
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Chem., 13 August 2019
Sec. Organic Chemistry
Volume 7 - 2019 | https://doi.org/10.3389/fchem.2019.00568
 Carmine Ostacolo1†
Carmine Ostacolo1† Veronica Di Sarno2†Simona Musella3Tania Ciaglia2Vincenzo Vestuto2
Veronica Di Sarno2†Simona Musella3Tania Ciaglia2Vincenzo Vestuto2 Giacomo Pepe2Fabrizio Merciai2
Giacomo Pepe2Fabrizio Merciai2 Pietro Campiglia2,3
Pietro Campiglia2,3 Isabel M. Gomez Monterrey1*
Isabel M. Gomez Monterrey1* Alessia Bertamino2*
Alessia Bertamino2*Ultrasound-promoted N-aminomethylation of indoles can be achieved in basic medium using sodium hydride and dichloromethane (DCM) as C1 donor source. This innovative amino methylation protocol results in good to excellent yields of multifunctional indole derivatives. The procedure is also applicable to other aza-heterocyclic compounds and, interestingly, affords direct access to aminomethyl-substituted aryl alcohols.
The indole nucleus is present in a wide range of bioactive natural products and it is considered as privileged structure in the fields of pharmaceutical and material chemistry (Barden, 2010). Research of new synthetic metal- or organo-catalyzed methodologies for the rapid construction of functionalized indole has seen relevant progress in recent years (Patil and Yamamoto, 2008; Bandini and Eichholzer, 2009; Bartoli et al., 2010; Cacchi and Fabrizi, 2011; Dalpozzo, 2015; Leitch et al., 2017). Indole amino methylation, one of the most important methods for the direct formation of C–C and C–N bonds (Hwang and Uang, 2002; Ibrahem et al., 2004; Murai et al., 2012; Fujii et al., 2014; Nagae et al., 2015; Xu et al., 2015; Kim and Hong, 2017; Mastalir et al., 2017; Mondal et al., 2017), continues to be a challenge for chemists, especially indole aminomethylation at N-1 position. Mannich and Mannich-type Friedel–Crafts reactions, the later using imines, N,O acetals or N,N aminals in the presence of a Lewis acid, constitute the most commonly used chemical approaches for the construction of aminomethylated indoles (Chart 1; Swaminathan and Narasimhan, 1966; Katritzky et al., 1990; Matsumoto et al., 1993; Arend et al., 1998; Saaby et al., 2000; Speckamp and Moolenaar, 2000; Sakai et al., 2003, 2010, 2014; Jiang and Huang, 2005; Lindquist et al., 2006; Wang et al., 2006; Kang et al., 2007; Rowland et al., 2007; Alonso et al., 2008; Zou et al., 2015; Xie et al., 2018). However, both approaches are limited by the well-known regioselectivity toward the C-3 position when 1,3-non-substituted indoles are used (Sakai et al., 2014). The classic Mannich reaction at low temperatures (0–5°C) resulted in the high-yield synthesis of isogramines and derivatives (Katritzky et al., 1990). Under tBuOK-promoted basic conditions, Love and Nguyen (1998) and Love (2007) described the regioselective formation of the N-1 derivative using the reaction of unprotected indole with 1-(N,N'–dialkylaminomethyl) benzotriazoles as alkylating agents. Sakai et al. (2010) showed that, in the reaction of indoles with N,O acetals, the use of Hf(OTf)4 as Lewis acid regioselectively promoted N-aminomethylation. Mastalir et al. (2017) obtained an N1 derivative in basic medium by reaction of indole with a secondary amine using a manganese-based catalyst and methanol as C1 donor source. However, all these methods require highly controlled conditions or the presence of specific catalysts.
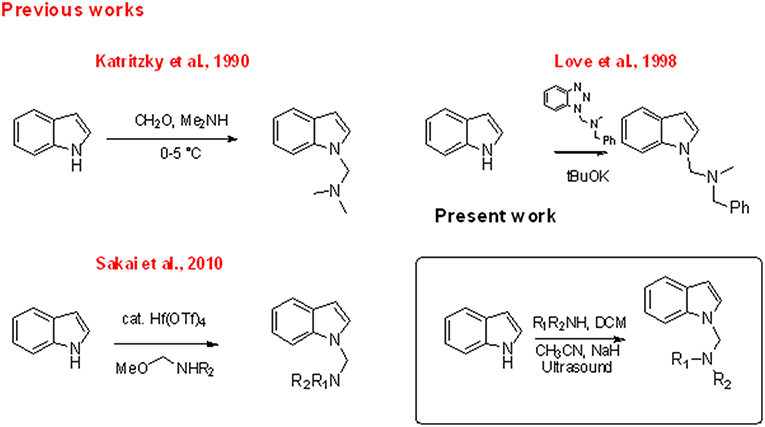
CHART 1. Aminomethylation of N-1 indole position.
Reagents, starting materials, and solvents were purchased from Sigma-Aldrich (Milan, Italy) and used as received. Reactions were carried out with magnetic stirring in 25 mL round-bottomed or in falcon tubes (10 mL). Ultrasonication was performed in a Bandelin Sonorex Digital 10P ultrasonic bath with a frequency of 60 Hz and power of 240 W. Microwave assisted closed vessel reactions were performed in a Biotage Initiator+ reactor, using 10 mL vials type and external temperature sensor. Analytical thin layer chromatography (TLC) was performed on pre-coated glass silica gel plates 60 (F254, 0.25 mm, VWR International). UHPLC analyses were performed on a Nexera UHPLC system (Shimadzu, Kyoto, Japan) consisting of a CBM-20A controller, two LC-30AD dual-plunger parallel-flow pumps, a DGU-20 AR5 degasser, an SPD-M20A photo diode array detector (equipped with a 2.5 μL detector flow cell volume), a CTO-20A column oven, a SIL-30AC autosampler. The chromatographic profile was obtained on a Kinetex™ C18 150 × 2.1 mm × 2.6 μm (100 Å) column (Phenomenex, Bologna, Italy). The optimal mobile phase consisted of 0.1% TFA/H2O v/v (A) and 0.1% TFA/ACN v/v (B). Analysis was performed in gradient elution as follows: 0–13.00 min, 5–65% B; 13–14.00 min, 65–95% B; 14–15.00 min, isocratic to 95% B; 15–15.01 min, 95–5% B; then 3 min for column re-equilibration. Flow rate was 0.5 mL min−1. Column oven temperature was set to 45°C. Injection volume was 2 μL of sample. The following PDA parameters were applied: sampling rate, 12.5 Hz; detector time constant, 0.160 s; cell temperature, 40°C. Data acquisition was set in the range 190–800 nm and chromatograms were monitored at 254 nm. For the quantification of main chromatographic peaks, indole was selected as external standard. Stock solution (1 mg mL−1) was prepared in methanol, the calibration curve was obtained in a concentration range of 250–10.0 μg mL−1 with six concentration levels and triplicate injection of each level were run. Peak areas of indole derivatives were plotted against corresponding concentrations (μg mL−1) and the linear regression was used to generate calibration curve (y = 0.00024x−1.39094) with R2 values was ≥ 0.9999. Purifications were conducted on the Biotage Isolera One flash purification system, using pre-packed KP-sil columns (Biotage, Uppsala, Sweden). 1D and 2D NMR spectra were recorded with Bruker Avance (400 MHz) spectrometer, at room temperature. Spectra were referenced to residual chloroform (7.24 ppm, 1H; 77.23 ppm, 13C) or methanol (3.31 ppm, 1H; 49.15 ppm, 13C). Chemical shifts are reported in δ values (ppm) relative to internal Me4Si, and J values are reported in hertz (Hz). The following abbreviations are used to describe peaks: s (singlet), d (doublet), dd (double doublet), t (triplet), bs (broad singlet), and m (multiplet). HR-MS experiments were performed by an LTQ-Orbitrap-XL-ETD mass spectrometer (Thermo Scientific, Bremen, Germany), using electrospray ionization. Elemental analysis was performed by the FlashSmart Elemental Analyzer (Thermo Fisher Scientific, Waltham, MA USA).
Indole (1 mmol), base (2 mmol), piperidine (1.5 mmol) were mixed in different solvents (5 mL) under the conditions reported in Table 1. After the time indicated in Table 1, the reaction was quenched with 5 mL of a 10% citric acid solution and the organic solvents were evaporated in vacuo. The crude was dissolved in DCM (20 mL) and extracted with water (3 × 20 mL). Compounds 2, 3, and 4 were obtained after flash chromatography, using 1/4 ethyl acetate/n-hexane as eluent mixture.
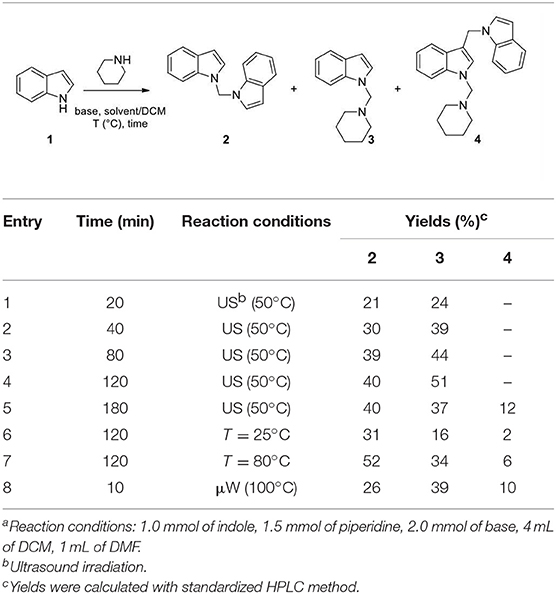
Table 1. Reaction of indole with piperidine using different approachesa.
Substrates (1 mmol) were dissolved in acetonitrile (5 mL) in a falcon tubes (10 mL) and sodium hydride (2 mmol), amines (1.5 mmol), and dichloromethane (3 mmol) were added. The mixture was introduced in an ultrasonic bath setting the temperature at 50°C and irradiating for 120 min. Then, the work up of the reaction and the purification of final compounds were performed as described above. The NMR spectra of synthesized compounds are depicted in Figures S1–S60.
Rf = 0.70 (ethyl acetate/n-hexane 1/4). (Yield = 81.2 mg, 33%). 1H NMR (CDCl3, 400 MHz): δ: 6.37 (s, 2H, CH2); 6.57 (s 2H, aryl); 7.15–7.19 (m, 4H, aryl); 7.27 (t, 2H, aryl, J = 6.9 Hz); 7.49 (d, 2H, aryl, J = 8.2 Hz); 7.65 (d, 2H, aryl, J = 7.9 Hz). 13C NMR (CDCl3, 100 MHz) δ: 56.4; 103.4; 109.2; 120.3; 121.3; 122.5; 127.0; 129.1; 135.8. Elemental analysis calcd (%) for C17H14N2: C 82.90, H 5.73, N 11.37; found: C 83.06, H 5.70, N 11.31.
Rf = 0.40 (ethyl acetate/n-hexane 1/4). (Yield = 130.6 mg, 61%). 1H NMR (CDCl3, 400 MHz): δ: 1.29 (bs, 2H, CH2 piperidin); 1.48–1.51 (m, 4H, CH2 piperidin); 2.45 (t, 4H, CH2 piperidin, J = 4.7 Hz); 4.78 (s, 2H, CH2); 6.43 (d, 1H, aryl, J = 3.0 Hz); 7.02 (t, 1H, aryl, J = 7.1 Hz); 7.07 (d, 1H, aryl, J = 3.0 Hz); 7.13 (t, 1H, aryl, J = 7.1 Hz); 7.40 (d, 1H, aryl, J = 8.2 Hz); 7.55 (d, 1H, aryl, J = 7.8 Hz). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 51.8; 68.6; 101.3; 110.1; 119.5; 120.7; 121.6; 128.5; 128.8; 137.1. HR-MS m/z: calcd for C14H19N2, [(M+H)+]: 215.1543; found 215.1550. Elemental analysis calcd (%) for C14H18N2: C 78.46, H 8.47, N 13.07; found: C 78.54, H 8.40, N 13.11.
Obtained from indole and piperidine at 180 min. Rf = 0.15 (ethyl acetate/n-hexane 1/4). (Yield = 41.2 mg, 12%). 1H NMR (CDCl3, 400 MHz): δ: 1.31–1.37 (m, 2H, CH2 piperidine); 1.51–1.57 (m, 4H, CH2 piperidine); 2.46 (bs, 4H, CH2 piperidine); 4.77 (s, 2H, CH2); 5.45 (s, 2H, CH2); 6.46 (d, 1H, aryl, J = 3.08 Hz); 7.02 (s, 1H, aryl); 7.04–7.13 (m, 3H, aryl); 7.16–7.21 (m, 2H, aryl); 7.42–7.46 (m, 3H, aryl): 7.62 (d, 1H, aryl, J = 7.8 Hz). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.7; 41.9; 51.8; 68.5; 101.1; 109.6; 110.4; 118.8; 119.3; 119.8; 120.9; 121.3; 122.2; 127.0; 127.7; 127.8; 128.8; 137.7. HR-MS m/z: calcd for C23H26N3, [(M+H)+]: 344.2121; found 344.2130. Elemental analysis calcd (%) for C23H25N3: C 80.43, H 7.34, N 12.23; found: C 80.51, H 7.29, N 12.25.
Rf = 0.55 (ethyl acetate/n-hexane 1/9). (Yield = 62.7 mg, 19%). 1H NMR (CDCl3, 400 MHz): δ: 1.52–1.54 (m, 2H, CH2 piperidin); 1.64 (t, 4H, CH2 piperidin, J = 5.0 Hz); 2.48 (t, 4H, CH2 piperidin, J = 5.0 Hz); 6.57 (d, 2H, aryl, J = 3.0 Hz); 6.85 (s, 1H, CH); 7.15 (t, 2H, aryl, J = 7.6 Hz); 7.23 (t, 2H, aryl, J = 7.6 Hz); 7.48 (d, 2H, aryl, J = 3.1 Hz); 7.53 (d, 2H, aryl, J = 8.2 Hz); 7.63 (d, 2H, aryl, J = 7.8 Hz). 13C NMR (CDCl3, 100 MHz) δ: 24.5; 25.7; 50.5; 82.4; 103.4; 109.9; 120.3; 121.1; 122.3; 124.9; 129.0; 135.9. HR-MS m/z: calcd for C22H24N3, [(M+H)+]: 330.1965; found 330.1977. Elemental analysis calcd (%) for C22H23N3: C 80.21, H 7.04, N 12.76; found: C 80.29, H 6.99, N 12.83.
Obtained from indole and morpholine. Rf = 0.35 (ethyl acetate/n-hexane 1/4). (Yield = 147.0 mg, 68%). 1H NMR (CDCl3, 400 MHz): δ: 2.57 (t, 4H, CH2 morpholine, J = 4.3 Hz); 3.72 (t, 4H, CH2 morpholine, J = 4.3 Hz); 4.81 (s, 2H, CH2); 6.56 (d, 1H, aryl, J = 3.0 Hz); 7.14–7.18 (m, 2H, aryl); 7.27 (t, 1H, aryl, J = 8.4 Hz); 7.50 (d, 1H, aryl, J = 8.2 Hz); 7.67 (d, 1H, aryl, J = 7.7 Hz). 13C NMR (CDCl3, 100 MHz) δ: 50.9; 66.7; 68.3; 101.9; 110.1; 119.8; 120.9; 121.8; 128.4; 128.8; 135.2. HR-MS m/z: calcd for C13H17N2O, [(M+H)+]: 217.1335; found 217.1339. Elemental analysis calcd (%) for C13H16N2O: C 72.19, H 7.46, N 12.95, O 7.40; found: C 72.16, H 7.51, N 13.04, O 7.47.
Obtained from indole and benzylamine. Rf = 0.25 (ethyl acetate/n-hexane 1/6). (Yield = 163.0 mg, 69%). 1H NMR (CDCl3, 400 MHz): δ: 3.59 (s, 2H, CH2 benzyl); 4.99 (s, 2H, CH2); 6.45 (d, 1H, aryl, J = 3.1 Hz); 7.04–7.08 (m, 2H, aryl); 7.12–7.31 (m, 7H, aryl); 7.59 (d, 1H, aryl, J = 7.8 Hz). 13C NMR (CDCl3, 100 MHz) δ: 50.0; 59.8; 101.5; 109.3; 119.7; 121.2; 121.7; 127.2; 127.9; 128.2; 128.5; 129.1; 135.8; 139.2. HR-MS m/z: calcd for C17H19N2, [(M+H)+]: 251.1543; found 251.1548. Elemental analysis calcd (%) for C16H16N2: C 81.32, H 6.82, N 11.85, found: C 81.41, H 6.90, N 11.87.
Obtained from indole and phenethylamine. Rf = 0.30 (ethyl acetate/n-hexane 1/4). (Yield = 177,6 mg, 71%). 1H NMR (CDCl3, 400 MHz): δ: 2.72 (t, 2H, CH2 ethyl, J = 6.8 Hz); 2.83 (t, 2H, CH2 ethyl, J = 6.8 Hz); 5.08 (s, 2H, CH2); 6.53 (d, 1H, aryl, J = 3.0 Hz); 7.11 (d, 2H, aryl, J = 7.2 Hz); 7.14–7.28 (m, 6H, aryl); 7.39 (d, 1H, aryl, J = 8.2 Hz); 7.68 (d, 1H, aryl, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz) δ: 36.2; 47.6; 60.7; 101.5; 109.2; 119.7; 121.1; 121.7; 126.3; 127.7; 128.5; 128.7; 129.0; 135.8; 139.4. HR-MS m/z: calcd for C17H19N2, [(M+H)+]: 251.1543; found 251.1548. Elemental analysis calcd (%) for C17H18N2: C 81.56, H 7.25, N 11.19; found: C 81.50, H 7.31, N 11.14.
Obtained from indole and 4-methoxyaniline. Rf = 0.50 (ethyl acetate/n-hexane 1/4). (Yield = 106.0 mg, 42%). 1H NMR (CDCl3, 400 MHz): δ: 3.75 (s, 3H, OCH3); 5.50 (s, 2H, CH2); 6.50 (d, 1H, aryl, J = 2.9 Hz); 6.66 (d, 2H, aryl, J = 8.8 Hz); 6.78 (d, 2H, aryl, J = 8.8 Hz); 7.17 (t, 1H, aryl, J = 7.3 Hz); 7.22 (d, 1H, aryl, J = 3.0 Hz); 7.27 (t, 1H, aryl, J = 7.6 Hz); 7.47 (d, 1H, aryl, J = 8.2 Hz); 7.66 (d, 1H, aryl, J = 7.8 Hz). 13C NMR (CDCl3, 100 MHz) δ: 55.7; 57.5; 101.8; 109.3; 115.0; 115.8; 119.8; 121.2; 121.8; 126.8; 129.2; 135.4; 139.7; 153.5. HR-MS m/z: calcd for C16H17N2O, [(M+H)+]: 253.1335; found 253.1341. Elemental analysis calcd (%) for C16H16N2O: C 76.16, H 6.39, N 11.10, O 6.34; found: C 76.18, H 6.42, N 11.03, O 6.40.
Obtained from 5-iodoindole and piperidine. Rf = 0.45 (diethyl ether/n-hexane 1/1). (Yield = 200.6 mg, 59%). 1H NMR (CDCl3, 400 MHz): δ: 1.36–1.41 (m, 2H, CH2 piperidine); 1.56–1.61 (m, 4H, CH2 piperidine); 2.50 (t, 4H, CH2 piperidine, J = 4.6 Hz); 4.82 (s, 2H, CH2); 6.44 (d, 1H, aryl, J = 2.5 Hz); 7.13 (d, 1H, aryl, J = 3.1 Hz); 7.28 (d, 1H, aryl, J = 8.1 Hz); 7.46 (d, 1H, aryl, J = 8.6 Hz); 7.97 (s, 1H, aryl). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 51.8; 68.8; 83.0; 100.6; 112.2; 129.5; 129.9; 131.1; 136.5. HR-MS m/z: calcd for C14H18IN2, [(M+H)+]: 341.0509; found 341.0514. Elemental analysis calcd (%) for C14H17IN2: C 49.43, H 5.04, I 37.30, N 8.23, found: C 49.47, H 5.08, I 37.20, N 8.28.
Obtained from 5-methylindole and piperidine. Rf = 0.55 (ethyl acetate/n-hexane 1/4). (Yield = 150.6 mg, 66%). 1H NMR (CDCl3, 400 MHz): δ: 1.37–1.42 (m, 2H, CH2 piperidine); 1.58–1.63 (m, 4H, CH2 piperidine); 2.50 (s, 3H, CH3); 2.55 (t, 4H, CH2 piperidine, J = 5.0 Hz); 4.86 (s, 2H, CH2); 6.47 (d, 1H, aryl, J = 3.0 Hz); 7.08 (d, 1H, aryl, J = 8.3 Hz); 7.15 (d, 1H, aryl, J = 3.0 Hz); 7.40 (d, 1H, aryl, J = 8.4 Hz); 7.45 (s, 1H, aryl). 13C NMR (CDCl3, 100 MHz) δ: 21.4; 23.9; 25.9; 51.8; 68.7; 100.7; 109.8; 120.4; 123.2; 128.7; 128.9; 135.6. HR-MS m/z: calcd for C15H21N2, [(M+H)+]: 229.1699; found 229.1705. Elemental analysis calcd (%) for C15H20N2: C 78.90, H 8.83, N 12.27; found: C 78.88, H 8.79, N 12.35.
Obtained from 5-methoxyindole and piperidine. Rf = 0.50 (acetate/n-hexane 2/3). (Yield = 183.0 mg, 75%). 1H NMR (CDCl3, 400 MHz): δ: 1.25–1.31 (m, 2H, CH2 piperidine); 1.46–1.52 (m, 4H, CH2 piperidine); 2.43 (t, 4H, CH2 piperidine, J = 5.2 Hz); 3.78 (s, 3H, CH3); 4.72 (s, 2H, CH2); 6.34 (d, 1H, aryl, J = 3.0 Hz); 6.79 (dd, 1H, aryl, J1 = 2,4 Hz; J2 = 6.5 Hz); 7.00 (d, 1H, aryl, J = 2.4 Hz); 7.03 (d, 1H, aryl, J = 3.0 Hz); 7.29 (d, 1H, aryl, J = 8.9 Hz). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 51.8; 55.8; 68.8; 100.8; 102.3; 110.9; 111.9; 128.9; 129.3; 132.4; 154.0. HR-MS m/z: calcd for C15H21N2O, [(M+H)+]: 245.1648; found 245.1655. Elemental analysis calcd (%) for C15H20N2O: C 73.74, H 8.25, N 11.47, O 6.55; found: C 73.77, H 8.19, N 11.53, O 6.59.
Obtained from tert-butyl ((1H-indol-5-yl)methyl)carbamate and piperidine. Rf = 0.35 (dichlorometane/acetate 9/). (Yield = 240.2 mg, 70%). 1H NMR (CDCl3, 400 MHz): δ: 1.28 (bs, 2H, CH2 piperidine); 1.40 (s, 9H, CH3); 1.49 (bs, 4H, CH2 piperidine); 2.43 (bs, 4H, CH2 piperidine); 4.33 (d, 2H, CH2, J = 2.9 Hz); 4.75 (s, 2H, CH2); 6.39 (bs, 1H, aryl); 7.05–7.07 (m, 2H, aryl); 7.35 (d, 1H, aryl, J = 8.2 Hz); 7.45 (s, 1H, aryl). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 28.4; 51.8; 68.7; 101.3; 110.4; 119.9; 121.8; 128.6; 129.4; 129.9; 136.5; 155.9. HR-MS m/z: calcd for C20H30N3O2, [(M+H)+]: 344.2333; found 344.2340. Elemental analysis calcd (%) for C20H29N3O2: C 69.94, H 8.51, N 12.23, O 9.32; found: C 69.99, H 8.45, N 12.18, O 9.37.
Obtained from tert-butyl (2-(1H-indol-3-yl)ethyl)carbamate and piperidine. Rf = 0.35 (ethyl acetate/n-hexane 2/1). (Yield% = 221.5 mg, 62%). 1H NMR (CDCl3, 400 MHz): δ: 1.37–1.40 (m, 2H, CH2 piperidine); 1.47 (s, 9H, CH3); 1.56–1.62 (m, 4H, CH2 piperidine); 2.54 (bs, 4H, CH2 piperidine); 2.97 (t, 2H, CH2, J = 6.2 Hz); 3.48 (bs, 2H, CH2); 4.64 (s, 1H, NH); 4.82 (s, 2H, CH2); 7.01 (s, 1H, aryl); 7.13 (t, 1H, aryl, J = 7.6 Hz); 7.24 (t, 1H, aryl, J = 8.0 Hz); 7.46 (d, 1H, aryl, J = 8.1 Hz); 7.61 (d, 1H, aryl. J = 7.7 Hz). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 28.4; 51.8; 68.5; 110.2; 111.9; 118.8; 119.1; 121.8; 126.8; 127.9; 137.6; 156.0. HR-MS m/z: calcd for C21H31N3O2, [(M+H)+]: 358.2489; found 358.2492. Elemental analysis calcd (%) for C21H31N3O2: C 70.55, H 8.74, N 11.75, O 8.95; found: C 70.43, H 8.69, N 11.74, O 9.01.
Rf = 0.40 (dichloromethane). (Yield = 50.4 mg, 22%). 1H NMR (CDCl3, 400 MHz): δ: 1.35 (bs, 2H, CH2 piperidin); 1.44–1.45 (m, 4H, CH2 piperidin); 2.39 (bs, 7H, CH2 piperidin and CH3); 4.53 (s, 2H, CH2); 6.17 (s, 1H, aryl); 7.00 (t, 1H, aryl, J = 7.5 Hz); 7.04 (d, 1H, aryl, J = 7.0 Hz); 7.32 (d, 1H, aryl, J = 8.0 Hz); 7.42 (d, 1H, aryl, J = 7.6 Hz). 13C NMR (CDCl3, 100 MHz) δ: 13.0; 24.4; 25.7; 51.9; 65.7; 100.7; 109.8; 119.3; 119.4; 120.4; 128.0; 137.5; 138.0. HR-MS m/z: calcd for C15H21N2, [(M+H)+]: 229.1699; found 229.1708. Elemental analysis calcd (%) for C14H18N2: C 78.90, H 8.83, N 12.27; found: C 78.81, H 8.85, N 12.45.
Rf = 0.70 (dichloromethane). (Yield = 85.0 mg, 31%). 1H NMR (CDCl3, 400 MHz): δ: 2.30 (s, 6H, CH3); 6.31–6.33 (m, 4H, CH2 and aryl); 7.10–7.12 (m, 4H, aryl); 7.19–7.21 (m, 2H, aryl); 7.54–7.56 (m, 2H, aryl). 13C NMR (CDCl3, 100 MHz) δ: 13.3; 52.7; 102.5; 109.2; 120.0; 121.4; 128.4; 136.4; 137.0. Elemental analysis calcd (%) for C19H18N2: C 83.18, H 6.61, N 10.21; found: C 83.256, H 6.48, N 10.55.
Obtained from pyrrole and piperidine. Rf = 0.40 (ethyl acetate/n-hexane 1/3). (Yield = 99.0 mg, 74%). 1H NMR (CDCl3, 400 MHz): δ: 1.38–1.41 (m, 2H, CH2 piperidine J = 4.1 Hz); 1.57–1.63 (m, 4H, CH2 piperidine); 2.49 (t, 4H, CH2 piperidine J = 4.8 Hz); 4.65 (s, 2H, CH2); 6.18 (bs, 2H, aryl); 6.70 (bs, 2H, aryl). 13C NMR (CDCl3, 100 MHz) δ: 23.8; 25.9; 51.3; 71.8; 107.8; 121.6. HR-MS m/z: calcd for C10H17N2, [(M+H)+]: 165.1386; found 165.1392. Elemental analysis calcd (%) for C10H16N2: C 73.13, H 9.82, N 17.06, O 8.95; found: C 73.08, H 9.90, N 17.10.
Obtained from carbazole and piperidine. Rf = 0.40 (ethyl acetate/n-hexane 1/3). (Yield = 235.1 mg, 89%). 1H NMR (CDCl3, 400 MHz): δ: 1.43 (bs, 2H, CH2 piperidine); 1.57–1.63 (m, 4H, CH2 piperidine); 2.64 (bs, 4H, CH2 piperidine); 4.97 (s, 2H, CH2); 7.28 (t, 2H, aryl, J = 7.2 Hz); 7.49 (t, 2H, aryl, J = 7.2 Hz); 7.59 (d, 2H, aryl, J = 8.2 Hz); 8.13 (d, 2H, aryl, J = 8.2 Hz). 13C NMR (CDCl3, 100 MHz) δ: 24.2; 25.8; 52.3; 66.1; 109.8; 119.2; 120.1; 123.1; 125.7; 141.5. HR-MS m/z: calcd for C18H21N2, [(M+H)+]: 265.1699; found 265.1706. Elemental analysis calcd (%) for C18H20N2: C 81.78, H 7.63, N 10.60; found: C 81.88, H 7.60, N 10.66.
Obtained from benzimidazole and piperidine. Rf = 0.30 (ethyl acetate/methanol 5/1). (Yield = 94.3 mg, 38%). 1H NMR (CDCl3, 400 MHz): δ: 6.43 (s, 2H, CH2); 7.24–7.29 (m, 4H, aryl); 7.36–7.38 (m, 2H, aryl); 7.74–7.77 (m, 2H, aryl); 8.09 (s, 2H, aryl). 13C NMR (CDCl3,100 MHz) δ: 53.5; 109.2; 121.1; 123.4; 124.3; 132.7; 142.1; 143.9. Elemental analysis calcd (%) for C15H12N4: C 72.56, H 4.87, N 22.57; found: C 72.48, H 4.93, N 22.54.
Obtained from phenol and piperidine. Rf = 0.40 (dichlorometane/methanol 9/1). (Yield = 105.0 mg, 55%). 1H NMR (CDCl3, 400 MHz): δ: 1.42 (bs, 2H, CH2 piperidine); 1.55–1.59 (m, 4H, CH2 piperidine); 2.43 (bs, 4H, CH2 piperidine); 3.59 (s, 2H, CH2); 6.69 (t, 1H, aryl, J = 7.4 Hz); 6.73 (d, 1H, aryl, J = 8.0 Hz); 6.88 (d, 1H, aryl, J = 7.2 Hz); 7.08 (t, 1H, aryl, J = 7.6 Hz). 13C NMR (CDCl3, 100 MHz) δ: 24.0; 25.9; 53.9; 62.2; 116.0; 118.9; 121.7; 128.4; 158.1. HR-MS m/z: calcd for C12H18NO, [(M+H)+]: 192.1383; found 192.1389. Elemental analysis calcd (%) for C12H17NO: C 75.35, H 8.96, N 7.32, O 8.36; found: C 75.40, H 9.02, N 7.27, O 8.30.
Obtained from o-cresol and piperidine. Rf = 0.40 (n-hexane/ethere 2/1). (Yield = 119.0 mg, 58%). 1H NMR (CD3OD, 400 MHz): δ: 1.46 (bs, 2H, CH2 piperidine); 1.57–1.61 (m, 4H, CH2 piperidine); 2.09 (s, 3H, CH3); 2.62 (bs, 4H, CH2 piperidine); 3.73 (s, 2H, CH2); 6.61 (t, 1H, aryl, J = 7.5 Hz); 6.82 (d, 1H, aryl, J = 7.3 Hz); 6.95 (d, 1H, aryl, J = 7.4 Hz). 13C NMR (CD3OD, 100 MHz) δ: 14.6; 23.0; 24.8; 53.2; 59.9; 118.8; 119.6; 124.4; 127.1; 130.3; 155.3. HR-MS m/z: calcd for C13H20NO, [(M+H)+]: 206.1539; found 206.1543. Elemental analysis calcd (%) for C13H19NO: C 76.06, H 9.33, N 6.82, O 7.79; found: C 76.00, H 9.39, N 6.75, O 7.87.
Obtained from p-cresol and piperidine. Rf = 0.40 (ethyl acetate/n-hexane 1/3). (Yield = 125.1 mg, 61%). 1H NMR (CDCl3, 400 MHz): δ: 1.42 (bs, 2H, CH2 piperidine); 1.54–1.58 (m, 4H, CH2 piperidine); 2.16 (s, 3H, CH3); 2.43 (bs, 4H, CH2 piperidine); 3.55 (s, 2H, CH2); 6.63 (d, 1H, aryl, J = 8.1 Hz); 6.69 (s, 1H, aryl); 6.88 (d, 1H, aryl, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz) δ: 20.4; 24.0; 25.9; 53.9; 62.2; 115.7; 121.3; 127.9; 128.9; 155.7. HR-MS m/z: calcd for C13H20NO, [(M+H)+]: 206.1539; found 206.1547. Elemental analysis calcd (%) for C13H19NO: C 76.06, H 9.33, N 6.82, O 7.79; found: C 76.14, H 9.36, N 6.87, O 7.70.
Obtained from 2,4-dichlorophenol and piperidine. Rf = 0.50 (ethyl acetate/n-hexane 1/3). (Yield = 108.8 mg, 42%). 1H NMR (CD3OD, 400 MHz): δ: 1.46–1.50 (m, 2H, CH2 piperidine); 1.59–1.64 (m, 4H, CH2 piperidine); 2.62 (bs, 4H, CH2 piperidine); 3.75 (s, 2H, CH2); 6.91 (d, 1H, aryl, J = 2.5 Hz); 7.16 (d, 1H, aryl, J = 2.5 Hz). 13C NMR (CD3OD, 100 MHz) δ: 22.9; 24.8; 52.9; 60.0; 121.6; 123.0; 127.2; 128.3; 154.6. HR-MS m/z: calcd for C12H16Cl2NO, [(M+H)+]: 260.0603; found 260.0608. Elemental analysis calcd (%) for C12H15Cl2NO: C 55.40, H 5.81, Cl 27.25, N 5.38, O 6.15; found: C 55.44, H 5.74, Cl 27.30, N 5.43, O 6.11.
Obtained from o-tert-butylphenol and piperidine. Rf = 0.50 (ethyl acetate/n-hexane 1/3). (Yield% = 163.0 mg, 66%). 1H NMR (CD3OD, 400 MHz): δ: 1.29 (s, 9H, CH3); 1.44–1.50 (m, 2H, CH2 piperidine); 1.60–1.66 (m, 4H, CH2 piperidine); 2.73 (bs, 4H, CH2 piperidine); 3.72 (s, 2H, CH2); 6.65 (d, 1H, aryl, J = 8.1 Hz); 6.95 (d, 1H, aryl, J = 8.1 Hz); 7.15 (s, 1H, aryl). 13C NMR (CD3OD, 100 MHz) δ: 22.5; 23.7; 28.5; 34.2; 52.9; 61.8; 115.8; 122.1; 129.1; 136.2; 157.0. HR-MS m/z: calcd for C16H26NO, [(M+H)+]: 248.2009; found 248.2013. Elemental analysis calcd (%) for C16H25NO: C 77.68, H 10.19, N 5.66, O 6.47; found: C 77.72, H 10.22, N 5.63, O 6.50.
Obtained from o-tert-butylphenol and piperidine. Rf = 0.65 (ethyl acetate/n-hexane 1/3). (Yield = 34.3 mg, 11%). 1H NMR (CDCl3, 400 MHz): δ: 1.47 (s, 18H, CH3); 5.87 (s, 2H, CH2); 7.00–7.04 (m, 2H, aryl); 7.22–7.26 (m, 2H, aryl); 7.32 (d, 2H, aryl, J = 7.2 Hz); 7.37 (dd, 2H, aryl, J1 = 6.4 Hz; J2 = 1.4 Hz). 13C NMR (CDCl3, 100 MHz) δ: 30.1; 34.9; 91.5; 114.8; 122.0; 126.9; 127.2; 138.6; 156.5. Elemental analysis calcd (%) for C21H28O2: C 80.73, H 9.03, O 10.24; found: C 80.59, H 9.12, O 10.28.
Obtained from 2,6-diisopropylphenol and piperidine. Rf = 0.55 (ethyl acetate/n-hexane 1/4). (Yield = 246.7 mg, 67%). 1H NMR (CDCl3, 400 MHz): δ: 1.24 (d, 24H, CH3, J = 6.8 Hz); 3.40–3.47 (m, 4H, CH); 5.26 (s, 2H, CH2); 7.18 (s, 6H, aryl). 13C NMR (CDCl3,100 MHz) δ: 23.9; 26.7; 100.8; 124.0; 125.2; 142.2; 151.0. Elemental analysis calcd (%) for C25H36O2: C 81.47, H 9.85, O 8.68; found: C 81.60, H 9.79, O 8.59.
Obtained from 1-naphthol and piperidine. Rf = 0.50 (ethyl acetate/n-hexane 1/5). (Yield = 217.0 mg, 90%). 1H NMR (CDCl3, 400 MHz): δ: 1.45 (bs, 2H, CH2 piperidine); 1.59–1.62 (m, 4H, CH2 piperidine); 2.50 (bs, 4H, CH2 piperidine); 3.74 (s, 2H, CH2); 6.98 (d, 1H, aryl, J = 8.2 Hz); 7.20 (d, 1H, aryl, J = 8.5 Hz); 7.35–7.37 (m, 2H, aryl); 7.67 (d, 1H, aryl, J = 5.2 Hz); 8.16 (d, 1H, aryl, J = 5.2 Hz). 13C NMR (CDCl3, 100 MHz) δ: 24.1; 25.9; 62.4; 113.9; 118.0; 122.0; 124.7; 125.0; 125.8; 126.5; 127.3; 133.8; 153.8. HR-MS m/z: calcd for C16H20NO, [(M+H)+]: 242.1539; found 242.1546. Elemental analysis calcd (%) for C16H19NO: C 79.63, H 7.94, N 5.80, O 6.63; found: C 79.70, H 8.03, N 5.72, O 6.59.
Obtained from 2-naphthol and piperidine. Rf = 0.50 (ethyl acetate/n-hexane 1/5). (Yield = 226.7 mg, 94%). 1H NMR (CDCl3, 400 MHz): δ: 1.46 (bs, 2H, CH2 piperidine); 1.61–1.65 (m, 4H, CH2 piperidine); 2.52 (bs, 4H, CH2 piperidine); 4.05 (s, 2H, CH2); 7.01 (d, 1H, aryl, J = 8.8 Hz); 7.21 (d, 1H, aryl, J = 7.8 Hz); 7.35 (t, 1H, aryl, J = 8.1 Hz); 7.60 (d, 1H, aryl, J = 8.8 Hz); 7.67 (d, 1H, aryl, J = 8.0 Hz); 7.72 (d, 1H, aryl, J = 8.6 Hz). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 57.1; 111.0; 119.3; 120.9; 122.3; 126.2; 128.4; 128.9; 129.1; 132.7; 156.8. HR-MS m/z: calcd for C16H20NO, [(M+H)+]: 242.1539; found 242.1548. Elemental analysis calcd (%) for C16H19NO: C 79.63, H 7.94, N 5.80, O 6.63; found: C 79.68, H 8.00, N 5.83, O 6.67.
Obtained from isoquinolin-5-ol and piperidine. Rf = 0.40 (ethyl acetate/n-hexane 1/3). (Yield = 210.7 mg, 87%). 1H NMR (CDCl3, 400 MHz): δ: 1.47 (bs, 2H, CH2 piperidine); 1.61–1.63 (m, 4H, CH2 piperidine); 2.51 (bs, 4H, CH2 piperidine); 3.78 (s, 2H, CH2); 7.10 (d, 1H, aryl, J = 8.0 Hz); 7.31 (d, 1H, aryl, J = 8.0 Hz); 7.91 (d, 1H, aryl, J = 8.0 Hz); 8.41 (d, 1H, aryl, J = 8.0 Hz); 9.07 (s, 1H, aryl). 13C NMR (CDCl3, 100 MHz) δ: 23.9; 25.8; 62.3; 115.1; 117.4; 118.1; 127.6; 127.8; 128.9; 142.0; 151.8; 153.2. HR-MS m/z: calcd for C15H19N2O, [(M+H)+]: 243.1492; found 243.1500. Elemental analysis calcd (%) for C15H18N2O: C 74.35, H 7.49, N 11.56, O 6.60; found: C 74.41, H 7.62, N 11.48, O 6.65.
Obtained from thiophenol and piperidine. Rf = 0.40 (n-hexane). (Yield% = 92.8 mg, 40%). 1H NMR (CDCl3, 400 MHz): δ: 4.38 (s, 2H, CH2); 7.28 (t, 2H, aryl, J = 7.1 Hz); 7.33 (t, 4H, aryl, J = 7.2 Hz); 7.46 (d, 4H, aryl, J = 7.5 Hz). 13C NMR (CDCl3,100 MHz) δ: 40.7; 127.2; 129.0; 130.8; 135.0. Elemental analysis calcd (%) for C13H12S2: C 67.20, H 5.21, S 27.60; found: C 67.33, H 5.18, S 27.79.
When we performed N-methylation reactions of non-substituted indole using CH3I in sodium hydride/DCM/DMF solution assisted by ultrasound irradiation (US), we observed the almost exclusive formation of 1-diindolylmethane (86% of yield), which suggested that DCM is a bridging agent in the formation of this N-aminomethylated compound (Mills et al., 1987, 2009; Matsumoto et al., 1993; Souquet et al., 2006; Rudine et al., 2010; Zhou et al., 2011). In an attempt to capitalize on DCM behavior, we introduced a secondary amine, specifically piperidine, in the reaction. In this case, we observed the formation of 1-diindolylmethane 2 and -(piperidin-1-ylmethyl)-1H-indole 3, which were isolated in yields of 40 and 51%, respectively. Here we describe an efficient approach to the synthesis of 1-indolyl methanamines, starting from different indole substrates and amines under basic conditions.
An initial analysis of time, temperature, and irradiation conditions (Table 1) for the reaction of indole with piperidine, in DMF/DCM (1/4; 5 mL) using 2.0 equiv. of NaH, confirmed the efficacy of operating ultrasound (Baig and Varma, 2012; Cravotto et al., 2013) as reaction catalyst, and 2 h at 50°C as optimal time reaction and temperature conditions (Table 1). However, under these conditions we did not observe any selectivity vs. the formation of 3 (2/3 ratio in the range 1–2, entries 1–8, Table 1 and entry 1, Table 2). When we increased the reaction time to 3 h (entry 5, Table 1) we observed the progressive formation of the 3-((1H-indol-1-yl)methyl)-1-(piperidin-1-ylmethyl)-1H-indole (4), probably as a result of the attack of 1-methylene-1H-indol-1-ium at C-3 position of compound 3 (entry 5, Table 1). An increase of temperature in batch and μW irradiation also favored the formation of 4 (entries 6–8, Table 1).
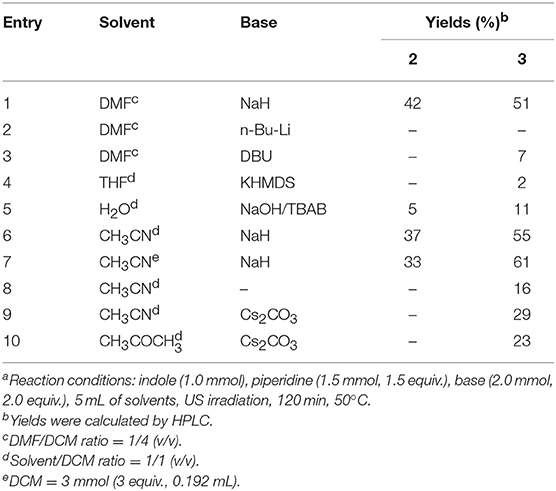
Table 2. Optimization of the conditions for the reaction of 1 with piperidinea.
As shown in Table 2 (entries 2–10), the substitution of NaH by different bases (entries 2–5, 9, and 10) resulted in a strong decrease of 3 yields, while acetonitrile was the solvent of choice (entries 6 and 7). Interestingly, the absence of NaH in CH3CN (entry 8) resulted in the aminomethylation of N-1 in a yield of 16%, while Cs2CO3 did not improve the reaction performance in terms of either CH3CN or acetone (entries 9 and 10).
We also found that the 3-(piperidin-1-ylmethyl)-1H-indole regioisomer was not formed under any of the conditions used, which suggests that 1-methylene-1H-indol-1-ium, but not 1-methylenepiperidine-1-ium, is the active intermediate of this reaction (Mills et al., 1987). In fact, 1-methylindole was recovered unchanged when it replaced indole as starting material. The greater reactivity of N-1 vs. C-3 in indoles under the above reaction conditions has been also observed replacing dichloromethane with chloroform. In fact, the reaction of indole in CH3CN/3 eq. CHCl3 with piperidine, gives both indole-3-aldheyde (5) and 1,1'-(piperidin-1-ylmethylene) bis(1H-indole) (6) in yields of 11 and 19%, respectively (Scheme 1).
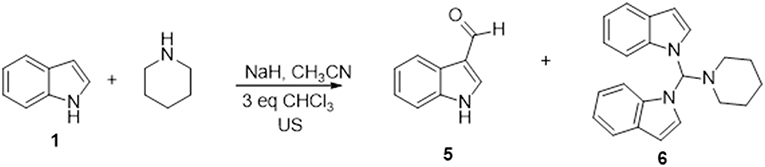
Scheme 1. Reaction of 1 with piperidine in the presence of CHCl3.
The formation of these products can be explained considering the dichlorocarbene generated from chloroform in basic conditions as electrophilic species (Hine et al., 1953; Saunders and Murray, 1960; Kirmse et al., 1990; Wynberg and Meijer, 2005). The addition of the dichlorocarbene to positions 2 and 3 of indole leads to the well-known Reimer-Tiemann (Wynberg and Meijer, 2005) formylated derivative 5, while, according to literature (Hine et al., 1953; Saunders and Murray, 1960; Kirmse et al., 1990), compound 6 could be obtained from a halogenated alkyl adduct, which quickly undergoes β-elimination leading to a reactive chloromethylene indolinium intermediate, as shown in Scheme S1. Addition of a nucleophile and regeneration of the indolium species followed by a second nucleophilic attack leads to the major compound 6.
Besides these results confirm the halogenated solvents as appropriate C1 sources, the low yields obtained using CHCl3 discouraged further investigations. Therefore, we next explored the scope of the reaction using DCM as C1 source under the optimized reaction conditions (entry 7, Table 2), by varying the amine partners, using alkyl, and aryl amines as the second reaction component. Given the incidence of nitrogen heterocycles in chemistry and pharmaceuticals (Vitaku et al., 2014; Blakemore et al., 2018), we used various substituted indoles and other N-heterocycles in combination with piperidine (Chart 2). The reactions of indole with another secondary amine, morpholine, or with primary alkyl and aryl amines such as benzyl and phenylethyl amines resulted in N-((1H-indol-1-yl)methyl) derivatives 7–9 in high yields (68–71%, Chart 2).
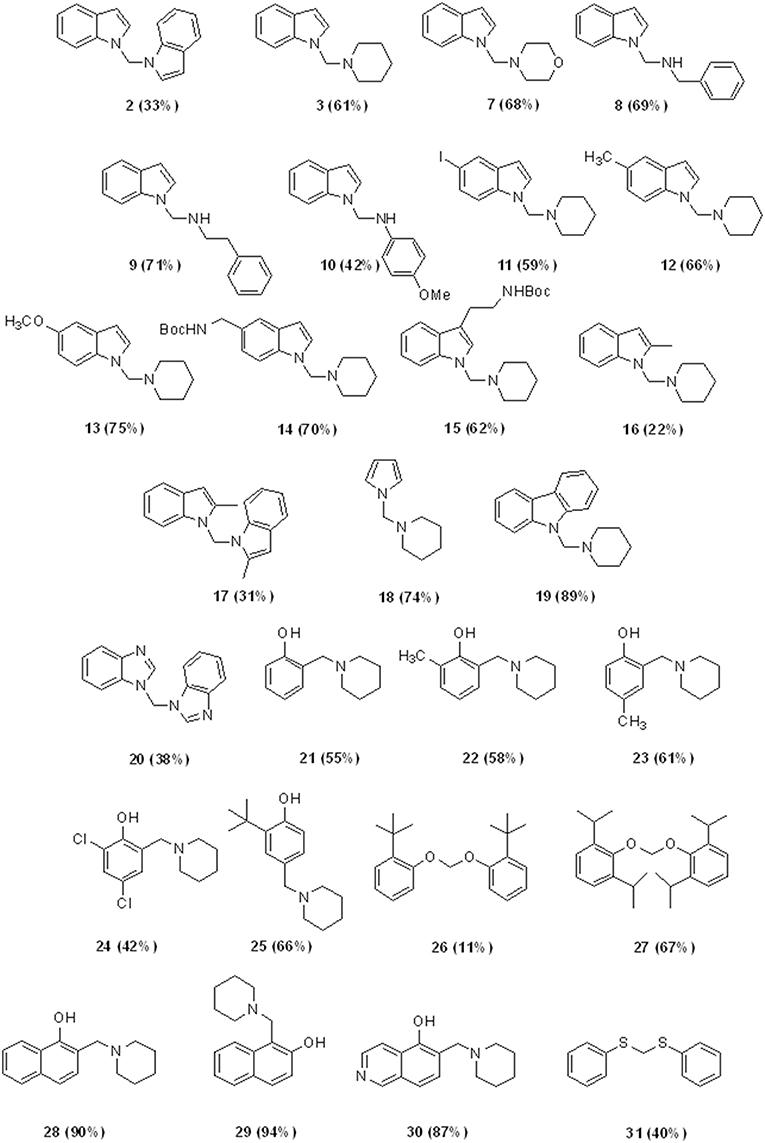
CHART 2. Aminomethylation of Aromatic Compounds with DCM and Aminesa,b. aReaction conditions: 3.0 mmol of CH2Cl2, 1.0 mmol of substrate, 1.5 mmol of amine, 2.0 mmol of NaH, 5 mL of CH3CN, under ultrasound irradiation at room temperature for 120 min. bIsolated yield.
However, the reaction with anilines can only be performed with anilines containing an electron donor group. Therefore, using 4-methoxy aniline, we obtained the amino methylene derivative 10 in a yield of 42%. Biologically relevant 3- or 5- substituted indoles (Bertamino et al., 2016; Musella et al., 2016) reacted with piperidine to provide the N-aminomethyl derivatives 11–15 in a yield range of 59–75% and high selectivity, especially in the case of indoles substituted with electron donor groups (Table 3).
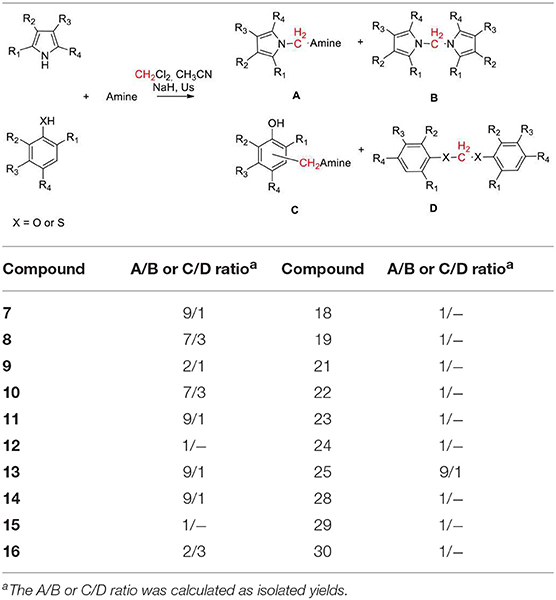
Table 3. Selectivity of the aminomethylation reaction.
Compounds 14 and 15 containing a Boc-protecting group are stable under classical acid deprotection conditions, thus becoming effective intermediates in the synthesis of more complex derivatives. Using 2-methyl indole as starting material, we also obtained the aminomhetylated product (16) in 22% of yield, and its related dimeric compound (17) in 31% of yield. Next, the reaction of piperidine with pyrrole and carbazole generated a high yield of the corresponding aminomethyl derivatives 18 (74%) and 19 (89%), which were also obtained with high selectivity (Table 3). However, benzoimidazole yielded only bis-benzoimidazolylmethane (20, 38%) while pyridine and pyridinol derivatives did not react in our conditions (Mastalir et al., 2017).
Given the chemist community's interest in the chemistry of phenol and its derivatives, in particular for the activation of C-H bonds to generate new C-C bonds (Nair et al., 1994; Joshi et al., 2004; Roman, 2015; Dai et al., 2017; Mastalir et al., 2017), we applied the above described methodology to phenols as well as to other heterocycles namely, 1- and 2-napthol, 5-hydroxyisoquinoline, thiophene, and thiophenol, again using piperidine and DCM as the other two reaction components (Chart 2).
Phenol, 2- and 2,4- electron-donating substituted phenols selectively produced the 2-(piperidin-1-ylmethyl) phenol derivatives 21–23 in a yield range of 55–61%. Reaction with 2,4-dichlorophenol led to 42% of 2,4-dichloro-6-(piperidin-1-ylmethyl)phenol (24, Table 3 for the selectivity ratio). Reaction from bulky substituted phenol orto t-butyl phenol gave the expected 2-(tert-butyl)-6-(piperidin-1-ylmethyl)phenol 25 in a yield of 66% and also bis(2-(tert-butyl)phenoxy)methane 26 in a yield of 11%, while with 2,6-di-isopropylphenol the diphenoxymethane derivative 27 was exclusively obtained (Bauerle and Brodbelt, 1995). 1- or 2-naphtol, and 5-isoquinolinol produced selectively the 2-(piperidin-1-ylmethyl)naphthalen-1-ol (28), 1-(piperidin-1-ylmethyl)naphthalen-2-ol (29), and 8-(piperidin-1-ylmethyl)isoquinolin-5-ol (30), in very high yields (90, 94, and 87%, respectively). As we expected, the reaction with thiophene led to degradation products, while the same conditions applied to thiophenol led to bis(phenylthio)methane 31 in a yield of 40%.
In conclusion, we have developed a practical and sustainable three-component aminomethylation method using different N-heterocycles in combination with a wide range of amines and DCM as C1 source. Thanks to the full N- vs. C- regioselectivity observed in this reaction, this method is an attractive alternative approach to the synthesis of 1-aminomethyl indole derivatives, a class of compounds hitherto poorly accessible. This atom-efficient reaction exploits the potential of ultrasound waves to provide new highly functionalized indoles that are stable both over time and in common synthetic transformations thereby increasing the molecular diversity of this important template. The methodology may also be suitable for other aza-heterocycles, phenols, and some of its derivatives as aryl alcohols, which suggests its potential in the chemistry of materials and medicines, as well as in the life sciences.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
CO and VD synthesized and characterized compounds. SM, TC, and VV contributed to synthesis and data analysis. GP and FM performed HPLC analysis. PC analyzed data and contributed in conceptualization and writing. IG and AB designed the experiments, analyzed and organized data, and wrote the article.
This work was supported by a grant from Regione Campania-PON Campania FESR 2014–2020 Combattere la resistenza tumorale: piattaforma integrate multidisciplinare per un approccio tecnologico innovativo alle oncoterapie-Campania Oncoterapie (Project No. B61G18000470007).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00568/full#supplementary-material
Alonso, I., Esquivias, J., Gomez-Arrayas, R., and Carretero, J. C. (2008). Understanding the behavior of N-tosyl and N-2-pyridylsulfonyl Imines in Cu-II-Catalyzed aza-friedel-crafts reactions. J. Org. Chem. 73, 6401–6404. doi: 10.1021/jo800986g
Arend, M., Westermann, B., and Risch, N. (1998). Modern variants of the Mannich reaction. Angew. Chem. Int. Ed. 37, 1044–1070. doi: 10.1002/(Sici)1521-3773(19980504)37:8<1044::Aid-Anie1044>3.0.Co;2-E
Baig, R. B., and Varma, R. S. (2012). Alternative energy input: mechanochemical, microwave and ultrasound-assisted organic synthesis. Chem. Soc. Rev. 41, 1559–1584. doi: 10.1039/c1cs15204a
Bandini, M., and Eichholzer, A. (2009). Catalytic functionalization of indoles in a new dimension. Angew. Chem. Int. Ed. Engl. 48, 9608–9644. doi: 10.1002/anie.200901843
Barden, T. C. (2010). “Indoles: industrial, agricultural and over-the-counter uses,” in Heterocyclic Scaffolds II: Reactions and Applications of Indoles, ed G. W. Gribble (Berlin: Springer, 31–46
Bartoli, G., Bencivenni, G., and Dalpozzo, R. (2010). Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 39, 4449–4465. doi: 10.1039/b923063g
Bauerle, G. F., and Brodbelt, J. S. (1995). Evaluation of steric and substituent effects in phenols by competitive reactions of dimethyl ether ions in a quadrupole ion-trap. J. Am. Soc. Mass Spectr. 6, 627–633. doi: 10.1016/S1044-0305(05)80051-0
Bertamino, A., Ostacolo, C., Ambrosino, P., Musella, S., Di Sarno, V., Ciaglia, T., et al. (2016). Tryptamine-based derivatives as transient receptor potential melastatin type 8 (TRPM8) channel modulators. J. Med. Chem. 59, 2179–2191. doi: 10.1021/acs.jmedchem.5b01914
Blakemore, D. C., Castro, L., Churcher, I., Rees, D. C., Thomas, A. W., Wilson, D. M., et al. (2018). Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394. doi: 10.1038/s41557-018-0021-z
Cacchi, S., and Fabrizi, G. (2011). Update 1 of: synthesis and functionalization of indoles through palladium-catalyzed reactions. Chem. Rev. 111, PR215–PR283. doi: 10.1021/cr100403z
Cravotto, G., Gaudino, E. C., and Cintas, P. (2013). On the mechanochemical activation by ultrasound. Chem. Soc. Rev. 42, 7521–7534. doi: 10.1039/c2cs35456j
Dai, J. L., Shao, N. Q., Zhang, J., Jia, R. P., and Wang, D. H. (2017). Cu(II)-catalyzed ortho-selective aminomethylation of phenols. J. Am. Chem. Soc. 139, 12390–12393. doi: 10.1021/jacs.7b06785
Dalpozzo, R. (2015). Strategies for the asymmetric functionalization of indoles: an update. Chem. Soc. Rev. 44, 742–778. doi: 10.1039/c4cs00209a
Fujii, S., Konishi, T., Matsumoto, Y., Yamaoka, Y., Takasu, K., and Yamada, K. (2014). Radical aminomethylation of imines. J. Org. Chem. 79, 8128–8133. doi: 10.1021/jo501332j
Hine, J., Pollitzer, E. L., and Wagner, H. (1953). The dehydration of alcohols in the presence of haloforms and alkali1. J. Am. Chem. Soc. 75, 5607–5609. doi: 10.1021/ja01118a039
Hwang, D. R., and Uang, B. J. (2002). A modified Mannich-type reaction catalyzed by VO(acac)(2). Org. Lett. 4, 463–466. doi: 10.1021/ol017229j
Ibrahem, I., Casas, J., and Córdova, A. (2004). Direct catalytic enantioselective alpha-aminomethylation of ketones. Angew. Chem. Int. Ed. 43, 6528–6531. doi: 10.1002/anie.200460678
Jiang, B., and Huang, Z. G. (2005). Synthesis of α-(3-indolyl)glycine derivatives via spontaneous Friedel-Crafts reaction between indoles and glyoxylate imines. Synthesis 2005, 2198–2204. doi: 10.1055/s-2005-869978
Joshi, N. S., Whitaker, L. R., and Francis, M. B. (2004). A three-component Mannich-type reaction for selective tyrosine bioconjugation. J. Am. Chem. Soc. 126, 15942–15943. doi: 10.1021/ja0439017
Kang, Q., Zhao, Z. A., and You, S. L. (2007). Highly enantioselective Friedel-Crafts reaction of indoles with imines by a chiral phosphoric acid. J. Am. Chem. Soc. 129, 1484–1485. doi: 10.1021/ja067417a
Katritzky, A. R., Lue, P., and Chen, Y. X. (1990). An alternative route to 2-substituted indoles via N-aminal-directed lithiation. J. Org. Chem. 55, 3688–3691. doi: 10.1021/jo00298a064
Kim, S., and Hong, S. H. (2017). Ruthenium-catalyzed aminomethylation and methylation of phenol derivatives utilizing methanol as the C-1 source. Adv. Synth. Catal. 359, 798–810. doi: 10.1002/adsc.201601117
Kirmse, W., Kilian, J., and Steenken, S. (1990). Carbenes and the oxygen-hydrogen bond: spectroscopic evidence for protonation of diarylcarbenes to give diarylcarbenium ions. J. Am. Chem. Soc. 112, 6399–6400. doi: 10.1021/ja00173a039
Leitch, J. A., Bhonoah, Y., and Frost, C. G. (2017). Beyond C2 and C3: transition-metal-catalyzed C-H functionalization of indole. ACS Catal. 7, 5618–5627. doi: 10.1021/acscatal.7b01785
Lindquist, C., Ersoy, O., and Somfai, P. (2006). Parallel synthesis of an indole-based library via an iterative Mannich reaction sequence. Tetrahedron 62, 3439–3445. doi: 10.1016/j.tet.2006.01.047
Love, B. E. (2007). Facile synthesis of N-dialkylaminomethyl-substituted heterocycles. J. Org. Chem. 72, 630–632. doi: 10.1021/jo061723p
Love, B. E., and Nguyen, B. T. (1998). A general synthesis of 1-(dialkylaminomethyl)indoles. Synlett 1998, 1123–1125.
Mastalir, M., Pittenauer, E., Allmaier, G., and Kirchner, K. (2017). Manganese-catalyzed aminomethylation of aromatic compounds with methanol as a sustainable C1 building block. J. Am. Chem. Soc. 139, 8812–8815. doi: 10.1021/jacs.7b05253
Matsumoto, K., Uchida, T., Hashimoto, S., Yonezawa, Y., Iida, H., Kakehi, A., et al. (1993). Mannich reactions of indoles with dichloromethane and secondary-amines under high-pressure. Heterocycles 36, 2215–2219. doi: 10.3987/Com-93-6451
Mills, J. E., Maryanoff, C. A., Cosgrove, R. M., Scott, L., and McComsey, D. F. (2009). The reaction of amines with methylene chloride. A brief review. Org. Prep. Proced. Int. 16, 97–114. doi: 10.1080/00304948409356172
Mills, J. E., Maryanoff, C. A., Mccomsey, D. F., Stanzione, R. C., and Scott, L. (1987). The reaction of amines with methylene-chloride - evidence for rapid aminal formation from N-methylenepyrrolidinium chloride and pyrrolidine. J. Org. Chem. 52, 1857–1859. doi: 10.1021/Jo00385a038
Mondal, S., Samanta, S., Singsardar, M., and Hajra, A. (2017). Aminomethylation of imidazoheterocycles with morpholine. Org. Lett. 19, 3751–3754. doi: 10.1021/acs.orglett.7b01594
Murai, N., Miyano, M., Yonaga, M., and Tanaka, K. (2012). One-pot primary aminomethylation of aryl and heteroaryl halides with sodium phthalimidomethyltrifluoroborate. Org. Lett. 14, 2818–2821. doi: 10.1021/ol301037s
Musella, S., di Sarno, V., Ciaglia, T., Sala, M., Spensiero, A., Scala, M. C., et al. (2016). Identification of an indol-based derivative as potent and selective varicella zoster virus (VZV) inhibitor. Eur. J. Med. Chem. 124, 773–781. doi: 10.1016/j.ejmech.2016.09.014
Nagae, H., Shibata, Y., Tsurugi, H., and Mashima, K. (2015). Aminomethylation reaction of ortho-pyridyl C-H bonds catalyzed by group 3 metal triamido complexes. J. Am. Chem. Soc. 137, 640–643. doi: 10.1021/ja511964k
Nair, S. K., Ludwig, P. A., and Christianson, D. W. (1994). 2-Site binding of phenol in the active-site of human carbonic-anhydrase-ii - structural implications for substrate association. J. Am. Chem. Soc. 116, 3659–3660. doi: 10.1021/Ja00087a086
Patil, N. T., and Yamamoto, Y. (2008). Coinage metal-assisted synthesis of heterocycles. Chem. Rev. 108, 3395–3442. doi: 10.1021/cr050041j
Roman, G. (2015). Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 89, 743–816. doi: 10.1016/j.ejmech.2014.10.076
Rowland, G. B., Rowland, E. B., Liang, Y., Perman, J. A., and Antilla, J. C. (2007). The highly enantioselective addition of indoles to N-acyl imines with use of a chiral phosphoric acid catalyst. Org. Lett. 9, 2609–2611. doi: 10.1021/ol0703579
Rudine, A. B., Walter, M. G., and Wamser, C. C. (2010). Reaction of dichloromethane with pyridine derivatives under ambient conditions. J. Org. Chem. 75, 4292–4295. doi: 10.1021/jo100276m
Saaby, S., Fang, X., Gathergood, N., and Jørgensen, K. A. (2000). Formation of optically active aromatic alpha-amino acids by catalytic enantioselective addition of imines to aromatic compounds. Angew. Chem. Int. Ed. 39, 4114–4116. doi: 10.1002/1521-3773(20001117)39:22<4114::Aid-Anie4114>3.0.Co;2-V
Sakai, N., Hirasawa, M., Hamajima, T., and Konakahara, T. (2003). The aminomethylation of electron-rich aromatics with an N-silyl-N,O-acetal catalyzed by a metal triflate-TMSCl system: Facile synthesis of aromatic primary amines, 1-aryl-trichloroethylamines. J. Org. Chem. 68, 483–488. doi: 10.1021/jo026465k
Sakai, N., Okano, H., Shimamura, K., and Konakahara, T. (2014). N-Aminomethylation vs. C-aminomethylation of indole and pyrrole with an N,O-acetal controlled by the hardness of a counter ion of an iminium compound. Chem. Lett. 43, 501–503. doi: 10.1246/cl.131116
Sakai, N., Shimamura, K., Ikeda, R., and Konakahara, T. (2010). Hf(OTf)(4)-catalyzed regioselective N-aminomethylation of indoles and related NH-containing heterocycles. J. Org. Chem. 75, 3923–3926. doi: 10.1021/jo.1006043
Saunders, M., and Murray, R. W. (1960). The reaction of dichlorocarbene with amines. Tetrahedron 11, 1–10. doi: 10.1016/0040-4020(60)89001-1
Souquet, F., Martens, T., and Fleury, M.-B. (2006). Use of methylene chloride as a C1 unit in N,N-dialkylaminomethylation reaction. Synth. Commun. 23, 817–828. doi: 10.1080/00397919308009843
Speckamp, W. N., and Moolenaar, M. J. (2000). New developments in the chemistry of N-acyliminium ions and related intermediates. Tetrahedron 56, 3817–3856. doi: 10.1016/S0040-4020(00)00159-9
Swaminathan, S., and Narasimhan, K. (1966). N-isogramin und einige verwandte N-Mannich-basen des indols. Chem. Ber. 99, 889–894. doi: 10.1002/cber.19660990325
Vitaku, E., Smith, D. T., and Njardarson, J. T. (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274. doi: 10.1021/jm501100b
Wang, Y. Q., Song, J., Hong, R., Li, H., and Deng, L. (2006). Asymmetric Friedel-Crafts reaction of indoles with imines by an organic catalyst. J. Am. Chem. Soc. 128, 8156–8157. doi: 10.1021/ja062700v
Wynberg, H., and Meijer, E. W. (eds.). (2005). “The Reimer-Tiemann reaction,” in Organic Reactions (Hoboken, NJ: John Wiley & Sons), 1–36.
Xie, L. H., Cheng, J., Luo, Z. W., and Lu, G. (2018). Mannich reaction of indole with cyclic imines in water. Tetrahedron Lett. 59, 457–461. doi: 10.1016/j.tetlet.2017.12.069
Xu, J., Chen, X., Wang, M., Zheng, P., Song, B. A., and Chi, Y. R. (2015). Aminomethylation of enals through carbene and acid cooperative catalysis: concise access to beta(2)-amino acids. Angew. Chem. Int. Ed. 54, 5161–5165. doi: 10.1002/anie.201412132
Zhou, G., Wu, D., Snyder, B., Ptak, R. G., Kaur, H., and Gochin, M. (2011). Development of indole compounds as small molecule fusion inhibitors targeting HIV-1 glycoprotein-41. J. Med. Chem. 54, 7220–7231. doi: 10.1021/jm200791z
Keywords: aminomethylation, ultrasound assisted, dichloromethane, indole N-1 selectivity, aza-heterocyclic compounds
Citation: Ostacolo C, Di Sarno V, Musella S, Ciaglia T, Vestuto V, Pepe G, Merciai F, Campiglia P, Gomez Monterrey IM and Bertamino A (2019) An Efficient Approach to Aromatic Aminomethylation Using Dichloromethane as Methylene Source. Front. Chem. 7:568. doi: 10.3389/fchem.2019.00568
Received: 13 June 2019; Accepted: 26 July 2019;
Published: 13 August 2019.
Edited by:
Guigen Li, Texas Tech University, United StatesReviewed by:
Albert Moyano, University of Barcelona, SpainCopyright © 2019 Ostacolo, Di Sarno, Musella, Ciaglia, Vestuto, Pepe, Merciai, Campiglia, Gomez Monterrey and Bertamino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabel M. Gomez Monterrey, aW1nb21lekB1bmluYS5pdA==; Alessia Bertamino, YWJlcnRhbWlub0B1bmlzYS5pdA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.