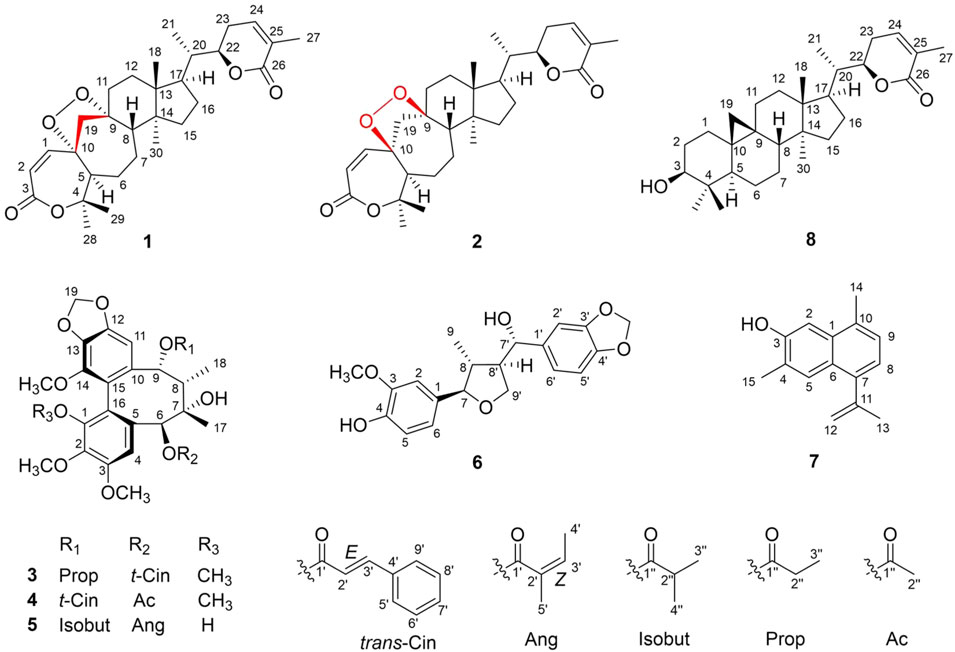
FIGURE 1. Structures of new compounds (1–7) and a natural product (8).
2 Materials and Methods
2.1 General Experimental Procedures
Optical rotations were recorded on a Perkin–Elmer 341-MC digital polarimeter at room temperature. A TU-1900 spectrophotometer (Shimadzu Europa GmbH, Duisburg, Germany) was used for obtaining the UV/Vis spectrum; IR spectra were scanned using a Hitachi 260–30 spectrometer. A Jasco J-810 circular dichroism spectropolarimeter was used to measure the ECD spectra at room temperature. 1D- and 2D-NMR spectra were obtained on a Bruker ARX-600 spectrometer (Bruker Technology Co., Ltd., Karlsruhe, Germany). The HRESIMS spectra were acquired using the UPLC/xevo G2 Qtof spectrometer (Waters Corporation, Milford, MA, United States). Semi-preparative HPLC was conducted on an Agilent 1,260 liquid chromatography (Santa Clara, CA, United States) with an Agilent C18 column (250 mm × 34 mm). Silica gels (80–100 and 300–400 meshes) were obtained from Qingdao Marine Chemical Inc. (Qingdao, China). All analytical-grade solvents were obtained from Shanghai Titan Scientific Co., Ltd., Shanghai, China. HPLC-grade methanol and acetonitrile were purchased from Merck KGaA (Darmstadt, Germany).
2.2 Plant Material
The leaves of Kadsura heteroclita (Schisandraceae) were collected in Shimen county, Changde city, Hunan province, China, during March 2014 and identified by Prof. Wei Wang, School of Pharmacy, Hunan University of Chinese Medicine. The voucher specimen number (KH-shimen-201403) has been deposited in the School of Pharmacy, Hunan University of Chinese Medicine, Changsha city, Hunan province, P. R. China.
2.3 Extraction and Isolation
The air-dried leaves of K. heteroclita (8 kg) were powdered and extracted thrice with 90% EtOH (24.0 L) for 1.5 h each using ultrasonic extraction. Then, all the extract solvents were evaporated under reflux condition to obtain the crude EtOH extract (750.1 g). The crude extract was then suspended in H2O (3.2 L) and successively partitioned with dichloromethane (DCM) and ethyl acetate (EtOAc) to yield DCM-soluble (70.1 g) and EtOAc-soluble (55.9 g) fractions, respectively.
The DCM fraction was then subjected to silica gel column chromatography (CC), which was eluted with petroleum ether (PE)–ethyl acetate (EA) (1:0, 100:1, 50:1, 20:1, 10:1, 5:1, 2:1, 1:1, and 0:1 gradient systems) to obtain six fractions (Fr. A–Fr. F). Fraction B (9.7 g) was further subjected to CC over silica gel, eluting with PE–EA (1:0–0:1) to yield four sub-fractions (Fr. B1–Fr. B4). Fr. B2 (2.6 g) after successive separation on a silica gel column, a Sephadex LH-20 column, and preparative TLC afforded the pure compounds 16 (33.8 mg), 28 (1.5 mg), 40 (80.2 mg), 45 (45.7 mg), and 48 (1.0 mg). Fr. B3 (3.2 g) was repeatedly purified on a silica gel column and a Sephadex LH-20 column to obtain the pure compounds 7 (2.1 mg), 13 (5.5 mg), 17 (6.1 mg), 18 (4.0 mg), 19 (14.1 mg), and 20 (7.0 mg). Fraction C (12.8 g) was separated on a silica gel CC using PE–EA (1:0–0:1) as elution solvents to afford six fractions (Fr. C1–Fr. C6). Fr. C3 (3.8 g) was subjected to successive separations, and ultimately compounds 4 (3.2 mg, retention time = tR 26.21 min), 21 (39.6 mg, tR 27.71 min), 23 (4.1 mg, tR 29.66 min), and 25 (8.6 mg, tR 39.28 min) were separated by semi-preparative HPLC with 72% MeOH/H2O at a flow rate of 2 ml/min. Fr. C4 (4.1 g) after successive chromatography on silica gel and a Sephadex LH-20 column yielded pure compounds 22 (2.9 mg, tR 15.74 min), 5 (1.8 mg, tR 18.10 min), 24 (5.5 mg, tR 21.01 min), and 3 (6.5 mg, tR 25.03 min) on semi-prep HPLC with the 65% ACN/H2O solvent system at a flow rate of 2 ml/min. Furthermore, compound 26 (12.1 mg) was also separated from the same sub-fraction on a silica gel CC with PE–EA (in a gradient manner from 1:0 to 0:1). Fraction D (9.6 g) was then isolated through a silica gel CC eluted with PE–EA (from 1:0 to 0:1) to obtain four sub-fractions (Fr. D1–Fr. D4). Fr. D2 (2.3 g) was further separated on a silica gel CC eluted with PE–EA (1:0–0:1) to afford eight sub-fractions (Fr. D2.1–Fr. D2.8). Compounds 30 (3.9 mg, tR 18.13 min), 31 (1.0 mg, tR 18.96 min), 6 (1.7 mg, tR 21.60 min), and 27 (2.0 mg, tR 22.22 min) were isolated from Fr. D2.3 (157.2 mg) by semi-prep HPLC with 65% MeOH/H2O. Compound 12 (1.6 mg, tR 27.39 min) was purified from Fr. D2.4 (135.3 mg) by semi-prep HPLC with ACN-H2O (55: 45). Compounds 49 (1.6 mg, tR 8.84 min) and 46 (1.3 mg, tR 9.82 min) were obtained from Fr. D2.5 (95.3 mg) by semi-prep HPLC with ACN-H2O (45: 55). Compounds 1 (5.8 mg) and 2 (6.1 mg) were purified from Fr. D2.5 (216.7 mg) by CC over silica gel eluted with hexane–acetone (from 9:1 to 7:3). Fr. D3 (3.7 g) yielded compounds 15 (3.5 mg), 29 (100.4 mg), and 41 (50.0 mg) by a series of silica gel CC, Sephadex LH-20 CC, and preparative TLC. Fraction E (16.9 g) was further separated on a silica gel CC eluted with DCM–MeOH (from 1:0 to 0:1) to afford six fractions (Fr. E1–Fr. E6). Fr. E3 (2.6 g) was purified by a silica gel column, a Sephadex LH-20 column, and preparative TLC method to obtain pure compounds 9 (4.1 mg), 10 (3.0 mg), 11 (11.0 mg), 38 (1.0 mg), and 39 (37.7 mg). Fr. E4 (5.7 g) was then subjected to successive silica gel CC, Sephadex LH-20 CC, ODS CC, and preparative TLC to obtain compounds 14 (30.0 mg), 32 (3.6 mg), 33 (7.3 mg), 34 (1.2 mg), 35 (4.7 mg), 36 (2.0 mg), 37 (1.2 mg), 42 (8.1 mg), 43 (3.3 mg), 44 (11.1 mg), and 47 (1.0 mg).
The EA fraction was then separated through silica gel CC using a gradient system of PE/EA (1:0, 50:1, 20:1, 10:1, 5:1, 2:1, 1:1, and 0:1) for elution to yield 10 fractions (Fr. A–Fr. J). Fraction C (860.8 mg) was isolated through a series of CC experiments over silica gel by gradient elution of PE–EA (1:0–0:1) to obtain five sub-fractions (Fr. C1–Fr. C5). Compound 8 (13.8 mg) was obtained from Fr. C2 (286.4 mg) and Fr. C3 (101.2 mg), which was subjected to silica gel CC using DCM/MeOH (from 1:0 to 0:1). Fraction F (2.4 g) was isolated through a silica gel CC eluted with PE–EA (from 1:0 to 0:1) to afford compound 51 (21.5 mg). Fraction J (44.33 g) after successive chromatography on a silica gel column using a gradient elution of DCM–MeOH (from 1:0 to 0:1) afforded three sub-fractions (Fr. J1–Fr. J3). Fr. J3 (40.3 g) was eluted on ODS CC with a gradient solvent system of MeOH–H2O (0:1–1:0) to yield compound 50 (10.0 g).
2.3.1 Xuetonin A
White amorphous powder;
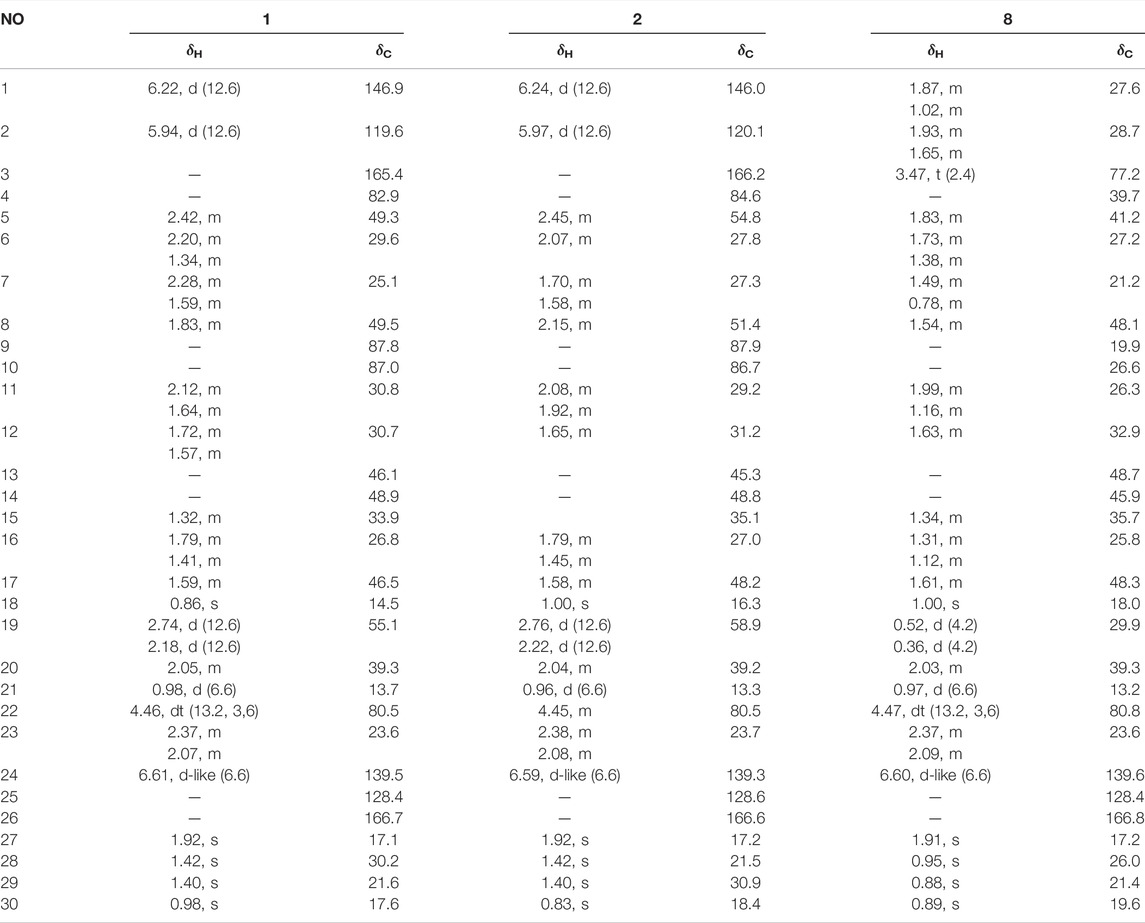
TABLE 1. 1H (600 MHz) and 13C NMR (150 MHz) data of compounds 1, 2, and 8 in CDCl3 (J in Hz).
2.3.2 Xuetonin B
White amorphous powder;
2.3.3 Xuetonlignan A
White amorphous powder;
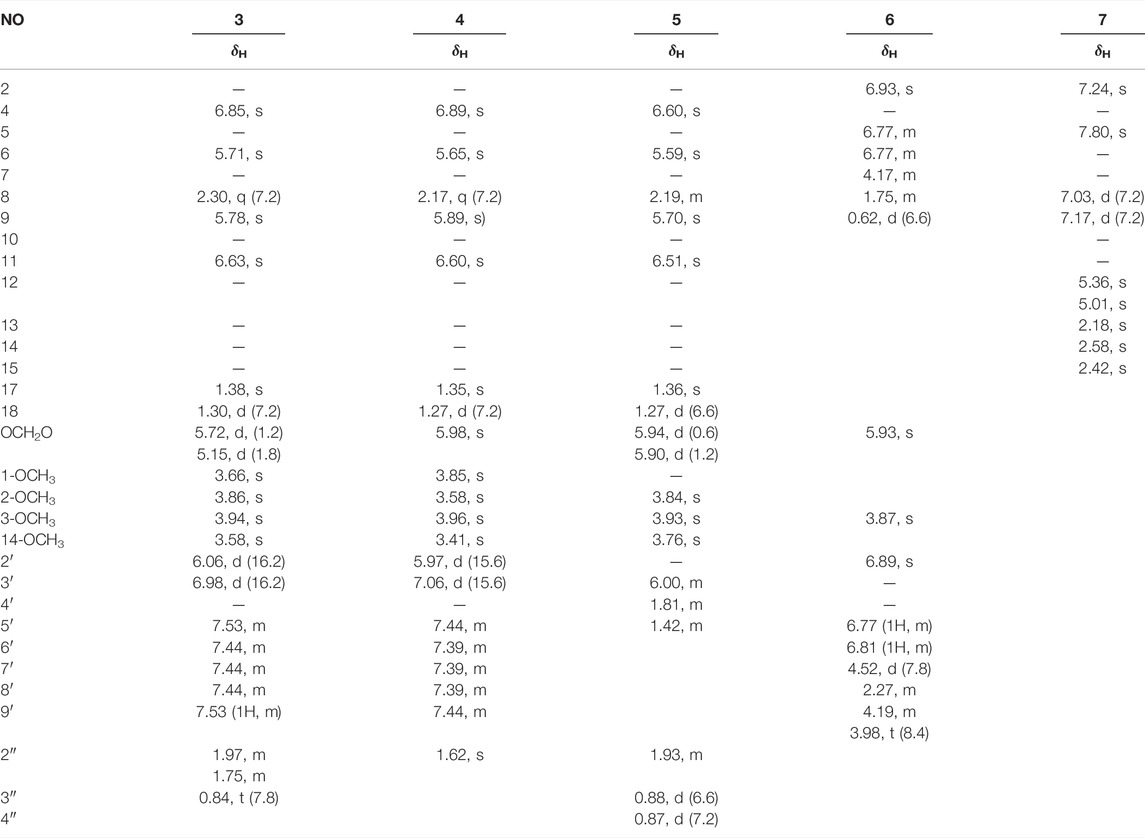
TABLE 2. |1H NMR (600 MHz) data of compounds 3–6 in CD3OD and 7 in CDCl3 (J in Hz).
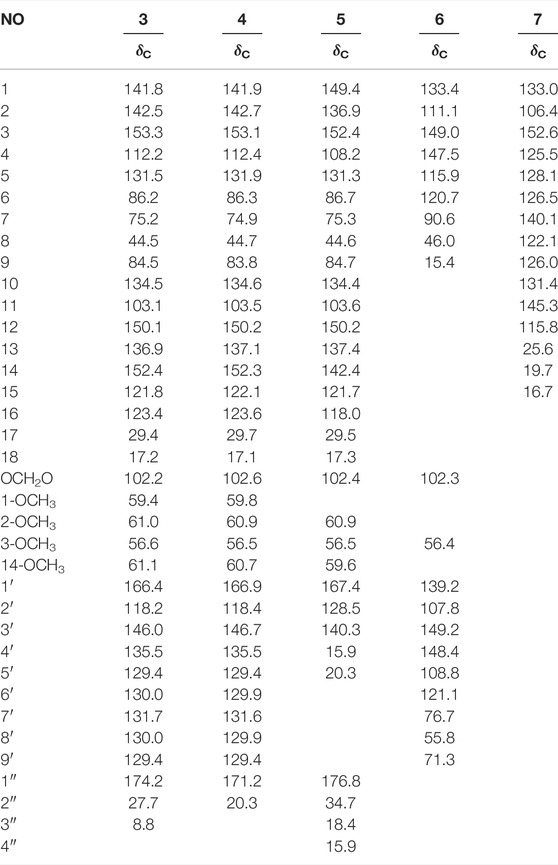
TABLE 3. |13C NMR (150 MHz) data of compounds 3–6 in CD3OD and 7 in CDCl3.
2.3.4 Xuetonlignan B
White amorphous powder;
2.3.5 Xuetonlignan C
White amorphous powder;
2.3.6 Xuetonlignan D
White amorphous powder;
2.3.7 Xuetonpene
Yellow oily matter; UV (CH2Cl2) λmax (log ε): 204 (3.34), 287 (2.19) nm; IR νmax: 3,385, 2,925, 1714, 1,489, 1,443, 1,248, 1,038, 935, and 703 cm−1; (+) HRESIMS: m/z 213.1276 [M + H]+, calcd for C15H17O, 213.1279; 1H and 13C NMR data: see Tables 2, 3.
2.3.8 Xuetonin C
2.4 X-Ray Crystallographic Analysis
Colorless crystals were obtained from methanol at room temperature by slow evaporation. The X-ray crystallographic data of the compound were obtained using a SuperNova, Dual, Cu at zero, AtlasS2 diffractometer. The structures were determined by direct methods and refined anisotropically with a full-matrix least-squares based on F2 using the SHELXL-2018 procedure via Olex2 software (Zhao et al., 2020). Crystallographic data for 21 have been deposited at the Cambridge Crystallographic Data Center (CCDC: 2102216).
2.4.1 Crystallographic Data of 21
C32H34O11 (M = 594.59 g/mol): monoclinic, space group P21 (no. 4), a = 9.8206 (2) Å, b = 16.2506 (2) Å, c = 10.6303 (2) Å, α = 90°, β = 117.374 (3)°, γ = 90°, V = 1,506.55 (6) Å3, Z = 2, T = 149.99 (10) K, μ (Cu–Kα) = 0.829 mm−1, ρcalc = 1.311 g/cm3, 11,564 reflections measured (9.368° ≤ 2Θ ≤ 147.24°), 5,370 unique (Rint = 0.0190, Rsigma = 0.0200), which were used in all calculations. The final R1 was 0.0294 (I > 2σ(I)) and wR2 was 0.0767 (all data). The goodness of fit on F2 was 1.056. Flack parameter: 0.05 (4).
2.5 Anti-Rheumatoid Arthritis Fibroblast-Like Synoviocyte Activity Assay
Human HFLS-RA cells were cultured in DME/F-12 with 10% fetal calf serum at 37°C in a constant temperature incubator with 5% CO2. The cells were then digested by 0.25% trypsin in 0.02% EDTA. HFLS-RA cells were seeded into each well of 96-well multiplates. After 12 h of incubation at 37°C, the cells were administrated with different doses of compounds (0, 2.5, 5, 7.5, 10, 12.5, 15, and 20 µM) and incubated for another 48 h. The cells were subjected to the MTT assay. Methotrexate was used as the positive control substance (Ding et al., 2019).
2.6 Hepatoprotective Activity Assay
Human HepG2 hepatoma cells were cultured in DMEM supplemented with 10% fetal calf serum at 37°C in a humidified atmosphere of 5% CO2. HepG2 cells were seeded into 96-well cell culture plates. After overnight incubation, 10 μM test samples and APAP (final concentration of 5 mm) were added into the wells and incubated for another 24 h. The cell viability was determined by the MTT assay. Bicyclol was used as the positive control (Hao et al., 2012).
2.7 Molecular Docking Study
The crystal structure of the receptor activator of nuclear factor κ-B ligand (RANKL) (PDB ID: 3urf) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/) (Ganesan and Rasool, 2019). The structures of compounds 13 and 15 were drawn by Chemdraw and generated to 3D structures with energy minimization using the MM2 minimize. Docking was performed using Autodock, and structure visualization was performed with Pymol and Discovery Studio software.
3 Results and Discussion
Compound 1 (xuetonin A) was isolated as a white amorphous powder and was shown to have a molecular formula of C30H42O6 by a positive HRESIMS peak at m/z 499.3066 ([M + H]+, calcd. 499.3060). The absorption maximum (209 nm) in the UV spectrum was attributed to the α,β-unsaturated ester system. The 1H NMR data of 1 showed three olefinic protons at δH 6.61 (1H, d-like, J = 6.6 Hz), 6.22 (1H, d, J = 12.6 Hz), and 5.94 (1H, d, J = 12.6 Hz) that were attributed to two double bonds. An oxygenated methine signal appeared at δH 4.46 (1H, dt, J = 13.2, 3.6 Hz) along with six methyl singlets (3H each, δH 1.92, 1.42, 1.40, 0.98, 0.98, and 0.86). The 13C NMR and DEPT-135° data displayed 30 carbon signals, including two conjugated carbonyl carbons at δC 166.7 and 165.4, four olefinic carbons at δC 146.9, 139.5, 128.4, and 119.6, three oxygenated quaternary carbons at δC 87.8, 87.0, and 82.9, one oxygenated methenyl carbon at δC 80.5, and six methyl carbons at δC 30.2, 21.6, 17.6, 17.1, 14.5, and 13.7. The NMR data of 1 resembled those of schisanlactone A (Liu et al., 1983a), except for the presence of a peroxy bridge between C-9 and C-10. This was confirmed by the HRESIMS. Moreover, two doublets for the C-19 methylene group resonance signals occurred at δH 2.74 (1H, d, J = 12.6 Hz) and 2.26 (1H, d, J = 12.6 Hz) due to the effect of the peroxy bridge. This was further evidenced by HMBC correlations of H-2 (δH 5.94)/H-5 (δH 2.42) with C-10 (δC 87.0) and of H-19b (δH 2.18)/H-12b (δH 1.57) with C-9 (δC 87.8) (Figure 2). Thus, the planar structure of 1 was determined as a 3,4-seco-cycloartane with a rare peroxy bridge by the 1D-NMR, 1H–1H COSY, HSQC, and HMBC spectral analyses. The β-configuration of H-19 was deduced by the ROESY cross peaks between H-19a (δH 2.74), H-8 (δH 1.63) and CH3-29 (δH 1.40). Conversely, the peroxy bridge was deduced to be in the α-orientation. Moreover, the absolute configuration of 1 was determined to be 5S, 8S, 9S, 10S, 13R, 14S, 17R, 20S, and 22R by comparing the experimental and calculated ECD spectra (Figure 3). Thus, compound 1 was established and named as xuetonin A.
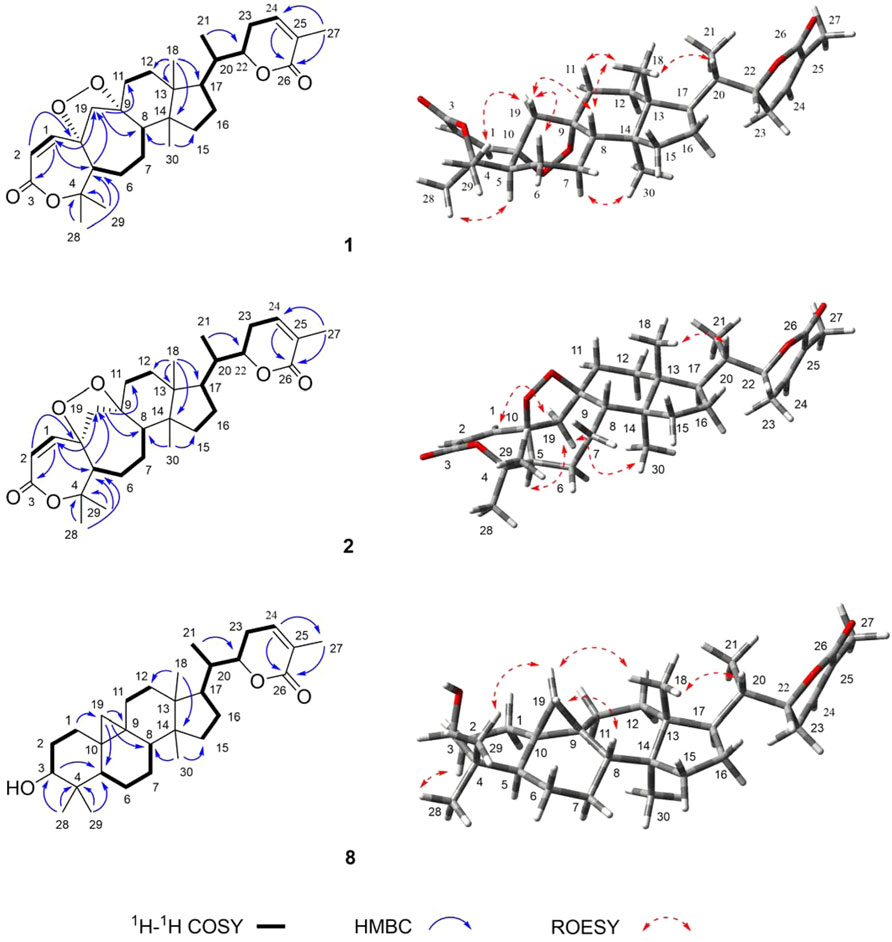
FIGURE 2. Key 1H–1H COSY, HMBC, and ROSEY correlations of 1, 2, and 8.
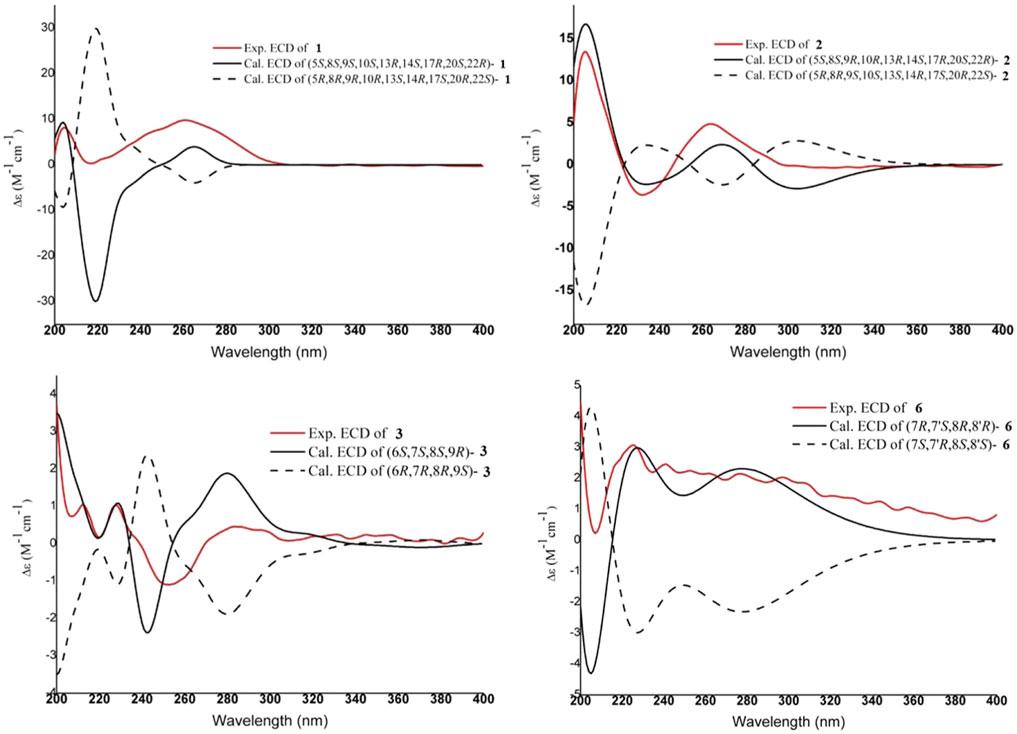
FIGURE 3. Experimental and calculated ECD spectra of compounds 1–3 and 6
Compound 2 (xuetonin B) was isolated as a white amorphous powder with the molecular formula C30H42O6, as determined by HRESIMS from the peak at m/z 499.3068 ([M + H]+, calcd. 499.3060). Comparison of the HRESIMS, UV, 1D-, and 2D-NMR spectra of 2 with those of 1 suggested that they are a pair of 3,4-seco-cycloartane isomers with the same planar structure. The differences are the configurations of C-9 and C-10. In compound 2, the peroxy bridge on C-9 and C-10 was found to be in the β-orientation, deduced from ROESY correlations of H-19 (δH 2.76) with H-5α (δH 2.45) (Figure 2). Consequently, the absolute configuration of 2 was determined to be 5S, 8S, 9R, 10R, 13R, 14S, 17R, 20S, and 22R based on the comparisons of the experimental ECD curves and calculated ones (Figure 3). Thus, compound 2 was established, and it was named xuetonin B. Compounds 1 and 2 were identified as new 3,4-seco-cycloartane triterpenoids with a rare peroxy bridge between C-9 and C-10. To date, only one cycloartane-derived triterpenoid (schinalactone A) containing the peroxy bridge has been found from Schisandra sphenanthera (He et al., 2010).
Compound 3 (xuetonlignan A), isolated as white amorphous powders, had the molecular formula C35H38O11 deduced from its HRESIMS analysis (m/z 652.2758, [M + NH4]+, calcd for 652.2758). The UV data of 3 provided a characteristic peak (λmax 218) of dibenzocyclooctadiene lignan (Luo et al., 2017). The 1H NMR data (Table 1) displayed two aromatic protons for a biphenyl moiety at δH 6.85 and 6.63, two characteristic signals of a methylenedioxy moiety at δH 5.72 and 5.15 ppm, and four singlets for methoxy moiety at δH 3.94, 3.86, 3.66, and 3.58 ppm. A cyclooctadiene ring was deduced. Furthermore, two oxymethine (δH 5.78 and 5.71 ppm), a methine (δH 2.30 ppm), and two methyl signals (δH 1.38 and 1.30 ppm) also appeared in the spectrum. The 13C NMR spectrum of 3 showed 35 carbon signals, including 12 aromatic carbons belonging to the biphenyl moiety (δC 153.3, 152.4, 150.1, 142.5, 141.8, 136.9, 134.5, 131.5, 123.4, 121.8, 112.2, and 103.1), a methylenedioxy signal (δC 102.2), three oxymethine carbons (δC 86.2, 84.5, and 75.2), four methoxy groups (δC 61.1, 61.0, 59.4, and 56.6), one methine carbon (δC 44.5), two methyl carbons (δC 29.4 and 17.2) and a trans-cinnamoyl group (δC 166.4, 146.0, 135.5, 131.7, 130.0, 130.0, 129.4, 129.4, and 118.2) and a propionyl group (δC 174.2, 27.7, and 8.8) (Dong et al., 2012). The above data indicated that 3 is a C18-dibenzocyclooctadiene lignan with a trans-cinnamoyl group and a propionyl group. The locations of groups were confirmed by 1H–1H COSY and HMBC data. The HMBC correlations from H-11 (δH 6.63) to C-12 and C-13 and from the four methoxy protons to C-1, C-2, C-3, and C-14 showed that the methylenedioxy moiety is connected to C-12 and C-13, and the four methoxy moieties are connected to C-1, C-2, C-3, and C-14. The presence of a trans-cinnamoyl group at C-6 and a propionyl group at C-9 was deduced by the HMBC correlations from H-6 (δH 5.71) to C-1' (δC 166.4) and C-4 (δC 112.2) and from H-9 (δH 5.78) to C-1'' (δC 174.2) and C-11 (δC 103.1). Furthermore, CH3-17 at C-7 and CH3-18 at C-8 can together be confirmed by the HMBC correlations between H3-17 (δH 1.38, s) and C-6, C-7, C-8, and H3-18 (δH 1.30, d) with C-9, C-8, and C-7; and the spin system of H3-18/H-8/H-9 in the 1H–1H COSY.
The absolute configuration of 3 was established with the help of ECD combined with ROESY data. The ECD experiment exhibited a negative cotton effect (CE) around 252 nm and a positive CE at 227 nm, suggesting the S-biphenyl configuration of 3 (Luo et al., 2017). The ROESY correlations between H-6/H-4, H-11/H-9/H-8, and H-8/H3-17 indicated that H-6 and CH3-18 were α-oriented, while H-8, CH3-17, and H-9 were β-oriented. The ROESY and ECD data of 3 were found to be similar to those of 21 (heteroclitalignan D) (Wang et al., 2006b). X-ray crystallographic analysis of 21 eventually established the stereochemistry of 3, especially at C-6, C-7, C-8, and C-9. Futhermore, based on the comparisons of the experimental and calculated ECD spectra, the absolute configuration of 3 was found to be 6S, 7S, 8S, and 9R (Figure 3). Therefore, the structure of 3 was established for xuetonlignan A.
Compound 4 (xuetonlignan B) possesses the molecular formula C34H36O11 through analysis of the HRESIMS (m/z 638.2596 [M + NH4]+). The UV, NMR, and ECD data of 4 indicated the presence of an S-biphenyl–configured dibenzocyclooctadiene lignan with almost identical data and the same planar structure to arisanschinin C (Liu et al., 2010). The only difference between them was in the configurations of C-7 and C-8. This can be confirmed by the ROESY correlations of H-4 with H-6, of H-11 with H-9 and H-8, and of H-8 with H-17. This was further confirmed from the similarity between ROESY and ECD spectra of 4 and 3. Based on the data, the absolute configuration of 4 was shown as 6S, 7S, 8S, and 9R. Accordingly, the structure of 4 was established for xuetonlignan B.
Compound 5 (xuetonlignan C) was determined to have the formula C31H38O11 by deducing from its HRESIMS at m/z 604.2754 [M + NH4]+ (calcd for 604.2758). The UV, 1D-NMR, and ECD data showed that 5 is an S-biphenyl–configured dibenzocyclooctadiene lignan. Comparison of the spectral data of 9 with kadsuphilol R (Cheng et al., 2011) exhibited the presence of the isobutyryl moiety instead of the angeloyl moiety at C-9 in 5. The HMBC correlations from H-9 (δH 5.70) to C-1'' (δC 176.8) and from H-2'' (δH 1.93), H-3'' (δH 0.88), and H-4'' (δH 0.87) to C-1'' (δC 176.8) in 5 established the locations of the isobutyryl group at C-9. The ROESY correlations of H-4 with H-6 and 3-OCH3, of H-11 with H-9 and H-8, and of H-8 with H3-17 indicated that H-6 and CH3-18 were α-oriented and that H-9, H-8, and CH3-17 were β-oriented. This was further evidenced from the lack of ROESY correlation between CH3-17 and CH3-18. Thus, the structure of xuetonlignan C (5) was established.
Compound 6 (xuetonlignan D) was obtained as white amorphous powders, having the molecular formula C20H22O11 inferred from its HRESIMS analysis (m/z 381.1310, [M + Na]+, calcd for 381.1314). The 1H NMR spectrum exhibited aromatic protons at δH 6.93 (1H, s), 6.89 (1H, s), 6.81 (1H, d, J = 7.8 Hz), and 6.77 (3H, m, overlapped) that were attributed to two 1,3,4-trisubstituted phenyl groups. A methylenedioxy group at 5.93 (2H, s), two oxygenated methenyls at δH 4.52 (1H, d, J = 7.8 Hz) and 4.17 (1H, m), an oxygenated methylene at δH 4.19 (1H, m) and 3.98 (1H, t, J = 8.4 Hz), a methoxyl at δH 3.87 (3H, s), two methenyls at δH 2.27 (1H, m) and 1.75 (1H, m), and a methyl at 0.62 (3H, d, J = 6.6 Hz) signals also appeared in the spectrum. These moieties were also identified based on the 13C and DEPT-135° NMR data analysis. Comparison of the 1D-NMR spectral data of 6 with the ones of 3-methoxy-3′,4′-methylenedioxy-7,9′-epoxylignane-4,7′,9-triol, isolated from Asiasarum heterotropoides, revealed both compounds to be quite similar structurally, except that 6 lacked a hydroxy group at C-9 (Lee et al., 2013). This was determined by the HMBC correlations from H3-9 (δH 0.62) to C-8 (δC 46.0), C-7 (δC 90.6), and C-8' (δC 55.8). The relative stereochemistry was confirmed by ROESY data. ROESY correlations of H-9 with H-7 (δH 4.17) and H-8' (δH 2.27) and of H-8 (δH 1.75) with H-7' (δH 4.52) exhibited that H-9, H-7, and H-8′ were of the same orientation; H-8 and H-7′ were of the same orientation. The absolute configuration of 6 was confirmed by comparing the experimental and calculated ECD spectra (Figure 3). Thus, compound 6 was confirmed to be (7R,8R,7′S,8′R)-3-methoxy-3′,4′-methylenedioxy-7,9′-epoxylignane-4,7′-diol and named xuetonlignan D.
Compound 7 (xuetonpene) had the molecular formula C15H16O on the basis of its HRESIMS data at m/z 213.1276 [M + H]+ (calcd 213.1279). The 1H NMR spectroscopic data showed two singlet signals and two double signals for aromatic protons in two phenyl moieties at δH 7.80, 7.24, 7.17, and 7.03, one pair of proton resonances at δH 5.36 and 5.01, and three methyl groups at δH 2.58, 2.42, and 2.18. The 13C NMR, DEPT-135°, and HSQC spectra of 7 showed 15 carbon resonances, including 10 aromatic carbons (δC 152, 140.1, 133.0, 131.4, 128.1, 126.5, 126.0, 125.5, 122.0, and 106.4), two olefinic carbons (δC 145.3 and 115.8), and three methyl carbons (δC 25.6, 19.7, and 16.7). The abovementioned data suggested that 7 was an analog of 7-hydroxycadalene, except for the addition of one terminal double bond at C-11 (Sankaram et al., 1981). This was confirmed by the HMBC correlations from H-12 (δH 5.36 and 5.01) to C-13 (δC 25.6) and C-7 (δC 140.1) (Supplementary Figure S3). Therefore, the structure of xuetonpene (7) was defined as shown in Figure 1.
Compound 8 (xuetonin C) was determined to have the molecular formula C30H46O3 from HRESIMS (m/z, 477.3335, [M + Na]+, calcd 477.3345) analysis. The 1H and 13C NMR data of 8 were the same as those of 3β-hydroxycycloart-24Z-ene-22(S)→26 lactone, which was an enzymatic hydrolysis compound derived from juncoside I (Greca et al., 1994). The structure of 8 was confirmed by the comprehensive analysis of its 2D NMR data. Thus, 8 has the same structure as 3β-hydroxycycloart-24Z-ene-22(S)→26 lactone and is a new natural product named xuetonin C.
Heteroclitalignan D (21) was obtained as colorless crystals. The X-ray diffraction data of 21 were reported for the first time in this study (Figure 4). Biosynthetically, mangiferolic acid might be the precursor of compounds 1, 2, 8–10, 13, 15, and 17 through a series of oxidative cleavage processes via esterification, the Baeyer–Villiger oxidation, ring expansion, hydroxylation, cyclization, and epoxidation steps obtained from compounds 1, 2, 8–10, 13, 15, and 17, respectively. A plausible biogenetic pathway for 1, 2, 8–10, 13, 15, and 17 is shown in Figure 5.
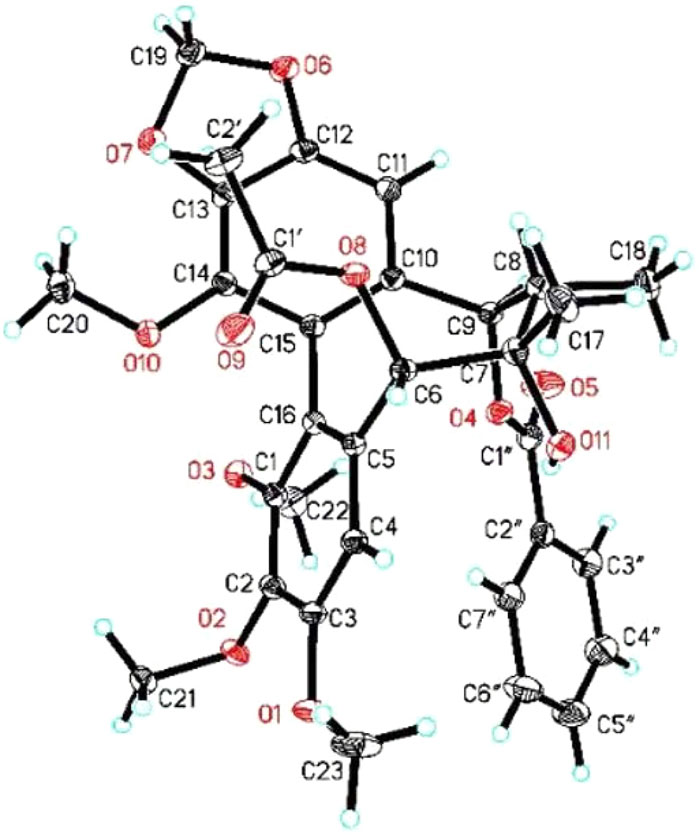
FIGURE 4. X-ray ORTEP drawing of 21.
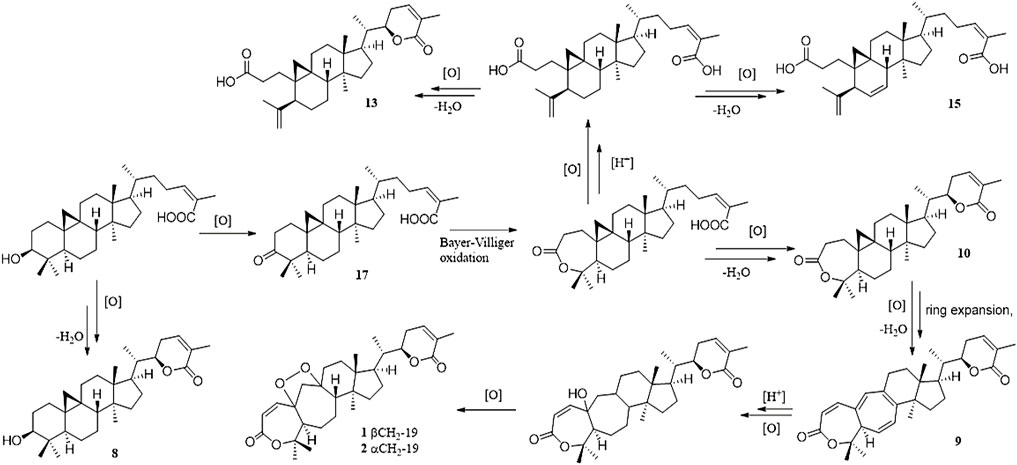
FIGURE 5. Plausible biosynthetic pathway for 1, 2, 8–10, 13, 15, and 17.
Forty-three known compounds isolated during this project were characterized as lancilactone B (9) (Chen et al., 1999), kadsudilactone (10) (Rui et al., 1991), schisanlactone B (11) (Liu et al., 1983b), kadsuphilactone B (12) (Shen et al., 2005), xuetongsu (13) (Shehla et al., 2020), heteroclitalactone A (14) (Wang et al., 2006a), changnanic acid (15) (Liu and Huang, 1991), cycloartenone (16) (Wang et al., 2006a), schizandronic acid (17) (Li et al., 2003), seco-coccinic acid F (18) (Minh et al., 2014), kadsuracoccinic acid B (19) (Li et al., 2008), sorghumol (20) (Cambie et al., 1992), heteroclitalignan D (21) (Wang et al., 2006b), kadsurarin (22) (Chen et al., 1973), kadsuphilol T (23) (Cheng et al., 2011), kadsuphilol R (24) (Cheng et al., 2011), kadsuphilol C (25) (Luo et al., 2017), kadsulignan N (26) (Gao et al., 1998), enshizhisu (27) (Huang et al., 1982), machilolin A (28) (Chen et al., 2009), (+)-pinoresinol (29) (Fan et al., 2020), (+)-2-(3,4-dimethoxyphenyl)-6-(3,4-dimethoxyphenyl)-3,7-dioxabicyclo [3,3,0] octane (30) (Latip et al., 1999), meso-dihydroguaiaretic acid (31) (Lu and Chen, 2008), 6α,9α-dihydroxycadinan-4-en-3-one (32) (Cao et al., 2019c), (4R)-4-hydroxy-1,10-seco-muurol-5-ene-1,10-dione (33) (Kiem et al., 2014), litseachromolaevane A (34) (Zhang et al., 2003), cryptomeridiol (35) (Ragasa et al., 2005), (-)-5β,11-dihydroxyiphionan-4-one (36) (Lin et al., 2019), aromadendrane-4β,10α-diol (37) (Goldsby and Burke, 1987), lochmolin F (38) (Tseng et al., 2012), loliolide (39) (Kim et al., 2004), β-sitosterol (40) (Luo et al., 2009), daucosterol (41) (Tezuka et al., 1998), stigmasterol (42) (Luo et al., 2009), schleicheol 2 (43) (Pettit et al., 2000), 7-hydroxy-β-sitosterol (44) (Chaurasia and Wichtl, 1987), stigmastan-3-one (45) (Brasil et al., 2010), mexoticin (46) (Chakraborty et al., 1967), pterosonin E (47) (Liu R. H et al., 2018), physcion (48) (Pang et al., 2016), 5-O-methylvisanninol (49) (Baba et al., 1981), shikimic acid (50) (Talapatra et al., 1989), and protocatechuic acid (51) (Guan et al., 2009) by comparing their NMR spectrum with the reported literature.
The anti-RAFLS activities of the isolated terpenoids (1–2, 7–20, and 32–39) were assessed on the RA fibroblast-like synoviocytes. Compounds 2, 10, 13–15, and 17–19 displayed evident inhibitory activities on the RA fibroblast-like synoviocytes with IC50 values of 19.81 ± 0.26, 12.73 ± 0.29, 5.70 ± 0.24, 9.25 ± 0.79, 5.66 ± 0.52, 11.91 ± 0.44, 13.22 ± 0.27, and 15.94 ± 0.36 μM, respectively, as shown in Table 4. The structure–activity relationship (SAR) study showed that the introduction of the carboxyl moiety enhances the activity. Furthermore, the results also showed that the orientation of C-19 affected the anti-RAFLS effects, as is evident from the data obtained for compounds 1 and 2. According to the abovementioned bioactivity results, it could be preliminarily deduced that triterpenoids may be the principal chemical constituents responsible for the anti-RAFLS effect of the leaves of K. heteroclita.

TABLE 4. |Effects of compounds 2, 10, 13–15, and 17–19 on rheumatoid arthritis fibroblast-like synoviocytes.
The hepatoprotective activities of the isolated lignans (3–6, 21–31) were evaluated in APAP-induced toxicity in HepG2 cells at 10 μM. Compounds 22, 25, and 31 showed significant hepatoprotective activity with increasing cell viability by 12.93%, 25.23%, and 13.91% compared with the model group (cf. bicyclol, 12.60%) at 10 μM, respectively, as shown in Figure 6. According to the abovementioned bioactivity results, it could be preliminarily deduced that lignans may be the principal components for the hepatoprotective effect of the leaves of K. heteroclita.
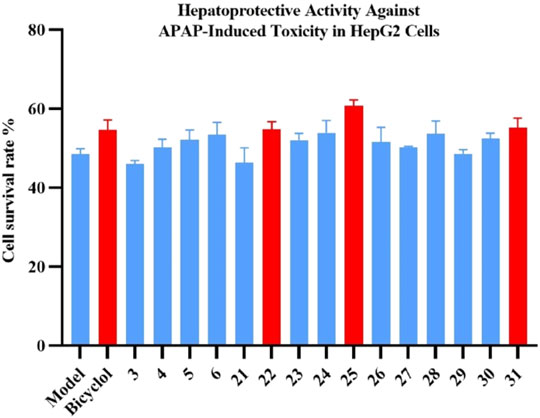
FIGURE 6. Effects of compounds 3–6 and 21–31 on N-acteyl-p-aminophenol (APAP)–induced toxicity in HepG2 cells. Data are presented as the mean ± SD (n = 3). Bicyclol was used as the positive control.
3.1 Molecular Docking
Compounds 13 and 15 exhibited lesser docking parameters (binding energy: −5.38 and −4.20 kcal/mol, respectively). As shown in Figure 6, compound 13 formed hydrogen bonds with LYS-267, PHE-272, SER-265, and ASN-267 residues and hydrophobic interactions with PHE-270, TRP-264, and HIS-271 residues. Similarly, compound 15 mainly interacted with LYS-38 by hydrogen bonds and with CYS-41, LYS-6, TPR-53, and PRO-24 by hydrophobic interactions. This docking simulation revealed the important role of the carboxyl moiety at C-3 in the structures of compounds 13 and 15 (Figure 7).
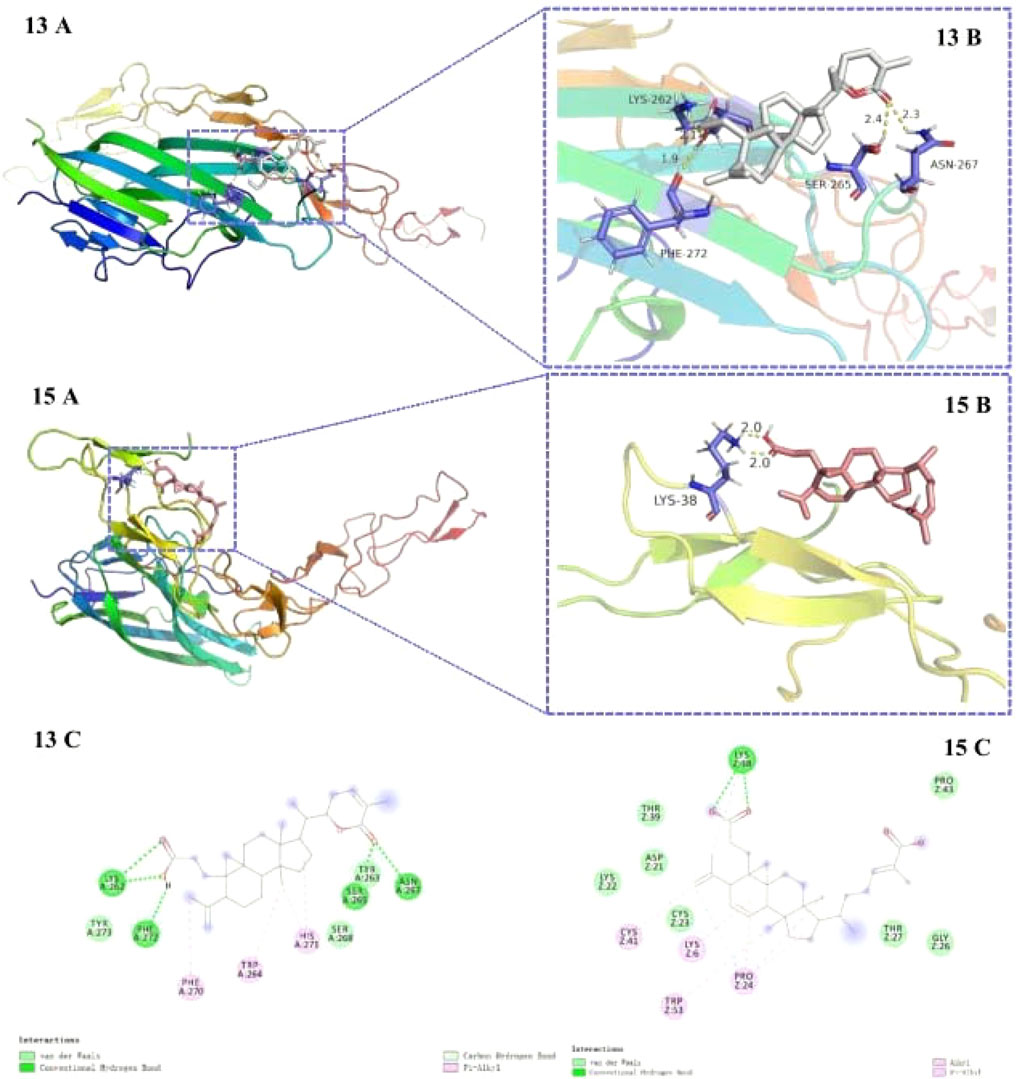
FIGURE 7. Docking poses (A) and interactions (B, C) of compounds 13 and 15 at the binding site of RANKL (receptor activator of nuclear factor k-B ligand). Hydrogen bonds and hydrophobic interactions are represented by the green and pink lines, respectively (c).
4 Conclusion
In summary, a total of 51 compounds, including two new highly oxidized cycloartane-type triterpenoids, four new lignans, one new sesquiterpene, and a new natural product, were obtained from the leaves of K. heteroclita. Among them, compounds 13–15 displayed potent anti-RAFLS activity with IC50 values of 5.70 ± 0.24, 9.25 ± 0.79, and 5.66 ± 0.52 μM, respectively, using methotrexate (IC50 = 3.10 ± 0.68 μM) as the positive control by the MTT method. In addition, the orientation of CH3-17 in dibenzocyclooctadiene lignans was determined by the direct ROE correlation of H-4 but not by the ROE correlation of H-6, even if they had ROE correlations, which were determined by X-ray diffraction of compound 21. This is the first phytochemical report of the leaves of K. heteroclita. It was observed that its main compound types are similar with those of the stem of K. heteroclita. It can, thus, be inferred that the leaves may also be used to treat relevant diseases. This study provides a bridge between traditional uses and modern biological studies and offers the experimental basis for the full development of K. heteroclita, which is of great significance in terms of scientific value.
Data Availability Statement
The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
WW and YJ conceived and designed the experiment. MW, SJ, and NH were responsible for compound isolation and writing. QX was responsible for structure identification. FH and LM evaluated activities of the compounds. SZ and BL revised the article. All authors have read and agreed to the final manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (Nos 82174078, 82074122, 81803708, and 81874369), the Changjiang Scholars Program in Ministry Education, People’s Republic of China (No. T2019133), the Natural Science Foundation of Hunan Province (No. 20JJ0502), and the Changsha Municipal Natural Science Foundation (No. kq2014092).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.878811/full#supplementary-material
References
Baba, K., Hata, K., Kimura, Y., Matsuyama, Y., and Kozawa, M. (1981). Chemical Studies of Angelica Japonica A. GRAY. I. On the Constituents of the Ethyl Acetate Extract of the Root. Chem. Pharm. Bull. 29, 2565–2570. doi:10.1248/cpb.29.2565
Brasil, D., Müller, A., Guilhon, G., Alves, C. N., Peris, G., Llusar, R., et al. (2010). Isolation, X-ray Crystal Structure and Theoretical Calculations of the New Compound 8-Eepicordatin and Identification of Others Terpenes and Steroids from the Bark and Leaves of Croton Palanostigma Klotzsch. J. Brazil. Chem. Soci. 21, 731–739. doi:10.1590/S0103-50532010000400021
Cambie, R. C., Lal, A. R., Rutledge, P. S., and Woodgate, P. D. (1992). Triterpenes from the Fruit of Elaeocarpus Chelonimorphus. Biochem. Syst. Ecol. 20, 708–709. doi:10.1016/0305-1978(92)90029-d
Cao, L., Li, B., Shehla, N., Gong, L.-m., Jian, Y.-Q., Peng, C.-Y., et al. (2020a). Triterpenoids from Stems of Kadsura Heteroclita. Fitoterapia 140, 104441. doi:10.1016/j.fitote.2019.104441
Cao, L., Shehla, N., Li, B., Jian, Y., Peng, C., Sheng, W., et al. (2020b). Schinortriterpenoids from Tujia Ethnomedicine Xuetong-The Stems of Kadsura Heteroclita. Phytochemistry 169, 112178. doi:10.1016/j.phytochem.2019.112178
Cao, L., Shehla, N., Tasneem, S., Cao, M., Sheng, W., Jian, Y., et al. (2019c). New Cadinane Sesquiterpenes from the Stems of Kadsura Heteroclita. Molecules 24, 1664–1673. doi:10.3390/molecules24091664
Chakraborty, D. P., Chowdhury, B. K., and Das, B. C. (1967). Mexoticin, a New Coumarin from L. Tetrahedron Lett. 8, 3471–3473. doi:10.1016/s0040-4039(01)89823-3
Chaurasia, N., and Wichtl, M. (1987). Sterols and Steryl Glycosides from Urtica Dioica. J. Nat. Prod. 50, 881–885. doi:10.1021/np50053a018
Chen, C.-Y., Cheng, M.-J., Chiang, Y.-J., Bai, J.-C., Chiu, C.-T., Lin, R.-J., et al. (2009). Chemical Constituents from the Leaves of Machilus Zuihoensis Hayata Var. Mushaensis (Lu) Y.C. Liu. Nat. Product. Res. 23, 871–875. doi:10.1080/14786410802401432
Chen, D.-F., Zhang, S.-X., Wang, H.-K., Zhang, S.-Y., Sun, Q.-Z., Cosentino, L. M., et al. (1999). Novel Anti-HIV Lancilactone C and Related Triterpenes from Kadsura Lancilimba. J. Nat. Prod. 62, 94–97. doi:10.1021/np980291d
Chen, Y.-P., Liu, R., Hsu, H.-Y., Yamamura, S., Shizuri, Y., and Hirata, Y. (1973). The Structures and Conformations of Two New Lignans, Kadsurin and Kadsurarin. Tetrahedron Lett. 14, 4257–4260. doi:10.1016/S0040-4039(01)87163-X
Cheng, Y.-B., Lin, Y.-C., Taha Khalil, A., Liou, S.-S., Lee, G.-C., Kuo, Y.-H., et al. (2011). Seven New Lignan Esters from Kadsura Philippinensis. Hca 94, 148–158. doi:10.1002/hlca.201000156
Ding, Y., Zhao, Q., and Wang, L. (2019). Pro-apoptotic and Anti-inflammatory Effects of Araloside A on Human Rheumatoid Arthritis Fibroblast-like Synoviocytes. Chem. Biol. Interact. 306, 131–137. doi:10.1016/j.cbi.2019.04.025
Dong, K., Pu, J.-X., Zhang, H.-Y., Du, X., Li, X.-N., Zou, J., et al. (2012). Dibenzocyclooctadiene Lignans from Kadsura Polysperma and Their Antineurodegenerative Activities. J. Nat. Prod. 75, 249–256. doi:10.1021/np200937h
Fan, H., Li, Y., Liang, X., Yan, S., Cui, Y., Zhang, H., et al. (2020). Chemical Constituents Isolated from Valeriana Officinalis L. Biochem. Syst. Ecol. 93, 104143. doi:10.1016/j.bse.2020.104143
Ganesan, R., and Rasool, M. (2019). Ferulic Acid Inhibits Interleukin 17‐dependent Expression of Nodal Pathogenic Mediators in Fibroblast‐like Synoviocytes of Rheumatoid Arthritis. J. Cel. Biochem. 120, 1878–1893. doi:10.1002/jcb.27502
Gao, X.-M., Pu, J.-X., Huang, S.-X., Yang, L.-M., Huang, H., Xiao, W.-L., et al. (2008). Lignans from Kadsura Angustifolia. J. Nat. Prod. 71, 558–563. doi:10.1021/np0705108
Goldsby, G., and Burke, B. A. (1987). Sesquiterpene Lactones and A Sesquiterpene Diol from Jamaican Ambrosia Peruviana. Phytochemistry 26, 1059–1063. doi:10.1016/S0031-9422(00)82350-X
Greca, M. D., Fiorentino, A., Monaco, P., and Previtera, L. (1994). Juncoside I, a New Cycloartanelactone Glucoside fromJuncus Effusus. Nat. Product. Lett. 4, 183–188. doi:10.1080/10575639408043903
Guan, S., Ge, D., Liu, T.-Q., Ma, X.-H., and Cui, Z.-F. (2009). Protocatechuic Acid Promotes Cell Proliferation and Reduces Basal Apoptosis in Cultured Neural Stem Cells. Toxicol. Vitro 23, 201–208. doi:10.1016/j.tiv.2008.11.008
Hao, Z.-Y., Liang, D., Luo, H., Liu, Y.-F., Ni, G., Zhang, Q.-J., et al. (2012). Bioactive Sesquiterpenoids from the Rhizomes of Acorus calamus. J. Nat. Prod. 75, 1083–1089. doi:10.1021/np300095c
He, F., Pu, J.-X., Huang, S.-X., Wang, Y.-Y., Xiao, W.-L., Li, L.-M., et al. (2010). Schinalactone A, a New Cytotoxic Triterpenoid from Schisandra Sphenanthera. Org. Lett. 12, 1208–1211. doi:10.1021/ol902974j
Huang, M. F., Fang, S. D., and Li, Y. H. (1982). The Structure of Enshizhisu-A New Lignan Isolated from Schisandra Henryi. Chin. Tradit. Herbal Drugs 13, 22.
Kiem, P. V., Minh, C. V., Nhiem, N. X., Cuc, N. T., Quang, N. V., Tuan Anh, H. L., et al. (2014). Muurolane-type Sesquiterpenes from Marine Sponge Dysidea Cinerea. Magn. Reson. Chem. 52, 51–56. doi:10.1002/mrc.4030
Kim, M.-R., Lee, S.-K., Kim, C.-S., Kim, K.-S., and Moon, D.-C. (2004). Phytochemical Constituents of Carpesium macrocephalum F(R). et S(AV). Arch. Pharm. Res. 27, 1029–1033. doi:10.1007/BF02975426
Latip, J., Hartley, T. G., and Waterman, P. G. (1999). Lignans and Coumarins Metabolites from Melicopehayesii. Phytochemistry 51, 107–110. doi:10.1016/S0031-9422(98)00720-1
Lee, J., Lee, Y., Oh, S.-M., Yi, J.-M., Kim, N., and Bang, O.-S. (2013). Bioactive Compounds from the Roots of Asiasarum Heterotropoides. Molecules 19, 122–138. doi:10.3390/molecules19010122
Li, H., Wang, L., Miyata, S., and Kitanaka, S. (2008). Kadsuracoccinic Acids A−C, Ring-A Seco-Lanostane Triterpenes from Kadsura Coccinea and Their Effects on Embryonic Cell Division of Xenopus laevis. J. Nat. Prod. 71, 739–741. doi:10.1021/np700739t
Li, R.-T., Han, Q.-B., Zhao, A.-H., and Sun, H.-D. (2003). Micranoic Acids A and B: Two New Octanortriterpenoids from Schisandra Micrantha. Chem. Pharm. Bull. 51, 1174–1176. doi:10.1002/chin.20041217910.1248/cpb.51.1174
Lin, Q.-M., Wang, Y., Yu, J.-H., Liu, Y.-L., Wu, X., He, X.-R., et al. (2019). Tyrosinase Inhibitors from the Leaves of Eucalyptus Globulus. Fitoterapia 139, 104418. doi:10.1016/j.fitote.2019.104418
Liu, C.-C., Abd El-Razek, M., Liaw, C.-C., Cheng, Y.-B., Chen, C.-K., Chien, C.-T., et al. (2010). Arisanschinins A-E, Lignans fromSchisandra arisanensisHay. Planta Med. 76, 1605–1610. doi:10.1055/s-0029-1241014
Liu, J.-S., Huang, M.-F., Arnold, G. F., Arnold, E., Clardy, J., and Ayer, W. A. (1983a). Schisanlactone A, a New Type of Triterpenoid from a. Tetrahedron Lett. 24, 2351–2354. doi:10.1016/S0040-4039(00)81923-110.1016/s0040-4039(00)81922-x
Liu, J.-S., Huang, M.-F., Ayer, W. A., and Bigam, G. (1983b). Schisanlactone B, a New Triterpenoid from a. Tetrahedron Lett. 24, 2355–2358. doi:10.1016/s0040-4039(00)81923-1
Liu, J. S., and Huang, M. F. (1991). Isolation and Structures of Schisanlactone E and Changnanic Acid. Acta Chim. Sinica 05, 502–506.
Liu, R. H., Lin, S., Zhang, P. Z., Chen, L. Y., Mei, D. Y., Shao, F., et al. (2018). Flavanoids from the Heartwood of Dalbergia Latifolia. J. Chin. Med. Mat. 41, 617–619. doi:10.13863/j.issn1001-4454.2018.03.023
Liu, Y.-B., Yang, Y.-P., Yuan, H.-W., Li, M.-J., Qiu, Y.-X., Choudhary, M. I., et al. (2018). A Review of Triterpenoids and Their Pharmacological Activities from Genus Kadsura. Digital Chin. Med. 1, 247–258. doi:10.1016/S2589-3777(19)30032-1
Lu, Y., and Chen, D.-F. (2008). Kadsutherin D, a New Dibenzocyclooctadiene Lignan fromKadsuraspecies. Nat. Product. Res. 22, 1344–1349. doi:10.1080/14786410601133525
Luo, Y.-Q., Liu, M., Wen, J., Wang, W.-G., Hu, K., Li, X.-N., et al. (2017). Dibenzocyclooctadiene Lignans from Kadsura Heteroclita. Fitoterapia 119, 150–157. doi:10.1016/j.fitote.2017.04.013
Luo, Y. P., Wang, S. J., Zhao, J. F., Yang, X. D., and Li, L. (2009). Study on the Chemical Constituents of Kadsura Heteroclita. J. Yunnan Univ. 31, 406–409. doi:10.3321/j.issn:0258-7971.2009.04.017
Minh, P. T., Lam, D. T., Tien, N. Q., Tuan, N. N., Nhung, V. P., Van Hai, N., et al. (2014). New Schiartane-type Triterpene from Kadsura Heteroclita and Their Cytotoxic Activities. Nat. Prod. Commun. 9, 373–374. doi:10.1177/1934578x1400900324
Pang, M.-j., Yang, Z., Zhang, X.-l., Liu, Z.-f., Fan, J., and Zhang, H.-y. (2016). Physcion, a Naturally Occurring Anthraquinone Derivative, Induces Apoptosis and Autophagy in Human Nasopharyngeal Carcinoma. Acta Pharmacol. Sin. 37, 1623–1640. doi:10.1038/aps.2016.98
Pettit, G. R., Numata, A., Cragg, G. M., Herald, D. L., Takada, T., Iwamoto, C., et al. (2000). Isolation and Structures of Schleicherastatins 1−7 and Schleicheols 1 and 2 from the Teak Forest Medicinal Tree Schleichera Oleosa. J. Nat. Prod. 63, 72–78. doi:10.1021/np990346r
Ragasa, C. Y., Kristin C. Co, A. L., and Rideout, J. A. (2005). Antifungal Metabolites from Blumea Balsamifera. Nat. Product. Res. 19, 231–237. doi:10.1080/14786410410001709773
Sankaram, A. V. B., Reddy, N. S., and Shoolery, J. N. (1981). New Sesquiterpenoids of Bombax Malabaricum. Phytochemistry 20, 1877–1881. doi:10.1016/0031-9422(81)84026-5
Shehla, N., Li, B., Cao, L., Zhao, J., Jian, Y., Daniyal, M., et al. (2020). Xuetonglactones A-F: Highly Oxidized Lanostane and Cycloartane Triterpenoids from Kadsura Heteroclita Roxb. Craib. Front. Chem. 7, 935–947. doi:10.3389/fchem.2019.00935
Shen, Y.-C., Lin, Y.-C., Chiang, M. Y., Yeh, S. F., Cheng, Y.-B., and Liao, C.-C. (2005). Kadsuphilactones A and B, Two Novel Triterpene Dilactones from Kadsura Philippinensis. Org. Lett. 7, 5348. doi:10.1021/ol052226i
Talapatra, B., Das, A. K., and Talapatra, S. K. (1989). Defuscin, a New Phenolic Ester from Dendrobium Fuscescens: Conformation of Shikimic Acid. Phytochemistry 28, 290–292. doi:10.1016/0031-9422(89)85065-4
Tan, R., Xue, H., and Li, L.-N. (1991). Kadsulactone and Kadsudilactone, Two New Triterpenoid Lactones fromKadsuraSpecies. Planta Med. 57, 87–88. doi:10.1055/s-2006-960031
Tezuka, Y., Kasimu, R., Li, J. X., Basnet, P., Tanaka, K., Namba, T., et al. (1998). Constituents of Roots of Salvia Deserta SCHANG. (Xinjiang-Danshen). Chem. Pharm. Bull. 46, 107–112. doi:10.1248/cpb.46.107
Tseng, Y.-J., Shen, K.-P., Lin, H.-L., Huang, C.-Y., Dai, C.-F., and Sheu, J.-H. (2012). Lochmolins A-G, New Sesquiterpenoids from the Soft Coral Sinularia Lochmodes. Mar. Drugs 10, 1572–1581. doi:10.3390/md10071572
Wang, M., Jiang, S., Yuan, H., Zafar, S., Hussain, N., Jian, Y., et al. (2021). A Review of the Phytochemistry and Pharmacology of Kadsura Heteroclita, an Important Plant in Tujia Ethnomedicine. J. Ethnopharmacology 268, 113567. doi:10.1016/j.jep.2020.113567
Wang, W., Liu, J., Han, J., Xu, Z., Liu, R., Liu, P., et al. (2006a). New Triterpenoids fromKadsura Heteroclitaand Their Cytotoxic Activity. Planta Med. 72, 450–457. doi:10.1055/s-2005-916263
Wang, W., Liu, J., Liu, R., Xu, Z., Yang, M., Wang, W., et al. (2006b). Four New Lignans from the Stems ofKadsura Heteroclita. Planta Med. 72, 284–288. doi:10.1055/s-2005-916213
Zhang, H.-J., Tan, G. T., Santarsiero, B. D., Mesecar, A. D., Hung, N. V., Cuong, N. M., et al. (2003). New Sesquiterpenes from Litsea Verticillata. J. Nat. Prod. 66, 609–615. doi:10.1021/np020508a
Keywords: Kadsura heteroclita, triterpenoids, lignans, anti-RAFLS activity, hepatoprotective activity
Citation: Wang M, Jiang S, Hussain N, Zafar S, Xie Q, Huang F, Mao L, Li B, Jian Y and Wang W (2022) Anti-RAFLS Triterpenoids and Hepatoprotective Lignans From the Leaves of Tujia Ethnomedicine Kadsura heteroclita (Xuetong). Front. Chem. 10:878811. doi: 10.3389/fchem.2022.878811
Received: 18 February 2022; Accepted: 11 April 2022;
Published: 10 May 2022.
Edited by:
Cheng-Peng Sun, Dalian Medical University, ChinaCopyright © 2022 Wang, Jiang, Hussain, Zafar, Xie, Huang, Mao, Li, Jian and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuqing Jian, Y3B1anlxMjAxMEAxNjMuY29t; Wei Wang, d2FuZ3dlaTQwMkBob3RtYWlsLmNvbQ==